CHAPTER 125
Cutaneous Reactions to Cold and Heat
Saqib J. Bashir1 and Ai-Lean Chew2
1King's College Hospital, London, UK
2Guy's and St Thomas' NHS Foundation Trust, London; and King's College Hospital, London, UK
PHYSIOLOGICAL REACTIONS TO COLD
The maintenance of a steady core body temperature is achieved by various thermoregulatory mechanisms, not least of which is control of the skin's blood flow. Exposure to cold causes constriction of the arterioles and veins by a direct mechanism mediated in part by endothelial synthesis of the vasoconstrictor peptide endothelin-1 [1]. A reflex increase in sympathetic tone is triggered by cold receptors in the skin and, if the blood temperature falls, by the hypothalamic heat-regulating centre. Heat conservation is further enhanced by a counter-current exchange system between the arteries and veins in the limbs. Cold-induced vasoconstriction causes shunting of blood to the deep venous system which allows heat to be transferred from the arteries to veins. Consequently, arterial blood passing into the limbs is cooler, venous blood returning to the body is warmer and less heat is lost to the outside environment.
Persistent exposure of fingers to cold leads to the ‘hunting reaction’ of Lewis in which there are repeated cycles of vasodilatation following periods of vasoconstriction [2]. Cold-induced vasoconstriction is a heat-preserving, protective mechanism but prolonged vasospasm may jeopardize the vitality of the skin. Therefore, a transient vasodilatory response, mediated by the opening of arteriovenous anastamoses, protects against skin necrosis. With continued cold exposure, there is a phasic increase and decrease in blood flow through the cutaneous microvasculature. However, when core temperature is under threat the hunting reaction stops and vasoconstriction persists [2].
Cold-induced vasoconstriction causes a rise in intracapillary pressure and increased filtration of fluid into the interstitium, resulting in haemoconcentration and a reduction in plasma volume. Other physiological effects of cold include increased blood viscosity, slowing of the dissociation of oxyhaemoglobin to haemoglobin, diminished conduction velocity in cutaneous nerves and changes in platelet adhesiveness [3].
DISEASES CAUSED OR AGGRAVATED BY COLD
The ambient temperature and duration of exposure will determine the type and degree of injury sustained by all people when exposed to severe cold. However, there is a variable endogenous susceptibility to cold; certain individuals suffer cold-related disorders on exposure to modest degrees of cold that would be tolerated without ill effect by other normal individuals. Hence, cold-induced diseases can be divided into two groups: (i) diseases of cold exposure; and (ii) diseases of abnormal susceptibility to cold (Box 125.1).
Frostbite
Definition and nomenclature
Frostbite is the term used to describe tissue damage caused by freezing. Skin and subcutaneous tissues are at risk of frostbite when exposed to cold air, liquids or metals.
Introduction and general description
Frostbite, and its precursor, frostnip, can lead to varying degrees of tissue damage, ranging from erythema to necrosis of soft tissue, muscle and bone. Most cases of frostbite are seen in winter sports enthusiasts and climbers who have been stranded in exposed sites in cold weather. Frostbite also occurs in soldiers, homeless people and those who work outdoors in cold climates.
Pathophysiology
Predisposing factors
The risk of frostbite increases with alcohol use and smoking [1].
Pathology
Freeze-induced damage results largely from the formation of ice in both the intracellular and extracellular compartments [2]. Fast freezing tends to produce intracellular ice, while slow freezing causes the formation of extracellular ice. Ice crystals not only injure cellular architecture but also disturb the flux of electrolytes and water across cell membranes [3]. The degree of cryodamage is also influenced by the rate of thawing. Slow rewarming causes the formation of larger, more destructive ice crystals and the development of greater osmotic stresses. As well as the direct effects on cells, tissue damage in frostbite is compounded by cold-induced vascular and haematological responses. Reflex vasoconstriction in the extremities results in decreased capillary perfusion, which is aggravated by cold-induced hyperviscosity and a tendency to thrombus formation [4].
Clinical features
The parts of the body that can be least protected from cold are affected – the toes, feet, fingers, ears, nose and cheeks. Frostnip involves the skin only and is characterized by painful erythema, which normalizes with rewarming. In superficial frostbite there is involvement of the skin and subcutis with erythema accompanied initially by pain and then a sense of warmth. The affected skin becomes waxy and white. The injury in deep frostbite extends to the subcutaneous tissues and may involve the nerves, major vessels, muscle and bone, resulting in joint immobility and paralysis [5]. The cold exposure is fairly evident at presentation, but the degree of damage may take many weeks to become apparent.
Classification of severity
A method of classifying frostbite severity has been proposed. In first-degree frostbite there is partial skin freezing, with erythema, oedema and hyperaemia, but no blisters or necrosis. Desquamation develops 5–10 days later, leading to complete recovery. Second-degree frostbite is a full-thicknesss skin freeze, with erythema, substantial oedema, vesiculation, blistering and a black eschar, requiring soft-tissue amputation only. Third-degree frostbite requires bone amputation. Fourth-degree frostbite necessitates major amputation and is complicated by systemic effects [6].
Complications and co-morbidities
The extent and severity of tissue damage become apparent on rewarming. Erythema and mild pain lasting for a few hours occur following mild frostbite. Blistering, full-thickness skin necrosis and gangrene can be seen in severe cases [7]. Damage to nerves and blood vessels can lead to parasthesiae, abnormal sensitivity to cold and hyperhidrosis, which may last for months to years [7].
Prognosis
The prognosis of frostbite injury only becomes apparent over a period of weeks, as the demarcation between viable and non-viable tissue becomes evident.
Investigations
Technetium-99 bone scintigraphy may be helpful in evaluating outcome in frostbite injury and indicating the level of amputation needed in severe cases [8]. Magnetic resonance angiography may also be beneficial, with the ability to visualize directly any vascular occlusion, and simultaneously image the surrounding soft tissues.
Management
First line
It is important that immediate warming techniques are instituted. Debridement surgery should be delayed until the full extent of the recovery/persisting damage is clear.
Rapid rewarming by immersion in water at 37–39°C is recommended. Use a thermometer to measure water temperature, or a caregiver's hand, to ensure that an additional thermal burn injury is not imparted. As the water will cool rapidly, it will need to be topped up to maintain the temperature, and circulated around the limb. Rapid rewarming has been shown to be more effective than slow rewarming and the tissue can be considered completely rewarmed when it takes on a red/purple appearance and is soft and pliable to the touch. Further additional rewarming does not add additional benefit. The area should be air dried or patted dry to avoid shearing forces damaging tissue [9]. Other authors have advocated a higher temperature rewarming for at least 30 min at 40–41°C until tissues are soft and pliable, with continuing twice daily warming.
Non-steroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen have been recommended although the evidence for their use is weak. The Wilderness Medicine Society recommends their use at 12 mg/kg body weight twice daily in order to reduce levels of prostaglandins and thromboxanes that may contribute to vasoconstriction, ischaemia and tissue damage. Also, NSAIDs may help alleviate the pain of rewarming. Topical aloe vera gel has been suggested on the basis that it may also reduce inflammatory prostaglandins; however there is a lack of strong clinical evidence or animal studies to confirm efficacy. It is recommended that non-haemorrhagic blisters should be aspirated as the blister fluid may also contain prostaglandins and thromboxanes. The blister roof should be kept intact and not debrided.
Second line
In the hospital setting, other pharmacological interventions can be beneficial. A retrospective study has indicated a reduction in the need for digital amputation if intra-arterial infusion of tissue plasminogen activator (tPA) is given within 48 h of injury [10]. In this study, the incidence of amputation reduced from 41% (untreated) to 10% in those treated within 24 h of injury. Vasodilator drugs may also improve outcome [11] – in addition to vasodilatation, they may also inhibit microvascular occlusion and platelet aggregation. In a study of aspirin with buflomedil, versus aspirin plus ilprost, or aspirin plus iloprost and tPA, no amputations were seen in the iloprost group (0/16) versus 3/16 patients when combined with tPA or 9/15 patients treated with buflomedil. The authors recommend aspirin with iloprost for third-degree frostbite, with consideration of tPA on a case-by-case basis for fouth-degree frostbite [12].
Third line
Surgical removal of gangrenous tissue should be delayed until there is a distinct demarcation between viable and non-viable tissue, a process that usually takes several weeks.
Resources
Further information
McIntosh SE, Hamonko M, Freer L et al. Wilderness Medical Society Practice Guidelines for the Prevention and Treatment of Frostbite, http://wemjournal.org/article/S1080-6032(11)00077-9/fulltext (last accessed June 215).
Trench foot
Definition and nomenclature
Trench foot describes a combination of stasis, non-freezing cold injury and wet conditions leading to numbness and skin changes on the distal legs and feet.
Introduction and general description
Trench foot and immersion foot are regarded as similar processes. Trench foot was a common problem suffered by soldiers in the early years of trench warfare in the First World War [1] but is now mostly encountered among the homeless population [2]. A similar entity, tropical immersion foot, is a warm water immersion injury and is less severe.
Pathophysiology
Predisposing factors
Smoking and peripheral vascular disease probably contribute to the severity of the tissue damage.
Pathology
The pathological changes are those of dependent oedema and lymphovenous stasis. Perivascular inflammation occurs and in severe cases arterial occlusion and ischaemic necrosis. There is damage to both myelinated and unmyelinated nerve fibres [3].
Clinical features
Affected individuals develop numbness of the feet and distal legs accompanied by skin changes caused by non-freezing cold injury. This is compounded by wet conditions and aggravated by leg dependency, immobility and constrictive footwear.
Affected legs and feet are cold and anaesthetic. There is erythema, oedema, tenderness and, in severe cases, areas of superficial gangrene. On rewarming there is worsening oedema, hyperaemia and painful parasthesiae.
Differential diagnosis
Other causes of limb ulceration should be considered, such as diabetes, arterial and venous insufficiency, autoimmune disease, inflammatory disease such as pyoderma gangrenosum and infections.
Prognosis
Initially the ability to sweat is lost but with time there is hyperhidrosis, cold sensitivity and vasomotor instability. Changes may persist for months or years.
Investigations
The investigations required depend on the clinical presentation. Usually there is a clear history of cold water immersions and no specific investigations are required.
Management
The best approach is prevention, but once the condition has occurred bed rest, elevation and analgesics are helpful. Conservative excision of necrotic tissue may be necessary and antibiotics should be given if there is evidence of infection.
Perniosis
Definition and nomenclature
Perniosis describes localized inflammatory lesions on the acral skin, which occur as an abnormal reaction to cold in susceptible individuals [1].
Introduction and general description
Perniosis lesions are cold induced and affect areas of the skin vulnerable to cold exposure, such as the digits, nose and ears.
Epidemiology
Age and sex
There is a female preponderance (79%) [2].
Associated diseases
Aetiological factors include poor nutrition, anorexia nervosa and systemic diseases, most typically lupus erythematosus (LE) and haematological malignancy [3, 4, 5].
Pathophysiology
In perniosis there is a persistent cold-induced vasoconstriction of the deep cutaneous arterioles with concomitant dilatation of the smaller, superficial vessels. This is in contrast with normal subjects, in whom cold exposure induces cutaneous vasoconstriction succeeded by vasodilatation, a homeostatic mechanism necessary for the maintenance of reperfusion. Investigation of the cutaneous nerves in patients with perniosis demonstrated no quantitative or qualitative difference in immunoreactivity for substance P, neuropeptide Y, calcitonin gene-related peptide or vasoactive intestinal peptide compared with controls [6]. However, in the affected skin of patients with acral perniosis, who also had a past history of very low body weight, immunohistochemistry revealed a great increase in nerve bundles in the papillary dermis, some with an abnormal morphology [6]. This indicates that in uncomplicated perniosis the neuronal supply to the microvasculature is normal and suggests that the pathology involves the microvessels themselves.
Pathology
The histopathology of perniosis usually demonstrates dermal oedema with superficial and deep dermal inflammation [7]. The mononuclear cell infiltration, mostly T cells, is primarily perivascular but also occurs in a peri-eccrine distribution [8]. Epidermal changes in chilblains consist mainly of necrotic keratinocytes and spongiosis. Perniosis occurring on the lateral thigh is characterized by an intense mononuclear cell infiltrate extending throughout the dermis and into the subcutaneous fat, with ‘fluffy oedema’ of the blood vessel walls [7]. In these cases of deep perniosis, dermal oedema is not a prominent feature. Inflammation occurring in the deeper dermis and subcutis may be explained by the combined effects of external cooling together with insulation from internal warming [7].
Genetics
There may be a genetic influence in perniosis since several generations within a family can be affected [1].
Environmental factors
Perniosis occurs in susceptible individuals during the autumn or winter in a climate that is both cold and damp.
Clinical features
The lesions are red-purple and usually macular, papular or nodular. Plaque-like chilblains also occur, as do lesions with an annular morphology. Perniosis develops symmetrically on the acral skin, in particular the fingers and toes, but other body extremities may also be involved including the heels, lower legs, nose and ears. Pruritus and burning pain are common, although some lesions may be asymptomatic. In severe cases blistering and ulceration may occur. Each lesion tends to undergo spontaneous resolution after 2–3 weeks; however, in some patients chilblains persist throughout the winter and occasionally continue to develop in the summer.
Chilblain LE (also known as Hutchinson lupus) is a form of cutaneous LE that presents in a similar way to idiopathic perniosis. Erythrocyanotic papules, located on the fingers and toes, develop in cold weather and tend to persist, in some cases becoming ulcerated [9]. Chilblain LE may be accompanied by discoid LE or other forms of cutaneous LE. Up to 20% of patients with chilblain LE develop systemic LE [10].
Perniosis may also complicate haematological malignancy, typically myelodysplastic syndrome and chronic myelomonocytic leukaemia (see Chapter 148). Cyanotic swelling of acral digital skin, particularly the toes, has been reported in these patients [11]. The onset of perniosis may coincide with a blast crisis, which can be demonstrated on skin biopsy by the presence of large, atypical mononuclear cells in the perivascular infiltrate [12].
Clinical variants
There are various less common variants. In papular perniosis, crops of small chilblains develop on the sides of the fingers often on a background of acrocyanosis [13]. Perniosis occurring on the outer thighs has been reported in women who are horse-riding enthusiasts (‘equestrian panniculitis’) [9]. Clinically, these lesions are composed of clustered papules or plaques, which may ulcerate. Tight-fitting trousers, such as riding breeches, have been implicated aetiologically [9]. In chronic perniosis, especially in the presence of arterial disease or prolonged cold exposure, irreversible changes of fibrosis, lymphoedema and hyperkeratosis may occur, altering the physical signs.
Differential diagnosis
This includes granuloma annulare, peripheral vascular insufficiency and vasculitis.
Prognosis
The prognosis depends on the underlying cause. Most patients will experience good disease control when steps are taken to prevent cold exposure.
Investigations
Investigations should be guided by the clinical presentation of the patient but may include a full blood count and/or blood film, protein electrophoresis, an autoimmune screen, cryoglobulins and cold agglutinins. Investigations were positive in 11/20 tested patients from a Mayo Clinic series [2].
Management
Warm clothing and central heating generally prevent perniosis. Susceptible individuals must avoid cold, damp conditions and should not smoke. A double-blind, placebo-controlled study demonstrated the efficacy of nifedipine in clearing existing chilblains and preventing the development of new lesions [14]. A study has demonstrated the successful treatment of chilblain LE with mycophenolate mofetil [15].
Acrocyanosis
Definition
Acrocyanosis is a persistent cyanotic or erythrocyanotic mottled discoloration of the hands and, less commonly, feet and face.
Introduction and general description
Acrocyanosis may be idiopathic or secondary to a number of systemic disorders, including an underlying malignancy (Box 125.2) [1]. Sometimes there is a family history, indicating a genetic basis. Rarely, it is drug induced.
Epidemiology
Age and sex
Presentation is typically in adolescence, with a reported female preponderance [2].
Pathophysiology
There is vasospasm of peripheral arterioles, aggravated by cold, and dilatation of the subpapillary venous plexus [3]. The condition is most probably a primary vascular defect since studies have not demonstrated a deficit of neuronal supply to the cutaneous vessels [4]. Decreased acral blood flow may be further compromised by plasma hyperviscosity. In ethylmalonic encephalopathy – a rare metabolic disorder with neuromotor delay, hyperlactic acidaemia and orthostatic acrocyanosis – a mutation has been demonstrated in ETHE1, a gene encoding a mitochondrial matrix protein [5].
Clinical features
Idiopathic acrocyanosis usually starts in adolescence and persists into adult life. The changes may be transient after cold exposure but usually persist during the winter and even throughout the summer months. Clinically there is a painless mottled duskiness of both hands in the presence of normal peripheral pulses. Similar changes may be seen on the feet and face. Trophic changes, such as ulceration, are absent. In some individuals acrocyanosis spontaneously remits. Acrocyanosis must be distinguished from the Raynaud phenomenon, which occurs episodically with triphasic colour changes and often involves just a few digits. Arterial and venous occlusion must be excluded. In cases of acrocyanosis developing for the first time in adult life a secondary cause should be sought (Box 125.2).
Differential diagnosis
This includes Raynaud phenomenon, arterial occlusion and venous occlusion.
Prognosis
The prognosis varies with the underlying disorder. Primary acrocyanosis in young adults may disappear spontaneously by middle age.
Investigations
Capillaroscopy has been used to demonstrate dilated and congested capillary loops at the base of the fingernail. However, the usefulness of this technique in distinguishing primary acrocyanosis from connective tissue disease has yet to be firmly established. Duplex vascular ultrasound can be used to exclude intravascular thrombosis.
Management
There is no effective medical treatment for acrocyanosis. Vasodilator therapies, such as the calcium-channel antagonists, do not appear to be beneficial. Drug-induced acrocyanosis will be improved by cessation of the culprit drug. Treatment of an underlying systemic disorder may improve the appearance in secondary acrocyanosis.
Erythrocyanosis
Definition
Erythrocyanosis is a persistent, dusky erythema occurring at sites with a thick layer of underlying subcutaneous fat, such as the thighs and lower legs. Erythrocyanosis is seen less commonly on the buttocks and arms.
Introduction and general description
Erythrocyanosis is a condition that predominately affects the lower legs, distinguishing it from acrocyanosis, which affects peripheral areas such as digits and appendages [1].
Epidemiology
Age and sex
Erythrocyanosis occurs most commonly in adolescent girls and middle-aged women.
Pathophysiology
It has been hypothesized that the subcutaneous fat in these sites acts to insulate the superficial vessels from the warmth of the underlying blood supply, thus rendering them susceptible to cold exposure. It is exacerbated by cold and therefore usually more prominent during the winter.
Clinical features
Erythrocyanosis is seen on the lower legs of adolescent girls, the thighs and buttocks of overweight boys and the thighs and lower legs of middle-aged women. Very occasionally it can occur on the forearms of infants. It is characterized by dusky discoloration of the skin and may be accompanied by keratosis pilaris, angiokeratomas and telangiectases. The area is cold to touch. Nodular perniotic lesions occurring after cold exposure may complicate erythrocyanosis. Oedema and fibrosis may be seen as chronic manifestations of erythrocyanosis.
Differential diagnosis
Whilst the diagnosis is often clinically apparent, other vascular disorders and livedo reticularis may be considered in the differential.
Prognosis
The disorder may persist indefinitely but spontaneous improvement can occur in adolescent patients.
Investigations
None are required.
Management
Warm clothing, exercise, weight reduction and elastic support hosiery may be helpful. Vasodilators, such as calcium-channel antagonists, are of limited value.
Livedo reticularis
Definition and nomenclature
Livedo reticularis is a mottled, cyanotic discoloration of the skin, which has a characteristic network pattern. It is accentuated by cold.
Introduction and general description
Livedo reticularis is a lace-like pattern on the surface of the skin created by low blood flow within anastomoic areas of the skin.
Pathophysiology
The blood supply of the skin is arranged in cones, the bases of which measure 1–4 cm in diameter and lie on the skin's surface [1]. Each cone is supplied by an arteriole, which passes through the dermis perpendicular to the surface. When blood flow through the feeding arterioles is diminished, deoxygenated blood at the anastamotic junctions produces a cyanotic network pattern on the skin which is livedo reticularis (Figure 125.1). Livedo reticularis may be physiological, idiopathic or secondary to intravascular obstruction or vessel wall disease (Box 125.3).
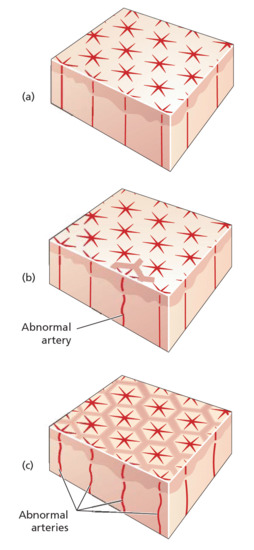
Figure 125.1 (a) Normal vasculature. (b) Livedo racemosa due to patchy arterial pathology. (c) Livedo reticularis due to diffuse arterial pathology .
Pathology
There is hyperkeratosis, red blood cell aggregates and vessel wall thickening in the deep dermis. If the livedo reticularis is caused by vasculitis, there will be vascular inflammation and arterial obliteration in the deep dermis and the subcutaneous tissue. There is no difference in the histological features between the blanched and erythematous areas [2].
Clinical features
Livedo reticularis most commonly occurs on the legs but the arms and trunk may also be affected. Cold usually exacerbates the cyanotic discoloration, while leg elevation tends to decrease the intensity of the colour changes. Diffuse arterial disease or hyperviscosity problems give rise to diffuse livedo reticularis; limited arterial disease leads to patchy mottling. In many cases livedo reticularis forms a complete network, in other cases there is a branching configuration known as livedo racemosa. Ulceration of the dark areas occurs rarely but, if present, suggests significant large-vessel vasculitis or intraluminal thrombosis. The appearance of livedo reticularis is initially reversible if the underlying cause is treated, but with chronic problems permanent telangiectases develop.
Clinical variants
Physiological livedo reticularis. Also known as cutis marmorata, this is a transient cyanotic mottling of the skin that occurs as a physiological response to cold exposure and disappears with warming. It is usually encountered in healthy infants and resolves during the first year of life. Involvement of the trunk as well as the limbs is common. Physiological livedo reticularis rarely occurs in adults, but in this situation is often associated with a disorder that causes stasis within blood vessels, for example paralysis.
Congenital livedo reticularis. Also known as cutis marmorata telangiectatica congenita, this is a rare developmental defect, present at birth, characterized by a red-purple vascular network [3, 4, 5]. Lesions are usually asymmetrical, typically on a limb, less often involving the torso or head. The reticulated area is persistent and enhanced by cold, crying and exercise. Skin atrophy may accompany the livedo. Usually congenital livedo reticularis occurs in isolation but it may be associated with a variety of other congenital abnormalities. In most cases the condition gradually resolves, with most improvement occurring during the first 2 years of life.
Acquired idiopathic livedo reticularis. This occurs predominantly in young adult and middle-aged women. Mild degrees are harmless, while more severe cases are associated with ulceration, usually in the winter. The mottling is at first transient and occurs on exposure to cold. Permanent livedo may develop with time. Tingling and numbness of the skin, and sometimes oedema, may be present. The diagnosis is clinical and can only be made once other disorders have been ruled out, including erythema ab igne, capillary naevi and angioma serpiginosum, as well as the causes of secondary livedo reticularis. Livedo reticularis occurs in 20–25% of migraine sufferers and in this subset stroke is more frequent, raising the possibility that livedo reticularis can be used as a clinical marker to identify those migraine sufferers with an increased risk of stroke [6].
Differential diagnosis
This includes erythema ab igne, capillary naevi and angioma serpiginosum.
Prognosis
Prognosis is related to the underlying cause, if one is present.
Investigations
Laboratory studies should be directed by the underlying medical presentation. Typical investigations would include a thrombophilia screen, autoimmune screen, full blood count, metabolic panel and serum protein electrophoresis. Further investigations such as ultrasound and echocardiography may be required to look for sources of emboli.
Skin biopsies should be performed from both the central blanched areas and the purplish livedo areas to increase the diagnostic yield as changes may be present in either or both areas.
Management
The management is directed to the underlying condition, if one is identified.
Raynaud phenomenon
Definition
The Raynaud phenomenon is defined as episodic digital ischaemia occurring in response to cold or emotional stimuli.
Introduction and general description
The Raynaud phenomenon is characterized by sequential colour changes: white (pallor), blue (cyanosis) and red (rubor). Pallor is essential for the diagnosis. However, in severe recalcitrant Raynaud phenomenon, particularly in association with a connective tissue disease, attacks of long duration may occur in which the initial pallor is short lived and succeeded by prolonged cyanosis. Primary Raynaud phenomenon (also called Raynaud disease) is idiopathic and occurs as an isolated innocuous disorder (Box 125.4). Secondary Raynaud phenomenon occurs in association with underlying diseases, or is caused by physical factors or drugs (Box 125.5).
The sequential colour changes were first described by Maurice Raynaud in 1862 [1]. In 1901, Jonathan Hutchinson reported that there were several causes for the phenomenon [2] and, subsequently, Allen and Brown established clinical criteria to distinguish innocent primary Raynaud phenomenon from secondary Raynaud phenomenon [3]. The advent of immunological tests for the connective tissue diseases has helped refine the diagnosis of primary Raynaud phenomenon (Box 125.4).
Epidemiology
Incidence and prevalence
Studies of prevalence in Raynaud phenomenon have been hampered by differences in diagnostic criteria and survey technique. Population-based surveys have reported a prevalence of between 6% and 20% in women, and between 3% and 12.5% in men [4]. There is some evidence for an increased prevalence of Raynaud phenomenon in family members of affected individuals, suggesting a genetic susceptibility [5]. A genome linkage study of affected individuals within a family indicated five candidate regions with possible linkage, of which three were potential candidate genes: the beta subunit of the muscle acetylcholine receptor and the serotonin 1B and 1E receptors [6].
Age
The age of onset is usually under 40 years, but it may occur over this age.
Sex
Primary Raynaud phenomenon is commoner in women, in the proportion of at least 5 : 1 [7].
Pathophysiology
The pathogenesis of Raynaud phenomenon is centred on a functional unit composed of vascular endothelium, smooth muscle cells and nerve endings. This integrated neurovascular system responds to a range of soluble mediators and physical stimuli, which determine the balance between vasoconstriction and vasodilatation. In primary Raynaud phenomenon the vascular changes are considered to be functional. In contrast, in secondary Raynaud phenomenon there are structural vascular changes, most clearly delineated in patients with systemic sclerosis. Severe intimal hyperplasia consisting of collagen deposits is often associated with intravascular thrombi, which can completely occlude the lumen [8]. In systemic sclerosis there is also distorted nail fold capillary architecture with dilated loops and areas of vessel drop-out.
Aberrant expression of endogenous vasodilatory substances (nitric oxide, prostacyclin, prostaglandin and leukotrienes) and vasoconstrictors (endothelin, angiotensin II and thromboxane A2) has been implicated in the pathogenesis of the Raynaud phenomenon [9]. A disturbance in vascular homeostasis may lead to uncontrolled vasoconstriction and studies have demonstrated down-regulation of nitric oxide and up-regulation of endothelin-1 in Raynaud phenomenon patients [10, 11]. Investigation into the innervation pathways of vascular smooth muscle have highlighted a prominent role for α2c-adrenoreceptors in cold-induced vasoconstriction [12]. Studies of Raynaud phenomenon patients have demonstrated that, compared to normal controls, the increased contractile response to cold and α2-adrenergic agonists is associated with increased activity of protein tyrosine kinase and tyrosine phosphorylation in vascular smooth muscle [13].
Clinical features
The Raynaud phenomenon affects the hands and, less often, the feet; changes elsewhere are exceptional, although the tongue can be involved [14].
A typical attack consists of sudden pallor of one or more digits, followed after a few minutes by cyanosis or sometimes by erythema. In primary Raynaud phenomenon the condition is usually symmetrical and involves several digits. In secondary Raynaud phenomenon only one or a few digits are affected and asymmetry is not unusual. Attacks are usually precipitated by cold, either local or of the whole body, or by psychological stimuli. Episodes may occur infrequently or many times each day. Severe cases, which are usually of the secondary type, may be complicated by telangiectases of the nail fold, thinning and ridging of the nail, and atrophy or sclerosis of the fingers (sclerodactyly). Skin necrosis is extremely rare in primary Raynaud phenomenon but not uncommon in secondary Raynaud phenomenon and may result in destruction of the digits.
The disease tends to run a variable course. In primary Raynaud phenomenon the outcome is good in 80% of cases, but some disability occurs in 20%. In secondary Raynaud phenomenon the prognosis is that of the underlying disease.
Differential diagnosis
This includes acrocyanosis, hand–arm vibration syndrome, heavy metal intoxication, ergot intoxication, thoracic outlet syndrome and cervical rib, and Buerger disease and other arterial diseases.
Prognosis
Prognosis varies with the underlying disease, if one is present.
Investigations
Investigations are directed towards detecting an underlying cause for the Raynaud phenomenon. A variety of vascular imaging studies have been employed but are not specifically useful in clinical diagnosis.
Management
Conservative management includes taking measures to keep the hands and feet warm and reducing cold exposure and also emotional stress. Hand and feet warmers may be useful. With drug treatment, the clinician must balance the beneficial effects versus drug-related adverse effects [15].
First line
Calcium-channel antagonists can be useful in decreasing the frequency, duration and severity of attacks. A meta-analysis of 18 randomized, placebo-controlled, double-blinded trials assessed the efficacy of calcium-channel blockers against placebo in patients with primary Raynaud phenomenon and found a decrease in attacks and reduction in symptom severity [16]. Recommended doses of nifedipine range from 30 to 180 mg daily and for amlodipine between 5 and 20 mg daily. Slow release or long-acting preparations are recommended to improve compliance and reduce side effects; nonetheless discontinuation occurs in approximately 15% of subjects because of headaches and leg oedema [17].
Second line
In a double-blind, placebo-controlled study of 16 patients with secondary Raynaud phenomenon, sildenafil 50 mg twice daily demonstrated significant improvement in mean attack rates and duration [18]. Sildenafil is a phosphodiesterase inhibitor and acts by increasing the vasodilatory effect of both nitric oxide and prostacyclin. A variety of newer phosphodiesterase inhibitors are being investigated that may increase perfusion but with variable efficacy on duration and severity of attacks.
Third line
Intravenous infusion of vasodilatory prostaglandins can reverse ischaemic complications in Raynaud phenomenon. Iloprost, a prostacyclin analogue, is commonly administered to patients with severe digital ulceration. In a randomized, placebo-controlled, double-blind study of 131 patients with systemic sclerosis, the mean weekly number of Raynaud phenomenon attacks significantly decreased on iloprost compared with placebo [19]. Repeated treatment with iloprost over 1 year was found to be more effective than nifedipine in reducing the severity score of Raynaud phenomenon in patients with systemic sclerosis [20].
A study of bosentan, an endothelin receptor antagonist, demonstrated a marked reduction in new digital ulcers in systemic sclerosis patients but did not decrease the frequency or severity of Raynaud's attacks [21]. A randomized, controlled trial demonstrated that the angiotensin II receptor antagonist losartan significantly reduced the frequency and severity of vasoconstrictive episodes in patients with primary Raynaud phenomenon and in Raynaud phenomenon secondary to systemic sclerosis [22].
Topical glyceryl trinitrate, a nitric oxide donor, significantly reduced the number and severity of Raynaud's attacks in individuals with primary Raynaud phenomenon and secondary Raynaud phenomenon compared with placebo [23]. Glyceryl trinitrate 2% was associated with headaches whereas glyceryl trinitrate 1% reduced the incidence of side effects but maintained a similar improvement in Raynaud phenomenon symptoms [23]. Botulinum toxin A, injected into the hand, can also cause vasodilatation; its role in the treatment of Raynaud phenomenon has yet to be established [24].
Cryoglobulinaemia
Definition
Cryoglobulinaemia refers to the presence of immunoglobulin complexes that precipitate in vitro when cooled below body temperature [1] (see also Chapters 101 and 102).
Introduction and general description
Cutaneous features in cryoglobulinaemia occur as a consequence of intravascular precipitation of cryoglobulins in the small vessels of the skin or as an immune complex disease (see Chapter 102). In the demonstration of cryoglobulins, venous blood is drawn into a warm syringe and allowed to clot at 37°C. The serum (or plasma if cryofibrinogen is suspected) is cooled to 4–5°C and any precipitate noted. This should redissolve on warming. The amounts of cryoglobulin reported to cause symptoms are very variable: less than 25 mg/dL may rarely be associated with symptoms, however much higher levels may be symptomless. Levels as high as 80 g/L have been recorded.
Epidemiology
Incidence and prevalence
There are few epidemiological data on the prevalence of cryoglobulinaemia. However, a population study of a small town in Italy, Origgio, demonstrated a prevalence of mixed cryoglobulinaema affecting 8.5/10 000 in the general population, or 26/10 000 in those aged over 50 years. In this study, the main underlying association was hepatitis C [2].
Age
The mean age of onset of symptoms in mixed cryoglobulinaemia is reported as being 53 years, with a mean age at diagnosis of 56 years [3].
Pathophysiology
Type I cryoglobulins are single monoclonal immunoglobulins usually associated with haematological disorders, such as multiple myeloma, macroglobulinaemia and lymphoma. These precipitate in blood vessels, leading to ischaemia from vascular occlusion.
Type II mixed cryoglobulins are composed of a monoclonal component (usually IgMκ) with rheumatoid factor activity against polyclonal IgG. In type III mixed cryoglobulins, all the components are polyclonal [3]. Mixed cryoglobulinaemia is most commonly associated with hepatitis C virus (HCV) infection and is mainly of type III [4]. Other infections are also implicated in mixed cryoglobulinaemia, including hepatitis B and HIV, and cases are also seen in conjunction with autoimmune diseases such as Sjögren syndrome, systemic LE and rheumatoid arthritis [5]. Mixed cryoglobulinaemia causes a systemic vasculitis with multiorgan involvement, mainly of the skin, joints, kidneys and peripheral nerves.
Clinical features
The most usual skin manifestation is purpura on the lower legs, which may develop after cooling of the extremities. In cryoglobulinaemia of all types, other skin signs are livedo reticularis, Raynaud phenomenon, atypical ulceration of the legs, digital skin necrosis and cold urticaria [6].
Differential diagnosis
Other causes of vascular occlusion such as hypercoagulable states, embolic disease and vasculitides need to be included in the differential diagnosis.
Investigations
Investigations include a skin biopsy to look for vascular occlusion or vasculitis, immunoglobulin titres and plasma protein electrophoresis, and hepatitis B and C serology. Serology for autoimmune disease should also be performed.
If cryoglobulinaemic vasculitis is suspected clinically, investigations should demonstrate circulating cryoglobulins, high rheumatoid factor titre and low C4 levels.
Histology may show a leukocytoclastic vasculitis of the small blood vessels on a skin biopsy. Histopathology of cryoglobulinaemia without vasculitis will reveal homogeneous eosinophilic material within the vascular lumina of dermal vessels, which corresponds to cryoglobulin deposits.
Management
Treatment of mixed cryoglobulinaemia is aimed at reducing immune complex activity by immunosuppression (with prednisolone and cyclophosphamide) and plasmapheresis. In the presence of HCV infection, therapy should also be directed at viral eradication with pegylated interferon and ribavirin. Recently reports have demonstrated benefit from the use of rituximab, a chimeric monoclonal anti-CD20 antibody, that exerts a selective B-cell control [7].
Cold agglutinins
Cold agglutinin disease is a disorder of autoimmune haemolysis in which cold-sensitive immunoglobulins react against erythrocyte surface antigens. In primary cold agglutinin disease, a bone marrow monoclonal CD20+ κ+ B-lymphocyte population is often demonstrated, while lymphoplasmacytic lymphoma may underlie the production of cold agglutinins in other patients [1]. Cases may also be secondary to a variety of diseases, notably Mycoplasma and Epstein–Barr virus infections. Cutaneous features occur mainly on acral sites and include Raynaud phenomenon, acrocyanosis and skin necrosis.
The results of therapy with corticosteroids, alkylating agents and interferon α have been poor. However, studies of the chimeric anti-CD20 antibody rituximab have produced good response rates [2].
