CHAPTER 105
Disorders of the Lymphatic Vessels
Peter S. Mortimer and Kristiana Gordon
St George's Hospital, London, UK
Introduction
Lymphatic dysfunction interferes with fluid homeostasis, tissue immunity and peripheral fat mobilization. Any chronic oedema represents lymphatic failure. If impaired lymph drainage is solely responsible, then lymphoedema results. This produces characteristic skin changes known as elephantiasis as well as increased fat deposition in the subcutaneous tissues. Impaired immune cell trafficking results in an increased risk of infection, particularly cellulitis (erysipelas), which often becomes recurrent. This chapter describes the clinical consequences of lymphatic dysfunction and in particular the impact on the skin and subcutaneous tissues. The sections are divided according to common clinical presentations.
CLINICAL PRESENTATIONS OF LYMPHATIC DYSFUNCTION
Chronic, venous and drug-induced oedema
Definition and nomenclature
Oedema is an excess of interstitial fluid. Any oedema, whatever the cause, is due to capillary filtration overwhelming the lymph drainage for a sufficient period of time [1]. Interstitial fluid is reabsorbed almost entirely by the lymphatic vessels. Contrary to popular belief, venous reabsorption of interstitial fluid cannot be maintained for any length of time in peripheral tissues. Therefore all peripheral oedema represents lymphatic failure. Most chronic oedemas arise from increased microvascular filtration overwhelming the lymph drainage (relative lymphatic failure). Examples are heart failure, venous disease and nephrotic syndrome. Oedema arising principally from a failure in lymph drainage is lympho-edema (absolute lymphatic failure).
Introduction and general description
It is important to determine if oedema (Greek oíde¯ma, a swelling) is due to fluid or another tissue component. Overgrowth syndromes, such as Klippel–Trenaunay syndrome, may have excessive growth of bone, fat or muscle (with or without additional fluid). A plexiform neurofibroma (neurofibromatosis) may cause tissue swelling from both the neural tumour and lymphoedema.
If swelling is fluid it is best not to approach a lower limb chronic oedema clinically by trying to pigeonhole the diagnosis into ‘heart failure’, ‘venous oedema’, ‘lymphoedema’, etc. A far better approach is to consider if the oedema represents pure lymphatic failure, or, as is most common, lymphatic failure due to the lymph drainage being overwhelmed by increased capillary filtration. Most cases of chronic oedema have more than one factor contributing to the impaired lymph drainage and increased capillary filtration (Table105.1). Consequently, treatment of chronic oedema should be to enhance the lymph drainage and address any factors causing increased filtration.
Table 105.1 Causes of chronic oedema.
| Increased capillary filtration | Reduced lymph drainage | |||
| ↑Capillary pressure | ↓Plasma proteins | ↑Capillary permeability | Primary lymphatic insufficiency | Secondary lymphatic insufficiency |
↑Venous pressure: Right heart failure DVT Venous obstruction Calcium channel antagonists Dependency Overtransfusion: Salt and water overload Advanced renal failure↑Blood flow: Inflammation Arteriovenous fistula |
↑Loss: Nephrotic syndrome Protein-losing enteropathy↓Synthesis: Cirrhosis Advanced cancer Malabsorption Malnutrition |
Inflammation: Varicose eczema Psoriasis Chronic infection Urticaria and angio-oedema Drugs |
Germline mutation: Genes known (Milroy disease, lymphoedema, distichiasis) Genes unknown (Meige disease) Mosaic mutation: Lymphatic malformation Overgrowth spectrum |
Iatrogenic Surgery Radiotherapy Cancer Infection Filanasis Cellulitis Accidental trauma Obesity Immobility Sustained lymph load: Venous disease Heart failure Venous obstruction DVT |
DVT, deep-vein thrombosis.
If lower limb oedema is unilateral or asymmetrical then local factors need to be considered such as pelvic lymph or venous obstruction, post-thrombotic syndrome or inflammation from dermatitis or infection. Bilateral lower limb oedema suggests systemic factors such as hypoproteinaemia or high central venous pressure (e.g. heart failure). In the obese person, several factors contribute. The weight of a huge abdominal apron causes obstruction of lymph and venous drainage in the thigh or groin when sitting [2]. Poor mobility results in no enhancement of lymph drainage. Sitting with legs dependent causes periods of high venous pressure and consequently high microvascular fluid filtration (falling asleep in a chair without leg elevation is particularly bad). Sleep apnoea syndrome leads to periods of arterial and pulmonary hypertension and fluid retention [3].
Epidemiology
Lymphoedema per se is perceived as uncommon, yet lymphatic insufficiency is a major contributing cause of chronic ankle oedema, which is considered common (particularly in the elderly). Lymphoedema can be a difficult diagnosis, particularly if mild or in the early stages, therefore it is frequently underdiagnosed. One survey, which determined the problem of chronic oedema (as a surrogate for lymphoedema) in the community, ascertained 823 patients in a catchment area of 619 000 in southwest London. This estimated the overall prevalence of chronic oedema as 1.33/1000 population; the prevalence increased with age and was 5.4/1000 in subjects aged over 65 years (i.e 1 in 200). In only a quarter did the oedema arise from cancer treatment [4].
Pathophysiology
Oedema develops when the microvascular (capillary and venular) filtration rate exceeds lymph drainage. Simply put, the blood vessel supplies tissue fluid and the lymphatic drains it away. For oedema to develop either the microvascular filtration rate is high, the lymph flow is low, or there is a combination of the two.
The filtration rate is governed by the Starling principle of fluid exchange. Microvascular filtration of fluid from capillary into interstitium is driven by the hydraulic (water) pressure gradient across the blood vessel wall (Pc – Pi) in which Pc indicates capillary pressure and Pi indicates interstitial pressure) and is opposed by the osmotic pressure gradient (πp – πi) in which πp indicates plasma osmotic pressure and πi indicates interstitial osmotic pressure from tissue proteins), which is the suction force retaining fluid within the vessel. This is the suction force retaining fluid within the vessel. The colloid osmotic pressures influencing filtration across both fenestrated and continuous capillaries are exerted across the endothelial glycocalyx; the osmotic pressure of the interstitial fluid does not directly determine transendothelial fluid exchange. There is substantial evidence that with important exceptions such as the renal cortex and medulla, downstream microvessels are not in a state of sustained fluid absorption as traditionally depicted. Although doggedly persistent in textbooks and teaching, the traditional view of a filtration–reabsorption balance has little justification in the microcirculation of most tissues. Tissue fluid balance thus depends critically on lymphatic function in most tissues [5].
Clinical features
History
Lymphoedema does not usually respond to elevation or diuretics, except in the early stages or when it is compounded by increased capillary filtration. Chronic oedema that does not reduce significantly after overnight elevation is likely to be lymphatic in origin. Chronic oedema associated with bacterial cellulitis indicates lymphatic insufficiency because of the important role the lymphatic plays in tissue immunosuveillance.
A drug history is important because a number of drugs can cause chronic oedema. Calcium channel antagonists are a common cause of peripheral oedema, with amlodipine one of the worst offenders. Other drugs described to cause oedema are corticosteroids, taxanes, non-steroidal anti-inflammatories, clonidine, morphine, gabapentin, olanzapine, pramipexole and thiazolidinediones.
Presentation
Interstitial fluid volume must increase by over 100% before oedema is clinically detectable through pitting or indentation of the skin from pressure. Dermal oedema manifests as ‘peau d'orange’ due to expansion of the interfollicular dermis, whereas subcutaneous oedema gives rise to pitting.
Lymphoedema differs from all other oedemas (in which increased capillary filtration is the major factor) in that cells, proteins and fat accumulate in addition to water. This results in a ‘solid’ as well as a ‘fluid’ component to the swelling, so giving rise to the brawny nature of the oedema, which does not readily pit except in the early stages. The lack of pitting is an unreliable sign in lymphoedema. Easy displacement of tissue fluid on pressure can often be demonstrated, particularly in the early stages. With time, the skin thickens in lymphoedema and becomes more warty. A positive Kaposi–Stemmer sign represents a failure to pinch a fold of skin at the base of the second toe and is pathognomonic of lymphoedema (Figure 105.1).

Figure 105.1 The Kaposi–Stemmer sign: an inability to pinch or pick up a fold of skin at the base of the second toe indicates lymphoedema.
In circumstances where the cause of the chronic oedema is not obvious a search for reasons for high microvascular fluid filtration should be pursued, for example raised jugular venous pressure in heart failure, local inflammation from dermatitis or infection, and low plasma proteins. In the lower limbs, signs of venous disease causing venous hypertension – for example varicose veins, haemosiderin deposition (particularly submalleolar), varicose eczema and atrophie blanche – should be carefully assessed. The reason severe venous disease may not exhibit oedema is because lymph drainage is maintained but in most cases it is the associated lymphatic insufficiency that causes the ‘venous oedema’.
Investigations
Management of chronic lower limb oedema requires treatment of the underlying cause. If increased microvascular filtration is suspected then the following should be considered and investigated.
- Causes of high central venous pressure (e.g. heart failure) should be pursued. B-type natriuretic peptide (BNP) estimation is a useful screen. The main clinical utility of either BNP or NT-proBNP (N-terminal pro-BNP) is that a normal level rules out acute heart failure.
- Plasma albumin should be measured and if low a search for loss (e.g. nephrotic syndrome) or failure (e.g. liver disease, malabsorption or malnutrition) of synthesis should be considered.
- Local inflammation should be looked for; this will increase microvascular filtration (e.g. infection, dermatitis or underlying arthritis).
There are limited methods available that permit reliable investigation of the lymphatics. Lymphoscintigraphy (isotope lymphography) involves the interstitial (dermis or subcutis) injection of a radiolabelled protein or colloid. Radioactivity, measured using a wide field-of-view gamma camera, is determined over the injection site depot and at regions of interest over vessels or nodes. Measurement of transit times and time activity curves permits a quantitative analysis of lymph drainage [6]. Measurement of tracer uptake within axillary or ilioinguinal lymph nodes at a specified time will discriminate lymphoedema from oedema of non-lymphatic origin (Figure 105.2).

Figure 105.2 (a) Normal lymphoscintigraphy. Images show patent lymph routes draining tracer from the feet to the ilio-inguinal nodes. (Courtesy of Professor A. M. Peters.) (b) Obstruction of lymph drainage at the groin leads to re-routing of tracer through the skin collaterals (dermal backflow).
Computed tomography (CT) of lymphoedematous limbs has demonstrated a characteristic ‘honeycomb’ pattern in the subcutaneous compartment which other oedemas do not show. CT not only provides information through cross-sectional area of volume change in a limb, but will also identify the compartment in which that change takes place. Thickening of the skin is also a characteristic feature of lymphoedema, although not specific. Magnetic resonance imaging (MRI) is potentially better than CT for distinguishing types of oedema. Magnetic resonance lymphangiography has recently been introduced to overcome the invasive nature of X-ray lymphography.
Management
Venous oedema results from venous hypertension causing excessive microvascular filtration. Varicose veins or post-thrombotic syndrome are the commonest causes. However, it must be remembered that it is the lymphatics and not the veins that are responsible for the drainage of tissue fluid. If the lymph drainage is compensating, no matter how severe the venous disease, there will be no oedema. By implication, oedema in the presence of venous disease indicates lymphatic failure and treatment needs to address improvements in lymph drainage as well as control of the venous disease. Surgical treatment of varicose veins will often not resolve the ‘venous’ oedema because lymph drainage is compromised and surgery will not improve it, indeed it may reduce it further. Therefore compression is the treatment of choice for venous oedema because compression garments (hosiery) have the advantage of reducing microvascular filtration (from venous hypertension) while at the same time improving lymph drainage.
In cases of drug-induced oedema (e.g. due to amlodipine), the drug should be replaced or measures introduced to control swelling. The empirical use of diuretics is to be discouraged as they are often ineffective over time.
Swelling of one leg points to a local cause such as venous obstruction from vein compression or a deep-vein thrombosis (DVT). Lymphatic obstruction usually produces whole-limb swelling that is worse proximally. Imaging is required in case of cancer.
Genital oedema occurring in isolation is usually a result of local inflammation, for instance due to infection, ano-genital granulomatosis (cutaneous Crohn disease), hidradenitis suppurativa or sarcoidosis. Genital oedema can be part of more widespread oedema from heart failure or nephrotic syndrome. Primary lymphoedema can affect the genitalia but not usually without lower limb involvement. Lymph or chylous reflux can produce genital oedema often with lymphangiectasia (weeping lymph blisters).
The same physiological principles apply to upper limb oedema as they do to lower limb oedema – that is, microvascular fluid filtration exceeding lymph drainage capacity for a sustained period. Upper limb oedema is much less common than lower limb oedema and usually results from either proximal venous obstruction (e.g. thoracic outlet syndrome) or an inflammatory disorder (e.g. due to infection, arthritis or eosinophilic fasciitis) or lymphoedema.
Chronically swollen leg
Definition and nomenclature
Swelling of the lower limb, due to oedema, is caused by increased microvascular fluid filtration overwhelming lymph drainage. Causes of increased filtration such as increased venous pressure, low plasma proteins and inflammatory states need to be considered as well as reasons for impaired lymph drainage.
Introduction and general description
Swelling of a leg may be caused by oedema, in which case pitting should be evident to some degree, or it may be caused by an increase in volume of other tissue elements, for example bone, muscle or fat. The cause of swollen legs is often multifactorial (Table105.2); therefore, the patient's individual history and an appropriate physical examination are important. Other differentials to consider are a ruptured Baker's cyst, infection, trauma and malignancy. Inflammation of a joint or periarticular structure may cause oedema that is not primarily vascular. MRI is useful in circumstances where the nature of the swelling is uncertain. A patient may perceive one leg to be swollen when in fact the other leg has become smaller, for example through atrophy of muscle or fat. This section addresses fluid-related swelling that is vascular in origin.
Table 105.2 Causes of a swollen leg.
| Genetic | Acquired | ||||||
| Vascular | Lymphatic | Other | Vascular | Lymphatic | Inflammatory | Musculoskeletal | Tumours |
Vascular malformation Diffuse phlebectasia Klippel–Trenaunay syndrome Parkes–Weber syndrome Maffucci syndrome |
Lymphoedema Lymphatic malformation Lymphangiomatosis |
Overgrowth spectrum: Fat hypertrophy Lipomatosis Lipoedema Proteus syndrome Muscle hamartoma/overgrowth Gigantism/hemihypertrophy |
DVT Post-thrombotic syndrome Chronic venous reflux Venous outflow obstruction Dependency syndrome Thrombophlebitis Venous injury, e.g. IV drug abuse Acute arterial ischaemia Idiopathic/cyclical oedema of women Drugs, e.g. calcium channel antagonists |
Lymphoedema: Cancer surgery DXT Filanosis Podoconiosis Trauma Reconstructive surgery Vein harvesting/vein stripping Immobility/armchair legs Factitial Chronic regional pain syndrome Obesity |
Cellulitis Pretibial myxoedema Varicose eczema Psoriasis Pompholyx Sarcoidosis Herpes simplex |
Rheumatoid arthritis Ruptured Baker's cyst Joint effusion Haematoma Torn muscle Pathological fracture Achilles tendonitis Myositis ossificans |
Lymphoma Sarcoma Metastases |
DVT, deep-vein thrombosis; DXT, radiotherapy; IV, intravenous.
Epidemiology
Chronic leg swelling is common but data are few. A recent report from Denmark indicated that of 595 hospitalizations of patients aged 75 years or above in the emergency department, 6.3% were due to suspected DVT or red swollen legs [1].
Pathophysiology
All oedema is caused by microvascular fluid filtration exceeding lymph drainage for a sufficient period of time. As lymph flow is responsible for the drainage of all tissue fluid, except for transient periods of venous reabsorption, a chronically swollen leg due to fluid indicates lymph drainage failure. This failure will be either due solely to lymphatic dysfunction (lymphoedema) or due to excessive microvascular fluid filtration overwhelming lymph drainage capacity [2]. Causes of increased microvascular fluid filtration are: (i) increased venous pressure due to chronic venous insufficiency, post-thrombotic syndrome, venous obstruction or heart failure; (ii) hypoproteinaemia from protein loss (e.g. nephrotic syndrome), malnutrition or a failure of protein synthesis (e.g. cirrhosis); (iii) increased vascular permeability (e.g. inflammatory states such as infection or dermatitis).
In cases of obesity and infirmity, lymph drainage routes in the leg may be patent but non-functional due to lack of mobility, for example in arthritis. In addition sitting for long periods without moving will cause sustained venous hypertension and increased fluid filtration into the legs, while the lack of movement will result in poor lymph drainage. Furthermore, a large obese abdominal apron pressing on the thighs when sitting will obstruct venous outflow. Sleep apnoea syndrome will result in systemic ‘fluid retention’ due to high heart chamber pressures.
Clinical features
History
Clinical features will depend upon the cause. Features that indicate primarily a lymphatic cause are: (i) persistent swelling, which can be intermittent at first; (ii) oedema that does not resolve with overnight elevation; (iii) a poor response to diuretics; and (iv) recurrent cellulitis.
Presentation
Chronic, non-inflammatory, asymmetrical lower limb oedema should always suggest a cause within the hindquarter (e.g. chronic venous hypertension or lymphoedema). Systemic causes of oedema including cardiac disease, renal disease or hypoproteinaemia should cause bilateral leg swelling. Calcium channel antagonists cause peripheral oedema in up to 30% of cases.
Lymphoedema characteristically produces a thickened skin. With increasing chronicity and severity, so skin changes of hyperkeratosis and papillomatosis (elephantiasis) supervene. A failure to pinch a fold of skin at the base of the second toe (Kaposi–Stemmer sign; see Figure 105.1) is pathognomonic of lymphoedema. In more advanced cases fat deposition and fibrosis lead to a more indurated or ‘brawny’ swelling that results in bulging folds of skin and subcutaneous tissue.
The chronically swollen red leg usually indicates lipodermatosclerosis. While usually attributed to chronic venous disease, oedema is always a feature and cases do occur with lymphoedema in the absence of venous reflux.
Investigations
Blood tests are important to exclude hypoproteinaemia and heart failure, where measurement of plasma albumin and BNP (or NT-proBNP) should be performed respectively. In cases of suspected venous and/or lymphatic obstruction, imaging such as ultrasound, CT or MRI of the root of the limb (e.g. the ilioinguinal region) should be undertaken. Lymphoscintigraphy is the investigation of choice to confirm a lymphatic aetiology. Venous duplex ultrasound will identify whether venous reflux is contributory to the fluid swelling. A skin biopsy may be necessary if pathologies such as Kaposi sarcoma, pretibial myxoedema or malignancy are considered.
Management
Treatment of a swollen leg is dependent on the cause. In circumstances where systemic causes for peripheral oedema (e.g. heart failure) have lead to, or coexist with, the lymphoedema, then treatment of the internal medical condition must be undertaken before embarking on specific lymphoedema therapy. The general principle for treating a swollen limb is to control for increased microvascular filtration and to enhance lymph drainage. Lymph drainage responds to exercise and movement while wearing compression [3]. This principle will also control venous hypertension.
Phleboedema and mixed lymphovenous disease
Definition and nomenclature
Phlebolymphoedema or mixed lymphovenous disease is a mixed aetiology swelling of the lower limb due to chronic venous insufficiency and lymphatic insufficiency. Phlebolymphoedema refers to chronic oedema arising from chronic venous hypertension causing increased microvascular fluid filtration overwhelming lymph drainage. Over time established lymphoedema results from the compromised lymphatics.
Introduction and general description
Veins and lymphatics are inextricably linked. Their endothelial parentage is identical as lymphatics and veins have a common embryological origin [1]. They have synergistic functions – the venous capillaries and venules filter fluid from circulation to tissue spaces and the lymphatics drain that fluid back (eventually) to the circulation.
Oedema is a common complication of venous insufficiency. It is assumed that venous oedema is the sole consequence of increased capillary filtration from venous hypertension. As lymph drainage is the main buffer against oedema, it is in fact the failure of local lymphatics to compensate for the increased lymph load from filtration that leads to oedema [2]. Lymphoedema associated with venous disease can give rise to the most gross swelling and skin changes owing to the combined effect of impaired lymph drainage in the face of increased lymph load (capillary filtration) (Figure 105.3).
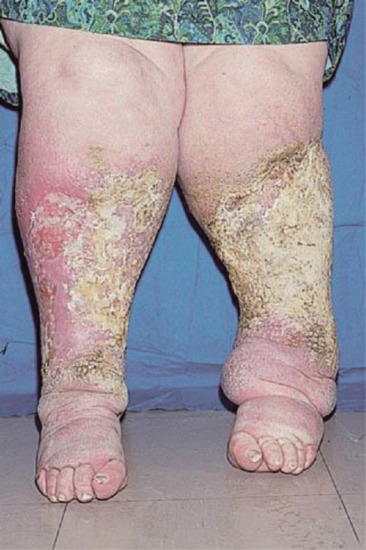
Figure 105.3 Lymphoedema associated with chronic venous disease.
Phlebolymphoedema most commonly occurs in the lower limb but can occur elsewhere in the body in circumstances of venous hypertension – for example a pendulous abdomen (hanging abdominal apron), large pendulous breast and upper limb venous outflow obstruction.
Epidemiology
Oedema is a common finding in chronic venous disease. The Bonn vein study identified up to 20% prevalence depending on age and severity of the venous disease [3].
Pathophysiology
Predisposing factors
Phlebolymphoedema requires raised venous pressure to increase fluid movement from the blood vessels (capillaries and venules) into the tissue spaces. In the lower limbs this may occur from varicose veins, DVT or venous obstruction (Figure 105.4). Because the calf muscle pump is important for venous drainage, long periods spent with the legs dependent and therefore subject to gravitational forces causes sustained periods of venous hypertension.
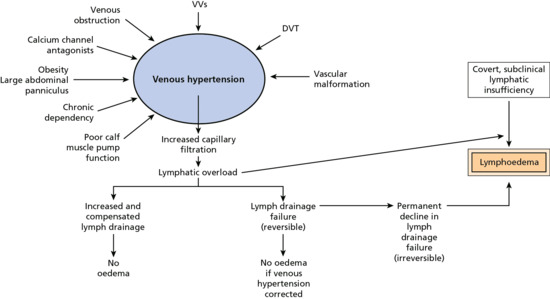
Figure 105.4 Causes of mixed lymphovenous disease and phlebolymphoedema.
Immobility tends to encourage swelling, particularly if gravitational forces (dependency syndrome) encourage ongoing fluid filtration. A common scenario is ‘armchair legs’, where patients sit in a chair day and night with their legs dependent (otherwise known as elephantiasis nostras verrucosis because of the severe lymphoedema skin changes that ensue). Patients at risk are those with neurological deficit in the legs preventing movement; those with chronic respiratory or cardiac disease, which requires them to sit upright in a chair day and night; and those with sleep apnoea syndrome who cannot lie flat. Obesity can also result in venous obstruction when a large abdomen compresses the thigh veins when sitting upright in a chair.
Treatment with calcium channel antagonists causes lower limb oedema in up to 30% of users [4]. Discontinuing the drug will often resolve the oedema.
Traditional surgical stripping of varicose veins or harvesting of the great saphenous vein for coronary artery bypass grafting could also damage leg lymphatics and lead to phlebolymphoedema. Fortunately, both procedures are rarely performed since the introduction of endovenous therapy and stenting.
Intravenous drug abuse can damage both the veins and lymphatics from both thrombosis and sepsis leading to phlebolymphoedema in the upper and lower limbs.
Pathology
Oedema is an excess of interstitial fluid. Any oedema, whatever the cause, is due to capillary filtration overwhelming the lymph drainage for a sufficient period of time [5]. Contrary to popular belief, venous reabsorption of interstitial fluid cannot be maintained for any length of time in peripheral tissues. Interstitial fluid is reabsorbed almost entirely by the lymphatic vessels. Venous hypertension causes an increase in microvascular fluid filtration that requires greater lymph drainage if oedema is to be avoided. In chronic venous disease the fluid load frequently overwhelms lymph drainage to produce oedema. Over time the high lymph load results in deteriorating lymph drainage and a permanent lymphoedema. Furthermore, high tissue fluid and venous pressure result in lipodermatosclerosis, an inflammatory condition of the most affected skin and subcutaneous tissues.
Genetics
Many primary lymphoedemas for which gene mutations are known also possess venous reflux because of a genetically determined venous valve failure. The best documented is lymphoedema distichiasis syndrome where the mutation is in the FOXC2 gene [6].
Clinical features
History
Phlebolymphoedema in the lower limb will start as a pitting oedema indistinguishable from any other chronic oedema. There may be a history of varicose veins including past venous surgery (stripping or endovenous therapy), DVT, heart failure, sleep apnoea syndrome, obesity or infirmity with long periods spent with the legs dependent.
Chronic venous disease may result in symptoms such as heaviness, aching, itching (from varicose dermatitis), skin pigmentation (from purpura or haemosiderin). Symptoms worsen towards the end of the day and are relieved by overnight elevation and are usually exacerbated by heat and alcohol. Lipodermatosclerosis will often produce severe pain and tenderness. Poor wound healing can result in a chronic ulcer.
When lymphoedema dominates, the skin becomes harder and swelling does not resolve as much with overnight elevation. Recurrent cellulitis always indicates lymphatic insufficiency.
Presentation
When chronic oedema is associated with symptoms and signs of chronic venous disease then phlebolymphoedema is likely. Oedema is usually confined to below the knee but severe cases can extend into the thigh. When signs of lymphoedema dominate then tissues will be indurated and pitting more difficult to elicite. The Kaposi–Stemmer sign will probably be positive (pathognomonic of lymphoedema) (see Figure 105.1).
Advanced cases develop elephantiasis skin changes with hyperkeratosis and papillomatosis [7]. Recurrent cellulitis can occur due to underlying lymphatic insufficiency and the effect lymphatic dysfunction has on local immune cell trafficking. Such infections will usually cause local signs of inflammation (i.e. pain, redness, heat and swelling), signs that can easily be confused with acute (on chronic) lipodermatosclerosis. Lipodermatosclerosis, however, does not cause systemic upset, an increased white cell count or C-reactive protein (CRP) or respond to antibiotics.
Chronic regional pain syndrome (reflex sympathetic dystrophy) can present like a phlebolymphoedema, but pain (particularly allodynia) and loss of function are distinctive features.
Differential diagnosis
When venous hypertension exists with lymphatic insufficiency then phlebolymphoedema is likely. However, there may be many causes for venous hypertension and more than one cause may coexist – for example, heart failure, obesity, chronic venous disease, dependency, etc.
Complications
Phlebolymphoedema may be complicated by lipodermatosclerosis, dermatitis, ulceration, lymphorrhoea and infection, especially cellulitis. Rarely, malignancy (e.g. lymphangiosarcoma) can complicate the phlebolymphoedema.
Prognosis
Prognosis is poor regarding long-term morbidity unless underlying causes are addressed. Compression therapy with exercise is the only satisfactory treatment.
Investigations
Venous duplex ultrasound is the investigation for chronic venous disease. It can detect venous reflux, thrombosis and some venous obstruction. In cases of mixed vascular malformations and iliac vein obstruction, more specialist imaging with venography may be necessary.
Lymphoscintigraphy is the investigation of choice for detecting lymphatic insufficiency. It is very sensitive but not that specific and can miss lymphoedema, particularly in the presence of venous disease.
Management
First line
Compression and exercise treat both venous disease and lymphoedema. Compression may be achieved through bandaging or compression garments. Bandaging is helpful initially to produce a reduction in swelling and improve limb shape so compression garments fit better. Standard ‘venous ulcer’ compression bandaging will adequately treat most cases but if there is marked forefoot involvement (swollen or papillomatous toes) or if swelling extends above the knee, then lymphoedema-style treatment in the form of decongestive lymphatic therapy (DLT) is preferred [8]. DLT involving toe and thigh bandaging can only be provided by trained therapists and is generally not available in the community.
Wounds, dermatitis and infection need to be treated before, or at the same time, as compression is applied.
Exercise is to be encouraged in preference to rest but when the patient is resting, the leg should be elevated to heart level. Patients should be discouraged from spending too long in a chair unless it is a reclining chair. In infirm patients pneumatic compression therapy may be helpful [9].
Second line
Superficial venous reflux may be amenable to endovenous therapy. Hopefully this will reduce the lymph load and so reduce oedema but it is often still necessary to wear compression garments afterwards [10].
Lipodermatosclerosis (chronic cellulitis)
Definition and nomenclature
The chronically swollen red leg is a common sight in medical practice. Often wrongly mistaken for bacterial cellulitis, it is frequently mismanaged. Lipodermatosclerosis (LDS) is an inflammatory condition of the skin and subcutaneous tissues affecting the lower third of the leg, and is commonly, although incorrectly, called chronic cellulitis. It is due to sustained ‘congestion’ – that is, high interstitial fluid and venous pressures. It is most usually described with chronic venous disease but the common denominator is chronic oedema and it can frequently be seen in lymphoedema without any venous reflux. While it resembles bacterial cellulitis there are no systemic symptoms or signs of infection. Bacterial infection (true cellulitis) can, however, frequently complicate LDS but antibiotics alone do not resolve it and the only proven treatment is compression therapy to ‘decongest’ the tissues.
Introduction and general description
Many patients diagnosed with bacterial cellulitis do not have infection and therefore antibiotic treatment is inappropriate. Distinguishing true cellulitis or erysipelas from its many mimics is challenging but critical if unnecessary use of antibiotics, unnecessary in-patient admissions and delays in treatment are to be avoided.
True bacterial cellulitis presents with local redness, heat, pain and swelling at one site (e.g. one leg), combined with systemic upset such as fever or flu-like symptoms. Inflammatory markers including white cell count and CRP are usually raised and there is a good response to antibiotic treatment.
LDS is usually bilateral with no systemic symptoms and no raised inflammatory markers. Its response to antibiotics is poor.
Epidemiology
Of 595 hospitalizations of patients aged 75 years or above in an emergency department in Denmark, 6.3% were due to suspected DVT or red swollen legs [1].
Pathophysiology
Predisposing factors
It is assumed to be chronic venous disease but chronic oedema is the only invariable predisposing factor. It is caused by lymphoedema, chronic venous disease, dependency and immobility and cellulitis.
Pathology
The pathology of LDS is not well understood but has always been considered secondary to chronic venous hypertension. mRNA and protein expression of matrix metalloproteinase 1 (MMP-1), MMP-2 and tissue inhibitors of metalloproteinase 1 (TIMP-1) have been shown to be significantly increased, indicating that LDS is characterized by elevated matrix turnover [2]. LDS is always accompanied by tissue iron overload. It has been suggested that patients with LDS are unable to counteract venous-induced skin iron overload [3].
Clinical features
History
Lipodermatosclerosis is usually bilateral. While redness and oedema are always present, warmth is usually, but not always, present. Induration indicates that the underlying subcutaneous tissues are involved with the inflammatory process (sclerosing panniculitis). Pain and tenderness are ever present but not itch; if itch occurs, varicose/stasis eczema probably coexists. Systemic symptoms and signs are absent as are raised inflammatory markers. Antibiotics have no effect, but because the patient is often admitted and confined to bed, or told to rest with legs elevated, there is an improvement in the inflammation because of a lessening of the congestion [4].
Presentation
There are two forms of LDS: acute and chronic (Figure 105.5). Acute LDS simulates acute cellulitis with a flare of local redness, heat and pain. With time the chronic form supervenes. The skin becomes ‘bound down’ and retracted as the subcutaneous tissues become more fibrotic and contracted. Eventually redness gives rise to brown pigmentation and the leg contour takes on an ‘inverted champagne bottle’ shape. Pitting oedema will continue to exist both above and below the area of LDS and is a common feature (and common denominator) throughout.
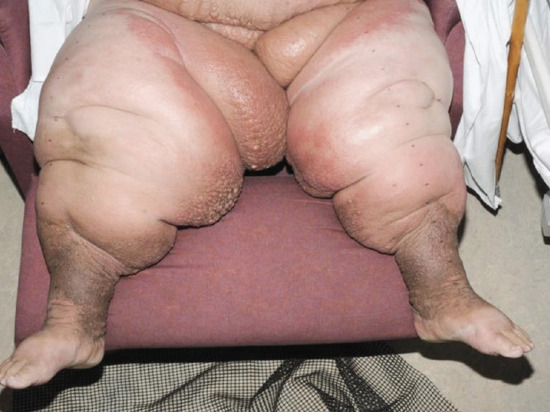
Figure 105.5 Acute and chronic lipodermatosclerosis; the bright red skin (acute) could be mistaken for bacterial cellulitis but it is an inflammatory response to the skin fluid congestion. The treatment is decongestive lymphatic therapy with or without antibiotic cover.
Differential diagnosis
The differential diagnosis is acute cellulitis.
Complications
Complications may include lymphorrhoea, infection and ulceration.
Investigations
Lipodermatosclerosis is a clinical diagnosis. Biopsy will reveal ‘stasis dermatitis’ changes together with a fibrotic panniculitis but there are no specific or diagnostic features. Furthermore, biopsy may induce ulceration if healing is poor.
Management
First line
Compression therapy is the only proven therapy. Compression bandaging will achieve quicker results than compression hosiery but may not be tolerated if the affected tissues are very inflamed and tender. In such circumstances it may be necessary to start with bed rest or even topical steroids before introducing gentle compression. In more chronic cases, where shape change exists, multilayer lymphoedema compression bandaging works better [5]. Bandaging may have to be continued until a more normal contour shape is obtained. Only then will compression hosiery fit and work satisfactorily.
Second line
If superficial venous reflux is proven on duplex ultrasound then endovenous therapy (e.g. radiofrequency vein ablation, laser vein ablation or foam sclerotherapy) could be administered. If endovenous therapy is considered unsuitable then traditional ligation and stripping of superficial veins could be undertaken [6].
Recurrent cellulitis (erysipelas)
Definition and nomenclature
Cellulitis (or erysipelas as it is more usually known in Europe) is one of the most common reasons for emergency admissions to hospital and up to half of patients have repeat attacks. Lymphoedema and leg ulcers provide the greatest risk for cellulitis, particularly recurrent cellulitis. Penicillin is effective in preventing subsequent attacks of cellulitis during prophylaxis, but the protective effect diminishes progressively once drug therapy is stopped [1]. Risk factors such as lymphoedema need to be addressed if recurrence is to be averted.
Introduction and general description
Cellulitis of the leg is a common infection of the skin and subcutaneous tissues. Most infections that affect intact skin are thought to be due to streptococci although other organisms may be responsible if the integrity of the skin is compromised [2]. Cellulitis is a common consequence of lymphoedema irrespective of the cause of the lymphoedema. In recurrent cellulitis, the damage to the lymphatics may make the lymphoedema worse and so predispose to yet further episodes of infection. There is evidence that covert lymphatic insufficiency may predispose to first time attacks of cellulitis [3].
In filarial lymphoedema, episodes of infection – referred to as acute dermatolymphangioadenitis, but to all intents and purposes the same as cellulitis – cause acute morbidity and increasingly severe lymphoedema [4].
In developed countries most patients with cellulitis are treated for the acute episode and discharged, yet the rate of recurrence is high, suggesting that underlying predisposing factors (e.g. lymphoedema) may not be sufficiently managed following the first attack.
Epidemiology
Recurrent cellulitis is common. In a recent study 53% of subjects with a history of cellulitis had at least one recurrence during the 3-year trial [1].
Pathophysiology
Predisposing factors
Several studies have determined risk factors for lower limb cellulitis. Dupuy et al., in a multivariate analysis, calculated an odds ratio of 71.2 for lymphoedema, 23.8 for breaks in the skin barrier (leg ulcer, toe-web intertrigo, dermatitis), 2.9 for venous insufficiency and 2.0 for obesity as independent risk factors associated with cellulitis [5]. In another series of 171 patients, 81 (47%) had recurrent episodes and 79 (46%) had chronic oedema. The concurrence of these two factors was strongly correlated (P <0.0002) [6].
Impaired lymph drainage leads to high rates of infection, particularly cellulitis, within the lymphatic basin. In a community-based survey, 29% of those with lymphoedema (64/218) had suffered cellulitis within the previous 12 months, of which 27% (16/64) required admission for intravenous antibiotics with a mean length of stay of 12 days at an estimated cost of £2300 per patient [7].
The afferent lymphatic vasculature provides the major exit route from the skin for soluble antigens and for immunologically active cells (e.g. lymphocytes, dendritic cells and macrophages) (Figure 105.6). It is likely that disturbances in immune cell trafficking compromise tissue immunosurveillance to predispose to infection, but the exact mechanism is not known [8].
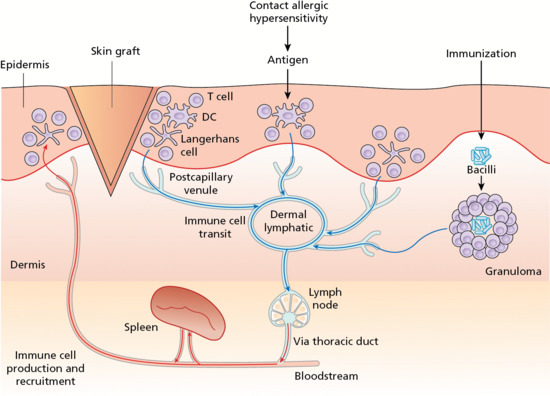
Figure 105.6 The lymphatic vessel sits centre stage for immune cell trafficking within the skin. DC, dendritic cell. (From Mortimer et al. 2014 [8].)
Causative organisms
Most episodes of cellulitis are believed to be caused by group A streptococci. However, microbiologists consider Staphylococcus aureus to be the cause in most patients [9, 10].
Clinical features
History
Cellulitis can vary from patient to patient and episodes can vary in presentation. Some episodes are accompanied by severe systemic upset, with high fever or rigors; others are milder, with minimal or no fever. Increased swelling of the affected area may occur. Inflammatory markers (CRP, erythrocyte sedimentation rate) are usually raised.
Presentation
When associated with established lymphoedema, clinical features may differ from classic cellulitis. Onset may be in minutes (as opposed to hours as in classic cellulitis). Toxicity may be severe with flu-like symptoms, nausea and vomiting, headache and high fever. Systemic symptoms may occur before local signs. The rash may be polymorphic with no defined border. In milder cases, inflammatory markers are not always raised. Episodes may be slow to resolve with oral antibiotics. Recurrence of infection is not unusual after only a week's course of antibiotics (Figure 105.7).
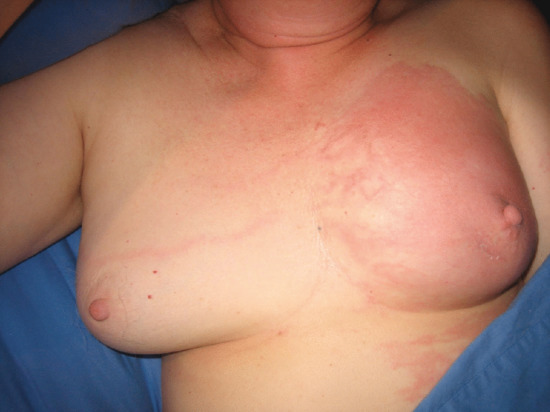
Figure 105.7 Recurrent cellulitis in lymphoedema following breast cancer treatment. Note lymphangitis crossing the watershed to the contralateral lymph node territory.
In some cases, symptoms and signs may grumble over a period of weeks. This is particularly so when associated with breast cancer-related lymphoedema (BCRL) of the breast. The patient may complain only of tiredness and of not feeling very well. Local signs may be redness and swelling with ‘flare-ups’ of redness from time to time. Inflammatory markers are usually negative and only a prompt response to a prolonged course of antibiotics confirms the diagnosis.
Differential diagnosis
The main differential diagnoses are DVT, necrotizing fasciitis and LDS. Other possibilities include gout, vasculitis, a ruptured Baker's cyst and pannicultis.
Investigations
Cellulitis is a clinical diagnosis supported by a blood neutrophilia and raised CRP. Blood cultures should be performed but may be positive in only 10% of cases [11]. Microbiology of any cuts or breaks in the skin or aspiration of blister fluid should be considered before antibiotics are started.
Management
First line
Cellulitis is best managed by dermatologists because up to a third of patients can be misdiagnosed and over a quarter of cases have associated skin disease, treatment of which is likely to reduce the chances of cellulitis recurrence [12].
Low-dose prophylactic penicillin, phenoxymethylpenicillin 250 mg twice daily, given for a period of 12 months almost halves the risk of recurrence during the intervention period compared with placebo [1]. However, although some level of protection appears to be sustained for several months after the end of prophylactic therapy, this effect is lost by 36 months, a finding that suggests that longer term prophylaxis may be required. Patients with a body mass index (BMI) of 33 or higher, multiple previous episodes of cellulitis or lymphoedema of the leg had a reduced likelihood of a response to prophylaxis. A high BMI, multiple attacks and lymphoedema would all be associated with lymphatic dysfunction, supporting the view that lymphoedema is a very strong risk factor for cellulitis.
In patients allergic to penicillin, or in whom penicillin prophylaxis fails, alternative antibiotics such as erythromycin should be considered although there are no data on safety or efficacy [13].
In all cases of recurrent cellulitis, risk factors such as lymphoedema, wounds, breaks in skin integrity (particularly interdigital), dermatitis and fungal infections should be treated.
Swollen arm
Definition and nomenclature
Swelling of the upper limb or extremity is invariably due to oedema but overgrowth of tissue can occur. Oedema is likely to be caused either from lymphatic insufficiency (e.g. breast cancer treatment) or from venous obstruction.
Introduction and general description
Swelling of an arm may be caused by oedema, in which case pitting should be evident to some degree, or it may be caused by an increase in the volume of other tissue elements, for example bone, muscle, fat or a tumour (Table105.3). A swollen arm may be normal but perceived to be bigger if the contralateral limb has shrunk. The commonest reason for upper limb swelling is lymphoedema following breast cancer treatment. Arm swelling can be a presentation of cancer with metastatic disease in the axilla.
Upper limb swelling may due to primary lymphoedema (usually associated with lymphatic abnormalities elsewhere) or with a lymphatic malformation. Secondary lymphoedema can be caused by rheumatoid arthritis, psoriatic arthropathy, hand dermatitis, yellow-nail syndrome, chronic regional pain syndrome (reflex sympathetic dystrophy), pretibial myxoedema, sirolimus treatment and following repeated infections such cellulitis and lymphangitis from herpes simplex.
Table 105.3 Causes of a swollen arm.
| Congenital/genetic | Acquired | |||||
| Vascular | Lymphatic | Other | Vascular | Lymphatic | Musculoskeletal | Tumours |
Vascular malformation Diffuse phlebectasia Klippel–Trenaunay syndrome Arteriovenous malformation |
Lymphoedema Lymphatic malformation Lymphangiomatosis |
Overgrowth spectrum: Proteus syndrome Fat hypertrophy Muscle hamartoma Gigantism/hemihypertrophy Lipoedema Dercum disease Madelung disease (benign symmetrical lipomatosis) |
Subclavian vein thrombosis: Effort thrombosis Venous catheterization Chemotherapy ports Chest radiotherapy Thoracic outlet syndrome Superior vena cava obstruction IV drug abuse |
Lymphoedema: Axillary surgery DXT Cancer Neurological deficit Chronic regional pain syndrome Lymphangitis (bacterial infection, herpes simplex, psoriasis, rheumatoid arthritis) Yellow-nail syndrome |
Rheumatoid arthritis Haematoma Torn muscle Pathological fracture Myositis ossificans osteomyelitis Septic arthritis |
Lymphoma Sarcoma Metastases |
DXT, radiotherapy; IV, intravenous.
Epidemiology
More than one in five women who survive breast cancer will develop arm lymphoedema [1].
Pathophysiology
Predisposing factors
Upper limb lymphoedema is most commonly caused by cancer treatment (i.e. axillary lymphadenectomy or radiation), but less commonly can be a presenting sign for advanced malignancy.
Venous outflow obstruction may be due to axillary/subclavian vein compression or stenosis (usually due to malignancy or radiation damage) or occlusion from thrombosis. Subclavian vein thrombosis is a rare condition that most often occurs in the context of central venous catheters, pacemakers, trauma, surgery immobilization, oral contraceptive pill use, pregnancy or malignancy. It occurs particularly in cancer patients receiving chemotherapy through central lines. It can also have primary causes such as anatomical anomalies, including thoracic outlet syndrome and Paget–Schröetter syndrome (so-called ‘effort thrombosis’). Arteriovenous fistulae for haemodialysis will increase arm size from an increased blood flow but arm oedema will only occur with thrombosis or if lymph drainage is compromised.
Pathology
Upper extremity swelling of vascular origin will be due to oedema or increased vascular volume (e.g. vascular malformation). All oedema is caused by microvascular fluid filtration exceeding lymph drainage for a sufficient period of time. As lymph flow is responsible for the drainage of all tissue fluid, except for transient periods of venous reabsorption, a chronically swollen arm due to fluid indicates lymph drainage failure. This failure will be either due solely to lymphatic dysfunction (lymphoedema) or due to excessive microvascular fluid filtration overwhelming lymph drainage capacity [2]. Increased filtration can be caused by high venous pressures or from enhanced vasular permeability from inflammation (e.g.dermatitis or infection).
Causative factors
Recurrent infections from herpes simplex can lead to upper limb lymphoedema [3]. Non-infective forms of inflammation due to chronic hand dermatitis [4], rheumatoid arthritis [5] or psoriatic arthropathy [6] can lead to lymphoedema.
Genetics
Gene mutations causing tissue overgrowth have recently been identified [7]. PIK3CA mutations are frequently associated with a lymphatic anomaly and are usually mosaic/somatic in nature. Primary lymphoedema of the upper limb can be caused by a CCBE1, FAT4 or GJC2 mutation.
Clinical features
History
In its mildest form BCRL may go unnoticed, even by the patient. Swelling of the hand or wrist may be observed. Alternatively, the patient may notice that their clothes are tight. Aching is frequently experienced.
Presentation
Upper limb BCRL should exhibit pitting oedema but in more advanced cases fat and fibrosis contribute more to the swelling, so the consistency of the swelling may be fatty or firm. The distribution of swelling along the arm varies between patients, and swelling may be confined to a specific region of the upper limb. In some patients the hand may be swollen, whilst in others the hand may be spared despite more proximal swelling of the forearm or upper arm. In cases of incipient or mild BCRL, when the arm is not obviously increased in size, inspection may reveal decreased visibility of subcutaneous veins on the ventral forearm and dorsal hand ipsilaterally (the skin is thickened in lymphoedema and therefore more opaque) with smoothing or fullness of the medial elbow and distal upper arm contours. By pinching up the skin and subcutis of each arm between finger and thumb, the thickened ipsilateral tissues can be palpated [8]. Skin colour is normal except in the presence of venous outflow obstruction when it is red to blue; or with infection when it is pink to red; or with LDS when it is deep to cherry red.
Differential diagnosis
Venous outflow obstruction due to axillary/subclavian vein compression or stenosis, or occlusion from thrombosis, will produce a discoloured (red/blue) painful swollen arm often with parasthesia.
A swollen arm due to overgrowth may be associated with lymphoedema or lymphatic malformation in which case there may be signs of a vascular birthmark often at the root of the limb. There may be overgrowth with fat either from lipohypertrophy, such as in lipoedema, or with lipomatosis, such as Madelung disease (Figure 105.8).
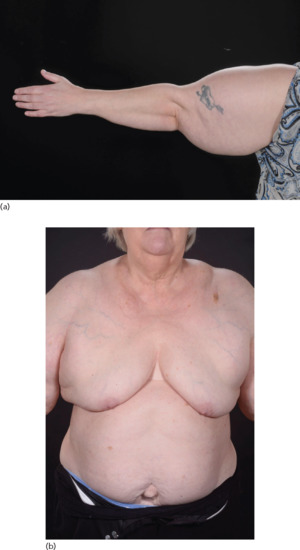
Figure 105.8 Madelung disorder. (a) Benign symmetrical lipomatosis (also known as benign symmetrical lipomatosis of Launois–Bensaude, Madelung disease, multiple symmetrical lipomatosis and cephalothoracic lipodystrophy) is a cutaneous condition characterized by extensive symmetrical fat deposits in the head, neck and shoulder girdle area. (b) Increased fat (lipomatosis) around the neck and shoulder girdle.
Other differentials to consider are musculoskeletal disorders (e.g. ruptured muscle, arthritis, myofascitis or polymyositis), infection, trauma, a neoplasm (including sarcoma and carcinoma), allergic reaction and factitious causes. Rarely, systemic causes such as heart failure, superior vena caval obstruction and hypoproteinaemia can produce arm swelling.
Complications
The main complication of lymphoedema anywhere is infection and, in particular, cellulitis. Tense lymphoedema can produce lymphangiectasia, that is, surface ‘lymph blisters’ that can weep lymph (lymphorrhoea). Rarely lymphangiosarcoma can complicate any form of lymphoedema.
Prognosis
Cellulitis complicating upper limb lymphoedema can on occasion be severe and life threatening.
Investigations
Lymphoedema is usually a clinical diagnosis in the context of past cancer treatment. However imaging of the axilla is necessary to exclude a relapse of cancer (e.g. breast cancer or melanoma).
In cases of suspected non-cancer lymphoedema, lymphoscintigraphy is the investigation of choice to confirm impaired lymph drainage. A venous duplex ultrasound examination is the first investigation of choice in suspected venous outflow obstruction, but CT or MRI venography may be necessary in neutral and stress postions to confirm a thoracic outlet obstruction. A thrombophilia screen is indicated in cases of thrombosis.
Where overgrowth is suspected, MRI comparing both upper limbs should identify enlarged muscle or fat. MRI can also help distinguish fat from fluid.
Management
Treatment of a swollen arm is dependent on the cause. In circumstances where systemic causes, for example cancer recurrence or heart failure, have lead to, or coexist with, the lymphoedema, then treatment of the medical condition must be undertaken before embarking on specific lymphoedema therapy. The general principle for treating a swollen limb is to limit increased microvascular filtration and enhance lymph drainage. Lymph drainage responds to exercise and movement done while wearing compression [9].
Where cellulitis, in particular recurrent cellulitis, occurs then prophylactic antibiotics may be indicated [10].
Although surgical decompression and venous angioplasty may be considered for thoracic outlet obstruction, the typical treatment for primary subclavian vein thrombosis is oral anticoagulation only [11]. Venous compression or stenosis may benefit from stenting.
Swollen face, head and neck
Definition and nomenclature
Facial swelling may be generalized or localized, for example to the eyelid(s), lips or one cheek. It may extend beyond the face to involve the head and neck. To be chronic it should persist for more than 3 months.
Introduction and general description
Chronic swelling of the face is most often due to fluid oedema but can arise due to an increase in other tissue components, such as blood vessels in a port wine stain (capillary malformation), acromegaly, overgrowth spectrum (hemihypertrophy) or tumours (Table105.4).
Table 105.4 Causes of head and neck swelling.
| Congenital/genetic | Acquired | ||||
| Vascular | Lymphatic | Overgrowth | Tumours | Inflammatory | Miscellaneous |
| Vascular malformation | Syndrome (neck webbing): Turner Noonan Generalized lymphatic dysplasia Mosaic with segmental lymphoedema Lymphangioma/lymphatic malformation |
Macrocephaly, e.g. macrocephaly capillary malformation syndrome | Metastatic head and neck cancer Angiosarcoma Radical neck lymphadenectomy Radiotherapy |
Rosacea/acne Cellulitis/erysipelas Oro-facial granulomatosis Tuberculosis Sarcoidosis Dental abscess Sinusitis Dermatomyositis Dermatitis/eczema, psoriasis, contact allergy Blepharochalasis Pediculosis Angio-oedema |
Acromegaly Accidental trauma (cauliflower ear) Cushing syndrome Graves disease |
Oedema may extend beyond the face to involve the head and neck, which occurs after surgery and/or radiotherapy for head and neck cancer or with recurrent cancer. Lymphoedema is a frequent late effect of head and neck cancer. Head and neck lymphoedema may be categorized as involving external structures (e.g. the skin and soft tissue of the face and neck) and internal structures, such as the mucosa and underlying soft tissue of the upper aerodigestive tract (e.g. the pharynx and larynx). In one study, the most common sites of external lymphoedema were the neck and submental area [1].
Oedema of the upper or lower lip (or both) may be from a vascular anomaly or result from recurrent angio-oedema, oro-facial granulomatosis (OFG), sarcoidosis and infective cheilitis or from the administration of lip fillers for cosmetic purposes.
Chronic oedema of the eyelids is common. Conditions that need to be considered include dermatomyositis, Graves disease and particularly rosacea/acne. Eyelid swelling may be quite simply due to acquired lax skin from photoageing and other processes that have undermined tissue compliance, such as blepharochalasis [2]. Contact allergy or angio-oedema, if persistent or recurrent, may slowly compromise lymphatic function. Equally, one severe attack of facial cellulitis may damage the lymphatics sufficiently to cause lymphoedema.
Chronic inflammatory disorders (e.g. rosacea, psoriasis, eczema), bacterial cellulitis, pediculosis, trauma and primary (congenital) lymphoedema can all lead to localized, lymphoedematous enlargement of the ear. Rosaceous enlargement is called otophyma [3].
Angiosarcoma or Kaposi sarcoma may infiltrate local lymph drainage, and manifest with eyelid oedema. Facial swelling can coexist with obvious primary lymphoedema of one or more limbs, suggesting that there is widespread congenitally determined lymphatic insufficiency.
Epidemiology
There are no data for facial lymphoedema from inflammatory disorder. The European literature reports that 46% of patients developed secondary lymphedema as a late effect of head and neck cancer treatment [4].
Pathophysiology
Oedema is an excess of interstitial fluid. Any oedema, whatever the cause, is due to microvascular (capillary) filtration overwhelming the lymph drainage for a sufficient period of time [5]. Interstitial fluid is reabsorbed almost entirely by the lymphatic vessels. Gravitational factors contributing to increased microvascular filtration do not play a part except overnight when the patient is lying down, hence facial swelling is often at its worst in the morning. However sustained increased venous pressures (e.g. superior vena caval obstruction) can produce facial oedema.
If primary facial lymphoedema occurs it is invariably present at birth. It is usually asymmetrical and associated with lymphoedema elsewhere. Head and neck oedema occurring in utero may regress by birth but can leave signs such as prominent medial epicanthic folds or neck webbing postnatally, as seen in Turner and Noonan syndromes. Congenital eyelid lymphoedema may be associated with conjunctival oedema.
A lymphatic malformation (lymphangioma) of the head and neck is more common than lymphoedema and gives rise to swelling from lymph fluid present within abnormally formed lymphatics (whereas lymphoedema is lymph fluid within the interstitial space). The mouth and particularly the tongue are common sites.
Facial lymphoedema may be secondary to other inflammatory pathologies of the skin such as rosacea or acne vulgaris [6]. The skin or subcutaneous initial lymphatics fail rather than the main regional collecting trunks, but in addition telangiectasia and inflammation contribute to oedema through increased fluid filtration. To what extent granulomatous rosacea, Morbihan disease and solid facial oedema (Figure 105.9) represent advanced versions of rosaceous lymphoedema remains unclear [7]. Other inflammatory disorders considered to cause facial oedema include eczema, psoriasis, infection, pediculosis [8] and trauma (cauliflower ears). Contact allergy or angio-oedema, if persistent or recurrent, may slowly compromise lymphatic function [9]. One severe attack of facial erysipelas or cellulitis may damage the lymphatics sufficiently to cause lymphoedema.

Figure 105.9 Solid facial oedema.
Chronic oedema of the eyelids is common and may just be due to acquired lax skin from photoageing and other processes that have undermined tissue compliance [2].
Medical conditions to be considered with periocular oedema are dermatomyositis, Cushing syndrome (moon face) and thyroid disease particularly Graves disease. Renal disease, contrary to expectations, does not cause oedema unless associated with hypoproteinaemia (i.e. nephrotic syndrome) or when advanced. Inflammation within underlying structures may manifest with facial oedema (e.g. dental root infection, chronic sinusitis and salivary duct obstruction).
Angiosarcoma or Kaposi sarcoma may infiltrate local lymph drainage routes and manifest with facial lymphoedema.
Oedema of the upper or lower lip (or both) may be congenital or result from chronic dermatitis, recurrent angio-oedema or OFG (Figure 105.10). In OFG it might prove difficult to identify granulomas on biopsy so their absence does not exclude the diagnosis. If present, a diagnosis of OFG (also called granulomatous cheilitis and Melkersson–Rosenthal syndrome) is made, but it remains unclear if the granulomas are cause or effect [10]. Granulomatous inflammation may exist only locally but a thorough search for gastrointestinal Crohn disease or systemic sarcoidosis should be made. Crohn disease of the bowel can become apparent some time after presentation of OFG. Granulomatous inflammation from administration of lip fillers for cosmetic purposes can also cause chronic swelling [11].
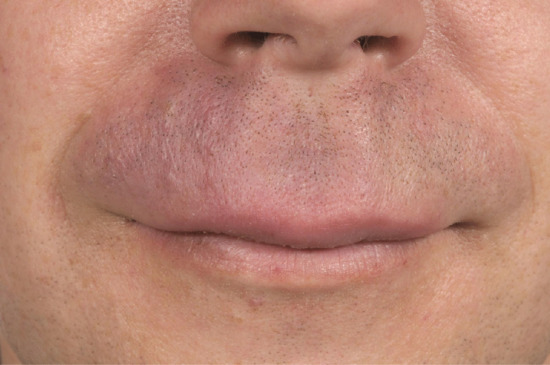
Figure 105.10 Oro-facial granulomatosis exhibiting redness and indurated swelling of the right upper lip.
Head and neck lymphoedema is becoming increasingly more common as more head and neck cancer is treated by lymph node neck dissection and radiotherapy (Figure 105.11). Fibrosis and secondary infection are frequent complications.
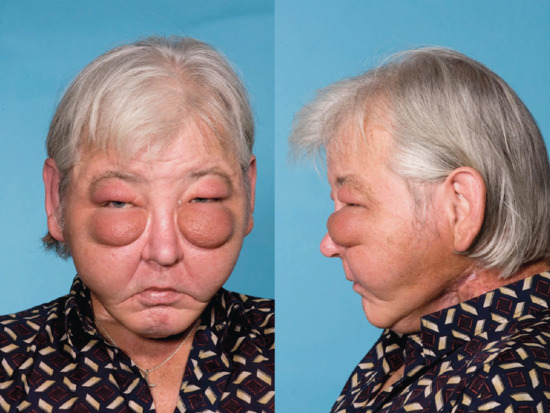
Figure 105.11 Severe facial lymphoedema following treatment for carcinoma of the tongue.
Clinical features
The clinical features of facial lymphoedema depend on the underlying aetiology. Swelling usually affects the central forehead, periocular skin and cheeks where it may be surprisingly asymmetrical. Erythema is always present in rosacea but inflammatory pustules and papules may be conspicuous by their absence. OFG starts with intermittent bouts of swelling resembling angio-oedema affecting the lips or cheeks, but with time the condition may become persistent. An extension of the oedema within the mouth is common and is the reason for the rugose changes on the buccal mucosal and tongue (scrotal tongue).
Lymphoedema may cause facial disfiguration and distress in patients with head and neck cancer [1].
Investigations
Skin biopsy may be helpful if granulomatous disease, rosacea, dermatomyositis, angiosarcoma or Kaposi sarcoma is suspected. Lymphoscintigraphy can be performed on the head and neck but is difficult to interpret. MRI or CT imaging may be useful if an underlying pathology such as cancer, sinusitis or dental root infection is suspected. Such imaging may also help distinguish between swelling from fluids and other tissue components such as fat.
Management
Treatment of facial lymphoedema will depend on the cause. Any inflammation will need to be treated to reduce the higher lymphatic load arising from increased vascular permeability and blood flow. Raising the head of the bed during overnight sleep helps to reduce venous pressure and therefore microvascular filtration. Otherwise the standard principles of enhancing lymph flow through massage techniques and facial exercises apply [12].
In rosaceous lymphoedema, antibiotic therapy appears disappointing in reducing swelling; low-dose isotretinoin has been advocated, but may need to be sustained for 1–2 years [13]. Laser ablation of the telangiectasia may reduce the fluid load on the lymphatics.
There are no data from clinical trials for the treatment of OFG. In one review of 45 patients who required treatment, 24 (53.3%) were treated with topical corticosteroids/immunosuppressants only, whereas 21 (46.7%) received a combined therapy (topical plus systemic corticosteroids/immunosuppressants and/or intralesional corticosteroids). The long-term outcome analysis showed complete or partial resolution of tissue swelling and oral ulceration in 78.8% and 70% of patients, respectively [14]. There are reports of therapeutic success with azathioprine, thalidomide, infliximab and mycophenylate mofetil.
Modified decongestive lymphoedema therapy can be successful in treating head and neck lymphoedema following cancer treatment [15].
Swollen genitalia and mons pubis
Definition and nomenclature
Genital lymphoedema may affect the shaft of penis and/or scrotum plus the mons pubis.
Introduction and general description
Genital lymphoedema may be primary or secondary (Table105.5). The genitalia have the option of bilateral lymph node drainage. For swelling to occur, drainage pathways to both inguinal regions must fail or local genital lymphatics must become occluded.
Table 105.5 Causes of lymphoedema of the genitalia and mons pubis.
| Primary (congenital/genetic) | Secondary |
Noonan syndrome Hennekam syndrome (CCBE1, FAT4) Generalized lymphatic dysplasia Chylous reflux Emberger syndrome (GATA2) Lymphoedema distichiasis (FOXC2) Yellow-nail syndrome |
Cancer (advanced primary, inflammatory cancer, pelvic relapse, skin infiltration) Lymphadenectomy (pelvic, bilateral, ilio-inguinal) Radiotherapy Accidental trauma Obesity Crohn disease/ano-genital granulomatosis Hidradenitis suppurativa Infections: Filanasis Cellulitis Lymphogranuloma venereum Donovanosis Systemic causes (heart failure, nephrotic syndrome) |
In primary genital lymphoedema gene mutations have been identified, including in GATA2 and FOXC2.
Secondary lymphoedema may be caused by advanced or local infiltration of cancer, extensive scarring from accidental or surgical trauma, obesity, granulomatous disease such as Crohn disease or ano-genital granulomatosis (Figure 105.12a), hidradenitis suppurativa (Figure 105.12b) and infections such as filariasis, lymphogranuloma venereum and donovanosis.
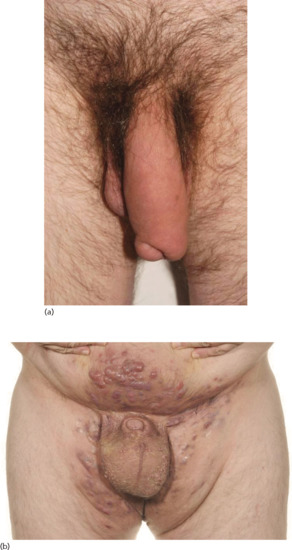
Figure 105.12 (a) Lymphoedema of the penis secondary to ano-genital granulomatosis. There may be no sign of inflammation. (b) Genital lymphoedema secondary to hidradenitis suppurativa. Note the cutaneous lymphangiectasia that predisposes to lymphorrhoea. (From Thomas et al. 2014 [13].)
Mons pubis swelling can develop in isolation but more often is associated with genital or lower limb lymphoedema.
Pathophysiology
Predisposing factors
For primary cases a genetic predisposition is likely. In both primary and secondary cases an infection or other forms of local inflammation (e.g dermatitis) may cause swelling. Compression of leg lymphoedema, through bandages or pneumatic compression pumps, can push fluid up to the trunk. This can result in genital oedema, especially if care is not taken to redirect the lymph through collateral drainage routes.
Pathology
In all forms of pure lymphoedema the pathology is the same, namely increased dermal and subcutaneous thickness through fluid, increased fat and fibrosis. A non-specific inflammatory infiltrate is invariably present [1]. Lymphatic vessels may be increased in number and expanded due to increased lymphatic pressure but they may also be reduced in number through genetically determined underdevelopment or if obliterated by fibrosis.
Causative factors
In primary lymphoedema a genetic cause is probable. To date there are at least four phenotypes for which mutations are known and which cause genital lymphoedema: Emberger syndrome, lymphoedema distichiasis syndrome, Hennekam syndrome and Noonan syndrome. Genital swelling may be a feature of congenital lymphoedema, particularly if part of a generalized lymphatic dysplasia. Chylous reflux into the scrotum may result from congenitally malformed retroperitoneal aortic and iliac lymphatics giving rise to megalymphatics or from intestinal lymphangiectasia.
Most cases (60%) of genital lymphoedema will be caused by obliteration of the upper thigh, inguinal and iliac lymph vessels for reasons not always apparent. One-quarter of cases will be caused by obliteration of outflow lymphatics from the scrotum and 15% caused by reflux [2].
The commonest cause of genital lymphoedema and hydrocele worldwide is filariasis [3]. Other secondary causes of genital lymphoedema include active cancer and its treatment (e.g. bilateral inguinal lymphadenectomy or radiotherapy), granulomatous disease (e.g. Crohn disease and ano-genital granulomatosis) and extensive local inflammation and scarring (e.g. hidradenitis suppurativa). Genital swelling may occur as part of extensive oedema below the waist in heart failure, hypoalbuminaemia and inferior vena cava obstruction. Less common causes include tuberculous lymphadenitis, lymphogranuloma venereum and dovovanosis [4].
Mon pubis lymphoedema is caused by local radiotherapy, obesity and by local inflammatory disorders such as Crohn disease and hidradenitis suppurativa.
Genetics
Mutations in GATA2 cause Emberger syndrome in which genital lymphoedema is one phenotypic feature [5]. Mutations in FOXC2 cause lymphoedema distichiasis syndrome; uncommonly genital lymphoedema and lymphangiectasia can also feature [6]. Mutations in CCBE1 and FAT4 cause Hennekam syndrome (lymphoedema–lymphangiectasia syndrome), a form of generalized lymphatic dysplasia; genital lymphoedema is variable as a feature [7]. In Noonan syndrome molecular genetic testing identifies a mutation in PTPN11 in 50% of affected individuals, in SOS1 in approximately 13%, in RAF1 in 3–17% and in KRAS in fewer than 5% [8].
Clinical features
History
The development of swelling will be dependent on the underlying cause. The onset may be insidious or sudden with no obvious trigger. Infection (e.g. cellulitis) may be a provoking factor. A history of exposure to filariasis with travel to endemic areas must always be considered. Primary cases invariably have one or both lower limbs swollen at the time of onset of genital lymphoedema.
Presentation
In primary lymphoedema swelling may be present at birth or develop later in life. Genital lymphoedema is much more common in men, probably because of anatomy and the dependent nature of male external genitalia. The various parts of the genitalia – the penis, scrotum and labia – are not always equally swollen.
Longstanding lymphoedema causes thickening and hyperkeratosis of the overlying skin with the production of papillomas. These probably arise from lymph congestion within the dermal lymphatics, which, in the early stages, can appear as ‘lymph blisters’ on the skin surface before the tissues become organized and fibrotic. This expansion of congested dermal lymphatics due to backpressure (dermal backflow) is called lymphangiectasia (and not lymphangioma, which strictly implies a lymphatic endothelial proliferation). When the ‘lymph blisters’ are filled with lymph they are translucent but when filled with chyle they are opaque and white. The ‘lymph blisters’ will rupture on occasion, resulting in a copious release of lymph (lymphorrhoea), mimicking incontinence or excessive sweating. Lymphorrhoea can seriously undermine quality of life and risk a contact dermatitis and cellulitis.
Cellulitis attacks are common with genital lymphoedema. Each attack further undermines lymph drainage routes, leading to worse swelling and a higher risk of infections, so establishing a vicious cycle. Offending organisms may be many and difficult to identify. Gram-negative infections should always be considered [9]. The inguinal lymph glands are often enlarged as a result of infection (filarial or bacterial).
Mons pubis lymphoedema presents as a dome-shaped swelling with peau d'orange skin changes. In the obese it can grow to epic proportions whereupon it resembles a pseudosarcoma and is called massive localized lymphoedema [10].
Differential diagnosis
The characteristic skin changes make a diagnosis of lymphoedema relatively straightforward. However, systemic causes of oedema such as heart failure and nephrotic syndrome should be considered when accompanied by more widespread oedema. Hydrocele and an inguinal hernia can be mistaken for oedema.
Complications
Genital lymphoedema can be complicated by infection (e.g. cellulitis) or the leakage of lymph or chyle with resulting contact dermatitis. Penile swelling may interefere with micturition and sexual function. Impotence may develop. A secondary balanoposthitis may occur.
Prognosis
Prognosis is dependent on the underlying aetiology but established lymphoedema is incurable. The risk of severe attacks of cellulitis is ever present.
Investigations
Filariasis must be excluded by a complement fixation test, or nighttime blood smears if active filarial infection is likely. A skin biopsy is essential to diagnose granulomatous disease or cancer infiltrating dermal lymphatics.
Imaging with a CT or MRI scan is necessary to exclude lymphatic obstruction within the pelvis or ilioinguinal glands from cancer or other pathologies (e.g. retroperitoneal fibrosis). Lymphoscintigraphy may be helpful to identify lower limb lymphatic abnormalities. It may demonstrate tracer within the scrotal lymphatics in cases of reflux.
Management
First line
Decongestive lymphatic therapy aims to reduce swelling through a combination of massage and compression [11]. This should only be undertaken if the underlying causes have been treated, such as cancer, granulomatous disease or infection.
Skin care should be scrupulous. Prophylactic antibiotics may be necessary to counter recurrent cellulitis [9]. Compression is easier on the female genitalia than the male. Custom-made tights or shorts are recommended. Foam inserts can increase local pressure comfortably. A scrotal sling or harness may provide support and compression in the male.
Second line
Surgical reduction may be straightforward and effective [12]. Circumcision may resolve preputial swelling and any redundant foreskin. Hyfrecation or diathermy is best for lymphangiectasia. Antibiotic cover is recommended in all cases.
Obesity-related lymphoedema
Definition
Obesity leads to, and exacerbates, lymphoedema at all sites but particularly in the lower limbs.
Introduction and general description
Obesity is a significant risk factor for lymphoedema of the arms, legs and abdomen. Obesity is the strongest risk factor for BCRL [1]. Furthermore, dieting improves arm lymphoedema beyond that possible through loss of subcutaneous fat alone (from weight loss irrespective of the diet used) [2]. The pathophysiology of lower limb lymphoedema can be complex, with increased fluid filtration from venous hypertension combined with impaired lymph drainage from an indirect effect of reduced mobility being the most important contributors (Box 105.1). The addition of obstructive sleep apnoea/sleep apnoea hypoventilation syndrome results in salt and water retention and heart failure.
Epidemiology
A crude estimate of approximately 15 000 patients attending a US clinic showed almost 75% of morbidly obese patients have chronic oedema of the legs [3].
Pathophysiology
Fat and lymphatics appear to have a close relationship. High-density lipoproteins require transport through the lymphatics to return to the bloodstream during reverse cholesterol transport [4]. In a model of hypercholesterolaemia, lymphatic function was severely compromised, including impaired dendritic cell migration. Removal of cholesterol from the peripheral tissues via reverse cholesterol transport requires lymph drainage [5]. Mice with a heterozygous Prox1-inactivating mutation have leaky lymphatic vessels and develop obesity and inflammation [6].
Fat deposition is a striking feature of lymphoedema swelling and the justification for liposuction as a treatment for lymphoedema. Obesity impairs lymphatic transport capacity and impaired lymphatic function promotes adipose deposition. How obesity predisposes to lymphoedema is not clear. Lymph drainage requires movement and exercise to promote flow. In a cross-sectional study, 33% of severely obese participants had lymphoedema and those participants had worse physical function than those without lymphoedema. This association was independent of BMI [7].
It is the lower limb that is most closely linked with lymphoedema. In one study all 10 patients with a BMI between 30 and 53 had normal lower extremity lymphatic function, whereas the five patients with a BMI greater than 59 had abnormal lymphatic drainage consistent with lymphoedema [8]. Using an isotope clearance technique, lymph drainage was found to be significantly lower in obese human subjects when compared with lean controls [9].
A large abdominal apron resting on the thighs during sitting obstructs venous drainage and probably interferes with lymph drainage as well. The pressure in the iliofemoral vein in morbidly obese patients is significantly higher than in non-obese subjects [10]. Abdominal adipose tissue potentially leads to elevated risk for both venous thromboembolism and chronic venous insufficiency. Excess body weight is also related to alterations in the coagulation system, including impaired fibrinolytic activity and elevated plasma concentrations of clotting factors [11].
Clinical features
History
Swelling is usually insidious in onset and progressive. Acute cellulitis may alert patient and carers to the swelling. More often a chronic redness with local pain and tenderness indicative of LDS may develop. Trivial trauma may result in the weeping of fluid from the skin (lymphorrhoea). Persistent weeping will irritate the surrounding skin to promote dermatitis, extensive erosion and even ulceration. Odour may result from bacterial colonization. The constant weeping can discourage the patient from going to bed (to avoid soiling the bed). Consequently, the patient sleeps in a chair, which further increases fluid filtration into the legs. The patient may choose to sleep in a chair anyway for reasons of comfort or sleep apnoea syndrome. As the legs swell more the extra weight further impairs mobility so reducing lymph drainage yet more.
Presentation
Skin changes of LDS and venous hypertension are invariable. Distinction from bacterial cellulitis can be difficult with acute flares of pain and redness. Elephantiasis skin changes are common.
Differential diagnosis
Systemic causes of oedema (including cardiac disease, hypoproteinaemia and abdominal–pelvic malignancy) should always be considered, particularly if bilateral leg swelling is present. Calcium-channel blocking antagonists can exacerbate peripheral oedema.
Complications and co-morbidities
Leg ulceration, heart failure and overwhelming sepsis are common complications. Co-morbidities such as diabetes, sleep apnoea syndrome and right-sided heart failure often coexist.
Prognosis
The prognosis is poor unless the patient loses weight and becomes more ambulant.
Investigations
Standard investigations such as venous duplex ultrasound and lymphoscintigraphy are probably unnecessary as they are unlikely to change management. More important are BNP to exclude heart failure, plasma protein estimation and d-DIMERS if thrombosis is considered likely.
Management
First line
There are a number of management options to pursue.
- Systemic conditions such as heart failure and sleep apnoea syndrome should be treated.
- Compression therapy in the form of multilayer lymphoedema bandaging is the treatment of choice [12]. Standard venous ulcer bandaging will not address toe swelling with skin changes or oedema extending into the lower thighs. Pneumatic compression devices or Velcro wraps can be useful adjunctive therapy. Compression garments should not be used until swelling is controlled and the skin is in good condtion.
- Exercise if logistically possible. This can be done through walking or static cycling, if safe. Active movements should always be encouraged but passive exercises are better than nothing.
- Elevation of legs when resting. Encourage sleeping in a bed and not in a chair unless it is a reclining chair.
- Treat active infection (e.g. cellulitis) [13]
- Wound care should be undertaken where necessary.
- Emollients such as 50/50 white soft and liquid paraffin should always be used. For the hyperkeratosis of elephantiasis, 10% salicylic acid is recommended.
Second line
Consider bariatric assessment and intervention.
Abdominal wall lymphoedema
Definition and nomenclature
Abdominal wall lymphoedema is skin and subcutaneous oedema of the lower abdominal wall generated when ilioinguinal lymph drainage is compromised bilaterally, or within a pendulous obese abdomen.
Introduction and general description
The abdominal wall is not considered a likely place for lymphoedema but is probably more common than generally realized. Diagnosis may be difficult because the soft tissues of the abdominal wall make pitting difficult to elicit. Furthermore, abdominal wall lymphoedema may manifest with more fat than fluid, making distinction from obesity demanding. Peau d'orange skin changes can be a helpful sign and palpation of fluid gives a more solid feel than fat.
Abdominal wall lymphoedema can develop following cancer treatment when ilioinguinal lymph drainage is compromised. In such circumstances lower limb lymphoedema would likely coexist. Cancer relapse must be considered if swelling develops some time after curative cancer treatment. Cancer relapse may be in the pelvis or with infiltration of the abdominal wall skin (e.g. carcinoma erysipeloides).
Non-cancer-related lymphoedema of the abdominal wall is invariably related to obese abdominal panniculus, or in circumstances of extensive oedema such as heart failure, nephrotic syndrome or yellow-nail syndrome. Pretibial myxedema has been described to cause abdominal wall oedema.
Pathophysiology
Compromised ilioinguinal lymph nodes affect all lymph drainage routes below the waist. This usually causes lower limb lymphoedema first, but in more severe cases the lower abdominal wall and genitalia are affected as well. External beam pelvic radiotherapy not only affects the lymph nodes but can also affect smaller collateral lymphatics within the skin and subcutaneous tissue of the mons pubis to cause lymphoedema within the radiation field.
Relapse of cancer with infiltration of the dermal and subcutaneous lymphatics of the lower abdomen can also produce lymphoedema. Advanced cancer can produce profound lymphoedema from the waist downwards, particularly if accompanied by iliac vein or inferior vena cava obstruction.
Obesity is known to cause lymphatic dysfunction and exacerbate lymphoedema. This is particularly so in abdominal panniculus when the abdomen becomes pendulous. This creates venous congestion so increasing fluid filtration into the abdominal wall. Intense oedema complicated by infection often leads to elephantiasis changes (elephantiasis nostras verrucosa). Obese patients often suffer sleep apnoea syndrome, which only further increases systemic salt and water retention.
Causative factors
Causative factors include cancer treatment (e.g. bilateral inguinal or pelvic lymphadenectomy) or radiation therapy for gynaecological, penile, bladder or prostate cancer, as well as relapsed cancer and cellulitis. Obesity where the abdominal panniculus is pendulous is also a major risk factor.
Clinical features
History
The patient may not perceive mild abdominal wall oedema. With progression there may be a sensation of getting fatter, or tightness and discomfort within the affected area. There may be a history of past cancer treatment, obesity or cellulitis.
Presentation
Pitting can usually be demonstrated only over the anterior superior iliac spine. Pinching a fold of lower abdominal skin will reveal thickening with a heavier, more solid feel than just fat. Peau d'orange skin changes may be observed. With time, and progressive accumulation of fat and fibrosis within the swollen area, the tissues will feel more indurated, particularly following an episode of cellulitis. More severe cases can develop marked hyperkeratosis, skin thickening and papillomatosis. These changes produce a warty, cobblestone appearance, referred to as elephantiasis nostras verrucosa (Figure 105.13) [1].

Figure 105.13 Lymphoedema of the pendulous abdomen and mons pubis from obesity.
Cellulitis is a frequent occurrence because of the compromised local tissue immunity. Recurrent attacks exacerbate the lymphoedema, leading to a vicious cycle. LDS may develop within the pendulous abdomen from venous congestion and past cellulitis [2].
Differential diagnosis
Abdominal wall oedema may develop in heart failure, hypoalbuminaemia (nephrotic syndrome, liver disease and malnutrition) and inferior vena cava obstruction.
Complications
Progression of abdominal wall lymphoedema can result in huge swelling, which may hang down as far as the patient's knees, so compromising mobility and risking overwhelming sepsis.
Prognosis
There is a high morbidity and life-threatening sepsis in the very obese.
Investigations
In cancer patients, restaging using abdominal ultrasound or a CT scan is recommended. Skin biopsy may be helpful if infiltrative cancer is considered to be possible. In the severely obese check for the following: (i) heart failure, through BNP estimation; (ii) low plasma albumin; and (iii) sleep apnoea syndrome, through sleep studies.
Management
First line
Correction of any underlying causes is the first priority wherever possible. Diuretics are usually of little help in lymphoedema but may be tried for short periods where physiology involves increased microvascular filtration.
In severe morbid obesity (BMI >50) bariatric intervention should be considered.
Antibiotics should be given if infection is present. Prophylactic antibiotics may be given if there is recurrent infection [3].
Decongestive lymphatic therapy (manual lymphatic drainage and abdominal compression using binding or compression garments) should be administered [4].
Second line
A melon slice surgical apronectomy may be considered but the risk of infection and wound dehiscence is high in obese patients [5].
Cancer-related lymphoedema
Definition and nomenclature
Lymphoedema is rarely a presenting feature of cancer unless the cancer is already advanced but it is a common consequence of cancer treatment and relapsed cancer.
Introduction and general description
Lymph flow is remarkably well maintained through malignant nodes, therefore cancer does not usually present with swelling. The few exceptions to this general rule are lymphophilic tumours, such as malignant eccrine poroma, Kaposi sarcoma, lymphangiosarcoma and inflammatory breast cancer (Table 105.6). Cancer- related lymphoedema usually results from cancer therapy. Surgical lymphadenectomy, radiation therapy and chemotherapy can all contribute.
Table 105.6 Causes of cancer-related lymphoedema.
| Cancers where lymphoedema is a presenting sign | Cancer treatment |
Inflammatory carcinoma Lymphangiosarcoma Kaposi sarcoma Malignant eccrine poroma Advanced primary cancer, e.g. axillary or pelvic lymph node metastases Relapsed cancer: Lymph node metastases Carcinoma erysipeloides Lymphangitis carcinomatosa Carcinoma en cuirasse |
Lymphadenectomy Radiotherapy Chemotherapy (taxanes) Breast reconstruction |
Advanced or relapsed cancer can present with lymphoedema. Extensive lymph node involvement can compromise lymph flow but other factors such as venous obstruction and hypoproteinaemia may also contribute to oedema formation. Recurrent cancer should always be considered as a cause of limb swelling, particularly if associated with pain. Full staging investigations should be undertaken in any cancer patient who develops new limb swelling.
Carcinoma erysipeloides (also called lymphangitis carcinomatosa, carcinoma telangiectatica or carcinoma en cuirasse) occurs when cancer cells infiltrate the dermal lymphatics (Figure 105.14). It represents metastatic disease. Bulk disease may be absent and therefore imaging may be normal. By obstructing collateral lymph drainage routes, dermal lymphatic infiltration by cancer is frequently associated with localized, or extensive limb, lymphoedema.
Figure 105.14 Carcinoma erysipeloides (carcinoma telangiectatica): the red vessels are dermal lymphatic vessels infiltrated with adenocarcinoma of the breast.
Epidemiology
Breast cancer-related lymphoedema. More than one in five women who survive breast cancer will develop arm lymphoedema [1]. Taxane chemotherapy significantly contributes to BCRL [2].
Lymphoedema related to cancers of the male and female uro-genital tract. The incidence of lower limb lymphoedema following radical hysterectomy alone was estimated at 5–10% [3] but can be as high as 49% by 10 years of follow-up in patients who have also received adjuvant radiation treatment [4]. The incidence after vulval cancer was reported at 28% [5]. For prostate cancer the rate was found to be 0–10% after extended pelvic lymphadenectomy [6]. With extended-field irradiation for carcinoma of the prostate an incidence of about 5% for genital and/or leg oedema has been noted and the oedema remained chronic in the majority of patients [7]. After penile cancer treatment the incidence of lymphoedema may be as high as 33% [8].
Melanoma-related lymphoedema. In a prospective study of lymphoedema after melanoma treatment, moderate lymphoedema (i.e. an increase in limb volume of >10%) occurred in 14.8% after sentinel lymph node biopsy but in 30.4% after therapeutic lymph node dissection [9].
Extremity soft-tissue sarcoma. The incidence of lymphoedema was 28.8% following limb salvage for extremity soft-tissue sarcomas. Nine percent of the cohort of 289 patients developed significant (grade ≥2) lymphoedema [10].
Pathophysiology
Predisposing factors
Extensive surgery (e.g. axillary lymph node dissection, greater number of lymph nodes dissected, mastectomy) and being overweight carried the highest risk for BCRL. Recent evidence suggests axillary radiation may convey less risk than axillary clearance [11]. Cellulitis may be a trigger as may an insult to the limb such as sterile or non-sterile skin puncture. Extreme resistance exercise such as carrying a heavy suitcase or shopping may trigger the swelling, as may a long haul flight.
Pathology
Breast cancer-related lymphoedema has been the most widely studied form of lymphoedema. The pathology consists of fat as well as fluid, hence the justification for liposuction treatment.
Carcinoma erysipeloides is a form of metastatic spread and occurs most commonly with breast cancer but can occur with melanoma, thyroid, lung, gastric, pancreatic, ovarian, prostate and colo-rectal cancer [12].
Clinical features
History
Lymphoedema following cancer treatment may occur immediately after lymphadenectomy, particularly if complicated by wound infection or ‘seroma’, or be delayed in onset for many years. Lymphoedema can cause aching but is predominantly painless unless associated with infection, thrombosis or active cancer.
Presentation
Carcinoma erysipeloides manifests clinically with a fixed erythematous patch or plaque resembling cellulitis, but without fever [13]. A network or lattice pattern of telangiectatatic vessels represents the infiltrated dermal lymphatics (Figure 105.14). Inflammatory breast cancer is a rare and very aggressive disease in which cancer cells block the lymph vessels in the skin of the breast. This type of breast cancer is called ‘inflammatory’ because the breast often looks swollen and red, or ‘inflamed’.
Investigations
If relapsed cancer is suspected, restaging investigations such as positron emission tomography (PET)/CT are indicated. Skin biopsy is the investigation of choice for skin metastases. MRI can determine if swelling is fluid and therefore likely to be lympho-edema.
Management
If active cancer is diagnosed then oncology treatment is the priority. If cancer is in remission or stable then lymphoedema treatment (decongestive lymphatic therapy) can be implemented.
Swollen breast and breast lymphoedema
Definition and nomenclature
Unilateral breast oedema is most often caused by breast cancer treatment but can be also caused by infection (e.g. cellulitis), malignancy (e.g. inflammatory breast cancer or angiosarcoma) and inflammatory mastitis. Rarely, it can occur with congestive cardiac failure, nephrotic syndrome and from treatment with mTor (mammalian target of rapamycin) inhibitors.
Introduction and general description
As changes to breast cancer treatment have led to more breast-conserving surgery and increased use of therapeutic radiation to the breast, so the incidence of lymphoedema localized to the breast has risen. The risk is higher in the obese and in women with larger breasts.
Epidemiology
Of 144 women enrolled into one study before cancer treatment, 38 developed breast lymphoedema (26%) [1].
Pathophysiology
Oedema is an excess of interstitial fluid. Any oedema, whatever the cause, is due to capillary filtration overwhelming the lymph drainage for a sufficient period of time [2]. Interstitial fluid is reabsorbed almost entirely by the lymphatic vessels. Inflammation will increase blood flow and vascular permeability, both of which amplify microvascular fluid filtration. Inflammation can be caused by radiation and infection. Lymph drainage may be unable to respond to higher filtration because of effects from axillary surgery in compromising lymph flow. Disturbances in the Starling principle of fluid exchange will be greatest in the most dependent regions of the breast.
Predisposing factors
Carcinoma erysipeloides refers to a red, swollen breast resulting from breast cancer infiltrating the dermal lymphatics overlying the breast. Removal of one or more axillary lymph nodes risks lymphoedema within the drainage basin, that is the ipsilateral upper limb and adjoining quadrant of the chest including the breast. Therapeutic radiation to the breast can also induce/exacerbate breast swelling. Obesity and size of breast increases risk, as may adjuvant taxane chemotherapy [3]. In multivariate analysis from one study, factors associated with the development of breast lymphoedema in the axillary surgery subgroup included baseline BMI (P = 0.004), surgical incision location (P = 0.009) and prior surgical biopsy (P = 0.01) [4].
Other systemic causes for breast lymphoedema include heart failure, low plasma proteins resulting from nephrotic syndrome or liver failure, axillary lymphadenopathy and central vein occlusion. There have been several reports of breast oedema associated with mTor inhibitors [5].
Genetics
Breast lymphoedema is a secondary lymphoedema for which no genetic factors have been yet identified.
Environmental factors
These include surgery, radiation and drugs.
Clinical features
History
Breast swelling can be observed immediately following axillary lymphadenectomy, particularly if a ‘seroma’ or wound infection has occurred (a seroma is a misnomer as it represents a collection of lymph not serum). The onset of swelling can be delayed for months or years, particularly when arising from radiation effects. Swelling may be triggered by an attack of cellulitis.
Symptoms are breast heaviness, swelling, indentations from a bra and sometimes pain and tenderness. Breast redness may feature, indicating inflammation usually secondary to cellulitis, radiation effects or malignancy.
Presentation
Pitting and peau d'orange skin changes are most noticeable on the undersurface of the breast. Inflammation can be present. Cellulitis can coexist and is difficult to distinguish from post-radiation changes (Figure 105.15).
Figure 105.15 Breast lymphoedema following cancer treatment. Note pitting from bra indentations in the skin of the right breast.
Differential diagnosis
Oedema can be determined clinically from indentation due to pressure (pitting). Other differential diagnoses include swelling from hormonal effects and fat hypertrophy.
Complications
Infection is a common complication of breast lymphoedema and further exacerbates oedema. Unexplained breast oedema should always be investigated in case of relapsed breast cancer or the development of (lymph)angiosarcoma.
Prognosis
Uncomplicated breast lymphoedema usually settles with treatment and resolves over time.
Investigations
Breast ultrasound is usually sufficient to confirm oedema and exclude malignancy. MRI is an alternative. A skin or breast biopsy may be necessary if malignancy is suspected.
Management
First line
An infection, if present, needs to be treated. Malignancy needs to be excluded. Obesity should be addressed and weight reduced to as near normal as possible.
Lymphoedema treatment should involve a supportive bra (a sports bra is often the best). It is recommended that the bra be worn both day and night in order to keep the breast uplifted, which overcomes gravitational factors. Massaging techniques are recommended, such as manual lymphatic drainage therapy, kinesiotaping and water immersion exercises (swimming aerobics), although the evidence base for their use is limited [6].
Second line
Mastectomy is a last resort.
Massive localized lymphoedema
Definition and nomenclature
Massive localized lymphoedema is a benign lymphoproliferative soft-tissue overgrowth in the morbidly obese patient. It represents gross lymphoedema usually confined to one area such as a thigh and appearing like a tumour.
Introduction and general description
Lymphoedema typically affects a limb but uncommonly it can present as a localized mass resembling a tumour. It is not unusual for an area of lymphoedema on the lower inner thigh, leg, inguinal region or abdominal apron to hypertrophy into an enlarged fold and then, under the influence of gravity, progress into a pendulous swelling (Figure 105.16). Obesity and repeat attacks of infection locally increase risk [1].

Figure 105.16 Massive localized lymphedema of the right thigh showing marked ‘cobblestone’ skin changes.
Pathophysiology
Predisposing factors
These include obesity, filariasis and recurrent infection.
Pathology
Solid or papillomatous plaques can mimic tumours but biopsy will reveal typical features of lymphoedema, namely oedema, dilated lymphatics, fibrosis, fat, epidermal acanthosis and hyperkeratosis, and inflammatory dermal infiltrate. In one series all 22 cases showed striking dermal fibrosis, expansion of the fibrous septa between fat lobules with increased numbers of stromal fibroblasts, lymphatic proliferation and lymphangiectasia. Multinucleated fibroblastic cells, marked vascular proliferation, moderate stromal cellularity and fascicular growth raised concern among referring pathologists for such conditions as atypical lipomatous tumour/well-differentiated liposarcoma, angiosarcoma and a fibroblastic neoplasm such as fibromatosis [2].
Clinical features
Presentation
An area of lymphoedema becomes raised like a tumour, then under the effects of gravity may become more polypoid-like and feels very heavy. Lesions most resemble a benign tumour such as a pedunculated lipoma although a soft-tissue sarcoma could also be suspected. The overlying skin is markedly thickened skin with a ‘cobblestone’ appearance with elephantiasis skin changes (Figure 105.16). The structure often appears lobulated and can grow to a considerable size [2].
Differential diagnosis
Differential diagnoses include lipoma, lymphatic malformation, lymphocele and sarcoma.
Complications
Weeping of lymph fluid and ulceration are common. Infection with septicaemia frequently occurs. When very large the ‘tumour’ can interfere with movement and mobility. A total of 65 cases of massive localized lymphoedema have been described in the literature, nine of which resulted in angiosarcoma (10.3% of all cases) [3].
Prognosis
The likelihood is that the lesion will continue to enlarge and suffer chronic sepsis unless treated.
Investigations
Magnetic resonance imaging typically demonstrates a sharply demarcated, pedunculated mass consisting of fat partitioned by fibrous septae surrounded by a thickened dermis. There is oedema both within the mass and tracking along the subcutaneous septae in a lace-like fashion outwards from the pedicle, outlining large lobules of fat [4].
Management
The only satisfactory treatment is surgical resection with reconstruction. However, this is not without hazard as wound dehiscence and overwhelming infection can present serious life-threatening risks. Intensive multilayer lymphoedema compression bandaging and IV antibiotics given first might reduce the surgical complications and improve prognosis.
Primary lymphoedema
Definition and nomenclature
Primary lymphoedema arises due to an intrinsic abnormality involving a genetically determined aplasia, hypoplasia, malformation or dysfunction of the lymphatic vessels, whereas secondary lymphoedema results from lymphatic damage due to extrinsic factors such as surgical lymphadenectomy, radiotherapy or chronic venous disease. Primary lymphoedema may occur as a non-syndromic Mendelian condition, or less commonly as part of a complex syndromic disorder [1].
Introduction and general description
Primary lymphoedema, or lymphoedema due to an underlying genetic abnormality, should always be suspected in a patient presenting with swelling and no obvious underlying medical cause. Suspicion of primary lymphoedema should be raised in those presenting during childhood or early adult years.
Historically, primary lymphoedema was classified into three categories depending on the age of onset of swelling: congenita (lymphoedema present at birth), praecox (lymphoedema developing after birth but before the age of 35 years) and tarda (lymphoedema developing after the age of 35 years). It became apparent that this classification system, based purely on age of onset, was oversimplified and redundant in clinical practice as it failed to facilitate categorization based on more specific phenotypes.
Mutations in several genes are known to cause primary lymphoedema. Some, but not all, of these factors have been shown to play a role in the regulating of lymphangiogenesis. The first two genes to be identified as causative for human lymphoedema were VEGFR3 (also known as FLT4) in Milroy disease [2, 3, 4,5] and FOXC2 in lymphoedema distichiasis syndrome [6, 7,8]. Since then several new genes for human lymphatic disease have been discovered. Mendola et al. recently screened 78 patients for mutations in known primary lymphoedema genes and detected mutations in just 36% of cases [9]. This suggests that other causal genes for primary lymphoedema have yet to be identified.
Developments in clinical phenotyping and identification of the genetic cause of a number of subtypes of primary lymphoedema demonstrate that primary lymphoedema is highly heterogeneous. A new classification system and diagnostic pathway has been developed in order to delineate specific primary lymphoedema phenotypes, and facilitate the discovery of new causative genes [1, 10]. Phenotyping and genotyping of patients with primary lymphoedema leads to a better understanding of the natural history and management of specific conditions and more accurate recurrence risks for future generations.
Epidemiology
Incidence and prevalence
Limited data exist on the prevalence of lymphoedema. The prevalence of primary lymphoedema was estimated as one in 6000 based on one UK clinic [11]. More recently, chronic lymphoedema (of primary and secondary causes) was estimated to affect as many as 1.33/1000 population in the UK [12]. A similar survey in Derby estimated 4/1000. Further epidemiological studies are needed to establish the true prevalence of primary lymphoedema. Estimates of prevalence have been difficult to calculate as it is presumed that many affected individuals do not come to medical attention.
Clinical features
Presentation
Primary lymphoedema may be present at birth or develop later in life. One or more limbs may be affected, or indeed there may be generalized/whole body swelling, depending on the subtype of lymphoedema.
The clinical signs of lymphoedema can range from mild swelling to that of grotesque enlargement in chronic, poorly managed patients. Protein-rich materials, lipids and debris accumulate in addition to water. This results in ‘solid’ and ‘fluid’ components to the swelling, giving rise to the ‘brawny’ nature of chronic oedema that resists pitting [13]. Skin changes may be present, including brawny fibrotic skin and the presence of the Kaposi–Stemmer sign (the failure to pinch/pick up a fold of skin at the base of the second toe, as a result of its thickness; see Figure 105.1). The Kaposi–Stemmer sign is pathognomonic of lymphoedema [14]. Papillomatosis (small flesh-coloured papules) occurs as a result of dilatation within the upper dermal lymphatics and subsequent fibrosis of the dermis. Lymphangiectasia appears as small blisters on the skin surface as a result of engorgement of lymphatic vessels. Lymph fluid frequently leaks as a result of minimal trauma and is termed lymphorrhoea.
Clinical variants
A number of subtypes of primary lymphoedema exist. They each present with differing age of swelling onset, distribution of lymphoedema and possible associated health problems. Connell et al.’s diagnostic pathway provides the simplest approach to classification of the various subtypes. However, the pathway remains a work in progress as new subtypes and causal genes are discovered [10]. The primary lymphoedema classification pathway is presented in the form of a colour-coded algorithm to illustrate the five main categories of primary lymphoedema, and the individual subtypes (including genotypes) within these categories (Figure 105.17). The main categories are:
- Syndromic associated with lymphoedema, but where lymphoedema is not the predominant feature) (blue).
- Localized or generalized lymphoedema associated with systemic/visceral lymphatic abnormalities (pink).
- Lymphoedema in association with disturbed growth and/or cutaneous/vascular anomalies (yellow).
- Congenital-onset primary lymphoedema, that is, lymphoedema that is present at birth or develops within the first year of life (green).
- Late-onset primary lymphoedema, that is, lymphoedema that develops after the first year of life (purple).
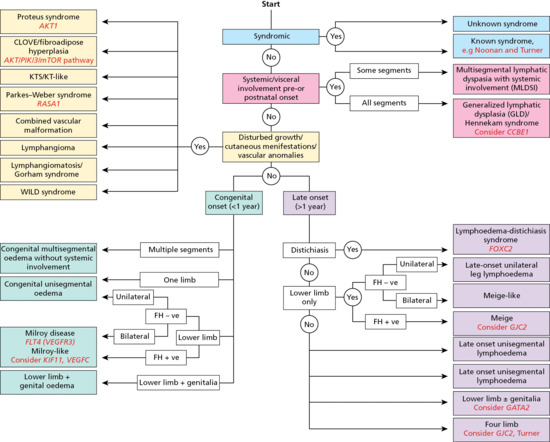
Figure 105.17 Classification pathway for primary lymphoedema. Text in red indicates the suggested genetic test for the subgroup. Please refer to Table105.7 for the definition of other terminology used in the figure [10]. FH, family history: +ve, positive; -ve, negative; KT, Klippel–Trenaunay; KTS, Klippel–Trenaunay syndrome. (From Connell et al. 2013 [10] and Mortimer and Rockson 2014 [54].)
These five categories are discussed in turn below, including the clinical phenotypes and causal gene mutations.
Syndromic
Lymphoedema is a recognized feature of many syndromes. Lymphoedema is not the primary problem in these conditions but is an associated feature. These include Turner and Noonan syndromes. Turner syndrome should always be suspected as the cause of congenital hand and/or foot swelling in female infants.
The genetic causes of many of these syndromes are known and testing is available. Table105.8 provides a list of syndromes that include lymphoedema as part of the phenotype.
Table 105.7 Definition of terminology used in the classification pathway.
| Term | Definition |
| Congenital onset | Onset of lymphoedema before the age of 1 year |
| Cutaneous manifestations | Naevi/pigmentation variations (e.g. epidermal naevi/vascular malformations) |
| Distichiasis | Presence of aberrant eyelashes arising from the meibomian glands |
| Disturbed growth | Hypertrophy (overgrowth) and hypotrophy of bone or soft tissue resulting in altered length of a limb or body part |
| KT/KT-like | Klippel–Trenaunay/Klippel–Trenaunay-like syndrome |
| Late onset | Swelling presenting after 1 year of age |
| Prenatal onset | Detection of lymphatic abnormality in the prenatal period. Isolated pedal oedema is excluded from this definition as this may be a presentation of Milroy disease |
| Segment | A region of the body affected by lymphoedema (i.e. face, conjunctiva, genitalia, upper limbs, lower limbs – each constitute one body part). Multisegmental refers to more than one segment affected by lymphoedema. Bilateral lower limb swelling is not considered to be multisegmental lymphoedema |
| Syndromic | A constellation of abnormalities, one of which is lymphoedema |
| Systemic involvement | Systemic lymphatic problems persisting beyond the newborn period or manifesting at any age thereafter. This includes hydrops fetalis, chylous ascites, intestinal lymphangiectasia, pleural and pericardial effusions and pulmonary lymphangiectasia |
| Vascular anomalies | Includes congenital vascular abnormalities |
Table 105.8 List of known syndromes associated with lymphoedema and the causative gene (or chromosomal abnormality) if known.
| Known syndrome | Chromosome/gene |
| Aagenaes syndrome | Not known |
| Carbohydrate-deficient glycoprotein types 1a, 1b, 1h | PMM2, PM1, ALG8 |
| Cardio-facio-cutaneous syndrome | RAS-MAP kinase pathway including KRAS, BRAF, MAP2K1, MAP2K2 |
| CHARGE syndrome | CDH7 |
| Choanal atresia-lymphoedema | PTPN14 |
| Ectodermal dysplasia, anhidrotic, immunodeficiency, osteopetrosis and lymphoedema (OLEDAID syndrome) | IKBKG (NEMO) |
| Fabry disease | GLA |
| Hennekam syndrome | CCBE1, FAT4 |
| Hypotrichosis-lymphoedema-telangiectasia | SOX18 |
| Irons–Bianchi syndrome | Not known |
| Lymphoedema-myelodysplasia (Emberger syndrome) | GATA2 |
| Macrocephaly-capillary malformation (MCM) | PIK3CA |
| Microcephaly with or without chorioretinopathy, lymphoedema and mental retardation (MCLMR) | KIF11 |
| Mucke syndrome | Not known |
| Noonan syndrome | RAS-MAP kinase pathway PTPN11, KRAS, SOS1 and others |
| Oculo-dento-digital syndrome (ODD) | GJA1 |
| Progressive encephalopathy, hypsarrhythmia and optic atrophy (PEHO) | Not known |
| Phelan–McDermid syndrome | 22q terminal deletion or ring chromosome 22 |
| Prader–Willi syndrome | 15q11 microdeletion or maternal uniparental disomy 15 |
| Thrombocytopenia with absent radius | 1q21.1 microdeletion and RBM8A |
| Turner syndrome | 45, X0 |
| Velo-cardio-facial syndrome | 22q11 microdeletion |
| Yellow-nail syndrome | Not known |
Adapted from Connell et al. 2013 [10].
Lymphoedema with systemic/visceral involvement
A widespread developmental abnormality of the lymphatic system leads to systemic/visceral involvement and swelling that may not be confined to the limbs.
Lymphatic dysfunction may present prenatally with hydrothoraces or hydrops fetalis. The development of in utero oedema may cause dysmorphic facial features such as epicanthic folds, a broad nasal bridge and neck webbing with low-set ears [15].
Systemic lymphatic abnormalities may present with pericardial and pleural effusions, chylous ascites and pulmonary and intestinal lymphangiectasia in the postnatal period. An individual with intestinal lymphangiectasia will complain of abdominal pain and diarrhoea following the ingestion of foods with a high fat content (as the intestinal lymphatics are responsible for fat absorption). Management of systemic lymphatic impairment is not straightforward and a multidisciplinary approach is key. Management includes the drainage of effusions and implementation of a medium-chain triglyceride diet to manage intestinal lymphangiectasia and chylous disorders [16].
Patients with systemic lymphatic abnormalities can be classified into one of two categories depending upon the clinical presentation: a multisegmental lymphatic dysplasia with systemic involvement (MLDSI) or a generalized lymphatic dysplasia (GLD).
Multisegmental lymphatic dysplasia with systemic involvement. Patients with MLDSI have a segmental pattern of lymphoedema. The swelling affects different body parts in association with a systemic lymphatic abnormality. For example, they may have lymphoedema of one or more limbs or body sites (including the face), in association with previous or current systemic lymphatic abnormalities (e.g. intestinal lymphangiectasia, recurrent chylous pleural effusions). The patient has no syndromic features and is of normal intelligence. The underlying mechanism is thought to be of somatic mosaicism, and no known causal genes have been identified to date. There is a low sibling and offspring recurrence risk.
Generalized lymphatic dysplasia. Patients with GLD have a more global pattern of lymphoedema. Swelling typically affects all body parts and often presents in utero with hydrops fetalis. A number of patients with GLD will have a family history of lymphoedema suggestive of autosomal recessive inheritance, inferring a higher recurrence risk than MLDSI. Hennekam syndrome is one type of autosomal recessive GLD and presents with lymphoedema of all four limbs, intestinal and/or pulmonary lymphatic dysplasia, a variable degree of learning difficulties and characteristic facies (a flat face, flat and broad nasal bridge and hypertelorism) [17]. Associated problems include hypothyroidism, glaucoma, seizures, hearing loss and renal abnormalities [18, 19]. Lymphoscintigraphy has rarely been undertaken in this condition but Bellini et al. demonstrated abnormal drainage in the upper and lower limbs and the thoracic duct in one patient [18].
Patients with suspected Hennekam syndrome may be screened for mutations in CCBE1 (collagen and calcium binding EGF domain 1) on chromosome 18q21 [20,21]. However, CCBE1 mutations only appear to account for 25% of cases, suggesting genetic heterogeneity [20]. This is supported by the recent identification of homozygous or compound heterozygous mutations of the FAT4 gene (a member of the protocadherin family, and not a recognized component of the lymphangiogenesis pathway) in four of 24 CCBE1 mutation-negative Hennekam syndrome patients [22].
The function of CCBE1 in humans has yet to be elucidated, but animal studies confirm that ccbe1 enhances the process of lymphangiogenesis induced by vascular endothelial growth factor C (VEGFC), and is also necessary for the budding and sprouting of lymphangioblasts [23, 24].
Lymphoedema associated with disturbed growth and/or cutaneous/vascular anomalies
Lymphoedema may develop in association with vascular abnormalities, disorders of growth and cutaneous abnormalities. Identifying the type of malformation and thereby the vessels involved is important for diagnosis and management.
These patients are challenging as there is considerable overlap in clinical findings and a clear-cut diagnosis based upon phenotype alone is not always possible. The recent identification of de novo somatic mutations as the underlying mechanism for some of these conditions may further understanding and improve management. For example, gene abnormalities within the AKT/PIK3/mTOR pathway have been found to cause Proteus and CLOVES syndromes [25,26]. This has allowed separation of these conditions within the diagnostic pathway, and has identified gene pathways that could be targeted by pharmacological therapy [27]. Conditions with gene abnormalities within the AKT/PIK3/mTOR pathway that are associated with lymphoedema include the following.
CLOVES syndrome. The spectrum of clinical signs include congenital lipomatous overgrowth, vascular malformations, epidermal naevi and skeletal abnormalities (CLOVES). Somatic mosaicism of activating mutations within PIK3CA have been reported [26]. The same gene and same mechanism have also been implicated in a few patients with Klippel–Trenaunay syndrome and with fibroadipose hyperplasia [26, 27]. Lymphatic malformations can arise as part of these conditions. Further molecular studies, together with careful phenotyping, will facilitate the understanding of this spectrum of disease. It is likely that combined vascular malformations may also arise as a result of somatic mosaicism.
Klippel–Trenaunay syndrome. Patients present with a combination of several or all of the following within a single limb: limb length hypertrophy of muscle, fat or bone (e.g. presenting as increased limb length or girth), varicose veins, vascular malformation (e.g. capillary malformations) and lymphoedema. Activating mutations within PIK3CA have also been reported [26].
Proteus syndrome. Proteus syndrome is a disease characterized by progressive, segmental overgrowth of the bones, skin and connective tissue. Lymphatic and capillary malformations are the most common vascular changes seen in this syndrome. Clinical signs of Proteus syndrome may not be present at birth but develop during infancy and progress throughout the individual's life. The diagnosis may be established using diagnostic clinical criteria and/or molecular analysis [28]. The majority of individuals with typical Proteus syndrome will have an activating mutation in AKT1, arising as a result of somatic mosaicism [25]. AKT acts within the molecular pathway that includes the mTOR and PIK3CA genes. This pathway is critical as a progrowth and antiapoptosis facilitator. The identification of the PIK3/AKT/mTOR signalling pathway underpinning disease processes gives rise to the potential for therapeutic targets through inhibition of this pathway [27].
Congenital multisegmental lymphoedema. Patients with congenital multisegmental lymphoedema without systemic lymphatic impairment have been included within the yellow section of the classification pathway in order to avoid confusion with multisegmental lymphoedema occurring in association with systemic lymphatic abnormalities (in the pink section of the pathway). These patients have an asymmetrical pattern of lymphatic failure with some limb sparing (i.e. some limbs are unaffected) but without overgrowth or cutaneous or vascular abnormalities. It is possible that somatic mosaicism in gene(s) involved in lymphangiogenesis could explain this subtype of congenital primary lymphoedema.
WILD syndrome. This syndrome is a condition comprising clinical signs of warts, immunodeficiency, lymphoedema and ano-genital dysplasia (WILD) [29]. These patients have extensive, asymmetrical, multisegmental lymphoedema comprising facial and conjunctival oedema, genital lymphoedema, and epidermal naevi and/or capillary malformations typically of the torso and upper limbs. These patients have a sporadic condition, suggesting probable somatic mosaicism [10]. The underlying genetic cause has yet to be identified.
Congenital-onset primary lymphoedema
Historically, all cases of congenital lymphoedema were classified as Milroy disease. However, several different types of congenital lower limb primary lymphoedema have been recognized.
Milroy disease. Milroy disease presents with congenital lymphoedema of the lower legs (usually symmetrical). The onset of swelling may occasionally be delayed but will occur within the first year of life. Lymphoedema is typically confined to the feet and ankles, but may progress up to the knees. Up-slanting ‘ski-jump’ toenails are present as a result of disturbance of the nail bed by oedema. Prominent large-calibre veins are frequently present on the feet and pretibial regions. Varicose veins, typically the long saphenous veins, are a common finding in adults with Milroy disease, but do not appear to affect the paediatric population. A third of affected males have hydroceles [30]. Milroy disease rarely presents in the antenatal period with hydrops fetalis but the outcome may be favourable (i.e. the swelling may regress and remain confined to the feet), or the hydrops may progress and result in intrauterine death [31, 32].
Lymphoscintigraphy in Milroy disease confirms failure of the initial lymphatic vessels to absorb fluid. The term ‘functional aplasia of lymphatic vessels’ has been used to describe the characteristic lymphoscintigraphy results [10]. The initial lymphatic vessels are present (confirmed on histological examination) but unable to absorb interstitial fluid [33].
Abnormalities within the gene that encodes vascular endothelial growth factor receptor type 3 (VEGFR-3) on chromosome 5q35 are causal of Milroy disease [2, 31]. Mutations in the tyrosine kinase domain of VEGFR3 are found in 70% of patients with congenital-onset primary lymphoedema affecting both lower limbs [34]. Inheritance is autosomal dominant but de novo cases may occur [2, 3].
Milroy-like lymphoedema. Individuals with congenital lower limb lymphoedema who do not have an underlying VEGFR3 mutation are classified as having Milroy-like lymphoedema. Mutations within the VEGFC gene have been identified as causal in several families within this category [35,36]. VEGFC is the ligand for VEGFR-3 and controls lymphatic sprouting during embryonic development [23, 37]. Affected individuals typically have all the clinical signs of Milroy disease (congenital lower limb lymphoedema, prominent large-calibre veins and hydroceles, inherited in an autosomal dominant pattern) yet their lymph scans are atypical.
MCLID syndrome. Microcephaly with or without chorioretinopathy, lymphoedema or intellectual disability (MCLID) syndrome is an autosomal dominant syndrome comprising congenital lower limb lymphoedema (mimicking Milroy disease) as well as microcephaly and variable degrees of learning difficulties that occur as a result of mutations in the KIF11 gene on chromosome 10q24 [38]. The presence of chorioretinopathy is variable but should always be excluded by an expert ophthalmology opinion. Lymphoscintigraphy demonstrates the same pattern of lymphatic functional aplasia as that seen in Milroy disease. The KIF11 protein product is involved in spindle formation during mitosis but is not clear how KIF11 relates to the lymphangiogenesis pathway [39]. MCLID should be considered in all patients with congenital bilateral lower limb lymphoedema and microcephaly.
Late-onset primary lymphoedema
The term late-onset lymphoedema is used to describe a primary lymphoedema that develops after the first year of life (i.e. non-congenital lymphoedema). This section contains a number of assorted conditions, some with life-threatening associated diseases (e.g. Emberger syndrome), but they all share the common finding of non-congenital limb swelling.
Lymphoedema distichiasis syndrome. This condition presents with pubertal onset of bilateral lower limb lymphoedema. Distichiasis (aberrant eyelashes arising from the meibomian glands) is present in 95% of affected individuals and is frequently present at birth but rarely causes symptoms until childhood [40]. Other clinical signs include ptosis, cleft palate and congenital heart disease. The majority of affected individuals have varicose veins [41]. Lymphoedema distichiasis syndrome occurs as a result of mutations in the FOXC2 gene on chromosome 16q24 and is inherited in an autosomal dominant manner [7]. FOXC2 encodes a transcription factor necessary for ensuring normal development of the lymphatic collecting vessels and valves [42]. Lymphoscintigraphy of affected individuals demonstrates reflux of lymph within the lower limbs as a result of valve failure within the lymphatic vessels [43]. Similarly, abnormal venous valves lead to early-onset venous reflux in all patients with FOXC2 mutations [44].
Meige disease. Meige disease is the most prevalent subtype of primary lymphoedema [10]. It typically presents with bilateral lower limb lymphoedema that rarely extends above the knee. The onset of symptoms is in adolescence or adulthood. There are no other associated features of the condition [45]. Lymphoscintigraphy frequently demonstrates abnormal deep rerouting of lower limb lymph drainage as evidenced by an increased uptake of tracer within the popliteal lymph nodes and impaired main superficial lymphatic tract filling [10]. Family history is consistent with an autosomal dominant pattern of inheritance yet the causal gene of classic Meige disease has not yet been identified.
Emberger syndrome. Emberger syndrome comprises late-onset (but in childhood) bilateral or unilateral lower limb with or without genital lymphoedema together with myelodysplastic syndrome and/or acute myeloid leukaemia [46]. It may also be associated with a high-frequency, progressive sensorineural deafness. Severe cutaneous warts occur as a result of the associated immune dysfunction. Myelodysplasia may develop at any stage and will progress to acute myeloid leukaemia with a high mortality [47]. Lymphoscintigraphy has not been routinely performed on patients with this condition.
Mutations in the GATA2 gene on chromosome 3q21are causal and are inherited in an autosomal dominant manner [48]. GATA2 is expressed in lymphatic, vascular and endocardial endothelial cells [49]. Mouse studies suggest the lymphoedema occurs as a result of abnormal lymphatic valve development [50]. GATA2 is also involved in the regulation of haematopoiesis, hence the association of lymphoedema with myelodysplasia in this rare, yet life-threatening, condition [51].
Late-onset four-limb lymphoedema. An autosomal dominant pattern of late-onset lymphoedema affecting either the lower limbs, or all four limbs, occurs as a result of mutations in the GJC2 gene [52]. Apart from lower limb varicose veins, no other associated conditions have been reported. Lymphoscintigraphy demonstrates lymphatic tracts that appear normal but with significantly reduced quantification uptake of tracer, reflecting reduced absorption from tissues by peripheral lymphatics in all four limbs [52]. The functional role of GJC2 within the lymphatic system is unclear as it encodes for the connexin 47 protein located on chromosome 1q42, and is not known to be involved in the lymphangiogenesis pathway.
Differential diagnosis
Secondary lymphoedema that has occurred as a result of an underlying medical cause needs to be excluded, for example lower limb lymphoedema as a result of venous insufficiency. A detailed history, examination (and possible investigation with lymphoscintigraphy or venous duplex imaging) should provide the underlying diagnosis.
Classification of severity
The severity of the lymphoedema may be classified according to the clinical features (Table105.9), and this may be useful in monitoring response to therapy over a prolonged period.
Table 105.9 Stages of lymphoedema adapted from the International Society of Lymphology Consensus Document [53].
| Stage | Clinical signs |
| Stage 0 | A latent/subclinical state where swelling is not evident despite impaired lymphatic transport. This stage may exist for many months or years before oedema becomes evident |
| Stage I | Early onset of lymphoedema where there is accumulation of tissue fluid that reduces with limb elevation; pitting may be present |
| Stage II | Accumulation of fluid that does not reduce on elevation; at later stages it may be non-pitting |
| Stage III | Fibrotic tissue and absent pitting; elephantiasis skin changes develop (e.g. acanthosis, fatty and fibrous tissue changes, and warty overgrowths) |
Complications and co-morbidities
See the section called Complications of lymphoedema later in this chapter.
Management
See the section called Lymphoedema management later in this chapter.
Resources
Patient resources
Lymphoedema Support Network: http://www.lymphoedema.org (last accessed August 2015).
Lipoedema
Definition and nomenclature
Lipoedema is a disorder of adipose tissue that occurs almost exclusively in women, usually at a time of hormonal change. Patients have progressive fatty swelling of the lower limbs, with associated easy bruising, skin tenderness and pain of the lower limbs.
Introduction and general description
Lipoedema is a condition characterized by abnormal adipose deposition within the lower limbs. It occurs almost exclusively in females, and is thought to be an inherited disorder. The onset of symptoms typically occurs at a time of hormonal change such as puberty or pregnancy. Patients complain of progressive fatty swelling of the lower limbs, with associated easy bruising, skin tenderness and pain of the lower limbs. Contrary to what the name suggests, oedema is not a key feature and the ‘swelling’ is caused by adipose tissue and not fluid. Lipoedema was first described by Allen and Hines in 1940 as ‘bilateral enlargement of the legs thought to be due to abnormal deposition of subcutaneous fat and accumulation of fluid in the lower legs’ [1]. The lack of a clear definition of the disorder has led to significant confusion regarding diagnosis and management. In fact, some clinicians consider it a physiological variant rather than a disease [2]. However, lipoedema recently received acceptance as a medical condition and obtained an MIM number in 2011.
Epidemiology
Incidence and prevalence
Limited prevalence data exist for lipoedema. Some clinicians believe it is a rare disorder [3], but many feel that it is more common than expected and is often misdiagnosed as simple obesity or lymphoedema [4, 5,6].
Age
Onset is typically at puberty or other times of hormonal change such as pregnancy or commencement of the oral contraceptive pill.
Sex
Lipoedema occurs almost exclusively in females. Only six cases of lipoedema affecting males have been reported in the literature [7,8,9]. Child et al. identified four unrelated isolated adult males with the condition. All were thought to have developed lipoedema secondary to hormonal disturbances, with reduced testosterone levels being a common factor [7].
Ethnicity
No ethnic differences have been reported.
Pathophysiology
Pathology
The aetiology of lipoedema is still unknown. It has not yet been determined whether the cause of the ‘fatty swelling’ is due to adipocyte hypertrophy or hyperplasia, or a combination of both. Histological examination of tissue biopsies and liposuction aspirates identified oedema of the adipocytes and/or interstitium, but no other abnormalities were detected [9, 10]. One study postulates that activated adipogenesis occurs in lipoedema tissue leading to hypoxia and subsequent adipocyte necrosis and recruitment of macrophages, as occurs in obesity [11]. Child et al. proposed that a hormonal influence was likely to underlie the condition as lipoedema appeared to be expressed most commonly at puberty or other times of hormonal change [7].
Genetics
Frequent observations of mother to daughter inheritance led to the hypothesis that lipoedema is a genetic disorder. Patterns of inheritance of the condition within families are consistent with either X-linked dominant inheritance or autosomal dominant inheritance with sex limitation [7].
Clinical features
History
Affected individuals develop bilateral and symmetrical ‘fatty’ non-pitting swelling, usually confined to the legs and hips [8]. The feet are spared, giving rise to an ‘inverse shouldering’ or ‘bracelet’ effect at the ankles (Figure 105.18). Patients frequently complain of tenderness and easy bruising of the affected areas. The onset of lipoedema is usually at or soon after puberty but can appear to develop at other times of hormonal change such as pregnancy or even menopause [7]. Over time, the patient may develop similar clinical signs in their upper arms. Wold et al. [8] have proposed a set of diagnostic criteria for lipoedema:
- Occurrence is almost exclusively in women.
- It has a bilateral and symmetrical nature with minimal involvement of the feet, resulting in an ‘inverse shouldering’ or ‘bracelet’ effect at the ankle.
- Minimal pitting oedema is seen.
- There is pain, tenderness and easy bruising.
- Persistent enlargement is found despite elevation of the extremities or weight loss.
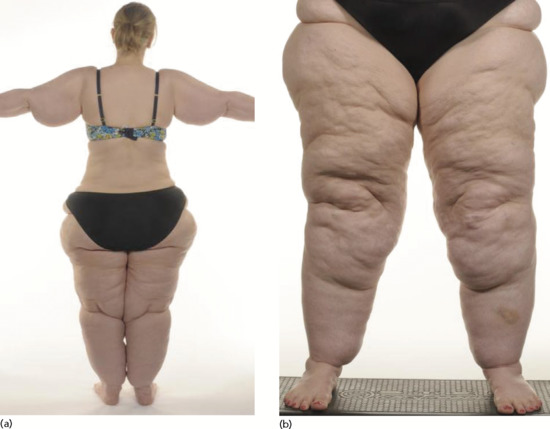
Figure 105.18 A patient with classic lipoedema. (a) Symmetrical fatty swelling of both lower limbs with sparing of the trunk. Increased adipose deposition of the upper arms is present. (b) Lipoedema features include sparing of the feet with ‘inverse shouldering’ of the ankles. Fat pads are developing on the medial aspect of both knees.
Presentation
In the teenager or young adult the lipoedema phenotype is characteristic; the classic disproportionate distribution of fat below the waist, the coexisting features of tissue tenderness and easy bruising and the lack of response to weight-reducing diets all argue against a form of obesity. However, later in life, lipoedema can be complicated by obesity and/or lymphoedema, rendering the diagnosis more difficult to make. In this case historical symptoms are key to the diagnosis [7]. Whilst lipoedema is confused with obesity by many clinicians, associated obesity has, nonetheless, been observed in 50% of patients [12].
Differential diagnosis
Lipoedema must be differentiated from lymphoedema, a more common cause of bilateral lower limb enlargement. However, lymphoedema typically results in asymmetrical oedematous swelling due to the accumulation of interstitial fluid within tissue spaces. Typical cutaneous findings of lymphoedema include brawny, hard and warty changes of the skin and subcutis. Pitting (where the skin remains indented for a few minutes after removal of firm finger pressure for 30 s) is often present during the initial stages of lymphoedema. In contrast, cutaneous changes and pitting are almost always absent in lipoedema [7].
Clinicians may be tempted to confuse lipoedema with Dercum disease, another ‘painful fat’ disorder. However, patients with Dercum disease typically have multiple, painful, diffuse or nodular lipomas (with chronic pain lasting more than 3 months) and a generalized obesity problem. Men may be affected, but there is a female preponderance with a ratio of 5–30 : 1 [13].
Classification of severity
Table105.10 contains the clinical stages of lipoedema as suggested by Schmeller and Meier-Vollrath [3].
Table 105.10 Stages of lipoedema.
| Stage | Clinical signs |
| Stage I | The skin is smooth and the subcutaneous layer is thickened, soft and with an even structure. This stage can last for several years |
| Stage II | The skin might be cool in certain areas as a result of functional vascular imbalance. Over time, subcutaneous nodules develop and the skin surface becomes uneven |
| Stage III | After several decades, patients may develop large amounts of tender subcutaneous tissue and bulging protrusions of fat, mainly at the inner side of the thighs or knees, which lead to an impairment of gait |
Adapted from Schmeller and Meier-Vollrath 2006 [3].
Complications and co-morbidities
Lipoedema is frequently complicated by the onset of a secondary lymphoedema, resulting in the clinical picture of lipo-lymphoedema [7]. The clinical features of lipo-lymphoedema range from mild pitting oedema of the feet to severe swelling of the entire lower limbs as a result of impaired lymphatic drainage. Allen and Hines proposed that the progressive oedema formation in lipoedema was the result of poor resistance of accumulated fat against the hydrostatic passage of fluid from the capillaries into the interstitium [1]. Lymphoscintigraphy performed after the onset of lipo-lymphoedema will confirm the presence of main tracts but lymphatic drainage will be impaired.
Disorder of gait, joint deformities (e.g. genu valgum) and pain may occur as a result of disproportionate adipose tissue of the legs. A number of women complain of knee pain prior to the onset of radiological changes, suggesting that knee pain is part of the lipoedema phenotype (possibly due to an associated disorder of connective tissue) or secondary to the strain put on the knee joint from increased limb volume.
Patients with lipoedema often develop psychological morbidity as a result of their chronic progressive disorder [14].
Disease course and prognosis
Poorly managed lipoedema will undoubtedly progress to lipo-lymphoedema.
Investigations
The diagnosis of lipoedema is currently clinical with no absolute phenotypic features or confirmatory test. Numerous investigations have been undertaken in patients with lipoedema, including lymphoscintigrams, venograms, arteriograms and magnetic resonance lymphangiography, all failing to demonstrate specific abnormalities but suggesting a subclinical reduction in lymphatic function in patients with lipoedema [4, 15–17]. None of these investigations demonstrated specific diagnostic signs of lipoedema.
High-resolution cutaneous ultrasonography has been proposed as a method for differentiating lipoedema from lymphoedema. Naouri et al. reported a significant difference in dermal thickness between the two groups – no dermal thickening detected in lipoedema compared with marked dermal thickness in lymphoedema [18]. However, the experienced clinician will be able to tell the two conditions apart by the soft ‘doughy’ consistency of excess subcutaneous adipose deposition in lipoedema compared with the pitting, firmer tissue consistency of lymphoedema.
Management
No curative therapies are available for lipoedema. Management goals are aimed at the improvement of subjective symptoms (e.g. pain), prevention of lipoedema progression and prevention of lipo-lymphoedema.
Conservative therapies including manual lymph drainage, intermittent pneumatic compression and multilayered short-stretch bandaging were shown to reduce the perception of pain in patients with lipoedema [19]. However, these treatments have no effect on the reduction of limb volume, unless there is coexistent lymphoedema. Compression therapy has been shown to prevent additional oedema formation [20, 21].
The prevention of lipoedema progression is key. Physical activity should be encouraged as it will reduce the risk of obesity and may have a positive impact on the oedema component. Dietary advice and advice on avoiding weight gain is a crucial part of the management plan. Patients should be made aware that obesity is a major exacerbating factor of lipoedema [22].
Liposuction has been accepted as a therapeutic option for lipoedema. Treatment aims to cause a reduction of limb volume/mass, a reduction of pain and improvement in mobility. Reliable criteria for liposuction timing do not exist. It would appear that treatment in the early stages yields a better outcome than when liposuction is used in advanced cases or in lipo-lymphoedema [22, 23]. The long-term data on liposuction are still weak but the immediate results appear promising. However, patients must be aware that liposuction does not offer a cure. If the patient does not maintain their weight in the postoperative period then they will return to the original disease state [24].
Resources
Patient resources
Lipoedema UK: www.lipoedema.co.uk.
Lymphoedema Support Network: www.lymphoedema.org.
(Both last accessed August 2015.)
Yellow-nail syndrome
Definition and nomenclature
Yellow-nail syndrome (YNS) comprises overcurved, smooth, translucent, slow-growing nails with a yellow discoloration along with respiratory ailments such as chronic sinusitis, bronchiectasis or pleural effusion, and lymphoedema. Although listed by McKusick [1] as a genetic condition it more commonly develops late in adulthood as a sporadic condition.
Introduction and general description
The first case series of 13 patients who were given a diagnosis of YNS was described by Samman and White [2]. Typically, the patient has: (i) lymphoedema; (ii) a respiratory disease such as pleural effusion; and (iii) yellow dystrophic nails. Two of these features are required for the diagnosis, since the complete triad is only observed in about one-third of patients.
Epidemiology
A recent systematic review of 150 patients revealed that their median age was 60 years (range: newborn to 88 years). YNS occurred in all age groups but in 78.7% (100/127) patients were aged between 41 and 80 years. The male : female ratio was 1.2 : 1. All cases had lymphoedema and 85.6% had yellow nails. Pleural effusions were bilateral in 68.3%. The appearance of the fluid was serous in 75.3%, milky in 22.3% and purulent in 3.5% [3].
Pathophysiology
The pathogenesis of this condition is unknown. Lymphangiogram abnormalities have been described [4] although a lymphoscintigraphic study demonstrated that a primary lymphatic abnormality is unlikely [5].
Pathology
The pleural effusion in YNS is an exudate in the vast majority of patients. There is a lymphocytic predominance in 96% of cases, with a low count of nucleated cells. In 61 of 66 (92.4%) patients, pleural fluid protein values were >3 g/dL.
Genetics
Wells described a family with eight cases in four sibships of two generations [6]. In the proband, who had yellow nails, lymphoedema began in the legs at the age of 51 years. At times oedema also affected the genitalia, hands, face and vocal cords. Lymphangiograms were interpreted as showing primary hypoplasia of the lymphatics [6].
Govaert et al. reported a girl who was born at 33 weeks’ gestation with non-immune hydrops and a recurrent left chylothorax to a mother with YNS. The non-immune hydrops in this case was diagnosed on a 29-week ultrasound examination [76]. Slee et al. reported a case of a newborn infant who, at 23 weeks’ gestation, was found to have hydrops on antenatal ultrasonography; bilateral chylothorax was found at delivery [8]. The mother had YNS with typical nail changes and bronchiectasis. The infant had a recurrent cough, possibly preceding an early onset of bronchiectasis.
Clinical features
History
The average age of onset reported is during the sixth decade. Patients are usually first aware of not needing to cut their nails. The nails then become thicker and harder to cut. The nail may discolour yellow but also may lift up and become opaque (due to onycholysis) or go green/black due to secondary haemorrhage or Pseudomonas infection. Picking up items may prove difficult due to tactile dysfunction. Nails may fracture or shed. Nail units may become painful, particularly if affected by a paronychia. Cough is the commonest respiratory symptom and may be productive. Shortness of breath may indicate pleural effusion. Peripheral oedema usually starts in the ankles bilaterally but may become very widespread and involve the trunk, genitalia and upper limbs if severe.
Presentation
Nail changes include slow growth, yellow-green discoloration, transverse and longitudinal overcurvature, onycholysis, shedding, cross-ridging and loss of lunulae and cuticles. The nail plate is yellow and overcurved, but remains translucent and smooth (Figure 105.19).
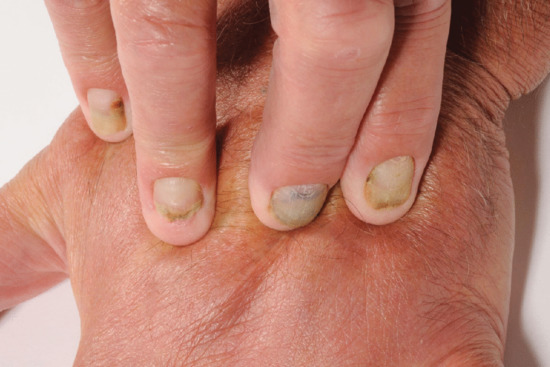
Figure 105.19 Yellow-nail syndrome showing slow-growing, overcurved, thickened nail plates and hand oedema.
Although lymphoedema is the most consistent association with yellow nails, other associations include bronchiectasis, sinusitis, chronic cough and pleural effusions. Rarely a pericardial effusion can occur.
Differential diagnosis
Nail changes are not unusual with lymphoedema due to secondary fungal infection, although fungus causes a softer and more friable nail plate. A diagnosis of YNS is difficult without the characteristic nail changes. Cor pulmonale can present with respiratory symptoms and peripheral oedema.
Complications
Recurrent chest infections are the commonest complication. Recurrent cellulitis frequently complicates lymphoedema. Acute paronychia can complicate the nail unit changes.
Prognosis
In one study four of the 11 patients had complete recovery of their nails over a mean period of 4–5 years [9]. There are some suggestions that life expectancy is reduced [10].
Investigations
Yellow-nail syndrome is a clinical diagnosis. There is no confirmatory diagnostic test.
Management
The most effective treatments for pleural effusions appear to be pleurodesis and decortication/pleurectomy [3]. The lymphoedema does not respond well to diuretics, which is predictable as diuretics do not improve lymph drainage. Nevertheless diuretics such as spironolactone may be necessary if oedema is widespread and severe. Vitamin E is the most common treatment prescribed for the nail changes but the evidence base for this is weak [11]. Treatment of the chronic paronychia with fluconazole has been claimed to be beneficial in anecdotal reports [12].
Lymphatic malformation and lymphangioma circumscriptum
Definition and nomenclature
Vascular tumours are endothelial neoplasms characterized by increased cellular proliferation. Haemangioma is the most common and is almost exclusive to infants. Vascular malformations, on the other hand, are the result of abnormal development of vascular elements during embryogenesis and fetal life. These may be single-vessel forms (capillary, arterial, lymphatic or venous) or a combination. Vascular malformations do not generally demonstrate increased endothelial turnover [1]. The same principles apply to lymphangioma as to lymphatic malformations.
Introduction and general description
The term lymphangioma circumscriptum has been in common usage by dermatologists to describe a lymphatic malformation. Strictly, a lymphangioma should be a ‘growing lymphatic tumour’ involving proliferating lymphatic endothelium. A lymphatic malformation once formed from an abnormal development of lymphatic vessels grows only by expansion/distension, not proliferation, and therefore lymphangioma is an inappropriate term in such circumstances. A lymphatic malformation results from an error in the embryonic development of the lymphatic system.
Lymphangiomatosis is a disorder of proliferative lymphatic vasculature. It is characterized by the progressive involvement of various body tissues and can involve the skeletal system, connective tissues and visceral organs.
Localized congenital lymphatic malformations can be divided into macrocystic (or deep) and microcystic (or superficial) lesions. Lymphatic malformations can have both microcystic and macrocystic components. Lymphatic malformations with few but large macrocystic swellings containing clear lymph are called cystic hygromas (hygroma = moist or watery tumour). Most occur in the neck, but they frequently extend into the upper mediastinum. The term tends to be reserved for those congenital lymphatic malformations that present at birth or are diagnosed by prenatal ultrasound. In the neck they presumably arise from an embryonic jugular lymph sac. Individuals with Turner syndrome are particularly prone to both hydrops and cystic hygroma. Exceptionally, a cystic hygroma occurs in the groin, presumably from an embryonic iliac lymph sac. Fetal cystic hygroma can give rise to severe abnormalities, leading to fetal death [2]. Deeper, larger and cavernous lymphatic malformations can be called macrocystic lymphatic malformations. Lymphangioma circumscriptum is a term best reserved for a lymphatic malformation that is localized to an area of skin, subcutaneous tissue and sometimes muscle [3] (Figure 105.20).
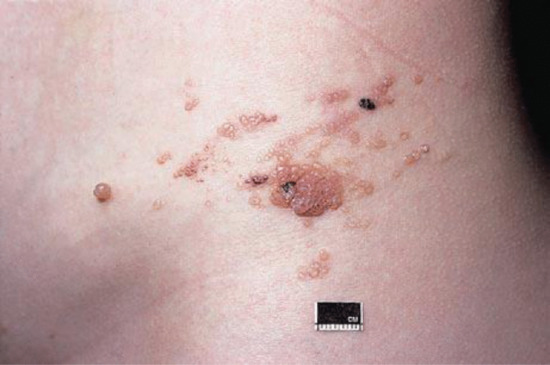
Figure 105.20 Lymphangioma circumscriptum showing fluid-filled vesicles resembling frogspawn. At times the vesicles can contain blood, weep clear fluid (lymphorrhoea) or become warty.
Epidemiology
Lymphatic malformations account for 4% of all vascular malformations, but comprise 25% of benign vascular growths in children [4]. Approximately 75% present at birth, while the remainder present by 2 years old. The anatomical location of a lymphatic malformation is most frequently the head and neck (48%). A study has demonstrated that lymphatic malformations involve the left side of the body more frequently (62%) [5].
Pathophysiology
Pathology
A lymphatic malformation is characterized by the size of the malformed channels: microcystic, macrocystic (often known as cystic hygroma if in the neck) or combined (microcystic/macrocystic) [6]. The contraction of thickened muscular linings may increase intramural pressure and cause cystic dilatation [7]. On light microscopy of biopsies, there are marked expanded channels in the dermis, which can extend into the subcutaneous and other deeper tissues. The channels are lined with flattened cells, which express CD31 and D2-40.
Lymphatic malformations can be isolated lesions disconnected from nearby normal lymphatic vessels (atruncular) or they can be connected (truncular) in which case lymphoedema will feature. In such a malformation, lymph fluid is trapped within the ectatic malformed lymph vessels, whereas in lymphoedema the lymph fluid is within the interstitial space of the tissues.
These malformations can be part of a syndrome such as Turner syndrome (due to monosomy X) or overgrowth syndromes, such as Proteus syndrome, Klippel–Trenaunay–Weber syndrome (capillary lymphaticovenous malformation) and CLOVES syndrome due to mutations in the AKT/PIK3 pathway [8].
Causative factors
Following regional lymph node excision or radiotherapy, cutaneous lymphangiectasia may occur [9]. In such circumstances, which commonly affect the external genitalia, the scrotum or vulva is studded with multiple clusters of tiny, translucent vesicles (see Figure 105.26). Previous surgery and/or radiation disrupt the deep collecting channels in the tissue, leading to lymph stasis, lymph backflow and, subsequently, distention of the upper dermal lymphatics. The mechanism for the skin ‘lymph blisters’ or vesicles is therefore the same as for congenital ‘lymphangioma circumscriptum’ but the cause is quite different.
Genetics
The lack of familial forms and the unifocality of the lesions suggest that the cause could be a somatic mutation restricted to cells of the lesion. Such a mutation would be too damaging (i.e. lethal) if it occurred germline [8].
Somatic mutations that activate PIK3CA have been identified in patients with CLOVES syndrome and in some with Klippel–Trenaunay–Weber syndrome [10]. All of these proteins are core elements of the AKT/PIK3/mTOR pathway, suggesting that sporadic lymphatic malformations as well as overgrowth syndromes may be caused by somatic changes in the components of this pathway. A recurrent somatic activating mutation has been discovered in the AKT1 pathway in patients with Proteus syndrome [11].
Clinical features
History
Lymphatic malformations manifest at birth or during infancy either as a localized subcutaneous swelling or as ‘frogspawn’ (groups of watery or haemorrhagic vesicles or lymph blisters) on the skin (see Figure 105.20). They grow proportionally with the affected child. Some show marked progression especially during puberty and may not be noticeable until this time. If the malformation is disconnected from normally draining lymphatic pathways/trunks there will be no lymphoedema (atruncular) (Figure 105.21). If connections exist then limb lymph drainage will be adversely affected and lymphoedema will be observed and may be the presenting feature (truncular) (Figure 105.22) [12].

Figure 105.21 Atruncular lymphatic malformation without lymphoedema.
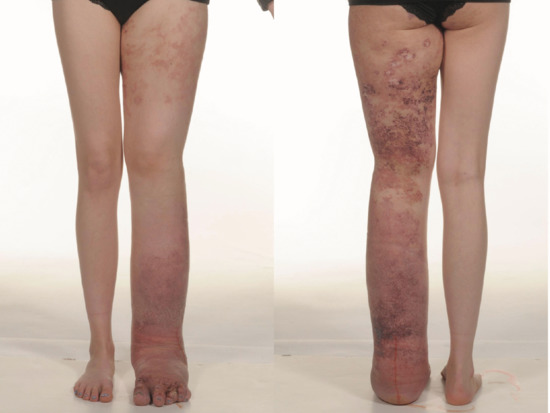
Figure 105.22 Truncular lymphatic malformation with blood-filled cutaneous lymphangiectasia and lymphoedema.
Lymphatic malformations may occur at any site, although they often appear in the mouth, on the neck or jaw or around the axilla or groin. The most common symptom is recurrent oozing, usually of clear fluid (lymph), known as lymphorrhoea. Infection (e.g. cellulitis) can be a presenting feature because of the dysfunctional immune cell trafficking associated with a lymphatic malformation.
Extensive lymphatic malformations can have a blood vascular component. Consumption coagulopathy and in particular low platelets can complicate them. This may be because the correct separation of the lymphatic and blood vasculatures during embryonic development is dependent on CLEC-2-mediated platelet activation [13].
Presentation
Lymphangioma circumscriptum may present at any age but is usually noted at birth or appears during childhood. The commonest sites are the axillary folds, shoulders, flanks, proximal parts of the limbs and perineum. Clinically, the condition manifests with fluid-filled vesicles (‘lymph blisters’), which bulge on the skin surface (see Figure 105.20). They may be translucent when the overlying epidermis is very thin, or they may vary in colour from red to blue-black when they contain blood, which is a frequent occurrence. The surface of the lymphangiomas may appear extremely warty and the lesions may be mistaken for viral warts. Weeping of lymph or blood-stained lymph (lymphorrhoea), or local cellulitis, may be other presentations.
Vascular birthmarks in the skin that look like blood capillary malformations may be mistaken for lymphatic capillary dermal malformations as seen in Proteus syndrome. Biopsy and staining with D2-40 may be necessary to make the distinction although the dermal vessels may be sufficiently undifferentiated to express both blood capillary and lymphatic phenotypes.
Differential diagnosis
A lymphatic malformation can be mistaken for other vascular malformations, hamartomas or tumours. The surface lymph vesicles can be confused with human papillomavirus warts.
Complications
Lymphatic malformations of the neck region often lead to obstruction of the upper airways. The volume of the malformation increases with infection or trauma. Intracystic haemorrhage is also common. Frequent discharge of lymph fluid (lymphorrhoea), ulceration and infection are the most frequent complications. Pain can be a problem but it is difficult to know if the cause is infection, lymph thrombosis or lymphatic vessel distension from intralymphatic pressure. Squamous cell carcinoma is described arising within lymphangioma circumscriptum [14].
Prognosis
Prognosis is usually excellent, providing there is no lymphangiomatosis. Infection can rarely be severe and life threatening.
Investigations
Biopsy will reveal angulated channels, which stain for CD31 and D2-40. Ultrasound should demonstrate fluid-filled channels, which on duplex will be slow flow. Distinction from a slow-flow venous malformation is difficult. MRI can demonstrate the extent of the lymphatic malformation – that is, extension into muscle or bone. Overgrowth of tissue elements (fat, muscle or bone) may also be seen, as may be any lymphoedema component. Lymphoscintigraphy should be normal in an atruncular lymphatic malformation but abnormal in a truncular lymphatic malformation.
Management
First line
The majority of lymphatic malformations are best managed conservatively. Sclerotherapy is the mainstay of treatment of macrocystic lymphatic malformations, but the response using traditional sclerosants is much less beneficial in microcystic lesions. Sclerotherapy of microcystic lymphatic malformations using bleomycin is effective and safe, giving a complete response in 38% and a partial response in 58% (n = 31) in one series [15].
It is generally not possible to excise a lymphatic malformation because of its extent and ill-defined infiltration of surrounding tissues. Indeed, attempted excision may result in further growth of the remaining malformation. Debulking procedures may be necessary for very large malformations causing complications such as the obstruction of vital organs and their functions.
Surface vesicles that weep or bleed should be destroyed by diathermy or laser. Since the vesicles have a tendency to reform, such procedures can be repeated as often as necessary.
Infection should be treated promptly with antibiotics, as in lymphoedema. If recurrent attacks of infection occur then prophylactic antibiotics should be considered [16].
For truncular lymphatic malformations with lymphoedema, compression garments are recommended. In atruncular lymphatic malformations compression may prove fruitless, as the lymph fluid is trapped within the closed vessel system of the malformation [17].
Second line
Sildenafil can reduce lymphatic malformation volume and symptoms in some children [18].
Third line
For those patients with a proven mutation in the AKT/PIK3/mTOR pathway, mTOR inhibitors such as sirolimus may prove helpful. Clinical trials are underway.
Lymphoedema as a result of amniotic band constriction
Definition and nomenclature
Lymphoedema of a limb may occur as a result of amniotic band constriction. This congenital disorder is caused by the in utero circumferential entrapment of fetal parts by fibrous amniotic bands. These bands cause constriction and fibrosis, with subsequent impairment of regional lymphatic drainage, resulting in lymphoedema. Early surgical release may prove beneficial, but affected individuals typically suffer lifelong problems with lymphoedema.
Introduction and general description
Lymphoedema of a digit or limb may occur as a result of constriction by amniotic bands. This congenital disorder, often termed amniotic band syndrome (ABS), is thought to occur as a result of in utero circumferential entrapment of fetal parts by fibrous amniotic bands formed during the rupture of the amnion during the first trimester of gestation. The rupture results in the formation of adhesions between the amnion and fetal skin. Small strands of amnion envelop the developing digits or limbs, leading to circumferential band-like constrictions.
Aside from circumferential limb constrictions, pseudosyndactyly, intrauterine amputation and umbilical cord constrictions have been reported. The reduction in amniotic fluid and/or limb tethering by amniotic bands may result in reduced fetal movements, scoliosis, limb deformity, lung hypoplasia and hydrops [1]. Examination of the placenta and membranes confirms the diagnosis when aberrant bands are detected.
Amniotic bands rarely present in the antenatal period on routine ultrasound imaging. Isolated amniotic bands are more likely to be detected at birth. If amniotic bands occur with limb deformities with or without necrosis, all attempts are made to salvage a necrotic limb, but amputation may be necessary.
Longer term sequelae of amniotic bands include the development of lymphoedema. The in utero circumferential entrapment of fetal parts by fibrous bands causes constriction and fibrosis, with subsequent impairment of regional lymphatic drainage. Impaired drainage of lymphatic fluid will result in lymphoedema, usually within the first few months of life. Patients with milder forms (e.g. amniotic bands that are not fully circumferential) may not develop lymphoedema until later life, if at all.
Epidemiology
Incidence and prevalence
The incidence is less than one in 1 000 000.
Age
Such lymphoedema is found in the neonatal period.
Sex
There is no gender discrepancy.
Ethnicity
There is no ethnic variation.
Associated diseases
There is a strong association between amniotic bands and clubfoot (talipes) [2]. Other associated abnormalities include clubhand, haemangioma, cleft lip and/or cleft palate [3].
Pathophysiology
Predisposing factors
A study conducted by the National Center on Birth Defects and Developmental Disabilities (Atlanta, USA) observed that maternal cigarette smoking and aspirin use increased the risk of limb reduction deficiencies accompanied by amniotic bands [4].
Pathology
The pathogenesis of amniotic bands is not yet fully understood. Two theories have been suggested, and most favour the first.
Amniotic band theory. This theory is that bands occur due to a partial rupture of the amniotic sac. The rupture involves only the amnion whilst the chorion remains intact. Fibrous bands of the ruptured amnion float within the amniotic fluid and encircle and constrict parts of the fetus. Subsequently, the fetus grows but the bands do not enlarge, causing constriction of the affected limb or digit. Constriction compromises the vascular supply, causing congenital abnormalities. One or more digits or limbs may require surgical amputation soon after birth if they are necrotic. A number of infants have undergone complete ‘natural’ amputation of an affected digit or limb prior to birth [5].
Vascular disruption theory. This suggests there must be an ‘intrinsic’ defect of the vascular circulation as the constricting mechanism of the amniotic band theory cannot fully explain the high incidence of cleft defects occurring with ABS. An underlying mechanism for this theory has not been reported [6].
Genetics
It is a sporadic condition, with rare exceptions: a few affected families have been described [7].
Clinical features
History
Amniotic bands rarely present in the antenatal period on ultrasound. Clinicians may be alerted to their presence if the fetus develops associated problems. Isolated amniotic bands are more likely to be detected at birth.
Presentation
Amniotic bands may affect digits or limbs. There is considerable variation in clinical presentation, depending upon the site of the amniotic band(s). Limb deformity and/or necrosis may be present [2]. Lymphoedema may develop in the antenatal period as a result of circumferential limb constriction by fibrous amniotic bands. The swelling will develop distal to the site of the band, enhancing the appearance of the narrow linear band around the affected limb (Figure 105.23). Milder cases (e.g. amniotic bands that are not fully circumferential) may not develop lymphoedema until later life, if at all.
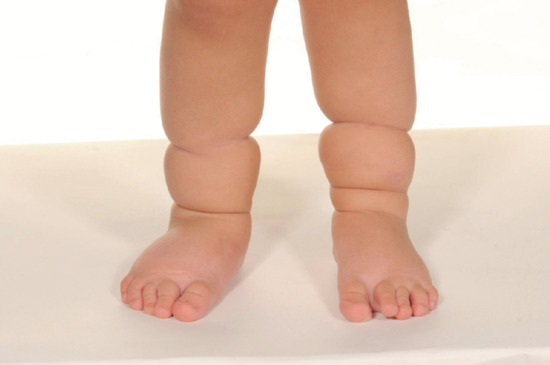
Figure 105.23 Amniotic bands and lymphoedema.
Differential diagnosis
The differentiation of amniotic bands from Adams–Oliver syndrome should be straightforward. The latter condition is an autosomal dominant disorder characterized by transverse limb reduction defects, cutis marmorata and aplasia cutis congenita over the posterior parietal region with an underlying bone defect [8].
Complications and co-morbidities
Affected individuals have an increased risk of infection (e.g. cellulitis) within the swollen limb due to impaired lymphatic drainage and subsequent reduced immune surveillance within it [9].
Disease course and prognosis
Prolonged constriction of a limb by an amniotic band will result in impaired lymphatic drainage, leading to lymphoedema distal to the band site. If untreated, this will deteriorate over time and cause a deformed, heavy limb with reduced function.
Investigations
Prenatal ultrasound scans may detect swelling of digits or limbs distal to the amniotic band constriction, but the band itself cannot be visualized [5]. Three-dimensional ultrasound and MRI may be used for more detailed imaging when the index of suspicion for ABS is high [10].
Management
First line
Treatment typically occurs after birth, but with the advancement of prenatal radiodiagnosis fetal surgery in utero has been attempted [11].
Children with circumferential or near-circumferential amniotic bands may undergo surgical release with a Z-plasty procedure [2]. The aim of surgery is to release the fibrous tissue, thereby relieving constriction of both the vascular supply and draining lymphatic vessels of the limb. Surgery cannot completely reverse the constriction so the child is left with a degree of impaired lymphatic drainage within the affected limb.
Second line
The child will benefit from the input of an experienced lymphoedema therapist to improve limb drainage. Provision of made-to-measure compression hosiery can improve lymphatic drainage if worn on a daily basis.
Lymphangiomatosis, lymphangioleiomyomatosis and non-malignant lymphatic tumours
Definition and nomenclature
Lymphangiomatosis refers to excessive growth of aberrant lymphatic vessels. The process may vary from overgrowth of a lymphatic malformation to a lymphatic tumour. A fundamental requirement is cellular proliferation of lymphatic endothelial cells.
Introduction and general description
There is no clear distinction between lymphatic malformations, lymphangiomatosis and some non-malignant lymphatic tumours. Lymphangiomatosis implies there is progression through proliferation of aberrant lymph vessels whereas in a lymphatic malformation there should be no progression other than expansion though distension from intralymphatic pressure. Most lymphatic malformations are stable and run a completely benign course. Conversely, some visceral thoracic and abdominal lymphatic malformations can relentlessly progress, infiltrating vital organs with fatal outcome and making distinction from lymphangiomatosis and frank neoplasia difficult.
Some non-malignant vascular tumours may exhibit a lymphangioma-like appearance with positive staining for lymphatic markers (e.g. tufted angioma, kaposiform haemangioendothelioma, multifocal lymphangioendotheliomatosis with thrombocytopenia, papillary intralymphatic angioendothelioma, retiform haemangioendothelioma and adult-type haemangioendotheliomas). However, it can be difficult to know if these are primarily lymphatic tumours. They can demonstrate locally aggressive behaviour and can be associated with life-threatening systemic complications such as Kasabach–Merritt syndrome.
The central nervous system seems to be spared, presumably because of its lack of lymphatic vessels. Lungs, retroperitoneal tissues and abdominal organs are most commonly affected by lymphatic malformations and lymphangiomatosis.
Chylous reflux may complicate visceral lymphangiomatosis. Disseminated intravascular coagulation can occur and may give rise to thrombosis and haemorrhage [1].
Gorham disease. Gorham disease (Gorham–Stout disease or disappearing/vanishing bone disease) is characterized by proliferation of lymphatic vessels resulting in progressive destruction and resorption of the osseous matrix. Overlying skin changes of lymphangioma may be present. The major distinguishing characteristic is the progressive osteolysis seen in this disease [2].
Generalized lymphatic anomaly. Generalized lymphatic anomaly (GLA) is a rare congenital disease resulting in malformation of the lymphatic vessels involving multiple sites of soft tissues, lungs, abdominal organs or bones but, unlike Gorham disease, is not usually progressive. Findings suggestive of GLA are more extensive involvement, particularly of the appendicular skeleton, the presence of discrete macrocystic lymphatic malformations and visceral organ lesions [3].
Benign lymphangioendothelioma. Acquired progressive lymphangioma (benign lymphangioendothelioma) is a benign tumour that differs from simple ‘acquired lymphangioma’ or simple cutaneous lymphangiectasia by its clinical behaviour and histopathology [4]. It presents as reddish or bruise-like plaques, which are usually located on the abdominal wall, thigh or calf. Typically, the condition affects young adolescents but may also arise in adults. It is usually localized, flat and grows slowly. Acquired progressive lymphangioma is considered to originate from lymphatic endothelium. The histopathological appearance can mimic a low-grade sarcoma or Kaposi sarcoma [5]. Anastomosing dilated channels, with a tendency to dissect the collagen bundles, are lined by swollen endothelial cells but without cellular atypia. It usually runs a long and benign course. D2-40 and Prox1 immunostains are positive [6].
Maffucci syndrome. Maffucci syndrome consists of diffuse haemolymphangiomatosis accompanied by severe, widespread deformities of bone and cartilage, notably enchondromas of the digits [7]. The lymphangiomas do not appear, on lymphography, to communicate with the main lymphatic pathways and often possess both blood vascular and lymphatic elements. Bony deformity may be gross; slowly uniting pathological fractures are common. The disease has high malignant potential including the development of lymphangiosarcoma [8].
Kaposiform lymphangiomatosis. Kaposiform lymphangiomatosis is a relatively newly described form of lymphangiomatosis, found mainly in children although adults can be affected. Skin involvement resembles diffuse Kaposi sarcoma and can lead to lymphoedema. It is distinguished from generalized lymphatic anomaly and diffuse pulmonary lymphangiomatosis in part by characteristic haematological abnormalities and haemorrhagic complications, including haemoptysis [9]. Characteristic clusters or sheets of spindled lymphatic endothelial cells (human herpesvirus 8 negative) accompany malformed lymphatic channels histologically (Figure 105.24).
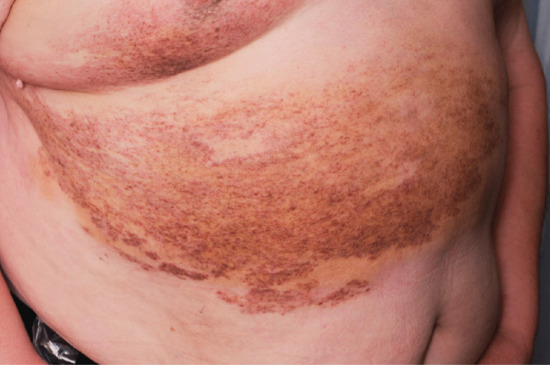
Figure 105.24 Kaposiform lymphangiomatosis showing a Kaposi sarcoma-like rash with localized lymphoedema of the breast and chest wall associated with haemoptysis.
Atypical vascular lesions. Cutaneous vascular proliferations that occur in the field of prior radiotherapy include angiosarcoma and small, cutaneous lesions with a pseudosarcomatous pattern that are reported as atypical vascular lesions or benign lymphangiomatous papules (Figure 105.25) [10]. Microscopically, the lesions are located mostly in the superficial/mid-dermis and are composed of expanded, irregularly jagged, vascular channels lined by a single layer of bland endothelial cells which are invariably D2-40 positive. Lesions can show additional cytological and/or architectural atypia but the prognosis is excellent [11,12].
Figure 105.25 Benign lymphangiomatous papules (atypical vascular lesions) post radiotherapy.
Lymphangioleiomyomatosis. Lymphangioleiomyomatosis (LAM) is a slowly progressive, low grade, metastasizing neoplasm, associated with cellular invasion and cystic destruction of the pulmonary parenchyma. LAM is almost exclusively seen in women, especially during childbearing age. LAM cells harbour mutations in the tuberous sclerosis genes. Lymphatic manifestations of LAM include thoracic duct wall invasion, lymphangioleiomyoma formation, chylous fluid collections in the peritoneal, pleural and pericardial spaces, chyloptysis, chyle leak from the vagina or umbilicus, chylous pulmonary congestion, and lower extremity lymphoedema. Skin and kidney involvement is frequently found in tuberose sclerosis–LAM. Dermatological manifestations in tuberose sclerosis include angiofibromas, hypomelanotic macules, shagreen patch and ungal fibromas. Serum VEGFD estimation and high-resolution CT can be helpful in diagnosis. mTor inhibitors (e.g.sirolimus) are effective treatment [13].
Lymphangiectasia
Definition and nomenclature
Lymphangiectasia (or lymphangiectasis) means distension, expansion or dilatation of a lymph vessel.
Introduction and general description
Cutaneous lymphangiectasia represents distended, but otherwise normal, dermal lymphatics engorged with lymph due to a failure of downstream drainage. The surface ‘lymph blisters’ or vesicles seen in cutaneous lymphangiectasia are not necessarily structurally or histologically any different from those seen in a lymphatic malformation or lymphangioma. Acquired or secondary lymphangioma is an alternative term, but confusing as there is no tumour or proliferative component.
Lower limb lesions usually arise in association with lymphoedema following either ilioinguinal block dissection or pelvic surgery and radiotherapy for cancer, or when cancer relapses. Lymphangiectasias/acquired lymphangiomas have been described in association with scarring processes, including recurrent or chronic infections (such as the scrofuloderma variant of tuberculosis), scleroderma, keloid and repeated trauma (Table105.11). They may also occur as a consequence of defective collagen or elastin, as documented in a report of penicillamine dermopathy [1].
Table 105.11 Causes of cutaneous lymphangiectasia.
| Congenital | Acquired (acquired lymphangioma) |
Lymphatic malformation (lymphangioma circumscriptum) Chylous reflux (intestinal lymphangiectasia) Noonan syndrome Lymphoedema distichiasis |
Post surgery Radiation therapy Scarring: Accidental trauma Inflammatory: Filariasis Crohn/ano-genital granulomatosis Hidradenitis suppurativa Tuberculosis adenitis/scrofuloderma Chylous reflux, e.g. post DXT |
DXT, radiotherapy.
Genital skin is particularly prone to lymphangiectasia. Lymphangiectasia/acquired lymphangioma of the vulva or scrotum is described following cancer treatment, tuberculous inguinal lymphadenitis, hidradenitis suppurativa and genital involvement with Crohn disease or ano-genital granulomatosis [2].
Pathophysiology
Cutaneous lymphangiectasia arises following damage to previously normal, deep lymphatic vessels [3]. The mechanism by which they form is identical to congenitally determined lymphatic malformations [4]. Obstruction to drainage leads to back pressure and dermal backflow, with subsequent congestion and expansion of the upper dermal lymphatics. Lymphangiectases are not true neoplasms or hamartomas, but represent simple expansion and engorgement (lymphangiectasia) of normal dermal initial lymphatic vessels.
Pathology
Histologically the dermis exhibits expanded, angular, lymphatic vessels, which are CD31 and D2-40 positive on staining. Endothelial cell atypia is usually absent.
Clinical features
History
For cutaneous lymphangiectasia, weeping and discharge of lymph or chyle is the commonest symptom. The discharge may be confusingly blood-stained as blood readily enters lymph.
Presentation
The clinical appearance of cutaneous lymphangiectases/acquired lymphangiomas may vary greatly, ranging from clear, fluid-filled blisters to smooth, flesh-coloured papules or nodules. Typically, lymphangiectasia can be seen as translucent, almost flat, papules or vesicles in the skin, which may ooze lymph spontaneously or after trauma. Blood content may make them look black and like angiokeratomas. Genital lesions may become hyperkeratotic on the surface and be mistaken for warts (Figure 105.26). Lesions may be solitary but scattered throughout a lymphoedematous limb, or they may be grouped, as seen in ‘lymphangioma circumscriptum’.
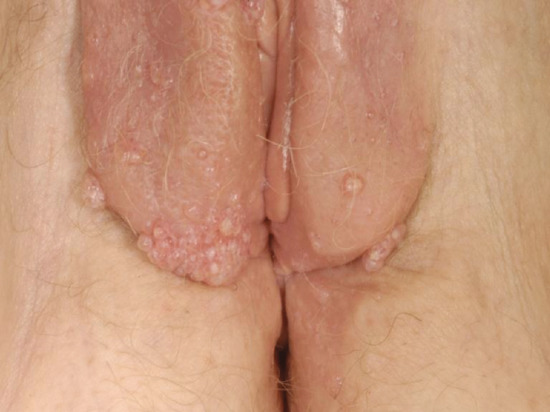
Figure 105.26 Vulval lymphangiectasia showing acquired lymphangiomas (lymphangiectasias) following cervical cancer treatment. The lymphangiomas were mistaken for genital warts.
Differential diagnosis
Differential diagnoses include human papillomavirus warts and molluscum contagiosum.
Clinical variants
Chylous reflux may manifest as lymphangiectasia. In such circumstances the lymph blisters/vesicles are white or cream coloured due to the milky chyle contained within. Fat is normally absorbed through the gut lacteals and drained through the cisterna chyli to the thoracic duct. Disturbances to this drainage route either within the gut wall (intestinal lymphangiectasia) or mesenteric lymphatics will result in a redistribution of chyle to other sites such as the pleural, pericardial or peritoneal cavities (chylous ascites). An incompetence of valves within the main abdominal lymphatic trunks results in gross reflux of chyle to the lower limbs, perineum and genitalia [5]. The reverse flow to skin or ‘dermal backflow’ will result in chylous cutaneous lymphangiectasia. Chylous ‘blisters’ may occur on the toes. Chylous reflux can occur through a fault of lymphatic development such as in Noonan syndrome or be acquired from damage to lymph drainage routes through accidental or surgical trauma, filariasis or malignancy.
Intestinal lymphangiectasia is an uncommon disorder and an important cause of protein-losing enteropathy [6]. The major symptoms are peripheral oedema, and abdominal pain and diarrhoea on dietary fat challenge. Hypoproteinaemia, low serum albumin and immunoglobulin levels are found on investigation. Biopsies of the small intestine showed variable degrees of dilatation of lymph vessels in the mucosa and submucosa. Capsule endoscopy is the investigation of choice [7]. Primary intestinal lymphangiectasia can be associated with the Turner, Noonan and Hennekam syndromes. In secondary intestinal lymphangiectasia, the dilatation of the lymphatics is caused by obstruction of the vessels or an elevated lymph pressure, secondary to elevated venous pressure. Obstruction can be seen in patients with inflammatory bowel disease, sarcoidosis or lymphoma or in patients who have had pelvic radiotherapy. Secondary intestinal lymphangiectasia is observed in children with congenital heart disease who have undergone a Fontan operation [8]. Dietary replacement of fat with medium-chain triglycerides (MCTs) is recommended as MCTs are absorbed and directed to the portal venous system rather than to the intestinal lacteals [9].
Complications
Recognition and appropriate treatment of cutaneous lymphangiectasia is important primarily because the lesions may act as portals of entry for infection. In addition, persistent leakage of lymphatic fluid may be mistaken for urinary incontinence in the case of vulval lymphangiectasia.
Prognosis
Even when treated, cutaneous lymphangiectasia often recurs.
Investigations
Biopsy with D2-40 stain is the definitive investigation.
Management
Treatment of cutaneous lymphangiectasia/acquired lymphangiomas is essentially the reduction of underlying lymphoedema and the control of infection. This may be relatively straightforward on the leg, but is not so easy on the genitalia, where compression is difficult to implement. Destruction of the ‘lymph blisters’ by laser or diathermy is helpful as palliative treatment [10].
Lymphocele, seroma and lymph fistula
Introduction and general description
Lymphoceles (lymphocysts) occur when afferent lymph vessels are disrupted and lymph fluid accumulates in a potential space without a distinct endothelial lining. When copious lymph fluid drains externally it is referred to as a lymph fistula. A seroma is a pocket of clear serous fluid that also collects in a tissue space usually after surgery. Seromas may be difficult to distinguish from lymphoceles.
Lymphocele
Lymphoceles usually occur following surgery or accidental injury. The wall of lymphoceles is ‘false’, in that no endothelial lining exists and instead a dense network of fibrin with lymphocytes is present [1]. Dermatologists might come across groin or axillary lymphoceles as they are superficial and frequently associated with infection and wound complications. In cases of groin lymphoceles, treatment options include observation, serial aspiration and compression, instillation of sclerosing agents, radiation therapy, negative pressure wound therapy, and operative resection of the cavity with or without muscle flap coverage.
Lymphoceles following varicose vein surgery or vein harvesting have become much less common with changes in vascular surgical practice. With increasing levels of plastic surgery, particularly abdominoplasties and thigh lifts, lymphatic complications are likely to become more common.
Seroma
Following lymphadenectomy it is not unusual for a localized swelling containing clear fluid to develop. Often referred to as a seroma, the fluid is not serum but an exudate, equivalent to lymph, that fills the tissue space [2]. Aspirated fluid is indistinguishable from lymph. Repeat aspiration is often necessary until collateral lymph drainage forms. A seroma, particularly if infected, may herald the onset of lymphoedema if alternative drainage routes are not established.
Lymph fistula
A lymph fistula occurs where a lymphatic vessel connects externally to the skin surface and weeps copious amounts of lymph. It usually occurs following trauma or surgery where lymph accesses the skin surface through a wound. It may be a feature of a lymphatic malformation. A chylous fistula drains fat rich lymph externally.
Lymphatic filariasis
Definition and nomenclature
The single largest cause of lymphoedema worldwide is lymphatic filariasis (LF) [1]. It is a parasitic disease caused by microscopic worms that are transmitted by mosquitos. The adult worms live within the human lymphatic system and disrupt drainage, resulting in lymphoedema and hydroceles. Worldwide, 120 million people are infected and 40 million of these have lymphatic problems. Access to health care and lymphoedema treatment is often limited, resulting in a severe burden of disease in endemic countries.
Introduction and general description
Infection with one of three parasitic filarial worms causes LF: Wuchereria bancrofti, Brugia malayi and B. timori. W. bancrofti infection accounts for 90% of LF worldwide [2] (see also Chapter 33). The adult worms reside within the afferent lymphatic vessels (and/or the lymph nodes) while their larvae, the microfilariae, circulate within the peripheral blood and are able to infect mosquito vectors as they feed, facilitating transmission to other human hosts (Figure 105.27). The adult female worm may survive for more than a decade and is able to release thousands of fully formed microfilariae into the lymphatic circulation of the host every day. Infected patients may be asymptomatic, or demonstrate acute or chronic manifestations. The clinical signs are related to the adult worms residing in the lymph vessels and are not due to the microfilariae. The filarial parasites specifically target the lymphatics and impair lymph flow, which is critical for the maintenance of fluid balance and physiological interstitial fluid transport [3,4].
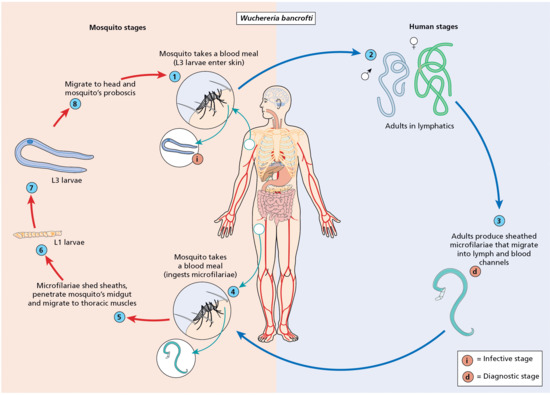
Figure 105.27 The life cycle of filarial nematodes in the human and mosquito hosts. Wuchereria bancrofti, Brugia malayi and B. timori have similar life cycles. The adult worms reside in the lymphatic system of humans and cause filarial disease. The female worm produces offspring (microfilariae), which leave the lymphatic system, enter the blood system of the human host and are taken up by mosquitoes during a blood-meal. The microfilariae undergo development within the mosquito, become infective larvae and subsequently migrate to the mosquito's mouthparts. These larvae may be transmitted to humans when the mosquito takes its next blood-meal. Once transmitted to humans, the larvae take approximately 6–12 months to mature into adult worms. (From Centers for Disease Control and Prevention 2015 [23].)
Epidemiology
Lymphatic filariasis is endemic in 73 countries and 1.2 billion people are at risk of transmission and subsequent infection. An estimated 120 million individuals are currently infected. Approximately 40 million have chronic lymphatic pathology: 13 million with lymphoedema/elephantiasis and an additional 27 million males have hydroceles as a result of LF [5, 6]. Areas where LF is endemic include parts of Africa, South-East Asia, the western Pacific, the Americas and the Middle East. Transmission and morbidity are highest in South-East Asia and sub-Saharan Africa [6].
Age
While infections are contracted throughout life, most individuals remain asymptomatic until symptoms emerge during adolescence and adulthood.
Sex
Males are more likely to be affected than females.
Pathophysiology
Pathogenesis of lymphatic disease
The pathogenesis of filarial disease remains poorly understood and has been a subject of great debate. The clinical consequences of lymphatic filariasis are believed to occur as a result of interaction between the pathogenic parasite, the immune response of the host and secondary bacterial and fungal infections that complicate the situation.
Lymphoedema may occur as a result of live adult worms within lymphatic vessels in the lower limbs and pelvic region. The live worms secrete irritant toxins that cause dilatation of the lymph vessels surrounding the worm [3,7]. This causes a reduction in lymphatic flow. The subsequent oedema promotes fibrosis. Lymphoedema is further aggravated by secondary bacterial and fungal infections that arise as a result of impaired immune surveillance within the lymphoedematous region [8, 9].
Lymphatic damage and subsequent lymphoedema may also occur as a direct result of dead adult worms within the lymphatic vessels (worm death due to old age or treatment). The presence of dead worms induces granuloma formation which leads to lymphatic outflow obstruction within the vessel and subsequent lymphoedema [10, 11].
Predisposing factors
People residing for prolonged periods in tropical or subtropical areas where lymphatic filariasis is endemic are at the greatest risk for infection. Repeated mosquito bites over several months are required in order to acquire LF. Visiting tourists have a very low risk of acquiring LF [6].
Causative organisms
Numerous parasitic filarial nematodes may infect humans but only W. bancrofti, B. malayi and B. timori species are responsible for LF. W. bancrofti is responsible for 90% of cases of LF worldwide [12].
Clinical features
Presentation
Lymphatic filariasis has a range of clinical manifestations, varying from clinically asymptomatic microfilaria-positive individuals to those with disfiguring chronic filarial disease (elephantiasis). Overlap exists between the different symptom groups.
Clinical disease occurs in the minority of those infected with LF. The majority of infected individuals have few manifestations, despite the large number of circulating microfilariae in the peripheral blood. However, most will have some degree of subclinical disease, including microscopic haematuria and/or proteinuria, dilated and tortuous lymphatic vessels seen on lymphoscintigraphy, and scrotal lymphangiectasia in affected males [10, 13].
The most common presentation of LF is with acute adenolymphangitis (ADL) in adolescence. ADL is characterized by sudden-onset fever, painful lymph nodes, lymphangitis and transient oedema. Involvement of the genitals appears to occur exclusively with W. bancrofti infection [14]. Previously asymptomatic individuals may experience symptoms lasting 4–7 days, with a tendency to develop recurrent episodes [15]. Individuals with established LF and lymphoedema develop more severe and prolonged episodes of ADL. However, filarial ADL is distinct from acute dermatolymphangioadenitis, a process characterized by plaque-like lesions of cutaneous or subcutaneous inflammation, accompanied by ascending lymphangitis and regional lymphadenitis. These features occur in association with systemic signs of inflammation including fever and chills. Dermatolymphangioadenitis is thought to result from secondary bacterial or fungal skin infections as a consequence of immunodeficiency due to the underlying lymphatic damage [16].
Chronic lymphatic obstruction as a result of filarial worms leads to the development of hydroceles, lymphoedema/elephantiasis skin changes (severe hyperkeratosis, papillomatosis and skin fissuring) and rarely chyluria. Hydroceles are the result of accumulation of clear, straw-coloured lymphatic fluid within the tunica vaginalis as a result of obstruction of lymphatic vessels draining the retroperitoneal and subdiaphragmatic areas. The diameter of the hydroceles may be significant, reaching up to 30 cm.
Lymphoedema occurs as a result of the accumulation of lymphatic fluid within tissues following lymphatic vessel damage. The sites most commonly affected are the lower limbs and scrotum. Other sites can be affected such as the upper limbs and trunk. Initially the lymphoedema is intermittent and pitting in nature, but over time it becomes persistent and fibrotic. It is accompanied by gross skin changes referred to as elephantiasis – profoundly thickened and fibrotic skin with severe papillomatosis and secondary microbial infections [6].
Chyluria is a rare complication of LF and is the result of the presence of chyle (intestinal lymph) within the urinary tract. It occurs as a result of impaired drainage of retroperitoneal lymph below the cisterna chyli with subsequent reflux and flow of the lymph directly into the renal lymphatic vessels, which may rupture and permit flow of chyle into the urinary tract. The urine appears milky white in colour. Serious nutritional deficiencies may occur as a result of the loss of fat and protein within the urine.
Tropical pulmonary eosinophilia syndrome may rarely affect an individual with LF due to W. bancrofti or B. malayi. They develop respiratory wheeze and a paroxysmal nocturnal cough, similar to asthma. Chest radiographs demonstrate nodular or diffuse pulmonary lesions. Other features of this syndrome include elevated peripheral blood eosinophilia and high levels of serum immunoglobulin E and specific antifilarial antibodies. Unlike other forms of lymphatic filariasis, patients with tropical pulmonary eosinophilia are hyperresponsive to filarial antigens, especially those derived from the microfilarial stage of the parasite. If untreated, the patient may develop restrictive lung disease with interstitial fibrosis. Treatment with diethylcarbamazine is effective [17, 18].
Differential diagnosis
The differential diagnosis includes podoconiosis and chronic, poorly managed lymphoedema due to any other cause.
Disease course and prognosis
The clinical course of untreated LF is of progressive skin changes, worsening lymphoedema and increased incidence of secondary infection. A poor quality of life is associated with untreated disease. Elephantiasis and subsequent deformity leads to social stigma, financial hardship from loss of income and increased medical expenses. The socio-economic burdens of isolation and poverty are immense.
Investigations
In endemic areas, adults with lower limb lymphoedema and/or male genital involvement are likely to have LF. A definitive diagnosis can be made by detection of the adult parasitic worm within the lymphatic vessels or accessible lymph nodes. Doppler ultrasound may detect motile adult worms within the scrotum. However, these diagnostic tests are not always suitable for use in developing countries.
Filarial parasites exhibit ‘nocturnal periodicity’ that restricts their appearance in the blood to the hours of 10 pm to 2 am. The diagnosis of LF has traditionally depended on the nocturnal laboratory examination for microfilaria in peripheral blood smears (stained with Giemsa or haematoxylin and eosin) between these hours to maximize the chances of detection.
In recent years, polymerase chain reaction (PCR) and rapid antigenic assays have been developed. Antigen testing is a simple, sensitive and specific tool for the detection of the Wuchereria bancrofti antigen and is being used widely by lymphatic filariasis elimination programmes. The test detects infection within minutes and can be carried out at any time of day, unlike previous tests [19].
Lymphoedema, elephantiasis skin changes and hydroceles may persist in individuals with burned-out infections. Therefore, it is impossible to exclude a diagnosis of filarial-induced disease in the absence of circulating antigens or parasites. This situation may occur in patients who have received multiple courses of treatment or who no longer live in the endemic area.
Management
Drug treatment
The World Health Organization (WHO) launched the Global Programme to Eliminate Lymphatic Filariasis (GPELF) which aims to stop the spread of LF. Pharmaceutical companies have pledged to donate the required drugs. The strategy proposed by WHO to achieve LF elimination comprises two components: (i) the interruption of transmission of filarial infection in all endemic countries by the drastic reduction of microfilariae prevalence levels; and (ii) the prevention and alleviation of disability and suffering in individuals already affected by LF.
The interruption of transmission of infection is only possible if the entire at-risk population is treated by mass drug administration for a prolonged period of time to ensure a reduction in the blood levels of microfilariae to a level where transmission can no longer be sustained. The following drug regimens have been recommended by the WHO to be administered once a year for at least 5 years, with a coverage of at least 65% of the total at-risk population: (i) 6 mg/kg diethylcarbamazine citrate (DEC) in combination with 400 mg albendazole; or (ii) 150 μg/kg ivermectin in combination with 400 mg albendazole (in areas where onchocerciasis is prevalent, in order to avoid adverse drug reactions with DEC) [6].
Lymphoedema, elephantiasis skin changes and acute inflammatory episodes are typically managed with simple measures of improved hygiene, skin care, exercise activities and elevation of affected limbs. The GPELF has pledged to provide access to a minimum package of care for every individual with chronic manifestations of LF in all areas where the disease is present, in a bid to alleviate suffering and improve their quality of life.
Surgical treatment
Males with hydroceles benefit from hydrocelectomy procedures to achieve volume reduction [20].
Lifestyle management
Lifestyle measures can reduce the bacterial and fungal load that contribute to worsening lymphoedema. These include regular washing with soap and water, use of footwear and access to antibiotics and lymphoedema treatment [21]. Prevention of infection can be achieved by avoidance of mosquito bites. Lifestyle measures include sleeping under a mosquito net, using mosquito repellent on exposed skin and wearing long sleeves and trousers.
Resources
Further information
Global Programme to Eliminate Lymphatic Filariasis: http://www.filariasis.org (last accessed August 2015).
Podoconiosis
Definition and nomenclature
Podoconiosis refers to the development of bilateral lower limb lymphoedema, thought to occur as a result of prolonged exposure to irritant mineral-rich soils present at high altitudes (Figure 105.28). These minerals appear to trigger an inflammatory response resulting in impaired lymphatic drainage and subsequent lymphoedema. Development of the condition is closely associated with barefoot living and working. A genetic susceptibility has also been postulated.
Introduction and general description
Podoconiosis is a leading cause of lower limb lymphoedema of young people in Africa, Central America and north India, yet it remains a neglected condition. It has been prevalent for centuries but was previously encompassed by the umbrella term ‘elephantiasis’ until the pathogenesis of filariasis was realized in the 19th century. All cases of elephantiasis were then assumed to be filarial in origin until the discrepancy between the widespread distribution of ‘elephantiasis’ cases and more focal distribution of filaria in North Africa, Central America and Europe prompted a review of this theory.
The term podoconiosis was proposed in the 1980s to describe non-filarial cases of elephantiasis skin changes, and has since gained widespread acceptance. It is derived from the Greek for foot, podos, and dust, konos [1].
Epidemiology
Incidence and prevalence
It is estimated that 4 million people are affected by podoconiosis, mainly in tropical Africa, Central and South America and South-East Asia. Ethiopia is the country with the highest reported prevalence, with an estimated 1 million people living with the disease [2,3]. Prevalence estimates are limited but have been made in Ethiopia, Cameroon and Uganda. Estimated prevalence is 4% in Ethiopia, 8% in Cameroon and 4.5% in Uganda [4,5, 6]. The variation in reported prevalence figures may be attributable to survey size and sampling methods.
Age
Onset is typically in the first or second decade of life, but may occur later.
Sex
Gender ratio results may be unreliable, especially in remote areas affected by podoconiosis. A recent Ethiopian survey reported that females are more likely to be affected than males [4].
Pathophysiology
Predisposing factors
Podoconiosis occurs in the highland areas of tropical Africa, Central and South America and South-East Asia. It has been suggested that podoconiosis was previously prevalent in North Africa (Algeria, Tunisia, Morocco and the Canary Islands) and Europe [7]. However, it has not affected these areas since the use of footwear became customary.
Certain occupations (i.e. those with prolonged contact with soil) are at higher risk of developing podoconiosis, especially farmers.
Podoconiosis is associated with lower levels of income, poor education, being unmarried and the delayed introduction of foot wear [8].
Pathology
The pathogenesis of podoconiosis is not yet fully understood. Current evidence suggests a pivotal role of mineral particles within the soil, in a genetically susceptible individual [9]. One possible theory is that irritant particles cause an inflammatory immune response with subsequent impaired lymphatic drainage of the lower limbs, perhaps as a result of intraluminal lymphatic vessel obstruction by inflammatory cells. Supportive evidence for this theory includes the detection of elemental particles present in irritant clays (e.g. aluminium, silicon, magnesium or iron) within lower limb lymph node macrophages of affected individuals who lived barefoot [10]. 9
Genetics
An association has been reported between podoconiosis and variants in HLA class II loci. Podoconiosis may be a T-cell-mediated inflammatory disease and could be a model for gene–environment interactions, and further research in this area is underway [11].
Environmental factors
The association between podoconiosis and exposure to irritant soil was established when maps of disease occurrence were superimposed onto geological survey maps, confirming a connection with clays derived from volcanic activity [12, 13]. A recent study indicated that soil particles including smectite, mica and quartz were associated with podoconiosis prevalence and underpin the process [9].
The climatic factors thought to be necessary for producing irritant clays are high altitude (greater than 1200 m above sea level) and high seasonal rainfall (over 1000 mm annually) [4]. These conditions contribute to the steady disintegration of volcanic ash and the reconstitution of the mineral components into irritant silicate clays.
Clinical features
History
The affected individual typically resides in a high-risk area and will have lived and worked barefoot. Podoconiosis presents with a prodromal phase of pruritus of the forefoot skin and a burning sensation of the feet and lower limbs prior to the onset of elephantiasis skin changes [14–16].
Presentation
Early changes of podoconiosis are similar to that of any other cause of lower limb lymphoedema. The affected individual develops bilateral lymphoedema of the foot and ankle regions. Lymphorrhoea (leakage of lymph), hyperkeratosis, papillomatosis, fibrosis and gross disfigurement of the below-knee regions develop if the condition is untreated. The toes develop a characteristic macerated and ‘mossy’ appearance, hence the pseudonym ‘mossy foot’. Recurrent lower limb cellulitis frequently complicates the clinical picture.
Clinical features of podoconiosis that differentiate it from filarial elephantiasis include the feet and ankles being the initial site of symptoms in podoconiosis, rather than the groin or proximal lower limbs. Podoconiosis affects both lower limbs, whereas filariasis typically presents with unilateral lower limb swelling that extends above the knee. Groin involvement in podoconiosis is extremely rare, unlike in filariasis.
Podoconiosis may be distinguished from leprotic lower limb lymphoedema by the preservation of sensation within the lower limbs, and the absence of trophic ulceration, thickened palpable nerves or involvement of other body sites.
Differential diagnosis
Differential diagnoses include filariasis, endemic Kaposi sarcoma and leprotic lymphoedema.
Complications and co-morbidities
Significant social stigma is attached to podoconiosis. Affected individuals are excluded from school, churches and mosques, and barred from marriage with unaffected individuals because of local beliefs that the condition is contagious [17]. An affected individual is less likely to work and this impacts the patient's financial status.
Disease course and prognosis
Without access to footwear and conventional lymphoedema treatment (skin care and compression), the condition is progressive and complicated by recurrent cellulitis.
Investigations
Typically, affected individuals do not have access to lymphoscintigraphy or other investigative techniques. Filariasis, leprosy and endemic Kaposi sarcoma should be excluded if suspected by the clinician.
Management
Primary prevention
Primary prevention of podoconiosis consists of avoiding prolonged contact between the skin of the feet and irritant soils by using good footwear.
Prevention of disease progression
Prevention of disease progression and recurrent infections may be possible if affected individuals are instructed in the use of foot hygiene (e.g. daily washing of feet with soap and water and the use of antiseptics), and the use of shoes and socks for life. Simple compression bandaging is effective in reducing limb volumes, assuming that elephantiasis changes have not yet developed. A change in occupation or relocation may be beneficial, but may not be feasible for the affected individual.
Advanced disease
Advanced cases of podoconiosis are managed with daily skin care, leg elevation and multilayer compression bandaging, assuming that the affected individual has access to health care providers. Debulking surgery was previously employed, but has since been abandoned because of disappointing results. The local belief that there is no effective medical treatment acts as a barrier to accessing local health care. Disappointingly, few organizations offer treatment for podoconiosis because of the lack of evidence-based treatment options compounded by patchy acknowledgement that the disease even exists.
Resources
Further information
Footwork (The International Podoconiosis Initiative): www.podo.org (last accessed August 2015).
Pretibial myxoedema
Definition and nomenclature
Pretibial myxoedema (PTM) is a form of cutaneous mucinosis that typically occurs in association with Graves disease, hence the synonym thyroid dermopathy. Deposition of hyaluronic acid within the dermis and subcutis causes the development of asymptomatic ‘swelling’, nodules and plaques, typically of the pretibal region of both lower limbs. The precise cause of this phenomenon remains uncertain.
Introduction and general description
Pretibial myxoedema is a form of cutaneous mucinosis that occurs in association with Graves disease (see also Chapter 149). Patients typically present with asymptomatic, pretibial nodules and plaques that may have a yellow-brown hue. Lower limb lymphoedema may develop in severe cases. PTM results from the dermal accumulation of hyaluronic acid and other glycosaminoglycans, secreted by fibroblasts.
Epidemiology
Incidence and prevalence
Pretibial myxoedema affects up to 5% of patients with Graves disease; it affects up to 13% of patients with severe eye disease [1,2].
Sex
It more commonly affects females, with a female:male ratio of 4 : 1 [3].
Associated diseases
Pretibial myxoedema occurs almost exclusively in Graves disease. The classic triad of Graves disease is: (i) pretibial myxoedema;, (ii) ophthalmopathy (e.g. exophthalmos and orbital myopathy); and (iii) thyroid acropachy (swelling of the digits and clubbing). PTM has rarely been reported in association with Hashimoto thyroiditis, primary hypothyroidism and in euthyroid patients [4, 5].
Pathophysiology
The hyperthyroidism of Graves disease occurs as a result of stimulation of the thyroid-stimulating hormone receptor (TSHR) on thyroid follicular cells by autoantibodies directed against the receptor. However, the pathogenesis of Graves ophthalmopathy and PTM is less clear. PTM occurs as a result of glycosaminoglycan (GAG) deposition within the dermis. The underlying mechanism of GAG deposition is not clearly understood, but is likely to occur as a result of several processes, including autoimmune, cellular and mechanical factors. For example, TSHR antibodies may bind to and stimulate dermal fibroblasts to increase the production of GAGs [6]. Dermal GAG accumulation (i.e. mucin deposition) results in the separation of collagen fibres, expansion of connective tissues and oedema formation [7]. The obstruction of dermal lymphatic vessels by mucin results in lymphoedema [8]. The involvement of mechanical factors in the development of PTM may explain the lower limb predilection: dependency and trauma have been proposed [9].
Pathology
Characteristic histopathological features of PTM consist of mucin deposition (i.e. GAGs including hyaluronic acid) within the reticular dermis. Alcian blue and periodic acid–Schiff stains demonstrate mucinous material between collagen fibres. With extensive deposition of mucin, the collagen fibres become frayed and fragmented. Stellate fibroblasts may be observed, but the actual number of fibroblasts is not increased. There may be hyperkeratosis of the overlying epidermis.
Mucin deposition restricted to an expanded papillary dermis, with nodular angioplasia, and haemosiderin deposition are more suggestive of stasis dermatitis rather than PTM [10].
Clinical features
History
Pretibial myxoedema occurs almost exclusively with Graves disease. The patient is likely to have a history of thyrotoxicosis (e.g. weight loss, palpitations and hyperhidrosis) and possible ophthalmopathy and acropachy. PTM may develop prior to, during or after the thyrotoxic state as it is not related to thyroid function. It usually occurs 12–24 months after the onset of Graves disease, but may occur up to 12 years after its development [11].
Presentation
Pretibial myxoedema is typically confined to the lower limbs. The shins are the classic site, but the toes may be the only affected site. PTM may occur within surgical scars or sites of trauma [12]. PTM may rarely affect other body sites, such as the head and neck region, torso and upper limbs, presumably triggered by trauma [13–15].
Patients typically present with firm, non-pitting indurated nodules and plaques on the pretibial regions and feet. The lesions may be flesh-coloured or have a yellowish brown hue. A peau d'orange appearance may develop as a result of expansion of the interfollicular dermis. Cutaneous lesions may be asymptomatic, occasionally pruritic, or cause discomfort. Mild cases are usually asymptomatic and only of cosmetic concern. In severe cases of PTM, lesions may coalesce to give the entire extremity an enlarged, verruciform appearance (Figure 105.29). Functional impairment (inability to wear shoes as a result of toe disfiguration) and/or the development of secondary lymphoedema may occur.
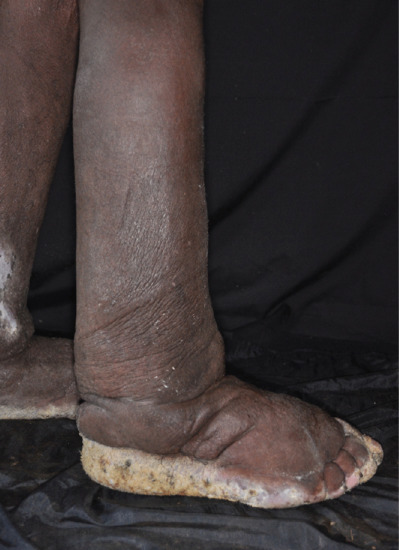
Figure 105.28 Lower limb lymphoedema due to podoconiosis. Note the presence of toe maceration and typical ‘mossy’ appearance of the foot. (Reproduced with permission from D. Markos.)
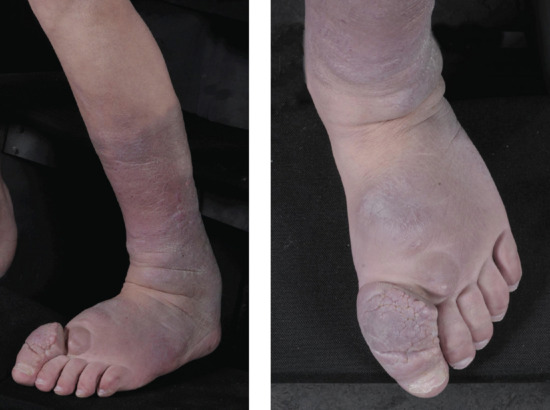
Figure 105.29 Pretibial myxoedema.
Differential diagnosis
Differential diagnoses include lymphoedema, stasis dermatitis, obesity-associated lymphoedematous mucinosis and lichen amyloidosis.
Complications and co-morbidities
Pretibial myxoedema rarely causes significant morbidity. Local discomfort and difficulty in finding footwear may be experienced. Severe forms of the disease may cause lower limb lymphoedema and subsequent complications of increased incidence of infection.
Disease course and prognosis
Few studies exist on the evaluation of PTM outcomes. However, the prognosis for mild cases appears to be favourable. In mild cases, 50% of patients achieve complete remission after several years [16]. The largest series reports after 25 years of follow-up that 70% of mild untreated and 58% of treated severe cases achieved complete or partial remission. Very severe forms appear to be persistent [3].
Investigations
The diagnosis of PTM is possible from the patient's history and characteristic clinical findings. It is rarely necessary to perform a skin biopsy, especially if there is a history of hyperthyroidism or Graves ophthalmopathy. If a biopsy is undertaken, the histopathological findings are characteristic.
The investigation of lymphatic function with lymphoscintigraphy is not routinely performed. However, lymphoscintigraphy and fluorescence microlymphography may confirm structural and functional alterations to the lymphatic drainage. Mucin deposition within the dermis may compress or occlude the initial lymphatic vessels, resulting in lymphoedema [8].
Management
Mild and asymptomatic cases of PTM may not require treatment. If symptomatic, treatment options include the use of potent topical corticosteroids under occlusion at night for several months. Prompt diagnosis and initiation of treatment appear to correlate with better outcomes [17]. The use of compression hosiery or multilayer bandages may be used to manage the associated lymphatic impairment/secondary lymphoedema seen in more extensive and chronic cases.
A small number of case reports suggest surgical excision may be of benefit in select cases [18]. However, surgery should be considered with caution as PTM may develop within areas of trauma.
Several therapeutic agents have been trialed in the treatment of PTM. Evidence of their efficacy is limited. These include intralesional steroids, systemic immunomodulators (e.g. prednisolone, plasmapheresis, intravenous immunoglobulin and rituximab) and octreotide [19,20, 21–23,24].
Trauma-induced lymphoedema
Introduction and general description
Lymphoedema and other lymphatic complications, such as lymphocele or lymph fistula, can develop after therapeutic interventions or accidental damage to lymph drainage pathways. The failure of lymphatics to regenerate and re-anastomose satisfactorily through scarred or irradiated tissue is probably responsible for lymphoedema development.
Epidemiology
The incidence of lymphoedema following varicose vein surgery was estimated to be 0.5% [1]. In another series the incidence of lymphatic complications from 5407 surgical procedures for varicose veins was 118 cases (2.2%); a lymphocele on the limb occurred in 1.3%, an inguinal fistula or lymphocele in 0.7% and lymphoedema in 0.2% [2].
Pathophysiology
Trauma to lymphatics, either from elective surgery or by accident, usually needs to be extensive to induce lymphoedema. Indeed, the experimental production of lymphoedema is extremely difficult to achieve owing to the excellent regenerative powers of lymphatics.
It remains a puzzle as to why most women who have a full axillary lymph node clearance following breast cancer surgery do not develop lymphoedema, yet 6% of women who have a single sentinel lymph node biopsy do develop arm swelling [3]. Furthermore, it is not known why breast cancer-related lymphoedema (BCRL) can manifest immediately postsurgery or be delayed for many years. A genetic predisposition may be relevant: mice with a heterozygous mutation in the adrenomedullin gene developed lymphoedema when subjected to surgery but wild-type mice did not [4].
Radiotherapy to lymph nodes can be as much a risk factor towards lymphoedema as surgery [5].
Genetic factors are likely to be important. Obesity increases the incidence of postsurgical lymphoedema, as has been demonstrated following breast cancer treatment [3]. Wound infections increase the incidence of postsurgical and accidental lymphoedema.
Clinical features
Lymphoedema can develop after varicose vein treatment. Varicose vein surgery may be undertaken for reasons of ‘venous oedema’. However, the chronic oedema may already reflect a compromised lymph drainage, in which case surgery may further undermine lymph drainage and make swelling worse. With the greater use of endovenous therapy using laser, radiofrequency or foam vein ablation (rather than traditional stripping and surgical ligation) the incidence of lymphoedema is likely to be reduced.
Lymphatics can be damaged and produce lymphoedema during saphenous vein harvesting for coronary artery bypass grafts. However, the increasing use of coronary stents has considerably reduced this risk. Similarly, great saphenous vein harvesting for critical limb ischaemia can result in lymphatic complications such as lymphocele [6].
Lymphoedema can develop at the donor site after reconstructive surgery such as a transverse upper gracilis free flap for breast reconstruction [7].
Resection of excess skin and soft tissue of the thighs after massive weight loss can also cause lymphoedema. The lymphatic collectors of the thigh sit superficial to the veins. Therefore, in a vertical medial thigh lift, choosing a dissection plane superficial to the great saphenous vein is unlikely to preserve the collectors of the ventromedial bundle [8]. Lymphoedema and other lymphatic complications (e.g. cellulitis, lymphocele) are a significant risk. An interruption of lymphatic pathways presumably results in a failure of adequate lymphangiogenesis and repair, resulting in lymphoedema.
Accidental trauma, such as a degloving injury to a limb, will produce lymphoedema distal to the injury if widespread circumferential scarring has occurred.
Self-inflicted injury, such as the repeated application of a tourniquet, will eventually cause permanent lymphatic damage and chronic swelling (Secrétan syndrome) [9]. The abrupt termination of the swelling often coincides with a skin contour change due to subcutaneous atrophy caused by a tight constricting band. Skin pigmentation may also coexist at the site. Factitious lymphoedema can be caused by tourniquets, blows to the arm or repeated skin irritation, usually in patients with known psychiatric conditions. Factitious lymphoedema results in symptoms and signs suggestive of chronic regional pain syndrome [10].
Intravenous drug abuse may cause lymphoedema due to a combination of infection and injected agents causing lymphangitis plus associated venous damage. Puffy hand syndrome is a long-term complication of intravenous drug abuse [11]. It can affect from 7% to 16% of intravenous drug users [12].
Investigations
Lymphoscintigraphy is the investigation of choice to determine lymphatic insufficiency.
Management
Decongestive lymphatic therapy is first line treatment [13]. Infection needs to be treated and prevented [14]. The use of additional therapies such as pneumatic compression therapy and Velcro wraps can be considered.
Lymphoedema due to immobility
Introduction and general description
Lymph drainage, unlike blood flow, requires intermittent changes in local tissue pressure generated by movement and exercise in order to produce initial lymphatic transport. Main limb collector lymphatic vessels pump the lymph supplied from the initial lymphatics. Lymphatic collector vessels rely on innervation and effective smooth muscle contraction for pumping. Consequently, immobility, by reducing initial lymphatic absorption and transport, reduces lymph flow to the collectors. Less pumping promotes swelling, particularly if gravitational forces (dependency syndrome) encourage ongoing fluid filtration into the tissues but without sufficient compensatory lymph drainage.
Lymphoedema is well recognized with certain neurological conditions that restrict movement. Cerebrovascular accident (CVA), spina bifida and multiple sclerosis (MS) are those that are best described.
Post-radiation brachial plexopathy following breast cancer treatment leads to severe lymphoedema with paralysis being the major contributing factor.
Epidemiology
In a review of 240 electronic medical records from an adult spina bifida clinic, 22 patients (9.2%) had lymphoedema [1]. Lower limb oedema is common in MS patients, especially in those with reduced mobility. In one study, 93 patients (45%) of a total of 205 patients with definite MS showed oedema with abnormal findings on lymphoscintigraphy [2].
Pathophysiology
Exercise and movement are essential for stimulating lymph drainage. Any reduction in movement lowers lymph drainage accordingly. Immobilizing any body part in the face of sustained microvascular filtration will result in (lymph)oedema despite patent, and otherwise normal, lymphatic vessels. The lower limb is most commonly affected because of the contribution from dependency/gravitational forces. Paralysis and neurological deficit cause lymphoedema for similar reasons.
Hand oedema after a stroke is not considered to be lymphoedema owing to normal lymphoscintigraphy. However, one would expect lymph drainage pathways to be patent but not functional owing to the lack of movement. Combine that fact with increased microvascular filtration (and high lymph load) due to dependency of the paralysed or spastic limb, and lymphoscintigraphy might be expected to be falsely normal. After all, if the lymphatics are working oedema should be avoided irrespective of cause [3].
It is likely, but unproven, that with neurological deficit a failure of lymphatic pumping due to denervation contributes most to oedema formation, but a lack of isotonic exercise (from muscle weakness or spasticity) will reduce initial lymph flow so making less lymph available for collector pumping.
With time, pathological changes within the failing, but hitherto normal, lymphatics occur. Lymphangiothrombosis or luminal fibrosis leads to an irreversible lymphoedema even if full mobility is restored.
Clinical features
Oedema associated with immobility will usually develop insidiously unless the immobility is sudden in onset (e.g. CVA). Hand or foot swelling is the usual story because of the effect of gravity being maximal in the distal part of the limb. Peripheral oedema can cause pain and tissue tenderness. Extreme oedema may result in the weeping of fluid from the skin (lymphorrhoea).
A common scenario is ‘armchair legs’, a term coined by Sneddon and Church [4] where patients sit in a chair day and night with their legs dependent (otherwise known as elephantiasis nostras verrucosis because of the severe lymphoedema skin changes that ensue). No premorbid abnormalities of the lymphatics exist, but the immobility results in minimal lymph drainage and a functional lymphoedema due to a lack of movement or exercise to stimulate normal lymph drainage. Dependency of the limb compounds the problem by increasing capillary filtration. The syndrome is not confined to the legs, but can affect any chronically dependent and immobile part, as demonstrated in the pendulous abdomen [5].
Physical examination reveals swelling in which pitting is marked, due to a mixed aetiology of reduced lymph drainage and increased microvascular fluid filtration. Neuropathic limbs take on a particular appearance with a bluish hue and background livedo.
Differential diagnosis
Deep-vein thrombosis should be considered if onset is acute.
Complications
As with all forms of lymphoedema, there is an increased risk of infection and lymphorrhoea.
Investigations
In cases of possible DVT, d-DIMERS and compression ultrasonography should be considered. In breast cancer patients PET/CT of the axilla should be undertaken to exclude axillary recurrence.
Management
Compression bandaging is the mainstay of treatment but needs to be undertaken with care if sensation is reduced. Oedema should reduce easily but refill occurs rapidly. Therefore, well-fitting compression garments are applied as soon as swelling is reduced. Pressure areas need to be carefully monitored. Skin care is of the utmost importance. The tendency of paralysed limbs to reswell means that bandaging may need to be continued or replaced with Velcro wraps (e.g. Farrow wraps). When resting, elevation of the lower extremities is desirable, either while sitting in a wheelchair or lying in the bed, with the back of the knees and calves supported by pillows. The oedematous limb or limbs should be positioned higher than the hips if possible.
Systems that encourage passive movements can prove helpful [6]. Pneumatic compression pumps may be helpful as they simulate the calf muscle pump.
Lymphangitis
Definition
Lymphangitis is an inflammation of lymphatic vessels. It is most often caused by infection from bacteria, virus or fungus or infiltration by cancer cells.
Introduction and general description
Lymphangitis represents inflammation of the lymphatic collectors and is clinically seen as tender red streaks up the limb corresponding to the inflamed vessels. In the lower limb, oedema is so often an accompanying feature that red streaks, such as are seen with lymphangitis of the arm, are rarely seen. A more diffuse erythema is seen extending up the medial side of the leg and thigh. Distinction from an ascending cellulitis becomes difficult. In some circumstances the inflammatory response may be profound and painful inflammation of the regional lymph glands (lymphadenitis) may arise.
Pathophysiology
Lymphangitis may occur without any demonstrable inflammation or may be recurrent, for example following relapsing herpes simplex infection [1]. Permanent obliteration of lymphatic collectors may follow severe or recurrent lymphangitis. In such cases, if reserve lymphatic capacity is limited, permanent swelling (lymphoedema) can result.
In immunocompetent individuals the commonest cause of lymphangitis is infection due to Group A beta-haemolytic streptococci (p. 26.20).
Lymphangitis occurs in filariasis [2]. Sporotrichoid spread (also known as nodular lymphangitis) describes a characteristic pattern of superficial cutaneous lesions that progress along the path of lymphatic drainage. Fungi, and classically sporotrichosis, exhibit this tendency but also nocardiosis, chromoblastomycosis and aspergillosis. Sporotrichoid spread through lymphangitis has been described with leishmaniaisis and atypical mycobacterium (e.g. Mycobacterium marinum). Cutaneous squamous cell carcinoma can rarely demonstrate sporotrichoid spread as can a hypersensitivity lymphangitis to an arthropod bite [3].
Snake toxin molecules are generally large and cannot directly enter the circulation, but are readily taken up by the lymphatics [4]. Snake bite venom will often cause lymphangitis [5]. Pressure bandaging with immobilization is the recommended first aid treatment with the intention of preventing lymph drainage [6].
Mondor disease is considered to be a form of superficial thrombophlebitis but it commonly complicates lymph node removal, so a form of lymphangitis or lymphatic thrombosis may be more likely [7[. Lymphatic cording or axillary web syndrome following axillary lymph node intervention may be similar. It is also described following a jellyfish sting [8,9]. Lymph can clot and lymphangiothrombosis may occur more often than realized [10].
Sclerosing lymphangitis of the penis is a condition related to vigorous sexual activity, manifesting as an asymptomatic, firm, cord-like swelling around the coronal sulcus of the penis [11].
Carcinoma erysipeloides (lymphangitis carcinomatosa, carcinoma telangiectatica, carcinoma en cuirasse) occurs when cancer cells infiltrate the dermal lymphatics. It is a form of metastatic spread and occurs most commonly with breast cancer but can occur with melanoma and thyroid, lung, gastric, pancreatic, ovarian, prostate and colo-rectal cancer [12]. Carcinoma erysipeloides manifests clinically with a fixed erythematous patch or plaque resembling cellulitis, but without fever [13]. The inflamed area may show a distinct raised periphery and oedema secondary to lymphatic obstruction. A network or lattice pattern of telangiectatatic vessels represents the infiltrated dermal lymphatics (Figure 105.14). Inflammatory breast cancer is a rare and very aggressive disease in which cancer cells block lymph vessels in the skin of the breast. This type of breast cancer is called ‘inflammatory’ because the breast often looks swollen and red, or ‘inflamed’.
COMPLICATIONS OF LYMPHOEDEMA
The major complications are swelling and infection.
Swelling
Limb swelling leads to discomfort, limb heaviness, reduced mobility and, on occasion, impaired function. The size and weight of affected limbs can result in secondary musculoskeletal complications such as back pain and joint problems, particularly in the case of asymmetrical lower limb swelling. Balance can be an issue with asymmetrical limb swelling affecting either the upper or lower limbs [1].
Skin changes
Elephantiasis refers to skin resembling elephant skin. The epidermis becomes hyperkeratotic and warty and the dermis markedly thickened and fibrotic. Distended dermal lymphatics can bulge on the skin surface and, with tissue organization, produce a cobblestone appearance. Elephantiasis skin changes occur in established chronic lymphoedema, particularly in the lower limb or in circumstances of profound lymphatic obstruction (Figure 105.30) [2]. Thickening of the skin impairs joint mobility. Leakage of lymph through the skin (lymphorrhoea) may occur from engorged dermal lymphatics (lymphangiectasia) (Figure 105.31).
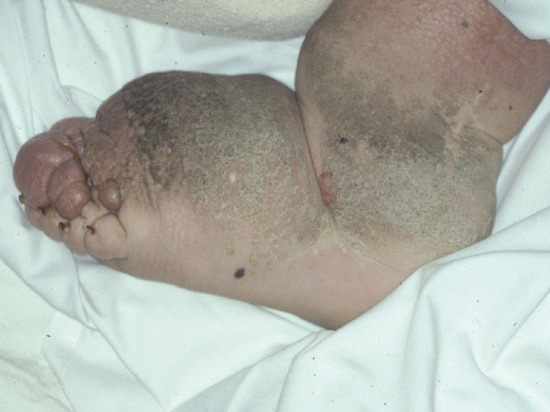
Figure 105.30 Elephantiasis verrucosis nostras showing marked hyperkeratosis and papillomatosis.
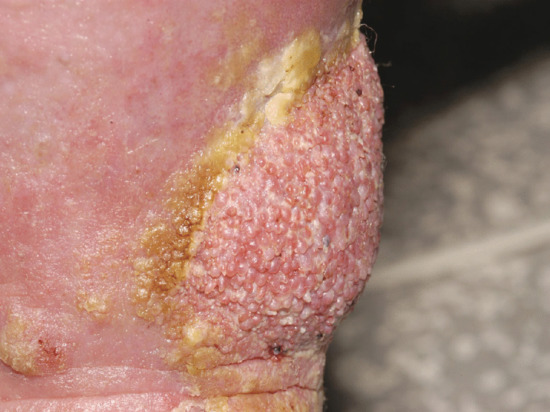
Figure 105.31 Acquired cutaneous lymphangiectasia leading to ulceration of the lower leg.
Infection
Episodes of secondary infection, particularly cellulitis, are a characteristic feature of lymphoedema. Patients with lymphoedema irrespective of cause are liable to these attacks. It is likely that disturbances in immune cell trafficking compromise tissue immunosurveillance, but the exact mechanism is not known [3].
Constitutional symptoms such as fever, rigors, headache or vomiting can be profound and sudden in onset. Within 24 h, redness appears within the lymphoedematous area but without an advancing border. Pain and heat also feature. Recurrent episodes may be frequent and further impair lymph drainage, so exacerbating the lymphoedema. Thus, a vicious cycle is established. Haemolytic streptococci of group A, B and particularly G have been demonstrated. Toxicity from infection can be extreme and even fatal [4]. It is not unusual for patients to comment that attacks of cellulitis can be induced by strenuous exercise or long car journeys. This suggests a mechanism not dissimilar to herpes simplex where the microorganism is always present but becomes reactivated.
Fungal infections, particularly tinea pedis, are difficult to avoid because of web-space skin maceration from swollen toes (Figure 105.32).
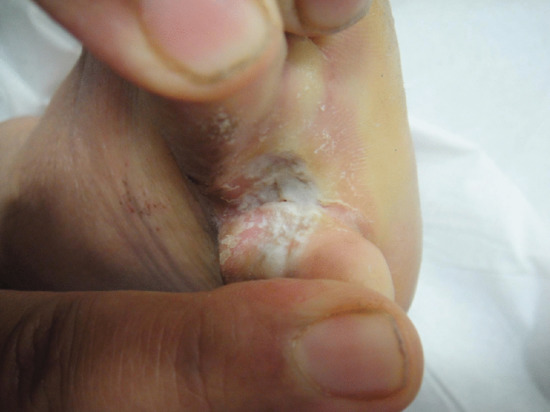
Figure 105.32 Macerated web-space skin leads to bacterial entry points and fungal infection.
Opportunistic infections are also described in lymphoedema, lending further support to the localized immunodeficiency theory [5].
Psychosocial issues
In one study over 80% of patients (188/217) had taken time off work due to their lymphoedema, with an estimated mean time off work of 10.5 days per year for medical appointments. Overall, 9% stated that the lymphoedema affected their employment status, with 4/209 (2%) respondents having to change jobs and 17/209 (8%) having to give up work because of it [6]. Relationships can also suffer.
The difficulty in finding clothes or shoes to fit creates social problems. Poor footwear will further compound the swelling by discouraging a normal gait or enough exercise.
Patients with arm swelling in relation to breast cancer experienced functional impairment, psychosocial maladjustment and increased psychological morbidity [7].
Malignancy
Chronic lymphoedema has a permissive effect with certain types of malignancies, particularly angiosarcomas, in what is known as Stewart–Treves syndrome [8]. The presumed mechanism is through immunodeficiency of the compromised drainage basin, in a similar way to malignancy complicating systemic immunodefiency, such as in renal transplant recipients [9]. The Stewart–Treves syndrome describes lymphangiosarcoma developing from well-established postmastectomy oedema. However, lymphangiosarcoma is now described as occurring with lymphoedema of any cause (Figure 105.33).
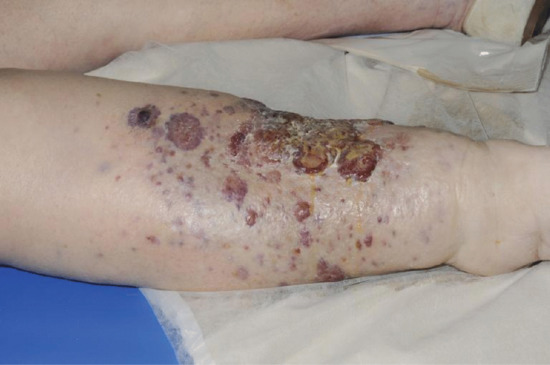
Figure 105.33 Lymphangiosarcoma arising in primary lymphoedema.
Other tumours that have been recorded to develop with lymphoedema include basal cell carcinoma [10], squamous cell carcinoma [11], lymphoma [12–14], melanoma [15, 16], malignant fibrous histiocytoma [17], Merkel cell tumour [18] and Kaposi sarcoma [19]. Certain uncommon benign tumours are reported associated with lymphoedema [20].
Miscellaneous conditions
A range of cutaneous conditions has been reported as occurring preferentially at sites of lymphoedematous involvement. These include xanthomatous deposits [21, 22], bullous pemphigoid [23], toxic epidermal necrolysis [24], atypical neutrophilic dermatosis [25] and severe necrotizing fasciitis [26].
IMAGING OF THE LYMPHATIC SYSTEM
Most patients with lymphoedema are diagnosed from the history and clinical findings. The use and value of imaging techniques is highly dependent on availability and expertise. Table105.12 compares the different techniques available for imaging the lymphatic system. The techniques used to investigate the lymphatic system are discussed below, but some patients will benefit from additional imaging in order to understand the cause of their swelling. For example, MRI and CT scanning may be indicated to assess the presence of intra-abdominal or pelvic pathology or masses obstructing lymphatic drainage. MRI may also be requested to assess tissue hypertrophy or signs of overgrowth (e.g. in Klippel–Trenaunay syndrome). Venous duplex imaging may be requested to investigate the possibility that a patient's lymphoedema is associated with venous incompetence.
Table 105.12 Lymphatic imaging techniques and their properties.
| Imaging technique | Contrast agent | Depth of imaging | Invasiveness | Availability |
| Lymphography/lymphangiography | Intralymphatic injection of a radio-opaque oil (e.g. Lipiodol) | Anatomical imaging of lymphatic collectors and lymph nodes | Very invasive; requires the identification of a lymphatic collecting vessel by exploratory surgery | Rarely performed |
| Lymphoscintigraphy | Simple intradermal/subcutaneous injection of a radiolabelled tracer protein (e.g. technetium-99-labelled colloid) | Lymphatic collectors and lymph nodes Poor resolution compared with lymphography technique Limited functional data acquired Not anatomical |
Minimal | Readily available in hospitals |
| Fluorescence microlymphangiography | Simple intradermal injection of fluorescent contrast agent (FITC-dextran) | Superficial dermal lymphatic vessels | Minimal | Research tool |
| Near infrared lymphangiography | Simple intradermal/subcutaneous injection of indocyanine green | Superficial dermal lymphatic vessels and superficial lymph nodes Contractility can be visualized but not quantified |
Minimal | Increasing availability in departments offering lymphatic microsurgery |
| Magnetic resonance lymphangiography | Simple intradermal/subcutaneous injection of a gadolinium-based contrast agent | Lymphatic collectors and lymph nodes Subcutaneous fat and oedema also visualized | Minimal | Research tool |
FITC, fluorescein isothiocanate.
Lymphography and lymphoscintigraphy
X-ray contrast lymphography (lymphangiography) remains the gold standard mode of demonstrating lymphatic collectors and lymph nodes. However, it is rarely used as the technique requires the invasive procedure of direct cannulation of the lymphatics [1].
Lymphoscintigraphy (isotope lymphography) is currently the investigation of choice for determining if chronic oedema may be due to lymphatic failure. It involves a simple intradermal/subcutaneous injection of a radiolabelled tracer protein, exclusively cleared by lymphatics. Measurement of tracer uptake and transit through the lymphatics permits qualitative and quantitative analyses of lymph drainage and may discriminate lymphoedema from lipoedema [2,3]. Lymphoscintigraphy has the potential to distinguish between different types of primary lymphoedema and their mechanisms of lymphatic failure, for example initial lymphatic dysfunction in Milroy disease, versus lymph collector reflux in lymphoedema distichiasis syndrome (Figure 105.34). However lymphoscintigraphy suffers from poor spatial resolution and cannot provide functional measurements of flow [4].
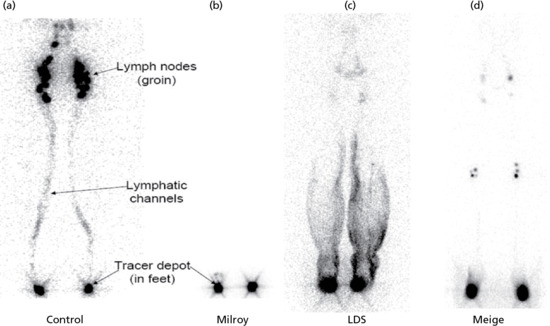
Figure 105.34 Lymphoscintigraphs showing the uptake of technetium-99 in the inguinal lymph nodes (ILNs) at 2 h after injection in the normal lymphatic system and in patients with differing types of lower limb primary lymphoedema. (a) Healthy control. (b) Milroy disease, demonstrating functional aplasia of the lymphatics (i.e. no uptake into the ILNs). (c) Lymphoedema distichiasis syndrome with reasonable uptake to the ILNs but evidence of marked reflux due to lymphatic valve incompetence. (d) Meige disease with poor uptake and evidence of rerouting via the deep lymphatic channels resulting in uptake within the popliteal lymph nodes. (Adapted from Connell et al. 2013 [11].)
Fluorescence microlymphangiography
Fluorescence microlymphangiography (FML) is a research tool that uses a fluorescent contrast agent (fluorescein isothiocanate (FITC) dextran) to provide information on superficial dermal lymphatic vessels. It has been used to improve understanding of lymphatic disease, including primary lymphoedema. For example, FML in conjunction with immunohistochemistry demonstrated that lymphatic dysfunction, not aplasia, underlies Milroy disease [5].
Near infrared lymphangiography
Near infrared lymphangiography (NIR) using indocyanine green (ICG) is a recently introduced and potentially useful technique used to demonstrate superficial lymphatic vessels [6]. NIR imaging techniques use tracers that fluoresce under the excitation of near infrared waves (750–1000 nm) and external fluorescent detectors. ICG is a dye with properties of NIR absorption and fluorescence emission. ICG-solution is injected intradermally or subcutaneously and enables real-time detection of the lymphatic vessels.
NIR has been used to assess lymphatic vessel structure and function. ICG is administered intradermally in order to map the lymphatic architecture [7]. Fluorescent images have been collated to describe patterns of abnormal lymphatic structure in lymphoedema (e.g. dilated vessels with proximal obliteration) and diffuse constellation-like patches of dye accumulation (termed the ‘Milky Way’ features). This method has also been used to quantify lymphatic function in the lower limb [7]. NIR is now widely used to assess lymphatic vessel patency prior to lymphatico-venous or lymphatico-lymphatic anastomosis surgery [8].
Magnetic resonance lymphangiography
The use of contrast-enhanced magnetic resonance lymphangiography (MRL) has been reported in the investigation of patients with lymphoedema. MRL has been used to demonstrate the presence of enlarged and tortuous lymphatic vessels in patients with unspecified lymphoedema [9]. It is still a research tool but could prove useful as it has the potential to provide high-quality images of anatomical abnormalities (Figure 105.35), and may be able to provide quantification of lymphatic function in the future.
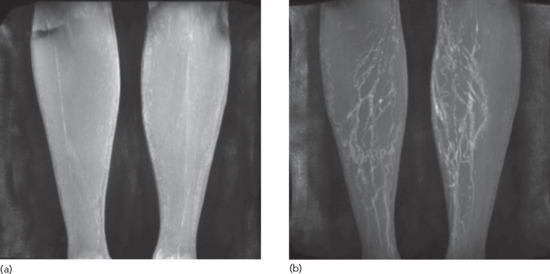
Figure 105.35 (a) Baseline magnetic resonance lymphangiography (MRL) images taken from a patient prior to contrast injection. His phenotype is of lymphoedema distichiasis syndrome (FOXC2 positive). Two main vessels can be seen within the lower limbs. These could be venous or lymphatic in origin. (b) Post-contrast MRL images demonstrating multiple, dilated, tortuous lymphatic vessels seen in the below-knee regions.
Histopathological investigation of the lymphatic system
Skin biopsies may be useful when investigating a suspected lymphatic malformation. The distinction between lymphatic and blood vessels in skin biopsy sections has been made more straightforward by specific lymphatic markers. D2-40 (podoplanin) is considered the most robust as it stains initial lymphatics, pre-collectors and collecting vessels but not blood vessels. LYVE-1 stains initial lymphatics but also macrophages and is down-regulated by inflammation. Prox-1 and VEGFR-3 are also lymphatic-specific and stain larger vessels. A panel of markers is recommended, particularly in vascular malformations where differentiation between venous and lymphatic phenotypes may not be clear-cut [10]. CD31 and CD34 stain both lymphatic and blood vascular endothelium.
LYMPHOEDEMA MANAGEMENT
Lymphatic failure results in the accumulation of protein as well as water within the swollen tissues. Treatment is difficult because of the presence of the ‘solid’ component in the swelling. The management of lymphoedema varies greatly around the world. In developed countries, the emphasis is more on physical forms of therapy involving massage, exercise and compression designed to stimulate lymph drainage. In poorer, hotter countries where hosiery and appropriate bandages are too costly and uncomfortable, surgery may be the mainstay of treatment. Two particular problems need to be overcome with lymphoedema: the swelling and the predisposition to infections, particularly recurrent cellulitis.
There is limited research to inform evidence-based guidelines on the treatment of lymphoedema. Nevertheless, robust guidelines developed through consensus by experts do exist [1].
Unfortunately there is no proven curative treatment for lymphoedema. Management is aimed at improving swelling through physical treatments designed to stimulate flow through existing or collateral drainage routes. Several surgical techniques have been implemented in recent years in a bid to improve lymphatic drainage, or achieve limb volume reduction via liposuction. However, in the absence of robust data to support most surgical techniques, so-called ‘conservative’ physical therapies are relied on for the majority of patients.
Medical assessment
Medical assessment aims to identify and exclude other causes of peripheral oedema. In circumstances where systemic causes for peripheral oedema, for example heart failure, have lead to or coexist with the lymphoedema, then treatment of the medical condition must be undertaken before embarking on specific lymphoedema therapy. Where necessary, appropriate investigations should be performed to confirm lymphoedema and to identify treatable underlying causes (e.g. active cancer) or co-morbidities (e.g. superficial venous incompetence).
Physical therapies
All patients with a diagnosis of lymphoedema should be referred to a local trained lymphoedema therapist. Management will focus on the needs of each individual patient. This may include: (i) risk reduction, for example in breast cancer patients; (ii) swelling reduction and improvement of shape; (iii) treatment and prevention of infection; (iv) treating skin problems such as elephantiasis, lymphorrhoea and wounds, as well as discouraging tissue fibrosis; (v) restoring functional independence and correcting posture imbalance; and (vi) pain and psychosocial management. Therapy assessment will include setting the benchmarks against which improvement can be judged, for example limb volume measurement, mobility and functional assessments. A treatment plan will depend on the site and severity of the lymphoedema and the need to engage other services, for example leg ulcer services, oncology or vascular surgeons.
Physical methods of treating lymphoedema have been practised in Europe for many years [2]. Therapy essentially aims to control lymph formation (capillary filtration), including treatment of inflammatory causes and/or venous hypertension; and to improve lymph drainage through existing lymphatics and collateral routes by applying normal physiological procedures that stimulate lymph flow. Physical treatment can, in the majority of cases, improve quality of life considerably. Central to management is getting patients to understand their condition and know what they can do for themselves. Only then can a high level of motivation and adherence to treatment be generated [3]. It is important to explain to patients that, unlike blood which is propelled by the heart, lymph drainage relies on local changes in tissue pressure generated by exercise and movement. Physical treatment exploits these principles, enhancing lymph flow as much as possible within the limits of a compromised drainage system. It should be appreciated that lymph flow still exists in lymphoedema, otherwise swelling would be a relentlessly progressive process.
The essential components of physical therapy include the following.
Care of the skin and prevention of infection
Elephantiasis skin changes are not only unsightly but lead to problems including infection, odour, lymphorrhoea, restricted movement from fibrosis (pseudoscleroderma) and poor wound healing. Regular application of an emollient is important for hydrating the hardened skin, so making it more supple and discouraging hyperkeratosis. Tinea pedis is almost invariable because of the closely apposed swollen toes – circumstances not improved by elastic hosiery. Modern antifungal creams unfortunately macerate skin further and therefore it is suggested that terbinafine cream is applied for 2 weeks followed by an alcohol wipe (assuming the skin is not broken). For deep cracks and crevices that bacteria may readily colonize, regular toilet is necessary followed by an antiseptic soak, for example potassium permanganate. Hyperkeratosis can often be improved through the regular application of 5% salicylic acid ointment, but the best treatment to reverse elephantiasis skin changes is long-term compression. Areas that constantly seep lymph should also respond to sustained compression.
Prevention of infection, particularly lymphangitis/cellulitis, is crucial to the control of lymphoedema. Care of the skin, good hygiene, control of tinea pedis and good antisepsis following abrasions and minor wounds are important in reducing the risk of cellulitis, as maintenance of skin integrity and an effective barrier will reduce the entry of microorganisms.
Exercise
Exercise and movement are crucial to lymph drainage [4]. Dynamic muscle contractions (isotonic exercises) encourage both passive (movement of lymph along tissue planes or through non-contractile lymphatics) and active (increased contractility and therefore propulsion of lymph within contractile lymphatics) phases of lymph drainage. Overexertion and excessive static (isometric, e.g. gripping) exercise increase blood flow, which tends to increase oedema.
External compression
External compression (hosiery, bandage or pneumatic compression) complements the exercise programme. Such compression is not intended to ‘squeeze’ oedema but to act as a counterforce to striated muscle activity and so generate higher tissue pressures during contractions. This provides the most powerful stimulus to lymph drainage. Compression also limits capillary filtration by opposing capillary pressure. Compression is much less effective without exercise. Multilayer bandaging can be used for limb reduction, but also has the advantage of restoring limb shape so that subsequent use of compression garments (hosiery) is more effective at controlling swelling [5].
Bandaging may be the only method suitable for huge misshapen limbs and for controlling lymphorrhoea. Layers of strong, non-elastic (short-stretch) bandages are applied to generate a high pressure during muscular contractions but low pressure at rest. The use of foam or soft padding helps to distribute pressure more evenly and to protect the skin. The digits are bandaged to control the swelling of fingers and toes. The strategic positioning of rubber pads ‘irons out’ pockets of swelling and deep skin folds (Figure 105.36). Multilayer bandaging is a skill that takes time to learn and should not be undertaken by any professional without appropriate training. The compression administered may have to be modified in circumstances such as cancer requiring palliative treatment, moderate limb ischaemia or if there is any neurological deficit. Hosiery (below-knee or full-length stockings, half or full tights and sleeves) usually requires high compression and double layers may occasionally be required. Most garments last no more than 6 months. Two garments (or pairs) should be provided, one to wear and one for the wash. Washing is necessary to maintain the compression properties of the garment. The patient's technique for the application, removal and care of garments is crucial for a successful outcome.
Figure 105.36 (a) Toe (or finger) bandaging with cotton crepe bandages. (b) Sub-bandage wadding over a tubular cotton bandage. (c) Short-stretch compression cotton bandages applied in layers in a figure of eight and/or spiral style.
Pneumatic compression therapy (intermittent/sequential pneumatic compression) should not be used in preference to exercise and compression but can be useful in mixed lymphovenous oedema and in infirm patients [6]. An inflatable boot, legging or sleeve is connected to a motor-driven pump and lymph is displaced proximally towards the root of the limb. If hosiery is not fitted immediately following compression therapy, the swelling readily recurs. Pneumatic compression softens the tissues and reduces limb volume during treatment, but it is doubtful that any long-term benefit is gained over hosiery and exercise alone.
Massage (manual lymphatic drainage therapy)
Massage is an important component of treatment, particularly for midline lymphoedema where there are few alternatives [2]. Manual lymphatic drainage (MLD) is a massage technique performed by lymphoedema therapists with the aim of re-routing the accumulation of lymph from the swollen region via collateral lymphatic pathways to lymphatic basins that are able to drain normally. The initial step in MLD is to decongest central/proximal areas before massaging the oedematous region. This facilitates the drainage of lymph via lymphatic vessels/pathways that have been stimulated by the massage technique. Tissue movement must be gentle if it is to stimulate lymph flow without increasing blood flow [7].
A number of MLD techniques are available but no single method appears to be superior. MLD is widely practised and many patients, therapists and physicians advocate the benefits. Continuous MLD delivered by a therapist is expensive and few health care providers will fund this long term. Simple lymphatic drainage is a good alternative to MLD, and can be delivered by a partner or carer trained in the technique.
Breathing, postural exercise, elevation and rest
Breathing and postural exercises are important, particularly for clearance of lymph from the thorax and abdomen. Without the dispersal of truncal lymph, more peripheral limb oedema will not drain. Elevation per se does nothing to improve lymph drainage, but lowering venous pressure (and therefore filtration) can help to reduce swelling. Rest and elevation alone, however, are not the correct treatment for lymphoedema!
Additional therapies that may of benefit include:
- Weight loss. Many patients with lymphoedema are overweight because of morbid obesity as well as fluid retention. Excessive weight gain is likely to impair lymph drainage in the same way as it impairs venous drainage, and obesity reduces mobility (and therefore exercise). Control of weight in combination with physical treatment may be sufficient to resolve oedema completely in some patients. Weight loss irrespective of type of diet has been shown to reduce arm volume over and above what would be expected from fat loss alone in BCRL [8,9].
- Hyperbaric oxygen (HBO), low-level laser therapy (LLLT) and kinesiotaping. There are data recommending the use of HBO and LLLT independently in BCRL [10, 11, 12] although a recent randomized controlled trial failed to demonstrate significant benefits from HBO treatment [13]. However, LLLT may reduce limb volumes and pain in patients with BCRL [14]. Kinesiotaping has been shown to increase lymph flow [15].
Intensive and maintenance treatment
Patients with mild limb lymphoedema, no fibrosis and no shape distortion can be started on maintenance treatment with compression hosiery. Intensive therapy, comprising a 2–4-week course of daily skin care, MLD, multilayer bandaging and exercises, is indicated for patients with moderate to severe limb swelling, poor limb shape or tissue changes such as fibrosis, elephantiasis or lymphorrhoea. Once intensive treatment is complete, maintenance treatment with hosiery is commenced immediately. While decongestive lymphatic therapy has become accepted first line therapy, evidence for best treatment is weak [16].
Pharmacological therapies
There is little use for drug therapy in the management of lymphoedema. Diuretics alone demonstrate minimal improvement in lymphoedema, as their mode of action is to reduce capillary filtration by a reduction in circulating blood volume.
Paroven (an oxerutin) and coumarin (a benzopyrone) have been trialled in lymphoedema and may create a small reduction in limb volume by reducing vascular permeability and thus the amount of fluid forming in the subcutaneous tissues. However, this has been shown to be of little clinical benefit to the patient [17].
Animal studies involving vascular endothelial growth factors has stimulated excitement and hope amongst the lymphatic community that successful forms of drug therapy may be possible in the future. Introduction of VEGFC into animal models of postsurgical lymphoedema (using mouse skin and rabbit ears) induced lymphatic vessel growth and a subsequent reduction in lymphoedema [18, 19]. A similar study demonstrated regeneration of functional cutaneous lymphatics in skin of Chy mice with primary lymphoedema by viral-mediated VEGFC gene transfer that caused overexpression of VEGFC [20]. More recent animal studies have incorporated growth factors with lymph node transfer surgical methods. Studies using transplanted lymph nodes that were transfected with VEGFC demonstrated that the lymphatic network in the defective area could be restored, with transplanted nodes and existing vessels becoming incorporated [21, 22]. Research is currently being undertaken to optimize growth factor delivery and human trials in patients with secondary lymphoedema should commence in the near future [23].
Surgical options
Surgery has a specific role in the management of lymphoedema [24]. It is of value in limb lymphoedema in a few patients in whom, even after conservative treatment, the size and weight of a limb inhibit its use or interferes with mobility. Surgery involves either removing excessive tissue or bypassing local lymphatic defects. Lifelong non-surgical measures such as hosiery must be continued postoperatively.
Excisional methods
Excisional surgical procedures for the management of lymphoedema, regardless of the underlying cause, have been employed for more than a century [25]. They are now rarely utilized as the postoperative complications can be disastrous. Reduction (excisional) operations remove a longitudinal ellipse of skin and the underlying abnormal subcutaneous tissue down to the deep fascia in a ‘melon slice’ that permits primary closure of the skin edges (Sistrunk procedure). Undercutting of the skin allows the removal of additional tissue (Homans procedure). This procedure is preferred to circumferential excision and skin grafting (Charles procedure) or to the addition of in-rolling of a skin flap (Thompson buried dermis flap operation). Postoperative complications include skin transplant necrosis, poor cosmetic results and worsening lymphoedema distal to the surgical site. None of the excisional procedures have curative potential as all chances of restoring effective lymphatic transport have been surgically removed. Volume reduction may be achieved through tissue reduction, but not through lymphatic drainage improvement. The indications for this type of surgery should be restricted to rare cases lacking alternative conservative or surgical treatment options. The exceptions to this rule are cases of genital or eyelid lymphoedema, where reduction/debulking surgery is used sooner rather than later [26].
Lymphatico-venous anastomosis surgery
Recently there has been much interest in lymphatico-venous anastomosis (LVA) surgery. LVA is a type of lymphovenous bypass, utilizing a supermicrosurgical technique to anastomose distal subdermal lymphatic vessels with adjacent venules less than 0.8 mm in diameter in an attempt to improve regional lymph drainage and potentially remove the need for use of hosiery (a scenario that many patients are desperate to achieve). It is popular amongst surgeons as it is performed under local anaesthetic and the surgical incisions are small. Postoperative complications include lymphatic vessel occlusion, possibly due to thrombus formation within the lumen [27]. Prior to LVA surgery patients undergo ICG fluorescent imaging to locate functional patent lymphatic vessels within the affected limb.
A review of the literature suggests that the efficacy of LVA surgery is questionable. Postoperative results (within 1 year of surgery) vary greatly between centres, with limb volume reductions of 4–67% [28, 29]. There is a lack of long-term data as the technique is relatively new. However, surgeons favour its use because of the low risk of complications. There is currently no method of determining its effect on lymphatic function. The development of imaging techniques could provide a tool to answer the question of its place in lymphatic treatment strategies.
Lymphatico-lymphatic anastomosis surgery
The rationale behind lymphatic grafting surgery is to avoid the problems inherent to LVA surgery caused by coagulation of blood within the lymphatics, by connecting the lymphatic system to itself. Again, there is a lack of robust long-term data regarding efficacy.
Lymph node transfer surgery
Autologous transplantation of normal lymphatic tissue within a local or free flap to a site deficient of lymph nodes and vessels has been performed. The rationale is that the transplant of normal lymph nodes could encourage and improve lymphatic drainage in a previously oedematous region. Few surgeons seem to be performing this type of surgery but reported results appear encouraging, with one case series suggesting 40% of patients were cured of their lymphoedema [30]. However, no long-term data are available on its use. One important concern to raise with this type of surgery is that it relies on the transfer of normal lymph nodes in order to improve lymphatic drainage. This may be possible in a patient with secondary lymphoedema, but may cause complications in patients with primary lymphoedema who may not have ‘normal’ lymph nodes, or ‘normal’ lymphatic vasculature, even at unaffected sites. This concern is supported by reports of donor site lymphatic vessel dysfunction in patients undergoing microvascular lymph node transfer surgery for cancer-related lymphoedema [31].
Liposuction
Chronic lymphoedema may be associated with fatty tissue deposition in some patients, although the mechanisms of this are not fully understood. Excess adipose tissue will not respond to decongestive lymphoedema treatments, anastomosis surgery or lymph node transfer procedures. Liposuction creates significant volume reduction in therapy-resistant lymphoedema of the extremities, when combined with lifelong compression therapy [32]. This adjunctive therapy is used in compliant patients willing to wear lifelong compression garments. If patients disregard regular use of their garments, a relapse or worsening of lymphoedema is observed as liposuction may cause injury to remaining subcutaneous lymphatics within the lymphoedematous extremities [32]. However, liposuction has been shown to create a long-term volume reduction of 100% in compliant patients followed for up to 17 years [33]. Liposuction techniques, although not curative, offer an effective symptomatic treatment.
Resources
Further information
Clinical Resource Efficiency Support Team. Guidelines on the Diagnosis Assessment and Management of Lymphoedema, 2008. http://www.gain-ni.org/images/Uploads/Guidelines/CrestGuidelines.pdf.
A list of trained therapists can usually be accessed through national professional bodies for lymphoedema, for example:
Australasian Lymphology Association: http://www.lymphoedema.org.au.
Lympoedema Support Network (UK): http://www.lymphoedema.org.
National Lymphedema Network (USA): http://www.lymphnet.org.
(All last accessed August 2015.)
References
Clinical presentations of lymphatic dysfunction
Chronic, venous and drug-induced oedema
- Levick JR. An Introduction to Cardiovascular Physiology, 5th edn. London: Arnold, 2010.
- Willenberg T, Schumacher A, Amann-Vesti B, et al. Impact of obesity on venous hemodynamics of the lower limbs. J Vasc Surg 2010;52(3):664–8.
- Dreifuss SE, Manders EK. Massive scrotal edema: an unusual manifestation of obstructive sleep apnea and obesity-hypoventilation syndrome. Case Rep Med 2013;2013:685–716.
- Moffatt CJ, Franks PJ, Doherty DC, et al. Lymphoedema: an underestimated health problem. Q J Med 2003;96(10):731–8.
- Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res 2010;87(2):198–210.
- Proby CM, Gane JN, Joseph AE, et al. Investigation of the swollen limb with isotope lymphography. Br J Dermatol 1990;123:29–38.
Chronically swollen leg
- Strømgaard S, Rasmussen SW, Schmidt TA. Brief hospitalizations of elderly patients: a retrospective, observational study. Scand J Trauma Resusc Emerg Med 2014;22:17.
- Levick JR. An Introduction to Cardiovascular Physiology, 5th edn. London: Arnold, 2010.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
Phleboedema and mixed lymphovenous disease
- Kaipainen A, Korhonen J, Mustonen T, et al. Expression of the fms-like tyrosine kinase FLT4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci USA 1995;92:3566–70.
- Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res 2010;87(2):198–210.
- Rabe E, Guex JJ, Puskas A, Scuderi A, Fernandez Quesada F; VCP Coordinators. Epidemiology of chronic venous disorders in geographically diverse populations: results from the Vein Consult Program. Int Angiol 2012;31(2):105–15.
- Telinius N, Mohanakumar S, Majgaard J, et al. Human lymphatic vessel contractile activity is inhibited in vitro but not in vivo by the calcium channel blocker nifedipine. J Physiol 2014;592(2):4697–714.
- Levick JR. An Introduction to Cardiovascular Physiology, 5th edn. London: Arnold, 2010.
- Mellor RH, Brice G, Stanton AW, et al; Lymphoedema Research Consortium. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation 2007;115(14):1912–20 .
- Dean SM, Zirwas MJ, Horst AV. Elephantiasis nostras verrucosa: an institutional analysis of 21 cases. J Am Acad Dermatol 2011; 64(6):1104–10.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
- Zaleska M, Olszewski WL, Durlik M. The effectiveness of intermittent pneumatic compression in long-term therapy of lymphedema of lower limbs. Lymphat Res Biol 2014;12(2):103–9.
- Brittenden J, Cotton SC, Elders A, et al. A randomized trial comparing treatments for varicose veins. N Engl J Med 2014;371(13):1218–27.
Lipodermatosclerosis (chronic cellulitis)
- Strømgaard S, Rasmussen SW, Schmidt TA. Brief hospitalizations of elderly patients: a retrospective, observational study. Scand J Trauma Resusc Emerg Med 2014;22:17.
- Herouy Y, May AE, Pornschlegel G, et al. Lipodermatosclerosis is characterized by elevated expression and activation of matrix metalloproteinases: implications for venous ulcer formation. J Invest Dermatol 1998;111(5):822–7.
- Caggiati A, Rosi C, Casini A, et al. Skin iron deposition characterises lipodermatosclerosis and leg ulcer. Eur J Vasc Endovasc Surg 2010;40(6):777–82.
- Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part II. Conditions that simulate lower limb cellulitis. J Am Acad Dermatol 2012;67(2):177.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
- National Institute for Health and Care Excellence (NICE). Varicose Veins in the Legs: the Diagnosis and Management of Varicose Veins. NICE Guidelines No. CG168. NICE, 2013. http://www.nice.org.uk/guidance/CG168 (last accessed August 2015).
Recurrent cellulitis (erysipelas)
- Thomas KS, Crook AM, Nunn AJ, et al; UK Dermatology Clinical Trials Network's PATCH I Trial Team. Penicillin to prevent recurrent leg cellulitis. N Engl J Med 2013;368(18):1695–703.
- Jeng A, Beheshti M, Li J, Nathan R. The role of beta-hemolytic streptococci in causing diffuse, nonculturable cellulitis: a prospective investigation. Medicine (Baltimore) 2010;89:217–26.
- Damstra RJ, van Steensel MA, Boomsma JH, et al. Erysipelas as a sign of sub-clinical primary lymphoedema: a prospective quantitative scintigraphic study of 40 patients with unilateral erysipelas of the leg. Br J Dermatol 2008;158:1210–15.
- McPherson T, Persaud S, Singh S, et al. Interdigital lesions and frequency of acute dermatolymphangioadenitis in lymphoedema in a filariasis-endemic area. Br J Dermatol 2006;154(5):933–41.
- Dupuy A, Benchikhi H, Roujeau JC, et al. Risk factors for erysipelas of the leg (cellulitis): case control study. BMJ 1999;318:591–4.
- Cox NH. Oedema as a risk factor for multiple episodes of cellulitis/erysipelas of the lower leg: a series with community follow-up. Br J Dermatol 2006;155:947–50.
- Moffatt CJ, Franks PJ, Doherty DC, et al. Lymphoedema: an underestimated health problem. Q J Med 2003;96:731–8.
- Mortimer PS, Rockson SG. New developments in clinical aspects of lymphatic disease. J Clin Invest 2014;124(3):915–21.
- Chira S, Miller LG. Staphylococcus aureus is the most common identified cause of cellulitis: a systematic review. Epidemiol Infect 2010;138(3):313–17.
- Chlebicki MP, Oh CC. Recurrent cellulitis: risk factors, etiology, pathogenesis and treatment. Curr Infect Dis Rep 2014;16(9):422.
- Tay EY, Thirumoorthy T, Pang SM, Lee HY. Clinical outcomes of bacteraemia in cellulitis of the leg. Clin Exp Dermatol 2014;39(6):683–8.
- Levell NJ, Wingfield CG, Garioch JJ. Severe lower limb cellulitis is best diagnosed by dermatologists and managed with shared care between primary and secondary care. Br J Dermatol 2011;164(6):1326–8.
- British Lymphology Society (BLS). Consensus Document on the Management of Cellulitis in Lymphoedema. BLS, 2013. http://www.thebls.com/docs/Cellulitis_Consensus_2013.pdf (last accessed August 2015).
Swollen arm
- DiSipio T, Rye S, Newman B, Hayes S. Incidence of unilateral arm lymphoedema after breast cancer: a systematic review and meta-analysis. Lancet Oncol 2013;14(6):500–15.
- Levick JR. An Introduction to Cardiovascular Physiology, 5th edn. London: Arnold, 2010.
- Fletcher PG, Sterling JC. Recurrent herpes simplex virus type 2 infection of the hand complicated by persistent lymphoedema. Australas J Dermatol 2005;46(2):110–13.
- Pearce VJ, Mortimer PS. Hand dermatitis and lymphoedema. Br J Dermatol 2009;161(1):177–80.
- Kiely PD, Bland JM, Joseph AE, et al. Upper limb lymphatic function in inflammatory arthritis. J Rheumatol 1995;22:214–17.
- Mulherin DM, Fitzgerald O, Bresnihan B. Lymphoedema of the upper limb in patients with psoriatic arthritis. Semin Arthritis Rheum 1993;22:350–6.
- Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, et al. Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. Am J Med Genet A 2014;164(7):1713–33.
- Stanton A, Modi S, Mellor R, Levick R, Mortimer P. Diagnosing cancer-related lymphoedema in the arm. J Lymphoedema 2006;1:12–15.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
- British Lymphology Society (BLS). What is Lymphoedema? www.thebls.com/cellulitis (last accessed August 2015).
- Bosma J, Vahl AC, Coveliers HME, Rauwerda JA, Wisselink W. Primary subclavian vein thrombosis and its long-term effect on quality of life. Vascular 2011;8:327–32.
Swollen face, head and neck
- Deng J, Murphy BA, Dietrich MS, et al. Impact of secondary lymphedema after head and neck cancer treatment on symptoms, functional status, and quality of life. Head Neck 2013;35(7):1026–35.
- Dozsa A, Karoli ZS, Degrell P. Bilateral blepharochalasis. J Eur Acad Dermatol Venereol 2005;19:725–8.
- Carlson JA, Mazza J, Kircher K, Tran TA. Otophyma: a case report and review of the literature of lymphedema (elephantiasis) of the ear. Am J Dermatopathol 2008;30(1):67–72 .
- Buntzel J, Glatzel M, Mucke R, et al. Influence of amifostine on late radiation-toxicity in head and neck cancer – a follow-up study. Anticancer Res 2007;27:1953–6.
- Levick JR. An Introduction to Cardiovascular Physiology, 5th edn. London: Arnold, 2010.
- Connelly MG, Winkelmann RK. Solid facial edema as a complication of acne vulgaris. Arch Dermatol 1985;121:87–90.
- Nagasaka T, Koyama T, Matsumura K, Chen KR. Persistent lymphoedema in Morbihan disease: formation of perilymphatic epithelioid cell granulomas as a possible pathogenesis. Clin Exp Dermatol 2008;33:764–7.
- Manzoon S, Azadeh B. Elephantiasis of external ear: a rare manifestation of pediculosis capitis. Acta Derm Venereol (Stockh) 1983;63:363–5 .
- Worm AM, Staberg B, Thomsen K. Persistent oedema in allergic contact dermatitis. Contact Dermatitis 1983;9:517–18.
- Nozicka Z. Endovasal granulomatous lymphangitis as a pathogenetic factor in cheilitis granulomatosa. J Oral Pathol 1985;14:363–5.
- Salles AG, Lotierzo PH, Gemperli R, et al. Complications after polymethylmethacrylate injections: report of 32 cases. Plast Reconstr Surg 2008;121:1811–20.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
- Odom R, Dahl M, Dover J, et al; National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. Standard management options for rosacea, part 2: options according to subtype. Cutis 2009;84(2):97–104.
- Al Johani KA, Moles DR, Hodgson TA, Porter SR, Fedele S. Orofacial granulomatosis: clinical features and long-term outcome of therapy. J Am Acad Dermatol 2010;62:611–20.
- Smith BG, Hutcheson KA, Little LG, et al. Lymphedema outcomes in patients with head and neck cancer. Otolaryngol Head Neck Surg 2015;152(2):284–91.
Swollen genitalia and mons pubis
- Daroczy j. Pathology of lymphoedema. Clin Dermatol 1995;13:433–44.
- Browse NL. Management of genital lymphoedema. In: Browse NL, Burnand KG, Mortimer PS, eds. Diseases of the Lymphatics. London: Arnold, 2003:217–30.
- Dandapat MC, Mohapatro SK, Patro SK. Elephantiasis of the penis and scrotum. A review of 350 cases. Am J Surg 1985;149:686–90.
- Narang T, Kanwar AJ. Genital elephantiasis due to donovanosis: forgotten but not gone yet … Int J STD AIDS 2012;23(11):835–6.
- Mansour S, Connell F, Steward C, et al; Lymphoedema Research Consortium. Emberger syndrome – primary lymphedema with myelodysplasia: report of seven new cases. Am J Med Genet A 2010;152A(9):2287–96.
- Brice G, Mansour S, Bell R, et al. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet 2002;39(7):478–83.
- Alders M, Al-Gazali L, Cordeiro I, et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome. Hum Genet 2014;133(9):1161–7.
- Smpokou P, Tworog-Dube E, Kucherlapati RS, Roberts AE. Medical complications, clinical findings, and educational outcomes in adults with Noonan syndrome. Am J Med Genet A 2012;158A(12):3106–11.
- British Lymphology Society (BLS). What is Lymphoedema? www.thebls.com/cellulitis (last accessed August 2015).
- Brewer MB, Singh DP. Massive localized lymphedema: review of an emerging problem and report of a complex case in the mons pubis. Ann Plast Surg 2012;68(1):101–4.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
- Garaffa G, Christopher N, Ralph DJ. The management of genital lymphoedema. BJU Int 2008;102(4):480–4.
- Thomas CL, Gordon KD, Mortimer PS. Rapid resolution of hidradenitis suppurativa after bariatric surgical intervention. Clin Exp Dermatol 2014;39(3):315–17.
Obesity-related lymphoedema
- Disipio T, Rye S, Newman S, Hayes S. Incidence of unilateral arm lymphoedema after breast cancer: a systematic review and meta-analysis. Lancet Oncol 2013;14(6):500–15.
- Shaw C, Mortimer P, Judd PA. A randomized controlled trial of weight reduction as a treatment for breast cancer-related lymphedema. Cancer 2007;110:1868–74.
- Todd M. Managing chronic oedema in the morbidly obese patient. Br J Nurs 2009;18(18):1120–4.
- Randolph GJ, Miller NE. Lymphatic transport of high-density lipoproteins and chylomicrons. J Clin Invest 2014;124(3):929–35.
- Lim HY, Thiam CH, Yeo KP, et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab 2013;17(5):671–84.
- Harvey NL, Srinivasan RS, Dillard ME, et al. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat Genet 2005;37(10):1072–81.
- O'Malley E, Ahern T, Dunlevy C, Lehane C, Kirby B, O'Shea D. Obesity related chronic lymphoedema-like swelling and physical function. Q J Med 2015;108(3):183–7.
- Greene AK, Grant FD, Slavin SA. Lower-extremity lymphedema and elevated body-mass index. N Engl J Med 2012;366:2136–7.
- Arngrim N, Simonsen L, Holst JJ, Bulow J. Reduced adipose tissue lymphatic drainage of macromolecules in obese subjects: a possible link between obesity and local tissue inflammation. Int J Obes (Lond) 2013;37(5):748–50.
- Arfvidsson B, Eklof B, Balfour J. Iliofemoral venous pressure correlates with intraabdominal pressure in morbidly obese patients. Vasc Endovascular Surg 2005;39:505–9.
- Willenberg T, Schumacher A, Amann-Vesti B, et al. Impact of obesity on venous hemodynamics of the lower limbs. J Vasc Surg 2010;52(3):664–8.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
- British Lymphology Society (BLS). What is Lymphoedema? www.thebls.com/cellulitis (last accessed August 2015).
Abdominal wall lymphoedema
- Buyuktas D, Arslan E, Celik O, Tasan E, Demirkesen C, Gundogdu S. Elephantiasis nostras verrucosa on the abdomen of a Turkish female patient caused by morbid obesity [Letter]. Dermatol Online J 2010;16(8):14.
- Bull RH, Mortimer PS. Acute lipodermatosclerosis in a pendulous abdomen. Clin Exp Dermatol 1993;18(2):164–6.
- British Lymphology Society (BLS). What is Lymphoedema? www.thebls.com/cellulitis (last accessed August 2015)..
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
- Ollapallil J, Koong D, Panchacharavel G, Butcher C, Yapo B. New method of abdominoplasty for morbidly obese patients. ANZ J Surg 2004;74(6):504–6.
Cancer-related lymphoedema
- DiSipio T, Rye S, Newman B, Hayes S. Incidence of unilateral arm lymphoedema after breast cancer: a systematic review and meta-analysis. Lancet Oncol 2013;14(6):500–15.
- Jung SY, Shin KH, Kim M, et al. Treatment factors affecting breast cancer-related lymphedema after systemic chemotherapy and radiotherapy in stage II/III breast cancer patients. Breast Cancer Res Treat 2014;148(1):91–8.
- Snijders-Keilholz A, Hellebrekers BW, Zwinderman AH, et al. Adjuvant radiotherapy following radical hysterectomy for patients with early-stage cervical carcinoma (1984–1996). Radiother Oncol 1999;51:161–7.
- Chatani M, Nose T, Masaki N, Inoue T. Adjuvant radiotherapy after radical hysterectomy of the cervical cancer. Prognostic factors and complications. Strahlenther Onkol 1998;174:504–9 .
- Gaarenstroom KN, Kenter GG, Trimbos JB, et al. Postoperative complications after vulvectomy and inguinofemoral lymphadenectomy using separate groin incisions. Int J Gynecol Cancer 2003;13:522–7 .
- Keegan KA, Cookson MS. Complications of pelvic lymph node dissection for prostate cancer. Curr Urol Rep 2011;12(3):203–8.
- Pilepich MV, Krall J, George FW, et al. Treatment-related morbidity in phase III RTOG studies of extended-field irradiation for carcinoma of the prostate. Int J Radiat Oncol Biol Phys 1984;10(10):1861–7.
- Bevan-Thomas R, Slaton JW, Pettaway CA. Contemporary morbidity from lymphadenectomy for penile squamous cell carcinoma: the M.D. Anderson Cancer Center Experience. J Urol 2002;167:1638–42.
- Hyngstrom JR, Chiang YJ, Cromwell KD, et al. Prospective assessment of lymphedema incidence and lymphedema-associated symptoms following lymph node surgery for melanoma. Melanoma Res 2013;23(4):290–7.
- Friedmann D, Wunder JS, Ferguson P, et al. Incidence and severity of lymphoedema following limb salvage of extremity soft tissue sarcoma. Sarcoma 2011;2011:289673.
- Donker M, van Tienhoven G, Straver ME, et al. Radiotherapy or surgery of the axilla after a positive sentinel node in breast cancer (EORTC 10981-22023 AMAROS): a randomised, multicentre, open-label, phase 3 non-inferiority trial. Lancet Oncol 2014;15(12):1303–10.
- Marneros AG, Blanco F, Husain S, et al. Classification of cutaneous intravascular breast cancer metastases based on immunolabeling for blood and lymph vessels. J Am Acad Dermatol 2009;60:633–8.
- Finkel LJ, Griffiths CEM. Inflammatory breast carcinoma (carcinoma erysipeloides), an easily overlooked diagnosis. Br J Dermatol 1993;129:324–6.
Swollen breast and breast lymphoedema
- Boughey JC, Hoskin TL, Cheville AL, et al. Risk factors associated with breast lymphedema. Ann Surg Oncol 2014;21(4):1202–8.
- Levick JR. An Introduction to Cardiovascular Physiology, 5th edn. London: Arnold, 2010.
- Jung SY, Shin KH, Kim M, et al. Treatment factors affecting breast cancer-related lymphedema after systemic chemotherapy and radiotherapy in stage II/III breast cancer patients. Breast Cancer Res Treat 2014;148(1):91–8.
- Boughey JC, Hoskin TL, Cheville AL, et al. Risk factors associated with breast lymphedema. Ann Surg Oncol 2014;21(4):1202–8.
- Hille U, Soergel P, Makowski L, Dörk-Bousset T, Hillemanns P. Lymphedema of the breast as a symptom of internal diseases or side effect of mTor inhibitors. Lymphat Res Biol 2012;10(2):63–73.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
Massive localized lymphoedema
- Lu S, Tran Ta, Jones DM, et al. Localized lymphedema (elephantiasis): a case series and review of the literature. J Cutan Pathol 2008;36:1–20.
- Manduch M, Oliveira AM, Nascimento AG, Folpe AL. Massive localised lymphoedema: a clinicopathological study of 22 cases and review of the literature. J Clin Pathol 2009;62(9):808–11.
- Chopra K, Tadisina KK, Brewer M, Holton LH, Banda AK, Singh DP. Massive localized lymphedema revisited: a quickly rising complication of the obesity epidemic. Ann Plast Surg 2015;74(1):126–32.
- Khanna M, Naraghi AM, Salonen D, et al. Massive localised lymphoedema: clinical presentation and MR imaging characteristics. Skeletal Radiol 2011;40(5):647–52 .
Primary lymphoedema
- Connell F, Brice G, Jeffery S, Keeley V, Mortimer P, Mansour S. A new classification system for primary lymphatic dysplasias based on phenotype. Clin Genet 2010;77(5):438–52.
- Ferrell RE, Levinson KL, Esman JH, et al. Hereditary lymphedema: evidence for linkage and genetic heterogeneity. Hum Mol Genet 1998;7(13):2073–8.
- Evans AL, Brice G, Sotirova V, et al. Mapping of primary congenital lymphedema to the 5q35.3 region. Am J Hum Genet 1999;64(2):547–55.
- Karkkainen MJ, Ferrell RE, Lawrence EC, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nature Genet 2000;25(2):153–9.
- Irrthum A, Karkkainen MJ, Devriendt K, Alitalo K, Vikkula M. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet 2000;67(2):295–301.
- Mangion J, Rahman N, Mansour S, et al. A gene for lymphedema-distichiasis maps to 16q24.3. Am J Hum Genet 1999;65(2):427–32.
- Fang JM, Dagenais SL, Erickson RP, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet 2000;67(6):1382–8.
- Brice G, Mansour S, Bell R, et al. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet 2002;39(7):478–83.
- Mendola A, Schlogel MJ, Ghalamkarpour A, et al. Mutations in the VEGFR3 signaling pathway explain 36% of familial lymphedema. Mol Syndromol 2013;4(6):257–66.
- Connell F, Gordon K, Brice G, et al. The classification and diagnostic algorithm for primary lymphatic dysplasia: an update from 2010 to include molecular findings. Clin Genet 2013;84(4):303–14.
- Dale RF. The inheritance of primary lymphoedema. J Med Genet 1985;22(4):274–8.
- Moffatt CJ, Franks PJ, Doherty DC, et al. Lymphoedema: an underestimated health problem. Q J Med 2003;96(10):731–8.
- Mortimer PS. Managing lymphoedema. Clin Exp Dermatol 1995;20(2):98–106.
- Stemmer R. A clinical sign for the early and differential diagnosis of lymphedema. Vasa 1976;5(3):261–2.
- Opitz JM. On congenital lymphedema. Am J Med Genet 1986;24(1):127–9.
- Jeffries GH, Chapman A, Sleisenger MH. Low-fat diet in intestinal lymphangiectasia. Its effect on albumin metabolism. N Engl J Med 1964;270:761–6.
- Hennekam RC, Geerdink RA, Hamel BC, et al. Autosomal recessive intestinal lymphangiectasia and lymphedema, with facial anomalies and mental retardation. Am J Med Genet 1989;34(4):593–600.
- Bellini C, Mazzella M, Arioni C, et al. Hennekam syndrome presenting as nonimmune hydrops fetalis, congenital chylothorax, and congenital pulmonary lymphangiectasia. Am J Med Genet A 2003;120A(1):92–6.
- Van Balkom IDC, Alders M, Allanson J, et al. Lymphedema-lymphangiectasia-mental retardation (Hennekam) syndrome: a review. Am J Med Genet 2002;112(4):412–21.
- Alders M, Hogan BM, Gjini E, et al. Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nature Genet 2009;41(12):1272–4.
- Connell F, Kalidas K, Ostergaard P, et al. Linkage and sequence analysis indicate that CCBE1 is mutated in recessively inherited generalised lymphatic dysplasia. Hum Genet 2010;127(2):231–41.
- Alders M, Al-Gazali L, Cordeiro I, et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome. Hum Genet 2014;133(9):1161–7.
- Hogan BM, Bos FL, Bussmann J, et al. ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nature Genet 2009;41(4):396–8.
- Bos FL, Caunt M, Peterson-Maduro J, et al. CCBE1 is essential for mammalian lymphatic vascular development and enhances the lymphangiogenic effect of vascular endothelial growth factor-C in vivo. Circ Res 2011;109(5):486–91.
- Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med 2011;365(7):611–19.
- Kurek KC, Luks VL, Ayturk UM, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet 2012;90(6):1108–15.
- Lindhurst MJ, Parker VER, Payne F, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nature Genet 2012;44(8).
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet 2006;14(11):1151–7.
- Kreuter A, Hochdorfer B, Brockmeyer NH, et al. A human papillomavirus-associated disease with disseminated warts, depressed cell-mediated immunity, primary lymphedema, and anogenital dysplasia – WILD syndrome. Arch Dermatol 2008;144(3):366–72.
- Brice G, Child AH, Evans A, et al. Milroy disease and the VEGFR-3 mutation phenotype. J Med Genet 2005;42(2):98–102.
- Gordon K, Spiden SL, Connell FC, et al. FLT4/VEGFR3 and Milroy disease: novel mutations, a review of published variants and database update. Hum Mutat 2013 2013;34(1):23–31.
- Ghalamkarpour A, Morlot S, Raas-Rothschild A, et al. Hereditary lymphedema type I associated with VEGFR3 mutation: the first de novo case and atypical presentations. Clin Genet 2006;70(4):330–5.
- Mellor RH, Hubert CE, Stanton AWB, et al. Lymphatic dysfunction, not aplasia, underlies Milroy disease. Microcirculation 2010;17(4):281–96.
- Connell FC, Ostergaard P, Carver C, et al. Analysis of the coding regions of VEGFR3 and VEGFC in Milroy disease and other primary lymphoedemas. Hum Genet 2009;124(6):625–31.
- Gordon K, Schulte D, Brice G, et al. Mutation in vascular endothelial growth factor-C, a ligand for vascular endothelial growth factor receptor-3, is associated with autosomal dominant Milroy-like primary lymphedema. Circ Res 2013;112(6):956–60.
- Balboa-Beltran E, Fernandez-Seara MJ, Perez-Munuzuri A, et al. A novel stop mutation in the vascular endothelial growth factor-C gene (VEGFC) results in Milroy-like disease. J Med Genet 2014;51(7):475–8.
- Kuchler AM, Gjini E, Peterson-Maduro J, Cancilla B, Wolburg H, Schulte-Merker S. Development of the zebrafish lymphatic system requires VEGFC signaling. Curr Biol 2006;16(12):1244–8.
- Leung AKC. Dominantly inherited syndrome of microcephaly and congenital lymphedema. Clin Genet 1985;27(6):611–12.
- Ostergaard P, Simpson MA, Mendola A, et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am J Hum Genet 2012;90(2):356–62.
- Kuhnt H. IV. Ueber Distichiasis (congenita) vera. Ophthalmologica 1899;2(1):46–57.
- Brice G, Mansour S, Bell R, et al. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet 2002;39(7):478–83.
- Norrmen C, Ivanov KI, Cheng J, et al. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J Cell Biol 2009;185(3):439–57.
- Petrova TV, Karpanen T, Norrmen C, et al. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nature Med 2004;10(9):974–81.
- Mellor RH, Brice G, Stanton AWB, et al. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation 2007;115(14):1912–20.
- Rezaie T, Ghoroghchian R, Bell R, et al. Primary non-syndromic lymphoedema (Meige disease) is not caused by mutations in FOXC2. Eur J Hum Genet 2008;16(3):300–4.
- Emberger JM, Navarro M, Dejean M, Izarn P. Deaf mutism, lymphedema of the lower limbs and hematological anomalies (acute-leukemia, cytopenia) with autosomal dominant transmission. J Genet Hum 1979;27(3):237–45.
- Mansour S, Connell F, Steward C, et al. Emberger syndrome – primary lymphedema with myelodysplasia: report of seven new cases. Am J Med Genet A 2010;152A(9):2287–96.
- Ostergaard P, Simpson MA, Connell FC, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nature Genet 2011;43(10):929–31.
- Khandekar M, Brandt W, Zhou Y, et al. A Gata2 intronic enhancer confers its pan-endothelia-specific regulation. Development 2007;134(9):1703–12.
- Kazenwadel J, Secker GA, Liu YJ, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood 2012;119(5):1283–91.
- Hahn CN, Chong C-E, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nature Genet 2011;43(10):1012–7.
- Ostergaard P, Simpson MA, Brice G, et al. Rapid identification of mutations in GJC2 in primary lymphoedema using whole exome sequencing combined with linkage analysis with delineation of the phenotype. J Med Genet 2011;48(4):251–5.
- International Society of Lymphology (ISL). The diagnosis and treatment of peripheral lymphedema: 2013 consensus document of the International Society of Lymphology. Lymphology 2013;46(1):1–11.
- Mortimer PS, Rockson SG. New developments in clinical aspects of lymphatic disease. J Clin Invest 2014;124(3):915–21.
Lipoedema
- Allen EV, Hines EA. Lipedema of the legs: a syndrome characterized by fat legs and orthostatic edema. Staff Meetings Mayo Clin 1940;15:184–7.
- Langendoen SI, Habbema L, Nijsten TE, Neumann HA. Lipoedema: from clinical presentation to therapy. A review of the literature. Br J Dermatol 2009;161(5):980–6.
- Schmeller W, Meier-Vollrath I. Tumescent liposuction: a new and successful therapy for lipedema. J Cutan Med Surg 2006;10(1):7–10.
- Beninson J, Edelglass JW. Lipedema – the non-lymphatic masquerader. Angiology 1984;35(8):506–10.
- Harwood CA, Bull RH, Evans J, Mortimer PS. Lymphatic and venous function in lipoedema. Br J Dermatol 1996;134(1):1–6.
- Fonder MA, Loveless JW, Lazarus GS. Lipedema, a frequently unrecognized problem. J Am Acad Dermatol 2007;57(Suppl. 2):S1–3.
- Child AH, Gordon KD, Sharpe P, et al. Lipedema: an inherited condition. Am J Med Genet A 2010;152A(4):970–6.
- Wold LE, Hines EA, Jr, Allen EV. Lipedema of the legs; a syndrome characterized by fat legs and edema. Ann Intern Med 1951;34(5):1243–50.
- Chen SG, Hsu SD, Chen TM, Wang HJ. Painful fat syndrome in a male patient. Br J Plast Surg 2004;57(3):282–6.
- Stallworth JM, Hennigar GR, Jonsson HT, Jr, Rodriguez O. The chronically swollen painful extremity. A detailed study for possible etiological factors. JAMA 1974;228(13):1656–9.
- Suga H, Araki J, Aoi N, Kato H, Higashino T, Yoshimura K. Adipose tissue remodeling in lipedema: adipocyte death and concurrent regeneration. J Cutan Pathol 2009;36(12):1293–8.
- Rapprich S, Dingler A, Podda M. Liposuction is an effective treatment for lipedema – results of a study with 25 patients. J Dtsch Dermatol Ges 2011;9(1):33–40.
- Hansson E, Svensson H, Brorson H. Review of Dercum's disease and proposal of diagnostic criteria, diagnostic methods, classification and management. Orphanet J Rare Dis 2012;7:23.
- Reich-Schupke S, Altmeyer P, Stucker M. Thick legs – not always lipedema. J Dtsch Dermatol Ges 2013;11(3):225–33.
- Boursier V, Pecking A, Vignes S. [Comparative analysis of lymphoscintigraphy between lipedema and lower limb lymphedema.] J Mal Vasc 2004;29(5):257–61.
- Lohrmann C, Foeldi E, Langer M. MR imaging of the lymphatic system in patients with lipedema and lipo-lymphedema. Microvasc Res 2009;77(3):335–9.
- Amann-Vesti BR, Franzeck UK, Bollinger A. Microlymphatic aneurysms in patients with lipedema. Lymphology 2001;34(4):170–5.
- Naouri M, Samimi M, Atlan M, et al. High-resolution cutaneous ultrasonography to differentiate lipoedema from lymphoedema. Br J Dermatol 2010;163(2):296–301.
- Szolnoky G, Varga E, Varga M, Tuczai M, Dosa-Racz E, Kemeny L. Lymphedema treatment decreases pain intensity in lipedema. Lymphology 2011;44(4):178–82.
- Wienert V, Gerlach H, Gallenkemper G, et al. Leitlinie Medizinischer Kompressionsstrumpf (MKS). Phlebologie 2006;35:315–20.
- Meier-Vollrath I, Schneider W, Schmeller W. Lipödem: Verbesserte Lebensqualität durch Therapiekombination. Dtsch Ärztebl 2005;102:A1061–7.
- Marshall M, Schwahn-Schreiber C. Das Lipödem – ein wenig beachtetes Krankheitsbild. Vasomed 2008;20:59–65.
- Schmeller W, Hueppe M, Meier-Vollrath I. Tumescent liposuction in lipoedema yields good long-term results. Br J Dermatol 2012;166(1):161–8.
- Peled AW, Slavin SA, Brorson H. Long-term outcome after surgical treatment of lipedema. Ann Plast Surg 2012;68(3):303–7.
- Schingale FJ. Lipödem und Lymphödem – komprimieren oder absaugen? Vasomed 2012;24:18–19.
Yellow-nail syndrome
- McKusick VA. Mendelian Inheritance in Man, 12th edn. Baltimore: Johns Hopkins University Press, 1998.
- Samman PD, White WF. The yellow nail syndrome. Br J Dermatol 1964;76:153–7.
- Valdés L, Huggins JT, Gude F, et al. Characteristics of patients with yellow nail syndrome and pleural effusion. Respirology 2014;19(7):985–92.
- Emerson PA. Yellow nails, lymphoedema and pleural effusions. Thorax 1966;21:247–53.
- Bull RH, Fenton DA, Mortimer PS. Lymphatic function in the yellow nail syndrome. Br J Dermatol 1996;134:307–12.
- Wells, GC. Yellow nail syndrome with familial primary hypoplasia of lymphatics, manifest late in life. Proc Roy Soc Med 1966;59:447.
- Govaert P, Leroy JG, Pauwels R, et al. Perinatal manifestations of maternal yellow nail syndrome. Pediatrics 1992;89:1016–18.
- Slee J, Nelson J, Dickinson J, Kendall P, Halbert A. Yellow nail syndrome presenting as non-immune hydrops: second case report. Am J Med Genet 2000;93:1–4.
- Hoque SR, Mansour S, Mortimer PS. Yellow nail syndrome: not a genetic disorder? Eleven new cases and a review of the literature. Br J Dermatol 2007;156(6):1230–4.
- Maldonado F, Tazelaar HD, Wang C, Ryu JH. Yellow nail syndrome: analysis of 41 consecutive patients. Chest 2008;134:375–81.
- Lambert EM, Dziura J, Kauls L, Mercurio M, Antaya RJ. Yellow nail syndrome in three siblings: a randomized double-blind trial of topical vitamin E. Pediatr Dermatol 2006;23(4):390–5 .
- Baran R, Thomas L. Combination of fluconazole and alpha-tocopherol in the treatment of yellow nail syndrome. J Drugs Dermatol 2009;8(3):276–8.
Lymphatic malformation and lymphangioma circumscriptum
- Marler JJ, Mulliken JB. Current management of hemangiomas and vascular malformations. Clin Plast Surg 2005;32(1):99–116.
- Chervanak FA, Isaacson G, Blakemore KJ, et al. Fetal cystic hygroma, cause and natural history. N Engl J Med 1983;309:822–5.
- Peachey RC, Lim CC, Whimster IM. Lymphangiomas of the skin. Br J Dermatol 1970;83:519–27.
- Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol 2009;48(12):1290–5.
- Hogeling M, Adams S, Law J, Wargon O. Lymphatic malformations: clinical course and management in 64 cases. Australas J Dermatol 2011;52(3):186–90.
- Greene AK, Perlyn CA, Alomari AI. Management of lymphatic malformations. Clin Plast Surg 2011;38:75–82.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol 1976;94:473–86.
- Brouillard P, Boon L, Vikkula M. Genetics of lymphatic anomalies. J Clin Invest 2014;124(3):898–904.
- Siew-Kiang Tan, Yong-Kwang Tay. Lymphangioma circumscriptum. N Engl J Med 2012;366:1724.
- Kurek KC, Howard E, Tennant LB, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet 2012;90(6):1108–15.
- Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med 2011;365(7):611–19.
- Lee BB, Kim YW, Seo JM, et al. Current concepts in lymphatic malformation. Vasc Endovasc Surg 2005;39(1):67–81.
- Navarro-Núñez L, Langan SA, Nash GB, Watson SP. The physiological and pathophysiological roles of platelet CLEC-2. Thromb Haemost 2013;109(6):991–8.
- Wilson GR, Cox NH, McLean NR, Scott D. Squamous cell carcinoma arising within congenital lymphangioma circumscriptum. Br J Dermatol 1993;129(3):337–9.
- Chaudry G, Guevara CJ, Rialon KL, et al. Safety and efficacy of bleomycin sclerotherapy for microcystic lymphatic malformation. Cardiovasc Intervent Radiol 2014;37(6):1476–81.
- British Lymphology Society (BLS). What is Lymphoedema? www.thebls.com/cellulitis (last accessed August 2015).
- Lee BB, Andrade M, Antignani PL, et al; International Union of Phlebology. Diagnosis and treatment of primary lymphedema. Consensus document of the International Union of Phlebology (IUP)-2013. Int Angiol 2013;32(6):541–74.
- Danial C, Tichy AL, Tariq U, et al. An open-label study to evaluate sildenafil for the treatment of lymphatic malformations. J Am Acad Dermatol 2014;70(6):1050–7.
Lymphoedema as a result of amniotic band constriction
- Yu VY. Neonatal consequences of placental and membrane dysfunction. Reprod Fertil Dev 1991;3(4):431–7.
- Das SP, Sahoo P, Mohanty R, Das S. One-stage release of congenital constriction band in lower limb from new born to 3 years. Indian J Orthop 2010;44(2):198–201.
- Doi Y, Kawamata H, Asano K, Imai YJ. A case of amniotic band syndrome with cleft lip and palate. Maxillofac Oral Surg 2011;10(4):354–6.
- Birth Defects Res A Clin Mol Teratol. 2009 January; 85(1): 52–57.
- Tadmor OP, Kreisberg GA, Achiron R, Porat S, Yagel S. Limb amputation in amniotic band syndrome: serial ultrasonographic and Doppler observations. Ultrasound Obstet Gynecol 1997;10:312–15.
- Sentilhes L, Verspyck E, Patrier S, Eurin D, Lechevallier J, Marpeau L. Amniotic band syndrome: pathogenesis, prenatal diagnosis and neonatal management. J Gynecol Obstet Biol Reprod 2003;32:693–704.
- Blyth M, Lachlan K. Amniotic bands in paternal half-siblings. Clin Dysmorphol 2010;19(2):62–4.
- Mempel M, Abeck D, Lange I, et al. The wide spectrum of clinical expression in Adams-Oliver syndrome: a report of two cases. Br J Dermatol 1999;140(6):1157–60.
- Mortimer PS, Rockson SG. New developments in clinical aspects of lymphatic disease. J Clin Invest 2014;124(3):915–21.
- Paladini D, Foglia S, Sglavo G, Martinelli P. Congenital constriction band of the upper arm: the role of three-dimensional ultrasound in diagnosis, counseling and multidisciplinary consultation. Ultrasound Obstet Gynecol 2004;23:520–2.
- Soldado F, Aguirre M, Peiró JL, et al. Fetoscopic release of extremity amniotic bands with risk of amputation. J Pediatr Orthop 2009;29:290–3.
Lymphangiomatosis, lymphangioleiomyomatosis and non-malignant lymphatic tumours
- Mazreeuw-Hautier J, Syed S, Leisner R, Harper J. Extensive venous/lymphatic malformations causing life-threatening haematological complications. Br J Dermatol 2007;157:558–63.
- Dellinger MT, Garg N, Olsen BR. Viewpoints on vessels and vanishing bones in Gorham-Stout disease. Bone 2014;63:47–52.
- Lala S, Mulliken JB, Alomari AI, Fishman SJ, Kozakewich HP, Chaudry G. Gorham-Stout disease and generalized lymphatic anomaly – clinical, radiologic, and histologic differentiation. Skeletal Radiol 2013;42(7):917–24.
- Wilson Jones E, Winkelmann RK, Zachary CB, et al. Benign lymphangio-endothelioma. J Am Acad Dermatol 1990;23:229–34.
- Guillou L, Fletcher CD. Benign lymphangioendothelioma (acquired progressive lymphangioma): a lesion not to be confused with well-differentiated angiosarcoma and patch stage Kaposi's sarcoma: clinicopathologic analysis of a series. Am J Surg Pathol 2000;24:1047–57.
- Wang L, Chen L, Yang X, Gao T, Wang G. Benign lymphangioendothelioma: a clinical, histopathologic and immunohistochemical analysis of four cases. J Cutan Pathol 2013;40(11):945–9.
- Carlton A, St Elkington JC, Greenfield JG, et al. Maffucci's syndrome. Q J Med 1942;11:203–28.
- Jermann M, Eid K, Pfammatter T, et al. Maffucci's syndrome. Circulation 2001;104:1693.
- Safi F, Gupta A, Adams D, Anandan V, McCormack FX, Assaly R. Kaposiform lymphangiomatosis, a newly characterized vascular anomaly presenting with hemoptysis in an adult woman. Ann Am Thorac Soc 2014;11(1):92–5.
- Diaz-Cascajo C, Borghi S, Weyers W, et al. Benign lymphangiomatous papules of the skin following radiotherapy: a report of five new cases and review of the literature. Histopathology 1999;35:319–27.
- Gengler C, Coindre JM, Leroux A, et al. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer 2007;109(8):1584–98.
- Flucke U, Requena L, Mentzel T. Radiation-induced vascular lesions of the skin: an overview. Adv Anat Pathol 2013;20(6):407–15.
- Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013;381(9869):817–24.
Lymphangiectasia
- Goldstein JB, McNatt NS, Hambrick GW. Penicillamine dermopathy with lymphangiectases. Arch Dermatol 1989;125:92–7.
- Harwood CA, Mortimer PS. Acquired vulval lymphangiomata mimicking genital warts. Br J Dermatol 1993;129: 34–6.
- Mallett RB, Curley RK, Mortimer PS. Acquired lymphangiomata: report of four cases and a discussion of the pathogenesis. Br J Dermatol 1992;126:380–2.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol 1976;94(5):473–86.
- Browse NL. Management of lymph and chyle reflux. In: Browse NL, Burnand KG, Mortimer PS, eds. Diseases of the Lymphatics. London: Arnold, 2003:259–92.
- Waldmann TA, Steinfeld JL, Dutcher TF, et al. The role of the gastrointestinal system in “idiopathic hypoproteinemia.” Gastroenterology 1961;41:197–207.
- Lee J, Kong MS. Primary intestinal lymphangiectasia diagnosed by endoscopy following the intake of a high-fat meal. Eur J Pediatr 2008;167:237–9.
- Braamskamp MJ, Dolman KM, Tabbers MM. Protein-losing enteropathy in children. Eur J Pediatr 2010;169(10):1179–85.
- Bliss CM, Schroy PC, III. Primary intestinal lymphangiectasia. Curr Treat Options Gastroenterol 2004;7:3–6.
- Makh DS, Mortimer P, Powell B. A review of the surgical treatment of vulval lymphangioma and lymphangiectasia: four case reviews. J Plast Reconstr Aesthet Surg 2006;59:1442–5.
Lymphocele, seroma and lymph fistula
- Ferguson JH, Maclure JG. Lymphocele following lymphadenectomy. Am J Obstet Gynecol 1961;82:783–92.
- Watt-Boolsen S, Nielsen VB, Jensen J, Bak S. Postmastectomy seroma. A study of the nature and origin of seroma after mastectomy. Dan Med Bull 1989;36(5):487–9.
Lymphatic filariasis
- Rockson SG. Lymphedema. Am J Med 2001;110:288–95.
- Lymphatic filariasis: the disease and its control. Fifth report of the WHO Expert Committee on Filariasis. World Health Organ Tech Rep Ser 1992;821:1–71.
- Nutman T. Insights into the pathogenesis of disease in human lymphatic filariasis. Lymph Res Biol 2013;11(3):144–8.
- Chakraborty S, Gurusamy M, Zawieja D, Muthuchamy M. Lymphatic filariasis: perspectives on lymphatic remodelling and contractile dysfunction in filarial disease pathogenesis. Microcirculation 2013;20(5):349–64.
- Bockarie MJ, Taylor MJ, Gyapong JO. Current practices in the management of lymphatic filariasis. Expert Rev Anti Infect Ther 2009;7:595–6.
- World Health Organization (WHO). Global programme to eliminate lymphatic filariasis. Wkly Epidemiol Rec 2014;89(38):409–18.
- Von Lichtenberg F. The Wellcome Trust lecture. Inflammatory responses to filarial connective tissue parasites. Parasitology 1987;94(Suppl.):S101–22.
- Olszewski WL, Jamal S, Manokaran G, et al. Bacteriologic studies of skin, tissue fluid, lymph, and lymph nodes in patients with filarial lymphedema. Am J Trop Med Hyg 1997;57:7–15.
- Shenoy RK, Kumaraswami V, Suma TK, Rajan K, Radhakuttyamma G. A double-blind, placebo-controlled study of the efficacy of oral penicillin, diethylcarbamazine or local treatment of the affected limb in preventing acute adenolymphangitis in lymphoedema caused by brugian filariasis. Ann Trop Med Parasitol 1999;93:367–77.
- Dreyer G, Noroes J, Figueredo-Silva J, Piessens WF. Pathogenesis of lymphatic disease in bancroftian filariasis: a clinical perspective. Parasitol Today 2000;16:544–8.
- Figueredo-Silva J, Noroes J, Cedenho A, Dreyer G. The histopathology of bancroftian filariasis revisited: the role of the adult worm in the lymphatic-vessel disease. Ann Trop Med Parasitol 2002;96:531–41.
- Bennuru S, Nutman TB. Lymphatics in human lymphatic filariasis: in vitro models of parasite induced lymphatic remodeling. Lymphat Res Biol 2009;7:215–19.
- Freedman DO, de Almeida Filho PJ, Besh S, et al. Lymphoscintigraphic analysis of lymphatic abnormalities in symptomatic and asymptomatic human filariasis. J Infect Dis 1994;170:927–33.
- Noroes J, Addis D, Amaral F, et al. Occurrence of living adult Wuchereria bancrofti in the scrotal area of men with microfilaremia. Trans R Soc Trop Med Hyg 1996;90:55–6.
- Pani SP, Yuvaraj J, Vanamail D, et al. Episodic adenolymphangitis and lymphoedema in patients with bancroftian filariasis. Trans R Soc Trop Med Hyg 1992;89:72–4.
- Dreyer G, Medeiros Z, Netto MJ, Leal NC, de Castro LG, Piessens WF. Acute attacks in the extremities of persons living in an area endemic for bancroftian filariasis: differentiation of two syndromes. Trans R Soc Trop Med Hyg 1999;93:413–17.
- Ottesen EA, Nutman TB. Tropical pulmonary eosinophilia. Annu Rev Med 1992;43:417–24.
- Rom WN, Vijayan VJ, Cornelius MJ, et al. Persistent lower respiratory tract inflammation associated with interstitial lung disease in patients with tropical pulmonary eosinophilia following conventional treatment with diethylcarbamazine. Am Rev Respir Dis 1990;142:1088–92.
- Weil GJ, Lammie PJ, Weiss N. The ICT filariasis test: a rapid-format antigen test for diagnosis of bancroftian filariasis. Parasitol Today 1997;13:401–4.
- Tandon V, Singh H, Dwivedi US, Mahmood M, Singh PB. Filarial chyluria: long-term experience of a university hospital in India. Int J Urol 2004;11(4):193–8.
- McPherson T. Impact on the quality of life of lymphoedema patients following introduction of a hygiene and skin care regimen in a Guyanese community endemic for lymphatic filariasis: a preliminary clinical intervention study. Filaria J 2003;2(1):1.
- Centers for Disease Control and Prevention (CDC). Lymphatic filariasis. http://www.cdc.gov/dpdx/lymphaticFilariasis/index.html (last accessed August 2015).
Podoconiosis
- Price EW. Non-filarial elephantiasis – confirmed as a geochemical disease and renamed podoconiosis. Ethiop Med J 1988;26:151–3.
- Davey G. Recent advances in podoconiosis. Ann Trop Med Parasitol 2009;103:377–82.
- Davey G. Podoconiosis, non-filarial elephantiasis, and lymphology. Lymphology 2010;43:168–77.
- Deribe K, Brooker SJ, Pullan RL, et al. Epidemiology and individual, household and geographical risk factors of podoconiosis in Ethiopia: results from the first nationwide mapping. Am J Trop Med Hyg 2015;92(1):148–58.
- Wanji S, Tendongfor N, Esum M, et al. Elephantiasis of non-filarial origin (podoconiosis) in the highlands of north-western Cameroon. Ann Trop Med Parasitol 2008;102(6):1–12.
- Onapa AW, Simonsen PE, Pedersen EM. Non-filarial elephantiasis in the Mt Elgon area (Kapchorwa District) of Uganda. Acta Trop 2001;78:171–6.
- Price EW. Podoconiosis: Non-filarial Elephantiasis. Oxford: Oxford Medical Publications, 1990.
- Molla YB, Le Blond JS, Wardrop N, et al. Individual correlates of podoconiosis in areas of varying endemicity: a case–control study. PLOS Negl Trop Dis 2013;7:e2554.
- Molla YB, Wardrop NA, Le Blond JS, et al. Modelling environmental factors correlated with podoconiosis: a geospatial study of non-filarial elephantiasis. Int J Health Geogr 2014;13:24.
- Price EW, Henderson WJ. The elemental content of lymphatic tissues in barefooted people in Ethiopia, with reference to endemic elephantiasis of the lower legs. Trans R Soc Trop Med Hyg 1978;72(2):132–6.
- Tekola Ayele F, Adeyemo A, Finan C, et al. HLA class II locus and susceptibility to podoconiosis. N Engl J Med 2012;366(13):1200–8.
- Price EW. The association of endemic elephantiasis of the lower legs in East Africa with soil derived from volcanic rocks. Trans R Soc Trop Med Hyg 1976;70(4):288–95.
- Price EW, Bailey D. Environmental factors in the etiology of endemic elephantiasis of the lower legs in tropical Africa. Trop Geog Med 1984;36(1):1–5.
- Cohen LB. Idiopathic lymphoedema of Ethiopia and Kenya. East Afr Med J 1960;37:53–74.
- Price EW. Endemic elephantiasis: early signs and symptoms, and control. Ethiop Med J 1983;21(4):243–53.
- Price EW. Pre-elephantiasic stage of endemic nonfilarial elephantiasis of lower legs: “podoconiosis”. Trop Doct 1984;14(3):115–19.
- Yakob B, Deribe K, Davey G. Health professionals' attitudes and misconceptions regarding podoconiosis: potential impact on integration of care in southern Ethiopia. Trans R Soc Trop Med Hyg 2009;104:42–7.
Pretibial myxoedema
- Fatourechi V, Pajouhi M, Fransway AF. Dermopathy of Graves disease (pretibial myxedema). Review of 150 cases. Medicine (Baltimore) 1994;73:1.
- Fatourechi V. Thyroid dermopathy and acropachy. Best Pract Res Clin Endocrinol Metab 2012;26:553.
- Schwartz KM, Fatourechi V, Ahmed DD, et al. Dermopathy of Graves' disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab 2002;98:438–46.
- Nair PA, Mishra A, Chaudhary A. Pretibial myxedema associated with euthyroid Hashimoto's thyroiditis: a case report. Clin Diagn Res 2014;8(6):YD01–2.
- Chen JJ, Ladenson PW. Euthyroid pretibial myxedema. Am J Med 1987;82(2):318–20.
- Cianfarani F, Baldini E, Cavalli A, et al. TSH receptor and thyroid-specific gene expression in human skin. J Invest Dermatol 2010;130:93–10.
- Douglas RD, Gupta S. The pathophysiology of thyroid eye disease: implications for immunotherapy. Curr Opin Ophthalmol 2011;22:385–90.
- Bull RH, Coburn PR, Mortimer PS. Pretibial myxoedema: a manifestation of lymphoedema? Lancet 1993;341:403–4.
- Davies TF. Trauma and pressure explain the clinical presentation of the Graves' disease triad. Thyroid 2000;10:629–30.
- Somach SC, Helm TN, Lawlor KB, Bergfeld WF, Bass J. Pretibial mucin. Histologic patterns and clinical correlation. Arch Dermatol 1993;129(9):1152–6.
- Fatourechi V. Thyroid dermopathy and acropachy. Best Pract Res Clin Endocrinol Metab 2012;26(4):553–65.
- Wright AL, Buxton PK, Menzies D. Pretibial myxedema localized to scar tissue. Int J Dermatol 1990;29:54–5.
- Akasu F, Takazawa K, Akasu R, et al. Localized myxedema on the nasal dorsum in a patient with Graves' disease: report of a case. J Endocrinol Invest 1989;12:717–21.
- Noppakun N, Bancheun K, Chandraprasert S. Unusual locations of localized myxedema in Graves' disease. Report of three cases. Arch Dermatol 1986;122:85–8.
- Rice SA, Peden NR, McGlynn S, et al. Atypical presentation of infiltrative thyroid dermopathy. Clin Exp Dermatol 2010;35:56–8.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol 2005;6(5):295–309.
- Takasu N, Higa H, Kinjou Y. Treatment of pretibial myxedema with topical steroid ointment application with sealing cover (steroid occlusive dressing technique: steroid ODT) in Graves' patients. Intern Med 2010;49(7):665–9.
- Pingsmann A, Ockenfels HM, Patsalis T. Surgical excision of pseudotumorous pretibial myxedema. Foot Ankle Int 1996;17:107–10.
- Deng A, Song D. Multipoint subcutaneous injection of long-acting glucocorticid as a cure for pretibial myxedema. Thyroid 2011;21:83–5.
- Wiersinga WM. Therapy: evidence-based treatment of Graves ophthalmopathy. Nature Rev Endocrinol 2009;5:653–4.
- Dandona P, Marshall NJ, Bidey SP, et al. Successful treatment of exophthalmos and pretibial myxoedema with plasmapheresis. BMJ 1979;1:374–6.
- Antonelli A, Navarranne A, Palla R, et al. Pretibial myxedema and high-dose intravenous immunoglobulin treatment. Thyroid 1994;4:399–408.
- Heyes C, Nolan R, Leahy M, Gebauer K. Treatment-resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol 2012;53(1):e1–4.
- Wemeau JL, Caron P, Beckers A, et al. Octreotide (long-acting release formulation) treatment in patients with Graves' orbitopathy: clinical results of a four-month, randomized, placebo-controlled, double-blind study. J Clin Endocrinol Metab 2005;90:841–8.
Trauma-induced lymphoedema
- Ouvry PA, Guenneguez H. Lymphatic complications from variceal surgery. Phlebologie 1993;46:563–8.
- Pittaluga P, Chastanet S. Lymphatic complications after varicose veins surgery: risk factors and how to avoid them. Phlebology 2012;27(Suppl. 1):139–42.
- Disipio T, Rye S, Newman S, Hayes S. Incidence of unilateral arm lymphoedema after breast cancer: a systematic review and meta-analysis. Lancet Oncol 2013;14(6):500–15.
- Nikitenko LL, Shimosawa T, Henderson S, et al. Adrenomedullin haploinsufficiency predisposes to secondary lymphedema. J Invest Dermatol 2013;133(7):1768–76.
- Kissin MW, della Rovere GO, Easton D, et al. Risk of lymphoedema following the treatment of breast cancer. Br J Surg 1986;73:580–4.
- Unno N, Yamamoto N, Suzuki M, et al. Intraoperative lymph mapping with preoperative vein mapping to prevent postoperative lymphorrhea following paramalleolar bypass surgery in patients with critical limb ischemia. Surg Today 2014;44(3):436–42.
- Buchel EW, Dalke KR, Hayakawa TE. The transverse upper gracilis flap: eefficiencies and design tips. Can J Plast Surg 2013;21(3):162–6.
- Tourani SS, Taylor GI, Ashton MW. Understanding the three-dimensional anatomy of the superficial lymphatics of the limbs. Plast Reconstr Surg 2014;134(5):1065–74.
- Reading G. Secrétan's syndrome: hard edema of the dorsum of the hand. Plast Reconstr Surg 1980;65:182–7.
- Nwaejike N, Archbold H, Wilson DS. Factitious lymphoedema as a psychiatric condition mimicking reflex sympathetic dystrophy: a case report. J Med Case Rep 2008;2:216.
- Andresz V, Marcantoni N, Binder F, et al. Puffy hand syndrome due to drug addiction: a case–control study of the pathogenesis. Addiction 2006;101(9):1347–51.
- Vollum DI. Skin lesions in drug addicts. BMJ 1970;2(7):647–50.
- Lymphoedema Framework. Best Practice for the Management of Lymphoedema. International Consensus. London: Medical Education, 2006. http://www.lympho.org/mod_turbolead/upload/file/Lympho/Best_practice_20_July.pdf (last accessed August 2015).
- British Lymphology Society (BLS). What is Lymphoedema? www.thebls.com/cellulitis (last accessed August 2015).
Lymphoedema due to immobility
- Garcia AM, Dicianno BE. The frequency of lymphedema in an adult spina bifida population. Am J Phys Med Rehabil 2011;90(2):89–96.
- Solaro C, Ucelli M, Brichetto G, et al. Prevalence of oedema of the lower limbs in multiple sclerosis patients: a vascular and lymphoscintigraphic study. Mult.Scler 2006;12:659–61.
- Geurts AC, Visschers BA, van Limbeek J, Ribbers GM. Systematic review of aetiology and treatment of post-stroke hand oedema and shoulder-hand syndrome. Scand J Rehabil Med 2000;32(1):4–10.
- Sneddon I, Church R. Practical Dermatology, 4th edn. London: Arnold, 1983:166.
- Bull RH, Mortimer PS. Acute lipodermatosclerosis in a pendulous abdomen. Clin Exp Dermatol 1993;18:164–6.
- Dirette D, Hinojosa J. Effects of continuous passive motion on the edematous hands of two persons with flaccid hemiplegia. Am J Occup Ther 1994;48(5):403–9.
Lymphangitis
- Gill MJ, Arlette J, Tyrrell DL, Buchan KA. Herpes simplex virus infection of the hand. Clinical features and management. Am J Med 1988;85(2A):53–6.
- Taylor MJ, Hoerauf A, Bockarie M. Lymphatic filariasis and onchocerciasis. Lancet 2010;376(9747):1175–85.
- Kennedy JL, Jeffus SK, Platts-Mills TA, et al. Seventeen-year-old girl hospitalized for localized swelling, pruritus, tenderness, and lymphatic streaking with eosinophilia. J Allergy Clin Immunol Pract 2013;1(3):299–301.
- Barnes JM, Trueta J. Absorption of bacteria and snake venoms from the tissues: importance of the lymphatic circulation. Lancet 1941;1:623–6.
- Pozio E. Venomous snakebites in Italy: epidemiological and clinical aspects. Trop Med Parasitol 1988;39(1):62–6.
- Anker RL, Straffon WG, Loiselle DS. First aid for snake bite. Med J Aust 1982;1:103.
- Marsch W, Haas N, Stuttgen G. Mondor's phlebitis: a lymphovascular process. Dermatologica 1986;172:133–8.
- Moskovitz AH, Anderson BO, Yeung RS, et al. Axillary web syndrome after axillary dissection. Am J Surg 2001;181:434–9.
- Ingram DM, Sheiner HJ, Ginsberg AM. Mondor's disease of the breast resulting from jellyfish sting. Med J Aust 1992;157(11–12):836–7.
- Lippi G, Favaloro EJ, Cervellin G. Hemostatic properties of the lymph: relationships with occlusion and thrombosis. Semin Thromb Hemost 2012;38(2):213–21.
- Babu AK, Krishnan P, Andezuth DD. Sclerosing lymphangitis of penis – literature review and report of 2 cases. Dermatol Online J 2014;20(7):ii.
- Marneros AG, Blanco F, Husain S, et al. Classification of cutaneous intravascular breast cancer metastases based on immunolabeling for blood and lymph vessels. J Am Acad Dermatol 2009;60:633–8.
- Finkel LJ, Griffiths CEM. Inflammatory breast carcinoma (carcinoma erysipeloides), an easily overlooked diagnosis. Br J Dermatol 1993;129:324–6.
Complications of lymphoedema
- Angin S, Karadibak D, Yavuzşen T, Demirbüken I. Unilateral upper extremity lymphedema deteriorates the postural stability in breast cancer survivors. Contemp Oncol (Pozn) 2014;18(4):279–84.
- Daróczy J. Pathology of lymphedema. Clin Dermatol 1995;13(5):433–44.
- Mortimer PS, Rockson SG. New developments in clinical aspects of lymphatic disease. J Clin Invest 2014;124(3):915–21.
- Nei T, Akutsu K, Shima A, et al. A case of streptococcal toxic shock syndrome due to group G streptococci identified as Streptococcus dysgalactiae subsp. equisimilis. J Infect Chemother 2012;18(6):919–24.
- Soria X, Bielsa I, Ribera M, et al. Acute dermal abscesses caused by Serratia marcescens. J Am Acad Dermatol 2008;58(5):891–3.
- Moffatt CJ, Franks PJ, Doherty DC, et al. Lymphoedema: an underestimated health problem. Q J Med 2003;96(10):731–8.
- Tobin MB, Lacey HJ, Meyer L, Mortimer PS. The psychological morbidity of breast cancer-related arm swelling. Psychological morbidity of lymphoedema. Cancer 1993;72(11):3248–52.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphoedema. Cancer 1948;1:64–81.
- Lee R, Saardi KM, Schwartz RA. Lymphedema-related angiogenic tumors and other malignancies. Clin Dermatol 2014;32(5):616–20.
- Benson PM, Pessoa CM, Lupton GP, et al. Basal cell carcinomas arising in chronic lymphoedema. J Dermatol Surg Oncol 1988;14:781–3.
- Epstein JL, Mendelsohn G. Squamous carcinoma of the foot arising in association with longstanding verrucous hyperplasia in a patient with congenital lymphoedema. Cancer 1984;54:943–7.
- Waxman M, Fatteh S, Elias JM, et al. Malignant lymphoma of skin associated with postmastectomy lymphoedema. Arch Pathol Lab Med 1984;108:206–8.
- Tatnall FM, Mann BS. Non-Hodgkin's lymphoma of the skin associated with chronic limb lymphoedema. Br J Dermatol 1985;113:751–6.
- Hills RJ, Ive FA. Cutaneous secondary follicular centre cell lymphoma in association with lymphoedema praecox. Br J Dermatol 1993;129:186–9.
- Sarkany I. Malignant melanomas in lymphoedematous arm following radical mastectomy for breast carcinoma. J R Soc Med 1972;65:253–4.
- Bartal AH, Pinsky CM. Malignant melanoma appearing in a postmastectomy lymphoedematous arm: a novel association of double primary tumours. J Surg Oncol 1985;30:16–18.
- Fergusson CM, Copeland SA, Horton L. Unusual sarcoma arising in lymphoedema. J R Soc Med 1985;78:1497–8.
- Peterson CM, Lane JE, Guill MA. Merkel cell carcinoma after postmastectomy lymphedema. J Am Acad Dermatol 2003;48(6):983.
- Merimsky O, Chaitchik S. Kaposi's sarcoma on a lymphoedematous arm following radical mastectomy. Tumori 1992;78:407–8.
- Pearson IC, Cox NH, Ostlere LS, Marsden RA, Mortimer PS. Porokeratosis in association with lymphoedema. Clin Exp Dermatol 2005;30(2):152–4.
- Tatnall FM, Sarkany I. Primary focal lymphoedema with xanthoma. J R Soc Med 1988;81:113–14.
- Romaní J, Luelmo J, Sáez A, et al. Localized xanthomas associated with primary lymphedema. Pediatr Dermatol 2012;29(1):113–14.
- Callens A, Vaillant L, Machet MC, et al. Localized atypical pemphigoid on lymphoedema following radiotherapy. Acta Derm Venereol (Stockh) 1993;73:461–4.
- Wilkinson SM, Heagarty AH, Smith AG. Toxic epidermal necrolysis localized to an area of lymphoedema. Clin Exp Dermatol 1991;17:456–7.
- Demitsu T, Tadaki T. Atypical neutrophilic dermatosis on the upper extremity affected by post-mastectomy lymphoedema: report of 2 cases. Dermatologica 1991;183:230–3.
- Kaier T, Larsen J. Necrotizing fasciitis in congenital lymphoedema. Int J Dermatol 1990;29:41–4.
Imaging of the lymphatic system
- Kinmonth JB. Lymphangiography in man; a method of outlining lymphatic trunks at operation. Clin Sci (Lond) 1952;11(1):13–20.
- Stewart G, Gaunt JI, Croft DN, Browse NL. Isotope lymphography: a new method of investigating the role of the lymphatics in chronic limb oedema. Br J Surg 1985;72(11):906–9.
- Proby CM, Gane JN, Joseph AE, Mortimer PS. Investigation of the swollen limb with isotope lymphography. Br J Dermatol 1990;123(1):29–37.
- Modi S, Stanton AW, Mortimer PS, Levick JR. Clinical assessment of human lymph flow using removal rate constants of interstitial macromolecules: a critical review of lymphoscintigraphy. Lymph Res Biol 2007;5(3):183–202.
- Mellor RH, Hubert CE, Stanton AW, et al. Lymphatic dysfunction, not aplasia, underlies Milroy disease. Microcirculation 2010;17(4):281–96.
- Xiong L, Engel H, Gazyakan E, et al. Current techniques for lymphatic imaging: state of the art and future perspectives. Eur J Surg Oncol 2014;40(3):270–6.
- Unno N, Inuzuka K, Suzuki M, et al. Preliminary experience with a novel fluorescence lymphography using indocyanine green in patients with secondary lymphedema. J Vasc Surg 2007;45(5):1016–21.
- Yamamoto T, Yamamoto N, Azuma S, et al. Near-infrared illumination system-integrated microscope for supermicrosurgical lymphaticovenular anastomosis. Microsurgery 2014;34(1):23–7.
- Lohrmann C, Foeldi E, Bartholoma JP, Langer M. Interstitial MR lymphangiography – a diagnostic imaging method for the evaluation of patients with clinically advanced stages of lymphedema. Acta Trop 2007;104(1):8–15.
- Costa da Cunha Castro E, Galambos C. Prox-1 and VEGFR3 antibodies are superior to D2-40 in identifying endothelial cells of lymphatic malformations – a proposal of a new immunohistichemical panel to differentiate lymphatic from other vascular malformations. Pediatr Dev Pathol 2009;12:187–94.
- Connell F, Gordon K, Brice G, et al. The classification and diagnostic algorithm for primary lymphatic dysplasia: an update from 2010 to include molecular findings. Clin Genet 2013;84(4):303–14.
Lymphoedema management
- Medical Education Partnership (MEP). Best Practice for the Management of Lymphoedema: International Consensus. London: MEP Ltd, 2006.
- Foldi E, Foldi M, Weissleder H. Conservative treatment of lymphoedema of the limbs. Angiology 1985;36:171–80.
- Mortimer P, Todd J. Lymphoedema: Advice on Self-management and Treatment, 3rd edn. Beaconsfield: Beaconsfield Publishers, 2007.
- Ikomi F, Schmid-Schonbein GW. Lymph transport in skin. Clin Dermatol 1995;13:419–27.
- Badger CMA, Peacock JL, Mortimer PS. A randomized, controlled, parallel group clinical trial comparing multi-layer bandaging followed by hosiery versus hosiery alone in the treatment of patients with lymphedema of the limb. Cancer 2000;88:2832–7.
- Anonymous. Compression for lymphoedema. Lancet 1986;i:896.
- Mortimer PS, Simmonds R, Rezvani M, Robbins M, Hopewell JW, Ryan TJ. The measurement of skin lymph flow by isotope clearance – reliability, reproducibility, injection dynamics, and the effect of massage. J Invest Dermatol 1990;95(6):677–82.
- Shaw C, Mortimer P, Judd PA. A randomised controlled trial of weight reduction as a treatment for breast cancer-related lymphedema. Cancer 2007;110:1868–74.
- Shaw C, Mortimer P, Judd PA. Randomised controlled trial comparing a low-fat diet with a weight-reduction diet in breast cancer-related lymphedema. Cancer 2007;109:1949–56.
- Gothard L, Stanton A, MacLaren J, et al. Non-randomised phase II trial of hyperbaric oxygen therapy in patients with chronic arm lymphoedema and tissue fibrosis after radiotherapy for early breast cancer. Radiother Oncol 2004;70:217–24.
- Carati CJ, Anderson SN, Gannon BJ, Piller NB. Treatment of postmastectomy lymphedema with low-level laser therapy: a double blind, placebo-controlled trial. Cancer 2003;98:1114–22. (Erratum in Cancer 2003;98:2742.)
- Kozanoglu E, Basaran S, Paydas S, Sarpel T. Efficacy of pneumatic compression and low-level laser therapy in the treatment of postmastectomy lymphoedema: a randomized controlled trial. Clin Rehabil 2009;23:117–24.
- Gothard L, Haviland J, Bryson P, et al. Randomised phase II trial of hyperbaric oxygen therapy in patients with chronic arm lymphoedema after radiotherapy for cancer. Radiother Oncol 2010;97(1):101–7.
- Smoot B, Chiavola-Larson L, Lee J, Manibusan H, Allen DD. Effect of low-level laser therapy on pain and swelling in women with breast cancer-related lymphedema: a systematic review and meta-analysis. J Cancer Surviv 2015;9(2):287–304.
- Tsai HJ, Hung HC, Yang JL, et al. Could Kinesio tape replace the bandage in decongestive lymphatic therapy for breast-cancer-related lymphedema? A pilot study. Support Care Cancer 2009;17:1353–60.
- Rockson S, Miller L, Senie R. Workshop III: diagnosis and management of lymphedema. Cancer 1998;83(Suppl.):282–5.
- Loprinzi CL, Kugler JW, Sloan JA, et al. Lack of effect of coumarin in women with lymphedema after treatment for breast cancer. N Engl J Med 1999;340(5):346–50.
- Saaristo A, Tammela T, Timonen J, et al. Vascular endothelial growth factor-C gene therapy restores lymphatic flow across incision wounds. FASEB J 2004;18(14):1707–9.
- Szuba A, Skobe M, Karkkainen MJ, et al. Therapeutic lymphangiogenesis with human recombinant VEGF-C. FASEB J 2002;16(14):1985–7.
- Karkkainen MJ, Jussila L, Ferrell RE, Finegold DN, Alitalo K. Molecular regulation of lymphangiogenesis and targets for tissue oedema. Trends Mol Med 2001;7(1):18–22.
- Tammela T, Saaristo A, Holopainen T, et al. Therapeutic differentiation and maturation of lymphatic vessels after lymph node dissection and transplantation. Nature Med 2007;13(12):1458–66.
- Sommer T, Buettner M, Bruns F, Breves G, Hadamitzky C, Pabst R. Improved regeneration of autologous transplanted lymph node fragments by VEGF-C treatment. Anat Rec 2012;295(5):786–91.
- Honkonen KM, Visuri MT, Tervala TV, et al. Lymph node transfer and perinodal lymphatic growth factor treatment for lymphedema. Ann Surg 2013;257(5):961–7.
- Browse NL, Burnand KG, Mortimer PS. Diseases of the Lymphatics. London, Arnold, 2003.
- Weinstein M, Roberts M. Elephantiasis and the Kondoleon operation; a 20-year postoperative follow-up. Am J Surg 1950;79(2):327–31.
- Garaffa G, Christopher N, Ralph DJ. The management of genital lymphoedema. Br J Urol Int 2008;102:480–4.
- Nielubowicz J, Olszewski W. Surgical lymphaticovenous shunts in patients with secondary lymphoedema. Br J Surg 1968;55(6):440–2.
- Campisi C, Bellini C, Campisi C, Accogli S, Bonioli E, Boccardo F. Microsurgery for lymphedema: clinical research and long-term results. Microsurgery 2010;30(4):256–60.
- Damstra RJ, Voesten HG, van Schelven WD, van der Lei B. Lymphatic venous anastomosis (LVA) for treatment of secondary arm lymphedema. A prospective study of 11 LVA procedures in 10 patients with breast cancer related lymphedema and a critical review of the literature. Breast Cancer Res Treat 2009;113(2):199–206.
- Becker C, Assouad J, Riquet M, Hidden G. Postmastectomy lymphedema: long-term results following microsurgical lymph node transplantation. Ann Surg 2006;243(3):313–15.
- Sulo E, Hartiala P, Viitanen T, Mäki M, Seppänen M, Saarikko A. Risk of donor-site lymphatic vessel dysfunction after microvascular lymph node transfer. J Plast Reconstr Aesthet Surg 2015;68(4):551–8.
- Warren AG, Brorson H, Borud LJ, Slavin SA. Lymphedema: a comprehensive review. Ann Plast Surg 2007;59(4):464–72.
- Brorson H. From lymph to fat: complete reduction of lymphoedema. Phlebology 2010;25(Suppl. 1):52–63.