CHAPTER 102
Cutaneous Vasculitis
Nick J. Levell and Chetan Mukhtyar
Norwich Medical School, Norfolk and Norwich University Hospital, Norwich, UK
Overview
Definition
Cutaneous vasculitis is inflammation of the blood vessel walls, usually resulting in palpable purpura. Nomenclature of the vasculitides, revised at the 2012 Chapel Hill Consensus conference [1], is based upon the size of blood vessel affected. Some conditions have been renamed, and eponyms have been dropped (Box 102.1).

Figure 102.1 Cutaneous small-vessel vasculitis producing palpable purpura. (Courtesy of Andrew Carmichael.)
Introduction and general description
Vasculitis is usually a multisystem disorder that presents in a myriad of ways. Diagnosis is based on a detailed history and careful examination. Patients may present to different specialties and their care should be led by a multidisciplinary team involving physicians with a specialist interest in vasculitis.
Vasculitis can be classified by aetiology or by calibre of the vessel involved. The accepted nomenclature was defined at an international consensus meeting held in 2012 in Chapel Hill, USA. The meeting did not involve dermatologists, and all cutaneous vasculitides could be considered under the umbrella of ’single-organ vasculitis’. It should be recognized that names and classifications will change in the future with greater understanding of the underlying disease mechanisms.
The treatment of primary vasculitis involves immunosuppression. The balance between disease severity and adverse effects of therapy requires expertise and experience in the management of these rare conditions. Secondary vasculitis can be due to infection, drugs, malignancy or inflammatory disease; treatment of the underlying condition may resolve the vasculitis.
Epidemiology
See the specific diseases.
Pathophysiology
The pathophysiology and histopathology varies according to the specific disease.
Clinical features
History
The management of patients presenting with cutaneous vasculitis should begin with a full history. Questions about systemic disease to consider include: (i) complications of vasculitis; (ii) potential malignant and infectious triggers; and (iii) systemic features of systemic vasculitides (Box 102.2). The history should consider diseases that may present with secondary vasculitis including rheumatological diseases (such as systemic lupus erythematosus), thrombo-occlusive disorders and other inflammatory dermatoses.
A full drug history, taken from the patient, the case notes and other relevant clinicians, should focus on medication changes in the days, weeks and months prior to the onset of vasculitis. Occasionally, drugs taken for many years may precipitate reactions. Drugs purchased from pharmacies or borrowed from relatives, herbal treatments, tonics and vitamins should also be considered. Patients may be unwilling to reveal recreational drugs, drugs causing addiction or drugs taken for bodybuilding or sexual purposes. The reason for any drug change should be established.
Vasculitis may be secondary to infection. A history should be taken of infections, both acute and chronic, and their treatments.
Presentation
On general examination, establish if the patient is acutely unwell; patients with systemic vasculitis may have life-threatening internal organ involvement requiring prompt management. All the skin should be examined. Cutaneous vasculitis often results in painful, palpable purpura (Figure 102.1). Leakage of blood from the vasculature into the interstitium causes purpura, which is identified by a failure to blanch on diascopy (pressure with glass). Increased pressure in the venous circulation increases blood vessel leakage and may worsen damage to the vessel walls. Purpura is therefore most apparent on the lower limbs. Prolonged standing exaggerates venous hypertension and thus increases blood leakage and purpura.
The physical signs are determined to some extent by the size of vessel involved (Table 102.1). Severe cutaneous vasculitis will result in painful ischaemia of the skin. Lesional skin will become haemorrhagic (Figure 102.2) and then necrotic and will eventually detach, leaving erosions or ulcers (Figure 102.3), most commonly on the lower limbs. These ulcers may then become secondarily infected. The ulcers may be slow to heal, even after resolution of the vasculitis, due to venous stasis, malnutrition, anaemia, lymphoedema, prolonged infection or old age.

Figure 102.2 Cutaneous small-vessel vasculitis demonstrating a haemorrhagic vesicle. (Courtesy of Andrew Carmichael.)
Table 102.1 Physical signs may give clues as to the predominant vessel size involved in the vasculitis.
| Blood vessel size | Physical signs |
| Small blood vessels | Purpuric macules and papules, haemorrhagic vesicles, urticarial plaques Necrosis not usually a major feature |
| Medium-sized blood vessels | Broken livedo (net/reticulate) pattern, infarction, ulceration, deep nodules |
The extent of systemic examination will depend on the history and on the overall assessment of the patient. Systemic examination may reveal an underlying infection or malignancy acting as a trigger for the vasculitis. Signs of a systemic primary vasculitis may be found (Box 102.3). Other underlying diseases that may cause secondary vasculitis such as rheumatoid arthritis or lupus erythematosus may be apparent.
Clinical variants
The size of purpuric lesions varies according to the disease. Areas of purpura less than 5 mm in diameter are called petechiae, those larger than 1 cm are ecchymoses. A reticulate (like a net) livedo pattern is seen in some vasculitides and thrombo-occlusive disorders (Table 102.2). A broken livedo (incomplete net) (Figure 102.3) is said to be a feature of vasculitis disorders but may also be seen in thrombo-occlusive disease.

Figure 102.3 Ulcerated necrotic lesions in a livedo distribution suggestive of medium-vessel disease. (Courtesy of Andrew Carmichael.)
Table 102.2 The purpura pattern may give clues as to the disease.
| Pattern of purpura | Diseases to consider |
| Pinpoint, cayenne pepper macular purpura, typically <5 mm | Capillaritis, exercise-induced purpura (’runners’ legs’), coughing, ligatures Purpura in contact dermatitis (e.g. rubber), venous hypertension, suction induced, fixed drug eruptions, cutaneous T-cell lymphoma |
| Macular purpura of any size | Purpura due to infections, platelet disorders and thrombocytopenia, other clotting disorders, mild small vessel vasculitis Trauma/ artefact |
| Large macular purpura, typically over 2 cm | Purpura of old age, topical, inhaled or systemic corticosteroid-induced purpura, scurvy |
| Painful palpable purpura of any size | Cutaneous vasculitis of all types, pityriasis lichenoides, thrombo-occlusive disorders of all types, secondary purpura in tuberculosis or leprosy reactions, neutrophilic disorders, atypical benign or malignant cutaneous growths (e.g. haemangiosarcoma, Kaposi sarcoma, amelanontic melanoma with haemorrhage) |
| Livedo pattern purpura | Antiphospholipid syndromes, vasculitis in medium-sized blood vessels (e.g. polyarteritis, ANCA vasculitides), thrombo-occlusive disorders of all types including cryoglobulins, chilblains |
ANCA, antineutrophil cytoplasmic antibody.
Differential diagnosis
Thrombo-occlusive disorders, trauma, inflammatory dermatoses with disordered clotting, purpura due to prolonged running, neutrophilic disorders, cellulitis (particularly in the elderly with oedematous legs), insect and snake bites are often confused with vasculitis.
Disease course and prognosis
See above and specific diseases.
Investigations
The investigation of vasculitis is dependent on the history and examination findings. A thorough assessment may clarify the likely cause and limit the need for extensive investigations. However, investigations are necessary for two main purposes. First, it is important to establish if the vasculitis is primary or secondary. Investigations should be directed to identify underlying rheumatological disease, malignancy, infection or a primary vasculitis. Second, investigations should be carried to demonstrate the presence of vasculitis involving internal organs. Urinalysis to exclude renal disease is useful in most patients.
Skin biopsy in vasculitis, if needed, should be taken from a fresh lesion less than 48 h old. Older vasculitic lesions may develop secondary thrombosis making difficult differentiation from a thrombo-occlusive disorder. Older lesions of thrombo-occlusive disorders may develop secondary vasculitis. A skin biopsy for direct immunofluoresence should be taken if IgA vasculitis is suspected. A ’lupus band’ of IgG and complement at the dermal–epidermal junction is of little value as a diagnostic test and is no longer recommended. A vasculitis screen may be used by inexperienced clinicians as a substitute for taking a history and examination and then applying logic. A list of tests is given in Table 102.3 but these should be used in support of clinical findings and intelligent thought.
Table 102.3 Vasculitis investigations. The ’vasculitis screen’ is dependent on the history and examination findings. In acute vasculitis with an obvious infection or drug trigger, investigations may be minimal. The purposes of investigation are threefold: to look for (i) complications of vasculitis; (ii) causes of vasculitis; and (iii) differential diagnoses of vasculitis, such as thrombo-occlusive disorders.
| Investigation | Notes |
| Blood and urine tests | |
| Urinalysis | Haematuria and proteinuria in renal involvement |
| Urea and electrolytes | Raised creatinine and urea in renal involvement |
| Full blood count | Raised white cells in infection/cryoglobulinaemia Thrombocytopenia may cause purpura |
| Liver function | Low albumin in renal disease |
| Erythrocyte sedimentation rate | May be raised in systemic vasculitis, infection and malignancy |
| C-reactive protein | May be raised in infections |
| Antineutrophil cytoplasmic antibody (ANCA) | May be present in systemic vasculitides – see text |
| Antinuclear antibodies | May be present in autoimmune connective tissue disease |
| Specialist haematological tests for thromboembolic disease such as lupus anticoagulant and anticardiolipin antibodies | If thrombo-occlusive disease is possible from the history, examination or histology |
| Cryoglobulins | If there is skin, kidney and joint vasculitis. Not necessarily triggered by cold |
| Tissue tests | |
| Skin biopsy from early lesion (less than 48 h old) for histopathology | Indicate if vasculitis or thromboembolic disorder Indicate size of blood vessel involvement and predominant cell type Indicate presence of granulomas Indicate certain infections, e.g. mycobacteria |
| Skin biopsy from early lesion for direct immunofluorescence | Indicate if IgA vasculitis |
| Skin biopsy for culture | May be useful for chronic infections, e.g. TB |
| Infection | |
| Infection screen: cultures, serology and radiology | Depends on age, history of travel and country of residence, history and examination. Screen for acute and/or chronic infections |
| Malignancy | |
| Malignancy screen: blood tests and radiology for malignancy | Relevant tests depend on the age of patient, history and examination |
| Inflammatory disease | |
| Investigations for other systemic inflammatory disease | If diseases (e.g. inflammatory bowel disease, rheumatoid arthritis) are suspected from history and examination |
Management
The management of vasculitis is dependent on the diagnosis and the severity and presence of systemic vasculitis. Triggering drugs should be stopped. Underlying infections should be treated. Malignancy or associated rheumatological diseases should be managed. If systemic vasculitis or vasculitic disease is identified then the treatment is described under the specific diseases in this book. The correction of venous stasis by elevation of the legs, treatment of secondary infection, appropriate dressings in ulcerated areas and pain relief are required.
In systemic vasculitis a multidisciplinary team approach is appropriate. Specialists may be required to deal with disease in almost any organ. In the UK, rheumatologists with an interest in vasculitis often lead or coordinate teams of other specialists to manage complex patients. Early referral is desirable to avoid potentially treatable disease in other organs causing irreversible damage.
SINGLE-ORGAN SMALL-VESSEL VASCULITIS
Cutaneous small-vessel vasculitis
Definition and nomenclature
Cutaneous small-vessel vasculitis (CSVV) is a single-organ vasculitis producing leucocytoclastic angiitis of cutaneous vasculature [1, 2].
Introduction and general description
The American College of Rheumatology (ACR) have produced classification criteria for CSVV. The presence of three of the following five criteria have 84% specificity for CSVV: (i) age greater than 16 years at disease onset; (ii) history of taking a medication at onset that may have been a precipitating factor; (iii) the presence of palpable purpura; (iv) the presence of a maculopapular rash; and (v) a biopsy demonstrating granulocytes around an arteriole or venule [3].
CSVV is a single-organ vasculitis and therefore by definition does not have systemic manifestations. However, the diagnosis should prompt ongoing surveillance because it may be a first manifestation of a more generalized vasculitis. CSVV is a clinical syndrome that encompasses vasculitis due to a variety of causes.
Epidemiology
Incidence and prevalence
The annual incidence of CSVV is reported to be between 15 and 30 per million [4, 5].
Age
The mean age at onset is between 36 and 56 years [4, 6, 7]. However, the age range is wide and extends from the second to the eighth decade of life.
Sex
In a Spanish cohort, men were more commonly affected with a ratio of 1.6 : 1 [4], but in a Singapore cohort, there was a female dominance of 2.1 : 1 [6].
Associated diseases
By definition the condition is localized to the skin, but it may be a precursor for other systemic vasculitides.
Pathophysiology
Predisposing factors
There are many causes of cutaneous vasculitis, but most CSVV is idiopathic [6]. This is a consequence of vasculitis classification systems. For example, if patients with CSVV are found to have viral hepatitis and cryoglobulinemia, they are no longer classifiable as CSVV. There are factors that are thought to contribute to a pure CSVV, which are listed in Box 102.4.
Pathology
Leukocytoclastic vasculitis with segmental inflammation in an angiocentric pattern, swelling of the endothelium, fibrinoid necrosis of vessel walls, extravasation of erythrocytes, and an infiltrate of neutrophils with karyorrhexis of the nuclei (i.e. leukocytoclasia) are major features of CSVV (Figures 102.4 and 102.5). In superficial dermal papillary vessels, IgM or complement C3 perivascular deposits are demonstrated in up to 80% of fresh lesions [8]. Some studies state lower proportions, but this may depend on the timing of the biopsy and also because IgM is relatively poor at fixing complement. IgG is found less often.

Figure 102.4 Leukocytoclastic vasculitis. (a) Low-magnification photomicrograph showing perivascular infiltrates and fibrinoid deposits within the vessels of the upper dermis. (b) Higher magnification demonstrating nuclear dust, fibrinoid deposits, vascular alteration and collagen degeneration. (Courtesy of Dr Omar Sangueza, Wake Forest University School of Medicine, Winston-Salem, NC, USA.)

Figure 102.5 Leukocytoclastic (small vessel) vasculitis at (a) lower and (b) higher magnifications. There is visible vascular wall damage with evidence of red blood cell extravasation, fibrinoid change and extensive leukocytoclasis (nuclear debris from polymorphonuclear leukocytes). (Courtesy of Dr Laszlo Igali, Norfolk and Norwich University Hospital, Norwich, UK.)
Causative organisms
In most patients, CSVV is idiopathic, but a small number of cases (4/138 in [9]) may be related to a bacterial infection.
Genetics
The genetics are not known.
Clinical features
History
The skin lesions of CSVV typically arise as a simultaneous ’crop’, resulting from exposure to an inciting stimulus. New lesion formation can continue for several weeks. They usually resolve within several weeks or a few months although approximately 10% of patients will have recurrent disease. There are no known risk factors to predict relapses.
Presentation
Lesions typically occur in areas prone to stasis, commonly including the ankles and lower legs (Figures 102.1 and 102.6a), and typically sparing intertriginous regions. CSVV is often asymptomatic, although pruritus, pain or burning may be experienced, as well as systemic symptoms including fever, arthralgia, myalgia and anorexia. The major cutaneous manifestation of CSVV is palpable purpura, ranging in size from 1 mm to several centimetres (Figure 102.6b, c). Sometimes macular in the early stages, such purpura may progress to a wide array of lesions including papules, nodules, vesicles, plaques, bullae or pustules, with secondary findings of ulceration, necrosis and post-inflammatory hyperpigmentation (Figure 102.7).

Figure 102.6 Cutaneous small-vessel vasculitis (CSVV). (a) Vesicles in a dependent area on the foot. (b) Purpura on the thighs; there was similar involvement on the lower legs. (c) CSVV progressing to blistering.

Figure 102.7 Vasculitis due to sepsis, in a patient with impaired level of consciousness. The necrotic lesion in (a) and the reticulate pattern on the leg in (b) are clues to the involvement of deeper vessels. Histologically, vasculitis due to infection may involve vessels at all levels of the dermis.
Clinical variants
Other cutaneous findings include oedema, livedo reticularis and urticaria. The presence of the latter two should prompt consideration of cutaneous polyarteritis nodosa and urticarial vasculitis, respectively.
Differential diagnosis
The differential diagnosis of CSVV includes many more specifically defined disorders, which are discussed in this chapter and listed in references [2, 3, 9–11, 12, 13]. CSVV is a diagnosis of exclusion. Cutaneous vasculitis should prompt a search for a wide array of differential diagnoses, including systemic vasculitides, cancer, infections, allergies, chemical exposures, etc.
Classification of severity
There is no validated biomarker for quantifying disease severity in patients with CSVV. Histopathology is not a good surrogate for severity of disease [10]. The Birmingham Vasculitis Activity Score (BVAS) v3 has been validated to quantify the activity of systemic vasculitis [11]. The validation cohort of BVAS v3 included patients with cutaneous vasculitis and it can be used to create a tangible activity score.
Complications and co-morbidities
Hyperpigmentation and haemosiderosis can take months to resolve. Ulcerated lesions may become infected adding to the morbidity.
Investigations
Investigation of CSVV is guided by the history and examination findings; the range of tests chosen will range from nothing to extensive blood testing, scanning and organ biopsies. The purpose of investigation is twofold: firstly to look for evidence of vasculitis in other organ systems, and secondly to look for evidence of a disease that is predisposing towards CSVV, such as infection or malignancy.
Management
Treatment of CSVV is often unnecessary, as the disease may be self-limiting. The evidence for efficacy of therapy is derived from clinical experience rather than controlled trials. If a triggering agent is identified, such as a drug or infection, it should be removed or treated. Efforts to minimize stasis, such as use of compression hosiery and the elevation of dependent areas, as well as the use of non-steroidal anti-inflammatory drugs (NSAIDs) and antihistamines, may reduce symptoms [10], although not altering the course of the disease.
First line
Oral prednisolone 30–80 mg once daily, tapered over 2–3 weeks, often gives symptom control, although no controlled trials have been carried out to evaluate the treatment of CSVV with oral corticosteroids. Corticosteroid use may be of particular benefit in cases with painful progressive cutaneous lesions. No data support the use of topical corticosteroids or antibiotics in CSVV, although such therapies are commonly used.
Second line
Colchicine 0.6 mg twice daily has been shown to be of benefit by anecdotal evidence and open-label studies [15–17]. Dapsone 50-150 mg daily may be advantageous in the treatment of CSVV [18–21].
Third line
In patients with disease refractory to the above therapies, cytotoxic agents may be considered. Such agents include azathioprine (1–2 mg/kg/day) and methotrexate (15–25 mg/week). The use of ciclosporin (2.5–4 mg/kg/day) and cyclophosphamide is almost never indicated for purely cutaneous disease.
Erythema elevatum diutinum
Definition and nomenclature
Erythema elevatum diutinum (EED) is a rare, chronic, cutaneous eruption. The first descriptions were by Hutchinson and Bury in the 1880s, and the condition was later named in 1894 by Radcliffe-Crocker and Williams. EED is characterized by fibrosing plaques with histological evidence of leukocytoclastic vasculitis.
Epidemiology
Incidence and prevalence
Erythema elevatum diutinum is very rare with only a few hundred cases described. There are small case series published but most reports are of single cases only. Few dermatologists have looked after more than a handful of cases.
Age
Erythema elevatum diutinum is most commonly seen in adults in the fourth to seventh decade although occasional childhood cases are reported [1].
Sex
It occurs equally in males and females.
Ethnicity
There has been no description of predilection in any ethnic groups.
Associated diseases
Erythema elevatum diutinum has been associated with autoimmune diseases such as rheumatoid arthritis, coeliac disease, inflammatory bowel disease and type 1 diabetes. Associations with infections, including Streptococcus, hepatitis and syphilis, have also been suggested [2–5, 6, 7, 8, 9, 10, 11]. Lesions characteristic of EED have been induced by injection of streptococcal antigen into the dermis [12–15], and have occurred at sites of mosquito bites [10]. EED has been associated with human immunodeficiency virus (HIV) infection. As lesions of EED have responded to antiretroviral and dapsone treatment in HIV-positive patients, it is now recognized as one of the defined reactive dermatoses associated with HIV [16]. EED has been associated with hypergammaglobulinaemia and IgA monoclonal gammopathies, as well as with myelodysplasia, pyoderma gangrenosum and relapsing polychondritis. The association with haematological abnormalities, such as multiple myeloma, is strong; however, EED may precede the haematological disease by several years [17].
Pathophysiology
Although the exact aetiology is unknown, EED is thought to be related to an Arthus-type reaction with immune complex deposition and subsequent inflammation.
Predisposing factors
See the associated diseases.
Pathology
Acute lesions of EED are characterized by leukocytoclastic vasculitis, with little fibrin deposition (Figure 102.8). Eosinophils may also be present in the upper and mid dermis. Depending on the degree of oedema and infiltration into the dermis, unaffected collagen may be present just under the epidermis. Chronic lesions demonstrate angiocentric eosinophilic fibrosis, capillary proliferation and infiltration of macrophages, plasma cells and lymphocytes. Cholesterol deposits in histiocytes and in the extracellular tissue (the latter in a pattern that has been termed ’extracellular cholesterolosis’) may be present in older lesions [18]. Dermal nodules of EED contain spindle cells and fibrosis [19].

Figure 102.8 Erythema elevatum diutinum at (a) lower and (b) higher magnifications. There is a perivascular infiltrate containing neutrophils, with leukocytoclasis and some perivascular fibrin deposition. (Courtesy of Dr Laszlo Igali, Norfolk and Norwich University Hospital, Norwich, UK.)
Clinical features
History
Although they are generally asymptomatic, the lesions of EED may be painful.
Presentation
Lesions of EED most commonly appear chronically in a symmetrical fashion over the dorsa of the hands, knees, buttocks and Achilles tendons (Figure 102.9). They are red-violaceous, red-brown or yellowish papules, plaques or nodules. Occasionally, the face and ears are also affected by EED. Initially, the lesions are soft, but eventually they fibrose and later leave atrophic scars.

Figure 102.9 Erythema elevatum diutinum. (a) On the hands. (b) Early non-fibrotic lesions at a typical site on the knee. This patient also had EED on the hands, and pyoderma gangrenosum.
Differential diagnosis
Erythema elevatum diutinum may be difficult to distinguish from CSVV on histology but the clinical presentation enables accurate diagnosis. EED and Sweet syndrome are both described as neutrophilic dermatoses. However, EED differs from Sweet syndrome by the character of the lesions and their distribution, as well as by histopathological features. The classic assumption that lesions from patients with Sweet syndrome lack histopathological fibrinoid necrosis of the vessel walls has been challenged. In one series, 29% of patients had biopsy specimens showing leukocytoclastic vasculitis [20], although this may have been secondary changes in older lesions. Clinically, the lesions in Sweet syndrome are acute, more often asymmetrical and located on the arms, face and neck [21]. By contrast, EED lesions are chronic, symmetrical and classically located over the dorsum of the hands and knees, buttocks and Achilles tendons. Although leukocytoclastic vasculitis has now been reported as a possible feature of Sweet lesions [2], it is not always present and the fibrosis seen in lesions of EED correlates with the clinical chronicity. Granuloma faciale has been considered in the same group of disorders as EED, but may have features of the IgG4-related sclerosing diseases, such as storiform fibrosis, which is histologically absent in EED [22].
Complications and co-morbidities
Although the lesions can be painful and heal with scarring, complications are rare.
Disease course and prognosis
Erythema elevatum diutinum may last from 5 to 35 years, with crops of new lesions developing every few weeks to months.
Investigations
The demonstration of IgA antineutrophil cytoplasmic antibodies (ANCA) (with various specificities) in six of 10 cases of EED has been suggested to be of some diagnostic value [23]; in this series, seven of the 10 patients had raised IgA levels (monoclonal in three).
Management
Treatment of an associated disorder such as HIV infection or paraproteinaemia may be effective [24].
First line
Dapsone is usually effective in EED [15], although relapse on stopping may occur.
Second line
Niacinamide has also been used with good effect [25]. High potency topical, or intralesional, corticosteroids may minimize the size of lesions in patients with limited disease; 5% topical dapsone gel has been described as effective [26].
Third line
Other therapies used for CSVV may also be effective in treating patients with EED.
Recurrent cutaneous necrotizing eosinophilic vasculitis
Definition and nomenclature
This is a relatively recently described and rare vasculitis consisting of a predominantly centripetal purpuric papular rash, angio-oedema, peripheral blood eosinophilia and an eosinophilic necrotizing vasculitis of small vessels [1, 2]. It can be argued that this is a pathological subdivision of CSVV rather than a distinct entity.
Epidemiology
Incidence and prevalence
This is a very rarely described disease with only a few cases in the literature.
Age
The condition has been described in adults aged 17–81 years.
Sex
Either sex may be affected.
Ethnicity
There is no known association.
Associated diseases
Association with connective tissue diseases and with rheumatoid arthritis has been reported [3, 4].
Pathophysiology
The cause is unknown. As in other strongly eosinophilic disorders, eosinophil cytokines such as interleukin 5 (IL-5), and toxic eosinophil granule proteins such as the major basic protein, have been demonstrated in serum and tissues, respectively, and presumably play a part in the tissue damage. Neutrophil elastase is prominent around vessels, and mast cell degranulation occurs. Eosinophilic vasculitis has also been reported in a patient with the hypereosinophilic syndrome; in this patient, CD40 (a glycoprotein of the tumour necrosis factor (TNF) receptor family) was considered to be important in pathogenesis [5].
Predisposing factors
There are no known predisposing factors.
Pathology
Histopathology shows fibrinoid deposition and necrosis of small dermal vessels with an infiltrate of eosinophils and absent or minimal leukocytoclasis. Small epidermal vesicles containing eosinophils may be present. Immunoglobulin deposition is not a feature. This eosinophilic small vessel vasculitis may be distinct from other vasculitides such as eosinophilic granulomatosis with polyangiitis (previously known as Churg–Strauss syndrome), in which predominantly medium vessels are affected; and from most drug-induced vasculitis in which eosinophils are generally less prominent.
Causative organisms
There is no known association with causative organisms.
Genetics
The genetics of the condition are unknown.
Environmental factors
There are no reported environmental triggers.
Clinical features
History
Patients may initially present with pruritic papules over the lower limbs. The course is long and recurrent, but fever, arthralgia and visceral involvement are absent.
Presentation
Recurrent pruritic papules and urticarial lesions occur at any site, especially the lower limbs, head and neck, with angio-oedema of the face and extremities. Digital occlusions manifesting as the Raynaud phenomenon or digital gangrene have been reported in patients with cutaneous eosinophilic vasculitis associated with the hypereosinophilic syndrome [5, 6], but they can also occur in the hypereosinophilic syndrome in the absence of cutaneous eosinophilic vasculitis [7, 8].
Clinical variants
An eosinophilic vasculitis, typically with hypocomplementaemia, also occurs in connective tissue diseases [9].
Differential diagnosis
This condition was recently distinguished from other eosinophilic vasculitides that affect medium-sized vessels (eosinophilic granulomatosis with polyangiitis; see separate section this chapter) and from eosinophilic disorders in which pruritic papules and/or angio-oedema may occur, such as hypereosinophilic syndrome, episodic angio-oedema with eosinophilia, dermatitis herpetiformis, Wells syndrome, polymorphic eruption of pregnancy or drug eruptions.
Complications and co-morbidities
Ulceration and secondary infection of necrotic lesions may occur. By contrast with eosinophilic granulomatosis with polyangiitis, systemic features are not reported.
Disease course and prognosis
A good response to corticosteroids is reported.
Investigations
Investigations are guided by history and clinical examination and will be needed to exclude the differential diagnoses, listed above.
Management
First line
The few cases described have been treated with oral corticosteroids with good effect, intermittently or as prolonged maintenance therapy depending on response [10].
Second line
Secondary infection of ulcerated lesions may require topical or systemic antibiotics according to sensitivities.
Granuloma faciale
Definition and nomenclature
Granuloma faciale is an uncommon condition typified by asymptomatic cutaneous nodules occurring primarily on the face, with occasional extrafacial involvement. Granuloma faciale is limited to the skin, without any systemic manifestations.
Introduction and general description
Granuloma faciale is an uncommon condition of unknown aetiology that is characterized by the presence of benign, purely CSVV. In 1945, Wigley described a 46-year-old woman with recurrent, multiple, raised, discrete, smooth, greyish brown, facial lesions. The histology demonstrated pleomorphic infiltrate with predominant eosinophils, but also polymorphs and plasma cells. In the absence of any bony involvement, this was diagnosed as an eosinophilic granuloma [1]. The term ’granuloma faciale’ and ’facial granuloma with eosinophilia’ was first used by Boersma in 1951 [2].
Epidemiology
Incidence and prevalence
Granuloma faciale is a rare condition.
Age
It is seen most commonly in 40–60 year olds [3, 4].
Sex
Granuloma faciale is commoner in males [3].
Ethnicity
Granuloma faciale has been reported from various parts of the world and does not seem to have any ethnic predilection.
Associated diseases
As noted by Wigley [1], the dermal infiltrate consists of eosinophils and plasma cells. Cesinaro et al. reported a storiform fibrotic pattern and the presence of large amounts of IgG4-staining deposits [5]. This raises the possibility that some patients with granuloma faciale may have IgG4-related disease.
Pathophysiology
Although the aetiology is unclear, this disease is considered to be a histological variant of leukocytoclastic vasculitis with a prominent eosinophilic infiltrate and confined to the skin [4]. The presence of plasma cells and IgG deposition in and around the dermal vasculature has been demonstrated, indicating that granuloma faciale may be immune complex mediated [5]. The reporting of T cells in the tissue is variable. Smoller and Bortz reported large numbers of CD4+ cells that stain strongly for IL-2R antibodies [6], and Cesinaro et al. found T-cell subsets to be variable but with a predominance of GATA-3 lymphocytes [5].
Pathology
Granuloma faciale is a misnomer. The one pathological finding that is almost always absent is a granuloma [7]. It is characterized by a mixed inflammatory infiltrate with a predominance of eosinophils and plasma cells as part of a pleomorphic infiltrate, mainly in the upper half of the dermis but with occasional spread into the lower dermis and subcutaneous tissue (Figure 102.10). A band of normal collagen referred to as a ’Grenz’ zone typically separates the inflammatory infiltrate from the epidermis and pilosebaceous appendages. Nuclear dust (fragmented neutrophil nuclei) may be observed near capillaries. The vascular changes may be mild (perivascular distribution of inflammatory cells) to florid (leucocytoclastic vasculitis with fibrinoid necrosis). Perivascular storiform fibrosis and obliterative venulitis have been observed.

Figure 102.10 Granuloma faciale. (a) Low-magnification view showing perivascular nodular infiltrates within the dermis. (b) At a higher magnification the infiltrate shows lymphocytes, eosinophils and a few neutrophils. (Courtesy of Dr Omar Sangueza, Wake Forest University School of Medicine, Winston-Salem, NC, USA.)
Clinical features
History
Lesions of granuloma faciale commonly occur on the face (Figure 102.11); multiple lesions are present in about a third of cases but extrafacial involvement is uncommon, occurring in five of 66 patients in one study [4]. They are almost always asymptomatic, although some patients may describe itching, burning or pain associated with the lesions.

Figure 102.11 Granuloma faciale. (a) Reddish brown plaque on the nose (courtesy of Dr G. Dawn, Monklands Hospital, UK). (b) Close up of a facial plaque.
Presentation
The nodules or plaques are soft and red-brown . They are smooth, with prominent follicular orifices and telangiectatic surface changes or scaling. The lesions never ulcerate. Dermoscopy shows parallel, arborizing blood vessels, brown dots and globules and dilated follicular openings [8].
Clinical variants
Extrafacial granuloma faciale is rare, but it has been reported on the scalp, back, shoulders, arms, breast and trunk [9–12]. In a study of 66 patients, only five patients had extrafacial lesions [4]; all these lesions coexisted with facial lesions. Intranasal lesions have been reported.
Eosinophilic angiocentric fibrosis is thought to be a mucosal variant of granuloma faciale that may occur in the nasal passages or upper airways in conjunction with skin lesions of granuloma faciale [13, 14]. Eosinophilic angiocentric fibrosis may cause fibrotic stenosis of the affected site with localized extension and damage [15]. For example, epiphora and proptosis have been reported in patients with obstructive sinonasal eosinophilic angiocentric fibrosis [16].
Differential diagnosis
Granulomatous rosacea does not have vasculitis on histology. Sarcoid, TB, cutaneous lupus erythematosus and rarely EED may present with solitary cutaneous lesions.
Disease course and prognosis
Granuloma faciale is a chronic disease with intermittent acute flares that is notoriously resistant to treatment.
Investigations
A definitive diagnosis of granuloma faciale requires clinically consistent lesions and a confirmatory biopsy. Although most laboratory studies are normal, mild peripheral blood eosinophilia may be present [10].
Management
Management of granuloma faciale may be challenging. Topical tacrolimus may be effective but the condition is difficult to treat.
SMALL-VESSEL IMMUNE COMPLEX-ASSOCIATED VASCULITIS
IgA vasculitis
Definition and nomenclature
IgA vasculitis, previously called Henoch–Schönlein purpura (HSP), is an immune complex vasculitis characterized by IgA1-dominant immune deposits affecting small vessels (predominantly capillaries, venules or arterioles). It often involves the skin and gastrointestinal tract, and frequently causes arthritis. Glomerulonephritis indistinguishable from IgA nephropathy may occur [1].
Introduction and general description
William Heberden, in the 1780s, described two children with petechiae, purpura and ecchymosis in conjunction with arthritis. One of the two boys also had abdominal pain, melena and haematuria [2]. In the 19th century, Johann Schönlein and Eduard Henoch independently characterized the condition, which bore their name until the renaming of eponymous vasculitides in 2012 [1].
For the purposes of homogeneity and classification, there are two sets of classification criteria in use. The ACR proposed classification criteria in 1990: if any two of the following four criteria were satisfied, the case could be classified as IgA vasculitis: (i) palpable purpura; (ii) bowel angina; (iii) age <20 years at onset; and (iv) the presence of granulocytes in the vessel wall on biopsy [3]. These criteria were modified in a combined effort by the European League Against Rheumatism and the Paediatric Rheumatology Society for classifying childhood-onset vasculitis. The presence of any one of the following four features in the presence of palpable purpura satisfies a classification of IgA vasculitis: (i) diffuse abdominal pain (ii) any biopsy demonstrating predominant IgA deposition; (iii) any acute arthritis or arthralgia; and (iv) renal involvement in the form of haematuria or proteinuria. [4].
Epidemiology
Incidence and prevalence
The annual incidence of IgA vasculitis is 10–20/100 000 in children [5–7, 8], and about 1–1.5/100 000 in adults [9, 10]. The incidence of nephritis in conjunction with IgA vasculitis is lower in children at about 3.5/100 000 [11]. A peak incidence of 70.3/100 000 children between 4 and 6 years of age has been observed [8].
Age
The peak incidence is between the ages of 4 and 6 years [8]. Children developing nephritis are typically slightly older. In one study, children >8 years of age had an odds ratio of 2.7 for developing nephritis [12]. Adult-onset IgA vasculitis can occur at any age.
Sex
There may be a mild male preponderance with reported ratios of 1.8 : 1 [13].
Ethnicity
There is a higher incidence of IgA vasculitis reported from Scotland (20.3-26.7/100 000) [5] than in Taiwan (12.9/100 000) [6] or the Czech Republic (10.2/100 000) [7]. IgA-related nephritis has been more commonly reported in American Indians as compared to Hispanics [14].
Associated diseases
Pathologically, IgA vasculitis in the kidney is indistinguishable from IgA nephropathy. Patients with IgA vasculitis are typically younger and have more extrarenal manifestations [15].
There may be an association of IgA vasculitis with familial Mediterranean fever (FMF). MEFV (familial Mediterranean fever) gene mutations have been observed in IgA vasculitis more commonly than the general population [16, 17]. The clinical syndrome of IgA vasculitis has been observed more commonly in patients with FMF than in the general population, and some consider it to be a feature of FMF [17–19].
Pathophysiology
Predisposing factors
IgA vasculitis appears to be commoner in the spring, autumn and winter as compared with the summer months [6, 20–22]. Respiratory infections may be a precursor in a small number of cases and may be the second hit in patients with a genetic predisposition [21, 23]. Streptococcal infections are the most commonly observed predisposing infections [24, 25].
Pathology
IgA is thought to be key in the pathogenesis of HSP. Increased levels of IgA in the serum (in 50% with active disease), circulating immune complexes containing IgA, and the deposition of IgA in blood vessel walls and in the renal mesangium are associated with IgA vasculitis (Figure 102.12). In IgA vasculitis, IgA1 rather than IgA2 is the main IgA subclass deposited in skin lesions [26, 27]. Diminished glycosylation of the proline-rich hinge region of the IgA1 heavy chain is thought to be an important factor in allowing the IgA to be deposited in the mesangium and in activating the alternative pathway of complement in IgA, as it makes such IgA1 molecules more prone to forming macromolecular complexes [28]. Other IgA antibodies that occur in HSP include IgA ANCA, although this finding is very variable between studies. IgA rheumatoid factor and IgA anticardiolipin antibodies are also sometimes present, as are IgA antiendothelial cell antibodies (AECAs).

Figure 102.12 IgA vasculitis at (a) lower and (b) higher magnifications. There is perivascular leukocytoclasis and fibrin deposition, and eosinophils are present. (Courtesy of Dr Laszlo Igali, Norfolk and Norwich University Hospital, Norwich, UK.)
The activation of several cytokines is documented, although these are unlikely to be a primary cause. TNF-α levels are increased, and TNF-α can be detected in skin lesions. IL-6, IL-8, transforming growth factor β (TGF-β) and vascular endothelial growth factor (VEGF) levels are all increased in active HSP. TGF-β is of particular interest as blood levels of T cells that produce this cytokine are increased in HSP, and it is known to enhance IgA1 responses. Neutrophil activation, elevated nitric oxide levels, reactive oxygen species and increased urinary leukotriene are all documented.
Causative organisms
There are no definitely causative organisms for IgA vasculitis. Streptococcal infections predispose to IgA vasculitis, and antistreptolysin O titre positivity confers a 10-fold risk of IgA vasculitis [25] but the exact role of the bacteria is unknown. It is likely to be a complex interplay between genetic predisposition, bacterial infection and perhaps other environmental factors. Bartonella and Haemophilus have been implicated [29, 30]; Helicobacter antibodies have been reported in adults with IgA vasculitis [31].
Genetics
There is no definite association of any genes with IgA vasculitis. HLA class I genes may be of relevance [32]. MEFV gene mutations have been observed to be commoner in patients with IgA vasculitis than in the general population [16, 17].
Clinical features
History
Most commonly, IgA vasculitis manifests at the outset with the classic findings of purpura, arthralgia and abdominal pain. Individual lesions usually fade within 5–7 days but crops of lesions can recur for a few weeks to several months.
Presentation
The cutaneous findings are typically erythematous, urticarial papules, which may evolve within 24 h into palpable purpura with haemorrhage. Urticaria, vesicles, bullae (Figure 102.13a) and necrotic ulcers (Figure 102.13b) may develop. A retiform pattern within lesions is characteristic, but not always present. The presentation may be identical to CSVV (see Figure 102.1). Although it typically involves the extensor aspects of the limbs (especially the elbows and knees) and buttocks in a symmetrical fashion, IgA vasculitis may also affect the trunk and face. Renal involvement with IgA vasculitis is common, occurring in approximately 40–50% of patients; 25% have gross haematuria and the remainder microscopic haematuria. Proteinuria occurs in 60% of these, but is uncommon in the absence of haematuria. Gastrointestinal involvement is common (65%), with frank gastrointestinal bleeding in 30% of patients with IgA vasculitis. Painful arthritis is seen in about 75% of patients, most frequently affecting the knees and ankles. Less common manifestations of IgA vasculitis include orchitis (in 10–20% of boys), intussusception, pancreatitis, neurological abnormalities, uveitis, carditis and pulmonary haemorrhage.
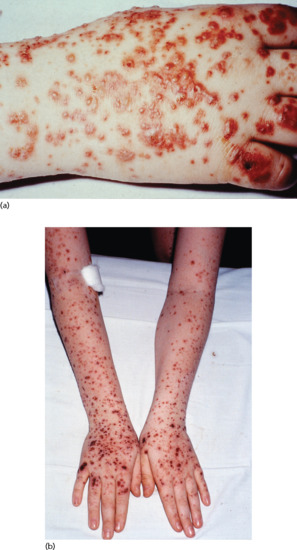
Figure 102.13 IgA vasculitis. (a) Haemorrhagic vesicles present on the hand. (Courtesy of Andrew Carmichael, South Tees Hospitals NHS Trust, UK.) (b) Vasculitis extending onto the arms.
Clinical variants
Rarely, gastrointestinal involvement and arthritis can occur in the absence of skin disease.
Differential diagnosis
This includes IgA nephropathy, idiopathic thrombocytopenic purpura, septic shock, acute abdomen and systemic lupus erythematosus (SLE).
Classification of severity
The BVAS (v3) system has been validated to assess the severity of IgA vasculitis alongside several other forms of systemic vasculitides. The tool is able to describe the nature of the clinical involvement, with higher scores suggestive of more severe disease, and is responsive to changes in clinical activity [33].
Complications and co-morbidities
End-stage renal disease is uncommon but, if it occurs, may need renal transplantation. Renal transplant survival is over 80% at 5 years. Uncommonly, IgA vasculitis may recur in the graft and cause graft rejection [34].
Disease course and prognosis
About 25% of patients will relapse, and typically the relapse is mild and easily treated [35]. IgA vasculitis can become chronic in 5–10% of patients, the cutaneous involvement usually lasting between 6 and 16 weeks. Only 1–3% of these patients progress to end-stage renal disease, although one-third to one-half of patients have renal abnormalities on long-term follow-up [13, 36].
Investigations
IgA vasculitis is a clinical diagnosis, with confirmation by direct immunofluorescence and routine histology. Perivascular IgA deposits are characteristic of IgA vasculitis and can help to distinguish it from other vasculitides including CSVV, granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis and microscopic polyangiitis. IgA immune complexes are not specific to IgA vasculitis, but can be seen in a variety of patients including those with SLE, endocarditis, dermatitis herpetiformis, alcoholism, IgA nephropathy, inflammatory bowel disease, ankylosing spondylitis, Sjögren syndrome, rheumatoid arthritis, some cancers and in some drug hypersensitivity reactions. No laboratory tests are specific for IgA vasculitis.
Management
The treatment of IgA vasculitis is supportive. The condition is usually self-limiting. There are no controlled studies of any drugs used in IgA vasculitis. Glucocorticoid agents may be of value in children with renal involvement and in most adults. Pulsed intravenous methylprednisolone, ciclosporin A, cyclophosphamide, azathioprine and mycophenolate mofetil have all been tried in open label fashion. There are no convincing data for any of them [37]. Systemic glucocorticoid treatment may be effective in the treatment of abdominal pain, arthritis and nephritis [38]; in this study the dose was prednisolone 1 mg/kg/day for 2 weeks, tapering over a further 2 weeks.
Cryoglobulinaemic vasculitis
Definition and nomenclature
Cryoglobulins are abnormal immunoglobulins that precipitate spontaneously when serum is cooled to a temperature below 37°C. Cryoglobulinaemia is the condition characterized by the presence of circulating cryoglobulins; the accompanying vasculitis that affects the small vessels is due to cryoglobulins deposited as immune complexes. It is mainly the skin glomeruli and peripheral nerves that are affected. Not all cryoglobulinaemia is associated with symptoms. This chapter only deals with the vasculitis manifestations; details of the history, aetiology and pathogenesis of cryoglobulinaemia are provided in Chapter 0.
Introduction and general description
Cryoglobulinaemic vasculitis is a small-vessel vasculitis affecting the skin, joints, peripheral nerves and kidneys. About 80% of cases are secondary to hepatitis C infection [1]. Other causes include B-cell lymphoproliferative disorders, autoimmune diseases like Sjögren syndrome, other viral disorders (hepatitis B, HIV) and essential mixed cryoglobulinaemia.
Cryoglobulins may be divided into three main subtypes:
- Monoclonal immunoglobulin, usually IgG or IgM, accounts for about 10–25% of cases and is usually associated with lymphoproliferative disease, especially multiple myeloma or Waldenström macroglobulinaemia.
- Mixed polyclonal (usually IgG) immunoglobulin and monoclonal (usually IgM-κ) immunoglobulin, the latter having rheumatoid factor activity, accounts for about 25% of cases.
- Polyclonal IgM with rheumatoid factor activity and polyclonal IgG with antigenic activity account for about 50–65% of cases.
Epidemiology
Incidence and prevalence
Cryoglobulinaemic vasculitis is a rare disease and its incidence and prevalence are not known.
Age
The condition is seen usually in adults and is very rare in children.
Associated diseases
Hepatitis C is responsible for about 80% of cryoglobulinaemic vasculitis. The other common associations are Sjögren syndrome and B-cell lymphoproliferative disorders.
Pathophysiology
The mechanism for the production of cryoglobulins by a clonally expanded B-cell population is ill understood. Since most patients with hepatitis C do not develop vasculitis, but have circulating cryoglobulins, there may be a failure of a separate mechanism responsible for the disease manifestations. There are significantly lower circulating T-regulatory cells in patients who develop vasculitis as compared to those with just cryoglobulinaemia [2].
Typically, a polyvalent IgM rheumatoid factor binds to antigen (although other monovalent immunoglobulins can also be responsible) to produce immune complexes that activate complement resulting in endothelial activation and tissue damage.
Predisposing factors
Cold and immobility may precipitate acute episodes.
Pathology
Cryoglobulinaemic vasculitis affects capillaries, arterioles and venules. In the skin, it produces a pandermal leucocytoclastic vasculitis that may extend into the subcutis. Eosinophilic periodic acid–Schiff (PAS) positive globular immune complex deposits and PAS-negative intraluminal fibrin deposits can be visualized. More chronic lesions develop a mononuclear-predominant infiltrate and may become granulomatous. Changes in other organs include membranoproliferative glomerulonephritis, immune complex deposition in the lungs causing bronchiolitis obliterans organizing pneumonia, and vasa nervosa vasculitis causing a peripheral neuropathy.
Causative organisms
The main aetiological factor in mixed cryoglobulinaemia is hepatitis C virus (HCV) infection, which accounts for about 80% of cases. However, although cryoglobulinaemia can be detected in about 50% of subjects with HCV, immune complex vasculitis occurs in less than 5% [3].
Environmental factors
Exposure to cold and immobility can trigger gelling in cryoglobulinaemia, resulting in cutaneous necrosis.
Clinical features
History and presentation
The classic ’Meltzer's triad’ of arthralgia, purpura and weakness was described in 1966, but is seen in less than a third of patients [4]. Myalgia, headache, fever and weight loss are common. Palpable purpura, the commonest presenting feature, is nearly universal (Figure 102.14). Sensorimotor neuropathy and mononeuritis multiplex are both commonly documented. Pulmonary involvement is rare. Renal involvement is usually in the form of membranoproliferative glomerulonephritis, and presents with nephrotic range proteinuria [5]. A smaller proportion of patients may present with proliferative mesangial lesions or thrombotic lesions [5]. The Raynaud phenomenon may be seen in patients with associated connective tissue disease.

Figure 102.14 Cryoglobulinaemic vasculitis.
Differential diagnosis
Cryoglobulinaemic vasculitis should be distinguished from other causes of CSVV because corticosteroid therapy, although sometimes necessary in the short term, may in the longer term worsen the underlying infection that is present in the majority of cases. Most other causes of leukocytoclastic vasculitis cause a more superficial vasculitis on biopsy specimens, and if cryoglobulin deposits are seen histologically then the diagnosis is usually suspected (although this is much commoner in type I cryoglobulinaemia). Clinically, head and neck involvement, significant livedo, acrocyanosis, Raynaud phenomenon or larger vessel occlusion are all more suggestive of type I cryoglobulinaemia.
Complications and co-morbidities
In patients with hepatitis C-induced disease, the complications are those of liver involvement. Associated glomerulonephritis is common and important and may be more frequent in those with hepatitis C [6]. There is an increased risk of myeloproliferative disorders, particularly a B-cell non-Hodgkin lymphoma [7]. This appears to be fourfold greater in patients without hepatitis C [8]. Sjögren syndrome has been reported in up to 20% of patients [4].
Disease course and prognosis
Cryoglobulinaemic vasculitis per se does not confer a significant mortality risk. In patients with hepatitis C-induced disease, the viral disease will determine the prognosis. In patients without hepatitis C, renal involvement is associated with greater morbidity. There are no large cohort studies to predict outcomes.
Investigations
A pivotal consideration when testing for cryoglobulinaemic vasculitis is transport of the specimen. The greatest care should be taken to ensure that the blood is transported to the laboratory at 37°C to ensure that the tests are not falsely negative. The demonstration of cryocrit in patients with clinical evidence of small-vessel vasculitis is probably the gold standard for the diagnosis (Figure 102.15). A low complement C4 level with a near normal C3 is nearly universal. Rheumatoid factor is positive in high titres. Evidence of viral hepatitis should be looked for. Inflammatory markers will be elevated.

Figure 102.15 Serum protein electrophoresis. (a) Demonstrating a clear band in lanes 1, 2 and 5. Lanes 3 and 4 are negative controls and lane 5 is a urine control from a nephrotic specimen. The sample was run on an enhanced urine program and gel for greater demonstration of the migration of low protein concentrations. (b) Demonstrating the same sample as an IgM-κ band. (Courtesy of Ian Thirkettle and Karen Ashurst.)
Urine analysis can range from normal to nephrotic range proteinuria. A renal biopsy will often be positive in the presence of significant renal disease, although occasionally the lesions may be due to minimal change disease [5]. A cutaneous biopsy will demonstrate leucocytoclastic vasculitis.
Management
Treatment will depend on the underlying cause. In patients with hepatitis C infection, the treatment should be coordinated with a hepatologist and will need a combination of glucocorticoids, antiviral therapy and immunomodulatory agents [9]. The HCV genotype will dictate the exact choice and duration of the antiviral agent. Interferon a in combination with ribavirin is beneficial, but relapses are common, necessitating long-term treatment [9]. Rituximab may be of benefit in this group of patients [10].
There is little evidence for the management of non-hepatitis C cryoglobulinaemic vasculitis. Consensus from the European League Against Rheumatism recommends a combination of immunomodulatory agents and glucocorticoid treatment based on the model of treating the other small-vessel vasculitides, such as the ANCA-associated vasculitides [9].
Hypocomplementaemic urticarial vasculitis
Definition and nomenclature
This section deals with hypocomplementaemic urticarial vasculitis (HUV); other forms of urticarial vasculitis are discussed in Chapter 0. HUV is defined as vasculitis affecting small vessels (i.e. capillaries, venules or arterioles), accompanied by urticaria and hypocomplementaemia and associated with anti-C1q antibodies. Glomerulonephritis, arthritis, obstructive pulmonary disease and ocular inflammation are common [1].
Introduction and general description
The condition is very rare and is characterized by persistant urticarial lesions lasting for longer than 24 h with circulating anti-C1q autoantibodies. Arthritis, glomerulonephritis, pulmonary and ocular disease, leukocytoclastic vasculitis, ocular inflammation and abdominal pain are associated.
Epidemiology
Incidence and prevalence
Hypocomplementaemic urticarial vasculitis is very rare with only a few hundred cases described.
Age
The most common presentation is when a person is in their thirties but childhood cases have been described.
Sex
It is commoner in females [2, 3].
Ethnicity
There are no associations.
Associated diseases
It may be associated with SLE [3], and may be linked with increased susceptibility to pyogenic infections.
Pathophysiology
The sera of patients with HUV contains polyclonal IgG with C1q precipitin activity contained within the Fab fragments [4]. These IgG antibodies are directed against the collagen-like region of C1q, resulting in a reduction of C1q in the serum with subsequent activation of the complement pathway [5].
Predisposing factors
These are unknown.
Pathology
Lesions of urticarial vasculitis are typically viewed as showing a leukocytoclastic vasculitis. HUV shows a large number of interstitial neutrophils rather than the pleomorphic infiltrate of normocomplementaemic vasculitis [6]. The deposition of immune complexes is present in normal and lesional skin in HUV, as opposed to only lesional skin in normocomplementaemic vasculitis [3, 6].
Clinical features
History
Hypocomplementaemic urticarial vasculitis is characterized by weals, which are characteristically painful but can be itchy, and persist for more than 24 h. In HUV, weals resolve with areas of discoloration. Angio-oedema is common and may be a presenting feature.
Presentation
Cutaneous lesions of both the hypocomplementaemic and normocomplementaemic forms of UV are erythematous indurated weals that may contain purpuric foci (Figure 102.16). Angio-oedema and macular erythema may also occur. Livedo reticularis, nodules and bullae may be evident, and may also contain purpuric foci. Patients with the hypocomplementaemic form may have constitutional symptoms.

Figure 102.16 Urticarial vasculitis.
Differential diagnosis
Hypocomplementaemic urticarial vasculitis has features similar to SLE and may overlap. Signs such as ocular inflammation, angio-oedema and chronic obstructive pulmonary disease may help distinguish the two processes. Pre-bullous pemphigoid, erythema multiforme, Sweet syndrome, other causes of vasculitis and urticaria coexisting with various forms of eczema should be considered, as should mixed cryoglobulinaemia, Muckle–Wells syndrome, Cogan syndrome and Schnitzler syndrome.
Classification of severity
There is no classification.
Complications and co-morbidities
Cough or dyspnoea may indicate pleural and pericardial effusions, emphysema or chronic obstructive pulmonary disease (seen in 20–50%). Proteinuria or haematuria may indicate glomerulonephritis, which may progress to end-stage renal failure, particularly in those with childhood onset. Gastrointestinal symptoms (abdominal discomfort, nausea, vomiting and diarrhoea), arthritis, episcleritis, uveitis, conjunctivitis, aseptic meningitis, nerve palsies and transverse myelitis may occur.
Investigations
If urticarial lesions last for longer than 24 h (which can be determined by drawing around their margin), then they are not ordinary urticaria (except delayed pressure urticaria) and a skin biopsy should be considered. Pain rather than itch, or the presence of purpura, also suggests urticarial vasculitis. History, physical examination and laboratory studies, including C3, C4 and antinuclear antibody, should help to establish the extent of disease and to exclude underlying disease (e.g. hepatitis C) and to evaluate for SLE. Some patients may demonstrate an elevated erythrocyte sedimentation rate (ESR), hypocomplementaemia, a low-titre positive antinuclear antibody and haematuria. A biopsy may help confirm the diagnosis and to exclude other disorders.
Management
The evidence for the treatment of HUV is anecdotal. Systemic corticosteroids are effective. Steroid-sparing agents should be considered, but the evidence for their use is restricted to case reports for cyclophosphamide [7], methotrexate [8], dapsone [9], colchicine [10] and hydroxychloroquine [11]. Some patients require oral antihistamines for the control of angio-oedema and urticaria-like lesions, in addition to therapies directed at the vasculitis.
First line
Although no single treatment is effective for all cases of urticarial vasculitis, the majority of patients respond to systemic corticosteroids.
Second line
Drugs that have been shown to be effective for the treatment of urticarial vasculitis include dapsone (100–200 mg once daily), colchicine (0.6 mg twice to three times daily) and hydroxychloroquine (200 mg once to twice daily).
Third line
For patients with refractory disease, there is limited evidence for the use of cyclophosphamide or mycophenolate mofetil [7, 12].
Antiglomerular basement membrane vasculitis disease
Definition and nomenclature
Antiglomerular basement membrane (anti-GBM) vasculitis affects glomerular capillaries or pulmonary capillaries, or both, with GBM deposition of anti-GBM autoantibodies. Lung involvement causes pulmonary haemorrhage, and renal involvement causes glomerulonephritis with necrosis and crescents [1].
Introduction and general description
In 1919, Ernest Goodpasture described two men who he thought had a viral influenza. One of them had the classic changes that came to bear the eponymous diagnosis of Goodpasture syndrome – alveolar haemorrhage and glomerulonephritis with arteriolar vasculitis [2]. The name was formally changed by international consensus to ’anti-GBM disease’ in 2012 [1]. The consensus name is flawed because the antibodies bind to pulmonary alveolar capillary basement membranes. Cutaneous involvement is not common, although there are anecdotal reports of non-vasculitic skin changes with immune deposition. [3]. Patients may present to dermatologists with pallor.
Epidemiology
Incidence and prevalence
There are about 0.5 cases/million population per year [4]. The prevalence is unknown.
Age
The disease is known to occur in children [5]; but the peaks seem to occur in the third and seventh decades [6].
Sex
In children the male to female ratio is 1 : 2 [7], whereas this is reversed in those older than 65 years with a M : F ratio of 1.9 : 1 [8].
Ethnicity
The relative ethnic differences are unknown.
Pathophysiology
The condition is due to immune complexes composed of autoantibodies directed against the NC1 domain of the a3 chain of type IV collagen [9]. The distribution of this molecule restricts the condition to the lung and kidney.
Pathology
Perivasculitis and anti-IgM and C3 antibodies at the basement membrane zone in cutaneous lesions have been described in one case [3].
Genetics
Familial instances of the disease have been described including in a pair of identical twins with exposure to hydrocarbon fumes [10].
Environmental factors
It is most common in late spring and early summer.
Clinical features
History and presentation
Haemoptysis, fatigue, dyspnoea and cough may be presenting features. Pallor, oedema, chest signs, heart murmurs and hepatomegaly may be present. Discrete, erythematous, macular lesions on the instep of the foot have been described [3].
Clinical variants
Respiratory features predate renal disease by up to a year in two-thirds of cases, and there may be a gap of up to 12 years.
Differential diagnosis
Granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis, IgA vasculitis and microscopic polyangiitis may all present with renal failure and pulmonary haemorrhage. Identification of anti-GBM antibodies helps diagnosis.
Disease course and prognosis
Untreated outcome is very poor, with near 100% mortality. With treatment, 1-year survival depends on early renal function: there is 100% survival if the serum creatinine is <500 μmol/L, but 65% survival in dialysis-dependent cases [11].
Investigations
Investigations should exclude other systemic vasculitides. ANCA may be positive but antinuclear antibody is usually negative. Specific testing is to anti-GBM antibodies.
Management
First line
Treatment is with corticosteroids, cyclophosphamide and plasma exchange.
Second line
Patients may require renal dialysis if in renal failure or respiratory support if there is severe pulmonary haemorrhage.
SMALL-VESSEL ANCA-ASSOCIATED VASCULITIS
Microscopic polyangiitis
Definition and nomenclature
Microscopic polyangiitis (MPA) is a necrotizing vasculitis, with few or no immune deposits, predominantly affecting small vessels (i.e. capillaries, venules or arterioles). Necrotizing arteritis involving small and medium arteries may be present. Necrotizing glomerulonephritis is very common and pulmonary capillaritis often occurs. Granulomatous inflammation is absent [1]. Historically, MPA was grouped with polyarteritis nodosa, and the two terms were often used interchangeably. It was defined and classified as a separate condition in 1994 at the first Chapel Hill consensus conference [2].
Introduction and general description
Friedrich Wohlwill described two cases with glomerulonephritis and non-granulomatous inflammation of small vessels in 1923 [3]. However, it was not until 1994 that MPA was formally defined by an international consensus [2]. MPA is part of a group of conditions termed ANCA-associated vasculitis (AAV). The other two conditions in this group are granulomatosis with polyangiitis (GPA) and eosinophilic granulomatosis with polyangiitis (EGPA). They are united by their association with antibodies directed against proteinase 3 (PR3) and myeloperoxidase (MPO). PR3 and MPO are proteins that serve as antigens inside the azurophilic granules in the cytoplasm of a neutrophil. Phenotypically, MPA and GPA are very similar in presentation, with the prime difference being the absence of granulomatous inflammation in MPA.
Epidemiology
Incidence and prevalence
The annual incidence of MPA is 2.5–10/million [4, 5]. However, this rises to 45/million per year in the population over the age of 65 [6]. The point prevalence figures vary from 25 to 100/million population [7–9].
Age
The peak incidence is in populations aged over 65 [6].
Sex
No predilection is known.
Ethnicity
White people are more commonly affected [10]. In Japan, the overall incidence of AAV is similar to that in the western European population, but the AAV is exclusively MPA, with almost no GPA [6].
Associated diseases
No associations are known.
Pathophysiology
The exact mechanism of production of vascular inflammation is not known, but ANCA has a prime role in the pathogenesis. Murine models have demonstrated the pathogenicity of MPO ANCA in producing glomerulonephritis and pulmonary haemorrhage [11, 12]. ANCA can induce degranulation of neutrophils primed by TNF-α [13]. Primed neutrophils exhibit MPO on their surface. The MPO ANCA can induce a respiratory burst leading to degranulation and the release of toxic oxygen radicals and intracytoplasmic enzymes, which may lead to vascular inflammation [13, 14].
Predisposing factors
Farming may predispose to pANCA positivity and MPA [15].
Pathology
Histological specimens from MPA lesions demonstrate segmental vascular necrosis. Neutrophils and monocytes permeate vessel walls, causing leukocytoclasis, the accumulation of fibrin and haemorrhage. Biopsy specimens from lesions of palpable purpura demonstrate leukocytoclastic vasculitis. Focal segmental glomerulonephritis with extracapillary crescents are a characteristic finding in renal biopsies. The presence of glomerulosclerosis is suggestive of the duration of disease and dictates the renal impairment.
Genetics
No culprit genes have been definitely identified, but the geographical distribution of the disease is suggestive of a genetic influence.
Environmental factors
Farming may be associated with MPA [15].
Clinical features
History
Many patients with MPA initially experience constitutional symptoms, including fever, weight loss, myalgia and arthralgia. These may be present for several weeks before the onset of the pulmonary and renal disease that often occurs in patients with MPA.
Presentation
About 40% of patients have palpable purpura on dependent skin sites upon presentation [16]. Mouth ulcers, necrotic lesions on the fingers or toes, splinter haemorrhages and livedo reticularis can all be present. The presence of nodules and livedo reticularis is commoner in polyarteritis nodosa; 80% of patients will have renal involvement. The presentation may be explosive with rapidly progressive glomerulonephritis or pulmonary haemorrhage (Figure 102.17). Peripheral neuropathy is common and is usually sensorimotor. Mononeuritis multiplex and even cranial nerve involvement are not unusual.

Figure 102.17 Computed tomography of the thorax demonstrating pulmonary haemorrhage affecting both lung fields in microscopic polyangiitis.
Differential diagnosis
Microscopic polyangiitis should be distinguished from other ANCA-associated vasculitides and polyarteritis nodosa. The classification of these patients should be set out as suggested by Watts et al. (Figure 102.18) [17]. The diagnosis of MPA can be made only in the absence of cardinal features of EGPA and GPA. Significant peripheral eosinophilia, extravascular eosinophils, nasal or paranasal sinus involvement, endobronchial involvement, granulomas on a biopsy, fixed pulmonary infiltrates, cavitating nodules on a chest X-ray, asthma and mastoidal or retro-orbital disease all lead away from a diagnosis of MPA.

Figure 102.18 Classification of antineutrophil cytoplasmic antibody vasculitides and polyarteritis nodosa (PAN). ACR, American College of Rheumatology; CHCC, Chapel Hill Consensus Conference; cPAN, cutaneous PAN; EGPA, eosinophilic granulomatosis with polyangiitis; GPA, granulomatosis with polyangiitis; MPA, microscopic polyangiitis; MPO, myeloperoxidase; PR3, proteinase 3; PSV, primary systemic vasculitides. (From Watts et al. 2007 [17]. Reproduced with permission of BMJ Publishing Group Ltd.)
Similarly the absence of blood, protein or red cell casts in the urine leads away from the diagnosis of MPA.
Classification of severity
The European Vasculitis Society has classified disease severity in AAV as below [18]:
- Localized. Upper and/or lower respiratory tract disease without any other systemic involvement or constitutional symptoms.
- Early systemic. Any, without organ-threatening or life-threatening disease.
- Generalized. Renal or other organ-threatening disease; serum creatinine <500 μmol/L.
- Severe. Renal or other vital organ failure; serum creatinine >500 μmol/L.
- Refractory. Progressive disease unresponsive to glucocorticoids and cyclophosphamide.
In practice, there are almost no patients without renal involvement in MPA, and therefore localized and early systemic disease is almost non-existent in this condition.
Complications and co-morbidities
Severe MPA can be associated with renal failure or life-threatening pulmonary haemorrhage. Pulmonary haemorrhage occurs in about 10% of patients and carries a high risk of death [19, 20]. In the long term, there is a raised risk of coronary artery disease and hypertension. At 5 years, there is a 16% incidence of cardiovascular events (myocardial infarctions, cerebrovascular accidents or coronary revascularization procedures) [21]. MPO ANCA positivity confers a higher risk of cardiovascular events compared with PR3 ANCA positivity.
Disease course and prognosis
Relapse is common and increases with time. In separate studies it has been documented to be 8% at 18 months [22] and 34% at 70 months [16]. Survival at 12 months is 82–92%, falling to 45–76% at 5 years.
Investigations
Anaemia of chronic disease and laboratory markers of an acute phase response predominate. Urine analysis provides big clues. Most patients will have a blood and protein leak to varying extents. ANCA directed against PR3 or MPO are present in nearly all patients. Imaging helps to establish the extent and severity of disease in patients with lung involvement. Chest X-ray followed by computed tomography (CT) scanning identifies alveolar haemorrhage or pulmonary fibrosis. Histopathology is the gold standard for diagnosis and when possible the kidneys should be biopsied on suspicion of MPA. Other tissues that may provide an answer are the sural nerve, muscle and skin.
Management
Recommendations for the management of MPA have been proposed by the European League Against Rheumatism [18] and the British Society for Rheumatology [23]. Immunosuppressive therapies, including oral or intravenous glucocorticoids, are the mainstay of treatment.
Remission induction
Pulsed intravenous cyclophosphamide (15 mg/kg every 2–3 weeks) or daily oral cyclophosphamide (2 mg/kg/day) form the mainstay of treatment for most patients with MPA. Intravenous cyclophosphamide has the advantage of lower cumulative dose and lower risk of adverse events, but carries a greater risk of relapse [24, 25]. Oral prednisolone in a dose of 1 mg/kg/day, to a maximum of 60 mg/day, is commonly used as an adjunct to cyclophosphamide, with the aim of reducing the dose to 15 mg/day at 3 months [18]. At the physician's discretion, intravenous methylprednisolone can be added to speed up the induction of remission at the commencement of cyclophosphamide. Standard practice would be to add 1 g intravenously per day for 3 days. In patients with severe renal disease, plasmapheresis may have a role in saving the kidney [26].
Relapsing and refractory disease
For relapsing disease, rituximab 375 mg/m2 per week for 4 weeks is superior to pulsed intravenous cyclphosphamide [27, 28]. For disease refractory to cyclophosphamide and glucocorticoid, rituximab 375 mg/m2 per week for 4 weeks has been shown to be of value [29].
Remission maintenance
- Post-cyclophosphamide. Due to the cumulative toxicity of cyclophosphamide, azathioprine (2 mg/kg/day) is preferred to maintain remission [22]. The switch over can happen either at the end of six pulses of intravenous cyclophosphamide or after 6 months of oral cyclophosphamide.
- Post-rituximab. In patients where rituximab is used to induce remission, current thinking suggests that rituximab should be used at 4–6-monthly intervals over the long term as a remission maintenance agent [30, 31].
Granulomatosis with polyangiitis
Definition and nomenclature
Granulomatosis with polyangiitis is a necrotizing granulomatous inflammation usually involving the upper and lower respiratory tract, and necrotizing vasculitis affecting predominantly small to medium vessels (e.g. capillaries, venules, arterioles, arteries and veins). Necrotizing glomerulonephritis is common [1].
Introduction and general description
Friedrich Wegener published three cases in 1937 of patients in their thirties who had a 4–7-month history of spiking temperatures with negative septic screens, raised ESR, predominant upper respiratory tract inflammation with nasal septal involvement and active urinary sediment, resulting in death [2]. Sven Johnsson first used the term Wegener granulomatosis as a distinct diagnosis [3]. The name was changed to ’granulomatosis with polyangiitis’ by international consensus in 2013 [1].
The ANCA-associated vasculitides are a group of conditions characterized by their association with the presence of antibodies directed against PR3 and MPO. PR3 and MPO are intracytoplasmic enzymes of neutrophils. GPA is the archetypal ANCA-associated vasculitis, incorporating most of the clinical features of the other two AAVs – MPA and EGPA.
Epidemiology
Incidence and prevalence
The incidence of GPA is 3–10/million per year [4–6, 7]. There may be a distinct latitudinal divide, with GPA being commoner in the northern latitudes than in the southern latitudes [6]. The point prevalence of GPA is 24–112/million [8–10].
Age
In children, the median age of onset is 14 years [11, 12]. In adulthood, the median age of onset is 50–59 years [10, 13, 14].
Sex
The condition affects males and females equally.
Ethnicity
The majority of cases occur in white people. GPA is rare in Japan [15], and may be rare in Inuits [16].
Pathophysiology
The exact mechanism of production of vascular inflammation and granulomas is not known, but ANCA has a prime role in the pathogenesis. Murine models have demonstrated the pathogenicity of MPO ANCA in producing glomerulonephritis and pulmonary haemorrhage [17, 18]. There is some evidence of in vitro pathogenicity of PR3 ANCA [19]. ANCA can induce the degranulation of neutrophils primed by TNF-α [20]. Primed neutrophils exhibit PR3 on their surface. The PR3 ANCA can induce a respiratory burst leading to degranulation and release of toxic oxygen radicals and intracytoplasmic enzymes, which may lead to vascular inflammation [20, 21].
There may be a role for a bacterial infection (e.g. Staphylococcus aureus), which stimulates autoreactive PR3 producing B cells within granulomas [22, 23]. In the autoinflammatory process, granuloma formation occurs prior to vasculitis. The granulomas in GPA have a high proportion of granulocytes, which serve as a source of PR3 [24], which in turn help to continue driving a Th1 cytokine response to further the inflammatory process.
Predisposing factors
Farming and occupational solvent exposure may predispose to GPA [25].
Pathology
Skin histology in GPA may show perivascular lymphocytic infiltrates; however, such non-specific infiltrates may not be related to disease pathogenesis. More specific findings such as leukocytoclastic vasculitis and/or granulomatous inflammation may be present in up to 50% of skin biopsy specimens (Figure 102.19). Granulomatous inflammation around vessels and palisading necrotizing granulomas are uncommonly demonstrated in skin lesions.

Figure 102.19 Granulomatosis with polyangiitis. (a) There is extensive leukocytoclastic vasculitis involving the entire dermis. (b) Note the extensive area of collagen degeneration, destruction of vessels and mixed inflammatory infiltrate. (c, d) There is a necrotizing vasculitis with fibrin deposition, red blood cell extravasation and a granulomatous reaction, at lower (c) and higher (d) magnifications. (Parts a and b, courtesy of Dr Omar Sangueza, Wake Forest University School of Medicine, Winston-Salem, NC, USA; c and d, courtesy of Dr Laszlo Igali, Norfolk and Norwich University Hospital, Norwich, UK.)
Causative organisms
Clinical observation points to Staphylococcus aureus as a potential trigger in some patients. The incidence of chronic nasal carriage of S. aureus is significantly higher in patients with GPA compared with healthy individuals and constitutes a risk factor for disease relapse [26, 27].
Genetics
There is evidence that GPA has an association with HLA DP, although this may be an association with PR3 ANCA rather than the syndrome of GPA [28, 29].
Clinical features
History
Features of the classic triad of GPA, including the skin, respiratory tract and kidneys, are not always present early in the course of the disease, making the diagnosis sometimes difficult. In up to 80% of patients, symptoms involving the upper or lower respiratory tract are present, and at presentation approximately 73% of patients will have nasal, sinus, tracheal or ear involvement.
Less than half present with pulmonary infiltrates or nodules. Renal disease is initially present in only 18%, although approximately 77% will eventually develop glomerulonephritis. Although 40% will eventually manifest skin findings, cutaneous manifestations and oral ulcers are only found in 13% and 6% of patients at initial presentation, respectively.
Presentation
The most common cutaneous manifestation of GPA is palpable purpura on dependent skin sites (Figure 102.20a); digital infarcts (Figure 102.20b), tender subcutaneous nodules, papules, vesicles and petechiae, as well as non-specific ulcers or pyoderma gangrenosum-like lesions may occur. Cases previously diagnosed as having ’malignant pyoderma’ may actually have had lesions secondary to GPA. Nodular, or papulonecrotic, lesions occur on the extremities and sometimes on the face and scalp. These may be differentiated from rheumatoid nodules in that they ulcerate, whereas rheumatoid nodules do not.

Figure 102.20 Granulomatosis with polyangiitis. (a) Cutaneous infarction. (b) Digital infarction.
Oral ulcers are the second most common mucocutaneous sign of GPA. The upper respiratory tract is commonly affected, with otitis, epistaxis, rhinorrhoea and sinusitis as frequent presenting features. A saddle nose deformity may result from necrotizing granulomas of the nasal mucosa. Lower respiratory signs and symptoms include cough, dyspnoea, chest pain and haemoptysis. Nodules, which may be cavitating, may be visible on imaging (Figure 102.21). Ulcerative lesions in GPA are shown in Figure 102.22.

Figure 102.21 Granulomatosis with polyangiitis. (a) X-ray showing bilateral nodules.(b) Computed tomography of the thorax demonstrating a thick-walled cavity in the right lung field.

Figure 102.22 Granulomatosis with polyangiitis. (a) Ulcerated lesions of cutaneous small-vessel vasculitis. (b) Larger ulcerated lesions with background vasculitis. (c) Deep skin ulcer in a person with nasal symptoms.
Differential diagnosis
Granulomatosis with polyangiitis must be differentiated from the other types of AAV. Destructive upper respiratory involvement and severe glomerulonephritis are unusual in EGPA, which typically has asthma and eosinophilia, along with paranasal polypoidal involvement. MPA has predominant renal involvement and is more likely to be associated with MPO ANCA (see Figure 102.18).
Classification of severity
The European Vasculitis Society has classified disease severity in AAV as below [30]:
- Localized. Upper and/or lower respiratory tract disease without any other systemic involvement or constitutional symptoms.
- Early systemic. Any, without organ-threatening or life-threatening disease.
- Generalized. Renal or other organ-threatening disease; serum creatinine <500 μmol/L.
- Severe. Renal or other vital organ failure; serum creatinine >500 μmol/L.
- Refractory. Progressive disease unresponsive to glucocorticoids and cyclophosphamide.
Complications and co-morbidities
Granulomatosis with polyangiitis or its treatment (especially cyclophosphamide) has a twofold increase in the risk of cancer including acute myeloid leukaemia, bladder cancer and non-melanoma skin cancers [31, 32]. GPA also predisposes to increased cardiovascular morbidity [33].
Disease course and prognosis
Remission can be achieved in up to 90% of patients [34]. At 2 years, the relapse rate is between 18% and 40% [34]. Untreated GPA has a 1-year mortality of 83%; the survival of treated disease at 1 year is >80% [35]. End-stage renal disease occurs in 7% at 12 months, rising to 14% at 5 years and 23% at 10 years [36]. Cancer risk is as discussed above.
Investigations
Investigations are as in MPA.
Management
Recommendations for the management of GPA have been proposed by the European League Against Rheumatism [30] and the British Society for Rheumatology [37]. Immunosuppressive therapy including oral or intravenous glucocorticoid is the mainstay of treatment.
First line
Patients with localized disease can be treated with methotrexate (15–25 mg/week) in conjunction with oral prednisolone 1 mg/kg/day tapering to about 15 mg/day at 3 months [30, 38]. The use of methotrexate in preference to cyclophosphamide is associated with a higher risk of relapse [39].
In patients with non-localized disease, cyclophosphamide forms the mainstay of treatment. Cyclophosphamide can be used in a daily oral 2 mg/kg/day dose for 6 months or as pulsed intravenous 15 mg/kg/pulse every 2–3 weeks for six pulses. Intravenous cyclophosphamide has the advantage of a lower cumulative dose and a lower risk of adverse events, but carries a greater risk of relapse [40, 41]. Oral prednisolone in a dose of 1 mg/kg/day, to a maximum of 60 mg/day, is commonly used as an adjunct to cyclophosphamide, with the aim of reducing the dose to 15 mg/day at 3 months [30]. At the physician's discretion, intravenous methylprednisolone can be added to speed up the induction of remission at the commencement of cyclophosphamide. Standard practice would be to add 1 g intravenously per day for 3 days.
In patients with severe renal disease, plasmapheresis may have a role in saving the kidney [42].
Second line
Patients with contraindications to cyclophosphamide can be treated with rituximab 375 mg/m2 per week for 4 weeks instead [43, 44].
Third line
Patients with refractory disease to cyclophosphamide and glucocorticoids can be treated with rituximab 375 mg/m2 per week for 4 weeks [45]. 15-Deoxyspergualin and intravenous immunoglobulin have a role in refractory and persistent disease [46, 47].
Remission maintenance
Due to the cumulative toxicity of cyclophosphamide, azathioprine (2 mg/kg/day) is preferred to maintain remission [48]. The switch-over can happen either at the end of six pulses of intravenous cyclophosphamide or after 6 months or oral cyclophosphamide. Methotrexate [49], leflunomide [50] and mycophenolate mofetil [51] are alternative remission maintenance agents. In patients where rituximab is used to induce remission, current thinking suggests that this should used at 4–6-monthly intervals long term as a remission maintenance agent [52, 53].
Eosinophilic granulomatosis with polyangiitis
Definition and nomenclature
Eosinophilic granulomatosis with polyangiitis (EGPA) is a very rare systemic disorder characterized by an eosinophil-rich and necrotizing granulomatous inflammation, often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium vessels. There is an association with asthma and eosinophilia. ANCA is more frequent when glomerulonephritis is present [1].
Introduction and general description
This is a rare systemic vasculitis characterized by asthma, peripheral blood and tissue eosinophilia (especially in the respiratory tract) and necrotizing vasculitis with extravascular granulomas. The majority of patients have cutaneous findings in the active phase of the disease. It was originally described by Rackemann and Greene in 1939 as an allergic disease and not classified as periarteritis nodosa; Churg and Strauss later described the syndrome and its histopathological characteristics in 1951 [2]. The condition was renamed EGPA in 2012 by the Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis [1]. EGPA is associated with antibodies directed against MPO and PR3, and, along with GPA and MPA, forms the group of conditions termed ’ANCA-associated vasculitis’.
Epidemiology
Incidence and prevalence
The incidence is 1–2.5 per million [3, 4, 5] with a prevalence of 10–15 per million [6, 7]. The incidence of EGPA in known asthma sufferers may be up to 67/million per year [8].
Age
It is most common in those aged 15–70 years with a peak incidence around the age of 50. It is very rare in children.
Sex
There may be a slight male predilection.
Ethnicity
No connection is recognized.
Associated diseases
It is associated with atopy, particularly asthma and allergic rhinitis.
Pathophysiology
The pathogenesis of EGPA is not precisely known. Allergy probably plays a central role, but the disease is almost certainly multifactorial. As the allergy association might suggest, the inflammatory response is primarily Th2 in nature, although Th1 and Th17 responses are seen [9, 10, 11]. The Th2 response has been thought to be responsible for eosinophilic activation, and prolonged eosinophil survival. The products of eosinophilic and neutrophilic degradation have been observed in inflamed tissues and are probably responsible for tissue injury [12, 13]. The association with ANCA probably suggests a B-cell involvement as well. The role of ANCA in producing vasculitis has been discussed in the GPA and EGPA sections.
Predisposing factors
None are known.
Pathology
EGPA has three key histopathological features: eosinophilic infiltration of tissue, formation of extravascular granulomas in visceral and cutaneous tissues, and vasculitis involving both arteries and veins. The histology of a cutaneous lesion in EGPA may demonstrate any one, if not all, of these features [14]. The granulomas contain necrotic polymorphonuclear leukocytes, eosinophils, severe fibrinoid and fibrillar collagen degeneration, and a proliferation of granulomatous tissue.
Causative organisms
None are known.
Genetics
HLA-DRB4 may be a risk factor for the development of EGPA [15].
Environmental factors
Environmental allergens are associated with more severe asthma.
Clinical features
History
Three phases of EGPA are recognized:
- The first phase, which may continue for years, consists of asthma with allergic rhinitis and nasal polyps. The asthma typically begins in adulthood in contrast to allergic asthma.
- The second phase is that of rising peripheral and tissue eosinophilia.
- The third phase is the predominately vasculitic phase of EGPA, which may affect almost all organ systems, including the cutaneous, cardiac, pulmonary, nervous, gastrointestinal, renal, genito-urinary and musculoskeletal systems. Cardiac involvement is the primary cause of death in patients who do not respond to conventional corticosteroid therapy.
Presentation
In all phases of the disease there may be cutaneous manifestations, with approximately 5% demonstrating cutaneous vasculitis [14]. Palpable purpura and infiltrated nodules (typically located on the scalp or limbs) are the most common skin manifestations, but livedo reticularis, necrotizing livedo (i.e. retiform purpura), migratory erythema, new-onset Raynaud phenomenon, aseptic pustules or vesicles, or infiltrated papules may also be present.
In the vasculitis phase of EGPA, 50–70% of patients have vasculitic skin lesions, most commonly on the lower limbs (Figure 102.23).

Figure 102.23 Relatively subtle vasculitis on the legs in eosinophilic granulomatosis with polyangiitis. The patient also had eosinophilia and rapidly developed a mononeuritis multiplex.
Differential diagnosis
The ACR 1990 criteria for the classification of EGPA from other vasculitides needed four of the following six features: (i) asthma; (ii) eosinophilia greater than 10% on a differential white blood cell count; (iii) mononeuropathy (including multiplex) or polyneuropathy; (iv) non-fixed pulmonary infiltrates on chest X-ray; (v) a paranasal sinus abnormality (Figure 102.24); and (vi) a biopsy demonstrating extravascular eosinophils [16].
Figure 102.24 Computed tomography scan of paranasal sinuses demonstrating opacification of the maxillary sinuses and bone erosion in eosinophilic granulomatosis with polyangiitis.
The Lanham criteria require asthma, peripheral eosinophilia and systemic vasculitis in two or more extrapulmonary organs [17].
The latest in a long line of algorithms to assist classification of the AAV and PAN recommends using either the Lanham criteria or the ACR criteria for classifying EGPA [18] (see Figure 102.18).
Disease course and prognosis
Remission is common in EGPA and achieved in >90% of patients [19]. Relapse rates rise from 10% at 12 months to 20% at 4 years [19]. Survival is better than in the other AAVs. In a pooled analysis, survival at 1 and 5 years was 94% and 60–97%, respectively [20]).
Investigations
Peripheral blood eosinophilia is a requisite for the diagnosis of EGPA. Inflammatory markers will be raised and IgE is often elevated. ANCA directed against MPO or PR3 is positive in 30% of patients [21, 22]. A urine microscopy demonstrates active urinary sediment in patients with glomerulonephritis. Renal involvement is more likely in ANCA-positive patients [21]. Chest radiographs demonstrate infiltrates. CT scans of the paranasal sinuses are often abnormal and demonstrate mucosal thickening, but rarely bone involvement. The latter is more a feature of GPA. The gold standard remains a biopsy of the affected organ.
Management
There are no randomized controlled trials of any treatment for EGPA. Our knowledge of its treatment comes from open labelled trials and from international consensus-based recommendations [23].
Remission induction
- Localized disease. Prednisolone 1 mg/kg/day may be enough to induce remission for most patients with EGPA [24]. However, even in patients without adverse prognostic features, there will be about 10% of patients where this strategy will not result in remission. A further 35% will suffer a relapse. There is currently no way of identifying those who will not do well with steroids alone. International recommendations therefore recommend that in patients without organ- or life-threatening involvement the addition of methotrexate 20–25 mg/week should be considered [23].
- Systemic disease. Intravenous pulsed cyclophosphamide 15 mg/kg/pulse at 2–3-weekly intervals (six pulses) with oral prednisolone 1 mg/kg/day (maximum 60 mg) is the currently preferred regimen for these patients [23].
- Refractory disease. Rituximab has been reported to have a beneficial outcome in several case series [9] and the authors have used it beneficially in refractory EGPA.
Remission maintenance
- Post-cyclophosphamide. By convention and international consensus, patients are switched to azathioprine 2 mg/kg/day [23].
- Post-methotrexate. Remission is maintained by continuing methotrexate long term.
- Post-rituximab. There is very little information on post-rituximab maintenance therapy. Current advice would be to use rituximab according to clinical need rather than every 6 months. These patients should be cared for by specialist centres.
MEDIUM-VESSEL VASCULITIS
Polyarteritis nodosa and cutaneous polyarteritis nodosa
Definition and nomenclature
Polyarteritis nodosa (PAN) is a rare necrotizing arteritis of medium or small arteries without glomerulonephritis and without vasculitis in the arterioles, capillaries or venules. It is not associated with ANCAs [1]. Cutaneous PAN (cPAN) is a single-organ vasculitis affecting the skin. It is better termed cutaneous arteritis. It can be considered a limited expression of PAN and does not exhibit systemic involvement [1, 2].
Introduction and general description
A condition described as periarteritis nodosa was described by Adolf Kussmaul and Rudolf Maier in 1866. The first use of the phrase ’polyarteritis nodosa’ denoting the pathological extent of the disease involving the arterial wall may have been in 1945 [3]. PAN was used as a term to cover a large variety of vasculitides. In 1994, the label was uniquely applied to a condition that spared small-calibre vessels [4].
This chapter considers cutaneous arteritis as a variant of PAN that is limited to the skin [5]. It may progress to become classic PAN [1], but conversion is exceedingly rare [2]. Although a distinct entity as described by Lindberg in 1931, the diagnosis of cutaneous arteritis should not be made until systemic disease is excluded.
Epidemiology
Incidence and prevalence
The annual incidence of classic PAN is 1–2.5/million [6, 7]. The point prevalence has been reported to be 30 per million [8, 9]. Cutaneous arteritis is much rarer and there are no formal reports on the incidence and prevalence of this variant.
Age
The peak age is between 40 and 60 years of age. However, it can be observed in all ages, including in children.
Sex
There is no specific predilection.
Ethnicity
No racial predilection has been described.
Associated diseases
Hepatitis B was described as an association in 1970 [10]. PAN can be the first manifestation of hepatitis B and occurs in most cases within 6 months of infection. Successful treatment of hepatitis B results in cure of PAN with disappearance of any aneurysms [11].
Pathophysiology
Predisposing factors
Viral infections have been implicated in provoking classic PAN. Besides hepatitis B virus, as indicated above, Epstein–Barr virus [12], HIV [13], erythrovirus (parvovirus B19) [14] and cytomegalovirus [15] have been reported in new cases of PAN. Other microbial associations reported have been streptococcal infections [16] and coxsackie B4 [17].
Streptococcal infections [18], erythrovirus (parvovirus B19) [19] and Mycobacterium fortuitum [20] have been reported in association with cutaneous arteritis. Minocycline has been reported to induce classic PAN-like vasculitis [21] as well as cutaneous arteritis [22].
Pathology
Early in the disease course, there is a predominantly neutrophilic inflammatory infiltrate in the walls of medium-sized arteries and arterioles of septae in the upper portions of the subcutaneous fat. The involved vessels classically demonstrate a target-like appearance resulting from an eosinophilic ring of fibrinoid necrosis. Later in the disease process, the infiltrate becomes less neutrophilic, consisting predominantly of lymphocytes and histiocytes. Complement and IgM deposits in vessel walls of lesions of cutaneous arteritis from some patients may be demonstrated by direct immunofluorescence. Unlike those of systemic PAN, lesions of cutaneous arteritis do not typically involve arterial bifurcations.
Genetics
Recessively inherited missense mutations in CECR1 (cat eye syndrome chromosome region, candidate 1), encoding adenosine deaminase 2 (ADA2) have been observed in nine unrelated cases from eight families [23]. Similar mutations were found in a separate study of 19 patients of Georgian Jewish descent [24].
Clinical features
History
Although some patients with cutaneous arteritis may report constitutional symptoms, along with mild involvement of the muscles and nerves, cutaneous manifestations are the most striking feature of the disease.
Presentation
Dermal or subcutaneous nodules are most commonly located on the distal lower extremities near the malleoli (Figure 102.25a) and may extend proximally to the thighs, buttock, arms or hands. Patients may report tenderness associated with the nodules, which may ulcerate (Figure 102.25b, c) or more commonly demonstrate necrotizing livedo reticularis, also referred to as retiform purpura (see Figure 102.3). Gangrene of the digits can ultimately occur, most commonly in children with cutaneous arteritis, but this finding should trigger an aggressive search to exclude systemic features of PAN.

Figure 102.25 Cutaneous polyarteritis nodosa. (a) Erythematous lesions on the leg. (b) Nodules and ulceration on the leg. (c) Ulcerating lesions on the leg. (d) Livedo of the leg.
Differential diagnosis
Recurrent spiking fevers, polyarthralgia and a macular upper extremity eruption are symptoms shared by both PAN and adult-onset Still disease (AOSD), and can sometimes create a diagnostic challenge. The presence of livedo reticularis (Figure 102.25d) and the finding of a characteristic skin biopsy appearance with PAN help to differentiate it from AOSD.
Those with necrotizing lesions of livedo reticularis must be evaluated for vasculitis or vasculopathy (e.g. antiphospholipid antibody syndrome, cholesterol emboli or other factors that can produce non-vasculitic vessel occlusion; see Chapter 0).
ANCA-associated vasculitis should also be considered as a differential diagnosis (see Figure 102.18) [25].
If nodules are present, they should be biopsied by incisional biopsy methods to assess for a pan-arteritis of muscular arteries which would confirm a diagnosis of PAN.
Cutaneous arteritis is best considered a variant of PAN, so evaluation by history, physical examination, screening laboratory tests and ongoing follow-up for systemic features is required. A multidisciplinary team approach helps accurate diagnosis, and limits the chances of missing or undertreating potentially life-threatening systemic features of PAN.
Classification of severity
Cutaneous arteritis is considered to have a more benign prognosis than PAN with systemic features.
Disease course and prognosis
Gastrointestinal tract, renal, heart and central nervous system involvement are associated with higher mortality [26].
Investigations
Laboratory investigations are usually non-specific, revealing an acute phase response. Screening for potential infective triggers should be undertaken. Diagnosis of PAN requires histological evidence of medium-sized artery vasculitis if possible. Biopsies should be from symptomatic organs. Skin, muscle and nerve histology offer higher diagnostic yield and may be safer. If biopsies are unsupportive, visceral angiography may identify multiple microaneurysms suggesting PAN.
Management
There should be screening for infection (see the section on predisposing factors) and consideration should be given to a trial of discontinuing medication that predates disease. If associated with hepatitis B infection, antiviral therapies form the focus of treatment in combination with immunosuppressive treatment.
First line
Non-steroidal anti-inflammatory drugs and salicylates can be an effective treatment for symptoms of cPAN. High-dose corticosteroids followed by tapering of the dosage over 3–6 months may occasionally be necessary for some patients. Also without evidence from controlled trials, but based on a strong association with streptococcal infection, penicillin is often used for treatment and prophylaxis in children with cPAN. Screening for recent streptococcal infection with anti-DNAse B or other tests may guide this decision.
Other treatments documented in anecdotal reports include the use of dipyridamole, sulfapyridine, pentoxifylline and dapsone in patients with cPAN. Low-dose weekly methotrexate (7.5–20 mg/week) has been successful in some patients with skin lesions unresponsive to corticosteroids given topically, intralesionally and orally [27]. Chronic leg ulcers resistant to treatment with high-dose corticosteroids have been successfully treated with granulocyte–macrophage colony-stimulating factor (GM-CSF) [28].
Second line
There is an international consensus that PAN requires treatment with a combination of cyclophosphamide and corticosteroids [29]. This combination achieves sustained remission but probably does not alter survival. For patients with hepatitis B-associated PAN, the recommendation is to start with high-dose corticosteroids for 2 weeks, followed by antiviral treatment and plasma exchange [29]. This treatment should be supervised at a specialist centre in conjunction with a hepatologist.
Third line
There are case reports for the use of rituximab in refractory disease [30, 31].
Kawasaki disease
Definition and nomenclature
The 2012 Chapel Hill Consensus defined Kawasaki disease as an arteritis associated with the mucocutaneous lymph node syndrome and predominantly affecting medium and small arteries. Coronary arteries are often involved; the aorta and large arteries may be involved. It is usually occurs in infants and young children [1].
Introduction and general description
Kawasaki disease occurs typically in infants and children less than 5 years of age. It was first recognized in 1967 and thought to be a benign, febrile illness associated with mucocutaneous inflammation and lymphadenopathy, until the demonstration of coronary arteritis in 1975 [2]. It is thought to be the commonest cause of acquired heart disease in children. Prompt diagnosis with treatment with aspirin and intravenous immunoglobulins reduces heart complications [3, 4].
Epidemiology
Incidence and prevalence
The annual incidence per 100 000 children aged under 5 years is 8.4 in the UK [5]. In a series of epidemiological surveys, Nakamura et al. have established that the incidence is rising in Japan every year and had peaked at 240/100 000 in 2010 [6].
Age
The disease almost always occurs in children.
Sex
There is a mild male predilection.
Ethnicity
The disease is much more common in Asia, particularly in Japan.
Associated diseases
Associated diseases include coronary vessel aneurysms and myocardial infarction.
Pathophysiology
Kawasaki disease is thought to be due to an intense inflammatory response to an unidentified infectious agent in genetically susceptible hosts.
Pathology
The angiitis of Kawasaki disease affects nearly all organs, with a very high frequency of cardiac involvement. It is predominantly a vasculitis of medium-sized arteries but can involve any smaller and larger calibre blood vessels. Initially, there is medial oedema associated with neutrophilic infiltration. The inflammatory processes result in the breakdown of internal and external elastic laminae, resulting in aneurysms and thrombosis. The inflammatory processes heal with scarring and resultant stenosis of the affected blood vessel.
Causative organisms
The disease is thought to be triggered by as yet unidentified infectious agents.
Genetics
A functional polymorphism of the ITPKC (inositol-1,4,5-trisphosphate 3-kinase C) gene on chromosome 19q13.2 is significantly associated with a susceptibility to Kawasaki disease and coronary artery aneurysms [7]. Single nucleotide polymorhphisms of interest have been found on the CD40LG gene [8] and CASP3 gene [9]. Genome-wide association studies have identified further loci of interest around the FAM167A-BLK region at 8p22-23, the HLA region at 6p21.3 and the CD40 region at 20q13 and the IgG receptor gene FCGR2A [10, 11].
Clinical features
History
Patients present with at least 5 days of fever, irritability, vomiting, anorexia, cough, diarrhoea, runny nose, weakness and abdominal and joint pain.
The disease typically has an initial acute, febrile stage lasting up to 2 weeks, a second phase lasting 4–6 weeks when the risk of death from coronary aneurysms is greatest, followed by a convalescent phase lasting up to 3 months and characterized by a reduction in the ESR and C-reactive protein (CRP) levels to normal. Larger aneurysms may expand leading to myocardial infarction in the convalescent phase. Those with established heart disease may enter a chronic phase with a risk of late aneurysm rupture even in adult life.
Presentation
The fever is typically spiking and unresponsive to paracetamol. There is acral and perianal erythema and acral oedema, bilateral conjuncitivitis with anterior uveitis, fissured lips and a strawberry tongue, and cervical lymphadenopathy, typically a single large cervical node.
Clinical variants
Incomplete Kawasaki disease should be considered in children who do not have all the features of the full disease. Typical echocardiographic features should lead to consideration of treatment as Kawasaki disease in the absence of external clinical features. [12].
Differential diagnosis
Scarlet fever, systemic-onset juvenile idiopathic arthritis and erythema multiforme can mimic Kawasaki disease, as can other localized and systemic infections. The diagnosis should be suspected in a child with prolonged fever.
Classification of severity
The Harada score has been used in some countries as an indication for intravenous immunoglobulin therapy [13]. Four of following seven criteria are needed: (i) white blood count >12 000/mm; (ii) platelet count <35 × 104/mm; (iii) CRP >3; (iv) haematocrit <35%; (v) albumin <3.5 g/dL; (vi) age <12 months; and (vii) male sex.
Complications and co-morbidities
There may be hepatic, renal and gastrointestinal dysfunction, myocarditis and pericarditis.
Disease course and prognosis
Deaths may occur due to myocarditis, dysrhythmias, pericarditis, rupture of aneurysms and occlusion of coronary arteries; there is also an increased risk of atherosclerosis due to endothelial cell dysfunction. Coronary aneurysms are demonstrated in around 20% of patients (and in 90% of those who die); some will regress (potentially with stenosis) but giant aneurysms (>80 mm) may require bypass surgery. Clinical factors that predict a higher risk of coronary artery arteritic lesions or aneurysms, or that predict a poor response to treatment, include age below 1 year, low serum albumin, low haemoglobin, high CRP, abnormal liver function and, especially, duration of fever before treatment. Peripheral blood eosinophilia (>4%) after treatment is also associated with treatment resistance. Early intravenous immunoglobulin (IVIg) reduces the coronary aneurysm risk from around 25% to less than 5%. Delaying IVIg beyond day 10 of fever increases the risk of death, particularly in boys under 1 year old.
Investigations
There are no diagnostic tests and Kawasaki disease remains a clinical diagnosis.
Management
Patients should be treated in a specialist paediatric unit. Aspirin and IVIg are the mainstay of treatment.
First line
Intravenous immunoglobulin and aspirin should be given early. Aspirin 100 mg/kg/day is given initially until the fever has settled and is then reduced to 3–5 mg/kg/day for 6–8 weeks in those with no cardiac abnormality, but longer in those with coronary aneurysms. All children should receive IVIg, usually given as a single dose of 2 g/kg over 12 h [4].
Second line
For children who remain febrile 36 h after the first dose of IVIg, a further dose of 2 g/kg can be given. Patients who are unresponsive to IVIg [14] can be treated with high-dose prednisolone 2 mg/kg/day, which should be tapered after normalization of the CRP [15].
LARGE-VESSEL VASCULITIS
Giant cell arteritis
Definition and nomenclature
Giant cell arteritis (GCA) is an arteritis, often granulomatous, usually affecting the aorta and/or its major branches, with a predilection for the branches of the carotid and vertebral arteries. The disease often involves the temporal artery. The onset is usually in patients older than 50 years and is often associated with polymyalgia rheumatica [1].
Introduction and general description
This is a disease of the elderly, often associated with polymyalgia rheumatica, which presents with headaches and tender palpable arteries, usually the temporal artery. It can rarely cause cutaneous infarction, so presenting to dermatologists [2].
Epidemiology
Incidence and prevalence
The highest mean annual incidence of GCA in people over the age of 50 is recorded at 32.8/100 000 in southern Norway [3]. The age-adjusted (>50 years) annual incidence per 100 000 population is 18.8 in Olmsted county, Minnesota, USA [4] and 22.0 in the UK [5].
Age
This affects people over the age of 50. The age-specific incidence per 100 000 population rises from 2.2 in the sixth decade to 51.9 in the ninth decade [4].
Sex
The disease is 2–3 times more common in women than men [4].
Ethnicity
Giant cell arteritis is considered a disease that particularly targets white people of northern European origin, being less common in African Americans, native Americans and Asians.
Associated diseases
Symptoms of polymyalgia rheumatica are commonly seen in GCA and it is hypothesized that the two conditions may have a similar aetiology, or lie on a disease spectrum.
Pathophysiology
The disease is thought to represent an inflammatory response in predisposed individuals towards an environmental factor. Local dendritic cells recruit and activate CD4 cells in the adventitia. Cytokine cascades involving Th1 and Th17 pathways dominate in the early phase, followed by a chronic smouldering arteritis led by chronic Th1 activation. The end result is a stenosing arteritis.
Predisposing factors
These are unknown.
Pathology
Lesions can be found in all layers of the affected branches of the aorta, particularly the carotid branches. These show segmental and focal pan-arteritis with polymorphic cell infiltrates along with T cells, macrophages and multinucleated giant cells, as well as intimal hyperplasia with a fragmented internal elastic lamina (Figure 102.26).
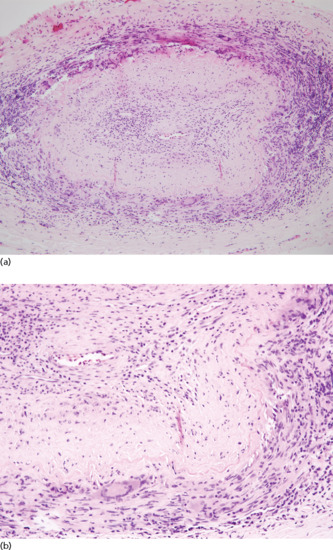
Figure 102.26 Giant cell arteritis at (a) lower and (b) higher magnifications. There are obliterative vascular changes with a lymphocytic and multinucleated giant cell reaction in the vessel wall. (Courtesy of Dr Laszlo Igali, Norfolk and Norwich University Hospital, Norwich, UK.)
Causative organisms
None known.
Genetics
There is no known genetic association.
Clinical features
History
Fever and weight loss may occur, and GCA is associated with polymyalgia rheumatica in about 50% of patients. Headache may be localized to the area of the affected artery, that is temporal with temporal arteritis and occipital with vertebrobasilar arteritis. The headache may start abruptly. Facial pain may occur on chewing due to claudication of the jaw muscles. Sudden, permanent visual loss related to ocular or orbital artery involvement may occur. Transient monocular loss of vision (amaurosis fugax) may precede permanent loss. Vertebrobasilar artery involvement may cause ataxia, vertigo or deafness.
Presentation
Patients may present with a new headache on the background of feeling generally unwell or with painless loss of vision. Very rarely, GCA may present with skin infarction. The temporal arteries may be tender, thickened and pulseless. A bruit may be heard over affected arteries (e.g. axillary).
Clinical variants
Isolated aortic inflammation without cranial artery involvement is well recognized.
Differential diagnosis
The temporal artery can be involved in other vasculitides, such as ANCA-associated vasculitides [6, 7]. Cancer should always be looked for when the diagnosis of GCA cannot be established beyond doubt [8].
Complications and co-morbidities
Permanent visual loss can be a presenting feature. There is a risk of aortic aneurysms developing as a late complication [9].
Disease course and prognosis
Following a diagnosis of GCA there is a slight excess mortality over 2 years (standard mortality rate 1.52; 95% confidence interval 1.20–1.85)), but not with longer follow-up [10]. The excess mortality was greater in women and in those aged ≤70 years.
Investigations
Histological diagnosis following biopsy of an affected artery, usually a temporal artery, is the gold standard for diagnosis. The biopsy should ideally comprise a 2 cm length of vessel and, for the greatest chance of obtaining a tissue diagnosis, the biopsy should be performed within 1–2 weeks of commencing corticosteroid therapy. A negative biopsy does not exclude the diagnosis because timing is important and the disease may have skip lesions. The ESR or CRP are almost always elevated. A normochromic, normocytic anaemia, thrombocytosis and raised alkaline phosphatase may all be present. Colour Doppler ultrasound of the temporal and axillary arteries (see Video 102.28 online at www.rookdermatology.com) in steroid-naive patients commonly reveals intramural inflammatory change – the halo sign (Figure 102.27) [11]. Positron emission tomography with 18-fluorodeoxyglucose is of value to demonstrate aortitis [12].

Figure 102.27 Colour Doppler ultrasound of the temporal artery demonstrating vessel wall oedema (the halo sign).
Management
Treatment should be started as soon as the diagnosis is suspected in order to avoid complications; if the diagnosis turns out to be incorrect, the corticosteroids can be withdrawn. Intravenous methylprednisolone 500–1000 mg daily for 3 days, followed by oral prednisolone, is commonly used in those with visual loss, although the visual loss is irreversible. Aspirin is recommended providing there are no contraindications.
First line
Corticosteroids, for example prednisolone 40–60 mg daily, are used for GCA [13]. The dose can usually be reduced slowly in small steps every month, providing that the CRP/ESR levels remain controlled. Treatment is usually for about 2 years [14].
Second line
There is some evidence of the steroid-sparing ability of methotrexate, which can be used in doses of 15–20 mg/week [15].
Takayasu arteritis
Definition and nomenclature
Takayasu arteritis is often granulomatous and predominantly affects the aorta and/or its major branches. The onset is usually in those younger than 50 years [1].
Introduction and general description
Makito Takayasu, a Japanese ophthalmologist, is credited with the eponym, but the earliest convincing clinical and pathological description in literature of this disease was provided by William Savory's account of a 22-year-old woman's 13-month hospital stay and postmortem examination that demonstrated widespread large arterial inflammation [2].
Epidemiology
Incidence and prevalence
The annual incidence of Takayasu arteritis in Europe and the USA is 0.5–2.5/million [3, 4, 5]. The incidence in Japan is believed to much higher [6].
Age
The disease is seen in younger people, typically below the age of 50, and is found also in children.
Sex
The disease is commoner in females than males.
Ethnicity
It is found in all populations but is commoner in Asians.
Pathophysiology
Pathology
The aorta and its branches are targeted and skip lesions can occur. During the acute phase, a pan-arteritis is present. The inflammatory infiltrate may be predominantly around the vasa vasorum, and fibrosis gradually replaces the inflammatory infiltrates. The vessel lumen may be narrowed secondary to the fibrosing stenotic lesions and/or by intraluminal thrombosis. In older patients there may be superimposed atherosclerosis, and calcification in the wall may occur as a late feature.
Genetics
IL-12B on chromosome 5, MLX on chromosome 17, FCGR2A/FCGR3A on chromosome 1 and HLA-B*52:01 are known associations [7, 8]. Two independent susceptibility loci have been identified in the HLA region (HLA-DQB1/HLA-DRB1 and HLA-B/MICA) [8].
Clinical features
History
Headache, malaise and fever are common presenting symptoms in children. Cutaneous lesions are present in around one-third of patients.
Presentation
Hypertension, pyrexia and pulseless disease are common findings in children. Skin lesions have been reported in up to a third of cases and may comprise erythema nodosum, erythema induratum and pyoderma gangrenosum, as well as ulcerated subacute nodular lesions, papulonecrotic eruptions, papular erythematous lesions of the hands and fingers, facial lupus-like rashes and panniculitis [9]. Cutaneous necrotizing vasculitis has been described resembling nodular vasculitis/erythema induratum. The skin lesions do not appear to relate to the distribution of vascular involvement in any way.
Complications and co-morbidities
Renal artery stenosis, increased arterial stiffness and increased sensitivity of the carotid sinus reflex all contribute to the hypertension. Involvement of the renal arteries can also cause renal dysfunction, and abdominal pain, bleeding or perforation may result from ischaemia or infarction of a viscus. Involvement of the aortic arch and its branches can lead to the ’aortic arch syndrome’ with arm claudication, absent radial or brachial pulses (hence ’pulseless disease’) or subclavian artery bruits. Aortic regurgitation, coronary artery ischaemia with angina or myocardial infarction, pulmonary hypertension, stroke, syncope and visual disturbances can occur.
Disease course and prognosis
Most patients with Takayasu arteritis will need vascular surgery [10], although restenosis is common [11]. The disease and its treatment both lead to an impairment in quality of life even for patients believed to be in remission [10].
Investigations
Positron emission tomography using 18-fluorodeoxyglucose has replaced conventional angiography as the gold standard for the diagnosis of Takayasu arteritis. However, due to the high radiation dose, magnetic resonance angiography could be used for follow-up monitoring. The ESR and CRP are usually elevated but this may be modest.
Management
There are no proven treatments in Takayasu arteritis.
First line
Prednisolone 1 mg/kg/day is the usual favoured first line treatment [12]. There is some evidence for the addition of azathioprine as an adjunct to corticosteroid therapy [13].
Second line
Cyclophosphamide, infliximab and tocilizumab have been reported to be of anecdotal value.
References
Single-organ small-vessel vasculitis
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
Cutaneous small-vessel vasculitis
- Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994;37(2):187–92.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Calabrese LH, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of hypersensitivity vasculitis. Arthritis Rheum 1990;33(8):1108–13.
- Garcia-Porrua C, Gonzalez-Gay MA. Comparative clinical and epidemiological study of hypersensitivity vasculitis versus Henoch-Schonlein purpura in adults. Semin Arthritis Rheum 1999;28(6):404–12.
- Watts RA, Jolliffe VA, Grattan CE, Elliott J, Lockwood M, Scott DG. Cutaneous vasculitis in a defined population – clinical and epidemiological associations. J Rheumatol 1998;25(5):920–4.
- Chua SH, Lim JT, Ang CB. Cutaneous vasculitis seen at a skin referral centre in Singapore. Singapore Med J 1999;40(3):147–50.
- Martinez-Taboada VM, Blanco R, Garcia-Fuentes M, Rodriguez-Valverde V. Clinical features and outcome of 95 patients with hypersensitivity vasculitis. Am J Med 1997;102(2):186–91.
- Boom BW, Mommaas AM, Vermeer BJ. Presence and interpretation of vascular immune deposits in human skin: the value of direct immunofluorescence. J Dermatol Sci 1992;3(1):26–34.
- Garcia-Porrua C, Gonzalez-Gay MA. Bacterial infection presenting as cutaneous vasculitis in adults. Clin Exp Rheumatol 1999;17(4):471–3.
- Cribier B, Couilliet D, Meyer P, Grosshans E. The severity of histopathological changes of leukocytoclastic vasculitis is not predictive of extracutaneous involvement. Am J Dermatopathol 1999;21(6):532–6.
- Mukhtyar C, Lee R, Brown D, et al. Modification and validation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis 2009;68(12):1827–32.
- Aouam K, Gassab A, Khorchani H, et al. Acenocoumarol and vasculitis: a case report. Pharmacoepidemiol Drug Saf 2007;16(1):113–14.
- Arbiser JL, Dzieczkowski JS, Harmon JV, Duncan LM. Leukocytoclastic vasculitis following staphylococcal protein A column immunoadsorption therapy. Two cases and a review of the literature. Arch Dermatol 1995;131(6):707–9.
- Bourelly PE, Grossman ME. Leukocytoclastic vasculitis following staphylococcal protein A column immunoadsorption therapy for idiopathic thrombocytopenic purpura. Cutis 1999;64(4):250–2.
- Deodhar A, Allen E, Daoud K, Wahba I. Vasculitis secondary to staphylococcal protein A immunoadsorption (Prosorba column) treatment in rheumatoid arthritis. Semin Arthritis Rheum 2002;32(1):3–9.
- Bucknall C, Darley C, Flax J, Vincent R, Chamberlain D. Vasculitis complicating treatment with intravenous anisoylated plasminogen streptokinase activator complex in acute myocardial infarction. Br Heart J 1988;59(1):9–11.
- Businco L, Falconieri P, Bellioni-Businco B, Bahna SL. Severe food-induced vasculitis in two children. Pediatr Allergy Immunol 2002;13(1):68–71.
- Casis FC, Perez JB. Leukocytoclastic vasculitis: a rare manifestation of propylthiouracil allergy. Endocr Pract 2000;6(4):329–32.
- Cohen BA, Greenberger PA, Saini S. Delayed occurrence of a severe cutaneous reaction in a multiple sclerosis patient taking interferon beta-1b. Allergy Asthma Proc 1998;19(2):85–8.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis 2001;67(4):303–7.
- Halevy S, Giryes H, Avinoach I, Livni E, Sukenik S. Leukocytoclastic vasculitis induced by low-dose methotrexate: in vitro evidence for an immunologic mechanism. J Eur Acad Dermatol Venereol 1998;10(1):81–5.
- Hsu CY, Chen WS, Sung SH. Warfarin-induced leukocytoclastic vasculitis: a case report and review of literature. Intern Med 2012;51(6):601–6.
- Jain KK. Cutaneous vasculitis associated with granulocyte colony-stimulating factor. J Am Acad Dermatol 1994;31(2 Pt 1):213–15.
- Knight D, Bangs MJ. Cutaneous allergic vasculitis due to Solenopsis geminata (Hymenoptera: Formicidae) envenomation in Indonesia. Southeast Asian J Trop Med Public Health 2007;38(5):808–13.
- Knox JP, Welykyj SE, Gradini R, Massa MC. Procainamide-induced urticarial vasculitis. Cutis 1988;42(5):469–72.
- McIlwain L, Carter JD, Bin-Sagheer S, Vasey FB, Nord J. Hypersensitivity vasculitis with leukocytoclastic vasculitis secondary to infliximab. J Clin Gastroenterol 2003;36(5):411–13.
- Oakley AM, Hodge L. Cutaneous vasculitis from maprotiline. Aust NZ J Med 1985;15(2):256–7.
- Odeh M, Lurie M, Oliven A. Cutaneous leucocytoclastic vasculitis associated with omeprazole. Postgrad Med J 2002;78(916):114–15.
- Prins M, Veraart JC, Vermeulen AH, Hulsmans RF, Neumann HA. Leucocytoclastic vasculitis induced by prolonged exercise. Br J Dermatol 1996;134(5):915–18.
- Rachid B, Rabelo-Santos M, Mansour E, de Lima Zollner R, Velloso LA. Type III hypersensitivity to insulin leading to leukocytoclastic vasculitis. Diabetes Res Clin Pract 2010;89(3):e39–40.
- Reiner MR, Brunetti VA. Imipenem-cilastatin-induced leukocytoclastic vasculitis. J Am Podiatr Med Assoc 1997;87(5):245–7.
- Sahin S, Comert A, Akin O, Ayalp S, Karsidag S. Cutaneous drug eruptions by current antiepileptics: case reports and alternative treatment options. Clin Neuropharmacol 2008;31(2):93–6.
- Tamir A, Wolf R, Brenner S. Leukocytoclastic vasculitis: another coumarin-induced hemorrhagic reaction. Acta Derm Venereol 1994;74(2):138–9.
- Wolf R, Ophir J, Elman M, Krakowski A. Atenolol-induced cutaneous vasculitis. Cutis 1989;43(3):231–3.
- Podjasek JO, Wetter DA, Pittelkow MR, Wada DA. Cutaneous small vessel vasculitis associated with solid organ malignancies: the Mayo Clinic experience, 1996 to 2009. J Am Acad Dermatol 2012;62(2): e55–65.
Erythema elevatum diutinum
- Tomasini C, Seia Z, Dapavo P, et al. Infantile erythema elevatum diutinum: report of a vesiculo-bullous case. Eur J Dermatol 2006;16:683–6.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum – a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol 1990;15:394–5.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum – an unusual association with ulcerative colitis. Clin Exp Dermatol 1991;16:204–6.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn's disease. J Am Acad Dermatol 1990;22:948–52.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol 1992;26:312–15.
- Planagumá M, Puig L, Alomar A, et al. Pyoderma gangrenosum in association with erythema elevatum diutinum: report of two cases. Cutis 1992;49:201–6.
- Cordier JF, Faure M, Hermier C, et al. Pleural effusions in an overlap syndrome of idiopathic hypereosinophilic syndrome and erythema elevatum diutinum. Eur Respir J 1990;3:115–18.
- Creus L, Salleras M, Sola MA, et al. Erythema elevatum diutinum associated with pulmonary infiltrate. Br J Dermatol 1997;137:652–3.
- Tasanen K, Raudasoja R, Kallioinen M, et al. Erythema elevatum diutinum in association with coeliac disease. Br J Dermatol 1997;136:624–7.
- Sangüeza OP, Pilcher B, Sangüeza JM. Erythema elevatum diutinum: a clinicopathological study of eight cases. Am J Dermatopathol 1997;19:214–22.
- Orteu C, McGregor JM, Whittaker SJ, et al. Erythema elevatum diutinum and Crohn disease: a common pathogenic role for measles virus? Arch Dermatol 1996;132:1523–5.
- Weidman FD, Bensacçon JH. Erythema elevatum diutinum. Role of streptococci and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol 1929;20:593–620.
- Wolff HH, Scherer R, Maciejewski W, et al. Erythema elevatum diutinum. II. Immunelekroneumikroskopische Untersuchung der leukocytocklastishew vaskulitis in einer Intrakutanreaktion mit Streptokokkenantigen. Arch Dermatol Res 1978;261:17–26.
- Cream JJ, Leven GM, Calnan CD. Erythema elevatum diutinum: an unusual reaction to streptococcal antigen and response to dapsone. Br J Dermatol 1971;84:393–9.
- Katz SI, Gallin JI, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestation, immunologic studies, and successful treatment with dapsone. Medicine 1977;56:443–55.
- Rover PA, Bittencourt C, Discacciati MP, et al. Erythema elevatum diutinum as a first clinical manifestation for diagnosing HIV infection: case history. Sao Paulo Med J 2005;123:201–3.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol 1992;26:38–44.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol 1986;8:392–402.
- Shanks JH, Banerjee SS, Bishop PW, et al. Nodular erythema elevatum diutinum mimicking cutaneous neoplasms. Histopathology 1997;31:91–6.
- Malone JC, Slone SP, Wills-Frank LA, et al. Vascular inflammation (vasculitis) in Sweet syndrome: a clinicopathologic study of 28 biopsy specimens from 21 patients. Arch Dermatol 2002;138:345–9.
- Cohen PR. Sweet's syndrome – a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis 2007;2:34.
- Cesinaro AM, Lonardi S, Faccheti F. Granuloma faciale: a cutaneous lesion sharing features with IgG4-associated sclerosing diseases. Am J Surg Pathol 2013;27:66–73.
- Ayoub N, Charuel J-L, Diemerte M-C, et al. Antineutrophil cytoplasmic antibodies of IgA class in neutrophilic dermatoses with emphasis on erythema elevatum diutinum. Arch Dermatol 2004;140:931–6.
- Chow RKP, Benny WB, Coupe RL, et al. Erythema elevatum diutinum associated with IgA paraproteinaemia successfully controlled with intermittent plasma exchange. Arch Dermatol 1996;132:1360–4.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol 1980;116:693–5.
- Frieling GW, Williams NL, Sim SJ. Novel use of topical 5% dapsone gel in erythema elevatum diutinum: safer and effective. J Drugs Dermatol 2013;12:381–4.
Recurrent cutaneous necrotizing eosinophilic vasculitis
- Chen K-R, Su WPD, Pittelkow MR, Leiferman KM. Eosinophilic vasculitis syndrome: recurrent cutaneous eosinophilic necrotizing vasculitis. Semin Dermatol 1995;14:106–10.
- Chen KR, Pittelkow MR, Su D, et al. Recurrent cutaneous eosinophilic necrotizing vasculitis: a novel eosinophil-mediated syndrome. Arch Dermatol 1994;130:1159–66.
- Chen K-R, Su WPD, Pittelkow MR, et al. Eosinophilic vasculitis in connective tissue disease. J Am Acad Dermatol 1996;35:173–82.
- Yomoda M, Inoue M, Nakama T, et al. Cutaneous eosinophilic vasculitis associated with rheumatoid arthritis. Br J Dermatol 1999;140:754–5.
- Jang KA, Lim YS, Choi JH, et al. Hypereosinophilic syndrome presenting as cutaneous necrotizing eosinophilic vasculitis and Raynaud's phenomenon complicated by digital gangrene. Br J Dermatol 2000;143:641–4.
- Kim SH, Kim TB, Yun YS, et al. Hypereosinophilia presenting as eosinophilic vasculitis and multiple peripheral artery occlusions without organ involvement. J Korean Med Sci 2005;20:677–9.
- Oppliger R, Gay-Crosier F, Dayer E, Hauser C. Digital necrosis in a patient with hypereosinophilic syndrome in the absence of cutaneous eosinophilic vasculitis. Br J Dermatol 2001;144:1087–90.
- Hamada T, Kimura Y, Hayashi S, et al. Hypereosinophilic syndrome with peripheral circulatory insufficiency and cutaneous microthrombi. Arch Dermatol 2007;143:812–13.
- Chen KR, Su WP, Pittelko MR, et al. Eosinophilic vasculitis in connective tissue disease. J Am Acad Dermatol 1996;35:173–82.
- Li W, Cao W, Song H, et al. Recurrent cutaneous necrotizing eosinophilc vasculitis: a case report and review of the literature. Diagn Pathol 2013;8:135.
Granuloma faciale
- Wigley JE. Eosinophilic granuloma. Sarcoid of Boeck. Proc R Soc Med 1945;38(3):125–6.
- Boersma D. Facial granuloma with eosinophilia (granuloma faciale). Am Med Assoc Arch Derm Syphilol 1951;63(4):520–1.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol 2004;51(2):269–73.
- Ortonne N, Wechsler J, Bagot M, Grosshans E, Cribier B. Granuloma faciale: a clinicopathologic study of 66 patients. J Am Acad Dermatol 2005;53(6):1002–9.
- Cesinaro AM, Lonardi S, Facchetti F. Granuloma faciale: a cutaneous lesion sharing features with IgG4-associated sclerosing diseases. Am J Surg Pathol 2013;37(1):66–73.
- Smoller BR, Bortz J. Immunophenotypic analysis suggests that granuloma faciale is a gamma-interferon-mediated process. J Cutan Pathol 1993;20(5):442–6.
- Ziemer M, Koehler MJ, Weyers W. Erythema elevatum diutinum – a chronic leukocytoclastic vasculitis microscopically indistinguishable from granuloma faciale? J Cutan Pathol 2011;38(11):876–83.
- Lallas A, Sidiropoulos T, Lefaki I, Tzellos T, Sotiriou E, Apalla Z. Photoletter to the editor: Dermoscopy of granuloma faciale. J Dermatol Case Rep 2012;6(2):59–60.
- Leite I, Moreira A, Guedes R, Furtado A, Ferreira EO, Baptista A. Granuloma faciale of the scalp. Dermatol Online J 2011;17(4):6.
- Nasiri S, Rahimi H, Farnaghi A, Asadi-Kani Z. Granuloma faciale with disseminated extra facial lesions. Dermatol Online J 2010;16(6):5.
- Rossiello L, Palla M, Aiello FS, Baroni A, Satriano RA. Granuloma faciale with extrafacial lesions. Skinmed 2007;6(3):150–1.
- Verma R, Das AL, Vaishampayan SS, Vaidya S. Keloidal granuloma faciale with extrafacial lesions. Indian J Dermatol Venereol Leprol 2005;71(5):345–7.
- Roberts PF, McCann BG. Eosinophilic angiocentric fibrosis of the upper respiratory tract: a mucosal variant of granuloma faciale? A report of three cases. Histopathology 1985;9(11):1217–25.
- Thompson LD, Heffner DK. Sinonasal tract eosinophilic angiocentric fibrosis. A report of three cases. Am J Clin Pathol 2001;115(2):243–8.
- Narayan J, Douglas-Jones AG. Eosinophilic angiocentric fibrosis and granuloma faciale: analysis of cellular infiltrate and review of literature. Ann Otol Rhinol Laryngol 2005;114(1 Pt 1):35–42.
- Jain R, Robblee JV, O'Sullivan-Mejia E, et al. Sinonasal eosinophilic angiocentric fibrosis: a report of four cases and review of literature. Head Neck Pathol 2008;2(4):309–15.
- Dinehart SM, Gross DJ, Davis CM, Herzberg AJ. Granuloma faciale. Comparison of different treatment modalities. Arch Otolaryngol Head Neck Surg 1990;116(7):849–51.
- Apfelberg DB, Maser MR, Lash H, Flores J. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol 1983;9(2):145–51.
- Ludwig E, Allam JP, Bieber T, Novak N. New treatment modalities for granuloma faciale. Br J Dermatol 2003;149(3):634–7.
- Zacarian SA. Cryosurgery effective for granuloma faciale. J Dermatol Surg Oncol 1985;11(1):11–13.
- Van de Kerkhof PC. On the efficacy of dapsone in granuloma faciale. Acta Derm Venereol 1994;74(1):61–2.
- Caldarola G, Zalaudek I, Argenziano G, Bisceglia M, Pellicano R. Granuloma faciale: a case report on long-term treatment with topical tacrolimus and dermoscopic aspects. Dermatol Ther 2011;24(5):508–11.
IgA vasculitis
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Rook A. William Heberden's cases of anaphylactoid purpura. Arch Dis Child 1958;33(169):271.
- Mills JA, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schonlein purpura. Arthritis Rheum 1990;33(8):1114–21.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis 2006;65(7):936–41.
- Penny K, Fleming M, Kazmierczak D, Thomas A. An epidemiological study of Henoch-Schonlein purpura. Paediatr Nurs Dec;22(10):30–5.
- Yang YH, Hung CF, Hsu CR, et al. A nationwide survey on epidemiological characteristics of childhood Henoch-Schonlein purpura in Taiwan. Rheumatology (Oxford) 2005;44(5):618–22.
- Dolezalova P, Telekesova P, Nemcova D, Hoza J. Incidence of vasculitis in children in the Czech Republic: 2-year prospective epidemiology survey. J Rheumatol 2004;31(11):2295–9.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002;360(9341):1197–202.
- Garcia-Porrua C, Gonzalez-Gay MA. Comparative clinical and epidemiological study of hypersensitivity vasculitis versus Henoch-Schonlein purpura in adults. Semin Arthritis Rheum 1999;28(6):404–12.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995;25(1):28–34.
- Kawasaki Y. The pathogenesis and treatment of pediatric Henoch-Schonlein purpura nephritis. Clin Exp Nephrol Oct;15(5):648–57.
- Jauhola O, Ronkainen J, Koskimies O, et al. Renal manifestations of Henoch-Schonlein purpura in a 6-month prospective study of 223 children. Arch Dis Child Nov;95(11):877–82.
- Trapani S, Micheli A, Grisolia F, et al. Henoch Schonlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and review of literature. Semin Arthritis Rheum 2005;35(3):143–53.
- Smith SM, Tung KS. Incidence of IgA-related nephritides in American Indians in New Mexico. Hum Pathol 1985;16(2):181–4.
- Calvo-Rio V, Loricera J, Martin L, et al. Henoch-Schonlein purpura nephritis and IgA nephropathy: a comparative clinical study. Clin Exp Rheumatol 2013;31(1 Suppl 75):S45–51.
- Altug U, Ensari C, Sayin DB, Ensari A. MEFV gene mutations in Henoch-Schonlein purpura. Int J Rheum Dis Jun;16(3):347–51.
- Bayram C, Demircin G, Erdogan O, Bulbul M, Caltik A, Akyuz SG. Prevalence of MEFV gene mutations and their clinical correlations in Turkish children with Henoch-Schonlein purpura. Acta Paediatr 2011;100(5):745–9.
- Flatau E, Kohn D, Schiller D, Lurie M, Levy E. Schonlein-Henoch syndrome in patients with familial Mediterranean fever. Arthritis Rheum 1982;25(1):42–7.
- Ozdogan H, Arisoy N, Kasapcapur O, et al. Vasculitis in familial Mediterranean fever. J Rheumatol 1997;24(2):323–7.
- Atkinson SR, Barker DJ. Seasonal distribution of Henoch-Schonlein purpura. Br J Prev Soc Med 1976;30(1):22–5.
- Chen O, Zhu XB, Ren P, Wang YB, Sun RP, Wei DE. Henoch Schonlein purpura in children: clinical analysis of 120 cases. Afr Health Sci 2013;13(1):94–9.
- Singh S, Aulakh R. Kawasaki disease and Henoch Schonlein purpura: changing trends at a tertiary care hospital in north India (1993–2008). Rheumatol Int 2010;30(6):771–4.
- Spyridis N, Salapata M, Fessatou S, Kontaxaki C, Spyridis P. Simultaneous presentation of Henoch Schonlein purpura in monozygotic twins. Scand J Infect Dis 2005;37(9):703–4.
- Al-Sheyyab M, el-Shanti H, Ajlouni S, Batieha A, Daoud AS. Henoch-Schonlein purpura: clinical experience and contemplations on a streptococcal association. J Trop Pediatr 1996;42(4):200–3.
- Al-Sheyyab M, Batieha A, el-Shanti H, Daoud A. Henoch-Schonlein purpura and streptococcal infection: a prospective case-control study. Ann Trop Paediatr 1999;19(3):253–5.
- Yang YH, Chuang YH, Wang LC, Huang HY, Gershwin ME, Chiang BL. The immunobiology of Henoch-Schonlein purpura. Autoimmun Rev 2008;7(3):179–84.
- Egan CA, Taylor TB, Meyer LJ, Petersen MJ, Zone JJ. IgA1 is the major IgA subclass in cutaneous blood vessels in Henoch-Schonlein purpura. Br J Dermatol 1999;141(5):859–62.
- Saulsbury FT. Henoch-Schonlein purpura. Curr Opin Rheumatol 2001;13(1):35–40.
- Ogura Y, Suzuki S, Shirakawa T, et al. Haemophilus parainfluenzae antigen and antibody in children with IgA nephropathy and Henoch-Schonlein nephritis. Am J Kidney Dis 2000;36(1):47–52.
- Ayoub EM, McBride J, Schmiederer M, Anderson B. Role of Bartonella henselae in the etiology of Henoch-Schonlein purpura. Pediatr Infect Dis J 2002;21(1):28–31.
- Novak J, Szekanecz Z, Sebesi J, et al. Elevated levels of anti-Helicobacter pylori antibodies in Henoch-Schonlein purpura. Autoimmunity 2003;36(5):307–11.
- Peru H, Soylemezoglu O, Gonen S, et al. HLA class 1 associations in Henoch Schonlein purpura: increased and decreased frequencies. Clin Rheumatol 2008;27(1):5–10.
- Demirkaya E, Ozen S, Pistorio A, et al. Performance of Birmingham Vasculitis Activity Score and disease extent index in childhood vasculitides. Clin Exp Rheumatol 2012;30(1 Suppl 70):S162–8.
- Samuel JP, Bell CS, Molony DA, Braun MC. Long-term outcome of renal transplantation patients with Henoch-Schonlein purpura. Clin J Am Soc Nephrol 2011;6(8):2034–40.
- Deng F, Lu L, Zhang Q, Hu B, Wang SJ, Huang N. Henoch-Schonlein purpura in childhood: treatment and prognosis. Analysis of 425 cases over a 5-year period. Clin Rheumatol 2010;29(4):369–74.
- Ronkainen J, Nuutinen M, Koskimies O. The adult kidney 24 years after childhood Henoch-Schonlein purpura: a retrospective cohort study. Lancet 2002;360(9334):666–70.
- Zaffanello M, Brugnara M, Franchini M. Therapy for children with Henoch-Schonlein purpura nephritis: a systematic review. Sci World J 2007;7:20–30.
- Ronkainen J, Koskimies O, Ala-Houhala M, et al. Early prednisone therapy in Henoch-Schonlein purpura: a randomized, double-blind, placebo-controlled trial. J Pediatr 2006;149(2):241–7.
Cryoglobulinaemic vasculitis
- Terrier B, Cacoub P. Cryoglobulinemia vasculitis: an update. Curr Opin Rheumatol 2013;25(1):10–18.
- Boyer O, Saadoun D, Abriol J, et al. CD4+CD25+ regulatory T-cell deficiency in patients with hepatitis C-mixed cryoglobulinemia vasculitis. Blood 2004;103(9):3428–30.
- Saadoun D, Delluc A, Piette JC, Cacoub P. Treatment of hepatitis C-associated mixed cryoglobulinemia vasculitis. Curr Opin Rheumatol 2008;20(1):23–8.
- Monti G, Galli M, Invernizzi F, et al. Cryoglobulinaemias: a multi-centre study of the early clinical and laboratory manifestations of primary and secondary disease. GISC. Italian Group for the Study of Cryoglobulinaemias. QJM 1995;88(2):115–26.
- Barbiano di Belgiojoso G, Bertoli S, Tarantino A, et al. Renal lesions in essential mixed IgG-IgM cryoglobulinemia. Study of 48 cases. Boll Ist Sieroter Milan 1981;60(4):316–27.
- Beddhu S, Bastacky S, Johnson JP. The clinical and morphologic spectrum of renal cryoglobulinemia. Medicine (Baltimore) 2002;81(5):398–409.
- Invernizzi F, Galli M, Serino G, et al. Secondary and essential cryoglobulinemias. Frequency, nosological classification, and long-term follow-up. Acta Haematol 1983;70(2):73–82.
- Saadoun D, Sellam J, Ghillani-Dalbin P, Crecel R, Piette JC, Cacoub P. Increased risks of lymphoma and death among patients with non-hepatitis C virus-related mixed cryoglobulinemia. Arch Intern Med 2006;166(19):2101–8.
- Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 2009;68(3):310–17.
- De Vita S, Quartuccio L, Isola M, et al. A randomized controlled trial of rituximab for the treatment of severe cryoglobulinemic vasculitis. Arthritis Rheum 2012;64(3):843–53.
Hypocomplementaemic urticarial vasculitis
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Dincy CV, George R, Jacob M, Mathai E, Pulimood S, Eapen EP. Clinicopathologic profile of normocomplementemic and hypocomplementemic urticarial vasculitis: a study from South India. J Eur Acad Dermatol Venereol 2008;22(7):789–94.
- Davis MD, Daoud MS, Kirby B, Gibson LE, Rogers RS, 3rd. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J Am Acad Dermatol 1998;38(6 Pt 1):899–905.
- Wisnieski JJ, Naff GB. Serum IgG antibodies to C1q in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum 1989;32(9):1119–27.
- Wisnieski JJ, Jones SM. IgG autoantibody to the collagen-like region of Clq in hypocomplementemic urticarial vasculitis syndrome, systemic lupus erythematosus, and 6 other musculoskeletal or rheumatic diseases. J Rheumatol 1992;19(6):884–8.
- Jones RR, Bhogal B, Dash A, Schifferli J. Urticaria and vasculitis: a continuum of histological and immunopathological changes. Br J Dermatol 1983;108(6):695–703.
- Worm M, Muche M, Schulze P, Sterry W, Kolde G. Hypocomplementaemic urticarial vasculitis: successful treatment with cyclophosphamide-dexamethasone pulse therapy. Br J Dermatol 1998;139(4):704–7.
- Stack PS. Methotrexate for urticarial vasculitis. Ann Allergy 1994;72(1):36–8.
- Cadnapaphornchai MA, Saulsbury FT, Norwood VF. Hypocomplementemic urticarial vasculitis: report of a pediatric case. Pediatr Nephrol 2000;14(4):328–31.
- Ashida A, Murata H, Ohashi A, Ogawa E, Uhara H, Okuyama R. A case of hypocomplementaemic urticarial vasculitis with a high serum level of rheumatoid factor. Australas J Dermatol 2013;54(3):e62–3.
- Lopez LR, Davis KC, Kohler PF, Schocket AL. The hypocomplementemic urticarial-vasculitis syndrome: therapeutic response to hydroxychloroquine. J Allergy Clin Immunol 1984;73(5 Pt 1):600–3.
- Enriquez R, Sirvent AE, Amoros F, Perez M, Matarredona J, Reyes A. Crescentic membranoproliferative glomerulonephritis and hypocomplementemic urticarial vasculitis. J Nephrol 2005;18(3):318–22.
Antiglomerular basement membrane vasculitis disease
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Goodpasture EW. Landmark publication from the American Journal of the Medical Sciences: the significance of certain pulmonary lesions in relation to the etiology of influenza. Am J Med Sci 2009;338(2):148–51.
- Ross JB, Cohen AD, Ghose T. Goodpasture's syndrome associated with skin involvement. Arch Dermatol 1985;121(11):1442–4.
- Li FK, Tse KC, Lam MF, et al. Incidence and outcome of antiglomerular basement membrane disease in Chinese. Nephrology (Carlton) 2004;9(2):100–4.
- Williamson SR, Phillips CL, Andreoli SP, Nailescu C. A 25-year experience with pediatric anti-glomerular basement membrane disease. Pediatr Nephrol 2011;26(1):85–91.
- Cui Z, Zhao MH, Xin G, Wang HY. Characteristics and prognosis of Chinese patients with anti-glomerular basement membrane disease. Nephron Clin Pract 2005;99(2):c49–55.
- Bayat A, Kamperis K, Herlin T. Characteristics and outcome of Goodpasture's disease in children. Clin Rheumatol 2012;31(12):1745–51.
- Cui Z, Zhao J, Jia XY, Zhu SN, Zhao MH. Clinical features and outcomes of anti-glomerular basement membrane disease in older patients. Am J Kidney Dis 2011;57(4):575–82.
- Turner N, Mason PJ, Brown R, et al. Molecular cloning of the human Goodpasture antigen demonstrates it to be the alpha 3 chain of type IV collagen. J Clin Invest 1992;89(2):592–601.
- D'Apice AJ, Kincaid-Smith P, Becker GH, Loughhead MG, Freeman JW, Sands JM. Goodpasture's syndrome in identical twins. Ann Intern Med 1978;88(1):61–2.
- Levy JB, Turner AN, Rees AJ, Pusey CD. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med 2001;134(11):1033–42.
Microscopic polyangiitis
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994;37(2):187–92.
- Wohlwill F. Über die nur mikroskopisch erkennbare Form der Periarteriitis nodosa. Virchows Arch Path Anat 1923;246(1):377–411.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995;25(1):28–34.
- Mohammad AJ, Jacobsson LT, Westman KW, Sturfelt G, Segelmark M. Incidence and survival rates in Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology (Oxford) 2009;48(12):1560–5.
- Fujimoto S, Uezono S, Hisanaga S, et al. Incidence of ANCA-associated primary renal vasculitis in the Miyazaki Prefecture: the first population-based, retrospective, epidemiologic survey in Japan. Clin J Am Soc Nephrol 2006;1(5):1016–22.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004;51(1):92–9.
- Mohammad AJ, Jacobsson LT, Mahr AD, Sturfelt G, Segelmark M. Prevalence of Wegener's granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford) 2007;46(8):1329–37.
- Gibson A, Stamp LK, Chapman PT, O'Donnell JL. The epidemiology of Wegener's granulomatosis and microscopic polyangiitis in a southern hemisphere region. Rheumatology (Oxford) 2006;45(5):624–8.
- Lane SE, Watts R, Scott DG. Epidemiology of systemic vasculitis. Curr Rheumatol Rep 2005;7(4):270–5.
- Heeringa P, Foucher P, Klok PA, et al. Systemic injection of products of activated neutrophils and H2O2 in myeloperoxidase-immunized rats leads to necrotizing vasculitis in the lungs and gut. Am J Pathol 1997;151(1):131–40.
- Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 2002;110(7):955–63.
- Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA 1990;87(11):4115–19.
- Hewins P, Williams JM, Wakelam MJ, Savage CO. Activation of Syk in neutrophils by antineutrophil cytoplasm antibodies occurs via Fcgamma receptors and CD18. J Am Soc Nephrol 2004;15(3):796–808.
- Lane SE, Watts RA, Bentham G, Innes NJ, Scott DG. Are environmental factors important in primary systemic vasculitis? A case-control study. Arthritis Rheum 2003;48(3):814–23.
- Guillevin L, Durand-Gasselin B, Cevallos R, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42(3):421–30.
- Watts R, Lane S, Hanslik T, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis 2007;66(2):222–7.
- Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 2009;68(3):310–17.
- Akikusa B, Sato T, Ogawa M, Ueda S, Kondo Y. Necrotizing alveolar capillaritis in autopsy cases of microscopic polyangiitis. Incidence, histopathogenesis, and relationship with systemic vasculitis. Arch Pathol Lab Med 1997;121(2):144–9.
- Lauque D, Cadranel J, Lazor R, et al. Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Groupe d'Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Medicine (Baltimore) 2000;79(4):222–33.
- Suppiah R, Judge A, Batra R,, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener's granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011;63(4):588–96.
- Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003;349(1):36–44.
- Ntatsaki E, Carruthers D, Chakravarty K, et al. BSR and BHPR guideline for the management of adults with ANCA-associated vasculitis. Rheumatology (Oxford) 2014;53(12):2306–9.
- Harper L, Morgan MD, Walsh M, et al. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis 2012;71(6):955–60.
- De Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009;150(10):670–80.
- Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 2007;18(7):2180–8.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010;363(3):221–32.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010;363(3):211–20.
- Jones RB, Ferraro AJ, Chaudhry AN, et al. A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2009;60(7):2156–68.
- Smith RM, Jones RB, Guerry MJ, et al. Rituximab for remission maintenance in relapsing antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2012;64(11):3760–9.
- Rhee EP, Laliberte KA, Niles JL. Rituximab as maintenance therapy for anti-neutrophil cytoplasmic antibody-associated vasculitis. Clin J Am Soc Nephrol 2010;5(8):1394–400.
Granulomatosis with polyangiitis
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Wegener F. On generalised septic vessel diseases. By Friedrich Wegener, 1937 (translation). Thorax 1987;42(12):918–19.
- Johnsson S. A case of Wegener's granulomatosis. Acta Pathol Microbiol Scand 1948;25(5):573–84.
- Gonzalez-Gay MA, Garcia-Porrua C, Guerrero J, Rodriguez-Ledo P, Llorca J. The epidemiology of the primary systemic vasculitides in northwest Spain: implications of the Chapel Hill Consensus Conference definitions. Arthritis Rheum 2003;49(3):388–93.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995;25(1):28–34.
- Watts RA, Gonzalez-Gay MA, Lane SE, Garcia-Porrua C, Bentham G, Scott DG. Geoepidemiology of systemic vasculitis: comparison of the incidence in two regions of Europe. Ann Rheum Dis 2001;60(2):170–2.
- Watts RA, Mooney J, Skinner J, Scott DG, Macgregor AJ. The contrasting epidemiology of granulomatosis with polyangiitis (Wegener's) and microscopic polyangiitis. Rheumatology (Oxford) 2012;51(5):926–31.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004;51(1):92–9.
- Gibson A, Stamp LK, Chapman PT, O'Donnell JL. The epidemiology of Wegener's granulomatosis and microscopic polyangiitis in a southern hemisphere region. Rheumatology (Oxford) 2006;45(5):624–8.
- Watts RA, Al-Taiar A, Scott DG, Macgregor AJ. Prevalence and incidence of Wegener's granulomatosis in the UK general practice research database. Arthritis Rheum 2009;61(10):1412–16.
- Akikusa JD, Schneider R, Harvey EA, et al. Clinical features and outcome of pediatric Wegener's granulomatosis. Arthritis Rheum 2007;57(5):837–44.
- Cabral DA, Uribe AG, Benseler S, et al. Classification, presentation, and initial treatment of Wegener's granulomatosis in childhood. Arthritis Rheum 2009;60(11):3413–24.
- Koldingsnes W, Nossent H. Epidemiology of Wegener's granulomatosis in northern Norway. Arthritis Rheum 2000;43(11):2481–7.
- Seck SM, Dussol B, Brunet P, Burtey S. Clinical features and outcomes of ANCA-associated renal vasculitis. Saudi J Kidney Dis Transpl 2012;23(2):301–5.
- Fujimoto S, Watts RA, Kobayashi S, et al. Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the UK. Rheumatology (Oxford) 2011;50(10):1916–20.
- Faurschou M, Helleberg M, Obel N, Baslund B. Incidence of granulomatosis with polyangiitis (Wegener's) in Greenland and the Faroe Islands: epidemiology of an ANCA-associated vasculitic syndrome in two ethnically distinct populations in the North Atlantic area. Clin Exp Rheumatol 2013;31(1 Suppl. 75):S52–5.
- Heeringa P, Foucher P, Klok PA, et al. Systemic injection of products of activated neutrophils and H2O2 in myeloperoxidase-immunized rats leads to necrotizing vasculitis in the lungs and gut. Am J Pathol 1997;151(1):131–40.
- Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 2002;110(7):955–63.
- Pfister H, Ollert M, Frohlich LF, et al. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood 2004;104(5):1411–18.
- Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA 1990;87(11):4115–19.
- Hewins P, Williams JM, Wakelam MJ, Savage CO. Activation of Syk in neutrophils by antineutrophil cytoplasm antibodies occurs via Fcgamma receptors and CD18. J Am Soc Nephrol 2004;15(3):796–808.
- Popa ER, Stegeman CA, Bos NA, Kallenberg CG, Tervaert JW. Staphylococcal superantigens and T cell expansions in Wegener's granulomatosis. Clin Exp Immunol 2003;132(3):496–504.
- Voswinkel J, Muller A, Lamprecht P. Is PR3-ANCA formation initiated in Wegener's granulomatosis lesions? Granulomas as potential lymphoid tissue maintaining autoantibody production. Ann NY Acad Sci 2005;1051:12–19.
- Braun MG, Csernok E, Gross WL, Muller-Hermelink HK. Proteinase 3, the target antigen of anticytoplasmic antibodies circulating in Wegener's granulomatosis. Immunolocalization in normal and pathologic tissues. Am J Pathol 1991;139(4):831–8.
- Lane SE, Watts RA, Bentham G, Innes NJ, Scott DG. Are environmental factors important in primary systemic vasculitis? A case-control study. Arthritis Rheum 2003;48(3):814–23.
- Popa ER, Tervaert JW. The relation between Staphylococcus aureus and Wegener's granulomatosis: current knowledge and future directions. Intern Med 2003;42(9):771–80.
- Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med 1994;120(1):12–17.
- Jagiello P, Gencik M, Arning L, et al. New genomic region for Wegener's granulomatosis as revealed by an extended association screen with 202 apoptosis-related genes. Hum Genet 2004;114(5):468–77.
- Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 2012;367(3):214–23.
- Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 2009;68(3):310–17.
- Faurschou M, Sorensen IJ, Mellemkjaer L, et al. Malignancies in Wegener's granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients. J Rheumatol 2008;35(1):100–5.
- Knight A, Askling J, Ekbom A. Cancer incidence in a population-based cohort of patients with Wegener's granulomatosis. Int J Cancer 2002;100(1):82–5.
- Faurschou M, Mellemkjaer L, Sorensen IJ, Svalgaard Thomsen B, Dreyer L, Baslund B. Increased morbidity from ischemic heart disease in patients with Wegener's granulomatosis. Arthritis Rheum 2009;60(4):1187–92.
- Mukhtyar C, Flossmann O, Hellmich B, et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008;67(7):1004–10.
- Mukhtyar C, Hellmich B, Jayne D, Flossmann O, Luqmani R. Remission in antineutrophil cytoplasmic antibody-associated systemic vasculitis. Clin Exp Rheumatol 2006;24(6 Suppl. 43):S93–8.
- Koldingsnes W, Nossent H. Predictors of survival and organ damage in Wegener's granulomatosis. Rheumatology (Oxford) 2002;41(5):572–81.
- Ntatsaki E, Carruthers D, Chakravarty K, et al. BSR and BHPR guideline for the management of adults with ANCA-associated vasculitis. Rheumatology (Oxford) 2014;53(12):2306–9.
- De Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005;52(8):2461–9.
- Faurschou M, Westman K, Rasmussen N, et al. Brief report: long-term outcome of a randomized clinical trial comparing methotrexate to cyclophosphamide for remission induction in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2012;64(10):3472–7.
- Harper L, Morgan MD, Walsh M, et al. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis 2012;71(6):955–60.
- De Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009;150(10):670–80.
- Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 2007;18(7):2180–8.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010;363(3):221–32.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010;363(3):211–20.
- Jones RB, Ferraro AJ, Chaudhry AN, et al. A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2009;60(7):2156–68.
- Flossmann O, Baslund B, Bruchfeld A, et al. Deoxyspergualin in relapsing and refractory Wegener's granulomatosis. Ann Rheum Dis 2009;68(7):1125–30.
- Jayne DR, Chapel H, Adu D, et al. Intravenous immunoglobulin for ANCA-associated systemic vasculitis with persistent disease activity. Q J Med 2000;93(7):433–9.
- Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003;349(1):36–44.
- Pagnoux C, Mahr A, Hamidou MA, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008;359(26):2790–803.
- Metzler C, Miehle N, Manger K, et al. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener's granulomatosis. Rheumatology (Oxford) 2007;46(7):1087–91.
- Hiemstra TF, Walsh M, Mahr A, et al. Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA 2010;304(21):2381–8.
- Smith RM, Jones RB, Guerry MJ, et al. Rituximab for remission maintenance in relapsing antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2012;64(11):3760–9.
- Rhee EP, Laliberte KA, Niles JL. Rituximab as maintenance therapy for anti-neutrophil cytoplasmic antibody-associated vasculitis. Clin J Am Soc Nephrol 2010;5(8):1394–400.
Eosinophilic granulomatosis with polyangiitis
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol 1951;27(2):277–301.
- Lane SE, Scott DG, Heaton A, Watts RA. Primary renal vasculitis in Norfolk – increasing incidence or increasing recognition? Nephrol Dial Transplant 2000;15(1):23–7.
- Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995;25(1):28–34.
- Ormerod AS, Cook MC. Epidemiology of primary systemic vasculitis in the Australian Capital Territory and south-eastern New South Wales. Intern Med J 2008;38(11):816–23.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004;51(1):92–9.
- Mohammad AJ, Jacobsson LT, Mahr AD, Sturfelt G, Segelmark M. Prevalence of Wegener's granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford) 2007;46(8):1329–37.
- Loughlin JE, Cole JA, Rothman KJ, Johnson ES. Prevalence of serious eosinophilia and incidence of Churg-Strauss syndrome in a cohort of asthma patients. Ann Allergy Asthma Immunol 2002;88(3):319–25.
- Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy 2013;68(3):261–73.
- Kiene M, Csernok E, Muller A, Metzler C, Trabandt A, Gross WL. Elevated interleukin-4 and interleukin-13 production by T cell lines from patients with Churg-Strauss syndrome. Arthritis Rheum 2001;44(2):469–73.
- Jakiela B, Sanak M, Szczeklik W, et al. Both Th2 and Th17 responses are involved in the pathogenesis of Churg-Strauss syndrome. Clin Exp Rheumatol 2011;29(1 Suppl. 64):S23–34.
- Drage LA, Davis MD, De Castro F, et al. Evidence for pathogenic involvementof eosinophils and neutrophilsin Churg-Strauss syndrome. J Am Acad Dermatol 2002;47(2):209–16.
- Schnabel A, Csernok E, Braun J, Gross WL. Inflammatory cells and cellular activation in the lower respiratory tract in Churg-Strauss syndrome. Thorax 1999;54(9):771–8.
- Davis MD, Daoud MS, McEvoy MT, Su WP. Cutaneous manifestations of Churg-Strauss syndrome: a clinicopathologic correlation. J Am Acad Dermatol 1997;37(2 Pt 1):199–203.
- Vaglio A, Martorana D, Maggiore U, et al. HLA-DRB4 as a genetic risk factor for Churg-Strauss syndrome. Arthritis Rheum 2007;56(9):3159–66.
- Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990;33(8):1094–100.
- Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) 1984;63(2):65–81.
- Watts R, Lane S, Hanslik T, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis 2007;66(2):222–7.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999;78(1):26–37.
- Mukhtyar C, Flossmann O, Hellmich B, et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008;67(7):1004–10.
- Sinico RA, Di Toma L, Maggiore U, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum 2005;52(9):2926–35.
- Sable-Fourtassou R, Cohen P, Mahr A, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005;143(9):632–8.
- Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 2009;68(3):310–17.
- Ribi C, Cohen P, Pagnoux C, et al. Treatment of Churg-Strauss syndrome without poor-prognosis factors: a multicenter, prospective, randomized, open-label study of seventy-two patients. Arthritis Rheum 2008;58(2):586–94.
Polyarteritis nodosa and cutaneous polyarteritis nodosa
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Daoud MS, Hutton KP, Gibson LE. Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol 1997;136(5):706–13.
- Anon. [Case history: polyarteritis nodosa with massive involvement of the kidney masqueraded as acute glomerular nephritis.] Calif Med 1945;65(4):186–8.
- Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994;37(2):187–92.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathological analysis and a review of the published work. J Dermatol;37(1):85–93.
- Mohammad AJ, Jacobsson LT, Westman KW, Sturfelt G, Segelmark M. Incidence and survival rates in Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology (Oxford) 2009;48(12):1560–5.
- Watts RA, Jolliffe VA, Carruthers DM, Lockwood M, Scott DG. Effect of classification on the incidence of polyarteritis nodosa and microscopic polyangiitis. Arthritis Rheum 1996;39(7):1208–12.
- Mohammad AJ, Jacobsson LT, Mahr AD, Sturfelt G, Segelmark M. Prevalence of Wegener's granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford) 2007;46(8):1329–37.
- Mahr A, Guillevin L, Poissonnet M, Ayme S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum 2004;51(1):92–9.
- Trepo C, Thivolet J. [Australia antigen, virus hepatitis and periarteritis nodosa.] Presse Med 1970;78(36):1575.
- Darras-Joly C, Lortholary O, Cohen P, Brauner M, Guillevin L. Regressing microaneurysms in 5 cases of hepatitis B virus related polyarteritis nodosa. J Rheumatol 1995;22(5):876–80.
- Caldeira T, Meireles C, Cunha F, Valbuena C, Aparicio J, Ribeiro A. Systemic polyarteritis nodosa associated with acute Epstein-Barr virus infection. Clin Rheumatol 2007;26(10):1733–5.
- Chiche L, Jean R, Cretel E, Figuarella-Branger D, Durand JM. [Central nervous system HIV-associated polyarteritis nodosa: long-term outcome.] Rev Med Interne 2006;27(8):625–8.
- Viguier M, Guillevin L, Laroche L. Treatment of parvovirus B19-associated polyarteritis nodosa with intravenous immune globulin. N Engl J Med 2001;344(19):1481–2.
- Doherty M, Bradfield JW. Polyarteritis nodosa associated with acute cytomegalovirus infection. Ann Rheum Dis 1981;40(4):419–21.
- David J, Ansell BM, Woo P. Polyarteritis nodosa associated with streptococcus. Arch Dis Child 1993;69(6):685–8.
- Corbeel L, Gewillig M, Baeten E, Casteels-Van Daele M, Eggermont E. Carotid and coronary artery involvement in infantile periarteritis nodosa possibly induced by coxsackie B4 infection. Favourable course under corticosteroid treatment. Eur J Pediatr 1987;146(4):441–2.
- Mader R, Schaffer I, Schonfeld S. Recurrent poststreptococcal cutaneous polyarteritis nodosa. Isr J Med Sci 1988;24(4-5):269–70.
- Durst R, Goldschmidt N, Ben Yehuda A. Parvovirus B19 infection associated with myelosuppression and cutaneous polyarteritis nodosa. Rheumatology (Oxford) 2002;41(10):1210–12.
- Chen HH, Hsiao CH, Chiu HC. Successive development of cutaneous polyarteritis nodosa, leucocytoclastic vasculitis and Sweet's syndrome in a patient with cervical lymphadenitis caused by Mycobacterium fortuitum. Br J Dermatol 2004;151(5):1096–100.
- Kermani TA, Ham EK, Camilleri MJ, Warrington KJ. Polyarteritis nodosa-like vasculitis in association with minocycline use: a single-center case series. Semin Arthritis Rheum;42(2):213–21.
- Tehrani R, Nash-Goelitz A, Adams E, Dahiya M, Eilers D. Minocycline-induced cutaneous polyarteritis nodosa. J Clin Rheumatol 2007;13(3):146–9.
- Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med;370(10):911–20.
- Navon Elkan P, Pierce SB, Segel R, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 2014;370(10):921–31.
- Watts R, Lane S, Hanslik T, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis 2007;66(2):222–7.
- Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996;75(1):17–28.
- Jorizzo JL, White WL, Wise CM, Zanolli MD, Sherertz EF. Low-dose weekly methotrexate for unusual neutrophilic vascular reactions: cutaneous polyarteritis nodosa and Behcet's disease. J Am Acad Dermatol 1991;24(6 Pt 1):973–8.
- Tursen U, Api H, Kaya TI, Cinel L, Ikizoglu G. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony-stimulating factor therapy in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol 2006;20(10):1341–3.
- Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 2009;68(3):310–17.
- Krishnan S, Bhakuni DS, Kartik S. Rituximab in refractory cutaneous polyarteritis. Int J Rheum Dis 2012;15(5):e127.
- Sonomoto K, Miyamura T, Watanabe H, et al. [A case of polyarteritis nodosa successfully treated by rituximab.] Nihon Rinsho Meneki Gakkai Kaishi 2008;31(2):119–23.
Kawasaki disease
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Kato H, Koike S, Yamamoto M, Ito Y, Yano E. Coronary aneurysms in infants and young children with acute febrile mucocutaneous lymph node syndrome. J Pediatr 1975;86(6):892–8.
- Baumer JH, Love SJ, Gupta A, Haines LC, Maconochie I, Dua JS. Salicylate for the treatment of Kawasaki disease in children. Cochrane Database Syst Rev 2006;Issue 4:CD004175.
- Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics 2004;114(6):1708–33.
- Harnden A, Mayon-White R, Perera R, Yeates D, Goldacre M, Burgner D. Kawasaki disease in England: ethnicity, deprivation, and respiratory pathogens. Pediatr Infect Dis J 2009;28(1):21–4.
- Nakamura Y, Yashiro M, Uehara R, et al. Epidemiologic features of Kawasaki disease in Japan: results of the 2009–2010 nationwide survey. J Epidemiol 2012;22(3):216–21.
- Onouchi Y, Gunji T, Burns JC, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet 2008;40(1):35–42.
- Onouchi Y, Onoue S, Tamari M, et al. CD40 ligand gene and Kawasaki disease. Eur J Hum Genet 2004;12(12):1062–8.
- Onouchi Y, Ozaki K, Buns JC, et al. Common variants in CASP3 confer susceptibility to Kawasaki disease. Hum Mol Genet 2010;19(14):2898–906.
- Onouchi Y, Ozaki K, Burns JC, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet 2012;44(5):517–21.
- Khor CC, Davila S, Breunis WB, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet 2011;43(12):1241–6.
- Ha KS, Jang G, Lee J, Lee K, Hong Y, Son C. Incomplete clinical manifestation as a risk factor for coronary artery abnormalities in Kawasaki disease: a meta-analysis. Eur J Pediatr 2013;172(3):343–9.
- Harada K. Intravenous gamma-globulin treatment in Kawasaki disease. Acta Paediatr Jpn 1991;33(6):805–10.
- Kobayashi T, Inoue Y, Takeuchi K, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation 2006;113(22):2606–12.
- Kobayashi T, Saji T, Otani T, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet 2012;379(9826):1613–20.
Giant cell arteritis
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Baum EW, Sams WM, Jr, Payne RR. Giant cell arteritis: a systemic disease with rare cutaneous manifestations. J Am Acad Dermatol 1982;6(6):1081–8.
- Haugeberg G, Paulsen PQ, Bie RB. Temporal arteritis in Vest Agder County in southern Norway: incidence and clinical findings. J Rheumatol 2000;27(11):2624–7.
- Salvarani C, Crowson CS, O'Fallon WM, Hunder GG, Gabriel SE. Reappraisal of the epidemiology of giant cell arteritis in Olmsted County, Minnesota, over a fifty-year period. Arthritis Rheum 2004;51(2):264–8.
- Smeeth L, Cook C, Hall AJ. Incidence of diagnosed polymyalgia rheumatica and temporal arteritis in the United Kingdom, 1990–2001. Ann Rheum Dis 2006;65(8):1093–8.
- Small P, Brisson ML. Wegener's granulomatosis presenting as temporal arteritis. Arthritis Rheum 1991;34(2):220–3.
- Lie JT, Nagpal S. Churg-Strauss syndrome with nongiant cell eosinophilic temporal arteritis. J Rheumatol 1994;21(2):366–7.
- Hedges TR, 3rd, Gieger GL, Albert DM. The clinical value of negative temporal artery biopsy specimens. Arch Ophthalmol 1983;101(8):1251–4.
- Evans JM, O'Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med 1995;122(7):502–7.
- Mohammad AJ, Nilsson JA, Jacobsson LT, Merkel PA, Turesson C. Incidence and mortality rates of biopsy-proven giant cell arteritis in southern Sweden. Ann Rheum Dis 2015;74(6):993–7.
- Schmidt WA, Kraft HE, Vorpahl K, Volker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med 1997;337(19):1336–42.
- Blockmans D, de Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients. Arthritis Rheum 2006;55(1):131–7.
- Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2009;68(3):318–23.
- Proven A, Gabriel SE, Orces C, O'Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum 2003;49(5):703–8.
- Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum 2007;56(8):2789–97.
Takayasu arteritis
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1–11.
- Savory WS. Case of a young woman in whom the main arteries of both upper extremities and of the left side of the neck were throughout completely obliterated. Med Chir Trans 1856;39:205–19.
- Dreyer L, Faurschou M, Baslund B. A population-based study of Takayasu s arteritis in eastern Denmark. Clin Exp Rheumatol 2011;29(1 Suppl. 64):S40–2.
- Hall S, Barr W, Lie JT, Stanson AW, Kazmier FJ, Hunder GG. Takayasu arteritis. A study of 32 North American patients. Medicine (Baltimore) 1985;64(2):89–99.
- Watts R, Al-Taiar A, Mooney J, Scott D, Macgregor A. The epidemiology of Takayasu arteritis in the UK. Rheumatology (Oxford) 2009;48(8):1008–11.
- Hotchi M. Pathological studies on Takayasu arteritis. Heart Vessels Suppl 1992;7:11–17.
- Terao C, Yoshifuji H, Kimura A, et al. Two susceptibility loci to Takayasu arteritis reveal a synergistic role of the IL12B and HLA-B regions in a Japanese population. Am J Hum Genet 2013;93(2):289–97.
- Saruhan-Direskeneli G, Hughes T, Aksu K, et al. Identification of multiple genetic susceptibility loci in Takayasu arteritis. Am J Hum Genet 2013;93(2):298–305.
- Frances C, Boisnic S, Bletry O, et al. Cutaneous manifestations of Takayasu arteritis. A retrospective study of 80 cases. Dermatologica 1990;181(4):266–72.
- Abularrage CJ, Slidell MB, Sidawy AN, Kreishman P, Amdur RL, Arora S. Quality of life of patients with Takayasu's arteritis. J Vasc Surg 2008;47(1):131–6; discussion 6–7.
- Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum 2007;56(3):1000–9.
- Mukhtyar C, Guillevin L, Cid MC, et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2009;68(3):318–23.
- Valsakumar AK, Valappil UC, Jorapur V, Garg N, Nityanand S, Sinha N. Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu's arteritis. J Rheumatol 2003;30(8):1793–8.