CHAPTER 132
Benign Melanocytic Proliferations and Melanocytic Naevi
Irene Stefanaki, Christina Antoniou and Alexander Stratigos
Department of DermatologyUniversity of Athens Medical School, Andreas Sygros Hospital, Greece
Freckle or ephelis
Definition
A freckle is a small reddish or pale to dark brown macule with a poorly defined border, on sun-exposed areas of the skin (Table 132.1). Freckles appear or darken during periods of UV exposure.
Table 132.1 Basic terminology and definitions used in benign melanocytic neoplasms
| Term | Description |
| Freckle (ephelis) | A pigmented macule on sun-exposed areas consisting of increased melanin pigmentation |
| Lentigo | A poorly demarcated area of uniform pigmentation consisting of increased melanin pigmentation, epidermal proliferation and replacement of basal cell keratinocytes by melanocytes |
| Café-au-lait macule | A well-circumscribed, uniformly light to dark brown macule or patch that spares mucous membranes and consists of increased melanin content in the basal cell layer |
| Nests of melanocytes | A group of melanocytes in contact with the basal layer of the epidermis but projecting downwards into the dermis |
| Junctional naevus | A pigmented melanocytic naevus in which the main histological feature is the presence of nests of melanocytes at the dermal–epidermal junction |
| Compound naevus | A pigmented melanocytic naevus in which the histological features include both junctional nests and the presence of naevus cells in the dermis |
| Intradermal/dermal naevus | A melanocytic lesion with naevus cells in the dermis. Melanin pigmentation is often absent and there is little or no abnormality of melanocytes in the epidermis. The deepest dermal cells tend to neural or fibroblastic differentiation |
Epidemiology
Age
Freckles are common during childhood.
Ethnicity
Freckles appear in all races, but are more frequently seen in individuals with light skin complexion, red hair and blue eyes.
Pathophysiology
Exposure to UV radiation leads to overproduction of melanin by melanocytes, which is subsequently transferred to neighbouring keratinocytes. Freckles could be considered a hyperplastic and hyperactive response of melanocytes to UV radiation in predisposed individuals [1].
Pathology
The basal cell layer appears hyperpigmented, without alteration of the epidermal architecture (Figure 132.1). In contrast to lentigines, the number of melanocytes is normal.
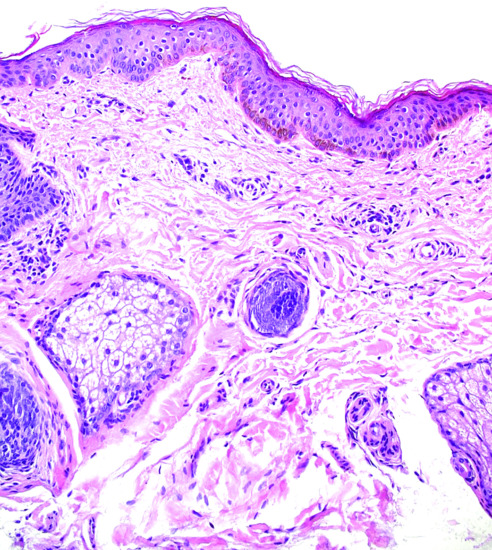
Figure 132.1 Epidermis of a freckle showing a hyperpigmented basal cell layer without elongated rete ridges or melanocytic hyperplasia. Magnification 20× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Genetics
The melanocortin 1 receptor gene (MCR1) has been characterized as the major freckle gene [2]. Variants of MC1R have been associated with freckling, possibly through the induction of phaeomelaninogenesis (compared to eumelaninogenesis), although other mechanisms may exist. The potential contribution of other pigmentation genes cannot be ruled out [3].
Environmental factors
UV exposure is responsible for the exacerbated pigment production by melanocytes that results in the development of freckles.
Clinical features
Presentation
Freckles typically appear after excessive sun exposure (either chronic or intermittent) in light-skinned red- or fair-haired individuals. They present as macular hyperpigmentations with a round or oval shape and ill-defined borders (Figure 132.2). In winter months freckles tend to lighten or even disappear.
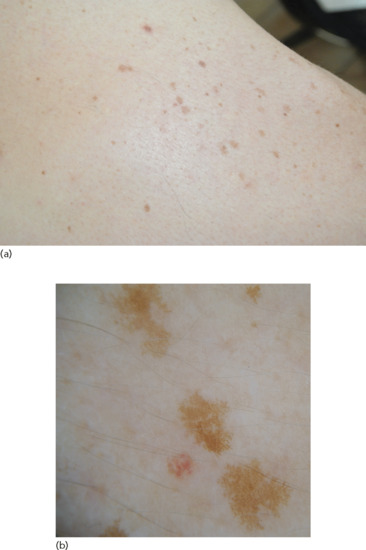
Figure 132.2 (a) Freckles. (b) Dermoscopic image showing hyperpigmented lesions with reticular pattern and moth-eaten edges.
Differential diagnosis
Freckles and solar lentigines are often grouped together in most studies, even though they are different. They are generally viewed as a response to sun exposure – solar lentigines to a greater extent – and both confer an increased risk for melanoma and epithelial skin cancers. They are distinguished from each other by the fact that lentigines persist even without UV exposure, and tend to appear more frequently in older ages. In addition, lentigines are histologically characterized by an increased number of melanocytes at the dermal–epidermal junction. Freckling can occur in neurofibromatosis type 1 in which it is more commonly located in non-exposed areas (trunk and axilla), while other manifestations of neurofibromatosis are present.
Complications and co-morbidities
Freckles are considered risk factors of melanoma. The estimated relative risk of melanoma based on the presence of freckling in a recent meta-analysis was 1.99, with a population-attributable fraction of 0.23 [4].
Disease course and prognosis
Freckles are benign lesions and often fade with age.
Investigations
In dermoscopy freckles present with a uniform pigmentation and a moth-eaten edge (Figure 132.2b).
Management
No treatment is required. Chemical peels, lasers, topical depigmenting drugs and dermocosmetic products can be used for cosmetic reasons [5]. Since they are induced by UV exposure, measures of photoprotection are indicated.
LENTIGINES
Definition
Lentigines are hyperpigmented macules that do not fade away in the absence of UV exposure. On microscopy they show increased melanin on the basal cell layer and increased numbers of singly arranged melanocytes, compared with the adjacent non-involved skin.
Introduction and general description
Lentigines are usually seen in light-skinned people, and represent proliferative responses of melanocytes to natural or artificial UV radiation. The different subtypes discussed below are mainly artificial distinctions based on the history of UV radiation exposure, anatomical location and the specific morphological characteristics of the lesion. The subtypes include simple lentigo, actinic (or solar) lentigo, psoralen and UVA (PUVA) lentigo and ink-spot lentigo.
Rarely, lentigines arise in the setting of potentially serious hereditary multisystem syndromes related to malignancies. These familial lentiginosis syndromes are characterized by autosomal dominant inheritance and include the Peutz–Jeghers syndrome, the PTEN (phosphate and tensin homologue) hamartomatous syndromes (Ruvalcaba–Myhre–Smith or Bannayan–Zonnana syndromes and Cowden disease), the Carney complex (and the closely related NAME (naevi, atrial myxoma, myxoid neurofibroma and ephelides) and LAMB (lentigines, atrial myxomas, mucocutaneous myxomas and blue naevi) syndromes and the LEOPARD/Noonan syndrome (lentigines, electrocardiogram anomalies, ocular anomalies, pulmonary stenosis, abnormal genitalia, retardation of growth and deafness) (Table 132.2) (see Chapter 70). Most of these syndromes are caused by mutations in the rat sarcoma–mitogen-actived protein (RAS-MAP) kinase and the mammalian target of rapamycin (mTOR) signalling pathway [6].
Table 132.2 Familial lentiginosis syndromes
| Disorder | Clinical manifestations | Inheritance | Related gene (chromosomal locus) |
| Peutz–Jeghers syndrome | Lentigines (lips, oral and bowel mucosa, palms, soles, eyes, nares, perianal region), hamartomatous GI polyps, neoplasms (GI tract, pancreas, breast, ovary, uterus, testis) | Autosomal dominant | LKB1/STK11 (19p13.3) |
| PTEN hamartomatous syndromes | Macrocephaly, lipomatosis, pigmentation of the glans penis, mental retardation, multiple hamartomas, neoplasms (breast cancer, follicular thyroid cancer, endometrial carcinoma) | Autosomal dominant | PTEN (10q23.31) |
| Carney complex | Lentigines (lips, conjunctiva, inner or outer canthi, genital mucosa), primary pigmented nodular adrenal cortical disease (PPNAD), cardiac and skin myxomas, schwannomas, acromegaly, breast and testicular tumours | Autosomal dominant | PRKAR1A (17q22–24) |
| Lentiginoses | Lentigines (centrofacial palmoplantar, trunk) Lentigines (centrofacial palmoplantar, trunk) plus mental retardation |
Autosomal dominant Autosomal dominant/sporadic |
Unknown Unknown |
| LEOPARD syndrome | Lentigines (mainly on face and upper trunk; rarely on oral mucosa, extremities, genitalia, conjunctiva), cardiac conduction abnormalities, aneurysms, pulmonic stenosis,cephalo-facial dysmorphism, short stature, sensorineural deafness, mental retardation, skeletal abnormalities | Autosomal dominant Autosomal dominant |
PTPN11 (12q24.1) – same as in Noonan syndrome RAF1 (3p25) |
Adapted from Guerrero 2012 [5].
GI, gastrointestinal; LEOPARD, lentigines, electrocardiogram anomalies, ocular anomalies, pulmonary stenosis, abnormal genitalia, retardation of growth and deafness; PTEN, phosphate and tensin homologue.
Lentiginosis profusa is a rare condition, with innumerable lentigines present at birth or arising early in life, without systemic abnormalities or mucosal involvement. The disorder has an autosomal dominant inheritance, but its exact genetic background is unknown. Agminated or segmental lentiginosis manifests as a circumscribed group of lentigines arranged in a segmental pattern that develop during childhood. They are presumed to represent mosaicism of an unidentified gene [7] and should be differentiated from neurofibromatosis type 1. Melanoma may occur in patients with segmental or generalized lentiginosis.
Simple lentigo
Definition and nomenclature
This is a light- to dark-brown or black macule that does not fade away once it appears, and is characterized histologically by increased melanocytes at the dermal–epidermal junction.
Epidemiology
Incidence and prevalence
Simple lentigos are very common, particularly in those with red hair and fair skin.
Age
They usually appear during childhood and increase in number until the age of 40. The majority of lentigines remain unchanged in adult life.
Associated diseases
Generalized lentiginosis has been rarely associated with the development of melanoma. There has been a single case report of a patient with lentiginoses and gastrointestinal stromal tumours harbouring a c-kit gene mutation [7].
Pathophysiology
Predisposing factors
There have been reports of lentigines developing after topical immunotherapy with tacrolimus, squaric acid dibutylester and diphencyprone [8, 9].
Pathology
There is a slight increase in the number of melanocytes along the dermal–epidermal junction, without nesting. Melanin hyperpigmentation is noted in melanocytes, adjacent keratinocytes and melanophages in the papillary dermis (Figure 132.3). The epidermal rete ridges are usually elongated and there might be a mild inflammatory infiltrate in the upper dermis.
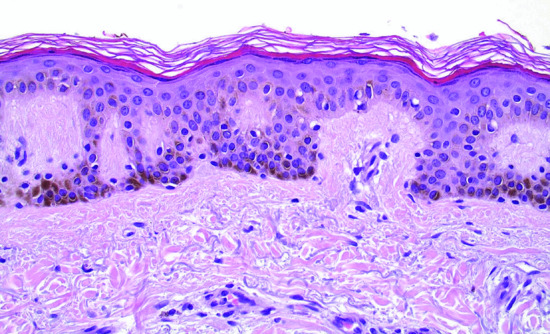
Figure 132.3 Lentigo simplex: hyperpigmentation is evident in the basal and squamous epidermal cells. There is a slight increase of non-atypical melanocytes between the epidermal basal cells. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Genetics
Multiple lentigines arising early in life on both exposed and non-exposed areas are usually a manifestation of inherited syndromes characterized by hyperplasias, hamartomas and neoplasia (Figure 132.4). Most of these syndromes are caused by mutations in the RAS-MAP kinase and the mTOR pathways.
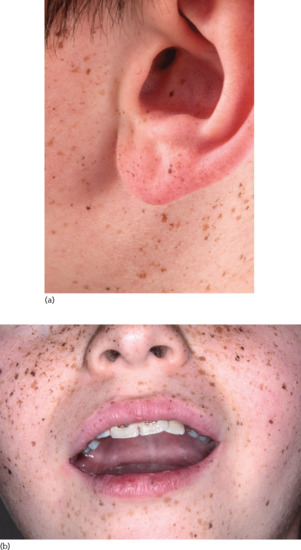
Figure 132.4 (a, b) Multiple lentigines in a patient with the Carney complex and a PRKAR1A mutation. Only about a third of patients with the complex have this classic pigmentation.
(Courtesy of Dr Constantine A. Stratakis, National Institutes of Health, Bethesda, MD, USA.)
In a recent study, simple lentigines, solar lentigines and melanocytic naevi were compared for mutations in the BRAF (common in melanocytic naevi), FGFR3 and PIK3CA genes (common in solar lentigines and seborrhoeic keratoses). Simple lentigines did not show mutations in any of these genes and thus are genetically differentiated from melanocytic naevi and solar lentigines. The BRAF mutations in simple lentigines contradict the proposed lentigo–naevus sequence of evolution, but do not exclude it.
Environmental factors
The role of sunlight is the most important environmental factor.
Clinical features
Presentation
Lentigines are poorly circumscribed, uniformly pigmented macules, with a round or oval shape and a diameter of up to 5 mm (Figure 132.5). There may be slight scaling of the surface, and several neighbouring lesions may coalesce. Their colour is pale to deep brown, depending on the skin colour of the individual. They are primarily located on photo-exposed areas as they are part of the spectrum of lesions (ephelides, simple lentigos, solar lentigos) resulting from excessive UV exposure.

Figure 132.5 (a) Simple lentigo. (b) Dermoscopic image of simple lentigo.
Differential diagnosis
The differential diagnosis of lentigines from freckles is made clinically by their comparatively darker colour, more scattered distribution and by their unchanged status in relation to sunlight exposure. In contrast to freckles, lentigines present histologically with an increased number of melanocytes.
The differentiation of lentigines from small junctional naevi is often impossible on clinical grounds. On histology, naevi show nests of naevus cells, while, in lentigines, melanocytes are separated from one another and do not typically form nests. However, there are cases of larger lesions that clinically appear as lentigines, but have small nests of naevus cells along the dermal–epidermal junction. This pattern is often referred to as the ‘jentigo’ pattern, meaning that the corresponding lesion combines features of both lentigo and junctional naevus. These transitional lesions may be regarded as precursors of future melanocytic naevi [1].
Lentigines may sometimes overlap clinically, or even histologically, with flat seborrhoeic keratoses (termed ‘liver spots’ when located at the dorsal hands). This overlap is clinically insignificant, as both are markers of light skin complexion, excessive sun exposure and a certain risk of skin cancer (see Chapter 143).
Distinction from lentigo maligna is made by dermoscopy or pathology143.
Disease course and prognosis
These are benign, relatively static lesions.
Investigations
With dermoscopy, lentigines show scalloped borders, a faint irregular network or pseudo-network and structureless areas (Figure 132.5b).
Management
There is no need for treatment. As the majority arise in sun-exposed areas, photoprotection could decrease the rate of new lesions developing. For cosmetic reasons, a variety of depigmenting topical agents and dermatological procedures such as chemical peels, lasers and photodynamic therapy reduces their pigmentation (see Chapters 159 and 160) [8]. In patients with multiple lentigines arising early in life on non-sun-exposed sites, the possibility of a hereditary multisystem syndrome (Table 132.2) should be considered. In the very rare cases of generalized lentiginoses, individuals may be at increased risk for melanoma, and thus should be educated on avoidance of sunburn and self skin examination.
Solar or actinic lentigo
Definition and nomenclature
A solar lentigo is a brown macule appearing after excessive sun exposure.
Epidemiology
Age
The number of solar lentigines increases with ageing.
Pathophysiology
The pathogenetic mechanism leading to the development of solar lentigines remains unclear. UVB stimulates keratinocytes to produce interleukin 1α (IL-1α), leading to secretion of keratinocyte growth factor (KGF). KGF has been found to increase pigment production in both pigmented epidermal equivalents and human skin grafts, suggesting a possible involvement of KGF in the molecular pathogenesis of solar lentigo [9]. Another scenario favours the role of fibroblasts, which – after UV exposure – release melanogenic growth factors (hepatocyte growth factor, KGF and stem cell factor) that subsequently act through keratinocytes and contribute to the hyperpigmentation of solar lentigines [10].
Predisposing factors
A history of occupational radon exposure, as well as recreational sun exposure, has been implicated in the development of multiple lentigines in a single case report [11].
Pathology
The histological features of solar lentigo are the same as for simple lentigo. Solar elastosis of the dermis and photoactivation features of melanocytes are usually present.
Genetics
FGFR3 and PIK3CA mutations have been observed in both solar lentigo and seborrhoeic keratosis, suggesting a common genetic basis [12].
Environmental factors
Solar lentigines are associated with both intermittent and chronic sun exposure [3]. Solar lentigines on the back have also been associated with a history of sunburns before the age of 20 years, while facial solar lentigines have been associated with cutaneous signs of photodamage [3].
Clinical features
Presentation
In younger patients, solar lentigines are most commonly seen on sun-exposed sites, such as the face in both sexes and the shoulders in males. They are macular, tan coloured and may be very large, with a striking irregular border. There is frequently a history of acute sunburn, followed by the sudden appearance of large numbers of these irregular macular lesions [13, 14]. In the UK, they are rare before the age of 12 years but in sunnier countries they may appear at a very young age.
Solar lentigines are also seen on older, fair-skinned patients who have had excessive sun exposure (Figure 132.6) [14]. The backs of the hands and the face are common sites. Once again the lesions are large and macular, have an irregular edge and are usually a uniform shade of brown. They are situated in an area of clinically evident sun-damaged epidermis and often manifest a clinical and pathological overlap with flat seborrhoeic keratoses.

Figure 132.6 (a) Solar lentigo in the middle of the left cheek. (b) Dermoscopic image of solar lentigo showing a uniform structureless macule with sharply demarcated borders.
Differential diagnosis
Differential diagnosis includes simple lentigo, seborrhoeic keratosis and, in some cases, lentigo maligna and melanoma in situ.
Disease course and prognosis
It has been proposed that solar lentigo may sometimes evolve into seborrhoeic keratosis.
Investigations
Dermoscopy is the same as in simple lentigo, while adjacent skin may show features of photodamage. Solar lentigo should be differentiated from seborrhoeic keratosis exhibiting milia-like cysts, cerebriform patterns and sharply demarcated borders. Pigmented actinic keratoses show a prominent pigmented pseudo-network and a background erythema with a red pseudo-network (‘strawberry pattern’). Lentigo maligna presents with specific dermoscopic feautures, such as asymmetrical follicular openings, ‘annular–granular’ pattern, pigmented rhomboidal structures and obliterated hair follicles, that are absent in solar lentigines.
In some cases, distinction between solar lentigo and lentigo maligna or melanoma in situ is very difficult when classic pathology is used. In such instances immunohistochemistry is a valuable diagnostic tool. Several stains have been used, and microphthalmia-associated transcription factor (MiTF) seems to be superior in the differential diagnosis of solar lentigo from melanoma in situ [15, 16].
Management
Patients often request treatment for these pigmented lesions located in visible body areas such as the face and back of hands.
Rigorous photoprotection with sun avoidance, use of a broad spectrum and high sun-protection-factor sunscreen, and appropriate clothing lowers the possibility of further lesions emerging in the future, and may also result in some degree of spontaneous resolution. A consensus has supported the use of cryotherapy as first line therapy, with topical therapy (e.g. topical retinoids) as an alternative or used for maintenance. There is, however, an extensive literature on the cosmetic treatment of lentigines using a variety of other treatments such as intense pulsed light and pigment-specific (Q-switched) laser systems (see Chapter 160).
Solar lentigines are benign lesions, but any pigmented lesion needs careful evaluation prior to laser treatment. There have been reports of cases that were referred for cosmetic laser therapy as solar lentigines and were actually melanomas [17]. Furthermore, non-invasive confocal imaging has revealed remaining melanocytes at the site of solar lentigines of all subjects after Q-switched ruby laser treatment, even though there was hardly any observable skin pigmentation on clinical examination [18].
Photochemotherapy (PUVA) lentigo
Definition
Lentigines can arise after long-term use of PUVA therapy.
Epidemiology
Incidence and prevalence
PUVA lentigines usually occur in patients who have received a high cumulative dose of PUVA treatment. They have been reported in 20% of 198 patients with a mean cumulative dose of UVA of 169.5 J/cm2 [19]. PUVA lentigines of any degree (slight, moderate or extensive) were also noted on the buttocks of 53% of 1380 psoriatic patients an average of 5.7 years after initiating PUVA therapy [20].
Pathophysiology
Predisposing factors
Fair-skinned individuals and patients who have received a greater number of PUVA treatments are at greater risk [21]. Lentigines appearing after narrow-band ultraviolet B (NB-UVB) radiation have also been reported [22].
Pathology
There are features resembling those of ephelides or lentigines. Melanocytes are increased in number and may have nuclear atypia. Ultrastructural studies have shown melanocytes with longer and more numerous dendrites as well as more active melanogenesis in PUVA lentigines, compared with solar lentigines and unexposed normal skin [23].
Clinical features
Presentation
PUVA lentigines are relatively large, pigmented macules that develop on the skin of patients receiving photochemotherapy (Figure 132.7).

Figure 132.7 PUVA-induced lentigines in a patient with psoriasis.
Differential diagnosis
As in solar lentigo.
Complications and co-morbidities
Follow-up of patients who had received PUVA in the USA revealed that PUVA lentigines are a marker of patients at increased risk of non-melanoma skin cancer [24]. Association with melanoma has not been firmly established. An increased risk was observed in a US study [20], but was not confirmed in a study from Scandinavia [25].
Disease course and prognosis
Some of the lesions persist for several months after completion of PUVA therapy.
Investigations
Dermoscopy helps to establish the diagnosis, revealing similar findings to solar lentigines.
Management
PUVA lentigines do not require treatment. Since they are considered potential markers for the development of non-melanoma skin cancer, patients with PUVA lentigines should have a regular follow-up.
Ink–spot lentigo
Definition
An ink-spot lentigo is a small, densely black macule resembling an ink spot displaying a markedly irregular outline, with a distribution similar to solar lentigo.
Pathophysiology
Pathology
There is lentiginous hyperplasia of the epidermis and prominent hyperpigmentation of the basal cells, with ‘skip’ areas involving the rete ridges (Figure 132.8). A minimal increase in the number of melanocytes is reported [26].
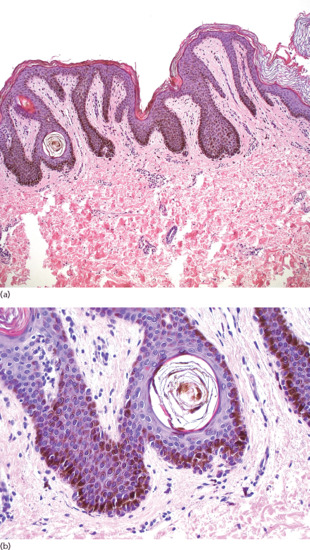
Figure 132.8 Ink-spot lentigo. (a) Epidermis with lentiginous hyperplasia and intensely hyperpigmented basal cells. Foci of less or no pigmentation are usually observed over the dermal papillae. Magnification 10× (H&E). (b) Hyperpigmentation is prominent to the lateral borders and the tips of the rete. There is a slight increase of melanocytes between the epidermal basal cells, and melanophages in the papillary dermis can be seen. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Clinical features
Presentation
Ink-spot lentigines are sharply demarcated macules with a jet-black colour and irregular shape (Figure 132.9) [26]. They usually arise on exposed areas, such as the upper back, and although the same patient may have multiple solar lentigines, ink-spot lentigines are commonly solitary lesions.
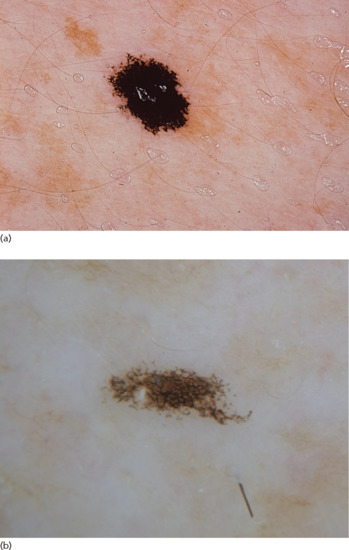
Figure 132.9 (a) Ink-spot lentigo. (b) Dermoscopic image of an ink-spot lentigo showing a bizarre-looking black pigment network which is typical.
(Courtesy of Dr S. Puig, Hospital Clinic Barcelona, IDIBAPS, Barcelona, Spain.)
Differential diagnosis
Due to their black colour, differential diagnosis mainly includes lentigo maligna and melanoma in situ.
Investigations
Dermoscopical features include a prominent black network with thin and/or thick lines.
Management
No treatment is required.
MUCOSAL MELANOTIC LESIONS
Pigmented melanotic macules
Definition and nomenclature
Mucosal melanotic macules (or lentigines) are benign pigmented patches of the mucosa. Although they occasionally have an alarming clinical appearance, histologically they are characterized by hyperpigmentation of basal keratinocytes and a normal or slightly increased number of melanocytes without atypical features or confluent proliferation.
Introduction and general description
Some degree of macular pigmentation of the mucosa in the mouth or on the genitalia is normal. More discrete, deeply pigmented macules are also common on the vulva and sometimes on the glans penis, where they are usually referred to as genital melanotic macules, or mucosal melanosis. These macules result from local excessive pigment production with normal numbers of melanocytes [27]. Rarely, these melanotic macules may expand to several centimetres in size and be patchy in distribution. They may develop irregular deep pigmentation, which resembles melanoma and causes concern. Since melanoma in the genital area may begin with a protracted in situ phase that clinically resembles a mucosal melanotic macule, a biopsy is often performed to exclude this possibility.
Epidemiology
Incidence and prevalence
The exact incidence is not known. However, they are the most common solitary pigmented melanocytic lesions found in the oral mucosa (representing 86% of solitary melanocytic lesions of the mouth) [28] or the genitalia.
Age
These macules involve a wide variety of ages, from childhood to old age. Congenital cases have rarely been reported.
Sex
There is a 2 : 1 female predilection.
Ethnicity
Pigmented melanotic macules occur in all races, but are more common in individuals with darker skin complexion. Macules of the buccal mucosa may be more frequent in black people [29].
Associated diseases
Pigmented macules in the female genitalia have been occasionally associated with lichen sclerosus [30]. Histological examination of these lesions reveals features of atypical melanocytic naevi.
Pathophysiology
Predisposing factors
In oral lesions, pigmentation is more pronounced at sites of trauma. Smoking may also result in melanosis. Post-inflammatory pigmentation can be observed, for example following oral lichen planus. Familial lentiginosis manifests as multiple lentigines in a familial context. Some patients with HIV present with oral macules, but it is unclear if these are induced by the virus or by the treatment [31]. The pathogenesis of genital macules remains unclear.
Pathology
A mild acanthosis of the epidermis, hyperpigmentation of basal layer keratinocytes and a slight increase in the number of melanocytes without nesting is observed (Figures 132.10 and 132.11). Scattered melanophages are seen in the upper dermis as a result of melanin incontinence.
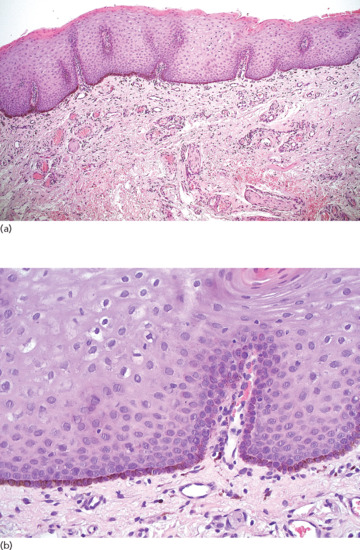
Figure 132.10 Mucosal melanosis. (a) Squamous epithelium of the oral cavity with hyperpigmentation of the basal cells. Magnification 10× (H&E). (b) Hyperpigmentation of the basal layer without melanocytic hyperplasia. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
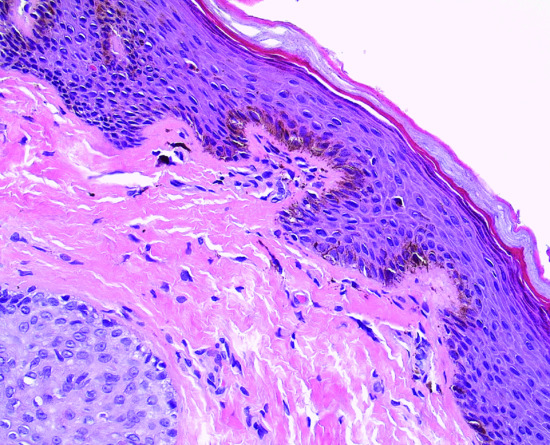
Figure 132.11 Penile lentiginosis: prominent melanocytic dendrites among the hyperpigmented basal layer cells. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Clinical features
Presentation
These are macules of uniform brown, black or grey pigmentation arising in the oral cavity (primarily on the lips and gingiva, followed by the palate and buccal mucosa) or the genitalia (mainly in the vulva, but also the penis or perineum) (Figure 132.12). Their diameter is relatively large, expanding slowly over time. They are usually single lesions, although multiple macules may arise occasionally. When located in the vulva, they are often asymmetrical with irregular borders and blue-black colour, simulating in situ melanoma.

Figure 132.12 Genital melanosis.
Differential diagnosis
Oral melanotic macules should be distinguished from other solitary pigmented melanocytic lesions, including oral melanocytic naevi, blue naevi, oral melanoacanthomas and oral melanomas. Oral melanoma is quite rare, occurring usually on the palate and accounting for less than 1% of oral malignancies [32, 33]. Exogenous pigmentation (e.g. amalgam tattoo), inflammatory hypermelanosis and smoker's melanosis should also be ruled out. Genital lentiginosis should be differentiated from lentigines, melanocytic naevi, clinically atypical naevi and in situ melanomas.
Disease course and prognosis
There are no reports of malignant transformation.
Investigations
Dermoscopic patterns of melanotic macules demonstrate a parallel, reticular, structureless pattern or globular pattern. A ring-like pattern, characterized by multiple white to tan structures with dark brown, well-defined regular borders, has been described in vulvar melanosis [34].
Management
If in clinical doubt, an incisional biopsy of an appropriately representative area is necessary to exclude melanoma. No treatment is needed if histology rules out malignancy. It is reasonable to follow up lesions with atypical clinical appearance in order to detect changes suggestive of melanoma.
If biopsy reveals significant melanocytic proliferation resembling lentigo maligna, then the lesion should be excised in its entirety and the tissue examined histologically. Topical imiquimod 5% followed by a new biopsy at the end of treatment may be considered in cases where surgical excision is technically difficult [35], although some patients may suffer a severe topical irritation.
Labial melanotic macules
Definition and nomenclature
A labial melanotic macule is a benign, hyperpigmented macule of the lip, quite similar to a freckle or simple lentigo.
Epidemiology
Incidence and prevalence
Such macules are encountered in approximately 3% of the general population.
Age
Labial melanotic macules usually appear around the age of 40, although in darker pigmented individuals it can present during adolescence [36].
Sex
They are more frequent in women.
Pathophysiology
Pathology
There is a linear increase in melanin pigment in the basal layer of the epidermis, with normal or slightly increased number of melanocytes, located singly between basal keratinocytes.
Environmental factors
Due to its common location in the middle of the lower lip, a UV-induced mechanism has been suggested.
Clinical features
Presentation
Such macules are brown to black and measure less than 6–7 mm (Figure 132.13). The most common location is the lower lip, especially the central third. It is usually a solitary lesion, developing rather rapidly in a young adult [36].
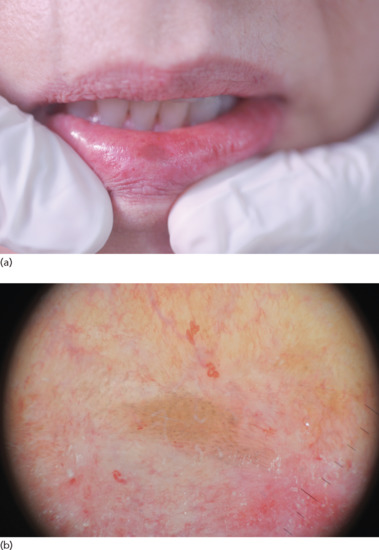
Figure 132.13 (a) Labial melanotic macule. (b) Dermoscopic image of a labial melanotic macule reveals a combination of grey-brown dots arranged in parallel lines and a background of light brownish homogeneous pigmentation.
Differential diagnosis
Labial melanotic macules are differentiated from melanoacanthoma of the lip by the histological presence of intraepithelial dendritic melanocytes in the latter. The banal histological features of a labial melanotic macule are easily distinguished from melanoma.
Disease course and prognosis
Malignant transformation has not been reported in these lesions. In a study with a mean follow-up of approximately 6 years, no alarming change, indicative of malignancy, was observed [37].
Investigations
Dermoscopy confirms that this is a benign lesion, revealing a uniform brown colour with parallel lines (‘fingerprint’ pattern) (Figure 132.13b).
Management
Patients should be reassured about the benign nature of labial melanocytic macules. Removal for cosmetic purposes can be achieved using a variety of methods including cryotherapy, infrared coagulation or laser therapy [38]. In the case of a newly formed lesion, or of changes in colour or size in a pre-existing lesion, a prompt evaluation of the patient is necessary.
DERMAL MELANOCYTIC LESIONS
Introduction and general description
Normally, during fetal life, melanocytes migrate from the neural crest to the dermal–epidermal junction. However, migrating melanocytes may occasionally remain entrapped in the dermis, not reaching their destination in the epidermis, and giving rise to dermal melanocytic lesions. These lesions have a bluish colour owing to the Tyndall effect.
Mongolian spot
Definition
Mongolian spots are congenital macular areas of blue-grey pigmentation of varying size and shape located on the sacral area in normal infants.
Epidemiology
Incidence and prevalence
Incidence varies among populations according to skin colour.
Age
The lesion develops in utero, increases in depth for a period during infancy and then diminishes.
Ethnicity
The Mongolian spot is uncommon in white people. In Europe it is more commonly observed in the Mediterranean region, while the highest incidence worldwide is documented in newborns with descent from the Far East. It has been observed in 13–26% of Turkish infants [39, 40] and 11–71% of Iranian newborns [41, 42]. Apart from genetic reasons, incidence differences among ethnicities could also be attributed to the amount of pigment produced in dermal melanocytes in darker individuals.
Associated diseases
There have been reports of association with Down syndrome, segmental café au lait macules and congenital haemangioma [43, 44, 45]. Extensive Mongolian spots have been associated with Hurler syndrome, GM1 gangliosidosis type 1 and mucolipidosis II [46, 47, 48, 49, 50], as well as with bilateral naevus of Ota [51].
Pathophysiology
Pathology
Elongated dendritic melanocytes are present around neurovascular bundles and in a ribbon-like pattern between collagen fibres of the middle and lower dermis distributed in parallel levels to the skin surface. No fibrosis or dermal melanophages are present, distinguishing a Mongolian spot from a blue naevus.
Clinical features
Presentation
The lesion is a diffuse macule with rather uniform, relatively faint blue to grey colour. It has a round or oval shape, with a diameter of a few – usually up to 10 – centimetres. Normally it presents as a single lesion, but multiple Mongolian spots may occasionally occur (Figure 132.14). The most common location is the lumbosacral region. In the case of generalized lesions, the buttocks, flanks or even shoulders and lower legs may be affected.
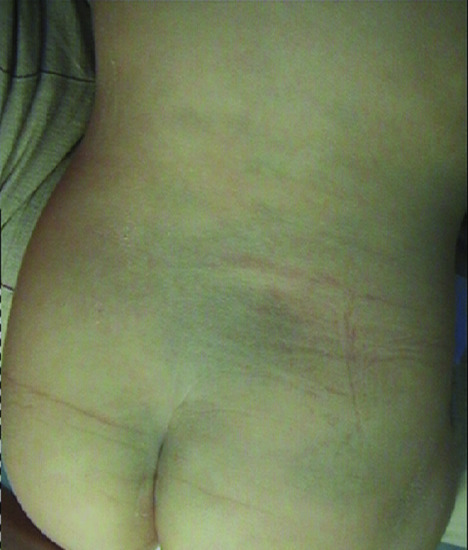
Figure 132.14 Mongolian spot in the lumbosacral area.
(Courtesy of Professor A. Katsarou-Katsari, Pediatric Dermatology Unit, Andreas Sygros Hospital, Athens, Greece.)
Differential diagnosis
Mongolian spots can be clinically differentiated from congenital naevi, which are also macular and present at birth, by their grey-blue colour. Histologically, Mongolian spots resemble blue naevi.
Disease course and prognosis
Mongolian spots typically resolve during childhood, but may occasionally persist into adult life.
Management
No treatment is required. Q-switched lasers, intense pulsed light and bleaching creams have been used in persistent cases [52, 53, 54]. Adults usually have a less favourable outcome [52, 53].
Naevus of Ota
Definition and nomenclature
A naevus of Ota is an extensive, bluish, patchy, dermal melanocytosis that affects the sclera and the skin adjacent to the eye, distributed along the first and the second branches of the trigeminal nerve. Extracutaneous lesions may also present in the uveal tract, dura, nasopharynx, tympanum and palate.
Epidemiology
Incidence and prevalence
Naevus of Ota usually presents in Asians, with an incidence between 0.014% and 0.034% [55]. It is very rare in other populations.
Age
Most lesions are present at birth or develop during the first year of life, increasing in size and number in subsequent years (Figure 132.15). A second peak of onset has been described in a minority of cases around puberty.
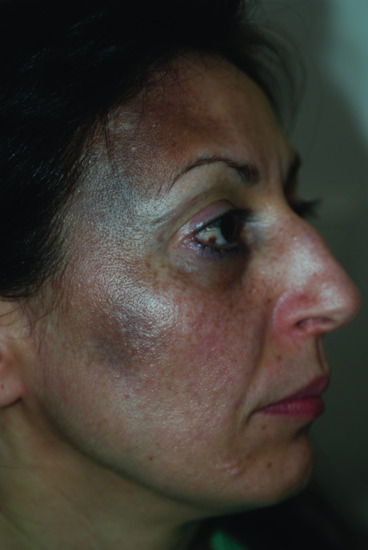
Figure 132.15 Naevus of Ota.
Sex
Lesions are more common in females.
Ethnicity
Naevus of Ota occurs more commonly in darkly pigmented individuals, particularly Asian and black people, although white people may also be affected. It is especially common in the Japanese (0.4–0.8% of dermatological patients).
Associated diseases
It is rarely associated with naevus of Ito. Bilateral cases of naevus of Ota are sometimes associated with extensive Mongolian spots. Sturge–Weber and Klippel–Trenaunay syndromes have been infrequently associated with naevus of Ota.
Pathophysiology
Pathology
Elongated dendritic melanocytes are scattered among collagen bundles mainly of the superficial dermis, and in larger numbers are compared to Mongolian spot (Figure 132.16). Occasionally they may extend deeper in the dermis or subcutaneous tissue. Melanophages are seldom present.
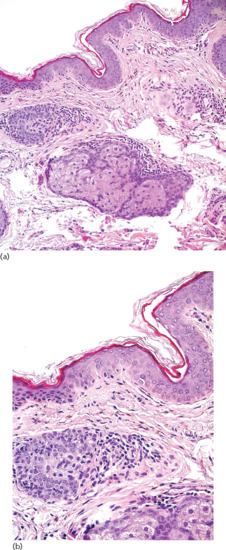
Figure 132.16 Naevus of Ota. (a) Skin with sparse dendritic melanocytes in the upper dermis and no melanocytic hyperplasia in the overlying epidermis. Magnification 20× (H&E). (b) Isolated dendritic melanocytes distributed among collagen bundles of the upper reticular dermis. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Genetics
GNAQ mutations have been reported in 6% of naevi of Ota, suggesting GNAQ is a genetic link between naevus of Ota and uveal melanoma [56].
Clinical features
History
The condition is named after Masao Ota, who in 1939 used the term ‘fuscocaeruleus ophthalmomaxillaris’.
Presentation
The lesion is often speckled, and is composed of blue and brown components that do not always coincide. This phenomenon can be better observed in the proximal eye, where the sclera appears blue and the conjunctiva brown. Brown pigmentation is patchy and superficial, following a reticular or geographical pattern; blue pigmentation is more diffuse and deeper.
Naevus of Ota is generally distributed along the ophthalmic and maxillary divisions of the trigeminal nerve. It presents in the periorbital region, involving the bulbar and palpebral conjunctiva and the sclera, as well as the temple, forehead, scalp, nose, ears, palate and malar area. It is usually unilateral, but bilateral lesions also exist.
Clinical variants
There have been several classifications based on clinical or histological features [57, 58, 59].
Differential diagnosis
Acquired bilateral naevus of Ota-like macules (Hori naevus) present as bilateral, blue-grey or brown, small, facial macules that are located in the same skin areas as naevus of Ota, but do not show mucous involvement [60]. They appear between the ages of 15 and 40 years in Asian women causing aesthetic problems. Their aetiopathogenesis is not clear [61], but oestrogens and UV radiation have been implicated.
Complications and co-morbidities
Meningeal melanocytomas of the brain, which are benign neoplasms, may complicate naevus of Ota [62]. The majority of melanomas associated with naevus of Ota occur in the meninges or in the choroid, iris or orbit [63, 64, 65, 66, 67]. There have been rare reports of cutaneous melanoma developing in a naevus of Ota.
Disease course and prognosis
Unlike Mongolian spots, it does not disappear with time.
Management
Q-switched lasers are the first line treatment, achieving a high rate of pigment clearing, depending on the age of the patient, the colour and histological depth of the lesion [68]. Post-treatment hypo- or hyperpigmentation, scarring and recurrence of the lesion can occur [55,69, 70, 71]. Cosmetic camouflage can also be used.
Despite the rare occurrence of malignant transformation in naevus of Ota, a close ophthalmological monitoring is essential in cases where the eye is involved. Any new subcutaneous nodule arising on a naevus of Ota should be further investigated histologically to exclude the possibility of melanoma.
Naevus of Ito
Definition and nomenclature
Naevus of Ito is a dermal melanocytosis involving the acromioclavicular region and the upper chest.
Epidemiology
Incidence and prevalence
Naevus of Ito is a rare disorder, presenting more commonly in Asians. It is less frequent than naevus of Ota.
Ethnicity
This naevus primarily occurs in Chinese and Japanese people.
Pathophysiology
Pathology
Histological features are identical to those of naevus of Ota.
Clinical features
History
This naevus was originally described by Minor Ito in 1954.
Presentation
Naevus of Ito presents as a unilateral, blue-greyish macular discoloration. It is distinguished from naevus of Ota by its location in the area innervated by the posterior supraclavicular and lateral cutaneous brachial nerves (Figure 132.17). Bilateral distribution has been reported occasionally [72, 73, 74, 75].

Figure 132.17 Naevus of Ito, showing a typical distribution over the shoulder area.
Differential diagnosis
A Becker naevus is a large, pigmented, often hairy, patch on the shoulder, chest or back of young males (see Chapter 75). Histologically it shows hyperpigmentation of the basal layer, mild acanthosis, elongation of the rete ridges and numerous melanophages in the upper dermis.
Complications and co-morbidities
Naevus of Ito is a benign lesion. There are only three cases of transformation to melanoma reported in the literature [76, 77, 78].
Disease course and prognosis
This is the same as in naevus of Ota.
Management
Pigment targeting Q-switched laser systems are the treatment of choice.
CONGENITAL MELANOCYTIC NAEVI
See Chapter 75 and Table 132.3.
Table 132.3 Basic clinical, pathological and dermoscopic patterns of acquired and congenital melanocytic naevi (see Chapter 75)
| Type of naevus | Subtype | Clinical presentation | Pathological characteristics | Dermoscopic presentation |
| Congenital naevi | Small (<1.5 cm) Medium (1.5–20 cm) Giant (>20 cm) |
Light to dark-brown colour, flat or elevated, with smooth to mammillated to verrucous surface and well-defined borders; may acquire coarse hair | Deeper naevus cells exhibit a tendency to extend deeply in relation to skin appendages | Reticular, globular or mixed pattern; milia-like cysts, perifollicular hypo-/hyperpigmentation, black or brown dots/globules and hypertrichosis may be present |
| Speckled lentiginous naevus | Multiple darkly pigmented macules or papules (representing junctional and compound naevi) arising on a lentiginous macule | Banal naevi arising in a macular lentigo with a subtle increase in melanocyte number | Globular, reticular, structureless brown and mixed patterns on a faint network | |
| Common acquired naevi | Junctional naevus | Uniformly pigmented brown macule, with a diameter of 2–10 mm | Benign proliferations of melanocytes in the epidermis | Globular, reticular, structureless brown and mixed patterns |
| Compound naevus | Slightly raised, oval or round papule with symmetrical shape; pigmented with variable shades of brown | Benign proliferations of melanocytes in the epidermis, showing evidence of migration of cells into the dermis and ‘maturation’ of those cells within the deeper dermis | Globular, reticular, structureless, brown, multicomponent and mixed patterns | |
| Intradermal/dermal naevus | Flesh-coloured, dome-shaped, exophytic papule or nodule | Benign tumours of melanocytes in which there is no longer epidermal proliferation; the naevus cells have migrated into the dermis and matured there | Symmetrical homogeneous pattern; may have a slight pigmented globular pattern or black dots. Comedo-like openings, crypts and comma vessels may be present | |
| Naevi in unusual sites | Naevus of the genital area | Usually hyperpigmented and larger in size compared with common acquired naevi | Atypical junctional proliferation of melanocytes may be present (large nests, discohesion of melanocytes) | Asymmetrical in colour and structure, often with irregular dots/globules or grayish-black blotches |
| Acral naevus | Macular or slightly elevated, uniformly pigmented lesion with irregular and sharp borders | Atypical junctional proliferation of melanocytes may be present | Parallel furrow, lattice-like or fibrillar pattern | |
| Nail-associated naevus | Longitudinal parallel and homogeneous light to dark brown to black pigmentation of the nail plate | Junctional or compound naevus with prominent hyperpigmentation and nuclear hyperchromasia | Brown, longitudinal parallel lines with regular spacing and thickness | |
| Naevi with unusual morphology | Combined melanocytic naevus | Bluish macule or papule surrounded by a macular brown area | Two different types of naevi (one of which is usually a blue naevus) within the same lesion | Usually a brownish reticular and/or globular pattern with central or eccentric structureless blue pigmentation |
| Recurrent melanocytic naevus | Macular area with hyper- or hypopigmentation, linear streaks and mottled pigmentation arising within a scar | Intraepidermal presence of melanocytes with abundant melanin and occasionally atypia, above the level of a scar | Irregular prominent network, globules and heterogeneous pigmentation, usually within the borders of the scar | |
| Halo naevus | A melanocytic naevus surrounded by a depigmented halo | Dense lymphocytic infiltrate in the early phase and subsequent elimination of naevus cells | Central naevus exhibits the globular and/or homogeneous patterns, surrounded by a white rim | |
| Meyerson naevus | A melanocytic naevus that develops an erythemato-squamous halo | Benign naevus with overlying spongiosis of the epidermis | Pattern of the involved naevus is often blurred by an overlying yellowish superficial crust | |
| Cockade naevus | A naevus with a target-like appearance | Central junctional or compound component, while the periphery consists of junctional nests | Darker central globular or homogeneous pattern, surrounded by a structureless inner ring and a more peripheral darker reticular ring | |
| Targetoid haemosiderotic naevus | Brown or red-brown or violaceous papule surrounded by a thin pale area and a peripheral ecchymotic ring | Naevus cells mingled with extravasated blood vessels | Red to purple colour haemorrhage surrounding a naevus | |
| Spitz naevus | Classic Spitz naevus | Solitary, firm, symmetrical, sharply demarcated, round or dome-shaped pink to red to reddish brown nodule (≤5–6 mm in diameter) | Symmetrical naevus of orderly architecture; consists of epithelioid/spindle cells arranged in nests showing zonation and maturation at the depth of lesion; intact epidermis, Kamino bodies, few superficial mitoses | Dotted vascular pattern with intersecting white lines (classic non-pigmented type); a ‘starburst’ pattern, aglobular pattern (blue-grey pigmentation surrounded by a rim of pigmented globules) or a multicomponent atypical pattern is seen in pigmented variants |
| Reed naevus | Solitary, densely pigmented, irregularly shaped, dark-brown or black papule or nodule | Similar to classic Spitz naevi; junctional melanocytic activity with large quantities of melanin pigment | ‘Starburst’ pattern (diffuse blue-black pigmentation with radial streaks in the periphery) | |
| Blue naevus | Common blue naevus | Blue-black or deep blue dome-shaped papule, with a diameter <1–2 cm | Spindle or dendritic melanocytes within the dermis, containing pigment even deeply in the dermis | Structureless, homogeneous, diffuse blue, blue-grey to steel blue pattern |
| Cellular blue naevus | Same as common blue naevus; usually larger diameter | Dermal naevus cells are more numerous and extend into the deep reticular dermis or to subcutaneous fat | Similar to blue naevus; may have pale or yellowish periphery | |
| Deep penetrating naevus | Larger than common blue naevus, may show diffuse and irregular lateral margin | Extension of naevus cells deep into the dermis with a wedge shape | Negative globular pattern with blue-brown homogeneous pigmentation | |
| Clinically atypical naevus | Larger than 5 mm with irregular borders and pigmentation; may contain a reddish hue. A central papular component is often surrounded by a macular periphery | Characteristic architectural and cytological atypia (see Table 132.4) | Multiple patterns; a common dermoscopic pattern shows central homogeneity and reticulated network or dots at the periphery |
Speckled lentiginous naevus
Definition and nomenclature
Speckled lentiginous naevus (SLN) is a congenital melanocytic naevus presenting as a lentiginous macule early in life, and subsequently developing multiple darkly pigmented macules or papules in a speckled distribution (Table 132.3).
Epidemiology
Incidence and prevalence
The estimated prevalence is about 1–2%.
Age
The naevus may be present at birth or appear during childhood.
Sex
There is no sex predilection.
Associated diseases
Speckled lentiginous naevi are occasionally part of complex disorders such as speckled lentiginous naevus syndrome (SLN syndrome), phakomatosis pigmentovascularis (PPV) or phakomatosis pigmentokeratotica (PPK) [79, 80, 81]. SLN syndrome is characterized by a papular SLN associated with ipsilateral neuromuscular, peripheral nerve and/or central nervous system defects. Phakomatosis spilorosea, which according to the new classification by Happle in 2005 is a type of PPV corresponding to previous types IIIa and IIIb, is characterized by the coexistence of a macular SLN with a pale-pink telangiectatic naevus [82]. PPK is characterized by the presence of a sebaceous naevus and a papular SLN, with or without skeletal and neurological disturbances.
Pathophysiology
Pathology
The background macule shows a subtle increase of melanocytes, whereas darkly pigmented speckles present a ‘jentigo’ pattern. Papular lesions correspond to superimposed dermal or compound melanocytic naevi.
Genetics
Phakomatosis pigmentovascularis was considered to be a didymosis with early postzygotic recombination [83]. However, Groesser and co-workers proposed that both naevi (sebaceous naevus and SLN) in PPK are caused by a postzygotic HRAS mutation in a multipotent progenitor cell, and should therefore be viewed as a mosaic RASopathy [84].
Clinical features
Presentation
A SLN comprises a flat macule on a tan background, often faintly appearing, and more darkly pigmented lentigo-like lesions or naevi (Figure 132.18). Other types of naevi, such as blue naevi, Spitz naevi or rarely congenital melanocytic naevi, can occur within the lesion. The sites more commonly involved are the trunk and the upper and lower extremities. Naevi present in various sizes, and occasionally show zosteriform or segmental distributions.
Figure 132.18 Speckled lentiginous naevi: numerous darker macules and papules (representing junctional and compound naevi) can be seen on a faintly visible tan macular background.
Clinical variants
Vidaurri de la Cruz et al. performed an extensive review of case reports on SLN and proposed two distinct subtypes of naevus spilus: macular (naevus spilus maculosus) and papular (naevus spilus papulosus) [85]. Macular SLN is characterized by a light brown macule with darker flat speckles, resembling a polka-dot pattern. In papular SLN, the light brown macule is superimposed with multiple papules or nodules with uneven distribution, reminiscent of a star map, while dark macules may also be present. Macular SLN is related to the phakomatosis spilorosea type of PPV, whereas papular SLN is present in phakomatosis pigmentokeratotica and SLN syndrome.
Differential diagnosis
Speckled lentiginous naevi can be confused with café-au-lait spot, congenital melanocytic naevus, Becker naevus, agminated naevomelanocytic naevus and segmental lentiginosis.
Disease course and prognosis
There have been a few reports of melanoma arising within SLNs of various sizes, but the relative risk is unknown. Considering the limited number of reported malignant transformation and the estimated 1–2% prevalence of SLN in the population, it is likely that this risk is quite low.
Management
There is no standardized management algorithm for SLN. Although they are not considered a major risk factor for melanoma or true melanoma precursors, patients should be instructed to monitor their naevi. Baseline photos and subsequent documenting of clinical changes could be useful. For patients with large naevi covering extensive body surface areas that are difficult for the patient to self-examine, clinical surveillance and sequential dermoscopic examination should be offered. Prophylactic excision is not justified, but suspicious lesions should be biopsied.
Excision, ablative and non-ablative lasers and dermabrasion for cosmetic reasons have been used with varying results [86, 87, 88].
COMMON ACQUIRED NAEVI
Acquired melanocytic naevi
Definition and nomenclature
These are common benign proliferations of uniform melanocytes that are located initially at the dermal–epidermal junction, and over time tend to migrate into the dermis and regress with subsequent morphological changes.
Introduction and general description
Acquired melanocytic naevi are benign neoplasms of melanocytic neavus cells that begin to proliferate at the dermal–epidermal junction (junctional naevus), and over time tend to migrate into the dermis while a component remains in contact with the basal layer (compound naevus) (Table 132.3). At the end stage of this process, all the naevus cells are completely detached from the overlying epidermis (intradermal naevus). The typical life cycle of a common melanocytic naevus follows the stages of initiation, promotion, senescence and involution.
Epidemiology
Incidence and prevalence
Acquired melanocytic naevi are the most common neoplasms among white people, and are usually multiple.
Age
The number of acquired melanocytic naevi increases from childhood to midlife, and decreases progressively thereafter. The occurrence of a new melanocytic lesion in a young individual is a common event, while the same phenomenon in a patient older than 60 years should raise concern of melanoma.
Sex
There is no sex predilection.
Ethnicity
These naevi present in all races, but their incidence and number is higher in white people.
Associated diseases
The presence of multiple acquired melanocytic naevi is a risk factor for melanoma. Patients with more than 100 naevi have a sevenfold increase in melanoma risk [89]. The risk of malignant transformation of an acquired melanocytic naevus is extremely low, ranging on an annual basis from one in 200 000 for men and women younger than 40 years, to one in 33 000 for men older than 60 years [90].
Pathophysiology
The exact mechanism of melanocytic naevus development is unknown, but probably involves an interplay of genetic and environmental factors. A common scenario proposes that naevi originate from a single precursor melanocyte of unknown nature.
Predisposing factors
Variations in naevus numbers have been observed among different groups of patients. Chemotherapy administered for leukemia in childhood [91] or after renal transplantation [92] has been associated with increased naevus count. Local trauma may trigger the development of eruptive naevi in predisposed patients [93]. Individuals with Turner syndrome have increased numbers of melanocytic naevi [94], while children with atopic eczema present fewer naevi compared with control subjects, suggesting that the pro-inflammatory cytokine milieu in the atopic skin might inhibit naevogenesis [95, 96].
Pathology
The pathology of acquired naevi consists of naevomelanocytes arranged partially in clusters or nests. They are usually round or cuboidal with intracytoplasmic granules of melanin pigment.
In junctional naevus, along the dermal–epidermal junction, single naevus cells are present in the basal layer. Nests of naevus cells are distributed mainly at the tips or, less commonly, between the rete ridges (Figure 132.19).
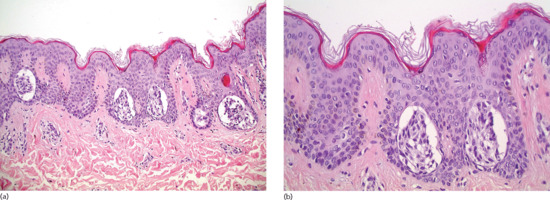
Figure 132.19 Junctional naevus. (a) Aggregates of naevomelanocytic cells in the rete ridges of a hyperplastic epidermis. There is no dermal component. Magnification 10× (H&E). (b) Naevomelanocytic nests are usually located in the tips and basilar area of the rete ridges and are often separated from the adjacent squamous cells by a clear space. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
A compound naevus has a junctional and an intradermal component with a variety of morphological types of naevomelanocytes (A, B and C cells) arranged in nests, cords or single units in the dermis (Figure 132.20a). Type A or epithelioid cells with intracytoplasmic melanin pigment in an abundant cytoplasm are typically found in the upper dermis. Type B or naevic cells, are smaller than type A, contain less melanin and lie in cords deeper in the dermis (Figure 132.20b). Type C or spindle cells rarely contain melanin and have a neurotized, schwannian morphology. They are located in the lower dermis, often in strands or in loose, fibrillar aggregates, called naevic corpuscles. Multinucleated cells may also be present at the upper dermis.
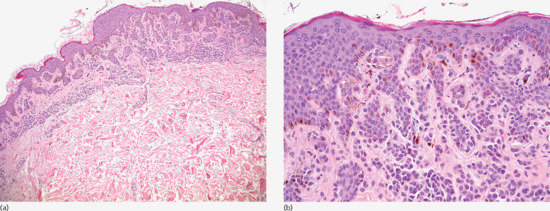
Figure 132.20 Acquired compound naevus. (a) Naevomelanocytic proliferation in the junctional area and the dermis. The dermal component extends beyond the boundaries of the junctional component. Magnification 10× (H&E). (b) Naevus cells type A (epithelioid) or type B (without significant atypia) present in the dermal–epidermal junction and in the dermis, in an isolated or nesting pattern of development. Melanin pigment is more pronounced in the upper portion of the lesion. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
An intradermal naevus has identical features to the dermal component of a compound naevus, with melanocytes gradually losing their ability to produce melanin as they progress from the upper to the deeper dermis (Figure 132.21). When the spindle-shaped type C cells prevail, the lesion mimics a neurofibroma and the term neural naevus is used for the variant without a type A or B cell component. The presence of fat cells between the naevus cells and of atypical senescence of naevomelanocytes are considered to indicate ageing of the lesion. The overlying epidermis may be hyperplastic, with hyperkeratosis and papillomatosis.
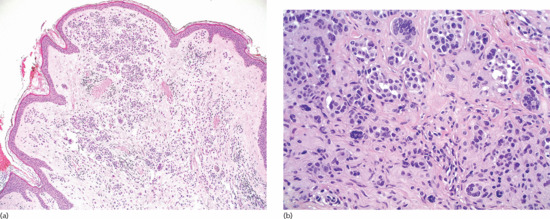
Figure 132.21 Aquired dermal naevus. (a) Nested pattern of development in the upper portion of the lesion. There is no junctional component. Magnification 10× (H&E). (b) All types of naevic cells (A, B, C and giant cells) are present in an isolated or nested pattern. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
In general, melanocytes gradually lose their ability to produce pigment as they progress from the epidermis to the dermis and ‘senesce’. These stages are not necessarily followed by every naevus.
The balloon cell naevus is a histological subtype of compound naevi. It consists of characteristic cells with swollen, vacuolated cytoplasm, containing none or a few small melanin granules, which are sometimes multinucleated. Regular melanocytes can be found at the periphery of the naevus. The biological significance of balloon cells is unknown, and they are not precursors to balloon cell melanoma. It is speculated that ‘ballooning’ is an intrinsic cellular degenerative process due to an arrest in the biosynthesis of melanin in melanosomes [97]. Rare cases of balloon cell naevi in the upper aerodigestive tract mucosa and conjunctiva have been reported [98, 99].
Genetics
Somatic BRAF mutations are present in the majority (c. 80%) of acquired melanocytic naevi, suggestive of the activation of the RAS/MAPK kinase pathway in the pathogenesis of naevi. BRAF mutations are an early initiating event in naevogenesis, leading to melanocytic proliferation and the formation of neoplastic clones [100]. In the absence of other genetic alterations these clones enter cell cycle arrest and senescence through the induction of p16INK4a and acidic β-galactosidase [101]. Genomewide association studies have identified several genetic loci on chromomes 9p21 and 22q13 that are potentially associated with naevus counts and melanoma development [102, 103].
Environmental factors
Although the prevalence of common naevi has not always been associated with UV exposure [104], international comparative studies have documented increased number of naevi in children living in sunnier climates compared with children of similar ethnicity residing in northern countries [105, 106]. Australian studies have demonstrated that ambient sunlight is associated with increased naevus counts in children [107, 108, 109].
Clinical features
Presentation
A newly formed melanocytic naevus is a junctional naevus, presenting as a uniformly pigmented brown macule, with a diameter of 2–10 mm (Figure 132.22). Naevus pigmentation is related to individual skin colour, with lighter phototypes typically presenting paler naevi. It is also associated with UV exposure, gaining a darker shade after exposure to sunlight (e.g. after a summer vacation) or artificial UV sources. It can be located anywhere in the body, but it is found more commonly on the trunk and the extremities.
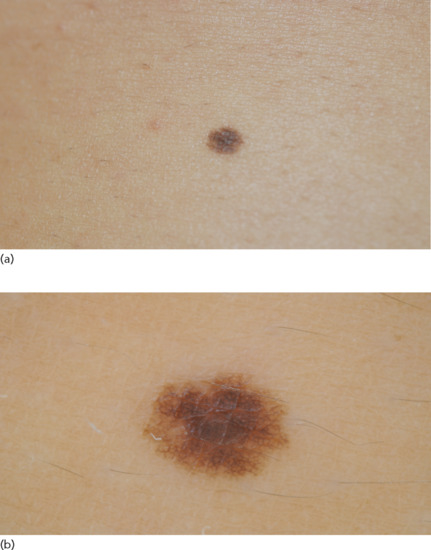
Figure 132.22 (a) Junctional naevus. (b) Dermoscopic image of a junctional naevus showing a reticular pattern with a smooth ending at the periphery.
A compound naevus is a slightly raised, oval or round papule with symmetrical shape (Figure 132.23). This naevus is also pigmented, with shades of brown according to the patient's skin colour.
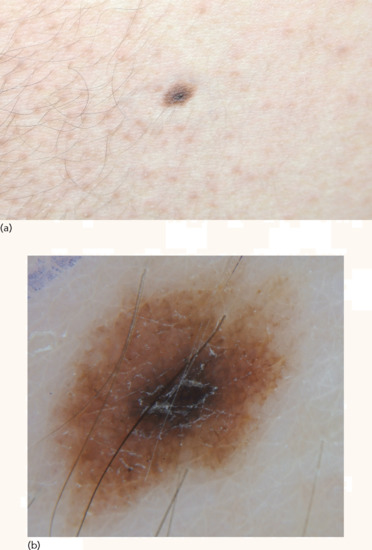
Figure 132.23 Compound naevus. (a) Hyperpigmented papule surrounded by symmetrical, lighter brown, macular area. (b) Dermoscopic image showing a structureless brown center with reticulated periphery.
Intradermal (or dermal) naevi are flesh-coloured, dome-shaped nodules that can be larger than junctional naevi (Figure 132.24). Their surface is usually smooth but can also appear papillomatous. They are located primarily on the head, neck and shoulders. One or a few hair shafts may project from the naevus surface. Acute inflammation of intradermal naevi, presenting as painful erythema and swelling, can occur due to mechanical friction or bacterial infection of the hair follicles inside the naevus and should not cause concern of malignant transformation.
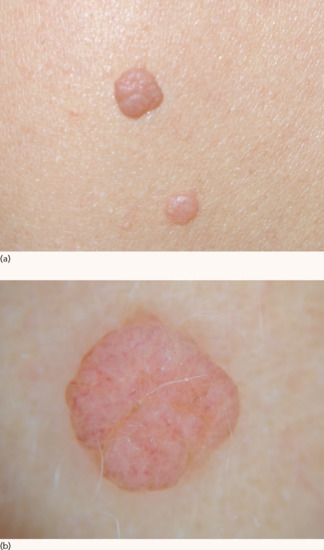
Figure 132.24 (a) Dermal naevi. (b) Dermoscopic image showing a symmetrical, flesh-coloured, homogeneous pattern with coma-shaped vessels.
Clinical variants
Agminated and eruptive melanocytic naevi can occasionally occur (see Chapter 146).
Differential diagnosis
The distinction between very small congenital and common acquired melanocytic naevi during the first years of life may be very difficult both clinically and dermoscopically.
Disease course and prognosis
Although 20–30% of melanomas arise in association with pre-existing naevi, malignant transformation of naevi is a very rare event.
Investigations
Dermoscopy (Figures 132.21b, 132.22b and 132.23b), digital dermoscopic follow-up and reflectance confocal microscopy (Figures 132.25 and 132.26) have allowed a detailed depiction of distinct morphological features. Upon dermoscopy, common acquired melanocytic naevi can be classified into globular, reticular, structureless brown and mixed patterns, which correlate to different histopathological features. Dermoscopic patterns are also related to patient's age and skin pigmentation, as well as anatomical location [110].

Figure 132.25 (a) Overview (3.5 × 5 mm) of a junctional lentiginous naevus by reflectance confocal microscopy showing a ringed pattern. (b) Detail (1.2 × 1.7 mm) where the ringed pattern is more evident. This pattern corresponds to lentiginous proliferation of melanocytes.
(Courtesy of Dr Giovanni Pellacani, Department of Dermatology, University of Modena and Reggio Emilia, Italy.)
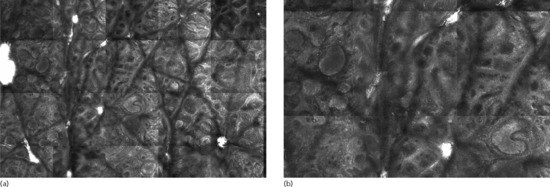
Figure 132.26 (a) Overview (1.8 × 2.6 mm) of a compound naevus by reflectance confocal microscopy. Some nests filling the papillae are clearly visible in the centre and left side of the image (compact dense nests). (b) Detail (1.2 × 1.7 mm) with the nests at a higher magnification.
(Courtesy of Dr Giovanni Pellacani, Department of Dermatology, University of Modena and Reggio Emilia, Italy.)
Management
No treatment is required. Surgical removal is performed only for aesthetic purposes, but has the potential risk of scarring. Superficial removal techniques, like curettage, dermabrasion, shave excision, electrosurgery and lasers do not destroy all naevus cells, so that the naevus can recur with an atypical presentation (‘pseudo-melanoma’). Scarring, inflammation, neovascularization and post-inflammatory hyper- or hypopigmentation are also frequent sequelae associated with these methods.
NAEVI IN UNUSUAL SITES
Definition
Naevi in certain locations include a group of benign melanocytic naevi with histological features resembling clinically atypical naevi or melanoma. They are located in distinct anatomical areas such as the scalp, ear, embryonic milkline, flexural sites, breast, genitalia and acral sites but comprise only a subset of naevi that present on these sites (see Table 132.3).
Introduction and general description
Although not broadly accepted as a unique entity, these lesions have a different morphological pattern from banal melanocytic naevi. This is partially due to anatomical factors, hormonal influences, trauma and epidermal thickness [111]. These naevi are clinically more atypical, presenting with a larger size and colour variegation. They exhibit distinct histological patterns such as pagetoid speading (acral naevi) [112], enlarged junctional nests with discohesion of melanocytes (flexural and genital naevi) [113] or large nests with bizarre shapes that extend down to the follicular epithelium (scalp) [114]. They can also present with atypical nesting patterns, stromal fibrosis and aytpical dermal cytology [115]. Their course is benign and, just as in naevi on other sites, they should be monitored clinically.
Melanocytic naevi of the genital area
Epidemiology
Incidence and prevalence
These hamaratomas are uncommon, found in 2.3% of patients undergoing routine gynaecological examinations [116].
Pathophysiology
Pathology
Most are compound naevi characterized by a florid junctional melanocytic proliferation, cellular dyscohesion and atypical cytology [117].
Clinical features
Presentation
These naevi are predominantly located in the vulva, but also occur in the perineum, mons pubis, penis and scrotum. A small subset of genital naevi (5%), termed atypical melanocytic naevi of the genital type, occurs mainly in young premenopausal women and is characterized by marked architectural and atypical cytology [118]. Clinically, these lesions are often hyperpigmented and larger in size compared with common acquired naevi.
Differential diagnosis
Despite their benign course, these lesions are often difficult to differentiate from vulvar melanoma which is, however, seen primarily in post-menopausal women [119].
Acral naevi
Pathophysiology
Pathology
Histologically, these naevi share the same features with common acquired naevi in other locations. They can be junctional, compound or intraepidermal (Figure 132.27a) [120, 121]. Lentiginous melanocytic proliferation and some degree of upward migration of naevus cells are seen (Figure 132.27b), the latter due to a transepidermal elimination effect from frequent trauma and friction on these sites. The dermal component of acral naevi shows maturation, bland cytology and lack of mitoses.

Figure 132.27 Compound acral naevus. (a) The junctional component predominates, with variable sized and shaped nests located mainly to the tips of the rete ridges. A dermal component is also present. Magnification 10× (H&E). (b) Single melanocytes present in the epidermis in a pagetoid spreading pattern may lead a pathologist to the erroneous diagnosis of melanoma. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Clinical features
Presentation
Acral naevi are usually macular or slightly elevated, uniformly pigmented lesions with irregular and sharp borders [120]. Their colour can range from brown to dark brown and they often present with a striated appearance, distributed along the parallel furrows of acral skin (Figure 132.28), in contrast to melanomas that are situated along the ridges of the palms and soles.
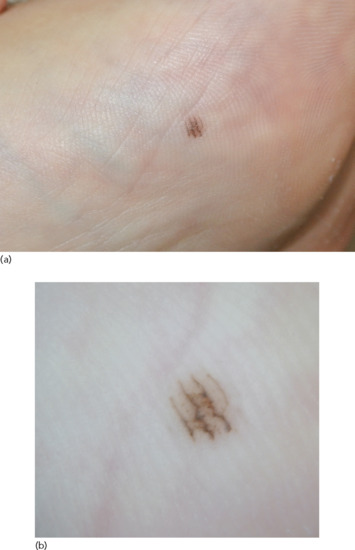
Figure 132.28 (a) Naevus on the sole of a foot. (b) Dermoscopic image of acral naevus showing a parallel furrow pattern.
Investigations
The most common dermoscopic pattern of acral naevi is the parallel furrow pattern (Figure 132.28b), followed by the lattice-like and fibrillar patterns [122].
Management
Despite their slightly atypical histology, the majority of acral naevi are clinically and histologically indistinguishable from benign naevi in other sites and should be managed similarly.
Conjunctival naevi
Epidemiology
Incidence and prevalence
Conjunctival naevi are the most common tumours of the conjunctiva and can be of the acquired or congenital type [123].
Patholphysiology
Pathology
Histologically, the compound type predominates although all histological variants have been reported, including a rapidly growing and often concerning lesion in children and adolescents [124]. The characteristic feature of conjunctival naevi is the histological presence of intralesional cysts (Figure 132.29).
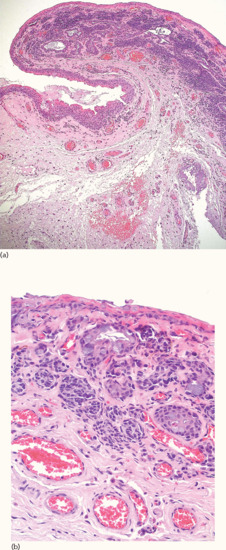
Figure 132.29 Compound conjunctival naevus. (a) Naevic aggregates are observed mainly beneath the conjunctival epithelium, in the substantia propria and partially around cystic epithelial inclusions. Magnification 10× (H&E). (b) Nests of fairly uniform naevomelanocytes showing a partial relationship to the epithelium of local cystic epithelial inclusions. There is also a small naevic component at the base of the surface epithelium. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Clinical features
Presentation
These naevi are similar to those occurring in the skin and present as circumscribed, flat or slightly raised macules or papules, occurring most commonly on the bulbar conjunctiva [125] (Figure 132.30). They are often amelanotic (30% of cases), particularly in children in whom they often acquire pigmentation after puberty.
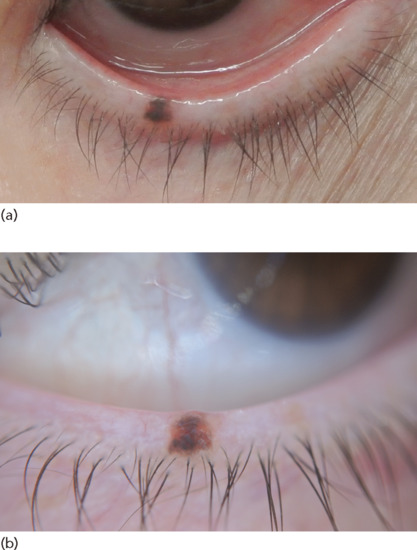
Figure 132.30 Conjunctival naevus. (a) A well-circumscribed papule of various shades of brown. (b) Dermoscopic image showing homogeneous brown-greyish pigmentation with a reddish hue.
(Courtesy of Dr D. Sgouros, Andreas Sygros Hospital, Athens, Greece.)
Disease course and prognosis
There are no specific clinical signs that predict the transformation of a conjunctival naevus to melanoma (presumed to occur in less than 1% of naevi). Attachment to the sclera, extension into the cornea and development of ‘feeder’ vessels upon slit lamp examination represent worrisome changes [126, 127].
Naevi of the nail matrix or nail bed
Epidemiology
Age
These naevi are usually seen in children or young adults [128].
Clinical features
Presentation
Nail-associated naevi may be congenital or acquired. Fingernails are more frequently affected than toenails. Their most common presentation consists of a longitudinal, parallel and homogeneous pigmentation ranging from light brown to dark brown to black on the underside of the nail plate (Figure 132.31) [129]. A pseudo-Hutchinson sign, presenting as visible pigmentation through a relatively translucent cuticle, is characteristically seen in nail matrix naevi.
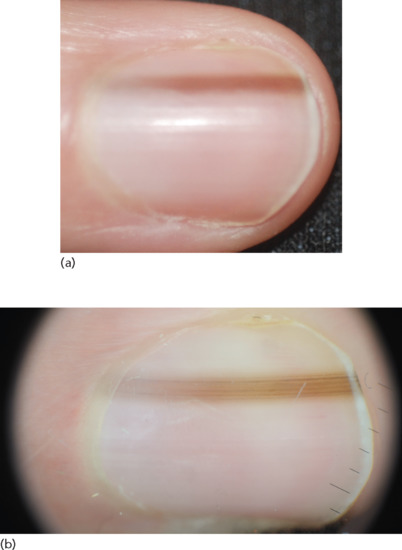
Figure 132.31 Nail matrix naevus. (a) A longitudinal pigmented band. (b) Dermoscopic image showing brown, longitudinal parallel lines with regular spacing and thickness.
Differential diagnosis
The distinction from early subungual melanoma is often difficult, requiring a complete longitudinal excision of the pigmented band for an accurate histological assessment.
Investigations
Dermoscopy can be used to improve the clinical diagnosis of such lesions, demonstrating brown, longitudinal, parallel lines with regular spacing and thickness compared with the irregular pattern observed in melanoma (Figure 132.31b) [130, 131].
NAEVI WITH UNUSUAL MORPHOLOGY
Introduction and general description
There is a distinct group of naevi that exhibit unusual clinicopathological and dermoscopic features. These naevi, designated as ‘special’ naevi, include those with clinically atypical presentation that simulate melanoma (e.g. combined and recurrent naevi) and those with targetoid morphology (e.g. halo, Mayerson, cockade and targetoid haemosiderotic naevi) (see Table 132.3).
Combined melanocytic naevi
Definition
Combined naevi present with two different types of benign melanocytic proliferations within the same naevus [132]. The most frequent combination is that of a blue naevus associated with a congenital, acquired or Spitz naevus.
Epidemiology
Incidence and prevalence
Combined naevi correspond to approximately 1% of excised naevi [133].
Pathophysiology
It is unclear whether pathogenetically they represent a mere coexistence of two different naevic populations or if they derive from a single cell proliferation differentiating into two different types of naevi.
Clinical features
Presentation
Clinically they present as a bluish macule or papule (representing a blue naevus) surrounded by a macular brown area (representing a compound naevus) (Figure 132.32) [134]. The back is the most common location.
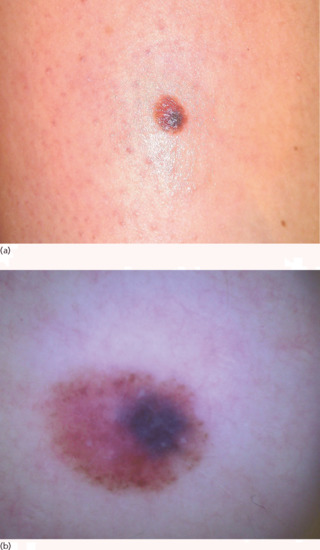
Figure 132.32 (a) Combined naevus. (b) Combined naevus with eccentric, structureless, bluish area and peripheral, brown, globular pattern.
(Courtesy of Dr D. Sgouros, Andreas Sygros Hospital, Athens, Greece.)
Differential diagnosis
Due to the presence of two different naevic components, combined naevi are characterized by colour variegations that raise the suspicion of melanoma. For this reason, excision and histopathological analysis of these lesions is generally recommended.
Investigations
Dermoscopically, the most frequent pattern is a typical brownish reticular and/or globular pattern with central or eccentric structureless blue pigmentation (Figure 132.32b) [135, 136].
Recurrent melanocytic naevi
Definition
Recurrent melanocytic naevi are benign melanocytic naevi that recur after incomplete surgical excision or trauma.
Epidemiology
Incidence and prevalence
They occur more frequently in women aged 20–30 years of age.
Pathophysiology
Various theories have been proposed such as regrowth from a residual dermal naevus, repopulation from seeded melanocytes after initial removal or from adnexal structures, and junctional stimulation from remaining hair roots or from the periphery of the lesion [132].
Pathology
Histopathologically, recurrent naevi have an intraepidermal presence of melanocytes above the level of a scar. Effacement of rete ridges and a lentiginous or nested proliferation of naevus cells is also seen. The naevomelanocytes have abundant melanin and uniform nucleoli, although low grade atypical cytology can be observed occasionally.
Clinical features
History
Recurrent naevi usually originate from acquired ordinary melanocytic naevi removed by shave biopsy for cosmetic reasons. Recurrences after incomplete removal of blue naevi, Spitz naevi and clinically atypical or dysplastic naevi have been also reported [137]. Recurrences typically arise in the centre of the scar, usually within a time frame from 6 weeks to 6 months after removal of the initial naevus [138].
Presentation
Clinically, they appear as a macular area with hyper- or hypopigmentation, linear streaks and mottled pigmentation (halo, stippled or diffuse) measuring 2–5 mm in diameter (Figure 132.33) [139]. Due to their atypical clinical and histopathological presentation, they are viewed as simulators of melanoma, hence the term ‘pseudo-melanoma’. Their most common location is the trunk (back), followed by the face and extremities [138].
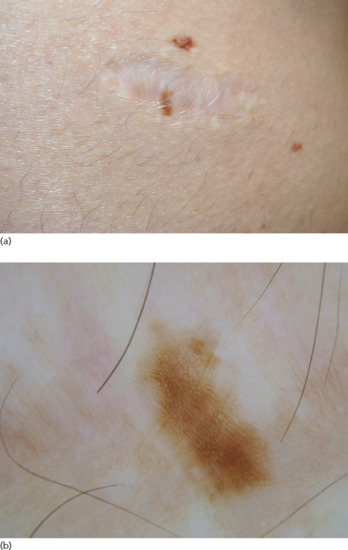
Figure 132.33 Recurrent naevus. (a) Macule developing within the scar of a previously excised melanocytic naevus. (b) Dermoscopic image showing a slightly asymmetrical macule consisting of a smooth reticular network of fine lines and brownish colour on a whitish scar.
(Courtesy of Dr D. Sgouros, Andreas Sygros Hospital, Athens, Greece.)
Differential diagnosis
The differential diagnosis of recurrent naevi is often difficult to make from recurrent melanoma and reactive pigmentation of scars. The history of occurrence after the removal of a pre-existing naevus, the distribution of pigmentation within the scar and specific dermoscopic findings are important parameters in setting the correct diagnosis. Their presentation within 6 months of excision and their confinement within the boundaries of the scar point more towards a recurrent naevus [140]. In contrast, a recurrent melanoma arises more slowly and tends to grow beyond the borders of the scar into the adjacent normal skin.
Investigations
Dermoscopy of recurrent naevi reveals an irregular prominent network, the presence of globules and a heterogeneous pigmentation (Figure 132.33b) [141]. According to a recent study examining the dermoscopic features of recurrent naevi versus recurrent melanomas, there is a more symmetrical and centrifugal growth confined within the area of the scar in recurrent naevi compared with the chaotic, often eccentric and non-continuous pigmentation of recurrent melanoma extending beyond the scar's edge [142].
Management
The management of these lesions is often not straightforward. Factors such as the patient's age, the anatomical site, the time from removal, the type of removal (surgical versus destructive modality) and the growth pattern are important clues, but the most important is the histopathology of the first excision. If the diagnosis of the primary lesion was that of a banal naevus then no further treatment is warranted. If there is a previous report of an atypical naevus or if the histopathology of the primary lesion is not available, then a thorough excision of the recurrent lesion and a histopathological evaluation is necessary.
Halo naevus
Definition and nomenclature
A halo naevus is a melanocytic naevus surrounded by a depigmented area resembling a halo.
Epidemiology
Incidence and prevalence
Halo naevi are relatively common, presenting in approximately 1% of the population. They may present as solitary or multiple lesions.
Age
These naevi are more commonly seen in children, adolescents and young adults.
Sex
There is no sex predilection.
Associated diseases
Halo naevi can be associated with autoimmune disorders like vitiligo, Hashimoto thyroiditis, alopecia areata and atopic eczema.
Pathophysiology
The exact pathophysiology of halo naevi is unknown. They are considered an autoimmune response against naevus cells. There is some laboratory evidence of local and circulating immunological T-cell activation in patients with unexcised halo naevi [143].
Predisposing factors
Stress and puberty are considered to be triggering factors [132]. Familial cases have been reported [144]. Regression of several melanocytic naevi in a patient with metastatic melanoma receiving ipilimumab has been observed [145].
Pathology
Halo naevi are usually compound melanocytic naevi, although junctional or dermal naevi are occasionally noted. At the time of halo appearance they demonstrate at the dermal component a heavy, lichenoid, lymphocytic infiltrate within the dermis, with naevus cells arranged in nests or singly among the inflammatory cells (Figure 132.34). In the intraepidermal component single lymphocytes are distributed among the naevomelanocytes, in a linear pattern, extending beyond the boundaries of the dermal component in cases of compound naevi. The use of dihydroxy-phenylalanine stains reveals a loss of epidermal melanocytes in the depigmented area.

Figure 132.34 Halo naevus. (a) Dense lymphocytic infiltrate with disruption of the naevomelanocytic aggregates, especially in the mid and deep portion of the dermal component of the naevus. Magnification 10× (H&E). (b) Small lymphocytes disrupt the naevic aggregates and intermingle with single naevomelanocytes in the dermal component of the naevus. Lymphocytes are also distributed between the basal cells of the overlying epidermis and the naevic cells of junctional nests. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Environmental factors
Halo naevi sometimes appear after intense sun exposure [146].
Clinical features
Presentation
Initially, a rim of depigmentation appears around a pre-existing melanocytic naevus (Figure 132.35). This white halo is particularly visible during the summer months when the unaffected adjacent skin acquires a tan. During the following months the naevus may gradually shrink or even disappear completely, leaving a white macule. Approximately half of halo naevi undergo total clinical and histological regression.

Figure 132.35 (a) Halo naevus. (b) Dermoscopic image of a halo naevus showing a symmetrical compound naevus (globular pattern) surrounded by a whitish halo.
Halo naevi are located primarily in the back. Multiple halo naevi may develop, while other adjacent naevi remain unchanged.
Differential diagnosis
In older patients presenting a single lesion, the possibility of a melanoma in regression should be excluded. In a case of melanoma, both the central pigmented area and the surrounding halo appear irregular, while the centre of the lesion presents dermoscopic features that are suggestive of melanoma. In suspicious cases, excisional biopsy should be performed.
Disease course and prognosis
The depigmented area usually persists for a decade or longer. A subgroup may progress through stages of involution with a return to normal colour, but even these lesions usually persist for several years (average of 7.8 years) [147].
Investigations
In dermoscopy, the central naevus exhibits the globular and/or homogeneous patterns characteristic of melanocytic naevi in young ages, surrounded by a rim of white regression-like depigmentation with a variable diameter (Figure 132.35b) [132,148].
Management
No treatment is required. Reassurance of patients, particularly in case of multiple lesions, and UV protection measures to avoid sunburn of the depigmented skin area are advised. A halo naevus presenting in an older patient should raise concern, especially in the absence of vitiligo and no history of halo naevi in the past. In such cases, a thorough skin and lymph node examination is recommended to exclude melanoma elsewhere.
Meyerson naevus
Definition and nomenclature
This is a melanocytic naevus that develops an eczematous-like inflammatory reaction.
Epidemiology
Incidence and prevalence
A Meyerson naevus is an unusual type of naevus.
Age
It usually presents in young individuals.
Associated diseases
Meyerson naevi have been associated with atopic eczema.
Pathophysiology
Aetiopathogenesis of this naevus remains unclear. Theories proposing an atypical pityriasis rosea-like reaction, subacute allergic dermatitis or hypersensitivity reaction have not been confirmed [149, 150, 151, 152].
Predisposing factors
Treatment with interferon α has been reported prior to the development of Meyerson naevi [153, 154].
Pathology
Histology reveals a common, usually compound, melanocytic naevus with associated spongiotic dermatitis of the overlying epidermis. Also seen is a dense, perivascular infiltrate in the upper dermis composed mainly of CD4+ lymphocytes and occasionally eosinophils.
Environmental factors
Severe sunburn has been implicated [155].
Clinical features
Presentation
A Meyerson naevus presents as a melanocytic naevus that develops an erythematous halo with overlying scales (Figure 132.36). The lesion resembles a naevus with superimposed discoid eczema and it may be slightly pruritic. It usually arises on the trunk and proximal extremities. Multiple lesions can occur.
Figure 132.36 Meyerson naevus. (a) A small-sized naevus with an eczematous component. (b) Dermoscopic image showing yellowish crusts covering a naevus with a faint brownish network.
(Courtesy of Dr I. Zalaudek, Skin Cancer Unit Arcispedale Santa Maria Nuova – IRCCS Reggio Emilia, Italy.)
Halo dermatitis has been reported around various benign skin lesions (e.g. common acquired and congenital naevi, Sutton naevi, Spitz naevi, clinically atypical naevi, sebaceous naevus, keloids, insect bites, seborrhoic keratoses, dermatofibromas) and malignant skin lesions (e.g. melanoma, basal and squamous cell carcinomas) [156].
Differential diagnosis
Single lesions could occasionally be confused with melanoma or halo naevus. In multiple Meyerson naevi, the differential diagnosis includes pityriasis rosea and roseola of secondary syphilis [132].
Disease course and prognosis
The eczematoid changes usually resolve spontaneously after a few months, leaving the involved naevus intact, although some degree of hypopigmentation or even complete resolution of the naevus has been described [157]. Meyerson naevus is a similar lesion to halo naevus and may coexist with this entity in the same patient. Occasionally, a Meyerson naevus can progress to a halo naevus or vice versa [158, 159].
Investigations
Dermoscopy reveals the benign pattern of the involved melanocytic naevus, often blurred by a yellowish, overlying, superficial serocrust (Figure 132.36b) [132].
Management
Normally, the eczematous reaction subsides after 1–2 weeks of treatment with a moderately potent topical steroid. Clinical re-evaluation and dermoscopic examination will confirm that the underlying naevus is benign.
Cockade naevus
Definition and nomenclature
A cockade naevus is a benign melanocytic lesion with a target-like appearance resembling a rosette.
Epidemiology
Incidence and prevalence
A cockade naevus is an uncommon type of naevus.
Age
It usually presents in children and adolescents.
Pathophysiology
It has been proposed that naevus cells in the central and peripheral areas produce melanin more actively [160], or that there is a lack of melanin synthesis involving the melanocytes of the non-pigmented rim [161].
Pathology
The central component of the naevus is that of a junctional or compound type, while the periphery of the lesion is composed of junctional nests and may present increased pigment in the dermis [162].
Clinical features
Presentation
The central component of the naevus is a dark, often papular area, which is surrounded by a non-pigmented circular zone and an outer pigmented ring (Figure 132.37). Lesions are usually multiple and located on the trunk, or on the scalp in young patients.
Figure 132.37 (a) Cockade naevus. (b) Dermoscropic image of a cockade naevus showing a darker, central homogeneous pattern, a lighter inner ring and a peripheral brown reticular ring.
(Courtesy of Dr I. Zalaudek, Skin Cancer Unit Arcispedale Santa Maria Nuova – IRCCS Reggio Emilia, Italy.)
Differential diagnosis
A target-like appearance has also been reported in association with blue naevi [163] and melanoma [164].
Investigations
Dermoscopy reveals a naevus with a darker, central globular or homogeneous pattern, surrounded by a structureless inner ring and a more peripheral darker reticular ring (Figure 132.37b) [165, 166].
Management
A cockade naevus is a benign lesion, thus no treatment is required.
Targetoid haemosiderotic naevus
Definition
A targetoid haemosiderotic naevus derives from mechanical trauma of a melanocytic naevus which is usually elevated or exophytic.
Epidemiology
Incidence and prevalence
It is common in children and young adults. The upper chest is the most common location [167, 168].
Pathophysiology
Pathology
Histopathologically, targetoid haemosiderotic naevus consists of naevus cells mingled with extravasated blood vessels and an increased number of irregular, ectatic, vascular channels. The peripheral halo demonstrates extensive haemorrhage with haemosiderin and fibrin deposits combined with slit-shaped vascular channels that dissect in between collagen bundles. A mild, inflammatory, primarily eosinophilic infiltrate is also seen.
Clinical features
History
Targetoid haemosiderotic naevus usually appears as a sudden change of pigmentation in a pre-existing naevus following mechanical irritation, usually from clothing, shaving or scratching [167, 168]. A history of trauma or irritation is not always apparent to the patient. Symptoms indicating trauma such as pain, tenderness or pruritus are common.
Presentation
Clinically, the lesion presents as a brown, red-brown or violaceous papule surrounded by a thin pale area and a peripheral ecchymotic area that tends to expand, become fainter and resolve while the central papule persists (Figure 132.38).
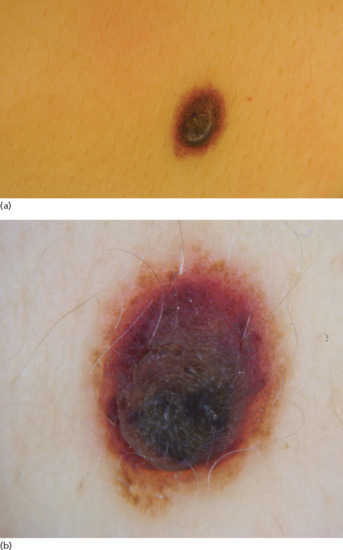
Figure 132.38 Targetoid haemosiderotic naevus. (a) Naevus acutely presenting with a haemorrhagic halo. (b) Dermoscopic image showing a peripheral, structureless, purple rim around a central pre-existing compound naevus.
Differential diagnosis
The differential diagnosis includes targetoid haemosiderotic haemangioma, cockade naevus, traumatized angiokeratoma and melanoma.
Investigations
Dermoscopy reveals haemorrhagic changes with a red to purple colour, surrounding a naevus with a usually globular pattern (Figure 132.38b) [169]. Jet-black areas with irregular shape and size as well as comma-shaped vessels are also seen. The periphery of the lesion representing the ecchymotic halo shows an ill-defined, pale area surrounded by a reddish zone with jagged edges.
Management
Management includes reassurance of the patient and the use of a topical antibiotic or steroid preparation until the inflammatory/haemorrhagic changes have resolved.
OTHER NAEVI
Spitz naevus
Definition and nomenclature
A Spitz naevus is a melanocytic lesion characterized by spindled and/or epitheloid naevomelanocytes.
Introduction and general description
Spitz naevus was initially described by Sophie Spitz as an unusual type of melanocytic proliferation in children, with features histologically indistinguishable from melanoma (hence the original term of ‘juvenile melanoma’), but with a favourable prognosis [170]. Currently the entity of Spitz naevi remains a subject of controversy due to their clinical and histological variability, their overlapping histological characteristics with melanoma and the uncertainty of their biological behaviour in certain cases. Refinement of the various clinical and histological subtypes of Spitz naevi has resulted in a highly complex morphological classification of these tumours [171,172]. At one end of the spectrum is the common or classic Spitz naevus (see Table 132.3), a benign proliferation frequently occurring in children; at the other end are lesions with extensive pleomorphic features sufficient for the diagnosis of melanoma (‘spitzoid melanoma’). In between lies a heterogeneous group of lesions with varying features and unknown malignant potential termed ‘atypical Spitz tumours’, ‘atypical spitzoid tumour of unknown malignant potential’ or ‘diagnostically controversial Spitzoid melanocytic tumours’ [173, 174, 175].
Epidemiology
Incidence and prevalence
Spitz naevi account for 1% of excised naevi in children [176].
Age
Spitz naevi are more commonly seen in children. Approximately 50% of lesions occur in patients under the age of 14 years, 25% between the ages of 15 and 30, and 25% over the age of 30 [177].
Sex
Both genders are equally affected, although age-dependent variations may be observed [178, 179, 180].
Ethnicity
Spitz naevi occur predominantly in white populations, but they have also been described in black, Hispanic and Asian groups [181, 182].
Associated diseases
Disseminated, eruptive Spitz naevi have been reported to occur with Addison disease, HIV infection, chemotherapy, puberty and pregnancy [163,184].
Pathophysiology
Most lesions are acquired, but up to 7% of Spitz naevi can occur congenitally [185].
Pathology
The ‘classic’ Spitz naevus typically is a symmetrical and well-defined compound naevus. Intraepidermal and intradermal forms have been also reported [186]. There is a degree of epidermal hyperplasia overlying the naevus, without evidence of malignant intraepidermal pagetoid spreading of naevic cells (Figure 132.39a). Naevomelanocytic nests are neatly located between keratinocytes, unlike their disorderly arrangement in melanoma. The naevomelanocytic nests at the dermal–epidermal junction are often separated from the surrounding keratinocytes by a cleft caused by a retraction artefact. The naevus cells may be either spindle shaped, streaming into the dermis in interlacing bundles, or epithelioid, arranged in clusters, with giant and multinucleated naevus cells seen among them.
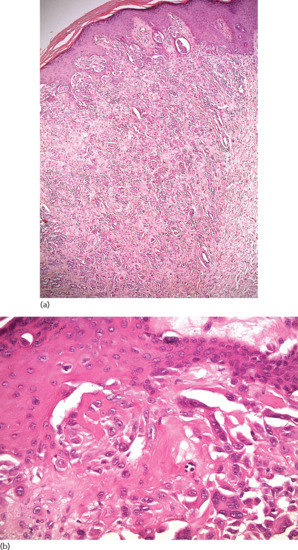
Figure 132.39 Compound naevus of Spitz. (a) Sparsly demarcated naevomelanocytic proliferation at the junctional area and mainly in the dermis. The dermal component has prominent cellular pleomorphism and evidence of maturation in the deeper portion of the lesion. Magnification 10× (H&E). (b) Epithelioid cells with eosinophilic cytoplasm and nuclear pleomorphism with prominent nucleoli. Multinucleated giant cells are common. Eosinophilic, partially coalescent, Kamino bodies are present in the junctional area and upper papillary dermis. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Kamino bodies (i.e. amorphous eosinophilic globules containing periodic acid–Schiff-positive basement membrane constituents) are noted intraepidermally and at the dermal–epidermal junction (Figure 132.39b) [187, 188]. Their presence is neither sensitive nor specific for Spitz naevi as they may also be present in early melanoma.
In classic Spitz naevi, dermal naevomelanocytic nests and fascicles exhibit a zonation phenomenon with depth, for example uniformity in size, shape and spacing along horizontal planes. Typical mitoses may occur, especially in the upper and mid-portion of the lesion. They usually do not exceed a rate of 2/mm2. Dermal vessels are dilated and the stroma is oedematous and infiltrated by lymphocytes [180].
Several histological features have been considered to indicate a more aggressive behaviour. A Spitz naevus thicker than 1 mm in depth, with asymmetry, poor circumscription, deeper extension and ulceration is considered to have increased metastatic potential [189]. In addition, single cell pagetoid spread beyond the epidermal nests, lack of zonation with depth and persistent expansile deep nests with more deeply seated mitotic activity, are considered to be atypical histological features. Cellular heterogeneity and atypical cytology (higher nuclear to cytoplasmic ratios, hyperchromatism and abnormal nuclear borders) are additional findings supportive of an atypical lesion [190].
Reed naevi, widely considered to be a pigmented subtype of Spitz naevi, presents with an organized symmetrical and uniform architecture, similar to classic Spitz naevi. A well-demarcated junctional melanocytic activity with large quantities of melanin pigment is noted (Figure 132.40a). Spindle-shaped melanocytes proliferate downwards towards the papillary dermis but rarely involve the reticular dermis (Figure 132.40b) [191].
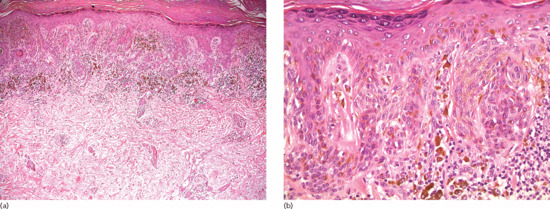
Figure 132.40 Pigmented spindle cell naevus of Reed. (a) An almost exclusively intraepidermal naevomelanocytic lesion with heavily pigmented spindle cells arranged in fascicles. There are aggregates of melanophages in the dermis beneath the lesion. Magnification 10× (H&E). (b) Epithelioid cells arranged in fascicles, often orientated vertically to the surface of the skin. A chronic inflammatory component with melanophages is prominent beneath the lesion. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Desmoplastic Spitz naevi are predominantly intradermal lesions, characterized by a relatively small number of mainly isolated large and bizarre cells with copious cytoplasm distributed among thick collagen fibres. Mitotic figures are rare [192].
Immunohistochemistry can be used to differentiate Spitz naevi from melanoma. MIB-1, which stains the proliferative marker Ki-67, has a lower expression in Spitz naevi compared with melanoma [193], while HMB-45 is more superficially expressed with diminished staining in the deeper dermal component [194]. S100 and Mart-1 are more weakly stained in Spitz naevi [195], whereas the S100A6 subtype shows more intense and diffuse expression in Spitz naevi compared with its weak and patchy staining in melanoma [196]. p16 reactivity is stained more intensely in desmoplastic Spitz naevi, while it is largely absent in melanomas [197].
Cytogenetic techniques, such as comparative genomic hybridization (CGH) and fluorescent in situ hybridization (FISH), reveal a subset of Spitz naevi (c. 15%) with amplifications in chromosome 11p (containing the HRAS gene) and a threefold increase in copy number alterations, respectively [198, 199]. Mutation analysis of Spitz naevi reveals a higher rate of HRAS mutations and a much lower rate of BRAF and NRAS mutations that are more frequently seen in melanoma and other melanocytic naevi [200].
Genetics
The rare incidence of NRAS and BRAF mutations in Spitz naevi compared with other melanocytic lesions, and the presence of HRAS mutations in a subgroup of Spitz naevi (11–15% of cases), suggests a distinct activation of the RAS pathway components in different melanocytic neoplasms [199,201,202]. A number of copy number aberrations have been described in atypical Spitz tumours, including isolated loss at 3p21 (in BAP1-associated Spitz tumours), 6q23 and 9p21 [203, 204].
In contrast to tumours with heterozygous 9p21 deletions on FISH, atypical Spitz tumours with homozygous 9p21 deletions have been associated with a considerably worse prognosis [204].
Multiple cutaneous, spitzoid, melanocytic tumours can occur in the context of a familial autosomal dominant syndrome caused by inactivating mutations of the BAP1 gene. Skin-coloured papules and nodules characterized histologically by a naevoid silhouette with large epithelioid melanocytes and an immunohistochemical loss of BAP-1 (termed naevoid melanoma-like melanocytic proliferations, or BAPomas) are typical features of this syndrome, along with a predisposition to cutaneous and uveal melanoma [205, 206].
Clinical features
History
Spitz naevi usually develop rapidly over a period of 3–6 months, reaching sizes of 1–2 cm. Following this rapid growth period, the lesions remain stable in size. Reports of spontaneous involution over time or conversion to more common types of melanocytic naevi have been reported [207].
Presentation
The classic Spitz naevus usually appears as a solitary, firm, symmetrical, sharply demarcated, round or dome-shaped nodule of equal or less than 5–6 mm diameter on average. The colour of the lesion is pink to red to reddish brown (Figure 132.41). Firm pressure or diascopy reveals the degree of melanin pigmentation. Ten per cent of Spitz naevi are pigmented lesions with colours that range from tan to brown to black (Figure 132.42). The surface of the lesion is smooth, with a thin, fragile epidermis, often causing bleeding and crusting after minor injury. The commonest sites for Spitz naevi are the head and neck area (37%), particularly the face and cheek in children, and the lower extremities (28%) in young adults [177,208]. Atypical Spitz tumours tend to be larger, more asymmetrical or ulcerated with irregular pigmentation and border outline (Table 132.4; Figure 132.43).
Table 132.4 Clinical, histological, immunohistochemical and molecular features of classic and atypical Spitz naevi
| Classic Spitz naevi | Atypical Spitz tumours | |
| Clinical features | ||
| Age | <10 years | >10 years |
| Location | Face, neck, extremities (thighs) | Trunk (back in men) |
| Diameter | <10 mm (usually 5–6 mm) | >10 mm |
| Symmetry | Usually symmetrical | Increasing asymmetry |
| Border | Well defined | Irregular |
| Surface | Smooth | Ulcerated |
| Colour | Pink/red | Irregular |
| Histology features | ||
| Architecture | Symmetrical, sharply demarcated, dome shaped, non-disruptive, orderly dispersion and regular spacing of nests and cells | Asymmetrical, poorly demarcated, infiltrating, irregular spacing and disorderly arrangement of nests and cells |
| Epidermal changes | Intact epidermis | Ulceration |
| Pagetoid spread | Limited pagetoid spread in lower epidermis | Single-cell pagetoid spread, beyond epidermal nests |
| Clefting | Presence of junctional clefting | Lack of junctional clefting |
| Kamino bodies | Aggregates of Kamino bodies | Absent or few Kamino bodeis |
| Zonation | Uniformity of cytological features across horizontal planes | Lack of zonation |
| Maturation | Maturation (smaller nests with depth), lack of deep involvement | Persistent expansile nests, subcutaneous involvement |
| Cell type | Spindle or epithelioid cell type | Hetererogeneous cell types |
| Mitoses | Mitoses <2 mm2, superficial, usually not atypical | Mitoses >2–6 mm2, deeply located, atypical |
| Cytology | Low nuclear to cytoplasmic ratio | High nuclear to cytoplasmic ratio |
| Ground glass or opaque cytoplasm | Granular or ‘dusty’ cytoplasm in epithelioid cells, scant in spindle cells | |
| Nuclear changes | Uniform nucleoli, delicate, dispersed chomatin | Large, eosinophilic nucleoli, hyperchromatism |
| Immunohistochemistry features | ||
| Immunohistochemistry | Weaker stain for S100 and Ki-67; superficial expression of HMB45, stronger expression of S100-A6 | Stronger expression of S100 and Ki-67; deeper stain for HMB45, weaker expression of S100-A6 |
| Molecular features | ||
| Cytogenetic techniques (CGH, FISH) and mutational profile | Amplifications in chromosome 11p; (+) HRAS (15% of Spitz naevi); low rates of NRAS and BRAF mutations | Multiple losses and gains; (+) BRAF/NRAS mutations |
Adapted from Luo et al. 2011 [172]; Spatz et al. 1999 [189]; Barnhill 2006 [272].
CGH, comparative genomic hybridization; FISH, fluorescent in situ hybridization.

Figure 132.41 Classic Spitz naevus. (a) A well-circumscribed pink nodule. (b) A round, symmetrical lesion with dotted vessels and lack of pigmentation.
(Courtesy of Dr I. Zalaudek, Skin Cancer Unit Arcispedale Santa Maria Nuova – IRCCS Reggio Emilia, Italy.)
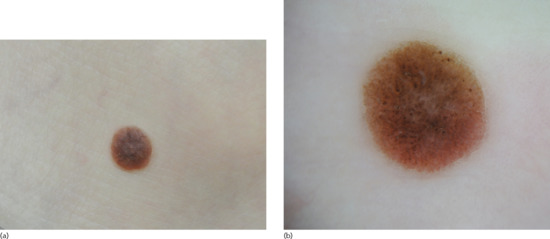
Figure 132.42 (a) Pigmented Spitz naevus. (b) Dermoscopic image of a pigmented Spitz naevus showing a well-circumscribed symmetrical nodule with a papillomatous surface and various shades of brown pigmentation. Also seen are locally distributed brownish dots and globules and white perpendicular lines in the centre of the lesion.
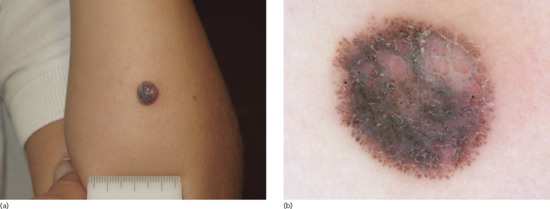
Figure 132.43 Atypical Spitz tumour. (a) Asymmetrical nodule with heterogeneous pigmentation. (b) Dermoscopic image of the symmetrical, peripheral distribution of pigmented streaks and heterogeneous pigmentation with bluish/black and whitish hue.
(Courtesy of Dr I. Zalaudek, Skin Cancer Unit Arcispedale Santa Maria Nuova – IRCCS Reggio Emilia, Italy.)
Clinical variants
A more deeply pigmented variant of Spitz naevus, called the spindle cell naevus of Reed, and initially described in 1975 [209], occurs mainly in young females and is most commonly seen on the thighs. It usually presents as solitary, densely pigmented, irregularly shaped, dark-brown or black nodule (Figure 132.44) [208].
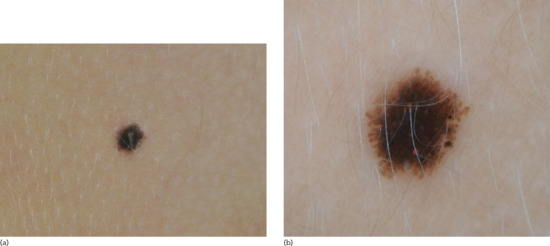
Figure 132.44 Reed naevus. (a) Darkly pigmented symmetrical macule. (b) Dermoscopic image showing a ‘starburst’ pattern with diffuse blue-black pigmentation and symmetrically distributed radial streaks in the periphery.
Rare accounts of multiple eruptive or disseminated Spitz naevi involving the entire cutaneous surface have been reported [210, 211, 212]. In agminated Spitz naevi, multiple Spitz naevi can also occur in a grouped fashion, resembling a speckled congenital naevus.
Desmoplastic Spitz naevus is a rare subtype of the Spitz naevus encountered most commonly in adults [178]. These lesions are usually pink or red, firm, raised nodules with little or no clinically visible melanocytic pigmentation. A desmoplastic Spitz is often the presenting type of a recurrent Spitz naevus following incomplete removal [213, 214].
Differential diagnosis
The differential diagnosis of Spitz naevi includes acquired, dysplastic and congenital variants of melanocytic naevi and melanoma. A number of non-melanocytic entities should also be considered such as dermatofibroma, molluscum contagiosum, pyogenic granuloma, haemangioma, mastocytoma, juvenile and adult xanthogranuloma, angiofibroma, histiocytoma and granuloma.
Disease course and prognosis
Even though the metastatic behaviour of atypical Spitz tumours is well established, their malignant potential is debated since they only rarely result in a fatal outcome [215, 216]. The use of sentinel lymph node biopsy in atypical Spitz tumours has clarified the rate of lymph node positivity (average of 38%) [217, 218, 219, 220, 221]. Nearly all sentinel lymph node biopsy positive cases resulted in negative nodes after complete lymphadenectomy and very few led to death in the immediate follow-up period. Risk stratification schemes using pathological criteria have attempted to classify Spitz tumours based on their metastatic potential. One study identified abnormal mitoses, mitotic counts of over 2/mm2 and deep mitoses as suggestive of spitzoid melanoma [222]. Spatz et al. have identified age over 10 years, diameter over 10 mm, ulceration, invasion to subcutaneous fat and mitotic activity of over 6/mm2 as suggestive of spitzoid melanoma rather than Spitz naevi [189]. Interobserver variations, even between expert pathologists, for spitzoid lesions are significant [171].
Investigations
Upon dermoscopy, Spitz naevi can demonstrate a diversity of patterns [223]. The classic, less pigmented variant (pink colour) shows a predominantly dotted vascular pattern with reticular depigmentation (see Figure 132.41b). Pigmented Spitz naevi or Reed naevi exhibit a ‘starburst’ pattern, a globular pattern or a multicomponent atypical pattern (see Figures 132.42b and 132.44b) [224]. Although there are no reliable dermoscopic features that characterize atypical Spitz tumours, these lesions often exhibit an asymmetrical peripheral distribution of pigmented streaks, as well as a heterogeneous pigmentation with bluish-black and whitish hue (see Figure 132.43b).
Management
There is a lack of consensus regarding management. Because of the clinical and histological overlap between Spitz naevi and melanoma some authors suggest that all spitzoid lesions should be surgically removed and examined histologically. Local excision of the lesion with a margin of 1–3 mm is usually sufficient to confirm the diagnosis [225]. Since local recurrences have been reported at a rate of 7–16% [226], often presenting with a more atypical histology and increased desmoplasia, incompletely removed tumours should be re-excised [176]. Due to the very low probability of melanoma in children with typical or classic Spitz naevi, some experts prefer clinical monitoring of these lesions rather than intervention, provided that a close clinical and dermoscopic follow-up is performed [227, 228]. Exceptions include large (>1 cm), nodular, ulcerated or rapidly growing lesions [224]. The histological diagnosis of an atypical Spitz tumour should be approached more aggressively and treated with a wide margin resection following the guidelines of melanoma resection. Patients can be reassured that the lesion may in fact be benign.
The significance of sentinel lymph node biopsy positivity and how to further manage the presence of nodal disease is currently unknown [215]. Given the low fatality rate of atypical Spitz tumours, the limited evidence of a survival benefit from selective lymphadenectomy in sentinel lymph node biopsy positive cases, and the potential morbidity associated with lymphadenectomy, a more judicious case-by-case use of sentinel lymph node biopsy is recommended before further evidence is available [229].
Resources
Patient resources
- Congenital Melanocytic Naevus Support Group: www.caringmattersnow.co.uk.
- Nevus Network: www.nevusnetwork.org.
- Nevus Support Australia: www.nevussupport.com. (All last accessed May 2014.)
Blue naevus and variants
Definition and nomenclature
This is a relatively common blue, blue-grey or blue-black benign melanocytic naevus comprised of dermal melanocytes, with several clinical and histological variants (see Table 132.3).
Epidemiology
Incidence and prevalence
Their estimated prevalence in white populations is 0.5–4%.
Age
Blue naevi usually present during childhood, puberty or early adulthood, but can occur at all ages.
Sex
They are more common in females.
Associated diseases
Multiple epithelioid blue naevi may be associated with the LAMB (lentigines, atrial and mucocutaneous myxomas and multiple blue naevi) syndrome.
Pathophysiology
The aetiopathogenesis is unclear. The most favoured hypothesis proposes that blue naevi originate from latent dermal dendritic melanocytes, which are remnants of the melanocyte migration from the neural crest to the epidermis during gestation.
Pathology
Common blue naevi consist of spindle-shaped, dendritic melanocytes admixed with ovoid or fusiform, elongated melanocytes, located in the dermis in an inverted wedge shape (Figure 132.45a). Naevus cells can extend into the lower dermis along appendages or in the perivascular and perineural areas. The dendritic melanocytes do not show significant mitotic activity or atypical cytology and stain positively for S100, HMB-45 and MART-1 (Figure 132.45b). An admixture of melanophages with intracytoplasmic coarse melanin granules, is often seen. With the exception of combined blue naevi, in which a blue naevus coexists with a congenital, acquired or Spitz naevus, blue naevi have no junctional component. A varying degree of stromal fibroplasia is also noted.

Figure 132.45 Common blue naevus. (a) Poorly circumscribed area of elongated, pigmented melanocytes in the dermis. The dendritic melanocytes are located between the collagen bundles of the reticular dermis. The overlying epidermis may be slightly hyperplastic. Magnification 10× (H&E). (b) A proliferation of elongated, dendritic melanocytes without atypia or mitoses and with finely distributed intracytoplasmic melanin can be seen among the collagen bundles of the reticular dermis. Round melanophages with heavily pigmented cytoplasm and coarse melanin intermingle with the melanocytes. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece)
The cellular blue naevus has a characteristic architecture related to a ‘dumb-bell’-shaped extension of the lesion to the deep reticular dermis or the subcutaneous fat (Figure 132.46). In common blue naevi there are nodules or fascicles of larger, oval to spindle naevomelanocytes with clear vacuolated, less pigmented cytoplasm. These cells stain positively for S100, HMB-4, and MART-1, and show rare mitotic activity or cytological atypia [230]. Variable numbers of melanophages with coarse melanin pigment are also present.
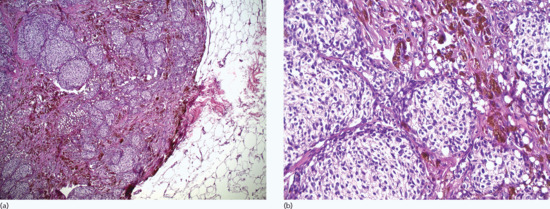
Figure 132.46 Cellular blue naevus. (a) Multiple amelanotic nodules of naevomelanocytes are characteristic. The cellular component of blue naevus extends deep to the reticular dermis of the skin and often projects into the adjacent subcutaneous fat. There is no necrosis. Magnification 10× (H&E). (b) The cellular nodules consist of almost uniform melanocytes with light eosinophilic or even clear cytoplasm. There is focal, finely distributed, intracytoplasmic melanin pigment. Between the cellular nodules there are aggregates of heavily pigmented dendritic melanocytes. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Cellular blue naevi with increased atypia and mitoses (3–4 mitoses/mm2) have been designated as atypical cellular blue naevi, a term used to denote a biological behaviour intermediate between cellular blue naevus and malignant blue naevus.
Epithelioid blue naevus (or pigmented epithelioid melanocytoma) can occur sporadically or in association with the Carney complex. The histological features that distinguish it from common blue naevi include vesicular rather than hyperchromatic nuclei of the dendritic cells and the presence of pigmented, polygonal and – more characteristicly – large epithelioid cells.
A deep penetrating naevus is characterized by loosely organized nests of pleomorphic, pigmented epithelioid cells that penetrate in an inverted wedge configuration deep into the dermis or subcutaneous fat [231]. Unlike the majority of blue naevi, the deep penetrating naevus does not harbour mutations in Gnaq and Gna11 proteins. The recent identification of HRAS mutations in these naevi suggest that they may be considered variants of Spitz rather than blue naevi [232].
Sclerosing (or desmoplastic) blue naevi show marked dermal fibrosis and hyaline sclerosis and should be differentiated from desmoplastic melanoma.
Occasionally, blue naevi can be amelanotic or hypomelanotic. Due to the lack of pigment, a diagnosis of blue naevus is based on the presence of dermal spindle cells that are HMB-45 positive on immunohistochemistry.
Genetics
Blue naevi do not show oncogenic mutations in the signalling components of the MAP kinase pathway (such as BRAF or NRAS) which are common in other types of melanocytic naevi as well as melanomas. Somatic mutations of the GNAQ gene (codon 209), a member of the G-protein a subunit, converting it into a dominant-acting oncogene are present in 46–83% of blue naevi, 50% of malignant blue naevi and 46% of ocular melanoma of the uvea. A smaller proportion of blue naevi (c. 7%) have mutations in the Gna11 gene, also a membrane bound guanosine triphosphatase (GTPase) [56].
Clinical features
Presentation
Blue naevi typically present as single, blue-black or deep blue, dome-shaped papules, with a diameter of less than 1–2 cm. Large or giant lesions, ulceration or the development of subcutaneous nodules can also be observed but are rare events. The blue colour is caused by the ‘Tyndall effect’: dermal pigment absorbs the longer wavelengths of light and scatters blue light. Locations more commonly involved are the face and scalp, the dorsal area of the distal extremities (Figure 132.47) and the buttocks.
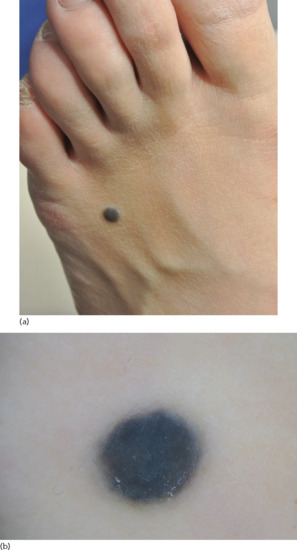
Figure 132.47 (a) Blue naevus. (b) Dermoscopic image of a blue naevus showing a homogeneous blue pattern.
Blue naevi can incidentally arise in the prostate, female genital tract, lymph nodes, conjunctiva, nasal and oral mucosa and lungs.
Clinical variants
A variety of clinical variants have been described, such as the large congenital blue naevus, large plaque blue naevus with subcutaneous cellular nodules, and agminate, eruptive and target blue naevi.
Some of the histological variants of blue naevi may also present with distinct clinical features. The combined blue naevus may have an unusual appearance, causing concern. A cellular blue naevus (Figure 132.48) may develop before birth, but frequently becomes apparent around puberty. The deep penetrating naevus is more commonly observed on the head and neck, presenting with a diffuse, irregular lateral margin.
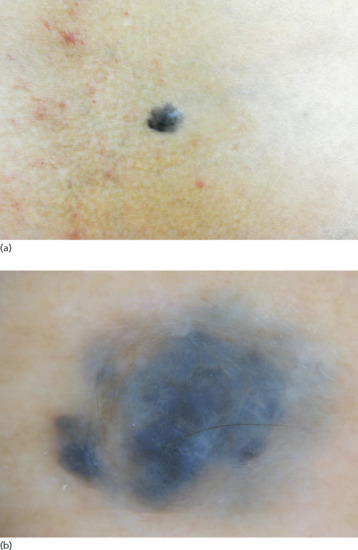
Figure 132.48 (a) Cellular blue naevus. (b) Dermoscopic image of a cellular blue naevus showing a well-circumscribed, protuberant nodule with homogeneous dark blue pigmentation and randomly distributed whitish zones corresponding histopathologically to fibrosis.
Differential diagnosis
The most important distinction is between the blue naevus and melanoma. Blue naevi have distinct dermoscopic features and are relatively static lesions. The histology of each is characteristic and can easily help separate the two entities. Immunohistochemistry and FISH assay may be useful in distinguishing challenging cases.
Satellite lesions of blue naevi resembling cutaneous metastases of melanoma have been reported [233].
Disease course and prognosis
Blue naevi are generally non-progressive proliferations. Enlargement of the lesion may raise concern of malignant transformation.
Investigations
In dermoscopy blue naevi lack the pigmented network of other melanocytic lesions, and show a distinct homogeneous, structureless, blue, blue-gray, blue-brown or blue-black colour (Figure 132.47b). However, the dermoscopy of blue naevi may vary and lesions with polychromatic pigmentation and local dermoscopic criteria (dots/globules, whitish, scar-like areas, peripheral streaks and vessels) are not uncommon, making their distinction from melanoma difficult (Figure 132.48b) [131].
Management
Blue naevi do not require treatment, except for cosmetic reasons (e.g. facial location). When in clinical doubt, an excisional biopsy should be performed to rule out melanoma.
Malignant blue naevus
Definition
This descriptive term corresponds to malignant melanomas that arise within a blue naevus or resemble a blue naevus.
Introduction and general description
A malignant blue naevus is in fact a melanoma that arises in association with a blue naevus (usually a cellular blue naevus), or at the site of a previously excised blue naevus, or is a melanoma arising de novo but with histological features resembling a blue naevus [234].
Epidemiology
Incidence and prevalence
Malignant blue naevus is extremely rare.
Age
It occurs in older ages compared with blue naevi, usually developing during the fourth decade of life.
Sex
Unlike blue naevi, malignant blue naevi are more common in men.
Pathophysiology
Pathology
Malignant blue naevi usually have the architectural features found in cellular blue naevi, with the addition of a malignant component. Abnormal mitotic figures, severe cytological atypia, foci of necrosis, vascular infiltration and destructive tumour invasion are histological findings that support the diagnosis of malignancy.
Clinical features
Presentation
A malignant blue naevus presents as an evolving deep-blue or black nodule or plaque which may become ulcerated. It is located in the same areas as blue naevi, with the scalp being the commonest site (Figure 132.49). Metastases can occur as in other melanoma subtypes.
Figure 132.49 Malignant blue naevus that has arisen in a previous cellular blue naevus. This lesion subsequently metastasized to the lymph nodes.
Differential diagnosis
Malignant blue naevi should be differentiated from blue naevi and the so-called atypical blue naevi.
Disease course and prognosis
The common belief that malignant blue naevi are more aggressive than other subtypes of melanomas was not confirmed in an Australian study. In this study no difference in survival or in the risk of metastases was observed compared with matched controls [235].
Management
Following excisional biopsy with a 2 mm margin and histological confirmation of malignancy, management should follow the established guidelines for melanoma (see Chapter 143).
Clinically atypical naevi
Definition and nomenclature
These are melanocytic naevi, 5 mm or larger in diameter, with a macular component, irregular and poorly defined borders, asymmetrical outline and variable pigmentation (see Table 132.3).
Introduction and general description
In 1978 Clark et al. described members of melanoma-prone families who had a personal history of melanoma and who exhibited high numbers of naevi with distinct clinical and histological characteristics [236]. These naevi were named B-K moles and the syndrome B-K mole syndrome after the initials of two probands. Their malignant potential was considered analogous to cervical dysplasia. During the same year, Lynch et al. used the term familial atypical multiple mole melanoma (FAMMM) syndrome in the same context [237]. In 1980, Elder et al. introduced the term dysplastic naevus syndrome (DNS) with familial and sporadic variants, stating that these naevi were precursors of melanoma due to their histological features [238]. The term dysplastic naevus was also proposed during the same year by Greene et al. as a distinction from banal naevi and an intermediate naevus phenotype in the spectrum of melanoma development [239]. Ever since, the confusion and controversy caused by differences in terminology, definitions and criteria (clinical and histological) used has not ceased.
Clinically atypical naevi are currently considered a distinct subgroup of naevi. Although they are benign lesions, they exhibit clinicopathological characteristics that may resemble early radial growth phase melanomas. They are also risk factors of melanoma and, to lesser extent, potential precursors of melanoma. In this way, they represent the intermediate part of the melanocytic neoplasm spectrum, with common (or banal) naevi at one end and melanoma at the other.
For the purposes of this chapter, the term ‘atypical naevus’ is used as a clinical description, while ‘dysplastic naevus’ is used as a histological one.
Epidemiology
Incidence and prevalence
The prevalence of atypical naevi varies considerably among different studies, ranging from 2% up to 50% [240]. Their frequency among melanoma patients is higher, ranging from 34% to 59% [241]. They are less frequent than banal naevi in the general population, their prevalence increasing in ‘high risk’ individuals with a personal or family history of melanoma [242].
Age
They are more prevalent in younger ages (less than 30–40 years of age), probably because a portion of them regress later in life. Typically, they appear during childhood and they become more prominent in puberty. Atypical naevi occur in older ages at a lower rate, and should be cautiously examined to rule out melanoma.
Sex
There is no sex predilection.
Ethnicity
Their prevalence among different ethnicities is variable.
Associated diseases
Clinically, atypical naevi have consistently been associated with melanoma risk in relevant studies. This risk seems to depend on the number of atypical naevi, as well as on the personal and family history of melanoma. In sporadic atypical naevi, the presence of one naevus confers a relevant risk (RR) of 1.45, rising up to 6.36 for five atypical naevi [89]. In the study by Tucker et al. [243], 10 or more atypical moles conferred a 12-fold risk of melanoma. Atypical naevi are also associated with a higher risk of multiple primary melanomas [244, 245, 246, 247, 248]. In the setting of melanoma kindreds with increased numbers of atypical naevi and multiple common naevi (FAMMM, DMS, AMS), the relative risk for melanoma is even greater, reaching 85-fold in melanoma-prone family members with dysplastic naevi (Figure 132.50) [249].

Figure 132.50 Dysplastic naevus syndrome: multiple clinically atypical naevi on the back. The lower surgical scar on the sacral area corresponds to a previously removed superficial spreading melanoma. The patient also had a second primary melanoma on the scalp.
Pathophysiology
Predisposing factors
Similar to common naevi, atypical naevi are also considered to be genetically determined, based on evidence from twin studies [250]. The role of environmental exposures is unknown although sunburn may be important [251].
Pathology
The main histological features of clinically atypical or dysplastic naevi include characteristic architectural and cytological features, as well as a fibrotic and inflammatory host response (Figure 132.51). These features were originally thought to be specific; however, there is a significant overlap with other types of naevi (e.g. acquired naevi smaller than 5 mm, some congenital naevi, atypical lentiginous naevi, naevi of special sites) [252] as well as with lentigo maligna and radial growth phase melanomas. Diagnostic criteria for dysplastic naevi have been proposed by several groups [253, 254, 255, 256] and are presented in Table 132.5. According to the World Health Organization, a histological diagnosis of dysplastic naevus requires the presence of both major and at least two minor criteria. The major criteria are: (i) the basilar proliferation of atypical naevomelanocytes (extending in at least three rete ridges or ‘pegs’ beyond any dermal naevocellular component); and (ii) the organization of this proliferation in a lentiginous or epithelioid cell pattern. The minor criteria include: (i) the presence of lamellar fibrosis or concentric eosinophilic fibrosis; (ii) neovascularization; (iii) an inflammatory response; and (iv) the fusion of rete ridges. The degree of mean concordance among the 10 members of the panel who examined 114 specimens of benign acquired naevi, dysplastic naevi and radial growth phase melanomas reached 92%. The National Institutes of Health has proposed that the term dysplastic naevus be abandoned and replaced by the term ‘naevus with architectural disorder’ accompanied by a statement describing the presence and degree of melanocyte atypia [257].
Table 132.5 Published criteria of the definition of clinically atypical or dysplastic melanocytic naevi
| Parameters | Clark's histological criteria [253] | WHO criteria [254]c | EORTC criteria [255]e | Duke University criteria/grading system [256]g |
| Architecture | Nests bridge rete ridges | Basilar proliferation of atypical melanocytes, extending at least three rete ridges beyond dermal component (major) | Marked junctional proliferation | Junctional component nested at both edges |
| Nests at the side of rete ridges | ||||
| Single cells between nests | Irregular naevus nests | Overall symmetry | ||
| Lentiginous elongation of rete ridges | Organization of proliferation in lentiginous or epithelioid cell pattern (major) | >5% of nests cohesive | ||
| Anastomosis of rete ridges | Suprabasal spread (prominent or at edge) | |||
| Little or no pagetoid spread | Fusion of rete ridges (minor) | >50% confluence of proliferation Single cell proliferation absent or focal |
||
| Cytological atypia | Variable slight to moderate atypia | Large nuclei | Nuclei round or oval and euchromatic | |
| Few (if any) mitoses | Nuclei larger than those of basal keratinocytes | |||
| Scattered epithelioid naevus cells and cells with finely granular melanin | ||||
| Nucleoli prominent | ||||
| Cell diameter twice that of basal keratinocytes | ||||
| Host response | Patchy lymphocytic infiltrate | Lamellar or concentric eosinophilic fibrosis (minor) | Lymphohistiocytic infiltrate | |
| Eosinophilic fibroplasia | ||||
| Lamellar fibroplasia | Neovascularization (minor) | |||
| Inflammatory response (minor) |
Adapted from Elder et al. 1982 [253]; Clement et al. 1991 [254]; de Wit et al. 1993 [255]; Shea et al. 1999 [256].
aThe diagnosis of dysplastic naevi requires fulfilment of both the major criteria and two of the minor criteria.
bThree of more of the listed features are required for the diagnosis of dysplastic naevus.
cFor each group (architectural disorder/cytology) a grading score is assigned as follows: mild, 0–1 criteria; moderate, 2 criteria; severe, 3–4 criteria.

Figure 132.51 Compound dysplastic naevus. (a) Hyperplastic epidermis with bridging of the adjacent rete ridges. Nests of melanocytes, of variable sizes and shapes, can be seen in the junctional area as well as lamellar fibrosis of the dermis beneath the rete ridges. There is focal lymphocytic infitrate of the dermis. Magnification 10× (H&E). (b) There are atypical or multinucleated giant melanocytes in the junctional portion of the naevus. Magnification 40× (H&E).
(Courtesy of Dr K. Frangia, HBD HistoBio Diagnosis, Athens, Greece.)
Although a clinically atypical naevus usually exhibits histological dysplasia, and vice versa, this is not always the case. This discorcodance between clinical and histological diagnosis may not pose a real problem, since the true value of the histological examination of a melanocytic lesion with clinically atypical features lies in the exclusion of melanoma.
Genetics
At a molecular level, atypical naevi exhibit features that place them in an intermediate position on a spectrum ranging from common naevi to overt melanoma [258]. CGH analysis has revealed that melanocytic naevi differ from melanoma as they show no chromosomal aberrations, or have a restricted set of alterations with basically no overlap with melanoma [259]. The BRAF mutation rate is high and comparable in both common (67%) and atypical (62%) naevi [260]. Although studies have not always been in accordance with each other, the most common molecular findings in atypical naevi include mutation/deletion of the p16 gene, altered expression of p53, increased microsatellite instability, alterations of pigmentation pathways, and mismatch repair gene expression [261, 262, 263].
Environmental factors
Painful sunburns before the age of 20 years have been associated with an increased risk for the development of melanocytic naevi (odds ratio (OR) 1.5; confidence interval (CI) 1.1–2.0) and atypical naevi (OR 1.4; CI 0.88–2.3) [264].
Clinical features
Presentation
Clinically, atypical naevi are larger than 5 mm with irregular borders and pigmentation (Figure 132.52). They sometimes present with a reddish hue that corresponds to a degree of inflammation. A central papular component is often surrounded by a macular periphery, so that the naevus resembles a ‘fried egg’ appearance. The well-known ABCDE rule of melanoma also applies to these naevi, albeit to a less pronounced extent.
Figure 132.52 (a–e) Clinically atypical naevi of variable shapes, sizes and pigmentary patterns (on the left) and their corresponding dermoscopic images (on the right).
(Parts c and e courtesy of Dr D. Sgouros, Andreas Sygros Hospital, Athens, Greece.)
Differential diagnosis
The critical distinction is between an atypical naevus and an in situ or early radial growth phase melanoma [265]. While the early detection of melanoma is of paramount importance in terms of prognosis, excessive prophylactic excision of benign naevi that are quite common in the general population is unjustified. Congenital melanocytic naevi and compound blue naevi sometimes can also exhibit atypical clinical features.
Classification of severity
Several studies have attempted to relate the grade of histological atypia (mild, moderate or severe) of these lesions to the risk of developing malignant melanoma [266]. Although patients with more severe histologically atypical naevi seem to have a higher risk of developing melanoma [242], the prognostic value of this classification is still limited due to a lack of uniform and objective criteria.
Disease course and prognosis
The usual natural history of acquired benign melanocytic naevi is that of a progression from junctional to compound and finally to dermal naevi, with subsequent termination of naevomelanocytic proliferation. Atypical naevi retain their ability to proliferate for an extended period before their maturation, resulting in a larger size and irregular shape and pigmentation compared with common naevi. Although the vast majority of naevi follow this course, there are some cases of both common and atypical naevi that evolve into radial growth phase melanomas. Histological examination of melanomas reveals that approximately one-quarter develop on pre-existing naevi. However, the rate of malignant transformation of naevi into melanomas is very low [90]. There are currently no data to support the notion that atypical naevi are more likely to progress to melanomas than common naevi, and there are no molecular markers for the identification of lesions at higher risk for transformation. Therefore, atypical naevi should mainly be viewed as risk markers – and occasionally simulants – for melanomas rather than true precursor lesions.
Investigations
Dermoscopy is always useful in assessing a melanocytic lesion with clinically atypical features. Different methods of analysis, such as qualitative pattern analysis, the ABCD rule and the seven-point checklist, yield different percentages of diagnostic accuracy for melanoma, yet they all add to the clinical differential diagnosis between atypical naevi and melanomas. Dermoscopic features that can be seen in atypical naevi vary [267, 268, 269, 270, 271]. It is usual to note an atypical or irregular pigmentation network, irregularly distributed and shaped brown globules and dots, as well as areas of regression (Figure 132.52, right hand images). In in vivo confocal microscopy, atypical naevi exhibit characteristics that correspond to their architectural and cytological features (Figure 132.53), yet its use is currently limited due to the high cost and limited availability.
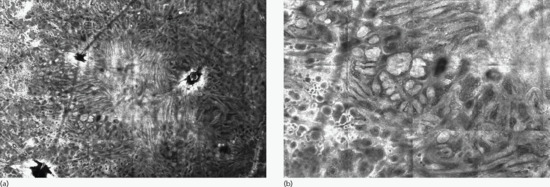
Figure 132.53 (a) Dysplastic nevus (4.2 × 6 mm) by reflectance confocal microscopy showing a more disarranged pattern with elongated junctional nests in the centre and some nests bulging from the side of the cristae. (b) Detail (1.4 × 2 mm) at a higher magnification where a few large (>40 μm) bright nucleated cells are also visible within the nests, corresponding to atypical melanocytes.
(Courtesy of Dr Giovanni Pellacani, Department of Dermatology, University of Modena and Reggio Emilia, Italy.)
Management
Since atypical naevi are risk markers rather than melanoma precursors, there is no need to excise for prophylactic reasons. The risk of melanoma conferred by the presence of atypical naevi remains even after their excision, since the majority of melanomas develop de novo. ‘Prophylactic’ excision is therefore unlikely to truly protect against future melanoma development, while it involves the patient with the additional cost and morbidity of the procedure, and possibly a false sense of safety. Patients with atypical naevi, especially those with high numbers of atypical and common naevi and/or a personal or family history of melanoma, are at high risk for melanoma development. These patients should be taught to self-examine their existing naevi as well as the rest of their skin for potentially suspicious lesions. They should also be counselled about sun protection strategies. Apart from a full skin examination, dermoscopy and photographic recording (either by digital dermoscopic imaging or by total body photography) should be used in patients with atypical naevi. There is no consensus on the frequency of follow-up visits or on the overall time period of surveillance for individual lesions. In the case of particularly atypical lesions, complete surgical excision with a 2–3 mm clinical margin and subsequent histological examination to rule out melanoma in situ or early melanoma should be performed.
Resources
Patient resources
- American Academy of Dermatology. Moles: who gets and types. http://www.aad.org/dermatology-a-to-z/diseases-and-treatments/m—p/moles/who-gets-types (last accessed May 2014).
Resources
Patient resources
- Congenital Melanocytic Naevus Support Group: www.caringmattersnow.co.uk.
- Nevus Network: www.nevusnetwork.org.
- Nevus Support Australia: www.nevussupport.com. (All last accessed May 2014.)
Patient resources
- Congenital Melanocytic Naevus Support Group: www.caringmattersnow.co.uk.
- Nevus Network: www.nevusnetwork.org.
- Nevus Support Australia: www.nevussupport.com. (All last accessed May 2014.)
References
- Elder DE, Elenitsas R, Murphy GF, Xu X. Benign pigmented lesions and melanoma. In: Elder DE, Elenitsas R, Johnson BL, Jr, Murphy GF, eds. Lever's Histopathology of the Skin, 9th edn. Philadelpia: Lippincott Williams and Wilkins, 2005:715–803.
- Bastiaens M, ter Huurne J, Gruis N, et al. The melanocortin-1–receptor gene is the major freckle gene. Hum Mol Genet 2001;10:1701–8.
- Bastiaens M, Hoefnagel J, Westendorp R, et al. Solar lentigines are strongly related to sun exposure in contrast to ephelides. Pigment cell research sponsored by the European Society for Pigment Cell Research and the International Pigment Cell Society. Pigment Cell Res 2004;17:225–9.
- Olsen CM, Carroll HJ, Whiteman DC. Estimating the attributable fraction for melanoma: a meta-analysis of pigmentary characteristics and freckling. Int J Cancer 2010;127:2430–45.
- Guerrero D. Dermocosmetic management of hyperpigmentations. Ann Dermatol Vénéréol 2012;139(Suppl. 4):S166–9.
- Lodish MB, Stratakis CA. The differential diagnosis of familial lentiginosis syndromes. Fam Cancer 2011;10:481–90.
- Toelle SP, Boltshauser E, Wirth MG, Itin P. Association of lentiginous mosaicism and congenital cataract in a girl. Eur J Dermatol 2006;16:360–2.
- Karrer S, Kohl E, Feise K, et al. Photodynamic therapy for skin rejuvenation: review and summary of the literature – results of a consensus conference of an expert group for aesthetic photodynamic therapy. J Deut Dermatol Gesellsch 2013;11:137–48.
- Chen N, Hu Y, Li WH, et al. The role of keratinocyte growth factor in melanogenesis: a possible mechanism for the initiation of solar lentigines. Exp Dermatol 2010;19:865–72.
- Kovacs D, Cardinali G, Aspite N, et al. Role of fibroblast-derived growth factors in regulating hyperpigmentation of solar lentigo. Br J Dermatol 2010;163:1020–7.
- Mauerer A, Betz RC, Pasternack SM, et al. Generalized solar lentigines in a patient with a history of radon exposure. Dermatology 2010;221:206–10.
- Hafner C, Stoehr R, van Oers JM, et al. FGFR3 and PIK3CA mutations are involved in the molecular pathogenesis of solar lentigo. Br J Dermatol 2009;160:546–51.
- Derancourt C, Bourdon-Lanoy E, Grob JJ, et al. Multiple large solar lentigos on the upper back as clinical markers of past severe sunburn: a case-control study. Dermatology 2007;214:25–31.
- Monestier S, Gaudy C, Gouvernet J, et al. Multiple senile lentigos of the face, a skin ageing pattern resulting from a life excess of intermittent sun exposure in dark-skinned caucasians: a case-control study. Br J Dermatol 2006;154:438–44.
- Kim J, Taube JM, McCalmont TH, Glusac EJ. Quantitative comparison of MiTF, Melan-A, HMB-45 and Mel-5 in solar lentigines and melanoma in situ. J Cutan Pathol 2011;38:775–9.
- Black WH, Thareja SK, Blake BP, et al. Distinction of melanoma in situ from solar lentigo on sun-damaged skin using morphometrics and MITF immunohistochemistry. Am J Dermatopathol 2011;33:573–8.
- Stankiewicz K, Chuang G, Avram M. Lentigines, laser, and melanoma: a case series and discussion. Laser Surg Med 2012;44:112–16.
- Yamashita T, Negishi K, Hariya T, et al. In vivo microscopic approaches for facial melanocytic lesions after quality-switched ruby laser therapy: time-sequential imaging of melanin and melanocytes of solar lentigo in Asian skin. Dermatol Surg 2010;36:1138–47.
- Abdullah AN, Keczkes K. Cutaneous and ocular side-effects of PUVA photochemotherapy – a 10–year follow-up study. Clin Exp Dermatol 1989;14:421–4.
- Stern RS; PUVA Follow-up Study. The risk of melanoma in association with long-term exposure to PUVA. J Am Acad Dermatol 2001;44:755–61.
- Rhodes AR, Stern RS, Melski JW. The PUVA lentigo: an analysis of predisposing factors. J Invest Dermatol 1983;81:459–63.
- Friedland R, David M, Feinmesser M, et al. NB-UVB (311–312 nm)-induced lentigines in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol 2012;26:1158–62.
- Nakagawa H, Rhodes AR, Momtaz TK, Fitzpatrick TB. Morphologic alterations of epidermal melanocytes and melanosomes in PUVA lentigines: a comparative ultrastructural investigation of lentigines induced by PUVA and sunlight. J Invest Dermatol 1984;82:101–7.
- Stern RS. Risks of cancer associated with long-term exposure to PUVA in humans: current status – 1991. Blood Cells 1992;18:91–7; discussion 8–9.
- Lindelof B, Sigurgeirsson B, Tegner E, et al. PUVA and cancer risk: the Swedish follow-up study. Br J Dermatol 1999;141:108–12.
- Bolognia JL. Reticulated black solar lentigo (‘ink spot’ lentigo). Arch Dermatol 1992;128:934–40.
- Lenane P, Keane CO, Connell BO, et al. Genital melanotic macules: clinical, histologic, immunohistochemical, and ultrastructural features. J Am Acad Dermatol 2000;42:640–4.
- Buchner A, Merrell PW, Carpenter WM. Relative frequency of solitary melanocytic lesions of the oral mucosa. J Oral Pathol Med 2004;33:550–7.
- Meleti M, Vescovi P, Mooi WJ, van der Waal I. Pigmented lesions of the oral mucosa and perioral tissues: a flow-chart for the diagnosis and some recommendations for the management. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;105:606–16.
- Carlson JA, Mu XC, Slominski A, et al. Melanocytic proliferations associated with lichen sclerosus. Arch Dermatol 2002;138:77–87.
- Ficarra G, Shillitoe EJ, Adler-Storthz K, et al. Oral melanotic macules in patients infected with human immunodeficiency virus. Oral Surg Oral Med Oral Pathol 1990;70:748–55.
- Meleti M, Leemans CR, Mooi WJ, et al. Oral malignant melanoma: a review of the literature. Oral Oncol 2007;43:116–21.
- Gondak RO, da Silva-Jorge R, Jorge J, et al. Oral pigmented lesions: clinicopathologic features and review of the literature. Med Oral Patol Oral Cirugia Bucal 2012;17:e919–24.
- Ferrari A, Buccini P, Covello R, et al. The ringlike pattern in vulvar melanosis: a new dermoscopic clue for diagnosis. Arch Dermatol 2008;144:1030–4.
- Rajpar SF, Marsden JR. Imiquimod in the treatment of lentigo maligna. Br J Dermatol 2006;155:653–6.
- Kaugars GE, Heise AP, Riley WT, et al. Oral melanotic macules. A review of 353 cases. Oral Surg Oral Med Oral Pathol 1993;76:59–61.
- Gupta G, Williams RE, Mackie RM. The labial melanotic macule: a review of 79 cases. Br J Dermatol 1997;136:772–5.
- Gupta G, MacKay IR, MacKie RM. Q-switched ruby laser in the treatment of labial melanotic macules. Laser Surg Med 1999;25:219–22.
- Ferahbas A, Utas S, Akcakus M, et al. Prevalence of cutaneous findings in hospitalized neonates: a prospective observational study. Pediatr Dermatol 2009;26:139–42.
- Egemen A, Ikizoglu T, Ergor S, et al. Frequency and characteristics of mongolian spots among Turkish children in Aegean region. Turk J Pediatr 2006;48:232–6.
- Reza AM, Farahnaz GZ, Hamideh S, et al. Incidence of Mongolian spots and its common sites at two university hospitals in Tehran, Iran. Pediatr Dermatol 2010;27:397–8.
- Moosavi Z, Hosseini T. One-year survey of cutaneous lesions in 1000 consecutive Iranian newborns. Pediatr Dermatol 2006;23:61–3.
- Bilgili SG, Akdeniz N, Karadag AS, et al. Mucocutaneous disorders in children with down syndrome: case-controlled study. Genet Counsel 2011;22:385–92.
- Wolf R, Wolf D, Davidovici B. Phacomatosis pigmentopigmentalis: aberrant Mongolian spots and segmental cafe au lait macules. Pediatr Dermatol 2009;26:228–9.
- Acebo E, Gardeazabal J, Gonzalez-Hermosa R, et al. Congenital hemangioma: a report of evolution from rapidly involuting to noninvoluting congenital hemangioma with aberrant Mongolian spots. Pediatr Dermatol 2009;26:225–6.
- Ashrafi MR, Shabanian R, Mohammadi M, Kavusi S. Extensive Mongolian spots: a clinical sign merits special attention. Pediatr Neurol 2006;34:143–5.
- Hackbart BA, Arita JH, Pinho RS, et al. Mongolian spots are not always a benign sign. J Pediatr 2013;162:1070.
- Grant BP, Beard JS, de Castro F, et al. Extensive mongolian spots in an infant with Hurler syndrome. Arch Dermatol 1998;134:108–9.
- Esterly NB, Weissbluth M, Caro WA. Mongolian spots and GM1 type 1 gangliosidosis. J Am Acad Dermatol 1990;22:320.
- Su F, Li F, Jin HZ. Extensive Mongolian spots in a child with mucolipidosis II. Int J Dermatol 2010;49:438–40.
- Hidano A, Kajima H, Ikeda S, et al. Natural history of nevus of Ota. Arch Dermatol 1967;95:187–95.
- Kagami S, Asahina A, Watanabe R, et al. Laser treatment of 26 Japanese patients with Mongolian spots. Dermatol Surg 2008;34:1689–94.
- Kagami S, Asahina A, Uwajima Y, et al. Treatment of persistent Mongolian spots with Q-switched alexandrite laser. Laser Medical Sci 2012;27:1229–32.
- Shirakawa M, Ozawa T, Ohasi N, et al. Comparison of regional efficacy and complications in the treatment of aberrant Mongolian spots with the Q-switched ruby laser. J Cosmetic Laser Ther 2010;12:138–42.
- Aurangabadkar S. QYAG5 Q-switched Nd:YAG laser treatment of nevus of Ota: an Indian study of 50 patients. J Cutan Aesthet Surg 2008;1:80–4.
- Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009;457:599–602.
- Chan HH, Lam LK, Wong DS, et al. Nevus of Ota: a new classification based on the response to laser treatment. Laser Surg Med 2001;28:267–72.
- Hori Y, Takayama O. Circumscribed dermal melanoses. Classification and histologic features. Dermatol Clin 1988;6:315–26.
- Hirayama T, Suzuki T. A new classification of Ota's nevus based on histopathological features. Dermatologica 1991;183:169–72.
- Hori Y, Kawashima M, Oohara K, Kukita A. Acquired, bilateral nevus of Ota-like macules. J Am Acad Dermatol 1984;10:961–4.
- Park JM, Tsao H, Tsao S. Acquired bilateral nevus of Ota-like macules (Hori nevus): etiologic and therapeutic considerations. J Am Acad Dermatol 2009;61:88–93.
- Rahimi-Movaghar V, Karimi M. Meningeal melanocytoma of the brain and oculodermal melanocytosis (nevus of Ota): case report and literature review. Surg Neurol 2003;59:200–10.
- Baroody M, Holds JB. Extensive locoregional malignant melanoma transformation in a patient with oculodermal melanocytosis. Plast Reconstr Surg 2004;113:317–22.
- Sharan S, Grigg JR, Billson FA. Bilateral naevus of Ota with choroidal melanoma and diffuse retinal pigmentation in a dark skinned person. Br J Ophthalmol 2005;89:1529.
- Wang J, Guo ZZ, Zhang SG, et al. Microsurgical treatment of meningeal malignant melanoma accompanied by nevus of Ota: two case reports and a literature review. Melanoma Res 2013;23:502–4.
- Radhadevi CV, Charles KS, Lathika VK. Orbital malignant melanoma associated with nevus of Ota. Indian J Ophthalmol 2013;61:306–9.
- Yang Q, Wei WB, Yang WL, et al. Choroidal malignant melanoma in patients with oculodermal melanocytosis: report of three cases. Chin Med J 2010;123:111–13.
- Watanabe S, Takahashi H. Treatment of nevus of Ota with the Q-switched ruby laser. New Engl J Med 1994;331:1745–50.
- Kar HK, Gupta L. 1064 nm Q switched Nd: YAG laser treatment of nevus of Ota: an Indian open label prospective study of 50 patients. Ind J Dermatol Venereol Leprol 2011;77:565–70.
- Lu Z, Fang L, Jiao S, et al. Treatment of 522 patients with nevus of Ota with Q-switched Alexandrite laser. Chin Med J 2003;116:226–30.
- Kono T, Nozaki M, Chan HH, Mikashima Y. A retrospective study looking at the long-term complications of Q-switched ruby laser in the treatment of nevus of Ota. Laser Surg Med 2001;29:156–9.
- Mukhopadhyay AK. Unilateral nevus of Ota with bilateral nevus of Ito and palatal lesion: a case report with a proposed clinical modification of Tanino's classification. Indian J Dermatol 2013;58:286–9.
- Seo SH, Jeong JT, Kim SN, Kye YC. A case of bilateral nevus of Ota associated with bilateral nevus of Ito.Korean J Dermatol 2001;39:106–8.
- Kim BH OY, Lee KW. Bilateral Ota nevus and bilateral Ito nevus: 2 cases of extensive dermal melanocytic nevi associated with vascular nevus. Korean J Dermatol 1981;19:503–7.
- Trindade F, Santonja C, Requena L. Bilateral nevus of Ito and nevus spilus in the same patient. J Am Acad Dermatol 2008;59:S51–2.
- Martinez-Penuela A, Iglesias ME, Mercado MR, Martinez-Penuela JM. [Malignant transformation of a nevus of Ito: description of a rare case.] Actas Dermo-sifiliograf 2011;102:817–20.
- Wise SR, Capra G, Martin P, et al. Malignant melanoma transformation within a nevus of Ito. J Am Acad Dermatol 2010;62:869–74.
- Van Krieken JH, Boom BW, Scheffer E. Malignant transformation in a naevus of Ito. A case report. Histopathology 1988;12:100–2.
- Happle R. Speckled lentiginous naevus: which of the two disorders do you mean? Clin Exp Dermatol 2009;34:133–5.
- Happle R. Speckled lentiginous nevi: no longer one single disorder. Arch Dermatol 2010;146:204.
- Torchia D. More on speckled lentiginous nevus syndrome. Dermatology 2010;221:324–5.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol 2005;141:385–8.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet 1996;65:363–5.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol 2013;133:1998–2003.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology 2006;212:53–8.
- Polder KD, Landau JM, Vergilis-Kalner IJ, et al. Laser eradication of pigmented lesions: a review. Dermatol Surg 2011;37:572–95.
- Moreno-Arias GA, Bulla F, Vilata-Corell JJ, Camps-Fresneda A. Treatment of widespread segmental nevus spilus by Q-switched alexandrite laser (755 nm, 100 nsec). Dermatol Surg 2001;27:841–3.
- Grevelink JM, Gonzalez S, Bonoan R, et al. Treatment of nevus spilus with the Q-switched ruby laser. Dermatol Surg 1997;23:365–9; discussion 9–70.
- Gandini S, Sera F, Cattaruzza MS, et al. Meta-analysis of risk factors for cutaneous melanoma: I. Common and atypical naevi. Eur J Cancer 2005;41:28–44.
- Tsao H, Bevona C, Goggins W, Quinn T. The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: a population-based estimate. Arch Dermatol 2003;139:282–8.
- Hughes BR, Cunliffe WJ, Bailey CC. Excess benign melanocytic naevi after chemotherapy for malignancy in childhood. BMJ 1989;299:88–91.
- Szepietowski J, Wasik F, Szepietowski T, et al. Excess benign melanocytic naevi in renal transplant recipients. Dermatology 1997;194:17–19.
- Navarini AA, Kolm I, Calvo X, et al. Trauma as triggering factor for development of melanocytic nevi. Dermatology 2010;220:291–6.
- Zvulunov A, Wyatt DT, Laud PW, Esterly NB. Influence of genetic and environmental factors on melanocytic naevi: a lesson from Turner's syndrome. Br J Dermatol 1998;138:993–7.
- Broberg A, Augustsson A. Atopic dermatitis and melanocytic naevi. Br J Dermatol 2000;142:306–9.
- Synnerstad I, Nilsson L, Fredrikson M, Rosdahl I. Fewer melanocytic nevi found in children with active atopic dermatitis than in children without dermatitis. Arch Dermatol 2004;140:1471–5.
- Martinez-Casimiro L, Sanchez Carazo JL, Alegre V. Balloon cell naevus. J Eur Acad Dermatol Venereol 2009;23:236–7.
- Lyall D, Srinivasan S, Roberts F. Balloon cell naevus of the conjunctiva: clinicopathological features and management. Clin Exp Ophthalmol 2011;39:271–3.
- Damm DD, White DK, Lyu PE, Puno P. Balloon cell nevus of the oral mucosa. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;105:755–7.
- Yeh I, von Deimling A, Bastian BC. Clonal BRAF mutations in melanocytic nevi and initiating role of BRAF in melanocytic neoplasia. J Natl Cancer Inst 2013;105:917–19.
- Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005;436:720–4.
- Nan H, Xu M, Zhang J, et al. Genome-wide association study identifies nidogen 1 (NID1) as a susceptibility locus to cutaneous nevi and melanoma risk. Hum Mol Genet 2011;20:2673–9.
- Falchi M, Bataille V, Hayward NK, et al. Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat Genet 2009;41:915–9.
- Rampen FH, Fleuren BA, de Boo TM, Lemmens WA. Prevalence of common “acquired” nevocytic nevi and dysplastic nevi is not related to ultraviolet exposure. J Am Acad Dermatol 1988;18:679–83.
- Green A, Sorahan T, Pope D, et al. Moles in Australian and British school children. Lancet 1988;2:1497.
- Fritschi L, McHenry P, Green A, et al. Naevi in schoolchildren in Scotland and Australia. Br J Dermatol 1994;130:599–603.
- Kelly JW, Rivers JK, MacLennan R, et al. Sunlight: a major factor associated with the development of melanocytic nevi in Australian schoolchildren. J Am Acad Dermatol 1994;30:40–8.
- English DR, Milne E, Simpson JA. Ultraviolet radiation at places of residence and the development of melanocytic nevi in children (Australia). Cancer Causes Control 2006;17:103–7.
- Mahe E, Beauchet A, de Paula Correa M, et al. Outdoor sports and risk of ultraviolet radiation-related skin lesions in children: evaluation of risks and prevention. Br J Dermatol 2011;165:360–7.
- Zalaudek I, Catricala C, Moscarella E, Argenziano G. What dermoscopy tells us about nevogenesis. J Dermatol 2011;38:16–24.
- Mason AR, Mohr MR, Koch LH, Hood AF. Nevi of special sites. Clin Lab Med 2011;31:229–42.
- Boyd AS, Rapini RP. Acral melanocytic neoplasms: a histologic analysis of 158 lesions. J Am Acad Dermatol 1994;31:740–5.
- Rongioletti F, Ball RA, Marcus R, Barnhill RL. Histopathological features of flexural melanocytic nevi: a study of 40 cases. J Cutan Pathol 2000;27:215–17.
- Fabrizi G, Pagliarello C, Parente P, Massi G. Atypical nevi of the scalp in adolescents. J Cutan Pathol 2007;34:365–9.
- Rongioletti F, Urso C, Batolo D, et al. Melanocytic nevi of the breast: a histologic case-control study. J Cutan Pathol 2004;31:137–40.
- Rock B, Hood AF, Rock JA. Prospective study of vulvar nevi. J Am Acad Dermatol 1990;22:104–6.
- Gleason BC, Hirsch MS, Nucci MR, et al. Atypical genital nevi. A clinicopathologic analysis of 56 cases. Am J Surg Pathol 2008;32:51–7.
- Friedman RJ, Ackerman AB. Difficulties in the histologic diagnosis of melanocytic nevi on the vulvae of premenopausal women. In: Ackerman AB, ed. Pathology of Malignant Melanoma. New York: Masson, 1981:119–27.
- Sugiyama VE, Chan JK, Shin JY, et al. Vulvar melanoma: a multivariable analysis of 644 patients. Obstet Gynecol 2007;110:296–301.
- LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol 2000;22:556–8.
- Evans MJ, Gray ES, Blessing K. Histopathological features of acral melanocytic nevi in children: study of 21 cases. Pediatr Dev Pathol 1998;1:388–92.
- Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol 2007;143:1423–6.
- Shields CL, Demirci H, Karatza E, Shields JA. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology 2004;111:1747–54.
- Thiagalingam S, Johnson MM, Colby KA, Zembowicz A. Juvenile conjunctival nevus: clinicopathologic analysis of 33 cases. Am J Surg Pathol 2008;32:399–406.
- Zembowicz A, Mandal RV, Choopong P. Melanocytic lesions of the conjunctiva. Arch Pathol Lab Med 2010;134:1785–92.
- Grin JM, Grant-Kels JM, Grin CM, et al. Ocular melanomas and melanocytic lesions of the eye. J Am Acad Dermatol 1998;38:716–30.
- Farber M, Schutzer P, Mihm MC, Jr. Pigmented lesions of the conjunctiva. J Am Acad Dermatol 1998;38:971–8.
- Tosti A, Baran R, Piraccini BM, et al. Nail matrix nevi: a clinical and histopathologic study of twenty-two patients. J Am Acad Dermatol 1996;34:765–71.
- Braun RP, Baran R, Le Gal FA, et al. Diagnosis and management of nail pigmentations. J Am Acad Dermatol 2007;56:835–47.
- Thomas L, Dalle S. Dermoscopy provides useful information for the management of melanonychia striata. Dermatol Ther 2007;20:3–10.
- Altamura D, Altobelli E, Micantonio T, et al. Dermoscopic patterns of acral melanocytic nevi and melanomas in a white population in central Italy. Arch Dermatol 2006;142:1123–8.
- Larre Borges A, Zalaudek I, Longo C, et al. Melanocytic nevi with special features: clinical-dermoscopic and reflectance confocal microscopic-findings. J Eur Acad Dermatol Venereol 2013, doi: 10.1111/jdv.12291.
- Scolyer RA, Zhuang L, Palmer AA, et al. Combined naevus: a benign lesion frequently misdiagnosed both clinically and pathologically as melanoma. Pathology 2004;36:419–27.
- Strungs I. Common and uncommon variants of melanocytic naevi. Pathology 2004;36:396–403.
- De Giorgi V, Massi D, Salvini C, et al. Dermoscopic features of combined melanocytic nevi. J Cutan Pathol 2004;31:600–4.
- Argenziano G. Dermoscopy of melanocytic neoplasms: targetoid combined blue nevi. Arch Dermatol 2004;140:1576.
- King R, Hayzen BA, Page RN, et al. Recurrent nevus phenomenon: a clinicopathologic study of 357 cases and histologic comparison with melanoma with regression. Mod Pathol 2009;22:611–17.
- Fox JC, Reed JA, Shea CR. The recurrent nevus phenomenon: a history of challenge, controversy, and discovery. Arch Pathol Lab Med 2011;135:842–6.
- Kornberg R, Ackerman AB. Pseudomelanoma: recurrent melanocytic nevus following partial surgical removal. Arch Dermatol 1975;111:1588–90.
- Marghoob AA, Changchien L, DeFazio J, et al. The most common challenges in melanoma diagnosis and how to avoid them. Australas J Dermatol 2009;50:1–13; quiz 4–5.
- Botella-Estrada R, Nagore E, Sopena J, et al. Clinical, dermoscopy and histological correlation study of melanotic pigmentations in excision scars of melanocytic tumours. Br J Dermatol 2006;154:478–84.
- Blum A, Hofmann-Wellenhof R, Marghoob AA, et al. Recurrent melanocytic nevi and melanomas in dermoscopy: results of a multicenter study of the International Dermoscopy Society. JAMA Dermatol 2014;150:138–45.
- Baranda L, Torres-Alvarez B, Moncada B, et al. Presence of activated lymphocytes in the peripheral blood of patients with halo nevi. J Am Acad Dermatol 1999;41:567–72.
- Herd RM, Hunter JA. Familial halo naevi. Clin Exp Dermatol 1998;23:68–9.
- Libon F, Arrese JE, Rorive A, Nikkels AF. Ipilimumab induces simultaneous regression of melanocytic naevi and melanoma metastases. Clin Exp Dermatol 2013;38:276–9.
- Pustisek N, Sikanic-Dugic N, Hirsl-Hecej V, Domljan ML. “Halo nevi” and UV radiation. Collegium Antropol 2010;34(Suppl. 2):295–7.
- Aouthmany M, Weinstein M, Zirwas MJ, Brodell RT. The natural history of halo nevi: a retrospective case series.J Am Acad Dermatol 2012;67:582–6.
- Kolm I, Di Stefani A, Hofmann-Wellenhof R, et al. Dermoscopy patterns of halo nevi. Arch Dermatol 2006;142:1627–32.
- Meyerson LB. A peculiar papulosquamous eruption involving pigmented nevi. Arch Dermatol 1971;103:510–12.
- Crovato F, Nazzari G, Gambini C, Massone L. Meyerson's naevi in pityriasis rosea. Br J Dermatol 1989;120:318–19.
- Weedon D, Farnsworth J. Spongiotic changes in melanocytic nevi. Am J Dermatopathol 1984;6(Suppl.):257–9.
- Brenan J, Kossard S, Krivanek J. Halo eczema around melanocytic nevi. Int J Dermatol 1985;24:226–9.
- Conde-Taboada A, de la Torre C, Feal C, et al. Meyerson's naevi induced by interferon alfa plus ribavirin combination therapy in hepatitis C infection. Br J Dermatol 2005;153:1070–2.
- Krischer J, Pechere M, Salomon D, et al. Interferon alfa-2b-induced Meyerson's nevi in a patient with dysplastic nevus syndrome. J Am Acad Dermatol 1999;40:105–6.
- Petit A, Viney C, Gaulier A, Sigal M. Coexistence of Meyerson's with Sutton's naevus after sunburn. Dermatology 1994;189:269–70.
- Tegner E, Bjornberg A, Jonsson N. Halo dermatitis around tumours. Acta Dermato-venereol 1990;70:31–4.
- Rolland S, Kokta V, Marcoux D. Meyerson phenomenon in children: observation in five cases of congenital melanocytic nevi. Pediatr Dermatol 2009;26:292–7.
- Ramon R, Silvestre JF, Betlloch I, et al. Progression of Meyerson's naevus to Sutton's naevus. Dermatology 2000;200:337–8.
- Fernandez-Flores A. Eponyms, morphology, and pathogenesis of some less mentioned types of melanocytic nevi. Am J Dermatopathol 2012;34:607–18.
- Guzzo C, Johnson B, Honig P. Cockarde nevus: a case report and review of the literature. Pediatr Dermatol 1988;5:250–3.
- James MP, Wells RS. Cockade naevus: an unusual variant of the benign cellular naevus. Acta Dermato-venereol 1980;60:360–3.
- Warin AP. Cockarde naevus. Clin Exp Dermatol 1976;1:221–4.
- Bondi EE, Elder D, Guerry DT, Clark WH, Jr. Target blue nevus. Arch Dermatol 1983;119:919–20.
- Champion RH. An unusual melanoma. Br J Dermatol 1964;76:347.
- Kessides MC, Puttgen KB, Cohen BA. No biopsy needed for eclipse and cockade nevi found on the scalps of children. Arch Dermatol 2009;145:1334–6.
- Tcheung WJ, Bellet JS, Prose NS, et al. Clinical and dermoscopic features of 88 scalp naevi in 39 children. Br J Dermatol 2011;165:137–43.
- Patrizi A, Giacomini F, Savoia F, et al. Targetoid hemosiderotic naevus. J Eur Acad Dermatol Venereol 2009;23:493–4.
- Tomasini C, Broganelli P, Pippione M. Targetoid hemosiderotic nevus. A trauma-induced simulator of malignant melanoma. Dermatology 2005;210:200–5.
- Kovacs S, Megahed M. [Targetoid hemosiderotic nevus. A melanoma simulator.] Hautarzt 2007;58:931–2.
- Spitz S. Melanomas of childhood. Am J Pathol 1948;24:591–609.
- Barnhill RL, Argenyi ZB, From L, et al. Atypical Spitz nevi/tumors: lack of consensus for diagnosis, discrimination from melanoma, and prediction of outcome. Hum Pathol 1999;30:513–20.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other Spitzoid lesions part I. Background and diagnoses. J Am Acad Dermatol 2011;65:1073–84.
- Smith KJ, Barrett TL, Skelton HG, 3rd, et al. Spindle cell and epithelioid cell nevi with atypia and metastasis (malignant Spitz nevus). Am J Surg Pathol 1989;13:931–9.
- Mones JM, Ackerman AB. “Atypical” Spitz's nevus, “malignant” Spitz's nevus, and “metastasizing” Spitz's nevus: a critique in historical perspective of three concepts flawed fatally. Am J Dermatopathol 2004;26:310–33.
- Barnhill RL, Gupta K. Unusual variants of malignant melanoma. Clin Dermatol 2009;27:564–87.
- Casso EM, Grin-Jorgensen CM, Grant-Kels JM. Spitz nevi. J Am Acad Dermatol 1992;27:901–13.
- Ferrara G, Argenziano G, Soyer HP, et al. The spectrum of Spitz nevi: a clinicopathologic study of 83 cases. Arch Dermatol 2005;141:1381–7.
- Cesinaro AM, Foroni M, Sighinolfi P, et al. Spitz nevus is relatively frequent in adults: a clinico-pathologic study of 247 cases related to patient's age. Am J Dermatopathol 2005;27:469–75.
- Dal Pozzo V, Benelli C, Restano L, et al. Clinical review of 247 case records of Spitz nevus (epithelioid cell and/or spindle cell nevus). Dermatology 1997;194:20–5.
- Weedon D, Little JH. Spindle and epithelioid cell nevi in children and adults. A review of 211 cases of the Spitz nevus. Cancer 1977;40:217–25.
- Miteva M, Lazova R. Spitz nevus and atypical spitzoid neoplasm. Semin Cutan Med Surg 2010;29:165–73.
- Berlingeri-Ramos AC, Morales-Burgos A, Sanchez JL, Nogales EM. Spitz nevus in a Hispanic population: a clinicopathological study of 130 cases. Am J Dermatopathol 2010;32:267–75.
- Bar M. Spitz and Reed nevi: acquired or congenital? Dermatol Pract Concept 2012;2:203–5.
- Ackerman AB, Elish D, Shami S. Spitz's Nevus: Reassessment Critical, Revision Radical. New York: Ardor Scribendi, 2007.
- Harris MN, Hurwitz RM, Buckel LJ, Gray HR. Congenital spitz nevus. Dermatol Surg 2000;26:931–5.
- Busam KJ, Barnhill RL. Pagetoid Spitz nevus. Intraepidermal Spitz tumor with prominent pagetoid spread. Am J Surg Pathol 1995;19:1061–7.
- Kamino H, Misheloff E, Ackerman AB, et al. Eosinophilic globules in Spitz's nevi: new findings and a diagnostic sign. Am J Dermatopathol 1979;1:323–4.
- Wesselmann U, Becker LR, Brocker EB, et al. Eosinophilic globules in spitz nevi: no evidence for apoptosis. Am J Dermatopathol 1998;20:551–4.
- Spatz A, Calonje E, Handfield-Jones S, Barnhill RL. Spitz tumors in children: a grading system for risk stratification. Arch Dermatol 1999;135:282–5.
- Berk DR, LaBuz E, Dadras SS, et al. Melanoma and melanocytic tumors of uncertain malignant potential in children, adolescents and young adults – the Stanford experience 1995–2008. Pediatr Dermatol 2010;27:244–54.
- Sade S, Al Habeeb A, Ghazarian D. Spindle cell melanocytic lesions – part I: an approach to compound naevoidal pattern lesions with spindle cell morphology and Spitzoid pattern lesions. J Clin Pathol 2010;63:296–321.
- Dhouib RS, Sassi S, Jbeli A, et al. Desmoplastic spitz nevus: report of a case and review of the literature. Pathologica 2008;100:181–4.
- Kanter-Lewensohn L, Hedblad MA, Wejde J, Larsson O. Immunohistochemical markers for distinguishing Spitz nevi from malignant melanomas. Mod Pathol 1997;10:917–20.
- Bergman R, Dromi R, Trau H, et al. The pattern of HMB-45 antibody staining in compound Spitz nevi. Am J Dermatopathol 1995;17:542–6.
- Rode J, Williams RA, Jarvis LR, et al. S100 protein, neurone specific enolase, and nuclear DNA content in Spitz naevus.J Pathol 1990;161:41–5.
- Ribe A, McNutt NS. S100A6 protein expression is different in Spitz nevi and melanomas. Mod Pathol 2003;16:505–11.
- Hilliard NJ, Krahl D, Sellheyer K. p16 expression differentiates between desmoplastic Spitz nevus and desmoplastic melanoma. J Cutan Pathol 2009;36:753–9.
- Bastian BC, Wesselmann U, Pinkel D, Leboit PE. Molecular cytogenetic analysis of Spitz nevi shows clear differences to melanoma. J Invest Dermatol 1999;113:1065–9.
- Bastian BC, LeBoit PE, Pinkel D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol 2000;157:967–72.
- Indsto JO, Kumar S, Wang L, et al. Low prevalence of RAS-RAF-activating mutations in Spitz melanocytic nevi compared with other melanocytic lesions. J Cutan Pathol 2007;34:448–55.
- Van Engen-van Grunsven AC, van Dijk MC, Ruiter DJ, et al. HRAS-mutated Spitz tumors: a subtype of Spitz tumors with distinct features. Am J Surg Pathol 2010;34:1436–41.
- Da Forno PD, Pringle JH, Fletcher A, et al. BRAF, NRAS and HRAS mutations in spitzoid tumours and their possible pathogenetic significance. Br J Dermatol 2009;161:364–72.
- Shen L, Cooper C, Bajaj S, et al. Atypical spitz tumors with 6q23 deletions: a clinical, histological, and molecular study. Am J Dermatopathol 2013;35:804–12.
- Yazdan P, Cooper C, Sholl LM, et al. Comparative analysis of atypical Spitz tumors with heterozygous versus homozygous 9p21 deletions for clinical outcomes, histomorphology, BRAF mutation, and p16 expression. Am J Surg Pathol 2014;38:638–45.
- Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 2011;43:1018–21.
- Wiesner T, Murali R, Fried I, et al. A distinct subset of atypical Spitz tumors is characterized by BRAF mutation and loss of BAP1 expression. Am J Surg Pathol 2012;36:818–30.
- Argenziano G, Agozzino M, Bonifazi E, et al. Natural evolution of Spitz nevi. Dermatology 2011;222:256–60.
- Schmoeckel C, Wildi G, Schafer T. Spitz nevus versus malignant melanoma: spitz nevi predominate on the thighs in patients younger than 40 years of age, melanomas on the trunk in patients 40 years of age or older. J Am Acad Dermatol 2007;56:753–8.
- Reed RJ, Ichinose H, Clark WH, Jr, Mihm MC, Jr. Common and uncommon melanocytic nevi and borderline melanomas. Semin Oncol 1975;2:119–47.
- Smith SA, Day CL, Jr, Vander Ploeg DE. Eruptive widespread Spitz nevi. J Am Acad Dermatol 1986;15:1155–9.
- Fass J, Grimwood RE, Kraus E, Hyman J. Adult onset of eruptive widespread Spitz's nevi. J Am Acad Dermatol 2002;46:S142–3.
- Paties CT, Borroni G, Rosso R, Vassallo G. Relapsing eruptive multiple Spitz nevi or metastatic spitzoid malignant melanoma? Am J Dermatopathol 1987;9:520–7.
- Sarin KY, Sun BK, Bangs CD, et al. Activating HRAS mutation in agminated Spitz nevi arising in a nevus spilus. JAMA Dermatol 2013;149:1077–81.
- Stern JB. Recurrent Spitz's nevi. A clinicopathologic investigation. Am J Dermatopathol 1985;7(Suppl.):49–50.
- Ludgate MW, Fullen DR, Lee J, et al. The atypical Spitz tumor of uncertain biologic potential: a series of 67 patients from a single institution. Cancer 2009;115:631–41.
- Sepehr A, Chao E, Trefrey B, et al. Long-term outcome of Spitz-type melanocytic tumors. Arch Dermatol 2011;147:1173–9.
- Cochran AJ, Binder S, Morton DL. The role of lymphatic mapping and sentinel node biopsy in the management of atypical and anomalous melanocytic lesions. J Cutan Pathol 2010;37(Suppl. 1):54–9.
- Murali R, Sharma RN, Thompson JF, et al. Sentinel lymph node biopsy in histologically ambiguous melanocytic tumors with spitzoid features (so-called atypical spitzoid tumors). Ann Surg Oncol 2008;15:302–9.
- Ghazi B, Carlson GW, Murray DR, et al. Utility of lymph node assessment for atypical spitzoid melanocytic neoplasms. Ann Surg Oncol 2010;17:2471–5.
- Urso C, Borgognoni L, Saieva C, et al. Sentinel lymph node biopsy in patients with “atypical Spitz tumors.” A report on 12 cases. Hum Pathol 2006;37:816–23.
- Su LD, Fullen DR, Sondak VK, et al. Sentinel lymph node biopsy for patients with problematic spitzoid melanocytic lesions: a report on 18 patients. Cancer 2003;97:499–507.
- Crotty KA, Scolyer RA, Li L, et al. Spitz naevus versus Spitzoid melanoma: when and how can they be distinguished? Pathology 2002;34:6–12.
- Peris K, Ferrari A, Argenziano G, et al. Dermoscopic classification of Spitz/Reed nevi. Clin Dermatol 2002;20:259–62.
- Ferrara G, Gianotti R, Cavicchini S, et al. Spitz nevus, Spitz tumor, and spitzoid melanoma: a comprehensive clinicopathologic overview. Dermatol Clin 2013;31:589–98, viii.
- Murphy ME, Boyer JD, Stashower ME, Zitelli JA. The surgical management of Spitz nevi. Dermatol Surg 2002;28:1065–9; discussion 9.
- Gambini C, Rongioletti F. Recurrent Spitz nevus. Case report and review of the literature. Am J Dermatopathol 1994;16:409–13.
- Nino M, Brunetti B, Delfino S, et al. Spitz nevus: follow-up study of 8 cases of childhood starburst type and proposal for management. Dermatology 2009;218:48–51.
- Brunetti B, Nino M, Sammarco E, Scalvenzi M. Spitz naevus: a proposal for management. J Eur Acad Dermatol Venereol 2005;19:391–3.
- Luo S, Sepehr A, Tsao H. Spitz nevi and other Spitzoid lesions part II. Natural history and management. J Am Acad Dermatol 2011;65:1087–92.
- Zembowicz A, Phadke PA. Blue nevi and variants: an update. Arch Pathol Lab Med 2011;135:327–36.
- Seab JA, Jr, Graham JH, Helwig EB. Deep penetrating nevus. Am J Surg Pathol 1989;13:39–44.
- Bender RP, McGinniss MJ, Esmay P, et al. Identification of HRAS mutations and absence of GNAQ or GNA11 mutations in deep penetrating nevi. Mod Pathol 2013;26:1320–8.
- Kang DS, Chung KY. Common blue naevus with satellite lesions: possible perivascular dissemination resulting in a clinical resemblance to malignant melanoma. Br J Dermatol 1999;141:922–5.
- Mones JM, Ackerman AB. “Atypical” blue nevus, “malignant” blue nevus, and “metastasizing” blue nevus: a critique in historical perspective of three concepts flawed fatally. Am J Dermatopathol 2004;26:407–30.
- Martin RC, Murali R, Scolyer RA, et al. So-called “malignant blue nevus”: a clinicopathologic study of 23 patients. Cancer 2009;115:2949–55.
- Clark WH, Jr, Reimer RR, Greene M, et al. Origin of familial malignant melanomas from heritable melanocytic lesions. ‘The B-K mole syndrome’. Arch Dermatol 1978;114:732–8.
- Lynch HT, Frichot BC, 3rd, Lynch JF. Familial atypical multiple mole-melanoma syndrome. J Med Genet 1978;15:352–6.
- Elder DE, Goldman LI, Goldman SC, et al. Dysplastic nevus syndrome: a phenotypic association of sporadic cutaneous melanoma. Cancer 1980;46:1787–94.
- Greene MH, Clark WH, Jr, Tucker MA, et al. Precursor naevi in cutaneous malignant melanoma: a proposed nomenclature. Lancet 1980;2:1024.
- Friedman RJ, Farber MJ, Warycha MA. The ‘‘dysplastic’' nevus. Clin Dermatol 2009;27:103–15.
- Naeyaert JM, Brochez L. Clinical practice. Dysplastic nevi. New Engl J Med 2003;349:2233–40.
- Duffy K, Grossman D. The dysplastic nevus: from historical perspective to management in the modern era: part I. Historical, histologic, and clinical aspects. J Am Acad Dermatol 2012;67:e1–16; quiz 7–8.
- Tucker MA, Halpern A, Holly EA, et al. Clinically recognized dysplastic nevi. A central risk factor for cutaneous melanoma. JAMA 1997;277:1439–44.
- Carey WP, Jr, Thompson CJ, Synnestvedt M, et al. Dysplastic nevi as a melanoma risk factor in patients with familial melanoma. Cancer 1994;74:3118–25.
- Burden AD, Newell J, Andrew N, et al. Genetic and environmental influences in the development of multiple primary melanoma. Arch Dermatol 1999;135:261–5.
- Titus-Ernstoff L, Perry AE, Spencer SK, et al. Multiple primary melanoma: two-year results from a population-based study. Arch Dermatol 2006;142:433–8.
- Marghoob AA, Slade J, Kopf AW, et al. Risk of developing multiple primary cutaneous melanomas in patients with the classic atypical-mole syndrome: a case-control study. Br J Dermatol 1996;135:704–11.
- Ferrone CR, Ben Porat L, Panageas KS, et al. Clinicopathological features of and risk factors for multiple primary melanomas. JAMA 2005;294:1647–54.
- Tucker MA, Fraser MC, Goldstein AM, et al. Risk of melanoma and other cancers in melanoma-prone families. J Invest Dermatol 1993;100:S350–5.
- Zhu G, Duffy DL, Eldridge A, et al. A major quantitative-trait locus for mole density is linked to the familial melanoma gene CDKN2A: a maximum-likelihood combined linkage and association analysis in twins and their sibs. Am J Hum Genet 1999;65:483–92.
- Carli P, Biggeri A, Nardini P, et al. Epidemiology of atypical melanocytic naevi: an analytical study in a Mediterranean population. Eur J Cancer Prev 1997;6:506–11.
- Braun-Falco M, Hein R, Ring J, McNutt NS. Histopathological characteristics of small diameter melanocytic naevi. J Clin Pathol 2003;56:459–64.
- Elder DE, Green MH, Guerry DT, et al. The dysplastic nevus syndrome: our definition. Am J Dermatopathol 1982;4:455–60.
- Clemente C, Cochran AJ, Elder DE, et al. Histopathologic diagnosis of dysplastic nevi: concordance among pathologists convened by the World Health Organization Melanoma Programme. Hum Pathol 1991;22:313–19.
- De Wit PE, van't Hof-Grootenboer B, Ruiter DJ, et al. Validity of the histopathological criteria used for diagnosing dysplastic naevi. An interobserver study by the pathology subgroup of the EORTC Malignant Melanoma Cooperative Group. Eur J Cancer 1993;29A:831–9.
- Shea CR, Vollmer RT, Prieto VG. Correlating architectural disorder and cytologic atypia in Clark (dysplastic) melanocytic nevi. Hum Pathol 1999;30:500–5.
- National Institutes of Health. Consensus Development Conference Statement on Diagnosis and Treatment of Early Melanoma, January 27–29, 1992. Am J Dermatopathol 1993;15:34–43; discussion 6–51.
- Duffy K, Grossman D. The dysplastic nevus: from historical perspective to management in the modern era: part II. Molecular aspects and clinical management. J Am Acad Dermatol 2012;67:19.e1–2; quiz 31–2.
- Bauer J, Bastian BC. Distinguishing melanocytic nevi from melanoma by DNA copy number changes: comparative genomic hybridization as a research and diagnostic tool. Dermatol Ther 2006;19:40–9.
- Uribe P, Andrade L, Gonzalez S. Lack of association between BRAF mutation and MAPK ERK activation in melanocytic nevi. J Invest Dermatol 2006;126:161–6.
- Hussein MR. Melanocytic dysplastic naevi occupy the middle ground between benign melanocytic naevi and cutaneous malignant melanomas: emerging clues. J Clin Pathol 2005;58:453–6.
- Hussein MR, Roggero E, Sudilovsky EC, et al. Alterations of mismatch repair protein expression in benign melanocytic nevi, melanocytic dysplastic nevi, and cutaneous malignant melanomas. Am J Dermatopathol 2001;23:308–14.
- Hussein MR, Sun M, Tuthill RJ, et al. Comprehensive analysis of 112 melanocytic skin lesions demonstrates microsatellite instability in melanomas and dysplastic nevi, but not in benign nevi. J Cutan Pathol 2001;28:343–50.
- Kennedy C, Bajdik CD, Willemze R, et al. The influence of painful sunburns and lifetime sun exposure on the risk of actinic keratoses, seborrheic warts, melanocytic nevi, atypical nevi, and skin cancer. J Invest Dermatol 2003;120:1087–93.
- Elder DE. Precursors to melanoma and their mimics: nevi of special sites. Mod Pathol 2006;19(Suppl. 2):S4–20.
- Arumi-Uria M, McNutt NS, Finnerty B. Grading of atypia in nevi: correlation with melanoma risk. Mod Pathol 2003;16:764–71.
- Grichnik JM. Dermoscopy of melanocytic neoplasms: subpatterns of dysplastic/atypical nevi. Arch Dermatol 2003;139:1238.
- Grichnik JM. Dermoscopy of melanocytic neoplasms: subpatterns of dysplastic/atypical nevi. Arch Dermatol 2003;139:970.
- Grichnik JM. Dermoscopy of melanocytic neoplasms: subpatterns of dysplastic/atypical nevi. Arch Dermatol 2004;140:142.
- Hofmann-Wellenhof R, Blum A, Wolf IH, et al. Dermoscopic classification of atypical melanocytic nevi (Clark nevi). Arch Dermatol 2001;137:1575–80.
- Annessi G, Bono R, Sampogna F, et al. Sensitivity, specificity, and diagnostic accuracy of three dermoscopic algorithmic methods in the diagnosis of doubtful melanocytic lesions: the importance of light brown structureless areas in differentiating atypical melanocytic nevi from thin melanomas. J Am Acad Dermatol 2007;56:759–67.
- Barnhill RL. The Spitzoid lesion: rethinking Spitz tumors, atypical variants, ‘Spitzoid melanoma’ and risk assessment. Mod Pathol 2006;19(Suppl. 2):S21–33.