CHAPTER 118
Benign Cutaneous Adverse Reactions to Drugs
Michael R. Ardern-Jones1 and Haur Yueh Lee2
1Dermatoimmunology, University of Southampton, Southampton General Hospital, UK
2Department of Dermatology, Singapore General Hospital; and DUKE-NUS Graduate Medical School, Singapore
Drug-induced exanthem
Definition and nomenclature
Exanthematic eruptions can be caused by a variety of drugs and resemble in appearance the classical rash of a viral infection, for which the paradigm is the morbilliform rash associated with measles. Typically, exanthematic drug reactions do not show the systemic involvement, such as fever, that would characterize a viral infection [1].
Introduction and general description
An exanthem is the most frequent of all cutaneous reactions to medicines (Box 118.1), and can occur after almost any drug at any time up to 3 weeks, but usually 1–2 weeks after administration. A variety of drugs can cause drug-induced exanthems, the commonest being penicillins, aromatic anticonvulsants, sulphonamides, non-steroidal anti-inflammatory drugs (NSAIDs) and allopurinol (listed in Box 118.1). It is not possible to identify the offending drug by the nature of the eruption.
Epidemiology
Exanthematous reactions are the most frequent presentation of non-immediate (non-immunoglobulin E (IgE)) drug allergy. The epidemiology of such reactions is not well characterized, but a French study identified 3.6 cutaneous allergic reactions per 1000 hospitalized patients, of which 56% showed exanthematous reactions [2] and a later study showed similar results [3].
Pathophysiology
The mechanism of cutaneous inflammation in drug-induced exanthems is mediated by drug-specific T cells (see Chapter 12).
Pathology
Histology is generally non-specific. However, some histological features are used to discriminate drug-induced exanthems from non-drug exanthems including: apoptotic keratinocytes, eosinophils within the inflammatory infiltrate, papillary oedema and vascular changes [4] (Figure 118.1).
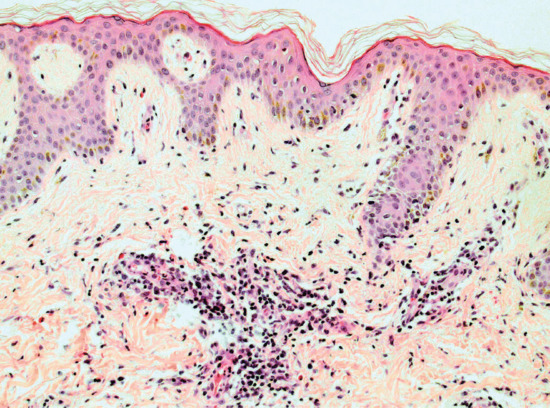
Figure 118.1 Histopathology of a drug-induced exanthem, H&E. Magnification 20×. This pigmented epidermis shows orthokeratosis and mild spongiosis. The papillary dermis is mildly oedematous. There is a brisk superficial perivascular lymphocytic infiltrate with many scattered interstitial eosinophils. Melanophages are also present. (Courtesy of Dr Marianna Philippidou, King's College Hospital, London, UK.)
Clinical features
The latency from drug initiation to onset of rash ranges from 5 to 21 days, but typically occurs at 7–10 days. A drug-induced exanthem may be accompanied by pruritus. The clinical features are variable; lesions may be scarlatiniform, rubelliform or morbilliform, or may consist of a profuse eruption of small pink papules showing no close resemblance to any infective exanthem (Figures 118.2 and 118.3). Less common are eruptions with large macules, polycyclic and gyrate erythema, reticular eruptions and sheet-like erythema. The trunk and extremities are usually involved, and not uncommonly intertriginous areas may be favoured, but the face is typically spared. Palmar and plantar lesions may occur, and sometimes the eruption is generalized. Purpuric lesions, especially on the legs, and erosive stomatitis may develop. There may be relative sparing of pressure areas.
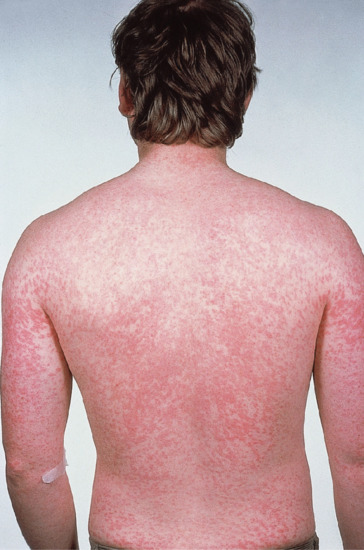
Figure 118.2 An exanthem caused by ampicillin.

Figure 118.3 A drug-induced exanthem composed of macular and papular lesions, becoming confluent on the chest.
Differential diagnosis
Viral infection is the most important differential diagnosis. Classical viral exanthems such as measles and chickenpox are readily identifiable [5]. In a recent series of atypical exanthems, morphology and laboratory investigations led to an aetiological diagnosis in 77% of cases [6]. Drugs were responsible for 25% of the exanthems (more commonly in adults) of which antibiotics and NSAIDs were most frequently implicated. It is useful in differentiating exanthematic drug eruptions from viral exanthems to remember that viral rashes tend to start on the face and acral sites with subsequent progression to involve the trunk, and are more often accompanied by fever, sore throat, gastrointestinal symptoms, conjunctivitis, cough and insomnia [6]. Pruritus is typically associated with drug causes in adults. Enanthems, which involve the mucous membranes, are more commonly associated with an infectious aetiology.
Drug reaction with eosinophilia and systemic symptoms (DRESS) (see Chapter 119) is important to exclude since the eruption in this systemic drug hypersensitivity syndrome can be exanthematous.
Complications and co-morbidities
If the administration of the drug is continued, an exfoliative dermatitis may develop, although occasionally the eruption subsides despite continuation of the medication.
Disease course and prognosis
The illness follows a benign course, and resolves without sequelae following cessation of the offending drug. Morbilliform drug eruptions usually, but not always, recur on rechallenge.
Investigations
Blood tests to exclude organ dysfunction and haematological abnormalities associated with DRESS are important. Viral serology/polymerase chain reaction (PCR) may be useful to exclude viral infections. Skin histology is not routinely undertaken as it is not diagnostic of a drug cause but may be useful to exclude other conditions where the clinical features are not characteristic.
Management
Cessation of the culprit drug is essential. On withdrawal of the drug, maculopapular drug eruptions usually fade with desquamation, sometimes with post-inflammatory hyperpigmentation. Generally, symptomatic treatment only is required; most cases benefit from emollients. Approximately 50% of exanthematous eruptions are pruritic and intermediate potency topical corticosteroids may be useful for these cases.
Resolution of drug-induced exanthems is faster than for infectious exanthems.
Drug-induced pruritus
Definition
Itch caused by a drug. Drug-induced pruritus may be localized or generalized.
Introduction and general description
Pruritus is generally a complication of systemic drugs but is also associated following the application of some topical agents, such as calcineurin inhibitors and β-adrenergic blockers [7, 8]. The systemic agents which can cause pruritus are listed in Box 118.2.
Epidemiology
Pruritus has been reported to arise in 13.3% of adverse reactions to prescribed drugs in general practice [9]. Opioid-induced pruritus is the most frequent and best recognized primary drug-induced pruritus and is reported to arise in 2–10% of patients treated with opiates [10].
Pathophysiology
Drug-induced pruritus may be primary, via neuronal/central nervous system interaction, or through secondary mechanisms. Secondary pruritus includes: (i) direct skin effects, e.g. induction of a hypersensitivity drug rash, other inflammatory skin disease or xerosis; (ii) alteration of biochemical profiles (e.g. renal or hepatic dysfunction); and (iii) other unexplained mechanisms.
Opioids can induce mast cell degranulation and histamine release causing an itchy urticarial rash, however most opioid pruritus appears to involve binding of the drug to central μ-opioid pain receptors in the medullary dorsal horn [11]. Serotonin and dopamine D(2) receptors, spinal inhibitory pathways and prostaglandins have also been implicated in opioid-induced pruritus [12].
Chloroquine can cause mast cell induced histamine pruritus. However, the precise cause of generalized pruritus seen in 60–70% of black Africans treated with antimalarials is unclear and likely to be multifactorial including μ-opioid receptor signalling as well as genetic (it is less common in white people) [13, 14]. Selective serotonin reuptake inhibitor (SSRI) induced pruritus is also well recognized and these drugs can induce itching when injected into the skin [15]. Interestingly, SSRIs can also be used to treat psychogenic itching [16].
Hydroxyethyl starch (HES) is used for colloid fluid replacement in some settings and has been associated with chronic pruritus in approximately one third of patients. HES-induced itch can arise with the administration of small volumes but the complication is more common with greater exposure. The symptoms typically arise after 1–6 weeks of HES infusion [17], can persist for 12–24 months [18] and are generally refractory to treatment [19].
Clinical features
The latency from drug initiation to onset of pruritus is usually just a few days. The patient complains of generalized itching often with signs of excoriation and/or lichenification. Dermographism is generally not present.
Differential diagnosis
Pruritus caused by other conditions must be excluded (see Chapter 83).
Disease course and prognosis
Although considered a mild adverse drug reaction, chronic pruritus can have a significantly negative effect on quality of life.
Investigations
Exclusion of other causes of itch is essential (see Chapter 83).
Management
Generally, cessation of the culprit drug brings rapid relief. However, some drug-induced pruritus, such as HES, can be long lived. Although antihistamine medications are frequently prescribed for drug-induced pruritus, they are rarely helpful. Cooling emollients such as 0.5% menthol in aqueous cream may be of benefit.
In view of the direct involvement of opioid receptors in opiate-induced pruritus, the most effective means of treatment is reduction in dose or cessation of opiate treatment. Alternatively introduction of naloxone, naltrexone (μ-receptor antagonists) or nalbuphine (partial μ-receptor agonist, μ-receptor antagonist) may be tried, but all of these approaches are likely to lead to loss of pain control. Other approaches include adding D2 receptor antagonists, serotonin (5-HT3) receptor antagonists (ondansetron, dolasetron), antihistamines and gabapentin [19]. Interestingly, μ-receptor antagonists have also been utilized in other forms of pruritus especially where endogenous endorphins are thought to be pathogenic, such as in cholestatic pruritus [20].
For more resistant cases of drug-induced pruritus, phototherapy and other therapeutic options may be of value (see Chapter 83).
Drug-induced eczema
Definition and nomenclature
Drug-induced eczema is a systemic allergic contact dermatitis (SACD) caused by drug hypersensitivity.
Introduction and general description
A patient sensitized to a drug may develop an eczematous reaction when the same drug, or a chemically related one, is subsequently administered systemically [21–23]. The medications which most commonly cause drug-induced eczema are listed in Box 118.3. Patients with a contact allergy to ethylenediamine may develop generalized or localized eczema following injection of aminophylline preparations containing ethylenediamine as a solubilizer for theophylline [24, 25]. Patients with contact allergy to parabens may develop systemic eczema on exposure to drugs containing parabens as a preservative [26]. Similarly, sensitized patients may develop eczema following oral ingestion of neomycin or hydroxyquinolines [27]. Diabetic patients sensitized by topical preparations containing p-amino compounds, such as p-phenylenediamine hair dyes, para-aminobenzoic acid (PABA) sunscreens and certain local anaesthetic agents (e.g. benzocaine), may develop a systemic contact dermatitis with the hypoglycaemic agents tolbutamide or chlorpropamide. Sulphonylureas may also induce eczematous eruptions in sulphanilamide-sensitive patients as a result of cross-reactivity. Phenothiazines can produce allergic contact dermatitis, photoallergic reactions and eczematous contact-type dermatitis, and may cross-react with certain antihistamines. Tetraethylthiuram disulphide (disulfiram, Antabuse®) for the management of alcoholism can cause eczematous reactions in patients sensitized to thiurams via rubber gloves. Drugs given systemically have also been identified as causing eczematous drug reactions without prior contact sensitization, and approximately 50% of cases of SACD are due to penicillins or β-lactam antibiotics. The term ‘endogenic contact eczema’ [28] refers to the occurrence of an eczematous contact drug reaction following primary sensitization by oral therapy, as in the case of a patient with a drug-related exanthem who later develops localized dermatitis due to topical therapy. Symmetrical drug-related intertriginous and flexural exanthem (SDRIFE) describes a clinically distinct form of drug-induced eczema (see later).
Epidemiology
Rare.
Pathophysiology
The eczematous response is mediated by drug-specific T-cell induced inflammation in the skin (see Chapter 12).
Pathology
The principal histological features are dermatitic changes with spongiosis and a superficial perivascular infiltrate, primarily composed of mononuclear cells.
Clinical features
The latency from drug initiation to onset of eczema typically occurs at 7–14 days. The eruption tends to be symmetrical, and may involve first, or most severely, the site(s) of the original dermatitis, before becoming generalized.
Differential diagnosis
- Idiopathic eczematous reactions.
- Allergic contact dermatitis.
- Irritant contact dermatitis.
Complications and co-morbidities
Following drug withdrawal, complications and co-morbidities are minimal.
Disease course and prognosis
On withdrawal of the offending drug, resolution of the clinical symptoms generally occurs in 1–3 weeks. Re-exposure to the culprit would be expected to reproduce the same clinical picture.
Investigations
Patch tests are commonly positive and usually vesicular, although histology of the eruption itself may show leukocytoclastic vasculitis. Oral challenge with the suspected antigen may be required to substantiate the diagnosis.
Management
The primary objective is cessation of the causative drug. Therapies utilized to treat eczematous dermatoses, such as topical corticosteroids, are usually effective. For severe reactions, systemic treatment with prednisolone may be appropriate.
Symmetrical drug-related intertriginous and flexural exanthem
Definition and nomenclature
A benign and self-limiting drug eruption characterized by symmetrical involvement of the gluteal and intertriginous areas, occurring in the absence of systemic involvement.
Introduction and general description
SDRIFE is a distinctive drug eruption characterized by symmetrical eruption of the gluteal region, thighs and other intertriginous areas such as the axillae, knees, elbows and neck. Previously, such cases were called drug-induced baboon syndrome – a form of systemic allergic contact dermatitis (see earlier). The term SDRIFE was proposed in 2004 as a culturally more neutral term, and also to reflect a spectrum of involvement beyond the gluteal region [37, 38]. The medications associated with SDRIFE are listed in Box 118.4.
Epidemiology
Age
Reported in all ages.
Sex
Male preponderance [37].
Pathophysiology
In SDRIFE, immunohistological findings on skin biopsies have shown an infiltration of CD4+ T cells in the dermis. The delayed latency and occasional positivity on patch testing to causative drugs support a type IV delayed hypersensitivity reaction [38]. Systemic drugs induce SDRIFE without prior skin sensitization and without cross-reactivity to known contact allergens. The reason for the peculiar distribution of the reaction is unknown. Postulated explanations include a preferential trafficking of activated memory T cells to these sites as a form of recall phenomenon from previous physical/inflammatory insult. Although excretion of drugs or their metabolites in the site has been postulated, there is no syringotropic inflammation which argues against a central role for sweat glands in the pathogenesis [39].
Pathology
There is significant heterogeneity in SDRIFE [37]. Features that have been described include: superficial perivascular lymphocytic infiltrate, which may also include neutrophils and eosinophils, spongiosis and vacuolar degeneration of the basal cells.
Clinical features
History
The latency from drug intake to the onset of SDRIFE ranges from hours to a few days [37, 38].
Presentation
This cutaneous adverse reaction is characterized by the following diagnostic criteria: (i) exposure to a systemically administered drug; (ii) sharply demarcated erythema of the gluteal/perianal area and/or V-shaped erythema of the inguinal area; (iii) involvement of at least one other intertriginous/flexural location; (iv) symmetry of the affected areas; and (v) absence of systemic symptoms and signs (Figures 118.4 and 118.5). Occasionally, the eruption may consist of small papules, pustules, vesicles and rarely bullae. Palmoplantar surfaces, face and mucosa are usually spared [37].
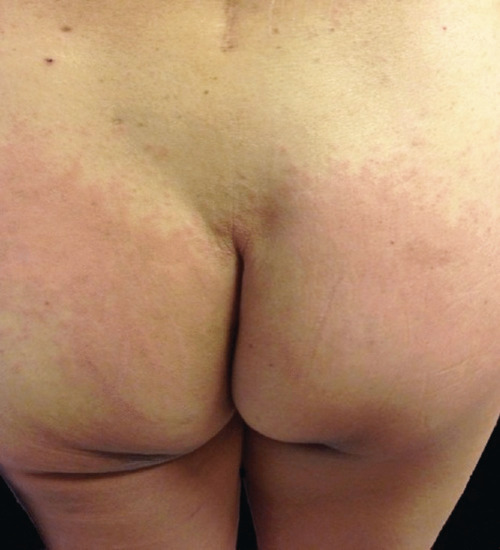
Figure 118.4 Symmetrical drug-related intertriginous and flexural exanthem (SDRIFE): well-defined patches of erythema involving the gluteal skin.

Figure 118.5 Symmetrical drug-related intertriginous and flexural exanthem (SDRIFE): intense erythema affecting the axillary skin.
Differential diagnosis
The differential diagnosis of intertriginous eruptions include inflammatory dermatoses such as contact dermatitis, inverse psoriasis, Hailey–Hailey disease, infective causes such as intertrigo and tinea cruris and other drug-induced reactions such as acute generalized exanthematous pustulosis (AGEP) and chemotherapy-associated toxic erythema. Although the sheets of pustules and constitutional symptoms of AGEP are usually obvious, the initial reaction may have a flexural predilection. Chemotherapeutic reactions such as neutrophilic eccrine hidradenitis/eccrine squamous metaplasia may also have a flexural predilection.
Disease course and prognosis
The reaction improves quickly on withdrawal of the culprit drug.
Investigations
Patch tests are positive in up to 50% with SDRIFE and oral provocation tests are positive in 80% of patients [38]. Prick testing and lymphocyte stimulation testing are negative in the majority of reported cases of SDRIFE.
Management
Treatment is symptomatic. The offending drug should be withdrawn and avoided in the future. No active therapy is required although topical or systemic glucocorticoids may hasten recovery [38].
Drug-induced urticaria, angio-oedema and anaphylaxis
Definition
Urticaria and angio-oedema are physical signs which involve circumscribed skin oedema and erythema (described in detail in Chapters 42 and 43). Anaphylaxis is a constellation of clinical findings which describes respiratory and cardiovascular compromise (bronchoconstriction and hypotension) in a life-threatening manner and is described in Chapter 42. This section is concerned with drug-induced causes of these reaction patterns.
Pseudoallergy (non-immune mediated) describes a presentation which is clinically indistinguishable from true allergy (IgE mediated). Pseudoallergic urticaria, angio-oedema and anaphylaxis are commonly mediated by drugs, as discussed later, and these reactions are sometimes referred to as ‘anaphylactoid’.
Introduction and general description
Drug-induced urticaria can be seen in isolation or in association with anaphylaxis and angio-oedema. Aspirin and NSAIDs are the most frequently implicated drugs in allergic reactions, most frequently causing urticaria [41] as well as exacerbating idiopathic urticaria [42].
The most frequent drug causes of anaphylaxis are antibiotics (usually β-lactams) and NSAIDs [43]. However, biologicals are increasingly common culprits. Anaphylaxis during general anaesthesia is an important problem and can be difficult to investigate if intraoperative exposures are not well recorded. Furthermore, distinction between true immune-mediated and non-immune-mediated reactions can be challenging. Common perioperative causes of anaphylaxis or anaphylactoid reactions include: anaesthetic agents (especially thiopental), neuromuscular blocking agents (may also be non-IgE mediated), analgesics (opioids are usually associated with non-IgE-mediated reactions), antibiotics, protamine and blood transfusions (mechanism unclear) [43]. Recently, anaphylaxis caused by drugs bioengineered onto medical equipment, such as chlorhexidine-coated central venous catheters, has been recognized [44]. β-blockers enhance anaphylactic reactions caused by other allergens, and may make resuscitation more difficult [45].
Epidemiology
Urticaria is the second most common type of adverse cutaneous drug eruption [46, 47]. Drug-induced urticaria is seen in 0.16% of medical in-patients and accounts for 9% of chronic urticaria or angio-oedema occurring in dermatology out-patient departments [46]. It has been estimated that 1 in 1500 of the population of England has experienced anaphylaxis at some point in their lives [48]. Of the deaths from anaphylaxis recorded each year in the UK (on average 20) at least half were due to drugs [49]. The incidence of anaphylaxis during general anaesthesia has been estimated to arise in 1 : 4000 to 1 : 25000 cases [43]. Infliximab is estimated to cause anaphylaxis or IgE-mediated reactions in 2–3% of patients [50] and reaction to other tumour necrosis factor (TNF) antagonists are also recorded [51].
Pathophysiology
The mechanisms underlying drug-induced vasodilation, oedema and itch in urticaria are identical to those seen in angio-oedema (deeper in skin) and anaphylaxis (systemic circulation). Classically, these reactions are mediated by the presence of drug-specific IgE. On exposure to the drug, cross-linking of IgE on the surface of mast cells (and possibly basophils) is followed by inflammatory mediator release (including histamine), which induces vasodilation, neuronal activation and smooth muscle contraction (see Chapter 8). Cyclo-oxygenase inhibitors, such as aspirin and indometacin, may also cause urticaria or angio-oedema by pharmacological mechanisms. Other drugs, such as radiocontrast media, local anaesthetics and dextrans (in plasma expanders) may release mast cell mediators directly. Angiotensin-converting enzyme (ACE) inhibitors can cause angio-oedema which is bradykinin mediated rather than histamine dependent and will therefore not respond to antihistamines [52].
Genetics
See Chapter 42.
Pathology
See Chapter 42.
Clinical features
Urticaria, angio-oedema and anaphylaxis arise within 24–36 h of drug ingestion at the first occasion. On re-challenge lesions may develop within minutes. Anaphylaxis usually develops on second exposure to a drug as it is thought that prolonged treatments may induce tolerization rather than allergy (in contrast to T-cell-mediated hypersensitivities). Anaphylaxis and anaphylactoid reactions usually develop within minutes to hours (the vast majority within the first hour) of drug administration.
Urticaria occurring alone is more common than urticaria arising with angio-oedema (Figure 118.6). Anaphylaxis and anaphylactoid reactions are often associated with skin or mucosal changes: in less severe cases, there may be premonitory dizziness or faintness, skin tingling and reddening of the bulbar conjunctiva, followed by urticaria, angio-oedema, bronchospasm, abdominal pain and vasomotor collapse. Intravenous administration is associated with more severe reactions and rapid progression, over minutes, to cardiac arrest. In cases of insect sting-related and food-induced anaphylaxis, the syndrome evolves more slowly [45].
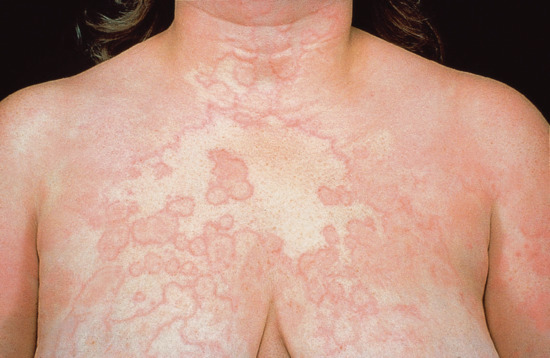
Figure 118.6 Urticaria induced by acetylsalicylic acid. (Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Differential diagnosis
Drug-induced urticaria needs to be distinguished from other causes of urticaria, especially infection. This distinction may require specialist testing. Drug-induced anaphylaxis needs to be distinguished from other causes of anaphylaxis.
Complications and co-morbidities
Anaphylaxis may result in death if untreated. With early intervention, treatment outcomes are good. However, it is critical that causation is addressed and investigated as necessary to prevent accidental recurrence.
Disease course and prognosis
On withdrawal of the offending culprit, clinical improvement in drug-induced urticaria and angio-oedema occurs within 24–48 h. It is important to recognize that after an initial improvement from anaphylaxis, late-phase reactions may arise 5–6 h afterwards. Deaths from drug-induced anaphylaxis are uncommon (<2%); the major predisposing risk factor for poor outcome is coexistent severe asthma [53, 54].
Investigations
The investigation of immediate drug allergy reactions (characterized by urticaria, angio-oedema or anaphylaxis) is well established and various guidelines represent consensus approaches to the investigation of the culprit drug [55]. The general approach is based upon a careful documentation of the exposure history in the hours preceding the reaction to establish the most likely causative drug. Careful record should be made of the timeline of clinical features. Testing involves an escalation of exposures which is stopped if any positive result is identified, thereby minimizing risk. In many circumstances, it is more useful to prove a negative through testing than confirm a drug allergy through testing, so as to establish what is safe for the patient to take. The testing process involves plasma sampling for drug-specific IgE, skin prick testing, intradermal testing and challenge testing.
Beta-lactam allergy reactions are due to IgE recognition of the drug bound to a carrier molecule. Skin testing is undertaken with benzylpenicilloyl polylysine ‘major antigenic determinant’ and ‘minor determinants’ (other penicillin antigens). Drug-specific IgE measured by Immunocap® (RAST®) has a high specificity but low sensitivity and therefore should only be used by specialists with access to the full range of skin testing [56]. Progression to intradermal testing and challenge if necessary is advocated by European and UK guidelines [55]. Multiple studies have shown the very low positive predictive value of labels of ‘penicillin allergy’ confirmed by negative testing including oral challenge. Although penicillin avoidance is not difficult for the majority of cases, adverse outcomes of mislabelling with penicillin allergy are increasingly recognized and confirmatory testing is recommended in specific groups [56].
Patch testing is not useful in the investigation of anaphylaxis, and caution should be employed when considering patch testing to allergens where the clinical reaction may have been IgE mediated. Anaphylaxis does occur after non-mucosal topical drug administration, especially to skin wounds or to skin with impaired barrier function [57].
Management
For evolving urticaria with or without angio-oedema or anaphylaxis, the key principal management step is to stop the offending drug and disease resolution occurs quickly.
Oral/IV antihistamines, oral/IV corticosteroids and s/c epinephrine/adrenaline may be required as per local guidelines [58]. Although cross-reactivity occurs throughout the NSAID class, selective COX-2 inhibitors are recommended in low-risk NSAID sensitive patients where anti-inflammatory therapy is required [56].
Drug-induced serum sickness-like reaction
Definition
Drug-induced serum sickness-like reactions (SSLR) are characterized by a clinical triad of fever, rash and arthralgias/arthritis.
Introduction and general description
SSLR is a drug reaction pattern, so named because of its similarity in clinical presentation to serum sickness. Classical serum sickness is a type III hypersensitivity reaction to foreign proteins resulting in the deposition of immune complexes in small blood vessels of various organs such as the skin, joints and other systems. In distinction, SSLR is typically due to medications and despite the clinical similarity, circulating immune complexes are not usually found [59].
Incidence and prevalence
SSLR accounts for about 2% of all cutaneous reactions attributed to β-lactams in a tertiary hospital [60]. Risk of SSLR is dependent on the underlying drug. Cefaclor is the most common reported trigger, occurring at 1 per 3000 cefaclor prescriptions compared to 1 per 120 000 in the case of amoxicillin.
Age
SSLR is more commonly reported in children and this may reflect the relatively frequent use of high-risk medications such as cefaclor in the young.
Pathophysiology
The pathophysiology of SSLR is not well studied. Prior in vitro studies on cefaclor suggest that drug metabolism and biotransformation of the parent drug to reactive metabolites is essential and inherited defects in the metabolism of these reactive intermediates may be a predisposing factor [61]. The downstream mechanism is unclear. However, the absence of circulating immune complexes and the lack of cross-reactivity between cases of cefaclor-induced SSLR and other cephalosporins suggest that a true hypersensitivity reaction is unlikely.
Pathology
The histological features of SSLR are consistent with an urticarial reaction pattern. Findings include dermal oedema with a superficial and deep perivascular infiltrate of lymphocytes, neutrophils and eosinophils. Vasculitis is usually absent [62].
Clinical features
History
A variety of drugs has been reported to cause SSLR eruptions and are listed in Box 118.5. The typical median latency from drug initiation to onset of rash ranges is 7 days with a range from 1 to 13 days [63, 64].
Presentation
The primary lesions of SSLR are urticarial weals which are migratory and pruritic (Figure 118.7). Some lesions may have purpuric or dusky centres and may be mistaken for erythema multiforme. Facial and periorbital oedema may coexist. No mucosal membranes are involved [64]. Other less common variants include morbilliform eruptions, urticarial or purpuric bullous plaques [62–67].
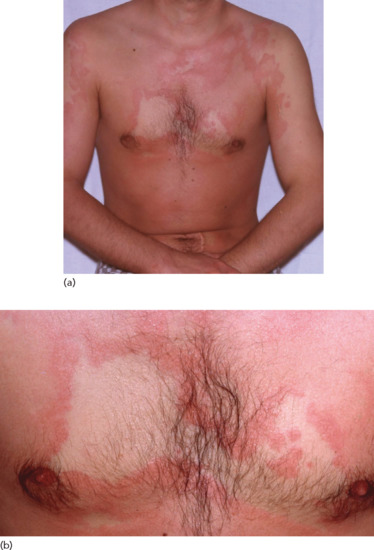
Figure 118.7 (a,b) Persistent urticarial lesions of a serum sickness-like reaction caused by penicillin.
Joints of the hands and feet are also often affected; associated symptoms include arthralgia, swelling, warmth and decreased range of movement [64]. Systemic involvement of the kidneys and liver is rare, in contrast to true serum sickness [68].
Differential diagnosis
Urticarial vasculitis, Still disease, Schnitlzer syndrome, human parvovirus B19 infection, Kawasaki disease and hereditary autoinflammatory diseases [69].
Disease course and prognosis
Cutaneous and musculoskeletal manifestations resolve on drug withdrawal. Typical median duration of rash and joint symptoms are 5 and 3 days, respectively. Long-term morbidity or sequelae are not associated with SSLR, although occasionally the duration of rash and joint symptoms may be prolonged [63, 64].
Investigations
Intradermal tests and patch tests are usually negative. Drug provocation testing is usually positive and is a safe procedure if the diagnosis needs to be confirmed [70].
Management
Withdrawal of the culprit drug is essential; the rash and joint symptoms usually resolve within 1 week of withdrawal [63]. Symptomatic treatment such as antihistamines, antipyretics and systemic corticosteroids have been prescribed, based on published series, however the impact of these medications in shortening the disease duration remains to be determined [64].
Lichenoid drug eruptions
Definition
Lichen planus (LP)-like drug eruptions are clinically indistinguishable from normal LP although are frequently more severe. Drug-induced lupus shows the clinical and immunological features of idiopathic lupus erythematosus (LE).
Introduction and general description
Lichenoid drug eruptions may present as a variety of clinical patterns including LP and LE. It has been suggested that the different clinical patterns reflect lymphocyte ‘cell rich’ (e.g. LP) and ‘cell poor’ (e.g. LE) lichenoid patterns [83]. There is no current evidence to indicate that a different disease mechanism underlies the contrasting conditions. The common causes of drug-induced LP are listed in Table 118.1. Drug-induced LE is classically caused by drugs such as procainamide, methyldopa, quinidine, minocycline and hydralazine [84–88], but many of these are no longer widely used. More recent reports of biologicals (especially anti-TNFα inhibitors) causing LE-type eruptions suggest that this may become an increasing problem [89–91]. The common causes of drug-induced LE are listed in Table 118.2.
Table 118.1 Drugs associated with drug-induced lichen planus.
| Well-established culprits | Less well-established culprits |
| Antimalarials | Angiotensin-converting enzyme inhibitors |
| Gold | β-blockers |
| Mercury amalgam | Lithium |
| Non-steroidal anti-inflammatory drugs | Methyldopa |
| Pencillamine | Quinidine |
| Thiazide diuretics | Other sulfonylureas |
Adapted from Ellgehausen et al. 1998 [93] and Halevy and Shai 1993 [94].
Table 118.2 Drugs associated with drug-induced lupus erythematosus.
| Important/widely used drugs | Less widely used drugs | Recent reports |
| Angiotensin-converting enzyme inhibitors (e.g. captopril) | Antithyroid drugs (e.g. propylthiouracil) | Biological anti-tumour necrosis factor monoclonal antibodies |
| β-blockers (e.g. atenolol)a | Chlorpromazine | Cimetidine |
| Calcium-channel blockers (e.g. diltiazem)b | Fluorouracil (systemic)b | Clobazam |
| Isoniazid | Hydralazine | Clopidogrel |
| Statinsa | Methyldopa | Clozapine |
| Sulfasalazine | Procanamide | Interferons |
| Terbinafineb | Quinidine | Interleukin 2 |
| Thiazide diuretics | Minocycline | Ticlopidine |
| Zafirlukast |
Adapted from Antonov et al. 2004 [95], Valeyrie-Allanore et al. 2007 [96] and Pretel et al. 2013 [97].
aMore typically associated with subacute cutaneous lupus erythematosus type reactions.
bMay be associated with both systemic lupus erythematosus and subacute cutaneous lupus erythematosus type reactions.
Epidemiology
Drug-induced LP reactions represent approximately 10% of all LP cases. Approximately 10% of all cutaneous LE cases are mediated by drugs.
Pathophysiology
The mechanisms underlying lichenoid drug eruptions are essentially unknown, but they may develop as a result of autoreactive T cells directed against a drug–major histocompatibility complex (MHC) antigen complex, such that keratinocytes and Langerhans cells are viewed by the immune system as ‘non-self’. Genetic factors have been strongly linked with drug-induced lupus such as hydralazine (73% HLA-DR4) [84] and minocycline (HLA-DQB1) [85], which support the prevailing concept of a T-cell-mediated disease. Lage et al. showed a significant positive correlation between CD8 frequency and perforin expression in skin biopsies from drug-induced lichenoid eruptions versus classical LP suggesting that cytotoxic CD8 T cells may be crucial for induction of keratinocyte apoptosis [86]. However, injection of autoreactive CD4 T-cell clones has been shown sufficient to induce a lichenoid skin reaction in a murine model reaching maximal severity at day 5 [83]. The presence of epidermotropic T cells correlates with that of class II MHC (HLA-DR)-expressing keratinocytes and Langerhans cells in lichenoid eruptions [83]. The cross-reactive nature of herpes simplex virus (HSV)-specific T cells with drug has been reported suggesting that antiviral skin-resident CD8 memory T cells may cross-react with drug antigens to induce lichenoid drug eruptions [83].
Pathology
Lichenoid drug eruptions show typical histological evidence of lichenoid change including hyperkeratosis, hypergranulosis, ‘saw tooth’ acanthosis, and a band-like infiltrate in the superficial dermis. A number of histological features have been attributed to a drug trigger, including a less dense but more pleomorphic infiltrate, cytoid bodies in the cornified and granular layers, the presence of focal parakeratosis, and focal interruption of the granular layer [87] (Figure 118.8). However, a recent study found that the only distinguishing histological characteristics of drug-induced LP, as opposed to non-drug LP, were a higher frequency of necrotic keratinocytes (in clusters), and infiltrating plasma cells and eosinophils [86].
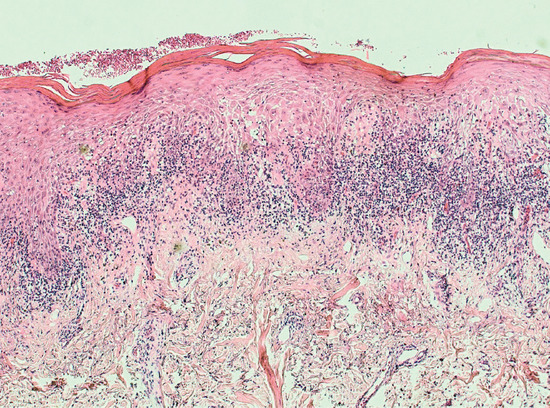
Figure 118.8 Histopathology of lichenoid drug eruption, H&E. Magnification 10×. There is compact hyperkeratosis overlying ‘sawtooth’ acanthosis of the epidermis with lymphocyte exocytosis. Basal vacuolar change and Civatte bodies accompany a band-like interface lymphocytic infiltrate. There are scattered interstitial eosinophils in the papillary dermis. (Courtesy of Dr Marianna Philippidou, King's College Hospital, London, UK.)
Clinical features
The onset of LP- and LE-like eruptions is usually months (even years) after the first exposure to the medication, but the precise reason for the typical delayed onset is not established. The skin changes of LP-like drug eruptions may be non-specific or may resemble the classical small shiny papules and plaques with Wickham's striae, as in idiopathic LP (Figures 118.9 and 118.10). Mucosal lichenoid reactions can also be caused by drugs. Photodistributed lichenoid lesions may occur with a number of drugs, including thiazide diuretics.
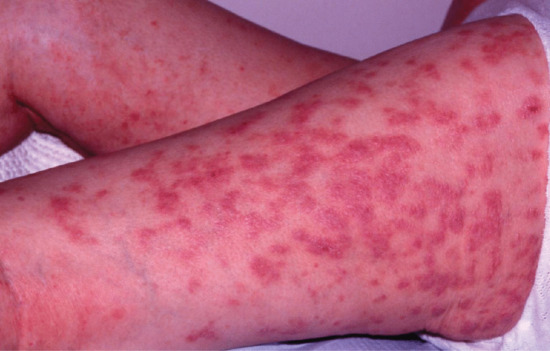
Figure 118.9 A lichenoid drug eruption triggered by therapeutic gold, presenting as multiple inflammatory plaques.
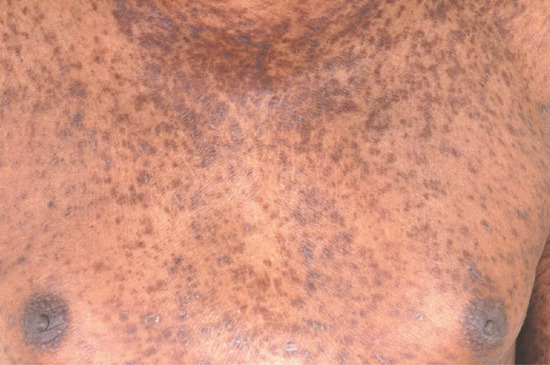
Figure 118.10 Pravastatin-induced lichenoid drug eruption with numerous hyperpigmented patches and plaques disseminated over the torso.
The clinical pattern of drug-induced LE is more commonly like SLE than subacute LE [88].
Differential diagnosis
- Idiopathic LP.
- Idiopathic cutaneous LE.
Complications and co-morbidities
If untreated, these are the same as the idiopathic conditions (see Chapters 37 and 51).
Disease course and prognosis
The diagnosis is proven by disease resolution on withdrawal of the offending drug. Disease resolution may take weeks to months.
Investigations
Skin histology is essential for the investigation of lichenoid drug eruptions. In drug-induced LE, immunological findings typically show positive antinuclear antigen and positive anti-Ro antibodies; 75% show positive antihistone antibodies (90% are ANA positive) [92].
Management
Withdrawal of the offending drug is essential. Use of a potent or super-potent topical corticosteroid is usually effective in clearing the eruption. In severe cases, therapy with oral glucocorticoids may be required.
Fixed drug eruption
Definition
Fixed drug eruption (FDE) is a cutaneous adverse drug reaction characterized by recurrent well-defined lesions occurring in the same sites each time the offending drug is taken [98, 99].
Introduction and general description
FDE is a drug eruption distinguished by its recurrence at the same sites on re-challenge, its short latency and benign nature.
History
FDE typically presents 30 min to 8 h after drug exposure. An extensive list of medications are known to cause FDE; however, the commonest culprit drugs are listed in Box 118.6. Co-trimoxazole, tetracyclines and NSAIDs have been frequently implicated as the top three causes of FDE [100, 101]. A survey of UK dermatologists yielded similar results with NSAIDs (25%), paracetamol (24%), co-trimoxazole (5%) and tetracycline (5%) being the most frequent triggers [102]. However, a recent French nationwide analysis of FDEs reports the common drugs as NSAIDs (25%), paracetamol (15%), carbocystine (7%) and amoxicillin (7%), which may be due to changing prescription practices [103]. Food and herbal medications have also been reported as aetiological agents [104–106].
Epidemiology
Incidence and prevalence
FDEs account for 4–39% of all drug eruptions [107–109].
Age
FDE has been reported in all ages. However, it is more common in adults, particularly in the range of 40–80 year olds [103].
Sex
Both genders are prone to the development of FDE, although a female preponderance may exist [103].
Pathophysiology
FDE is a form of classical delayed-type hypersensitivity reaction and skin resident T cells are believed to be the key mediators in eliciting FDE. Long after clinical resolution, ‘resting’ FDE lesions contain CD8+ T cells with an effector/memory phenotype. These cells are located at the dermal–epidermal junction and remain quiescent until drug re-challenge. On re-exposure to the drug, there is activation and expansion of these CD8+ lymphocytes with the release of interferon (IFN)-γ and cytotoxic granules resulting in keratinocyte apoptosis. At the end of the immune response, regulatory T cells are recruited into the lesions and limit further damage by inhibiting the cytotoxic T cells. Expanded and activated cytotoxic T cells are removed by apoptosis but a small population is prevented from apoptosis by keratinocyte derived interleukin (IL)-15 and remain as skin-resident memory T cells until the next activation cycle [98, 110, 111].
Genetics
A pharmacogenetic predisposition exists as drug-specific HLA associations have been reported for certain medications. These include febrazone-induced FDE and HLA-B22 and trimethoproim-sulfamethoxazole and HLA-A30 [112, 113].
Pathology
Early biopsy specimens show an interface dermatitis reaction pattern with vacuolar degeneration of basal keratinocytes, dermal oedema and a perivascular lymphocytic infiltrate of the upper dermis. Eosinophils may be present. Resolved or healing lesions are characterized by pigment-laden macrophages in the upper dermis [98, 114] (Figure 118.11).
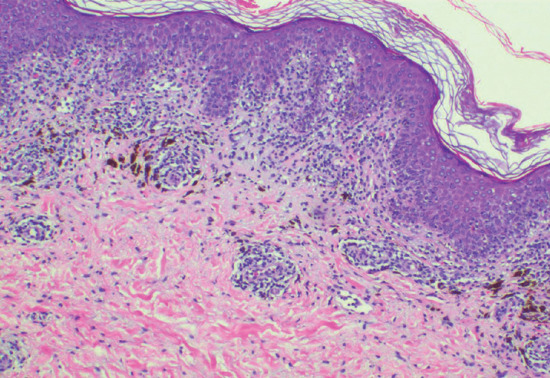
Figure 118.11 Histopathological features of fixed drug eruption: interface vacuolar degeneration with lymphocyte exocytosis and melanin incontinence; a perivascular lymphoid infiltrate is present around the superficial dermal vessels. (Courtesy of Dr Ronald Goh, Singapore General Hospital, Singapore.)
Clinical features
History
FDE usually develops 30 min to 8 h after drug exposure.
Presentation
Typically, FDE presents as a sharply-defined, round or oval erythematous and oedematous plaque which evolves to become dusky, violaceous and occasionally vesicular or bullous (Figure 118.12). Lesions are usually solitary or few in number although multiple lesions may be present or may develop as a consequence of repeated challenges [115] (Figure 118.13a,b). Commonly affected sites include the lips, genitals, palms and soles; 5% of cases may have an exclusive mucosal involvement [103].
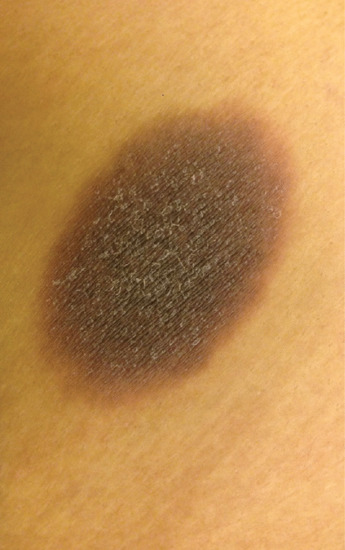
Figure 118.12 Fixed drug eruption: a well-defined, dusky and hyperpigmented oval plaque on the thigh.
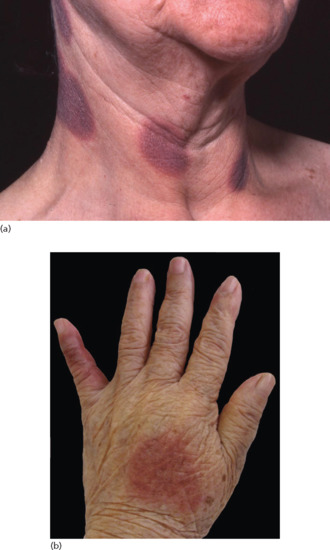
Figure 118.13 (a) Multiple discrete lesions of fixed drug eruption induced by aspirin (Anadin). (b) Well-defined erythematous plaque over the dorsum of the hand and little finger.
Drug-specific clinical patterns have been reported. These include: NSAID-induced FDE affecting the genitals and lips; tetracycline- and trimethoprim/sulfamethoxazole-induced FDE affecting the genitals; metamizole-induced FDE of the trunk and extremities; and carbocysteine-induced FDE of the face [103, 114, 115].
Generalized bullous FDE (GBFDE) is a form of extensive FDE which may be misdiagnosed as toxic epidermal necrolysis (TEN) [98, 116] (Figure 118.14). Differentiating features from TEN include: (i) prior history of similar episodes; (ii) mucosal surfaces are relatively uninvolved; (iii) the presence of large blisters with intact intervening skin; and (iv) the absence of multiple purpuric or target lesions.
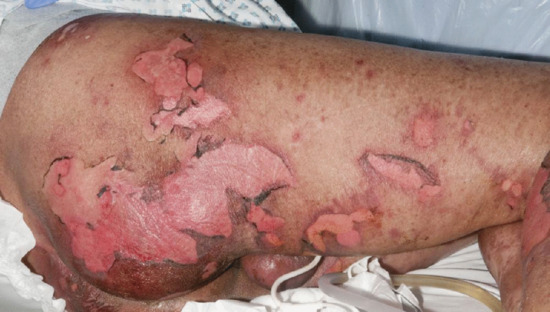
Figure 118.14 Generalized bullous fixed drug eruption: multiple bullous and eroded lesions induced by parecoxib.
Differential diagnosis
The differential diagnosis of FDE depends on the site, number of lesions and stage of evolution. Mucosal predominant lesions may mimic herpes simplex, aphthous stomatitis, pemphigus vulgaris and Stevens–Johnson syndrome (SJS). Extensive lesions in GBFDE may be diagnosed as SJS/TEN or bullous pemphigoid. In healed lesions, residual pigmentation may be reminiscent of erythema dyschromicum perstans [98, 99, 116].
Complications and co-morbidities
Patients with GBFDE generally lack visceral complications and are thought to have a benign clinical course. In contrast with SJS/TEN, GBFDE generally has modest or no mucosal involvement and is not associated with respiratory tract or intestinal involvement. However, in a case–control analysis of GBFDE versus SJS/TEN, in which cases were matched by age and extent of skin detachment, the mortality in GBFDE was not significantly lower that of SJS/TEN [117].
Disease course and prognosis
The majority of FDE is self-limiting with an excellent prognosis. Post-inflammatory hyperpigmentation can be prominent and persist for several months after the acute episode. In GBFDE, the mortality is approximately 20%. Patients with GBFDE require the same level of treatment and care as for SJS and TEN (see Chapter 119).
Investigations
Oral provocation of the implicated drug is the gold standard to confirm drug causality, however this should not be undertaken if the patient is at risk of GBFDE. Patch testing has been suggested as an alternative diagnostic method. Unlike conventional patch testing, the reagents should be placed at skin sites of previous lesions of FDE instead of the upper back. Use of non-lesional skin in patch testing for FDE usually yields a negative response [118, 119]. Patch tests on lesional skin are positive in about 50% of cases [95].
Management
Treatment involves stopping the offending drug and the use of topical corticosteroid. Systemic corticosteroids may be necessary in patients with multiple lesions. GBFDE should be treated in an intensive care centre with expertise in skin loss syndromes, or in a burns unit (see Chapter 119).
Drug-induced pityriasis rosea
Definition
A drug-induced dermatosis which clinically resembles pityriasis rosea (PR).
Introduction and general description
Drug-induced PR-like eruption is an uncommon cutaneous adverse reaction characterized by erythematous scaly papules and plaques that are typically located on the trunk [122]. A variety of drugs has been reported to cause PR-like eruptions and are listed in Box 118.7.
Epidemiology
Incidence and prevalence
Drug-induced PR-like reactions made up 2% of all cutaneous adverse reactions presenting at a drug surveillance centre [123].
Pathophysiology
The exact pathomechanism is unclear. However, some reports have shown that these reactions are dose dependent, suggesting that they may be due to the pharmacological effect of the medication (e.g. induction of increased levels of kinins by ACE inhibitors; inhibition of cyclo-oxygenase by NSAIDs) rather than a true hypersensitivity reaction [123, 124, 125].
Pathology
The histological features of such reactions are similar to classical PR, demonstrating parakeratosis and focal spongiosis with papillary dermal oedema and superficial perivascular infiltrate of lymphocytes. In contrast to classical PR eosinophils may be prominent [126, 127].
Clinical features
History
A variety of drugs have been reported to cause PR-like eruptions and are listed in Box 118.7.
Presentation
The typical latency from drug initiation to onset of rash ranges from 6 to 40 days [123, 128, 129]. Drug-induced PR-like eruptions present with erythematous, violaceous papules or plaques with a collarette of scale, arranged with their long axis parallel to the skin lines and are typically located on the trunk (Figure 118.15). Features that may distinguish drug-induced PR from its classical forms include the presence of fewer and larger lesions, variable fir-tree distribution, absence of the characteristic herald patch, persistence of rash beyond 2 months and larger bright red lesions accompanied by significant pruritus [123, 124, 128]. Oral lesions may also be present [130].
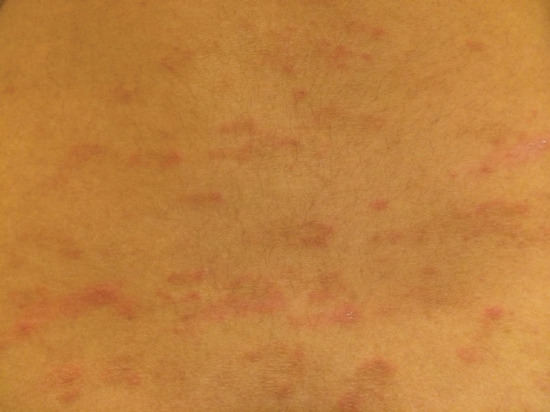
Figure 118.15 Drug-induced pityriasis rosea: erythematous papules over the lower back with some having a collarette of scales.
Differential diagnosis
Other papulosquamous eruptions such as classical pityriasis rosea, secondary syphilis, pityriasis lichenoides chronica, guttate psoriasis and nummular eczema.
Disease course and prognosis
Unlike the typical course of classical PR, drug-induced PR-like eruptions may last more than 8 weeks if the culprit drug is not withdrawn. Conversely, it resolves quickly within 1–2 weeks on drug withdrawal [124].
Investigations
Oral provocation tests, although not performed routinely, confirm the diagnosis.
Management
The cutaneous reaction often resolves without treatment on discontinuation of the culprit drug [124]. Topical corticosteroids may be useful in cases that are slow to respond [131].
Drug-induced erythema nodosum
Definition
A septal panniculitis induced by a medication.
Introduction and general description
Erythema nodosum (EN) is a cutaneous reactive septal panniculitis which may be triggered by a wide variety of stimuli including medications. Drug-induced EN is indistinguishable from that caused by other factors [146]. A number of drugs have been reported to cause EN and are listed in Box 118.8.
Epidemiology
Incidence and prevalence
The incidence of EN is estimated to be 1–5 cases per 100 000 persons per year and about 3–15% of cases can be attributed to medications [146–149].
Age
Reported in all ages but drug-induced EN is rare in children [150].
Sex
There is a female preponderance [149].
Pathophysiology
EN is believed to be a type IV delayed hypersensitivity reaction to various antigens, including medications [146, 151].
Pathology
EN is the prototypical septal panniculitis without the presence of vasculitis. Early lesions demonstrate oedema, haemorrhage and neutrophils within the septae, older lesions are characterized by fibrosis, periseptal granulation tissue, lymphocytes, histiocytes and multinucleated giant cells [146].
Clinical features
History
The latency from drug initiation to the onset of EN is usually a few weeks. However, a case of ciprofloxacin-induced EN, confirmed by repeated oral challenge had a latency of 1 day [152–154].
Presentation
The clinical features of EN are symmetrical, erythematous and tender subcutaneous nodules or plaques which are typically distributed over the anterior aspect of the limbs. Over a few days, these lesions become purplish before finally turning brown. Ulceration never occurs and the upper limbs are less often affected [146].
Occasionally, drug-induced EN may occur in the setting of a drug hypersensitivity disorder, particularly with azathioprine and minocycline, and is accompanied by fever and other visceral involvement [155, 156].
Differential diagnosis
This includes non-drug causes of EN, other forms of panniculitis and cutaneous polyarteritis nodosa.
Disease course and prognosis
The clinical course is self-limiting following drug withdrawal and usually resolves within 2–4 weeks [152–154].
Investigations
Exclusion of other causes of EN is essential. Relevant investigations include chest radiography, microbiological cultures and serological testing, such as anti-streptolysin titres, to exclude infective causes. Other tests and imaging studies may be warranted in the appropriate setting.
Management
The offending drug should be withdrawn and treatment is otherwise symptomatic. NSAIDs and compression hosiery may reduce pain and inflammation. Systemic corticosteroids are rarely indicated.
Drug-induced acneform eruptions
Definition and nomenclature
An acneform eruption which is induced by a medication.
Introduction and general description
Drug-induced acneform eruptions are inflammatory follicular reactions which resembles acne vulgaris. Clinically, monomorphic papules and pustules are seen and a variety of drugs have been implicated, including hormones, vitamins, halogens, neuropsychotherapeutic drugs, anti-tubercular treatments, immunomodulating agents, cytotoxic agents and newer targeted therapies [162–164]. Various drugs have been associated with acneform eruptions and the common drugs are summarized in Box 118.9.
Epidemiology
Incidence and prevalence
Acneform eruptions represent 1% of all drug-induced skin reactions [165].
Pathophysiology
Acneform eruptions are not hypersensitivity reactions. Pathological mechanisms vary according to the implicated agent. In acne vulgaris, Propionibacterium acnes facilitates inflammation through the binding of toll-like receptor 2 (TLR-2). Up-regulation of TLR-2 has been reported in human keratinocytes treated with glucocorticoids and this may explain why corticosteroid-associated acne consists of predominantly inflammatory lesions of papules and pustules [166]. Androgenic hormones stimulate follicular keratinocyte proliferation, promote sebaceous gland hyperplasia and increase sebum production [167, 168]. Sirolimus may induce acneform lesions via direct toxicity, chemical modification of sebum or its effects on epidermal growth factor receptors (EGFR) and testosterone synthesis [169].
Pathology
There is variation in the histopathology of drug-induced acneform eruptions dependent on the underlying drug. In steroid-induced acne initial lesions show features of focal necrosis in the infundibulum of the follicular epithelium with a localized intrafollicular and perifollicular neutrophilic inflammatory reaction [170]. EGFR-associated acneform eruptions are characterized by ectatic follicular infundibula with rupture of the epithelial lining associated with superficial neutrophilic folliculitis [171].
Clinical features
History
A drug-induced acneform eruption is to be suspected if the onset of acne is sudden and abrupt in the absence of past history of acne vulgaris. Other presentations include an unusually severe acne flare in a patient with a past history of mild acne vulgaris, or acne developing at an unusual age of onset [162, 163]. Various drugs have been associated with acneform eruptions and the commonest drugs are listed in Box 118.9. The latency between drug initiation and onset of acne varies between various types of drugs. Shorter latencies of 1 month or less have been reported with systemic corticosteroids [170], androgens [172–174] and vitamin B [175]; latencies of greater than 1 month are usually observed with ciclosporin [176], amineptine [177], lithium [178], anti-epileptics [179] and anti-tuberculosis treatment [180]. A characteristic papulopustular eruption may occur with EGFR inhibitors (see Chapter 120). The incidence ranges from 24 to 91%, being more common in monoclonal antibodies, such as cetuximab and panitumumab, compared to oral tyrosine kinase inhibitors such as erlotinib, gefitinib and lapatinib. Median latency from drug initiation to onset of acneform eruptions is 7–10 days [181, 182, 183], with the maximum severity being reached in the second week.
Presentation
The lesions of drug-induced acne are monomorphic papules and pustules which typically lack comedones or cysts. The dermatosis can be widespread and extend beyond the seborrhoeic areas such as the arms, lower back and genitalia. However the acneform eruption induced by EGFR inhibitors is usually distributed in the seborrhoeic areas, that is, the neck, chest, shoulders and upper back [182–184].
Differential diagnosis
This includes acne vulgaris, Gram-negative folliculitis and Pityrosporum folliculitis.
Disease course and prognosis
Improves on withdrawal of the offending drug.
Management
Drug withdrawal usually improves acneform eruptions, however such a decision needs to be balanced against the drug indication and/or if alternative agents are available. Topical and systemic medications used for acne vulgaris may be useful.
References
- Yawalkar N. Drug-induced exanthems. Toxicology 2005;209(2):131–4.
- Fiszenson-Albala F, Auzerie V, Mahe E, et al. A 6-month prospective survey of cutaneous drug reactions in a hospital setting. Br J Dermatol 2003;149(5):1018–22.
- Hernandez-Salazar A, Rosales SP, Rangel-Frausto S, Criollo E, Archer-Dubon C, Orozco-Topete R. Epidemiology of adverse cutaneous drug reactions. A prospective study in hospitalized patients. Arch Med Res 2006;37(7):899–902.
- Justiniano H, Berlingeri-Ramos AC, Sanchez JL. Pattern analysis of drug-induced skin diseases. Am J Dermatopathol 2008;30(4):352–69.
- Bialecki C, Feder HM Jr, Grant-Kels JM. The six classic childhood exanthems: a review and update. J Am Acad Dermatol 1989;21(5 Pt 1):891–903.
- Drago F, Paolino S, Rebora A, Broccolo F, Cardo P, Parodi A. The challenge of diagnosing atypical exanthems: a clinico-laboratory study. J Am Acad Dermatol 2012;67(6):1282–8.
- Rustin MH. The safety of tacrolimus ointment for the treatment of atopic dermatitis: a review. Br J Dermatol 2007;157(5):861–73.
- Jeck T, Edmonds D, Mengden T, et al. Betablocking drugs in essential hypertension: transdermal bupranolol compared with oral metoprolol. Int J Clin Pharmacol Res 1992;12(3):139–48.
- van der Linden PD, van der Lei J, Vlug AE, Stricker BH. Skin reactions to antibacterial agents in general practice. J Clin Epidemiol 1998;51(8):703–8.
- Swegle JM, Logemann C. Management of common opioid-induced adverse effects. Am Fam Phys 2006;74(8):1347–54.
- Reich A, Szepietowski JC. Opioid-induced pruritus: an update. Clin Exp Dermatol 2010;35(1):2–6.
- Ganesh A, Maxwell LG. Pathophysiology and management of opioid-induced pruritus. Drugs 2007;67(16):2323–33.
- Adebayo RA, Sofowora GG, Onayemi O, Udoh SJ, Ajayi AA. Chloroquine-induced pruritus in malaria fever: contribution of malaria parasitaemia and the effects of prednisolone, niacin, and their combination, compared with antihistamine. Br J Clin Pharmacol 1997;44(2):157–61.
- Ekpechi OL, Okoro AN. A pattern of pruritus due to chloroquine. Arch Dermatol 1964;89:631–2.
- Weisshaar E, Ziethen B, Gollnick H. Can a serotonin type 3 (5-HT3) receptor antagonist reduce experimentally-induced itch? Inflamm Res 1997;46(10):412–16.
- Stander S, Bockenholt B, Schurmeyer-Horst F, et al. Treatment of chronic pruritus with the selective serotonin re-uptake inhibitors paroxetine and fluvoxamine: results of an open-labelled, two-arm proof-of-concept study. Acta Derm Venereol 2009;89(1):45–51.
- Schneeberger R, Albegger K, Oberascher G, Miller K. [Pruritus – a side effect of hydroxyethyl starch? First report.] HNO 1990;38(8):298–303.
- Bork K. Pruritus precipitated by hydroxyethyl starch: a review. Br J Dermatol 2005;152(1):3–12.
- Reich A, Stander S, Szepietowski JC. Drug-induced pruritus: a review. Acta Derm Venereol 2009;89(3):236–44.
- Wolfhagen FH, Sternieri E, Hop WC, Vitale G, Bertolotti M, Van Buuren HR. Oral naltrexone treatment for cholestatic pruritus: a double-blind, placebo-controlled study. Gastroenterology 1997;113(4):1264–9.
- Rietschel RL, Fowler JF. Fisher's Contact Dermatitis, 6th edn. Ontario: BC Decker, 2007.
- Johansen JD, Frosch PJ, Lepoittevin J-P. Contact Dermatitis, 5th edn. Berlin: Springer-Verlag, 2010.
- Menne T, Veien NK, Maibach HI. Systemic contact-type dermatitis due to drugs. Semin Dermatol 1989;8(3):144–8.
- Berman BA, Ross RN. Ethylenediamine: systemic eczematous contact-type dermatitis. Cutis 1983;31(6):594, 596, 598.
- Hardy C, Schofield O, George CF. Allergy to aminophylline. BMJ (Clin Res Ed) 1983;286(6383):2051–2.
- Aeling JL, Nuss DD. Letter: Systemic eczematous “contact-type” dermatitis medicamentosa caused by parabens. Arch Dermatol 1974;110(4):640.
- Ekelund AG, Moller H. Oral provocation in eczematous contact allergy to neomycin and hydroxy-quinolines. Acta Derm Venereol 1969;49(4):422–6.
- Pirila V. Endogenic contact eczema. Allerg Asthma (Leipz) 1970;16(1):15–19.
- Thyssen JP, Maibach HI. Drug-elicited systemic allergic (contact) dermatitis – update and possible pathomechanisms. Contact Dermatitis 2008;59(4):195–202.
- Sans V, Jouary T, Hubiche T, Smith D, Milpied B, Taieb A. Baboon syndrome induced by cetuximab. Arch Dermatol 2008;144(2):272–4.
- Ferreira O, Mota A, Morais P, Cunha AP, Azevedo F. Symmetrical drug-related intertriginous and flexural exanthema (SDRIFE) induced by telmisartan-hydrochlorothiazide. Cutan Ocul Toxicol 2010;29(4):293–5.
- Allain-Veyrac G, Lebreton A, Collonnier C, Jolliet P. First case of symmetric drug-related intertriginous and flexural exanthema (SDRIFE) due to rivastigmine? Am J Clin Dermatol 2011;12(3):210–13.
- Lugovic-Mihic L, Duvancic T, Vucic M, Situm M, Kolic M, Mihic J. SDRIFE (baboon syndrome) due to paracetamol: case report. Acta Dermatovenerol Croat 2013;21(2):113–17.
- Culav I, Ljubojevic S, Buzina DS. Baboon syndrome/SDRIFE due to sulfamethoxazole-trimethoprim. Int J Dermatol 2013;52(9):1159–60.
- Veien NK. Systemic contact dermatitis. Int J Dermatol 2011;50(12):1445–56.
- Joly P, Benoit-Corven C, Baricault S, et al. Chronic eczematous eruptions of the elderly are associated with chronic exposure to calcium channel blockers: results from a case–control study. J Invest Dermatol 2007;127:2766–71.
- Hausemann P, Harr TH, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis 2004;51:297–310.
- Tan SC, Tan JW. Symmetrical drug-related intertriginous and flexural exanthema. Curr Opin Allergy Clin Immunol 2011;11:313–18
- Lee HY, Phillippidou M, Schey S, Selway R, Walsh S, Creamer D. Flexural eruption in two hospitalized patients. Clin Exp Dermatol 2013 May 20.
- Winnicki M, Shear NH. A systematic approach to systemic contact dermatitis and symmetric drug-related intertriginous and flexural exanthema (SDRIFE): a closer look at these conditions and an approach to intertriginous eruptions. Am J Clin Dermatol 2011;1:171–80.
- Dona I, Blanca-Lopez N, Torres MJ, et al. Drug hypersensitivity reactions: response patterns, drug involved, and temporal variations in a large series of patients. J Investig Allergol Clin Immunol 2012;22(5):363–71.
- Comert S, Celebioglu E, Karakaya G, Kalyoncu AF. The general characteristics of acute urticaria attacks and the factors predictive of progression to chronic urticaria. Allergol Immunopathol (Madr) 2013;41(4):239–45.
- Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol 2010;126(3):477–80, e1–42.
- Jee R, Nel L, Gnanakumaran G, Williams A, Eren E. Four cases of anaphylaxis to chlorhexidine impregnated central venous catheters: a case cluster or the tip of the iceberg? Br J Anaesth 2009;103(4):614–15.
- Rutkowski K, Dua S, Nasser S. Anaphylaxis: current state of knowledge for the modern physician. Postgrad Med J 2012;88(1042):458–64.
- Shipley D, Ormerod AD. Drug-induced urticaria. Recognition and treatment. Am J Clin Dermatol 2001;2(3):151–8.
- Mathelier-Fusade P. Drug-induced urticarias. Clin Rev Allergy Immunol 2006;30(1):19–23.
- Stewart AG, Ewan PW. The incidence, aetiology and management of anaphylaxis presenting to an accident and emergency department. QJM 1996;89(11):859–64.
- Pumphrey RS. Lessons for management of anaphylaxis from a study of fatal reactions. Clin Exp Allergy 2000;30(8):1144.
- Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn's disease. N Engl J Med 2003;348(7):601–8.
- Crayne CB, Gerhold K, Cron RQ. Anaphylaxis to etanercept in two children with juvenile idiopathic arthritis. J Clin Rheumatol 2013;19(3):129–31.
- Busse PJ, Buckland MS. Non-histaminergic angioedema: focus on bradykinin-mediated angioedema. Clin Exp Allergy 2013;43(4):385–94.
- Moneret-Vautrin DA, Morisset M, Flabbee J, Beaudouin E, Kanny G. Epidemiology of life-threatening and lethal anaphylaxis: a review. Allergy 2005;60(4):443–51.
- Bock SA, Munoz-Furlong A, Sampson HA. Fatalities due to anaphylactic reactions to foods. J Allergy Clin Immunol 2001;107(1):191–3.
- Mirakian R, Ewan PW, Durham SR, et al. BSACI guidelines for the management of drug allergy. Clin Exp Allergy 2009;39(1):43–61.
- National Institute for Health and Care Excellence (NICE) 2014. Drug allergy: diagnosis and management of drug allergy in adults, children and young people http://www.nice.org.uk/guidance/cg183 (last accessed 20 August 2015).
- Sachs B, Fischer-Barth W, Erdmann S, Merk HF, Seebeck J. Anaphylaxis and toxic epidermal necrolysis or Stevens–Johnson syndrome after nonmucosal topical drug application: fact or fiction? Allergy 2007;62(8):877–83.
- Dzingina M, Stegenga H, Heath M, et al. Assessment and referral after emergency treatment of a suspected anaphylactic episode: summary of NICE guidance. BMJ 2011;343:d7595.
- Vial T, Pont J, Pham E, et al. Cefaclor-associated serum sickness-like disease: eight cases and review of the literature. Ann Pharmacother 1992;26:910–14.
- Terrados S, Blanca M, Garcia J, et al. Nonimmediate reactions to betalactams: prevalence and role of the different penicillins. Allergy 1995;50:563–7.
- Kearns GL, Wheeler G, Childress SH, Letzig LG. Serum sickness-like reactions to cefaclor: Role of hepatic metabolism and individual susceptibililty. J Pediatr 1994;125:805–11.
- Tolpinrud WL, Bunick CG, King BA. Serum sickness-like reaction: Histopathology and case report. J Am Acad Dermatol 2011;65:e83–5
- King BA, Geelhoed GC. Adverse skin and joint reactions associated with oral antibiotics in children: The role of cefaclor in serum sickness-like reactions. J Paediatr Child Health 2003;39:677–81.
- Hebert AA, Sigman ES, Levy ML. Serum sickness-like reaction from cefaclor in children. J Am Acad Dermatol 1991;25:805–8.
- Venna S. Severe serum sickness-like type III reaction to insulin detemir. J Am Acad Dermatol 2011;64:128.
- Creamer JD, McGrath JA, Webb-Peploe M, Smith NP. Serum sickness-like illness following streptokinase therapy. A case report. Clin Exp Dermatol 1995;20:468–70.
- Landau M, Shachar E, Brenner S. Minocycline-induced serum sickness-like reaction. J Eur Acad Dermatol Venereol 2000;14:67–8.
- Katta R, Anusuri V. Serum sickness-like reaction to cefuroxime: A case report and review of the literature. J Drug Dermatol 2007;6:747–8.
- Cozzi A, Doria A, Gisondi P, Girolomoni G. Skin rash and arthritis: a simplified appraisal of less common associations. J Eur Acad Dermatol Venereol 2014;14:28:679–88.
- Blanco-Lopez N, Zapatero L, Alonson E, et al. Skin testing and drug provocation in the diagnosis of nonimmediate reactions to aminopenicillins in children. Allergy 2009;64:229–33.
- Park H, Knowles S, Shear NH. Serum sickness-like reaction to itraconazole. Ann Pharmacother 1998;32:1249.
- Swanson J, English JC 3rd. Serum sickness-like reaction to Pamabrom. J Drug Dermatol 2006;5:294–6.
- Parra F, Perez Eliase M, Cuevas M, Ferreira A. Serum sickness-like illness associated with rifampicin. Ann Allergy 1994;73:123–5.
- Ralph ED, John M, Rieder MJ, Bombassaro AM. Serum sickness-like reaction possibly associated with meropenem use. Clin Infect Dis 2003;36:e149–51.
- Beyens MN, Guy C, Mounier G, et al. Serious adverse reactions of bupropion for smoking cessation: analysis of the French Pharmacovigilance Database from 2001 to 2004. Drug Saf 2008;31:1017–26.
- Philips EJ, Knowles SR, Shear NH. Serum sickness-like reaction associated with clopidogrel. Ann Pharmacother 2003;56:583.
- Miller LG, Bowman RC, Mann D, Tripathy A. A case of fluoxetine-induced serum sickness. Am J Psychiatry 1989;146:1616–17.
- Sanchez G, Vila L, Pajaron M, Dieguez I. Skin manifestation of a case of phenylbutazone-induced serum sickness-like reactions. J Investig Allergol Clin Immunol 2000;10:170–2.
- Hamzaoglu H, Cooper J, Alsahli M, et al. Safety of infliximab in Crohn's disease: a large single-center experience. Inflamm Bowel Dis 2010;16:2109–16.
- Lecluse LLA, Piskin G, Mekkes JR, et al. Review and expert opinion on prevention and treatment of infliximab-related infusion reactions. Br J Dermatol 2008;159:527–36.
- Todd DJ, Helfgott. Serum sickness following treatment with rituximab. J Rheumatol 2007;34:430–3.
- Pilette C, Coppens N, Houssiau FA, Rodenstein DO. Severe serum sickness-like syndrome after omalizumab therapy for asthma. J Allergy Clin Immunol 2007;120:972–3.
- Shiohara T, Mizukawa Y. The immunological basis of lichenoid tissue reaction. Autoimmun Rev 2005;4(4):236–41.
- Batchelor JR, Welsh KI, Tinoco RM, et al. Hydralazine-induced systemic lupus erythematosus: influence of HLA-DR and sex on susceptibility. Lancet 1980;1(8178):1107–9.
- Dunphy J, Oliver M, Rands AL, Lovell CR, McHugh NJ. Antineutrophil cytoplasmic antibodies and HLA class II alleles in minocycline-induced lupus-like syndrome. Br J Dermatol 2000;142(3):461–7.
- Lage D, Juliano PB, Metze K, de Souza EM, Cintra ML. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol 2012;51(10):1199–205.
- Van den Haute V, Antoine JL, Lachapelle JM. Histopathological discriminant criteria between lichenoid drug eruption and idiopathic lichen planus: retrospective study on selected samples. Dermatologica 1989;179(1):10–13.
- Marzano AV, Tavecchio S, Menicanti C, Crosti C. Drug-induced lupus erythematosus. Giornal Ital Dermatol Venereol 2014;149(3):301–9.
- Asarch A, Gottlieb AB, Lee J, et al. Lichen planus-like eruptions: an emerging side effect of tumor necrosis factor-alpha antagonists. J Am Acad Dermatol 2009;61(1):104–11.
- Brazzelli V, Muzio F, Manna G, et al. Photoinduced dermatitis and oral lichenoid reaction in a chronic myeloid leukemia patient treated with imatinib mesylate. Photodermatol Photoimmunol Photomed 2012;28(1):2–5.
- Bakkour W, Coulson IH. GA101 (a novel anti-CD20 monoclonal antibody)-induced lichenoid eruption. Dermatol Ther (Heidelb) 2012;2(1):3.
- Hess E. Drug-related lupus. N Engl J Med 1988;318(22):1460–2.
- Halevy S, Shai A. Lichenoid drug eruptions. J Am Acad Dermatol 1993;29(2 Pt 1):249–55.
- Shiohara T, Mizukawa Y. Fixed drug eruption: a disease mediated by self-inflcited responses of intraepidermal T cells. Eur J Dermatol 2007;201–8.
- Antonov D, Kazandjieva J, Etugov D, Gospodinov D, Tsankov N. Drug-induced lupus erythematosus. Clin Dermatol 2004;22(2):157–66.
- Valeyrie-Allanore L, Sassolas B, Roujeau JC. Drug-induced skin, nail and hair disorders. Drug Saf 2007;30(11):1011–30.
- Pretel M, Marques L, Espana A. Drug-induced lupus erythematosus. Actas Dermosifiliogr 2014;1:18–30.
- Ellgehausen P, Elsner P, Burg G. Drug-induced lichen planus. Clin Dermatol 1998;16(3):325–32.
- Ozkaya E. Fixed drug eruption: state of the art. J Dtsch Dermatol Ges 2008;3:181–8.
- Thankappan TP, Zachariah J. Drug specific clinical pattern in fixed drug eruptions. Int J Dermatol 1991;30:867–70.
- Rahman MH. Fixed drug eruption in Bangladeshi population: confirmed by provocative test. Int J Dermatol 2014;53:255–8.
- Savin JA. Current causes of fixed drug eruption in the U.K. Br J Dermatol 2001;145:667–8.
- Brahumi N, Routier E, Raison-Peyron, et al. A three-year-analysis of fixed drug eruptions in hospital setting in France. Eur J Dermatol 2010;20:461–4.
- Volz T, Berner D, Weigert, et al. Fixed food eruption caused by asparagus. J Allergy Clin Immunol 2005;116:1390–2.
- Fukushima S, Kidou M, Ihn H. Fixed food eruption caused by cashew nut. Allergol Int 2008;57:285–7.
- Tan C, Zhu WY. Chinese herbal medicine: a neglected offender for fixed drug eruptions. Eur J Dermatol 2010;20:397–9.
- Lee HY, Tay LK, Thirumoorthy T, Pang SM. Cutaneous adverse drug reactions in hospitalised patients. Singapore Med J 2010;5:767–74.
- Apaydin R, Bilen N, Sokmeci S, et al. Drug eruptions: a study including all inpatients and outpatients at a dermatology clinic of a university hospital. J Eur Acad Dermatol Venereol 2000;14:513–22.
- Stubb S, Heikkila H, Kauppinen K. Cutaneous reactions to drugs: a series of inpatients during a five year period. Acta Derm Venereol 1994;74:289–91
- Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruptions. Br J Dermatol 2008;158:1230–8.
- Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol 2009;9:316–21.
- Ozkaya-Bayazit E, Akar U. Fixed drug eruption induced by trimethoprim-sulfamethoxazole: evidence for a link to HLA-A30 B13 CW6 haplotype. J Am Acad Dermatol 2001;45:712–17.
- Pellicano R, Lomuto M, Ciavarella G, et al. Fixed drug eruptions with feprazone are linked to HLA-B22. J Am Acad Dermatol 1997;36:782–3.
- Ramdial PK, Naidoo DK. Drug-induced cutaneous pathology. J Clin Pathol 2009;62:493–504.
- Mockenhaupt M. Severe drug-induced skin reactions: clinical pattern, diagnostics and therapy. JDDG 2009;7:142–62.
- Ozkaya-Bayazit E, Bayazit H, Ozarmagan G. Drug related clinical pattern in fixed drug eruption. Eur J Dermatol 2000;10:288–91.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens–Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol 2013;168:726–32.
- Barbaud A, Goncalo M, Bruynzeel D, et al. Guidelines for performing skin tests with drugs in the investigation of cutaneous adverse drug reactions. Contact Dermatitis 2001;45:321–8.
- Andrade P, Brinca A, Goncalo M. Patch testing in fixed drug eruptions – a 20-year review. Contact Dermatitis 2011;65:195–201.
- Gupta R, Thami GP. Fixed drug eruption caused by itraconazole: reactivity and cross reactivity. J Am Acad Dermatol 2008;58:521–2.
- Sehgal VN, Srivastava G. Fixed drug eruption (FDE): Changing scenario of incriminating drugs. Int J Dermatol 2006;45:897–908.
- Fox BJ, Odom RB. Papulosquamous diseases: a review. J Am Acad Dermatol 1985;12:597–624.
- Atzori L, Pinnal Al, Ferreli C, Aste N. Pityriasis rosea-like adverse reaction: review of the literature and experience of an Italian drug-surveillance center. Dermatol Online J 2006;12:1.
- Atzori L, Ferreli C, Pinna AL, et al. Pityriasis rosea-like adverse reaction to Lisinopril. Eur Acad Dermatol Venereol 2004;18:743–5.
- Wilkinson SM, Smith AG, Davis MJ, et al. Pityriasis rosea and discoid eczema: dose related reactions to treatment with gold. Ann Rheum Dis 1992;51:881–4.
- Ramdial PK, Naidoo DK. Drug-induced cutaneous pathology. J Clin Pathol 2009;62:493–504.
- Justiiano H, Berlingeri-Ramos AC, Sanchez JL. Pattern analysis of drug-induced skin diseases. Am J Dermatopathol 2008;30:352–69.
- Maize JC, Tomecki KJ. Pityriasis rosea-like drug eruption secondary to metronidazole. Arch Dermatol 1977;113:1458–9.
- Wilkin JK, Kirkendall WM. Pityriasis rosea-like rash from Captopril. Arch Dermatol 1982;118:186–7.
- Gonzalez LM, Allen R, Janniger CK, et al. Pityriasis rosea: An important papulosquamous disease. Int J Dermatol 2005;44:757–64.
- Buckley C. Pityriasis rosea-like eruption in a patient receiving omeprazole. Br J Dermatol 1996;135:660–1.
- Gupta AK, Lynde CW, Lauzon GJ, et al. Cutaneous adverse effects associated with terbinafine therapy: 10 case reports and a review of the literature. Br J Dermatol 1998;138:529–32.
- Helfman RJ, Brickman M, Fahey J. Isotretinoin dermatitis simulating acute pityriasis rosea. Cutis 1984;33:297–300.
- Makdisi J, Amin B, Friedman A. Pityriasis rosea-like drug reaction to Asenapine. J Drugs Dermatol 2013;12:1050–1.
- Evangelia P, Ionnis P, Michael M, et al. Pityriasis rosea-like eruption associated with lamotrigine. J Am Acad Dermatol 2013;68:180–1.
- Bangash HK, Finch T, Petronic-Rosi V, et al. Pityriasis rosea-like drug eruption due to nortriptyline in a patient with vulvodynia. J Low Genit Tract Dis 2013;17:226–9.
- Lai YW, Chou CY, Shen WW, Lu ML. Pityriasis rosea-like eruption associated with clozapine: a case report. Gen Hosp Psychiatry 2012;34:e5–7.
- Aydogan K, Karadogan SK, Adim SB, Tunali S. Pityriasis rosea-like eruption due to ergotamine: a case report. J Dermatol 2005;32:407–9.
- Brazzelli V, Prestinari F, Roveda E, et al. Pityriasis rosea-like eruption during treatment with imatinib mesylate: description of 3 cases. J Am Acad Dermatol 2005;53:S240–51.
- Sezer E, Erkek E, Cetin E, Sahin S. Pityriasis rosea-like drug eruption related to rituximab treatment. J Dermatol 2013;40:495–6.
- Guarneri C, Polimeni G, Nunnari G. Pityriasis rosea during etanercept therapy. Eur Rev Med Pharmacol Sci 2009;13:383–7.
- Raipara SN, Ormerod AD, Gallaway L. Adalimumab-induced pityriasis rosea. J Eur Acad Dermatol Venereol 2007;21:1294–6.
- Durusoy C, Alpsoy E, Yilmaz E. Pityriasis rosea in a patient with Behcet's disease treated with interferon alpha 2A. J Dermatol 1999;26:225–8.
- Sezer E, Erkek E, Cetin E, Sahin S. Pityriasis rosea-like drug eruption related to rituximab treatment. J Dermatol 2013;40:495–6.
- Sasmaz S, Karabiber H, Boran C, et al. Pityriasis rosea-like eruption due to pneumococcal vaccine in a child with nephrotic syndrome. J Dermatol 2003;30:245–7.
- Requena L, Sanchez Yus E. Erythema nodosum. Dermatol Clin 2008;26:425–38.
- Garcia-Porrua C, Gonzalez-Gay M, Vazquez-Caruncho M, et al. Erythema nodosum. Etiologic and predictive factors in a defined population. Arthritis Rheum 2000;43:584–92.
- Psychos DN, VoulgarI PV, Skopouli FN, et al. Erythema nodosum: the underlying conditions. Clin Rheumatol 2000;19:212–16.
- Papagrigoraki A, Gisondi P, Rosina P, et al. Erythema nodosum: etiologic factors and relapses in a retrospective cohort study. Eur J Dermatol 2010;20:773–7.
- Kakourou T, Drosatou P, Psychou F, et al. Erythema nodosum in children: a prospective study J Am Acad Dermatol 2001;44:17–21.
- Halevy S, Gold I, Cohen A, et al. In vitro interferon-gamma release test in the diagnosis of drug-induced erythema nodosum. Isr Med Assoc J 2004;6:59–60.
- Tan BB, Leart JT, Smith AG. Acne fulminans and erythema nodosum during isotretinoin therapy responding to dapsone. Clin Exp Dermatol 1997;22:26–7.
- Yang SG, Han KH, Cho KH, Lee AY. Development of erythema nodosum in the course of oestrogen replacement therapy. Br J Dermatol 1997;137:319–20.
- Bhalla M, Thami GP, Singh N. Ciprofloxacin-induced erythema nodosum. Clin Exp Dermatol 2007;32:115–16.
- De Fonclare A Khosrotehrani K, Aractingi S, et al. Erythema nodosum-like eruption as a manifestation of azathioprine hypersensitivity in patients with inflammatory bowel disease. Arch Dermatol 2007;143:744–8.
- Bridges AJ, Granziano FM, Calhoun W, Reizner GT. Hyperpigmentation, neutrophilic alveolitis and erythema nodosum resulting from minocycline. J Am Acad Dermatol 1990;22:959–62.
- Longueville C, Doffoel-Hantz V, Hantz S, et al. Garadil-induced erythema nodosum. Rev Med Interne 2012;33:e17–18.
- Di Giusto Ca, Bernhard JD. Erythema nodosum provoked by hepatitis B vaccine. Lancet 1986;1:2;1042.
- George S, George B, Gorak E. Erythema nodosum secondary to granulocyte colony-stimulating factor in a patient with Hodgkin lymphoma during CD34+ cell mobilization for autologous peripheral blood stem cell transplantation: a dose-mediated effect. Biol Blood Marrow Transplant 2005;11:816–17.
- Jhaveri K, Halperin P, Shin SJ, Vahdat L. Erythema nodosum secondary to aromatase inhibitor use in breast cancer patients: case reports and review of the literature. Breast Cancer Res Treat 2007;106:315–18.
- Soon SL, Crawford RI. Recurrent erythema nodosum associated with Echinacea herbal therapy. J Am Acad Dermatol 2001;44:298–9.
- Plewig G, Jansen T. Acneiform dermatoses. Dermatology 1998;196:102–7.
- Du-Thanh A, Kluger N, Bensalleh H, Guillot B. Drug-induced acneiform eruption. Am J Clin Dermatol 2011;12:233–45.
- Momin SB, Peterson A, Del Rosso JQ. A status report on drug-associated acne and acneiform eruptions. J Drugs Dermatol 2010;9:627–36.
- Valeyrie-Allanore, Sassolas B, Roujeau JC. Drug-induced skin, nail and hair disorders. Drug Saf 2007;30:1011–30.
- Shibata M, Katsuyama M, Onodera T, et al. Glucocorticoids enhance toll-like receptor 2 expression in human keratinocytes stimulated with Propionibacteria acnes or proinflammatory cytokines. J Invest Dermatol 2009;129:375–82.
- Scott MJ, Scott AM. Effects of anabolic-androgenic steroids on the pilosebaceous unit. Cutis 1992;50:113–16.
- Melnik B, Jansen T, Grabbe S. Abuse of anabolic-androgenic steroids and bodybuilding acne: an underestimated health problem. J Dtsch Dermatol Ges 2007;5:110–17.
- Mahe E, Morelon E, Lechaton S, et al. Acne in recipients of renal transplantation treated with sirolumus: Clinical, microbiologic, histologic, therapeutic and pathogenic aspects. J Am Acad Dermatol 2006;55:139–42.
- Fung MA, Berger TG. A prospective study of acute-onset acne associated with administration of intravenous corticosteroids. Dermatology 2000;200:43–4.
- Brodell LA, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol 2013;40:865–70.
- Fyrand P, Fiskaadal HJ, Trygstad O. Acne in pubertal boys undergoing treatment with androgens. Acta Derm Venereol 1992;72:148–9.
- Traupe H, Von Mijlendahl, Bramswig J, et al. Acne of the fulminans type following testosterone therapy in three excessively tall boys. Arch Dermatol 1988;124:414–17.
- Heydenreich G. Testosterone and anabolic steroids and acne fulminans. Arch Dermatol 1989;125:571–2.
- Dupre A, Albarel N, Bonafe JL, et al. Vitamin B-12 induced acnes. Cutis 1979;24:210–11.
- Bencini PL, Montagnino G, Sala F, et al. Cutaneous lesions in 67 cyclosporin-treated renal transplant recipients. Dermatologica 1986;172:24–30.
- Vexiau P, Gourmel B, Castot A, et al. Severe acne due to chronic amineptine overdose. Arch Dermatol Res 1990;282:103–7.
- Sarantidis D, Waters B. A review and controlled study of cutaneous conditions associated with lithium carbonate. Br J Psychiatr 1983;143:42–50.
- Gunwald MH, Ben-dor D, Livni E, et al. Acne keloidalis-like lesions on the scalp associated with antiepileptic drugs. Int J Dermatol 1990;29:559–61.
- Holdiness MR. Adverse cutaneous reactions to antituberculosis drugs. Int J Dermatol 1985;24:280–5.
- Robert C, Soria JC, Spatz A, et al. Cutaneous side-effects of kinase inhibitors and blocking antibodies. Lancet Oncol 2005;6:491–500.
- Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: An update. J Am Acad Dermatol 2008;58:545–70.
- Herbst RS, LoRusso PM, Purdom M, et al. Dermatologic side effects associated with gefitinib therapy: Clinical experience and management. Clin Lung Cancer 2003;4:366–9.
- Lacouture ME. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer 2006;6:803–12.
- Martin JM, Jorda E, Monteagudo C, et al. Follicular acneiform eruption induced by imatinib. J Eur Acad Dermatol Venereol 2006;20:1368–70.
Drug-induced generalized exfoliative dermatitis
- Khaled A, Sellami A, Fazaa B, et al. Acquired erythroderma in adults: a clinical and prognostic study. J Eur Acad Dermatol Venereol 2010;24:781–8.
- Li J, Zheng HY. Erythroderma: a clinical and prognostic study. Dermatology 2012;225:154–62.
- Nicolis GD, Helwig EB. Exfoliative dermatitis. A clinicopathologic study of 135 patients. Arch Dermatol 1973 Dec;108(6):788–97.
- Sigurdsson V, Toonstra J, Hazemans-Boer M, et al. Erythroderma – a clinical and follow-up study of 102 patients with special emphasis on survival. J Am Acad Dermatol 1996;35:53–7.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol doi:10.1111/BJD.12501.
- Fiszenson-Albala F, Auzerie V, Mahe E, et al. A 6-month prospective survey of cutaneous drug reactions in a hospital setting. Br J Dermatol 2003;149:1018–22.
- Lee HY, Tay LK, Thirumoorthy T, Pang SM. Cutaneous adverse drug reactions in hospitalized patients. Singapore Med J 2010;51:767–74.
- Fierro MT, Comessatti A, Quaglino P, et al. Expression pattern of chemokine receptors and chemokine release in inflammatory erythroderma and Sezary syndrome. Dermatology 2006;213:284–92.
- Barbaud AM, Bene MC, Reichert-Penetrat S, et al. Immunocompetent cells and adhesion molecules in 14 cases of cutaneous drug reactions induced with the use of antibiotics. Arch Dermatol 1998;134:1040–1.
- Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA 2005;102:4134–9.
- McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med 2011;364:1134–43.
- Walsh NMG, Prokopetz R, Tron VA, et al. Histopathology in erythroderma: review of a series of cases by multiple observers. J Cutan Pathol 1994;419–23.
- Zip C, Murray S, Walsh NMG. The specificity of histopathology in erythroderma. J Cutan Pathol 1993;20:393–8.
- Pal S, Haroon TS. Erythroderma: a clinic-etiologic study of 90 cases. Int J Dermatol 1998;37:104–7.
- Akhyani M, Ghodsi ZS, Toosi S, Dabbaghian H. Erythroderma: A clinical study of 97 cases. BMC Dermatol 2005;5:5.
- Hasan T, Jansen CT. Erythroderma: a follow-up of fifty cases. J Am Acad Dermatol 1983;8:836–40.
- Barbaud A, Goncalo M, Bruynzeel D, Bircher A. Guidelines for performing skin tests with drugs in the investigation of cutaneous adverse drug reactions. Contact Dermatitis 2001;45:321–8.
- Rym BM, Mourad M, Bechir Z, et al. Erythroderma in adults: a report of 80 cases. Int J Dermatol 2005;44:731–5.
- Sehgai VN, Srivastava. Exfoliative dermatitis: A prospective study of 80 patients. Dermatologica 1986;173:278–84.
- Zhang JC, Sun YT. Efavirenz-induced exfoliative dermatitis. Scand J Infect Dis 2013;45:70–2.
- Mahe E, Descamps V, Baikian B, et al. Acitretin-induced erythroderma in a psoriatic patient. J Eur Acad Dermatol Venereol 2006;20:1133–4.
- Cockayne SE, Glet RJ, Gawkrodger DJ, McDonagh AJG. Severe erythrodermic reactions to the proton pump inhibitors and lansoprazole. Br J Dermatol 1999;141:173–4.
- Reynolds NJ, Jonmes SK, Crossley J, Harman RRM. Exfoliative dermatitis due to Nifedipine. Br J Dermatol 1989;121:401–4.
- Vano-Galvan S, Fernandez-Guarino MM, Henriquez-Santana A, De Las Heras E. Imatinib-induced erythroderma mediated by an unusual non-dose-dependent mechanism. Eur J Dermatol 2007;17:538–9.
- Gallelli L, Ferraro M, Mauro GF, et al. Generalized exfoliative dermatitis induced by interferon alfa. Ann Pharmacother 2004;38:2173–4.
- Yuan XY, Guo JY, Dang YP, et al. Erythroderma: a clinical-etiological study of 82 cases. Eur J Dermatol 2010;20:373–7.
- Horiuchi Y. Propolis induced erythroderma. J Dermatol 2001;28:580–1.