CHAPTER 136
Cutaneous Histiocytoses
Thai Hoa Tran, Elena Pope and Sheila Weitzman
Hospital for Sick Children, Toronto, Ontario, Canada
Introduction
Skin involvement is common in all forms of histiocytic disorders and is seen at all ages. In general, for convenience, the histiocytoses can be divided into those involving dendritic cells (DC disorders) and those involving the macrophage–monocyte lineage (or non-DC disorders). It needs to be understood, however, that the cells of the immune system show considerable plasticity, and movement between the various cell types in response to demand is common. Langerhans cell histiocytosis (LCH) is the commonest DC disorder and haemophagocytic lymphohistiocytosis (HLH) the commonest non-DC disorder. Rarer disorders, the non-LCH histiocytoses, can also be divided according to their dendritic and non-dendritic cell origin. Moreover, they can be further subdivided into two clinical phenotypes: (i) those that mainly involve skin but occasionally have a systemic component; and (ii) those in which skin may be involved but the systemic component predominates.
This chapter will provide an overview of the histiocytic disorders including a brief discussion of possible pathogenetic mechanisms, followed by an in-depth discussion of the skin manifestations, pathology and therapy of the histiocytic disorders. The similarities and differences between childhood and adult forms of the diseases will be highlighted.
Ontogeny of histiocytes
Histiocytes represent the cells of the mononuclear phagocyte system (MPS). They all share a common bone marrow progenitor cell, the neutrophil–macrophage colony-forming unit (NM-CFU). Histiocytes can be broadly divided into two functionally distinct cell populations: the ‘professional’ phagocyte and the antigen-presenting cell (APC) of which the Langerhans cell (LC) is the primary example. In contrast to all other dendritic cell populations, LCs do not arise from adult bone marrow-derived myeloid progenitor cells, but originate from fetal liver-derived monocyte precursors that populate the skin prior to birth [1]. Postnatally, they repopulate directly from other skin LC cells during steady-state conditions, but when major inflammation occurs, they can be replaced by circulating monocytes [2]. Monocytes arise from NM-CFUs and following their release into the circulation, migrate to various tissues where they differentiate into macrophages and other histiocytic lines.
Function of histiocytes
The phagocytes include the majority of resident tissue macrophages and immature macrophages. Macrophages are specialized in particle uptake and degradation by phagocytosis. Phagocytosis starts with recognition of the foreign material via surface receptors, in particular the carbohydrate and lectin receptors, followed by endocytosis [1]. This mechanism is involved in the phagocytosis of bacteria and possibly tumour cells. Phagocytes also express the specific complement component receptors CR1 and CR3, which bind to C3b and C3bi, respectively, and Fc fragment of immunoglobulin (FcIgG) receptors, which bind to the Fc fragment of IgG. These receptors are important in the phagocytosis of material that has bound IgG and complement, which act as opsonins and augment phagocytosis. Although limited, phagocytes also possess some antigen-presenting capacity. They generally present antigen to sensitized T cells but not to naive or ‘memory’ T cells.
APCs are histiocytic cells, or in some instances other cell types, that have specialized functional activity in presenting antigen to T cells. These cells are represented in humans by the blood dendritic cell, epidermal LC, interdigitating reticulum cell of the lymph node paracortex and veil cell of the efferent lymph. These cells have no phagocytic activity and, unlike ‘professional’ phagocytes, are unable to adhere to surfaces. They are able to internalize antigen by endocytosis, process it by lysosomal digestion and re-express the antigen on their surface in association with MHC molecules (class II for external antigens and class I for internal antigens). These cells are potent antigen-presenting cells and are able to present antigen not only to sensitized T cells but also to memory and naive T cells [2].
Classification of histiocytoses
The classification of the histiocytoses has been challenging due to the heterogeneity of these conditions, which affect both children and adults. Histiocytic disorders are generally defined by their constitutive cells, on the basis of pathological and immunohistochemical criteria, but also by the correct clinical context. The Writing Group of the Histiocyte Society first attempted to classify the histiocytic disorders according to their relationship to normal histiocyte subsets in 1987 [1], and later revised this classification in 1997 [2] to take into account the biological behaviour of the various disorders. In this system, the major histiocytic disorders are divided into two broad groups, those of varying biological behaviour and those that are truly malignant. Within each category, the disorders are further subclassified according to their affiliation with either dendritic cells (DC disorders) or with the macrophage–monocyte pathway (also known as non-DC disorders). Although this classification is not perfect, it does provide a framework to standardize the nomenclature and establish a ‘universal histiocytic language’ [3, 4].
DISORDERS OF DENDRITIC CELLS
Langerhans cell histiocytosis
Definition and nomenclature
Langerhans cell histiocytosis is a proliferative disease characterized by excess accumulation of CD1a + Langerhans cells in various sites, leading to tissue damage.
Epidemiology
Incidence and prevalence
Langerhans cell histiocytosis is a rare disease affecting 2–5 children per million per year with an estimated prevalence of 1 : 50 000 in children under 15 years of age [1]. A more recent study from Sweden reported a higher incidence of 8.9 children per million per year [2]. However, these figures are most likely underestimated due to the heterogeneity of the disease manifestations in which many patients, in particular those with localized bone or skin LCH, may remain undiagnosed or undergo spontaneous resolution.
Age
Langerhans cell histiocytosis can present at any age, from the neonatal period until old age, but the disease is most common in the 0–4-year age group. The median age at diagnosis is 30.2 months as reported by the French LCH Study Group, which included 348 patients less than 15 years of age [1]. Single system (SS) disease constitutes 70% of paediatric LCH, with bone being the most commonly affected organ and skin being seen in around 10% of SS-LCH patients [3]. Unifocal bone disease occurs in 50% of children with SS bone LCH over the age of 5, while multifocal bone disease is found in children aged between 2 and 5 years of age [1]. Multisystem (MS) LCH usually occurs in children less than 2 years of age.
In adults, the mean age at diagnosis is 35 years, with 10% being older than 55 years. Sixty-nine per cent of adults with LCH from the Histiocyte Society Adult Registry had MS disease with skin and lung involvement in 51% and 62%, respectively [4]. Of the 31% of adult patients with SS disease, the lung was involved in 51%, most of whom were smokers, followed by bone and skin in 38% and 14 %, respectively.
Sex
Most studies consistently report a slight male to female predominance, although some series have reported a male predominance as high as 2 : 1 [1].
Pathophysiology
Pathology
The Langerhans cell is a unique APC found within the epidermis, bronchi and mucosa. LCs are characterized phenotypically by low levels of MCH-II, intermediate CD11c and high expression of Langerin (CD207) [5]. Langerin is associated with the formation of tennis-racket-shaped intracellular organelles known as Birbeck granules, which represent the structural hallmark of LCs [6]. Human LCs express myeloid markers CD13 and CD33; the leukocyte marker CD45; adhesion molecules such as CD40, CD44 and E-cadherin; and CD1a, an MCH class I-like protein that has the function of presenting lipid antigen to T cells [7].
The origins of LCs have been the matter of intense debate – whether LCs are blood-borne or derived from in situ localized epidermal LC precursors. However, recent evidence suggests a dual origin of the LC network. During steady state, LCs may originate from a localized LC precursor in the epidermis that maintains LC renewal, while LCs could source from a blood-borne precursor during inflammation [6, 8, 9, 10].
The clinical heterogeneity of LCH is surprisingly unified by the histopathology of one abnormal population of LCH cells. They represent pathological cells with the same immunophenotype as LCs, including a surface rim of CD1a and a granular cytoplasmic and surface staining for Langerin (Figure 136.1). Moreover, S100 immunostain will give a nuclear and cytoplasmic blush to the cells. Fascin is demonstrable in low to moderate amounts in only a few cells. HLA-II (LN3 antibody) and CD68 (KP-1) display punctate paranuclear Golgi-like intracytoplasmic staining [11]. Morphologically, LCH cells are round to oval in shape, measuring 20–25 μm in size (2–3 times as large as lymphocytes), most commonly found in aggregates and lacking the ‘dendritic’ morphology. Their nucleus is lobulated, coffee bean or boat shaped, but the most typical LCH nuclei have complex angular and elaborate folds. The cytoplasm is generous and homogeneously pink. A prototypical LCH lesion usually displays LCH cells interspersed with inflammatory cells, mainly eosinophils and lymphocytes. Macrophages, both phagocytic and giant cell forms, can be found in many sites. Osteoclast-like giant cells predominate in any lesion involving bone [11].

Figure 136.1 Langerhans cell histiocytosis (LCH) lesion in the skin showing (a) characteristic CD1a-positive staining of LCH cells and (b) skin LCH cells showing cytoplasmic and surface staining for langerin.
Genetics
The aetiology of LCH is unknown, but recent advances in molecular genetics have provided important insights into the pathophysiology of LCH and have deepened our understanding of the disease. For a prolonged period, immune dysregulation and abnormal cytokine expression have been stipulated as potential pathogenic mechanisms in LCH, given the immune and inflammatory nature of LCs and based on the observation of regulatory T-cell expansion in LCH [12, 13, 14]. However, no specific immune dysfunction has been demonstrated and patients do not have an increased risk of infection. More recently, advances in molecular and genomic technologies have supported the concept of LCH as a neoplastic disease. The clonality of pathological LCHs identified in non-pulmonary LCH provided the first argument supporting the idea of LCH as a neoplasm [15, 16]. The recent identification of a BRAF V600E mutation in 57% of LCH cases tested to date in addition to the demonstration of RAS pathway activation in all of the cases, even those without the BRAF mutation, suggest a common mechanism of activation of the signal transduction pathway and give strong support to the designation of LCH as a neoplasm, although not necessarily a malignancy [17, 18]. The BRAF V600E mutation was found in all risk groups and had no impact on overall survival, although its presence conferred a two times increased risk of reactivation (P = 0.04) [18]. Nonetheless, whether BRAF mutation in LCH cells is necessary or sufficient for the development of LCH remain unanswered, although next-generation technologies certainly represent an invaluable tool to address the molecular basis for the heterogeneous clinical entities of LCH.
Clinical features
Despite the identical histopathological appearance of various forms of LCH, the disease encompasses a heterogeneous clinical profile and affects many different organs. The classification of LCH is based on the number of organs systems involved with an initial subdivision into single system (SS) and multisystem (MS) disease. SS-LCH is further subdivided into unifocal and multifocal disease, while MS-LCH is divided into ‘low-risk’ disease and ‘risk’ disease, where risk represents the risk of death. For clarity, the latter group will be referred as ‘high risk’ in this chapter.
Presentation
SS bone LCH is the most common form of LCH. The skull vault is the most frequent site of disease; however, any bone can be involved except for the hands and feet. SS skin LCH (skin-only LCH) is the second most common site (Table 136.1). MS-LCH refers to the involvement of any organ, although gonads and kidneys are usually spared. The ‘risk’ designation is limited to involvement of the haematopoetic system, liver and spleen. In contrast to the adult smokers’ lung LCH, isolated paediatric lung LCH is no longer thought to give a major risk of death [19]. In this chapter, only skin LCH and its variants will be discussed in detail.
Table 136.1 Distribution of Langerhans cell histicytosis in 170 children with SS-LCH treated on the DAL-HX studies
| Sites | Patients (%) |
| Unifocal bone | 68 |
| Multifocal bone | 19 |
| Isolated skin | 11 |
| Isolated lymph node | 2 |
From Titgemeyer et al. 2001 [30]. Reproduced with permission of John Wiley & Sons.
Skin is the second most commonly involved organ, reported in 30–60% of paediatric cases with skin being the only affected site in 10% (skin-only LCH) [20]. Skin LCH can occur at any age. The appearance of the skin lesions is very variable and includes macules, papules, plaques, scales, vesicles, pustules, crusts, bullae and ulcerative lesions (Figure 136.2). The most characteristic cutaneous lesion of LCH in children consists of papulosquamous lesions with greasy scales, affecting the scalp, resembling seborrhoeic dermatitis (Figure 136.3). Other sites include skin folds such as the gluteal cleft and midline of the trunk, but any area can be involved including the nails. Persistent eruption on the scalp and in skin flexures outside of infancy should raise the suspicion of LCH even in the absence of other signs and symptoms. However, unusual persistence of ‘cradle cap’ or ‘nappy rash’ even in infancy should suggest the possibility of LCH and warrants a biopsy. Petechiae or areas of purpura can accompany skin lesions when the platelet count is reduced. Involvement of the external ear canal is usually considered part of skin LCH and may be associated with secondary infection with Pseudomonas aeruginosa. By contrast, LCH of the middle and inner ear is often associated with temporal bone disease. The mucous membranes of the mouth and genital tract may also be involved, the latter being seen more commonly in adult patients [19].

Figure 136.2 Langerhans cell histiocytosis showing a polymorphic eruption of papules, vesicles, crusts and telangiectasias affecting the nappy area, including the folds, in an infant.

Figure 136.3 Langerhans cell histiocytosis showing yellow crusted papules in a seborrhoeic dermatitis distribution in an older child.
Clinical variants
Skin LCH in adults
The International Histiocyte Society compiled a registry of adult patients with LCH and found that 14.3% of adults with SS-LCH and 62% with MS-LCH have skin involvement [4]. Skin lesions are often the presenting feature of the disease. The areas of involvement are similar to those seen in children but ulceration of the flexures, groin, perianal or vulvar area is common. Inframammary involvement may also occur. As with children, lesions may be papular, pustular, nodular, erythematous, poikilodermatous-like, xanthomatous, polypoid and peduncular. They may involve the nails and mucosa, including the genital mucosa. They may be asymptomatic or may be pruritic or burning [20, 21, 22].
Skin-only LCH
Langerhans cell histiocytosis localized to the skin occurs in approximately 10% of children and adults with SS-LCH. Skin-only LCH in children is seen in the very young child and may undergo spontaneous regression within weeks to many months (Figure 136.4). Alternatively, the disease may progress to MS ‘high-risk’ disease, which can be fatal. Of 12 skin-only infants in a series from the Hospital for Sick Children (SickKids, Toronto) [23], 10 were observed without therapy; four of the 10 progressed to MS disease 5 weeks to 5 months after diagnosis, resulting in two deaths. Additionally, one patient developed a single bone lesion almost 4 years after diagnosis with skin-only LCH. In infants from birth to 4 weeks of age, skin-only LCH is sometimes called Hashimoto–Pritzker disease or the congenital self-healing reticulohistiocytosis (CSHRH) variant (Figure 136.5). Skin-only LCH and CSHRH share the same pathology and immunostaining. Although some dermatologists claim to be able to distinguish CSHRH from skin-only LCH clinically, the literature does not support that unless myelin-dense bodies, thought to represent senescent mitochondria, are seen on electron microscopy. Battistella et al. described a series of 31 patients, 21 ‘self-regressive’ and 10 ‘non-self-regressive’, and found that mono-lesional forms, necrotic lesions and hypopigmented macules at presentation in addition to distal extremity involvement were seen only in patients with self-regressive cutaneous LCH [24]. Since both skin-only LCH and CSHRH require initial investigations to exclude systemic disease and both forms can be observed without therapy if limited to the skin, it is felt that the distinction is without real value. We emphasize that all young children with skin-only disease should be carefully observed and the diagnosis of spontaneously regressing disease should only be made retrospectively [23].

Figure 136.4 Skin-only Langerhans cell histiocytosis in an infant with spontaneous resolution.
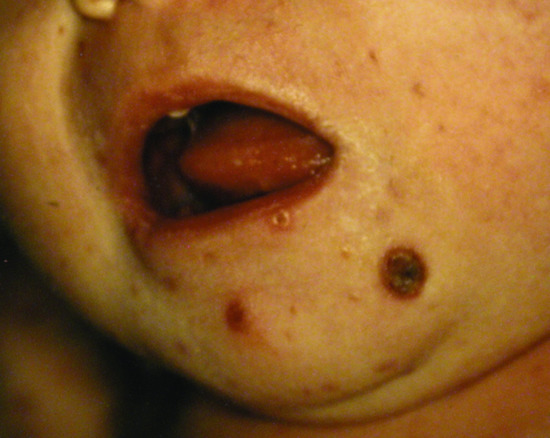
Figure 136.5 Congential self-healing reticulohistiocytosis (Hashimoto–Pritzker disease) showing ulcerated nodules in an infant.
A commonly asked question is whether patients with skin-only LCH have a risk of long-term complications such as diabetes insipidus. Unfortunately, no prospective studies have looked at this question. In two small published retrospective series, one consisting of 25 infants [25] and the other of 19 infants [26] with skin-only LCH, one patient in each series eventually developed diabetes insipidus. Prolonged follow-up is, therefore, recommended but in view of the generally good prognosis, radiological investigations should be limited and based on clinical suspicion.
Skin LCH as part of low-risk multisystem disease
This skin LCH clinical variant is formerly known as Hand–Schüller–Christian syndrome. This entity is a chronic, multifocal form of LCH. It is characterized by the presence of lytic bone lesions, exophthalmos, diabetes insipidus and skin lesions, although all these features are not required for diagnosis. The age of onset is usually later than the onset of skin-only LCH, typically between 2 and 6 years. Cutaneous manifestations include nodules and tumours that are yellow-brown in colour or with a seborrhoea-like picture, but any skin picture may be seen. Oral mucosa may be involved with gingival ulceration and haemorrhage. Premature tooth eruption may be the first manifestation of this variant in some cases [22].
Skin involvement as part of disseminated disease
This variant was previously known as Letterer–Siwe disease and represents the most extensive and severe form of LCH. It occurs in less than 15% of paediatric cases and is usually seen under the age of 2 years, often in the neonatal period. The skin is involved in 75–100% of cases, manifesting as a typical seborrhoea-like pattern in the scalp and nappy area; however, any part of the body may be affected. Extensive ulceration, superinfection, petechia and purpura may accompany skin lesions. Multiple organs are involved including the bones, liver, spleen, lungs, central nervous system (CNS) and bone marrow. This form carries the worst prognosis, is the least likely to resolve spontaneously and always requires systemic therapy [22].
Differential diagnosis
The differential diagnosis of skin LCH includes dermatophytoses, mastocytosis and scabies. If the lesions are vesicular, LCH needs to be distinguished from a larger number of vesiculobullous diseases including herpes simplex and herpes zoster. Papulonodular lesions may need to be differentiated from malignant nodules occurring in neuroblastoma, leukaemia and lymphoma. The nappy area and retroauricular involvement is often confused with seborrhoeic dermatitis. In adults, other conditions such as hydradenitis suppurative, Paget disease, keratosis follicularis and sexually transmitted diseases may have to be considered.
Disease course and prognosis
The natural history of paediatric LCH varies from spontaneous regression to a low-grade chronic disease with multiple reactivations and the possibility of significant long-term consequences, to a ‘high-risk’ disease that can be fatal. Its course also varies with different subgroups and depending on the extent of disease. The mortality from SS and low-risk MS-LCH is extremely low. The problem for these subgroups is the propensity for reactivations and for the development of significant long-term sequelae, of which diabetes insipidus is the commonest, and CNS disease the most serious.
Patients with SS disease tend to undergo spontaneous regression or respond well to limited therapy. In an earlier series from the Hospital for Sick Children (SickKids, Toronto) consisting of 180 patients with bone LCH, 12% of SS unifocal bone reactivated and there were no cases of diabetes insipidus. Of those with SS multifocal bone, 25% reactivated and the risk of diabetes insipidus was 12%; while in bone as part of low-risk MS disease, the reactivation rate was 50% and more than 25% developed diabetes insipidus [3]. In very young children with high-risk organ involvement, the mortality rate has been decreased to 10% in the most recent therapy protocols [27]. Historically, of those high-risk MS children who respond well to therapy, 50% reactivate in low-risk organs such as bone; these patients had a similar risk of long-term sequelae such as diabetes insipidus and CNS disease as the low-risk MS group. In the recently published LCH-III study, therapy was prolonged to 12 months for all high-risk MS patients, and the low-risk MS patients were randomized to receive 6 months versus 12 months of therapy. In both subgroups, prolongation of therapy to 12 months resulted in a reduction of the reactivation rate from 50% to about 30% but it has not yet been proven whether that results in a reduction in late sequelae [27]. The open LCH-IV study will randomize patients to between 12 and 24 months of therapy to see if the reactivation rate can be further reduced and will follow patients long term to evaluate the effect on serious late complications.
The natural history of LCH in adults is less clear but it is thought that spontaneous regression, even in SS disease, is less likely to occur and that adult patients typically require some form of therapy depending on the extent of organ involvement. In adult LCH, survival rates of patients with SS disease approach 100% [4]. The survival of adult patients with MS disease including skin is better than that seen in children due to the lower number of organs involved [22, 28].
Investigations
Diagnosis
Langerhans cell histiocytosis is diagnosed by the characteristic histology and immunostaining made on biopsy. Because LCs are normally found in the skin, nodes and lung, it is important to differentiate LCH from reactive LCs in these areas (see Figure 136.1). Thus LCH is a clinicopathological diagnosis; in other words, its diagnosis requires the correct histology and immunostaining in the correct clinical setting.
Evaluation
The aim of evaluation is to define disease extent in order to guide treatment planning and follow-up evaluation. Patients may present with skin, bone, lymph node, pituitary, lung or liver disease as the primary site, but need to be fully investigated before a diagnosis of SS disease can be made. Plain radiography, as part of the skeletal survey, is usually the first modality to assess bone involvement (Figure 136.6). Guidelines for diagnosis, work-up and treatment of paediatric LCH have been published [29]. Box 136.1 details the investigations that should be conducted on all suspected cases and the indication for more specialized investigations in some patients.

Figure 136.6 Radiograph of a humerus showing a typical punched-out lytic lesion of bone Langerhans cell histiocytosis.
Management
The modern treatment approach utilizes a risk stratification strategy based on the extent and severity of disease, which represent the main determinants of outcome. Paediatric patients with SS-LCH generally have a benign course with a high chance of spontaneous remission over a period of months to years, and a favourable outcome with no or limited treatment [30].
In unifocal bone disease, simple curettage or even biopsy can be curative. The intralesional instillation of steroids (75–150 mg of methylprednisolone) has been shown to be an effective and safe treatment modality, typically for symptomatic bone lesions [31]. Due to concerns of long-term sequelae and secondary malignancy, radiation at low dose (6–10 Gy) should be reserved only for emergency circumstances when vital structures such as the optic nerve and/or spinal cord are compromised [31].
Systemic therapy is reserved for multifocal SS disease and for MS disease. Many drugs have been used, from non-steroidal anti-inflammatory drugs and steroids to cytostatic drugs such as vincristine, vinblastine, etoposide, 6-mercaptopurine, methotrexate, cytarabine and cladribine, in addition to the recent immunotherapeutic agents (interferon α, anti-tumour necrosis factor α (anti-TNF-α), ciclosporin A, thalidomide, etc.) [7, 32, 33]. In the last 20 years, three large-scale, international, prospective, therapeutic trials conducted by the Histiocyte Society (LCH-I, LCH-II, LCH-III) yielded important insights into the management of MS-LCH [27, 34, 35]. Firstly, the most frequently used regimen consisting of vinblastine with a corticosteroid is a safe and effective regimen. The addition of either VP-16 or methotrexate to the vinblastine–steroid–6-MP backbone did not improve outcomes of patients with MS-LCH or with high-risk organ involvement [27, 36]. In patients with MS-LCH, a 12-month maintenance period reduces the rate of reactivation compared with a 6-month maintenance period [28]. The combination of vincristine, steroid and cytarabine, as reported by the Japanese cooperative group, represents an equivalent therapeutic alternative [37]. For refractory disease, cladribine monotherapy or in combination is an effective therapeutic option [38]. Haematopoietic stem cell transplantation can be considered in patients with high-risk organ involvement and refractory to salvage therapy [39].
Targeted therapy
The recent discovery that the majority of LCH cells carry the V600E BRAF mutation and that all have activation of the RAS pathway raises the possibility of BRAF and other inhibitors as promising novel therapies in LCH. A dramatic efficacy of vemurafenib, a newly approved BRAF inhibitor, has been shown in two patients with multisystemic and refractory Erdheim–Chester disease and LCH harbouring the BRAF V600E mutation [40]. Clinical trials utilizing vemurafenib and other BRAF inhibitors as first line therapy in adults with LCH and salvage therapy in paediatric patients with reactivated LCH, are open at several centres in the USA and Canada. At present, however, this therapy should be considered to be experimental.
Management of skin LCH
Paediatrics
With skin-only LCH, these young patients can be observed and therapy given only if there is pain, bleeding and/or ulceration. Physicians should try to avoid treating for cosmetic reasons alone. If treatment is required, topical therapy such as topical corticosteroids should be tried first. Other topical therapies that have been described, but that are not commonly used, include topical pimecrolimus or tacrolimus, narrow-band UVB and psoralen–UVA (PUVA) [22, 32].
Management usually consists of systemic chemotherapy for skin involvement as part of MS-LCH. The most commonly used protocol is the combination of vinblastine and prednisolone as per the standard arm of the LCH-II and LCH-IIII protocols of the Histiocyte Society [27, 34] with or without topical therapy (usually consisting of medium to high potency corticosteroid). Other possibilities are intravenous cytosine arabinoside alone or with vincristine and prednisolone. Progression of disease after only 6 weeks of therapy is a very poor prognostic sign and mandates a change to a salvage protocol and possibly even a reduced intensity conditioning haematopoetic stem cell transplant (RIC-HSCT).
Adults
By contrast to the young child, adult patients with skin-only LCH usually require some form of therapy. In general, prospective trials have been limited to paediatric MS disease and most of the published reports on therapy in adult LCH consist of single case reports or small series. The adult LCH study group of the Histiocyte Society recently published guidelines for the management of adult LCH [41] and these guidelines continue to be refined. Possible therapies for skin LCH in adults include surgery, topical, ultraviolet light, radiation or systemic therapy.
- Surgery. Surgical excision is a good option for limited skin lesions, but mutilating surgery such as vulvectomy or hemivulvectomy should not be performed.
- Topical therapy. First line topical therapy usually consists of high potency corticosteroids or intralesional steroids. Other options include topical tacrolimus, which has been successful in anecdotal reports, but systemic toxicity limits its use, especially for a large treatment area or ulcerated lesions. If utilized, tacrolimus levels should be measured. Topical imiquimod for 5 days/week for 2 months was used successfully in one adult patient with disease refractory to other therapy [41].
- Ultraviolet light therapy. Whole body UVB and PUVA have been utilized successfully in adult patients with skin LCH [42]. In addition, a recent report described the successful use of narrow-band UVB delivered locally by means of an Excimer laser [43].
- Radiation therapy. Localized radiation therapy and electron beam radiation therapy have been used but would not be the first choice of treatment [44].
- Systemic therapy. Adults do not tolerate or respond as well as their paediatric counterparts to vinblastine and prednisolone therapy. Nonetheless, a vinblastine and steroid combination remains the first line systemic therapy for adult LCH in many centres, particularly in Europe, including for skin LCH requiring systemic therapy. Other centres prefer to avoid these drugs and suggest starting with single agent cytosine arabinoside given for 5 days every month [45]. Other systemic therapy utilized in adult LCH patients and summarized by Girschikowsky et al. [46] include weekly oral methotrexate [47], daily oral etoposide [22] and oral chlorambucil (A. Chu, personal communication). Daily oral etoposide has been demonstrated to be less myelosuppressive and to carry much less risk of causing secondary acute myeloblastic leukaemia than the intravenous form of the drug. Interferon α has been successful in some refractory cases [21], similar to oral thalidomide [48] and oral isotretinoin [42]. The combination of interferonα and thalidomide proved successful in patients with skin disease refractory to either agent alone [49].
With all forms of therapy, a relapse of skin LCH following the discontinuation of therapy is common, but the disease often responds again to reinstitution of the same therapy and prolonged therapy and a slow taper may be required.
Resources
Further information
- Euro-Histio-Net: http://www.eurohistio.net/.
- Histiocyte Society: http://www.histiocytesociety.org.
- National Cancer Institute at the National Institutes of Health, Langerhans cell histiocytosis treatment: http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional. (All last accessed July 2014.)
DISORDERS OF NON-DENDRITIC CELLS
Haemophagocytic lymphohistiocytosis
Definition and nomenclature
Haemophagocytic lymphohistiocytosis is a hyperinflammatory condition resulting from uncontrolled ineffective immune response.
Introduction and general description
Haemophagocytic lymphohistiocytosis (HLH) is a rare histiocytosis characterized by widespread infiltration of multiple organs by lymphocytes and mature histiocytes showing prominent phagocytosis. HLH with an underlying genetic defect is regarded as ‘primary HLH’ while all other forms are reactive, such as macrophage activation syndrome, thus grouped as ‘secondary HLH’ [1]. Primary HLH can be divided into those inherited cases where HLH is the only clinical manifestation (e.g. familial haemophagocytic lymphohistiocytosis (FHL)), and those with other manifestations including oculocutaneous albinism where HLH is an important part of the clinical picture. In both subtypes, HLH is often due to gene mutations underlying a deficiency in the triggering of apoptosis. In secondary forms of HLH, haemophagocytosis occurs as a result of macrophage activation by a known stimulus, which can be infectious, malignant, autoimmune or physical. A number of infections, among which Epstein–Barr virus (EBV) is the commonest, have been shown to be associated with secondary HLH. Despite different aetiologies, the common pathway involves a production of high levels of proinflammatory cytokines by T-helper cells and excessive activation of monocytes and macrophages leading to phagocytosis of the blood cells.
Epidemiology
Incidence and prevalence
The estimated incidence of FHL is 1 in 50 000 live births [2], but this figure will almost certainly increase as more genetic defects are being discovered.
Age
The majority of FHL patients present before the age of 1 year, while most adult cases are due to secondary HLH. However, primary HLH can occur at any age.
Sex
The male to female ratio is 1 : 1 [2], although some studies demonstrate a slight male preponderance [3].
Ethnicity
There is an increased incidence in ethnic groups with higher rates of consanguinity [4].
Associated diseases
The association of secondary HLH and EBV infection has been well described, with more than 50% of patients coming from the Far East from Japan, China or Taiwan [5]. EBV may also trigger HLH in patients with primary HLH. More than half of EBV-associated HLH cases occur in children younger than 3 years of age [5]. Lymphoma, on the other hand, is the most common cause of adult-onset secondary HLH [6].
Pathophysiology
Pathology
Histologically, the involved tissue shows a diffuse infiltrate with lymphocytes and mature histiocytes. The histiocytes exhibit active phagocytosis, especially of erythrocytes but also of leukocytes and occasionally platelets (Figure 136.7). The histiocytes stain positively for acid phosphatase, non-specific esterase, lysozyme and α-antichymotrypsin. A striking histological finding is lymphocyte depletion of the lymph nodes, spleen and thymus [7]. Biopsy of the associated rash usually has non-specific findings with dermal perivascular infiltrates. Although haemophagocytosis is not usually found in the skin, one of the authors’ patients had HLH confirmed by haemophagocytosis within an infiltrative skin nodule (Sheila Weitzman, personal communication).

Figure 136.7 Haemophagocytosis in haemophagocytic lymphohistiocytosis.
Genetics
The pathophysiology of HLH is characterized by hypercytokinaemia (elevated serum levels of inflammatory cytokines) and a concomitant defect in effector lymphocytes, namely cytotoxic T lymphocytes and natural killer cells [4, 8, 9]. FHL is an autosomal recessive disease but certain X-linked conditions such as both forms of the X-linked lymphoproliferative syndrome (XLP) also result in the HLH phenotype [10]. Recent genetic studies have revealed that perforin gene mutations account for 20–40% of FHL cases [11]. Another 20–25% cases of FHL harbour mutations in MUNC13-4 [11]. Perforin is an apoptosis-triggering agent required for granzyme B to induce apoptosis in target cells [12] and MUNC13-4 mediates the exocytosis of the perforin-containing granules from effector lymphocytes [13]. Most of the genetic HLH cases are due to mutations of important genes in the same pathway. These underlying genetic defects provide a plausible explanation for the defect in cytotoxicity observed in HLH [14].
Clinical features
Presentation
The cardinal symptoms are prolonged high fever, hepatosplenomegaly and cytopenias. Fever is usually the first sign of the disease, with symptoms of an upper respiratory tract or gastrointestinal infection. Pallor, anorexia, vomiting, irritability, hepatosplenomegaly and lymphadenopathy are usually present at presentation. Around half of patients develop a transient, non-specific, maculopapular rash, which is often seen at times of high fever [15]. About 20% of patients have neurological symptoms, presenting with seizures or other signs of meningeal irritation.
The initial clinical picture of HLH often resembles a harmless viral illness; however, progression of clinical symptoms and worsening of laboratory values such as rising inflammatory markers or progressive cytopenias in a sick child should alert the physician to the possibility of HLH. Patients may improve initially with supportive care such as transfusions or antibiotics, but responses are usually short lived and the disease can rapidly be fatal.
Investigations
Diagnosis
The diagnosis of HLH is made by compiling a number of clinical and laboratory features that raise the possibility of HLH (Box 136.2). Laboratory tests should include a complete blood count that usually reveals cytopenia, initially with anaemia or thrombocytopenia. Up to 28% have leukocytosis, with either neutrophilia or lymphocytosis [15]. Neutropenia affects most children eventually and progressive pancytopenia is seen in untreated patients. Transaminitis with hyperbilirubinaemia is common. Some patients exhibit a consumptive coagulation picture with hypofibrinogenaemia, and d-dimers are elevated in most patients. Hypertriglyceridaemia is present in most children, and may reach levels >10 mmol/L. Hyperferritinaemia >500 μg/mL is found in the majority of patients, but serum ferritin levels above 10 000 μg/mL is more suggestive of HLH [10]. A positive Coombs’ test and platelet antibodies are present in some patients. Immunological testing shows abnormalities of both humoral and cellular components of the immune system. Cytotoxic T cell and natural killer cell activity is markedly reduced or absent in affected patients. Elevated levels of the α-chain of the soluble interleukin 2 receptor (sCD25) and of the soluble FAS (CD178) ligand reflect stimulation of histiocytes and T cells, while elevated soluble CD163 (sCD163) reflects activation of the alternative pathway of macrophage activation.
Evaluation
A bone marrow examination is mandatory to exclude underlying malignancy but only a minority of people show haemophagocytosis at presentation. The sole presence of haemophagocytosis does not make the diagnosis of HLH without the other clinical manifestations and laboratory criteria. A lumbar puncture should also be performed as more than 50% of patients will have asymptomatic neurological pathological abnormalities. Molecular genetic studies are now available to establish the diagnosis of a number of FHLs but not all the genes have as yet been identified (Table 136.2). It is important to perform a thorough history and diagnostic work-up to rule out the variety of underlying conditions leading to the same hyperinflammatory phenotype. Infections, systemic inflammatory rheumatic disorders, malignancies, inherited genetic defects and inborn errors of metabolism have been associated with the HLH phenotype.
Table 136.2 Genetic defects leading to haemophagocytic lymphohistiocytosis (HLH)
| Disease | Genea |
| FHL1 | Unknown |
| FHL2 | PRF1 |
| FHL3 | MUNC13.4 |
| FHL4 | STX11 |
| FHL5 | STXBP2 |
| Chediak–Higashi | LYST |
| Griscelli II | RAB27a |
| Hermansky–Pudlak II | AP3B1 |
| XLP1 | SH2D1A (SAP) |
| XLP2 | BIRC4 (XIAP) |
Adapted from Weitzman 2011 [21].
Except for XLP and some congenital immunodeficiencies, all known genetic HLH is due to mutations in genes important in the cytolytic secretory pathway that causes perforin and granzymes to induce apoptosis in target cells.
Diseases: FHL1–5, familial haemophagocytic lymphohistocytosis 1 to 5; XLP1–2, X-linked lymphoproliferative disease type 1 and 2. Genes: PRF1, perforin; STX11, syntaxin 11; STXBP2, syntaxin-binding protein 2; LYST, lysosomal trafficking regulator; RAB27A, RAS-associated protein 27A; AP3B1, adaptor 3 B1 subunit; SH2D1A, signalling lymphocyte activation molecule-associated protein; BIRC4, baculoviral IAP repeat-containing 4.
Management
Familial haemophagocytic lymphohistiocytosis is rapidly fatal if left untreated, with a reported median survival of less than 2 months from diagnosis, with 96% of patients dying within 12 months. Diagnosis can be challenging when there is no family history or no evidence of a molecular defect. Therefore, it should be emphasized that sometimes treatment must begin on a strong clinical suspicion even when diagnostic criteria are not completely fulfilled. Various cytotoxic regimens have been tried without success. Major improvements in survival were only observed when the combination of etoposide with corticosteroids was introduced into HLH therapy [16]. The HLH-94 treatment protocol consists of an induction phase with etoposide and corticosteroids for 8 weeks, followed by the addition of ciclosporin A (CSA) in the continuation phase. The 3-year probability of survival for the 113 eligible patients was 55% and the 3-year probability of survival for those who underwent HSCT was 62% [3]. All surviving HSCT patients are free of disease. A different regimen consisting of corticosteroids, CSA and antithymocyte globulin has been successful in inducing remission in patients in a French series but relapse is common [17, 18]. Therefore, allogeneic HSCT remains the only cure to date for FHL. Primary HLH patients slated for transplant should remain on maintenance therapy until transplant occurs. For patients who failed induction on the HLH-94 protocol, some responded to increased doses of the same drugs, in particular higher dose of dexamethasone. If that strategy fails, therapy with the anti-CD52 antibody alemtuzumab (Campath) has proven to be the best salvage therapy at present [19]. Campath is also an integral part of the reduced-intensity conditioning regimen which has successfully reduced the high transplant-related mortality associated with HLH-HSCT, although primary graft rejection remains a concern [20]. Experience in HLH-HSCT is necessary for centres undertaking these difficult transplants. Treatment of milder forms of HLH, as in secondary HLH, can be initiated with corticosteroids alone or in combination with intravenous immunoglobulin. If they fail to respond or progress, immunosuppressive agents such as etoposide or CSA can be added.
NON-LANGERHANS CELL HISTIOCYTOSES
The non-Langerhans cell histiocytoses (non-LCHs) are a diverse group of disorders defined by the accumulation of histiocytes that do not meet the phenotypical criteria for the diagnosis of LCH. The non-LCHs are generally benign proliferative disorders, which can be classified according to their dendritic or non-dendritic cell origin. Clinically, they can be further stratified into two major groups: (i) those that predominantly affect the skin but may have a systemic component (e.g. juvenile xanthogranuloma, reticulohistiocytoma); and (ii) those such as Erdheim–Chester disease and sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease) that are primarily systemic diseases where the skin may be involved.
DENDRITIC CELL ORIGIN
Disorders with mainly skin involvement with/without a systemic component
Juvenile xanthogranuloma
Definition and nomenclature
Juvenile xanthogramuloma (JXG) is a benign proliferative disorder of histiocytes occurring in early infancy and childhood that spontaneously regresses.
Epidemiology
Incidence and prevalence
Juvenile xanthogranuloma is the commonest of the non-LCH, non-HLH histiocytic disorders. The incidence of JXG is unknown and is likely underestimated due to its natural history of spontaneous involution.
Age
JXG occurs predominantly during infancy with median ages at onset ranging from 5 months to 1 year as reported in two large series [1, 2]. Lesions may occur at birth and very rarely in adults [3].
Sex
There is no sex predilection; although male preponderance was much higher (male : female 12 : 1) in children with multiple skin lesions [4].
Ethnicity
There is a racial predilection, being 10 times more common in white than in black children.
Associated diseases
JXG has been associated with neurofibromatosis type 1 (NF1) and juvenile myelomonocytic leukaemia (JMML) [5]. Patients with JXG and NF1 have a significantly higher risk of developing myeloid leukaemia than normal [6].
Pathophysiology
Pathology
JXG is characterized by a dense infiltrate of small histiocytes in the dermis, which stain positively for factor XIIIa, CD68, CD163, CD14 and fascin (Figure 136.8a, b). Stains for S100 and CD1a are negative. Touton giant cells, seen in 85% of JXG cases, can be distinguished by the characteristic wreath of nuclei around a homogenous eosinophilic centre and prominent xanthomatization in the periphery (Figure 136.8c) [1]. In extracutaneous JXG lesions, Touton cells are absent or reduced in numbers. In young infants less than 6 months old, JXG can present with mainly vacuolated histiocytes, without foamy histiocytes or giant cells.

Figure 136.8 Juvenile xanthogranuloma. (a) Small, slightly spindled histiocytes permeate the dermis, splaying collagen fibres. Touton cells are not represented. Magnification 200× (H&E). (b) This lesion has a high content of factor Xllla. Magnification 200× (diaminobenzidine). (c) Touton giant cells with CD68-positive immunoreactivity.
(Parts a and b, courtesy of Dr R. Jaffe, Children's Hospital of Pittsburgh, Pittsburgh, USA.)
The pathogenesis of JXG remains unclear. Lipid abnormalities do not play any role in JXG. It is suggested that dermal dendrocytes are the precursor cells of most non-LCHs, including JXG [4, 5, 7]. An origin from dermal dendrocyes was postulated based on the positive immunostaining for factor XIIIa, but derivation from an earlier circulating precursor cell has been suggested recently as being able to better explain the occurrence of extracutaneous JXG [6].
Genetics
Since neurofibromin, the protein product of the gene involved in NF1, negatively regulates the RAS oncogene, the association of JXG with NF1 and JMML has led to the speculation that the dysregulation of apoptosis via dysfunction of the RAS oncogene may lead to both leukaemia and JXG [6]. Interestingly, the recently described BRAF V600E mutation in LCH [8] and Erdheim–Chester disease [9], have not so far been identified in JXG [10]. This is despite many published case reports of JXG and LCH occurring in the same patient [11, 12, 13], either simultaneously or with LCH first and then JXG. Since it appears that some LCH lesions may become xanthomatous when inactive, this needs to be distinguished from true active JXG that can be seen with or following LCH. Furthermore, the mechanism for spontaneous resolution remains to be elucidated.
Clinical features
Presentation
Clinically, cutaneous lesions are the most common presentation of JXG. It presents with single to multiple papules or nodules with a predilection for the face, head and neck, followed by the upper torso and upper and lower extremities. Single lesions (unifocal JXG) are the most common, but multiple lesions, ranging from a few to hundreds, may occur – particularly in young male infants. They usually start as reddish yellow macules/papules, which may enlarge and evolve into yellow brown patches/plaques with surface telangiectasias (Figure 136.9). The consistency is generally firm and rubbery. Giant JXG, defined as a lesion greater than 2 cm in diameter, has been reported. It usually occurs in females, less than 14 months of age, on the proximal extremity or upper back, and may be misdiagnosed as haemangioma, particularly as it can be preceded by a congenital precursor lesion [14].

Figure 136.9 Juvenile xanthogranuloma showing multiple, disseminated, yellow papules.
Clinical variants
Systemic involvement occurs in 4% of children, mostly during infancy with a median age of 0.3 years [5]. Almost half of the patients have no skin lesions. The most common site is a solitary mass in the deep soft tissues (deep JXG) followed by the liver, spleen, lung and central nervous system (CNS). Most systemic lesions undergo spontaneous resolution, however ocular and CNS involvement lead to significant complications.
Ocular JXG occurs in up to 10% of cases, but in less than 1% of children with coexisting cutaneous JXG [15]. Eye involvement is usually, but not always unilateral, and commonly presents with erythema, irritation and photophobia, which may lead to acute hyphaema, glaucoma and blindness [16] (Figure 136.10). Thus, recognition of eye JXG is important for the prevention of vision loss.
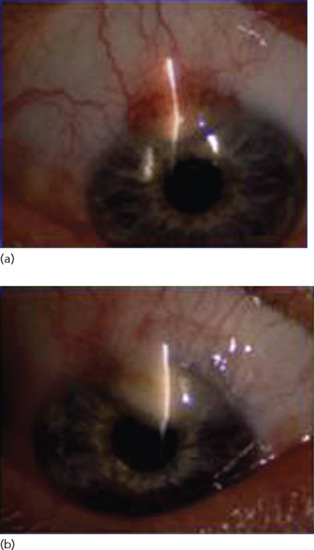
Figure 136.10 Juvenile xanthogranuloma of the iris. (a) Right eye, 4 November 2001. (b) Right eye, July 2003.
(Courtesy of Dr J. Donadieu, Trousseau Hospital, Paris, France.)
Disease course and prognosis
JXG, either cutaneous or systemic, usually follows a benign course with spontaneous resolution of lesions within 1 to 5 years. Long-term sequelae are rarely reported. However, CNS involvement may result in significant morbidity with seizures, ataxia, increased intracranial pressure, subdural effusions, developmental delay, diabetes insipidus and other neurological deficits [17]. Fatalities have been reported in cases of progressive CNS JXG [17, 18] and neonates with hepatic involvement [19].
Investigations
The diagnosis of JXG is made on immunohistochemistry. Extensive work-up should be reserved for those with clinical suspicion of systemic involvement.
Management
The management of JXG depends upon the site(s) of involvement. For patients with single and accessible lesions, surgical excision appears to be curative, although most childhood lesions will resolve spontaneously. As a result, cutaneous JXG does not require treatment, although parents need to be warned that occasionally resolution may result in a scar. For ocular JXG, therapy includes topical, intralesional and subconjunctival corticosteroids; surgery may be required to treat complications such as hyphaema and glaucoma; and systemic corticosteroids, chemotherapy or low-dose ‘non-cataractogenic’ radiation therapy (300–400 cGy) may be required for non-responders [20]. Ophthalmological surveillance is recommended for high-risk patients less than 2 years of age, who should undergo screening at diagnosis and every 3–6 months until aged 2 [20]. For systemic JXG, there is currently no established standard chemotherapeutic regimen. A variety of regimens have been tried with variable results; most of these regimens incorporate LCH-based agents such as vinblastine, prednisolone and/or methotrexate [21], but response to chemotherapy is unpredictable. Supportive care should be strongly emphasized due to the potential toxicity in these young infants. Multiagent chemotherapy, including cytarabine, methotrexate, vincristine and prednisolone, is reserved for life-threatening or progressive disease [22]. CNS involvement has been successfully treated with cladribine [23], but the disease does not always respond. Thalidomide and clofarabine have been reported to have activity in heavily pretreated, refractory patients [24, 25]. In the absence of BRAF mutations in JXG, BRAF inhibitors would likely not be relevant but other targets need to be sought.
Benign cephalic histiocytosis
Definition and nomenclature
Benign cephalic histiocytosis (BCH) is a rare, self-limiting histiocytic disorder. Many consider BCH as a clinical variant or a milder form of juvenile xanthogranuloma without systemic involvement [1, 2].
Pathophysiology
Pathology
Immunohistochemically, BCH cells are identical to JXG. On electron microscopy, they may show ‘worm-like’ cytoplasmic inclusions.
Clinical features
Patients are typically young children with a mean age of onset of 15 months (range 2–66 months) [3]. Asymptomatic, erythematous, brown macules/papules/nodules develop on the cheeks and spread to the forehead, earlobes and neck (Figure 136.11). Extension onto the trunk, upper limbs and rarely buttocks may be seen. There is no mucosal involvement.

Figure 136.11 Benign cephalic histiocytosis showing multiple, asymptomatic, reddish brown papules on the face of a toddler.
Management
Given the self-limiting course of the disease, no therapy is required.
Generalized eruptive histiocytosis
Introduction and general description
Generalized eruptive histiocytosis (GEH) is a rare cutaneous histiocytosis that mainly affects adults, although paediatric cases have been reported [1, 2]. It is characterized by asymptomatic, symmetrical papules on the face, trunk and arms, usually sparing the flexures. GEH must be distinguished from the eruptive histiocytomas associated with hyperlipidaemia.
Pathophysiology
Pathology
Histology shows a proliferation of monomorphic histiocytic cells in the upper and mid dermis. No giant cells or foam cells are present. Scattered lymphocytes may be present. Immunohistochemically, the cells are identical to JXG with positivity for CD68, Mac-387, lysozyme and factor XIIIa and negative for S100 and CD1a.
Clinical features
Presentation
The disease presents as multiple, symmetrical, small, red-brown papules on the face, trunk and arms, usually sparing the flexures (Figure 136.12). The lesions are asymptomatic and variable in numbers. The characteristic feature of GEH is the rapid appearance of a crop of lesions, which resolve spontaneously or leave a macular area of hyperpigmentation. Mucosal involvement is rare.

Figure 136.12 Generalized eruptive histiocytosis showing multiple, small, symmetrical, red-brown papules in an adult.
(Courtesy of Dr S. Walsh, Sunnybrook Hospital, Toronto, Canada.)
Management
The evolution of GEH to other non-LCHs has been reported. Patients need to be rebiopsied if lesions become xanthomatoid or flexural or if systemic symptoms develop [2]. The disease is generally self-limiting and often does not require treatment. One paediatric case demonstrated healing in sun-exposed areas, suggesting the value of ultraviolet light as a therapeutic option [3]. Twenty treatments with systemic PUVA were subsequently shown to be effective in a 32-year-old woman with widespread disease with no evidence of recurrence [4]. One patient treated for cosmetic and psychological reasons responded well to chloroquine, thalidomide and glucocorticoid therapy [5].
Papular xanthoma
Introduction and general description
This is a rare histiocytic disorder that was first described in adults and subsequently reported in children [1]. Whether it represents a separate clinicopathological entity or a variant of other xanthogranulomatous conditions is debatable.
Pathophysiology
Pathology
Histologically, there is an upper and mid dermal infiltrate of foamy histiocytes and giant cells. Few inflammatory cells are present. Histiocytic cells are positive for CD68 and factor XIIIa and negative for S100 and CD1a [1, 2]. More recent studies, however, have shown that the foamy cells in this disease are factor XIIIa negative [3, 4]. In such a rare condition, further studies are needed to confirm the dermal dendrocyte origin of the lesional cells. Electron microscopy shows similar changes to those seen in mature juvenile xanthogranuloma, with myeloid bodies filling the cytoplasm of the histiocytes with associated lysosomal inclusions, laminate bodies and lipid droplets.
Clinical features
Presentation
Clinically, it can resemble JXG, but has not been associated with systemic involvement or café-au-lait spots, and may resemble xanthoma disseminatum, but papules do not coalesce and there is no predilection for flexures. It is characterized by 2–15 mm yellow or reddish yellow papules/plaques affecting both the skin and mucous membranes. The back and head are the most common locations. Clinical presentation in adults is different from children. Mucosal involvement and risk for disease progression are features of adult presentation. In contrast, spontaneous resolution is the norm in children, with involution starting after weeks or months and being complete in 1–5 years, often leaving anetoderma-like scarring [2].
Management
No treatment is needed in children, while none has been shown to be effective in adults.
Progressive nodular histiocytosis
Definition and nomenclature
Progressive nodular histiocytosis (PNH) is a rare benign histiocytic disorder of unknown aetiology. It is characterized clinically by the development of multiple superficial skin-coloured or reddish orange papules and deep nodules distributed at random over the body.
Introduction and general description
Progressive nodular histiocytosis is a rare variant of non-LCH disorders that affects skin and mucosa. The skin manifestations consist of two types of lesions: superficial papules and deeper subcutaneous nodules, mainly consisting of spindle-shaped histiocytes [1]. The disease occurs most commonly in adults, although PNH in childhood has been occasionally reported [2, 3]. The major differential diagnoses are the other factor XIIIa-positive non-LCHs, which are distinguished on clinical grounds.
Pathophysiology
Pathology
Histologically, this is a dermal disease with neither epidermal involvement nor epidermotropism. Early lesions show an accumulation of xanthomatized and scalloped histiocytes with some infiltrating lymphocytes. In older lesions, the histiocytes are spindle shaped and arranged in a storiform pattern. Occasional giant cells may be present. Mitotic figures are absent. Cells are positive for CD68 and factor XIIIa. Stains for S100 and CD1a are negative [4].
Clinical features
Presentation
The disease is characterized by the progressive appearance of asymptomatic cutaneous lesions with no tendency to spontaneous involution and can result in severe disfigurement with time (Figure 136.13). Superficial papules are 2–10 mm in diameter and yellow-orange, while deep subcutaneous nodules are 1–5 cm in diameter and may be skin coloured or reddish orange due to overlying telangiectasia [5]. Both types of lesions can reach hundreds in numbers. Distribution is random with no predilection for the flexures. Lesions may occur in the oral cavity, larynx and conjunctival mucosa.

Figure 136.13 Progressive nodular histiocytosis in a 48-year-old man with nodular lesions in the posterior axillary fold.
(Courtesy of Professor J. M. Naeyaert, University Hospital, Gent, Belgium.)
Management
PNH is not generally associated with systemic involvement or other disorders. No treatment has yet been demonstrated to be effective in reducing the size of skin lesions or in inducing remission [6]. However, Chu suggests that the early stages of PNH may be more sensitive to chemotherapy and radiation therapy [7], but these modalities have generally not been shown to be useful. Large or painful lesions are usually excised.
Xanthoma disseminatum
Definition and nomenclature
Xanthoma disseminatum (XD) is a rare non-familial disease, characterized by proliferation of histiocytic cells in which lipid deposition is a secondary event.
Introduction and general description
Xanthoma disseminatum is characterized by proliferation of histiocytic cells in which lipid deposition is a secondary event, with involvement of the skin, mucous membranes of eyes, upper respiratory tract and meninges. Rarely, other organs may be affected, including the liver, spleen and bone marrow [1].
Epidemiology
The disease predominantly affects male children and young adults, but can occur at any age and in either sex. Fifty per cent of lesions appear before the age of 25 and 36% of patients are children [2].
Pathophysiology
XD is thought to be a reactive rather than a neoplastic process with a pathological immune response triggered by unknown inflammatory triggers. The lesional cell appears to be an inflammatory lipid-laden macrophage with a characteristic foamy appearance which could represent increased uptake, synthesis or decreased efflux of lipids [3].
Pathology
Histologically, XD is a dermal disease, characterized by early infiltration of the dermis with spindle-shaped mononuclear cells, foamy histiocytes, giant cells, lymphocytes, polymorphs and eosinophils (Figure 136.14). Lesional cells in XD have irregular scalloped borders with extensive cytoplasm and ovoid vesicular nuclei. Cells label strongly with factor XIIIa, CD68 and Ki-M1p and are negative for S100 and CD1a. Iron and lipid can be detected in the histiocytes. In older lesions, more foamy histiocytes are evident and Touton giant cells may be observed. At the ultrastructural level, histiocytic cells contain myeloid bodies and membrane-bound fat droplets.

Figure 136.14 Xanthoma disseminatum: similar histological picture to juvenile xanthogranuloma consisting of dermal infiltrate of small spindled histiocytes (H&E).
(Courtesy of Dr R. Jaffe, Children's Hospital of Pittsburgh, Pittsburgh, USA.)
The various members of the factor XIIIa-positive xanthogranuloma family, XD, PNH and GEH, may be indistinguishable from one another on histopathology and the diagnosis may depend on the clinical presentation. Some described differences in morphology of the cells, such as foamy xanthoma cells versus oncolytic or epitheliod cells, may be a function of when the biopsy was done in the course of evolution of the disease rather than a difference in diagnosis [4].
Clinical features
Presentation
The clinical lesions of XD are erythematous, yellow brown papules and nodules, which are symmetrically distributed on the trunk, scalp, face and proximal extremities. The lesions become confluent, especially in flexures, to form xanthomatous plaques, which may become verrucous (Figure 136.15). In 39–60% of patients, the mucous membranes are affected, with particular involvement of the lips, pharynx, larynx, conjunctivae and bronchus. XD also has the tendency to involve the upper respiratory tract (trachea, larynx) rather than lower respiratory tract, leading to stridor or respiratory compromise [5, 6]. Meningeal involvement is common, with infiltration at the base of the brain leading to diabetes insipidus in up to 40% of cases, which may, however, be transient. Other manifestations of meningeal involvement are seizures and growth retardation. Intracranial involvement presenting as a discrete mass simulating glioma has been reported [7] and progressive intracranial disease may be fatal in as many as 63% of patients [8]. Hepatic and bone involvement have been reported but represent rare complications of the disease.

Figure 136.15 (a, b) Xanthoma disseminatum showing large, yellow-brown plaques affecting the skin folds.
(Courtesy of Dr S. Walsh, Sunnybrook Hospital, Toronto, Canada.)
Disease course and prognosis
XD is a self-limiting disease but may be locally destructive and persist for years. Three clinical patterns have been identified: a rare self-healing form, a chronic, often progressive form and a progressive multiorgan form, which may be fatal.
Management
Skin lesions of XD are disfiguring and patients often request treatment. A carbon dioxide laser has been used with good results [9]. Other forms of surgical removal, excision, dermabrasion and electrocoagulation have been used with moderate effect [2, 9]. Localized conjunctival involvement can be treated with surgery. Surgery has also been used in CNS disease but recurrences may occur and not all CNS lesions are amenable to resection. Systemic involvement with lung, liver or CNS involvement requires active treatment but the response to therapy is not predictable and may not be long lasting. Glucocorticoids, chlorambucil, azathioprine and cyclophosphamide have been effective in some patients with cutaneous disease [3, 10]. Recently, cladribine was found to be useful in a small case series [11]. Interestingly, one patient had a partial response to a combination of three lipid-lowering agents, rosiglitazone, simvastin and the niacin analogue acipimox [3], and a second patient had a dramatic response after 5 months of simvastatin alone [12]. Anti-inflammatory and lipid-lowering drugs such as the statins need further evaluation in refractory XD patients, keeping in mind the possibility of spontaneous resolution, which may make assessment of response to therapy more difficult.
Diffuse plane xanthomatosis
Definition and nomenclature
In this disease, multiple flat xanthomatous macules and plaques develop in the skin in association with a monoclonal paraproteinaemia.
Introduction and general description
Diffuse plane xanthomatosis (DPX) is a rare, non-lipaemic disease in which xanthomatous lesions develop in the skin in association with paraproteinaemia or an underlying systemic disorder, usually of the haematological or lymphoproliferative type.
Epidemiology
The disease generally occurs in adults but rare paediatric cases have been reported [1].
Pathophysiology
The condition arises as a result of perivascular deposition of lipoprotein–immunoglobulin complexes. Antilipoprotein antibodies are formed in association with paraproteinaemia. Although serum lipid levels are usually normal, they may be raised, possibly due to reduced clearance of the lipoprotein–antibody complexes.
Pathology
The histological features include both xanthomatous and inflammatory elements. Accumulations of foamy macrophages infiltrate the dermis, with a distinct perivascular accentuation, and are associated with a variable degree with a mixed inflammatory cell reaction [2]. There are no reported immunohistochemical studies in literature.
Clinical features
Patients present with large, flat, plaque-like xanthomatous skin lesions involving the eyelids, neck, upper trunk, buttocks and flexures (Figure 136.16) [3]. Serum lipids are usually normal. Multiple myeloma and monoclonal gammopathy are the two most frequently associated diseases with DPX [4]. However, association with many other lymphoproliferative disorders has been reported, including chronic myeloid leukaemia, acute monoblastic leukaemia, chronic lymphatic leukaemia, chronic myelomonocytic leukaemia, lymphoma, Sezary syndrome and Castleman disease [3].

Figure 136.16 Diffuse plane xanthomatosis in a 73-year-old man of 7 years’ duration with associated IgG-κ paraprotein.
Management
Treatment of this condition is that of the underlying myeloproliferative disease or paraprotein. In patients with limited involvement, the individual lesions can be excised. Other options include chemoabrasion, dermabrasion and ablative laser therapy [5]. The erbium:YAG (yttrium aluminium garnet) laser has been used successfully to treat facial DPX in one patient [6].
Disorders in which skin may be involved but systemic component predominates
Erdheim-Chester disease
Definition and nomenclature
This disease is characterized by infiltration of viscera, bones, retroperitoneum and skin.
Introduction and general description
Erdheim–Chester disease (ECD) is a rare lipoid granulomatosis characterized by infiltration of the viscera, bones, retroperitoneum and skin. ECD may represent a variant of xanthogranuloma (XG) with mostly osseous and visceral involvement, which can be distinguished from XG clinically and radiographically [1].
Epidemiology
This is predominantly a disease of adults, with a mean age of 53 years (range 7–84 years) [2]. The male to female ratio was reported as 33 : 26 [2].
Pathophysiology
Pathology
Morphologically and immunohistochemically, ECD histiocytes are identical to those of JXG. They are positive for CD68 and factor XIIIa, but negative for CD1a and S100. Histological examination shows a xanthogranulomatous infiltration by lipid-laden histiocytes within a mesh and surrounded by fibrosis. Factor XIIIa-positive histiocytes are known to stimulate fibroblast proliferation, resulting in fibrosis, which is common in this disorder [3]. Touton giant cells and eosinophils may be prominent.
Genetics
The pathogenesis of ECD is poorly understood. Recent identification of a BRAF mutation in more than half of ECD patients suggests involvement of the RAS signalling pathway and has important therapeutic implications [4]. However, further studies are required to investigate this potential role of BRAF in the pathogenesis of ECD.
Clinical features
Presentation
The clinical presentation can range from asymptomatic to fulminant organ failure. Bone pain in the knees and ankles is the most common presenting symptom. Constitutional symptoms such as weakness, weight loss and fever are frequent. Bilateral symmetrical long bone involvement is nearly always present [2]. Diagnosis is made on the basis of radiological features of osteosclerosis in addition to the classic histology.
Complications and co-morbidities
In Veyssier-Belot's series of 59 patients, 86% of reported cases have involvement of the long bones, with the distal femur, proximal fibula and tibia being most commonly affected [2]. Up to 30% of patients show lytic lesions in the flat bones, which can be difficult to distinguish from LCH. Unlike JXG, skin involvement is less common in ECD, affecting approximately 20% of patients and presenting as xanthoma-like lesions despite normal plasma lipid levels, usually on the eyelids but occasionally on the trunk and submammary area [5]. Pulmonary involvement is seen in 25–50% of patients [6], and although mostly asymptomatic, the presence of cough and progressive dyspnoea carries a poor prognosis. However, pulmonary involvement was not found to be an independent predictor of decreased survival in one large series [7]. Cardiac involvement is relatively common in ECD, with periaortic fibrosis in 55.6%, pericardial involvement in 44.4% and myocardial involvement in 30.6% among 72 patients with ECD and cardiovascular involvement [8]. CNS involvement is seen in 15% of patients, presenting with ataxia, paraparesis, hemiparesis or change in mental state [9]. Neurological imaging can reveal thickening of the dura or more rarely intracerebral masses that can resemble meningioma-like tumours [9].
Disease course and prognosis
Erdheim–Chester disease appears to have a significantly worse prognosis compared with other histiocytoses, with an overall mortality of around 60% in one series [2], mainly from cardiorespiratory failure from lung fibrosis or renal failure from retroperitoneal fibrosis.
Management
Not all patients with ECD require treatment at the time of diagnosis. Active treatment is typically reserved for symptomatic patients. However, treatment for patients with asymptomatic CNS involvement is recommended since it represents an independent predictor of a worse outcome [10]. A variety of therapeutic options are available with varying degrees of success. These include surgical debulking, high-dose corticosteroids, ciclosporin, interferon α, systemic chemotherapy and radiation therapy [2, 11]. The commonest chemotherapy drugs utilized have been vinca alkaloids, anthracyclines and cyclophosphamide [12]. However, there is no clear consensus on the best therapeutic regimen given the small number of patients. Interferon α has been recently recommended as first line therapy because of the durable response seen in three patients with advanced disease [13, 14]. Several case reports have shown activity of imatinib in ECD [15, 16]. Although the exact role of imatinib in the treatment paradigm remains undefined, it offers an alternative therapeutic option for patients who fail to respond or are intolerant to interferon α. Finally, a recent report of the successful use of vemurafenib, a BRAF inhibitor, in refractory ECD patients harboring the BRAF V600E mutation is encouraging [17].
Non-dendritic cell origin
Disorders with mainly skin involvement with/without a systemic component
Reticulohistiocytoma
Definition and nomenclature
Reticulohistiocytoma is an uncommon, incompletely characterized histiocytic tumour of the skin and soft tissues.
Introduction and general description
Reticulohistiocytoma is the localized variant of multicentric reticulohistiocytosis. This uncommon tumour is generally solitary and asymptomatic. Lesions are less than 1 cm in diameter and present as papules or dome-shaped nodules. They may occur anywhere on the body including the genitalia. Oral mucosal lesions have been reported [1]. They are thought to represent a non-neoplastic reactive process.
Epidemiology
Reticulohistiocytoma tends to occur in young adult males. Rarely, it may appear in the newborn period.
Pathophysiology
Pathology
Histology shows nodules of large epithelioid histiocytes with abundant, glassy, eosinophilic cytoplasm extending from the papillary dermis to the mid-dermis associated with lymphoid cells and occasionally neutrophils. Cells may have a lacuna space-like clearing at the periphery and scalloped cytoplasm. Cells are CD68 and CD163 positive and generally CD1a and S100 negative. The presence of factor XIIIa is variable. In a study of five cases of solitary reticulohistiocytoma, factor XIIIa was found to be positive [1, 2].
Management
Treatment is surgical excision. Follow-up in 12 patients, with a median follow-up of 13 years, showed no recurrence following primary excision.
Familial sea–blue histiocytosis
Introduction and general description
This is a rare inherited abnormality of lipid metabolism in which characteristic histiocytic cells are found in the bone marrow and other tissues. The histiocytes are identified by the May–Gruenwald stain, which colours the cytoplasmic granules a deep azure blue, hence the name ‘sea-blue histiocytosis’. Sea-blue histiocytes have been described in a variety of disorders of lipid metabolism such as Niemann–Pick disease, in patients with prolonged use of intravenous fat emulsions, and in cases of partial sphingomyelinase deficiency and chronic myelogenous leukaemia [1].
Epidemiology
It usually presents in young adulthood with hepatosplenomegaly and thrombocytopenia, although the age at presentation ranges from 1 to 83 years [1].
Pathophysiology
The biological abnormality is poorly understood, but the condition probably represents a storage disease in which glycolipid, phospholipid or both accumulate in histiocytic cells in various organs including the bone marrow, liver and spleen.
Pathology
Histologically, it is characterized by micronodular infiltrates of large monomorphous histiocytes with cytoplasmic vacuoles and granules. The granules appear yellow-brown with H&E, dark blue with Giemsa and ‘sea-blue’ with May–Gruenwald staining [2].
Genetics
Familial sea-blue histiocytosis is an autosomal recessive disorder.
Clinical features
The skin, lungs, gastrointestinal tract, eye and nervous system may be involved. In the skin, patchy and irregular brownish grey pigmentation and/or nodules of the face, upper chest and shoulders have been reported and may cause disfigurement. In the eye, white stippled deposits may be observed at the margins of the fovea or macula, with discoloration of the macular region. Neurological symptoms occur early, with ataxia, epilepsy and dementia. Sea-blue histiocytosis is usually benign, but it may disseminate and lead to death from heart, liver or lung involvement.
Management
There is no specific treatment for sea-blue histiocytosis. Specific treatment of the associated disorder of lipid metabolism may induce resolution of the disease.
Hereditary progressive mucinous histiocytosis
Definition
Hereditary progressive mucinous histiocytosis is a rare autosomal dominant genodermatosis consisting of lesions affecting the nose, hands, forearms and thighs.
Epidemiology
This is a rare autosomal dominant genodermatosis, which was first described in 1988. All case reports to date have been in women, thus suggesting a link to hormones [1].
Pathophysiology
Histologically, the epidermis is normal but within the dermis there are small collections of epithelioid histiocytes with telangiectatic vessels in the upper dermis in early lesions. As tumours develop, the infiltrate changes to nodular mid-dermal aggregates of tightly packed, spindle-shaped cells. In both early and established lesions, there is moderate to extensive mucin production by the epithelioid histiocytes and spindle-shaped cells. On electron microscopy, the spindle-shaped cells are shown to be dendritic histiocytes with abundant lysosomal storage organelles, myelin bodies and zebra bodies. Immunohistochemically, these cells stain with CD68 and MS1 [2].
Clinical features
Skin lesions appear in the first decade of life and gradually increase throughout life. Lesions consist of skin-coloured to red-brown papules that characteristically affect the nose, hands, forearms and thighs.
Management
The condition is progressive, with a gradual increase in the numbers of tumours throughout life. These patients show no evidence of spontaneous resolution. No systemic involvement has been described and no treatment seems to have any impact on the disease [3, 4].
Malakoplakia
Definition
Malakoplakia is an immunodeficiency disease in which macrophages fail to phagocytose and digest bacteria adequately. The term ‘malakoplakia’, which means soft plaque, was adopted as a descriptive term.
Pathophysiology
Pathology
Histologically, sheets of large histiocytic cells with abundant cytoplasm are present in the skin, affecting any level from the epidermis to the subcutaneous fat. The cells have fine eosinophilic granules in their cytoplasm and are referred to as Hansemann cells. They also contain one or more round basophilic inclusion bodies (Michaelis–Gutmann bodies). Michaelis–Gutmann bodies are 5–15 μm in diameter and stain positively with periodic acid–Schiff stain (PAS), von Kossa stain (for calcium) and Perls ferrocyanide reaction (for ferric iron). They are considered pathognomonic for this disease and are thought to represent abnormal degradation of bacteria, with calcium and iron deposited on the remaining glycolipid [1].
Causative organisms
The aetiology of malakoplakia is still unclear. However, chronic infection has been suggested to play a role in the pathogenesis of the disease [2]. Common associated organisms include Escherichia coli, Proteus species, Mycobacterium tuberculosis and Staphylococcus aureus [2].
Clinical features
Presentation
Malakoplakia can affect many organs but most commonly affects the urinary and gastrointestinal tracts [3, 4]. Cutaneous lesions are rare, non-specific and variable. Draining abscesses, sinuses, ulcers, fluctuant masses, isolated tender nodules and grouped papules have been reported [5, 6]. Mucous membranes may be affected, including the tongue and cervix.
Disease course and prognosis
The disease generally runs a benign self-limiting course, but fatal cases have been reported [6].
Management
Management includes different combinations between surgical excision and/or use of antibiotics for the associated infections. Some case reports suggest higher cure rates with surgical excision when compared with antibiotic therapy [7]. In addition, when comparing antibiotics, quinolones seem to be superior [7].
Necrobiotic xanthogranuloma
Definition
Necrobiotic xanthogranuloma (NXG) is a rare, multisystem histiocytic disease in which widespread infiltrated xanthomatous nodules and plaques are strongly associated with haematological malignant conditions.
Epidemiology
Only about 100 cases have been reported in the literature since it was first recognized as a distinct dermatosis in 1980 [1].
Pathophysiology
The pathogenesis of NXG is unknown. Some believe that paraproteins function as autoantibodies leading to fibroblast proliferation and the deposition of dermal macrophages [2], while others suggest that the abnormal paraprotein becomes complexed with lipid and deposits in the skin, where it produces a foreign body giant cell granulomatous reaction [3]. However, this does not explain NXG cases without paraproteinaemia. The possibility of an infectious aetiology was raised by the finding of Borrelia species in some skin biopsy specimens [4]. Approximately 80–90% of patients have an underlying monoclonal gammopathy [5], among whom IgG-κ is the most frequent monoclonal gammopathy of unknown significance (MGUS) (65%), followed by IgG-λ (35%), and, much less commonly, IgA [1]. When a gammopathy is present, the underlying haematological condition is MGUS in half of the cases and myeloma in the other half [6], which can manifest years after the development of skin lesions. Because of the prolonged gap between the onset of the skin disease and these malignancies, their role in pathogenesis remains uncertain.
Pathology
Histologically, confluent granulomatous masses are present as either sheets or nodules, replacing much of the dermis and extending into the subcutaneous tissue. Hyaline areas of necrobiosis separate individual nodules. Numerous giant cells are present, with Touton cells and bizarre, angulated giant cells [7]. Cholesterol clefts, lymphoid nodules (some of which develop germinal centres) and perivascular aggregates of plasma cells are frequent features. Less common, but characteristic when present, are palisading cholesterol cleft granulomas and xanthogranulomatous panniculitis [8]. Granulomatous invasion of blood vessels with thrombosis has been described.
Clinical features
Presentation
The clinical picture of NXG consists of slowly progressive, reddish yellow, xanthomatous plaques/nodules that are infiltrative and destructive (Figure 136.17) [9]. More than 80% of the lesions are periorbital, but may occur on the trunk and limbs where subcutaneous nodules and xanthomatous plaques are present with atrophy and ulceration. The eyes are often affected with conjunctivitis, keratitis, uveitis, iritis and proptosis. Blindness has been reported in two affected patients [10].

Figure 136.17 Necrobiotic xanthogranuloma showing a reddish yellow, infiltrative plaque with evidence of necrosis.
(Courtesy of Dr S. Walsh, Sunnybrook Hospital, Toronto, Canada.)
Complications and co-morbidities
Association with haematological and lymphoproliferative malignant disorders such as myeloma, non-Hodgkin lymphoma and chronic lymphocytic leukaemia has been well described and typically occurs approximately 2 years from the onset of skin manifestations [1, 5]. The diagnosis of NXG should prompt a thorough evaluation to rule out the above conditions. Only one case report of a patient with typical cutaneous lesions and cerebral involvement presenting as tonic–clonic seizures has been published [11]. Systemic symptoms have been reported, including nausea, vomiting, fatigue, epistaxis, back pain and the Raynaud phenomenon. Atypical forms of NXG have been reported, including solitary tumours of the skin [12].
Management
Due to the rarity of the condition, there is no consensus regarding the optimal therapy. Treatment is generally directed to the associated paraproteinaemia. Alkylating agents such as melphalan, with or without prednisolone, have resulted in temporary clearing of the skin [9]. Other therapies included intralesional corticosteroids, high-dose systemic steroids, chlorambucil, interferon α, cyclophosphamide, methotrexate, hydroxychloroquine and azathioprine [9]. A recent review of reported cases treated with chlorambucil supported its use as a front line agent in selected cases [13]. In one patient where cytotoxic drugs had failed, plasmapheresis reduced the level of the circulating monoclonal IgG and resulted in clearing of the skin [10]. Radiotherapy was successful in one case involving the eye [12]. Cutaneous disease has also been successfully treated with carbon dioxide laser with no evidence of relapse after 12 months [14]. Two patients were successfully treated with IV immunoglobulin [2]. A single case report described one patient achieving long-term remission following the combination of thalidomide for 2 years and pulse dexamethasone for 9 months [9]. These authors felt that this combination should be tried as first line in patients who need systemic therapy but that high-dose and prolonged treatment is necessary.
Disorders in which skin may be involved but the systemic component predominates
Multicentric reticulohistiocytosis
Definition and nomenclature
Multicentric reticulohistiocytosis (MRH) is a rare non-LCH disorder characterized by the association of specific nodular skin lesions and destructive arthritis.
Introduction and general description
This is a rare multisystem disorder characterized by cutaneous and mucosal involvement in addition to a destructive arthropathy. The arthropathy usually precedes nodular skin involvement and mucosal infiltration. Other organs may be involved and about 28% of patients have an associated internal malignancy [1]. This must be differentiated from solitary or multiple reticulohistiocytomas that are restricted to skin with neither associated arthropathy nor internal malignancy. Moreover, differentiation from fibroblastic rheumatism (FR), which can also present with a destructive polyarthritis and skin lesions, is important as it differs both in prognosis and therapy. FR was originally classified as a class IIB histiocytic disorder but recent studies have suggested that FR more properly belongs to the fibroblastic disorders [2]. FR will not be discussed further in this chapter.
Epidemiology
Multicentric reticulohistiocytois is a disease of middle-aged adults, predominantly female [3], although rare paediatric cases have been reported [3, 4, 5]. The female to male ratio is 3 : 1, and 85% of reported adults were white [3].
Pathophysiology
Pathogenesis is unknown. The disease is generally considered to be a reactive histiocytosis. Inflammatory cytokines such as tumour necrosis factor α (TNF-α), interleukin 1 (IL-1) and IL-6 have been found to be highly expressed in MRH nodules [6]. IL-6 may also result in the osteoclastogenesis accounting for the multinucleated giant cells seen in the lesions.
Pathology
The characteristic pathological picture in the skin and mucous membranes is infiltration by mono- and multinucleated giant cells with voluminous ground-glass cytoplasm (Figure 136.18). In early lesions, the predominant infiltrating cells are histiocytes, lymphocytes and eosinophils, with few giant cells, but the giant cell infiltrate quickly follows. The giant cells are large (100 μm diameter) with 1–20 nuclei, which may be distributed randomly at the periphery or in the centre. The cells are PAS positive, contain diastase-resistant material and variable amounts of lipid and free or esterified cholesterol. In older lesions, fibrosis usually signals regression of the lesions, with a reduction in the inflammatory cell infiltrate. Ultrastructural studies have shown type IV collagen inclusions in MRH. These inclusions were both intracytoplasmic and extracytoplasmic. Such inclusions are usually found in lymphohistiocytic neoplasms, suggesting that MRH is a proliferative rather than an inflammatory disorder [7]. Immunocytochemical studies show a histiocytic phenotype of the cells, which are positive for acid phosphatase, adenosine triphosphatase, lysozyme and α1-antitrypsin. The cells are also positive for vimentin, CD45, CD68, CD11b and HAM56, but negative for CD1, S100, CD34 and factor XIIIa [8]. CD14 is usually positive in osteoclast-like macrophages and negative in true osteoclasts [9]

Figure 136.18 Reticulohistiocytoma/multicentric reticulohistiocytosis. (a) The dermis contains an infiltrate of large, eosinophilic, cytoplasm-rich histiocytes with an interspersed inflammatory cell component. Magnification 100× (H&E). (b) The large eosinophilic histiocytes generally have a single nucleus or two nuclei. Interspersed lymphocytes and eosinophils are prominent in this example. Some emperipolesis is noted. Magnification 400× (H&E).
(Courtesy of Dr R. Jaffe, Children's Hospital of Pittsburgh, Pittsburgh, USA.)
Causative organisms
No infective agent has been implicated, but in one study 33% of patients had evidence of tuberculosis exposure and 5% had active tuberculosis on examination [10].
Clinical features
Presentation
The disease is often limited to the skin, mucosa and joints but systemic involvement of the heart and other organs has been described. Two-thirds of patients present with symmetrical polyarthritis, followed by the appearance of skin lesions. The arthropathy typically affects the hands, but other joints may be involved as well, including the knees, shoulders, wrists, hips, ankles, feet, elbows, spine and temporo-mandibular joints. Although it often remits spontaneously, 15–50% of cases progress to mutilating osteoarthropathy with disabling deformities [1, 4]. In 20% of cases, skin nodules appear first while simultaneous skin and joint lesions emerge in the remainder [3]. The classic skin lesions are firm brown or yellow papules and plaques, which predominantly affect extensor surfaces, particularly on the hands and forearms (Figure 136.19). The face, scalp, hands and ears are often affected but involvement of the lower trunk and legs is rare. Coral bead-like lesions may occur around the nail folds, which may result in nail dystrophy. Skin lesions are of variable size and rarely ulcerate. Large nodular lesions in proximity to the affected joints and cystic swellings of the tendon sheaths may occur. About 25% of patients complain of pruritus associated with skin lesions. One-third of patients have symptoms of fever, weight loss and malaise. More than 50% of patients have mucosal involvement, characteristically of the lips and tongue, but it can also affect the mouth, gingiva, pharynx, larynx and sclera.

Figure 136.19 Multicentric reticulohistiocytosis nodules over the elbows (a) that shrank after an infusion of zoledronic acid (photograph taken 43 days after infusion) (b). (From Codriansky K et al. 2008 [22].
Reproduced with permission of John Wiley & Sons.)
Complications and co-morbidities
Around 15% of patients have an associated autoimmune disease such as systemic lupus erythematosus, diabetes or Sjogren syndrome, while 20% of patients have been found to have an associated internal malignancy. The commonest tumours are gastric, ovarian [11], breast and uterine carcinomas [12], myeloma, melanoma [13] and lymphomas. The diagnosis of MRH precedes that of the neoplasm in most cases, and the disease may relapse with recurrence of the neoplasm.
Disease course and prognosis
The prognosis is good, with the disease becoming quiescent in 7–8 years if there is no systemic malignancy. Fatal cardiac involvement may occur with widespread systemic involvement [14]. MRH may, however, leave considerable morbidity, with a crippling arthropathy and scarred skin [15].
Management
Treatment of the underlying disease is important, although it does not usually influence the disease course. Resolution of MRH was seen in a patient with renal cell carcinoma following nephrectomy. Most children have a self-limiting disease with non-deforming arthritis. However, this is not true for adults, in whom therapy is usually indicated. Gold, tamoxifen and d-penicillamine failed in all cases [16]. Several reports suggest that methotrexate alone or in combination may be effective [3, 4, 5, 17]. Cyclophosphamide alone induced complete remission in three cases [17]. Other immunosuppressive agents such as ciclosporin, leflunomide [18] and prednisolone and azathioprine [19] have resulted in complete remission in single case reports. Successful therapy with bisphosphonates alone or in combination with prednisolone and methotrexate have also been reported [20, 21]. More recently, anti-TNF agents such as etanercept, infliximab and adalimumab have been reported to be beneficial [15, 19] as has the anti-IL-6 antibody tocilizumab in patients resistant to methotrexate and prednisolone [6]. A reasonable approach in the past was to begin therapy with a combination of corticosteroid and low-dose methotrexate, and to add cyclophosphamide for poor responders. Consideration could be given to combining methotrexate with or without corticosteroid with one of these newer targeted agents as front line therapy in severely affected or resistant patients. As with all of the non-LCH disorders, different patients may respond differently and a switch from one agent to another may be needed in individual patients.
Sinus histiocytosis with massive lymphadenopathy
Definition and nomenclature
Sinus histiocytosis with massive lymphadenopathy (SHML) is a rare, non-neoplastic disorder of unknown aetiology that is usually self-limiting. It is characterized by abundant histiocytes in the lymph nodes throughout the body.
Epidemiology
Most patients are young adults (mean age 20.6 years), but with a wide age distribution from birth to 74 years [1]. Patients presenting with isolated intracranial disease appear to be older (mean age 37.5 years) [2]. The disease is slightly more common in males (58%) and in black people [3].
Pathophysiology
Predisposing factors
The aetiology of SHML is unknown. It is postulated that an infectious or malignant process may result in aberrant activation of the macrophage system with excessive cytokine release by the macrophages and T cells. An increased incidence of autoimmune disorders such as autoimmune haemolytic anaemia and various rheumatological disorders have been reported [4].
Pathology
Histologically, involved lymph nodes show massive sinus infiltration of large histiocytes admixed with lymphocytes and plasma cells (Figure 136.20). Immunohistochemicallly, SHML cells are positive for S100, CD68, CD163, HAM-56, α1-antichymotrypsin and α1-antitrypsin, but negative for CD1a [1]. SHML lesions show strong expression of IL-1 and TNF-α and moderate expression of IL-6 [5]. The latter may be related to the observed polyclonal plasmacytosis and hypergammaglobulinaemia. A hallmark of SHML is the presence of emperipolesis (phagocytosis of intact leukocytes, particularly lymphocytes) in histiocytes that express S100. In the skin, a dense, upper dermal, histiocytic infiltration with scattered multinucleate giant cells and plasma cells is seen [6]. Emperipolesis is less prominent in the skin than in the nodes.

Figure 136.20 Rosai–Dorfman disease. (a) The ‘lymph node-in-skin’ appearance is highlighted at a low-power magnification of 20× (H&E). (b) Rosai–Dorfman histiocytes have very abundant water-clear cytoplasm and one or two large hypochromatic nuclei. Emperipolesis of intact lymphocytes is notable in the cytoplasm. Magnification 400× (H&E).
(Courtesy of Dr R. Jaffe, Children's Hospital of Pittsburgh, Pittsburgh, USA.)
Clinical features
Presentation
Clinically, about 80% of patients present with bilateral, painless, cervical adenopathy, which may be associated with fever, malaise, night sweats, weight loss, leukocytosis and hypergammaglobulinaemia. Other nodal groups, such as axillary, mediastinal and inguinal, can be also involved. The lymph node enlargement can reach massive proportions. Extranodal involvement is described in 43% of patients, and skin represents the most common extranodal site [7].
Clinical variants
Skin is the most common extranodal site and is found in isolation in 50% of patients (Figure 136.21) [6]. Skin lesions are usually yellow, but may be violaceous or purple. Macular erythema, papules, nodules or infiltrated plaques have been reported. Scaling is often present and telangiectasias may be observed. Skin lesions may occur anywhere and there is no predilection for any site [8].

Figure 136.21 Rosai–Dorfman disease showing multiple nodules/tumours with superficial crusting and old scars on the nose.
Complications and co-morbidities
Other reported extranodal sites include the lung, uro-genital tract, breast, gastrointestinal tract, liver and pancreas. Head and neck involvement occurs in 22% of patients [7], with the nasal cavity and parotid gland as the commonest sites. Isolated intracranial disease can occur in SHML without extracranial lymphadenopathy, which makes the diagnosis challenging. Most intracranial lesions are attached to the dura and patients present with headaches, seizures, numbness or paraplegia [9].
Disease course and prognosis
The clinical course of SHML is unpredictable, with episodes of exacerbation and remission that may extend over many years. The outcome is usually good and the disease is often self-limiting. Nonetheless, about 5–11% of patients die from the disease. Patients with underlying immunological abnormalities at or prior to onset have a worse prognosis with more widespread nodal disease and higher fatality rate [7, 10].
Investigations
Laboratory investigations usually show a mild normochromic normocytic anaemia or hypochromic microcytic anaemia with an elevation of the erythrocyte sedimentation rate. Serum proteins are often abnormal, with a low serum albumin and polyclonal gammopathy. Serum lipids are normal. Chest computed tomography or head imaging should be considered depending on clinical symptomatology.
Management
Treatment is only necessary when a vital organ is being compromised or nodal enlargement leads to significant problems such as airway obstruction [11]. For intracranial dural-based lesions, surgical excision alone is successful in most cases [2]. There is currently no consensus on guidelines for systemic treatment when indicated. Antibiotics are not useful. Surgical debulking of resectable lesions achieved complete remission in eight of nine patients [11]. Systemic corticosteroids are useful in decreasing size and symptoms, but regrowth often occurs within a short period of discontinuation. Chemotherapy is generally unsuccessful. Complete remissions achieved with interferon α plus chemotherapy [12, 13], thalidomide or aciclovir have been reported in case reports [14]. Rituximab, a chimeric anti-CD20 antibody, has been successful in recent anecdotal reports including three patients treated by the author (Sheila Weitzman, personal communication), one of whom had severe liver involvement. Relapse after discontinuation of rituximab was seen in another anecdotal case. Retreatment and prolongation of therapy, similar to cases of follicular lymphoma, may be indicated in these refractory patients, but this approach is obviously experimental.
MALIGNANT HISTIOCYTOSES
Malignant histiocytoses are malignancies of the monocyte/macrophage series of cells. These diseases are separated into monocytic leukaemia, malignant histiocytosis, true histiocytic lymphoma and histiocytic sarcoma based on clinical criteria, but there is an enormous overlap and it may not always be possible to differentiate them.
In monocytic leukaemia, the malignancy primarily affects the bone marrow and blood but extramedullary involvement is common. This is classified as part of the myelomonocytic leukaemias and will be covered Chapter 0 on leukaemia cutis. In malignant histiocytosis, the histiocytes retain their ability to migrate through the body, which results in widespread involvement of the reticuloendothelial system. In true histiocytic lymphoma, the cells are derived from fixed tissue histiocytes and the tumours are localized, although they may disseminate. In histiocytic sarcoma, it involves mature histiocytes and represents a highly aggressive disease.
Malignant histiocytosis
Definition and nomenclature
Malignant histiocytosis (MH) is a widespread neoplastic proliferation of histiocytic cells that typically involves liver, spleen, lymph nodes and bone marrow.
Introduction and general description
The widespread increase in histiocytic cells in MH typically involves the liver, spleen, lymph nodes and bone marrow. The cells usually arise from sinusoidal histiocytes, although very rare cases of malignant histiocytosis of Langerhans cell phenotype have been reported. In addition, many cases previously reported as MH were eventually shown to be T- and B-cell lymphomas including anaplastic large cell lymphoma [1].
Epidemiology
Sex
Malignant histiocytosis is a rare disease with a male to female ratio of 3.5 : 1. It has been reported in all age groups, with a median age of 35 years [2]. Childhood disease is uncommon with few reported series [3]. The disease tends to occur earlier in women (second to third decades) than in men (third to fourth decades) [4].
Ethnicity
Reports have suggested an increased incidence of this disease in parts of tropical Africa, with reports from Malawi and Uganda [5, 6]. A recent review of deaths from ‘histiocytosis’ in the USA between 1979 and 2006 showed that LCH was significantly more common as a cause of death in people younger than 5 years of age irrespective of gender (P value <0.0001), whereas death rates from MH were significantly greater in ages >54 years (P value <0.00001) [7]. There were more MH deaths among males than females.
Pathophysiology
Malignant histiocytosis is a neoplastic proliferation of cells of the mononuclear phagocyte system.
Pathology
The histological picture in the skin and lymph nodes is similar and the diagnosis can be established in either site. Characteristically, there is an infiltrate of histiocytic cells showing varying degrees of atypia that are typically non-cohesive. Cells are large (up to 50 μm in diameter) with abundant cytoplasm and distinct cytoplasmic membranes. The histiocytic cells are heterogeneous. Some show more marked histiocytic differentiation, with pale cytoplasm, prominent vacuolation or even foamy cytoplasm, and exhibit phagocytosis of erythrocytes, leukocytes and cellular debris. Other cells are more ‘primitive’, with deeply eosinophilic or amorphous cytoplasm. Nuclei are usually lobulated, with finely granular or reticulated chromatin and prominent or bizarre nucleoli. Nuclear membranes tend to be thickened. Mitoses are common.
Immunohistochemistry showing a histiocytic origin and negative for myeloid, dendritic or other lymphoid markers is essential for the diagnosis of all the histiocytic malignancies. Cytochemical and immunohistochemical studies have shown that the cells in MH are negative for chloracetate esterase, Sudan black B, alkaline phosphatase and β-glucuronidase [8]. The presence of non-specific esterase, acid phosphatase and lysozyme is variable, with the better differentiated cells showing these enzymes [9]. The more differentiated phagocytosing cells usually stain for factor XIIIa and the antimonocyte monoclonal antibody MOI [10]. In some cases, epithelial membrane antigen (EMA), HLA-DR, CD25, CD30, CD68 and CD71 have been detected. In rare cases, CD1a or CD21/35 may be found. In lymph nodes, the architecture is disarranged but not effaced by the malignant cells.
In the skin, there is extensive perivascular and periappendageal infiltration of the dermis, with extension into the subcutaneous fat. In advanced lesions, fat necrosis may occur. The epidermis and papillary dermis are characteristically spared but in the more tumid lesions epidermal ulceration may be present.
Causative organisms
There is no evidence of a viral aetiology in this disease and no reported familial incidence.
Genetics
There have been reports of a characteristic chromosomal translocation t(5;6)(q35;p21) in MH [11, 12], but it is unclear whether those cases would be called MH by current criteria.
Clinical features
Presentation
Malignant histiocytosis is usually of acute onset, with fever, sweats, wasting, generalized painful lymphadenopathy and hepatosplenomegaly. As the disease progresses, jaundice, purpura, anaemia and leukopenia occur. In 50% of patients, extranodal extension of the disease is seen, most commonly affecting the skin, bone and gastrointestinal tract [13]. Cutaneous involvement occurs in 10–15% of cases, manifesting with single or multiple skin-coloured to violaceous papulonodular lesions [14]. These lesions tend to have a predilection for the lower extremities and buttocks, but may occur anywhere. Large lesions may ulcerate. A widespread, papulonodular eruption similar to that in acute monocytic leukaemia may also be seen. In the bone, the lesions are focal, destructive, lytic and may become widespread with associated hypercalcaemia. Gastrointestinal involvement is usually observed late in the disease. The small and large bowel may be involved, with infiltration of the lamina propria and local intraluminal masses. This presents with obstruction or haemorrhage or both. A rare presentation with multiple lesions is with malabsorption.
This disease was invariably lethal in the past, with death occurring within weeks to months of diagnosis. However, with aggressive management (radiotherapy or radiotherapy and chemotherapy) complete remission has been reported in up to 50% of cases, with a mean duration of complete remission of over 12 months [2]. Microscopic evidence of vascular invasion carries a poor prognosis [13].
Differential diagnosis
The major differential diagnosis is with large cell anaplastic lymphomas, in which the clinical and histological features may be similar. Other diseases that may be confused with malignant histiocytosis are familial haemophagocytic lymphohistiocytosis, virus-associated haemophagocytic syndrome, Hodgkin disease and SHML.
Investigations
Diagnosis can usually be established on clinicopathological features of the disease, although special stains may be needed to exclude large cell anaplastic lymphoma.
Management
Malignant histiocytosis is sensitive to both radiotherapy and chemotherapy but treatment must be started early, as many patients die before therapy can be started [3]. A review of the treatment of MH has been published [15], but similar to the other malignant histiocytic disorders, there are no recent large studies to inform therapy decisions. Conventional chemotherapy and radiotherapy remain the mainstays of treatment. In a study of 27 children with MH, complete remission was achieved in 22 children using a regimen of vincristine, cyclophosphamide, doxorubicin and prednisolone, with a 5-year survival of 81% [16]. There are anecdotal reports of success with LCH-type salvage therapy, such as cladribine and cytosine arabinoside as per the LCH-S-2005 protocol, as well as T-cell acute lymphoblastic leukaemia therapy. In patients who relapse after conventional chemotherapy, bone marrow transplantation has successfully achieved long-term remission [15]. Large skin tumours or ulcerated tumours, non-responsive to chemotherapy, can be treated with local radiotherapy.
True histiocytic lymphoma
Definition and nomenclature
True histiocytic lymphoma is a malignant histiocytic neoplasm that may disseminate.
Introduction and general description
The disease represents a malignant proliferation of non-Langerhans cell histiocytes or more rarely of Langerhans cells. Differentiation from malignant histiocytosis may be difficult. Many early cases of ‘histiocytic lymphoma’ have been reclassified as other lymphomas, including primary cutaneous B-cell lymphoma, and it has been recently suggested that histiocytic lymphomas should be classified and treated as histiocytic sarcomas [1].
Pathophysiology
The aetiology is unknown. True histiocytic lymphoma exhibits many of the features described in malignant histiocytosis, with infiltrating cells being predominantly dermal and non-cohesive. Nemes and Thomazy suggest that the cells in true histiocytic lymphoma are more differentiated than those in malignant histiocytosis and that the cell population is more homogeneous, showing phagocytosis and labelling for factor XIIIa [2]. These cells stain with macrophage markers CD11c and CD68 and are negative for T- and B-cell markers [3]. A rare spindle cell variant has been described that expressed CD163, CD68, CD45, lysozyme and NSE (neuron-specific enolase) [4].
Clinical features
This is a localized tumour of malignant histiocytes that may be nodal or extranodal. In 40% of patients, presentation is with the painless enlargement of one or more groups of superficial lymph nodes. Constitutional symptoms of malaise, anorexia, sweating and fever may be present. Extranodal presentation may be with bone, gastrointestinal tract or skin lesions. Bone and gastrointestinal tract lesions are as described in malignant histiocytosis. Skin lesions are localized bluish red tumours that can attain a large size. An isolated skin tumour of true histiocytic lymphoma in a 79-year-old patient has been described that reached 20 cm in diameter at presentation [5]. Hepatosplenomegaly occurs in only a minority of patients with true histiocytic lymphoma, and peripheral blood involvement is rare. In one case report, a 44-year-old man with true histiocytic lymphoma was treated with autologous bone marrow transplantation and subsequently developed histiocytic leukaemia classified as M5c monocytic leukaemia [6]. True histiocytic lymphoma is treatable, and the prognosis is probably better than in malignant histiocytosis.
Management
Review of the literature reveals no recent advances in therapy except for the testing of many new agents against a ‘histiocytic lymphoma’ cell line U937. From the earlier literature, it appears that true histiocytic lymphoma is both radiosensitive and chemosensitive. Complete remission has been achieved in localized skin disease using electron beam therapy [5]. Reports of therapeutic responses are difficult to evaluate because of doubt over the diagnosis in older series.
Histiocytic sarcoma
Definition
Histiocytic sarcoma (HS) is an extremely rare, non-LCH disorder of unknown cause that most commonly presents with symptoms due to unifocal or multifocal extranodal tumours.
Epidemiology
It mainly occurs in adults, but paediatric cases have been reported. HS can occur at any age (range 3 months to 89 years) without gender predilection [1].
Pathophysiology
Pathology
Histiocytic sarcoma is composed of cells that morphologically and immunohistochemically resemble mature tissue histiocytes. Histological examination demonstrates widespread infiltration of large, epitheloid cells with round to oval nuclei, large distinct nucleoli, finely to moderately dispersed chromatin, and abundant cytoplasm that is acidophilic with H&E and greyish with Giemsa staining [2]. Binucleated cells are common and multinucleated giant tumour cells can be found occasionally. The Ki67 index ranged from 5% to 40% with a mean of 18.8% in one analysis [3]. Tissue necrosis is not prominent. The immunohistochemical profile includes reactivity for CD45, CD68, lysozyme and CD163 with a variable expression of S100. Langerhans cell–dendritic cell markers such as CD1a, CD21 and CD35 should be negative.
Genetics
The pathogenesis of HS is unknown. Several studies have described cases of HS as well as other histiocytic disorders arising from lymphoid haematological malignancies, most commonly acute lymphoblastic laeukemia [4, 5] and lymphomas [5, 6, 7, 8, 9]. These histiocytic sarcomas share the same molecular or cytogenetic abnormality as the primary malignancy, suggesting a clonal relationship between the two entities. A recent case report described a patient with a long history of hairy cell leukaemia who subsequently developed histiocytic sarcoma. BRAF V600E mutation and the same clonal immunoglobulin rearrangement were detected in both leukaemic hairy cells and histiocytic sarcoma cells [10]. All cytogenetic abnormalities found in hairy cell leukaemia were also identified in HS. Furthermore, among genes involved in B lymphopoiesis, E2A/TCF3 was the only gene deleted in HS and not in hairy cell leukaemia for this patient [10]. Altogether, these support the hypothesis that HS and lymphoid malignancies could originate from a common B-cell precursor, and that secondary genetic events could have promoted the differentiation from a lymphoid neoplasm to HS [10]. In addition to lymphoid malignancies, HS has also been found in association with germ cell tumours and teratomas [11].
Clinical features
Presentation
The clinical presentation of HS varies depending on the site of involvement – most commonly affecting the intestinal tract, skin and soft tissues. In the largest series of HS, consisting of 18 cases [3], the three paediatric cases presented with multiple, isolated skin lesions, multiple lymph node involvement and disseminated disease, respectively. Among adults, sites of involvement can be diverse and have been reported in the thyroid gland, gastrointestinal tract, kidney, spleen, testes and CNS [3, 12]. The median age at presentation was 46 years.
Disease course and prognosis
Histiocytic sarcoma is usually aggressive. Among the 12 patients with available follow-up, six had no response to treatment and died of progressive disease; three of the five patients who went into remission experienced relapse within 3–5 years; only one survived but with a chemoresistant tumour [3]. Due to the rarity of the disease, prognostic factors remain unclear. However, sites of involvement, localized disease and tumour size may impact prognosis with small, localized, low-grade tumours that are amenable to surgical resection having better outcomes [13]. For cases of HS arising with/after lymphoid malignancies, the prognosis is usually poor, with three of four patients dying despite therapy in a recent review [4].
Management
Histiocytic sarcoma is a rare but highly aggressive disease. It generally shows poor response to treatment consisting of a combination of surgery, chemotherapy and radiotherapy. Chemotherapy is usually derived from non-Hodgkin lymphoma protocols such as ICE (ifosfamide/carboplatin/etoposide) or CHOP (cyclophosphamide/doxorubicin/vincristine/prednisolone) regimens, while surgery and radiotherapy is reserved for localized disease [14]. Optimal therapy in the refractory or relapsed setting is unknown. Single case reports have suggested efficacy for high-dose chemotherapy and autologous/allogeneic stem cell transplantation [15]. The anti-CD52 antibody alemtuzumab was successful in a refractory HS patient [16] and prolonged use of thalidomide appears to have been effective in four patients who did not respond to multiple other therapies [17, 18]. Recent identification of druggable targets in four cases of HS with an expression of platelet-derived growth factor receptor, vascular endothelial growth factor receptor and epidermal growth factor receptor [19] and one single case of BRAF V600E [10] suggest the possibility of novel targeted therapies in the future.
Resources
Further information
- Euro-Histio-Net: http://www.eurohistio.net/.
- Histiocyte Society: http://www.histiocytesociety.org.
- National Cancer Institute at the National Institutes of Health, Langerhans cell histiocytosis treatment: http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional. (All last accessed July 2014.)
Further information
- Euro-Histio-Net: http://www.eurohistio.net/.
- Histiocyte Society: http://www.histiocytesociety.org.
- National Cancer Institute at the National Institutes of Health, Langerhans cell histiocytosis treatment: http://www.cancer.gov/cancertopics/pdq/treatment/lchistio/HealthProfessional. (All last accessed July 2014.)
References
Ontogeny of histiocytes
- Hoeffel G, Wang Y, Greter M, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 2012;209(6):1167–81.
- Ginhoux F, Tacke F, Angeli V, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. 2006 Mar;7(3):265–73.
Function of histiocytes
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature 2003;422(6927):37–44.
- Nikolic T, Leenen P. Histiocyte function and development in the normal immune system. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:40–57.
Classification of histiocytoses
- Chu T, D'Angio GJ, Favara BE, Ladisch S, Nesbit M, Pritchard J. Histiocytosis syndromes in children. Lancet 1987;2(8549):41–2.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997;29(3):157–66.
- Egeler RM, Neglia JP, Arico M, et al. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am 1998;12(2):369–78.
- Weitzman S, Egeler RM. Histiocytic disorders of children and adults: introduction to the problem, overview, historical perspective and epidemiology. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:1–11.
Langerhans cell histiocytosis
- Weitzman S, Egeler RM. Histiocytic disorders of children and adults: introduction to the problem, overview, historical perspective and epidemiology. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:1–11.
- Stalemark H, Laurencikas E, Karis J, et al. Incidence of Langerhans cell histiocytosis: a population-based study. Pediatr Blood Cancer 2008;51(1):76–81.
- Lau LM, Stuurman K, Weitzman S. Skeletal Langerhans cell histiocytosis in children: permanent consequences and health-related quality of life in long-term survivors. Pediatr Blood Cancer 2008;50(3):607–12.
- Arico M, Girschikofsky M, Genereau T, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer 2003;39(16):2341–8.
- Berres ML, Allen CE, Merad M. Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv Immunol 2013;120:127–61.
- Chopin M, Nutt SL. Establishing and maintaining the Langerhans cell network. Semin Cell Dev Biol 2014, http://dx.doi.org/10.1016/j.semcdb.2014.02.001 (last accessed August 2014).
- Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: current concepts and treatments. Cancer Treat Rev 2010;36(4):354–9.
- Merad M, Manz MG, Karsunky H, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol 2002;3(12):1135–41.
- Ginhoux F, Merad M. Ontogeny and homeostasis of Langerhans cells. Immunol Cell Biol 2010;88(4):387–92.
- Hoeffel G, Wang Y, Greter M, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 2012;209(6):1167–81.
- Jaffe R. The diagnostic histopathology of Langerhans cell histiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:14–17.
- Ishii R, Morimoto A, Ikushima S, et al. High serum values of soluble CD154, IL-2 receptor, RANKL and osteoprotegerin in Langerhans cell histiocytosis. Pediatr Blood Cancer 2006;47(2):194–9.
- Senechal B, Elain G, Jeziorski E, et al. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLOS Med 2007;4(8):e253.
- Coury F, Annels N, Rivollier A, et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med 2008;14(1):81–7.
- Yu RC, Chu C, Buluwela L, Chu AC. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 1994;343(8900):767–8.
- Chen W, Wang J, Wang E, et al. Detection of clonal lymphoid receptor gene rearrangements in langerhans cell histiocytosis. Am J Surg Pathol 2010;34(7):1049–57.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116(11):1919–23.
- Badalian-Very G, Vergilio JA, Fleming M, Rollins BJ. Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol 2013;8:1–20.
- Donadieu J, Egeler RM, Pritchard J, et al. Langerhans cell histiocytosis: a clinical update. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:95–124.
- Holme SA, Mills CM. Adult primary cutaneous Langerhans' cell histiocytosis mimicking nodular prurigo. Clin Exp Dermatol 2002;27(3):250–1.
- Chang SE, Koh GJ, Choi JH, et al. Widespread skin-limited adult Langerhans cell histiocytosis: long-term follow-up with good response to interferon alpha. Clin Exp Dermatol 2002;27(2):135–7.
- Krafchik B, Pope E, Walsh S, et al. Histiocytosis of skin in children and adults. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:130–47.
- Lau L, Krafchik B, Trebo MM, Weitzman S. Cutaneous Langerhans cell histiocytosis in children under one year. Pediatr Blood Cancer 2006;46(1):66–71.
- Battistella M, Fraitag S, Teillac DH, Brousse N, de Prost Y, Bodemer C. Neonatal and early infantile cutaneous langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol 2010;146(2):149–56.
- Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer 2005;45(6):802–7.
- Stein SL, Paller AS, Haut PR, Mancini AJ. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med 2001;155(7):778–83.
- Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood 2013;121(25):5006–14.
- Kolde G, Schulze P, Sterry W. Mixed response to thalidomide therapy in adults: two cases of multisystem Langerhans' cell histiocytosis. Acta Derm Venereol 2002;82(5):384–6.
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 2013;60(2):175–84.
- Titgemeyer C, Grois N, Minkov M, Flucher-Wolfram B, Gatterer-Menz I, Gadner H. Pattern and course of single-system disease in Langerhans cell histiocytosis data from the DAL-HX 83- and 90-study. Med Pediatr Oncol 2001;37(2):108–14.
- Weitzman S, Egeler RM. Langerhans cell histiocytosis of bone. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:153–70.
- Gadner H, Ladisch S. The treatment of Langerhans cell histiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:227–49.
- Minkov M. Multisystem Langerhans cell histiocytosis in children: current treatment and future directions. Paediatr Drugs 2011;13(2):75–86.
- Gadner H, Grois N, Potschger U, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 2008;111(5):2556–62.
- Gadner H, Grois N, Arico M, et al. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr 2001;138(5):728–34.
- Moricke A, Reiter A, Zimmermann M, et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 2008;111(9):4477–89.
- Morimoto A, Ikushima S, Kinugawa N, et al. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer 2006;107(3):613–19.
- Weitzman S, Braier J, Donadieu J, et al. 2'-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). Results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer 2009;53(7):1271–6.
- Steiner M, Matthes-Martin S, Attarbaschi A, et al. Improved outcome of treatment-resistant high-risk Langerhans cell histiocytosis after allogeneic stem cell transplantation with reduced-intensity conditioning. Bone Marrow Transplant 2005;36(3):215–25.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim–Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121(9):1495–500.
- Taverna JA, Stefanato CM, Wax FD, Demierre MF. Adult cutaneous Langerhans cell histiocytosis responsive to topical imiquimod. J Am Acad Dermatol 2006;54(5):911–13.
- Singh A, Prieto VG, Czelusta A, McClain KL, Duvic M. Adult Langerhans cell histiocytosis limited to the skin. Dermatology 2003;207(2):157–61.
- Vogel CA, Aughenbaugh W, Sharata H. Excimer laser as adjuvant therapy for adult cutaneous Langerhans cell histiocytosis. Arch Dermatol 2008;144(10):1287–90.
- Moravvej H, Yousefi M, Barikbin B. An unusual case of adult disseminated cutaneous Langerhans cell histiocytosis. Dermatol Online J 2006;12(6):13.
- McClain K. Histiocyte Society 29th annual meeting abstracts, Washington, DC, USA. Pediatr Blood Cancer 2013;63, doi: 10.1002/pbc.25097.
- Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis 2013;8:72.
- Steen AE, Steen KH, Bauer R, Bieber T. Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br J Dermatol 2001;145(1):137–40.
- Broekaert SM, Metzler G, Burgdorf W, Rocken M, Schaller M. Multisystem Langerhans cell histiocytosis: successful treatment with thalidomide. Am J Clin Dermatol 2007;8(5):311–14.
- Montero AJ, Diaz-Montero CM, Malpica A, Ramirez PT, Kavanagh JJ. Langerhans cell histiocytosis of the female genital tract: a literature review. Int J Gynecol Cancer 2003;13(3):381–8.
Haemophagocytic lymphohistiocytosis
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997;29(3):157–66.
- Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand 1991;80(4):428–35.
- Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002;100(7):2367–73.
- Henter JI, Arico M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12(2):417–33.
- Janka G, Imashuku S, Elinder G, Schneider M, Henter JI. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12(2):435–44.
- Takahashi N, Chubachi A, Kume M, et al. A clinical analysis of 52 adult patients with hemophagocytic syndrome: the prognostic significance of the underlying diseases. Int J Hematol 2001;74(2):209–13.
- Jaffe R. The histopathology of hemophagocytic lymphohistiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:321–33.
- Sullivan KE, Delaat CA, Douglas SD, Filipovich AH. Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res 1998;44(4):465–8.
- Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood 2002;99(1):61–6.
- Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: clinical features, pathogenesis and therapy. Expert Rev Clin Immunol 2010;6(1):137–54.
- Ishii E, Ueda I, Shirakawa R, et al. Genetic subtypes of familial hemophagocytic lymphohistiocytosis: correlations with clinical features and cytotoxic T lymphocyte/natural killer cell functions. Blood 2005;105(9):3442–8.
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999;286(5446):1957–9.
- Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003;115(4):461–73.
- Wood SM, Ljunggren HG, Bryceson YT. Insights into NK cell biology from human genetics and disease associations. Cell Mol Life Sci 2011;68(21):3479–93.
- Janka G, Henter J-I, Imashuku S, et al. Clinical aspects and therapy of hemophagocytic lymphohistiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:353–73.
- Ambruso DR, Hays T, Zwartjes WJ, Tubergen DG, Favara BE. Successful treatment of lymphohistiocytic reticulosis with phagocytosis with epipodophyllotoxin VP 16-213. Cancer 1980;45(10):2516–20.
- Stephan JL, Donadieu J, Ledeist F, Blanche S, Griscelli C, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood 1993;82(8):2319–23.
- Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics 2007;120(3):e622–8.
- Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer 2013;60(1):101–9.
- Khandelwal P, Lawrence J, Filipovich AH, et al. The successful use of alemtuzumab for treatment of steroid-refractory acute graft-versus-host disease in pediatric patients. Pediatr Transplant 2014;18(1):94–102.
- Weitzman S. Approach to Hemophagocytic Syndromes. Washington, DC: American Society of Hematology Education Program, 2011,178–83.
Juvenile xanthogranuloma
- Dehner LP. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 2003;27(5):579–93.
- Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the Kiel Pediatric Tumor Registry. Am J Surg Pathol 2005;29(1):21–8.
- Isaacs H, Jr. Fetal and neonatal histiocytoses. Pediatr Blood Cancer 2006;47(2):123–9.
- Zelger BW, Cerio R. Xanthogranuloma is the archetype of non-Langerhans cell histiocytoses. Br J Dermatol 2001;145(2):369–71.
- Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-Langerhans cell histiocytoses. Pediatr Blood Cancer 2005;45(3):256–64.
- Newman B, Hu W, Nigro K, Gilliam AC. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007;56(2):302–16.
- Chu AC. The confusing state of the histiocytoses. Br J Dermatol 2000;143(3):475–6.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116(11):1919–23.
- Emile JF, Charlotte F, Amoura Z, Haroche J. BRAF mutations in Erdheim–Chester disease. J Clin Oncol 2013;31(3):398.
- Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012;120(13):2700–3.
- Hoeger PH, Diaz C, Malone M, Pritchard J, Harper JI. Juvenile xanthogranuloma as a sequel to Langerhans cell histiocytosis: a report of three cases. Clin Exp Dermatol 2001;26(5):391–4.
- Tran DT, Wolgamot GM, Olerud J, Hurst S, Argenyi Z. An ‘eruptive’ variant of juvenile xanthogranuloma associated with langerhans cell histiocytosis. J Cutan Pathol 2008;35(Suppl. 1):50–4.
- Bains A, Parham DM. Langerhans cell histiocytosis preceding the development of juvenile xanthogranuloma: a case and review of recent developments. Pediatr Dev Pathol 2011;14(6):480–4.
- Chang MW. Update on juvenile xanthogranuloma: unusual cutaneous and systemic variants. Semin Cutan Med Surg 1999;18(3):195–205.
- Chang MW, Frieden IJ, Good W. The risk intraocular juvenile xanthogranuloma: survey of current practices and assessment of risk. J Am Acad Dermatol 1996;34(3):445–9.
- Krafchik B, Pope E, Walsh S, et al. Histiocytosis of the skin in children and adults In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:130–47.
- Freyer DR, Kennedy R, Bostrom BC, Kohut G, Dehner LP. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr 1996;129(2):227–37.
- Orsey A, Paessler M, Lange BJ, Nichols KE. Central nervous system juvenile xanthogranuloma with malignant transformation. Pediatr Blood Cancer 2008;50(4):927–30.
- Hu WK, Gilliam AC, Wiersma SR, Dahms BB. Fatal congenital systemic juvenile xanthogranuloma with liver failure. Pediatr Dev Pathol 2004;7(1):71–6.
- Weitzman S, Whitlock J. Uncommon histiocytic disorder: the non-Langerhans cell histiocytoses. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:153–170.
- Stover DG, Alapati S, Regueira O, Turner C, Whitlock JA. Treatment of juvenile xanthogranuloma. Pediatr Blood Cancer 2008;51(1):130–3.
- Nakatani T, Morimoto A, Kato R, et al. Successful treatment of congenital systemic juvenile xanthogranuloma with Langerhans cell histiocytosis-based chemotherapy. J Pediatr Hematol Oncol 2004;26(6):371–4.
- Rajendra B, Duncan A, Parslew R, Pizer BL. Successful treatment of central nervous system juvenile xanthogranulomatosis with cladribine. Pediatr Blood Cancer 2009;52(3):413–15.
- Bailey KM, Castle VP, Hummel JM, Piert M, Moyer J, McAllister-Lucas LM. Thalidomide therapy for aggressive histiocytic lesions in the pediatric population. J Pediatr Hematol Oncol 2012;34(6):480–3.
- Simko SJ, Tran HD, Jones J, et al. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai–Dorfman disease. Pediatr Blood Cancer 2014;61(3):479–87.
Benign cephalic histiocytosis
- Zelger BW, Cerio R. Xanthogranuloma is the archetype of non-Langerhans cell histiocytoses. Br J Dermatol 2001;145(2):369–71.
- Sidwell RU, Francis N, Slater DN, Mayou SC. Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol 2005;22(1):40–3.
- Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol 2002;47(6):908–13.
Generalized eruptive histiocytosis
- Jang KA, Ahn SJ, Choi JH, Sung KJ, Moon KC, Koh JK. Histiocytic disorders with spontaneous regression in infancy. Pediatr Dermatol 2000;17(5):364–8.
- Wee SH, Kim HS, Chang SN, Kim DK, Park WH. Generalized eruptive histiocytoma: a pediatric case. Pediatr Dermatol 2000;17(6):453–5.
- Misery L, Kanitakis J, Hermier C, Cambazard F. Generalized eruptive histiocytoma in an infant with healing in summer: long-term follow-up. Br J Dermatol 2001;144(2):435–7.
- Lan Ma H, Metze D, Luger TA, Steinhoff M. Successful treatment of generalized eruptive histiocytoma with PUVA. J Dtsch Dermatol Ges 2007;5(2):131–4.
- Deng YJ, Hao F, Zhou CL, et al. Generalized eruptive histiocytosis: a possible therapeutic cure? Br J Dermatol 2004;150(1):171–3.
Papular xanthoma
- Caputo R, Gianni E, Imondi D, Carminati G, Gianotti R. Papular xanthoma in children. J Am Acad Dermatol 1990;22(6 Pt 1):1052–6.
- Fonseca E, Contreras F, Cuevas J. Papular xanthoma in children: report and immunohistochemical study. Pediatr Dermatol 1993;10(2):139–41.
- Chen CG, Chen CL, Liu HN. Primary papular xanthoma of children: a clinicopathologic, immunohistopathologic and ultrastructural study. Am J Dermatopathol 1997;19(6):596–601.
- Breier F, Zelger B, Reiter H, Gschnait F, Zelger BW. Papular xanthoma: a clinicopathological study of 10 cases. J Cutan Pathol 2002;29(4):200–6.
Progressive nodular histiocytosis
- Zelger BW, Cerio R. Xanthogranuloma is the archetype of non-Langerhans cell histiocytoses. Br J Dermatol 2001;145(2):369–71.
- Beswick SJ, Kirk JM, Bradshaw K, Sanders DS, Moss C. Progressive nodular histiocytosis in a child with a hypothalamic tumor. Br J Dermatol 2002;146(1):138–40.
- Kunimoto K, Uede K, Furukawa F. Progressive nodular histiocytosis. J Dermatol 2010;37(12):1071–3.
- Zelger BW, Staudacher C, Orchard G, Wilson-Jones E, Burgdorf WH. Solitary and generalized variants of spindle cell xanthogranuloma (progressive nodular histiocytosis). Histopathology 1995;27(1):11–19.
- Luftl M, Seybold H, Simon M, Jr, Burgdorf W. [Progressive nodular histiocytosis – rare variant of cutaneous non-Langerhans cell histiocytosis.] J Dtsch Dermatol Ges 2006;4(3):236–8.
- Nofal A, Assaf M, Tawfik A, et al. Progressive nodular histiocytosis: a case report and literature review. Int J Dermatol 2011;50(12):1546–51.
- Chu AC. The confusing state of the histiocytoses. Br J Dermatol 2000;143(3):475–6.
Xanthoma disseminatum
- Calverly DC, Wismer J, Rosenthal D, deSa D, Barr RD. Xanthoma disseminatum in an infant with skeletal and marrow involvement. J Pediatr Hematol Oncol 1995;17(1):61–5.
- Toberer F, Hartschuh W, Witzens-Harig M, Enk AH, Lonsdorf AS. Intertriginous orange-to-brownish papules and plaques: a quiz. Xanthoma disseminatum. Acta Derm Venereol 2013;93(4):493–4.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol 2008;58(Suppl. 2):S47–9.
- Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-Langerhans cell histiocytoses. Pediatr Blood Cancer 2005;45(3):256–64.
- Davies CW, Marren P, Juniper MC, Gray W, Wojnorowska F, Benson MK. Xanthoma disseminatum with respiratory tract involvement and fatal outcome. Thorax 2000;55(2):170–2.
- Ozcelik U, Dogru D, Akcoren Z, Coskun T, Kaya S, Gocmen A. Xanthoma disseminatum: a child with respiratory system involvement and bronchiectasis. Pediatr Pulmonol 2005;39(1):84–7.
- Chepuri NB, Challa VR. Xanthoma disseminatum: a rare intracranial mass. Am J Neuroradiol 2003;24(1):105–8.
- Zak IT, Altinok D, Neilsen SS, Kish KK. Xanthoma disseminatum of the central nervous system and cranium. Am J Neuroradiol 2006;27(4):919–21.
- Carpo BG, Grevelink SV, Brady S, Gellis S, Grevelink JM. Treatment of cutaneous lesions of xanthoma disseminatum with a CO2 laser. Dermatol Surg 1999;25(10):751–4.
- Seaton ED, Pillai GJ, Chu AC. Treatment of xanthoma disseminatum with cyclophosphamide. Br J Dermatol 2004;150(2):346–9.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol 2011;147(4):459–64.
- Lee EH, Kang TW, Kim SC. Successful treatment of xanthoma disseminatum with simvastatin. J Dermatol 2011;38(10):1015–17.
Diffuse plane xanthomatosis
- Hofmann M, Zappel K, Trefzer U, et al. Diffuse normolipemic plane xanthoma in a 9-year-old boy. Pediatr Dermatol 2005;22(2):127–9.
- Cambiaghi S, Boneschi V, Maffeis L, Gelmetti C. Diffuse normolipemic plane xanthoma in a child with common variable immunodeficiency. Pediatr Dermatol 2011;28(1):65–6.
- Marcoval J, Moreno A, Bordas X, Gallardo F, Peyri J. Diffuse plane xanthoma: clinicopathologic study of 8 cases. J Am Acad Dermatol 1998;39(3):439–42.
- Rosmaninho A, Fernandes I, Guimas A, Amorim I, Selores M. Diffuse plane xanthomatosis associated with monoclonal gammopathy. An Bras Dermatol 2011;86(4 Suppl. 1):S50–2.
- Bragg J. Diffuse plane xanthomata. Dermatol Online J 2005;11(4):4.
- Lorenz S, Hohenleutner S, Hohenleutner U, Landthaler M. Treatment of diffuse plane xanthoma of the face with the erbium:YAG laser. Arch Dermatol 2001;137(11):1413–15.
Erdheim–Chester disease
- Zelger B, Burgdorf WH. The cutaneous ‘histiocytoses’. Adv Dermatol 2001;17:77–114.
- Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim–Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996;75(3):157–69.
- Wilejto M, Abla O. Langerhans cell histiocytosis and Erdheim–Chester disease. Curr Opin Rheumatol 2012;24(1):90–6.
- Emile JF, Charlotte F, Amoura Z, Haroche J. BRAF mutations in Erdheim–Chester disease. J Clin Oncol 2013;31(3):398.
- Volpicelli ER, Doyle L, Annes JP, et al. Erdheim–Chester disease presenting with cutaneous involvement: a case report and literature review. J Cutan Pathol 2011;38(3):280–5.
- Arnaud L, Pierre I, Beigelman-Aubry C, et al. Pulmonary involvement in Erdheim–Chester disease: a single-center study of thirty-four patients and a review of the literature. Arthritis Rheum 2010;62(11):3504–12.
- Rush WL, Andriko JA, Galateau-Salle F, et al. Pulmonary pathology of Erdheim–Chester disease. Mod Pathol 2000;13(7):747–54.
- Haroche J, Amoura Z, Dion E, et al. Cardiovascular involvement, an overlooked feature of Erdheim–Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore) 2004;83(6):371–92.
- Lachenal F, Cotton F, Desmurs-Clavel H, et al. Neurological manifestations and neuroradiological presentation of Erdheim–Chester disease: report of 6 cases and systematic review of the literature. J Neurol 2006;253(10):1267–77.
- Arnaud L, Hervier B, Neel A, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim–Chester disease: a multicenter survival analysis of 53 patients. Blood 2011;117(10):2778–82.
- Devouassoux G, Lantuejoul S, Chatelain P, Brambilla E, Brambilla C. Erdheim–Chester disease: a primary macrophage cell disorder. Am J Respir Crit Care Med 1998;157(2):650–3.
- Gupta A, Kelly B, McGuigan JE. Erdheim–Chester disease with prominent pericardial involvement: clinical, radiologic, and histologic findings. Am J Med Sci 2002;324(2):96–100.
- Braiteh F, Boxrud C, Esmaeli B, Kurzrock R. Successful treatment of Erdheim–Chester disease, a non-Langerhans-cell histiocytosis, with interferon-alpha. Blood 2005;106(9):2992–4.
- Haroche J, Amoura Z, Trad SG, et al. Variability in the efficacy of interferon-alpha in Erdheim–Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum 2006;54(10):3330–6.
- Haroche J, Amoura Z, Charlotte F, et al. Imatinib mesylate for platelet-derived growth factor receptor-beta-positive Erdheim–Chester histiocytosis. Blood 2008;111(11):5413–15.
- Janku F, Amin HM, Yang D, Garrido-Laguna I, Trent JC, Kurzrock R. Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol 2010;28(31):e633–6.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim–Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121(9):1495–500.
Reticulohistiocytoma
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol 2006;30(4):521–8.
- Zelger B, Cerio R, Soyer HP, Misch K, Orchard G, Wilson-Jones E. Reticulohistiocytoma and multicentric reticulohistiocytosis. Histopathologic and immunophenotypic distinct entities. Am J Dermatopathol 1994;16(6):577–84.
Familial sea-blue histiocytosis
- Newman B, Hu W, Nigro K, Gilliam AC. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007;56(2):302–16.
- Bigorgne C, Le Tourneau A, Vahedi K, et al. Sea-blue histiocyte syndrome in bone marrow secondary to total parenteral nutrition. Leuk Lymphoma 1998;28(5–6):523–9.
Hereditary progressive mucinous histiocytosis
- Antoni-Bach N, Pfister R, Grosshans E, et al. [Hereditary progressive mucinous histiocytosis.] Ann Dermatol Venereol 2000;127(4):400–4.
- Schroder K, Hettmannsperger U, Schmuth M, Orfanos CE, Goerdt S. Hereditary progressive mucinous histiocytosis. J Am Acad Dermatol 1996;35(2 Pt 2):298–303.
- Wong D, Killingsworth M, Crosland G, Kossard S. Hereditary progressive mucinous histiocytosis. Br J Dermatol 1999;141(6):1101–5.
- Young A, Olivere J, Yoo S, Martins C, Barrett T. Two sporadic cases of adult-onset progressive mucinous histiocytosis. J Cutan Pathol 2006;33(2):166–70.
Malakoplakia
- Hyun KH, Shin HD, Kim DH. Malakoplakia in a healthy young female patient. Korean J Intern Med 2013;28(4):475–80.
- Lee WS, Chen WY, Ou TY, Chen FL, Lin YW. Malakoplakia in a patient with complicated urinary tract infection caused by extended-spectrum beta-lactamase-producing Escherichia coli. J Microbiol Immunol Infect 2014;Feb 13:1–2.
- Long JP, Jr, Althausen AF. Malacoplakia: a 25-year experience with a review of the literature. J Urol 1989;141(6):1328–31.
- Yousef GM, Naghibi B, Hamodat MM. Malakoplakia outside the urinary tract. Arch Pathol Lab Med 2007;131(2):297–300.
- Kohl SK, Hans CP. Cutaneous malakoplakia. Arch Pathol Lab Med 2008;132(1):113–17.
- Afonso JP, Ando PN, Padilha MH, Michalany NS, Porro AM. Cutaneous malakoplakia: case report and review. An Bras Dermatol 2013;88(3):432–7.
- Van der Voort HJ, ten Velden JA, Wassenaar RP, Silberbusch J. Malacoplakia. Two case reports and a comparison of treatment modalities based on a literature review. Arch Intern Med 1996;156(5):577–83.
Necrobiotic xanthogranuloma
- Rose A, Robinson M, Kamino H, Latkowski JA. Necrobiotic xanthogranuloma. Dermatol Online J 2012;18(12):30.
- Hallermann C, Tittelbach J, Norgauer J, Ziemer M. Successful treatment of necrobiotic xanthogranuloma with intravenous immunoglobulin. Arch Dermatol 2010;146(9):957–60.
- Balagula Y, Straus DJ, Pulitzer MP, Lacouture ME. Necrobiotic xanthogranuloma associated with immunoglobulin M paraproteinemia in a patient with Waldenstrom macroglobulinemia. J Clin Oncol 2011;29(11):e305–7.
- Zelger B, Eisendle K, Mensing C. Detection of spirochetal micro-organisms by focus-floating microscopy in necrobiotic xanthogranuloma. J Am Acad Dermatol 2007;57(6):1026–30.
- Inthasotti S, Wanitphakdeedecha R, Manonukul J. A 7-year history of necrobiotic xanthogranuloma following asymptomatic multiple myeloma: a case report. Dermatol Res Pract 2011;2011:1–5.
- Szalat R, Arnulf B, Karlin L, et al. Pathogenesis and treatment of xanthomatosis associated with monoclonal gammopathy. Blood 2011;118(14):3777–84.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol 2009;48(1):1–10.
- Wood AJ, Wagner MV, Abbott JJ, Gibson LE. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol 2009;145(3):279–84.
- Efebera Y, Blanchard E, Allam C, Han A, Lee S, Munshi N. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk 2011;11(3):298–302.
- Kossard S, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinemia. J Am Acad Dermatol 1980;3(3):257–70.
- Shah KC, Poonnoose SI, George R, Jacob M, Rajshekhar V. Necrobiotic xanthogranuloma with cutaneous and cerebral manifestations. Case report and review of the literature. J Neurosurg 2004;100(6):1111–14.
- Stork J, Kodetova D, Vosmik F, Krejca M. Necrobiotic xanthogranuloma presenting as a solitary tumor. Am J Dermatopathol 2000;22(5):453–6.
- Ryan E, Warren LJ, Szabo F. Necrobiotic xanthogranuloma: response to chlorambucil. Australas J Dermatol 2012;53(2):e23–5.
- Vieira V, Del Pozo, Martínez W, et al. Necrobiotic xanthogranuloma associated with lymphoplasmacytic lymphoma. Palliative treatment with carbon dioxide laser. Eur J Dermatol 2005;15(3):182–5.
Multicentric reticulohistiocytosis
- Gorman JD, Danning C, Schumacher HR, Klippel JH, Davis JC, Jr. Multicentric reticuuplohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum 2000;43(4):930–8.
- Trotta F, Colina M. Multicentric reticulohistiocytosis and fibroblastic rheumatism. Best Pract Res Clin Rheumatol 2012;26(4):543–57.
- Outland JD, Keiran SJ, Schikler KN, Callen JP. Multicentric reticulohistiocytosis in a 14-year-old girl. Pediatr Dermatol 2002;19(6):527–31.
- Candell Chalom E, Elenitsas R, Rosenstein ED, Kramer N. A case of multicentric reticulohistiocytosis in a 6-year-old child. J Rheumatol 1998;25(4):794–7.
- Havill S, Duffill M, Rademaker M. Multicentric reticulohistiocytosis in a child. Australas J Dermatol 1999;40(1):44–6.
- Pacheco-Tena C, Reyes-Cordero G, Ochoa-Albiztegui R, Rios-Barrera V, Gonzalez-Chavez SA. Treatment of multicentric reticulohistiocytosis with tocilizumab. J Clin Rheumatol 2013;19(5):272–6.
- Fortier-Beaulieu M, Thomine E, Boullie MC, Le Loet X, Lauret P, Hemet J. New electron microscopic findings in a case of multicentric reticulohistiocytosis. Long spacing collagen inclusions. Am J Dermatopathol 1993;15(6):587–9.
- Luz FB, Gaspar AP, Ramos-e-Silva M, et al. Immunohistochemical profile of multicentric reticulohistiocytosis. Skinmed 2005;4(2):71–7.
- Gomez-Mateo Mdel C, Monteagudo C. Nonepithelial skin tumors with multinucleated giant cells. Semin Diagn Pathol 2013;30(1):58–72.
- Campbell DA, Edwards NL. Multicentric reticulohistiocytosis: systemic macrophage disorder. Baillieres Clin Rheumatol 1991;5(2):301–19.
- Kishikawa T, Miyashita T, Fujiwara E, et al. Multicentric reticulohistiocytosis associated with ovarian cancer. Mod Rheumatol 2007;17(5):422–5.
- Malik MK, Regan L, Robinson-Bostom L, Pan TD, McDonald CJ. Proliferating multicentric reticulohistiocytosis associated with papillary serous carcinoma of the endometrium. J Am Acad Dermatol 2005;53(6):1075–9.
- Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and immunophenotypic study. Br J Dermatol 1995;133(1):71–6.
- Yee KC, Bowker CM, Tan CY, Palmer RG. Cardiac and systemic complications in multicentric reticulohistiocytosis. Clin Exp Dermatol 1993;18(6):555–8.
- Lovelace K, Loyd A, Adelson D, Crowson N, Taylor JR, Cornelison R. Etanercept and the treatment of multicentric reticulohistiocytosis. Arch Dermatol 2005;141(9):1167–8.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, Ramos-e-Silva M. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol 2001;15(6):524–31.
- Liang GC, Granston AS. Complete remission of multicentric reticulohistiocytosis with combination therapy of steroid, cyclophosphamide, and low-dose pulse methotrexate. Case report, review of the literature, and proposal for treatment. Arthritis Rheum 1996;39(1):171–4.
- Lonsdale-Eccles AA, Haworth AE, McCrae FC, Young-Min SA. Successful treatment of multicentric reticulohistiocytosis with leflunomide. Br J Dermatol 2009;161(2):470–2.
- Fedler R, Frantzmann Y, Schwarze EW, Frosch PJ. [Multicenter reticulohistiocytosis. Therapy with azathioprine and prednisolone.] Hautarzt 1995;46(2):118–20.
- Satoh M, Oyama N, Yamada H, Nakamura K, Kaneko F. Treatment trial of multicentric reticulohistiocytosis with a combination of predonisolone, methotrexate and alendronate. J Dermatol 2008;35(3):168–71.
- Mavragani CP, Batziou K, Aroni K, Pikazis D, Manoussakis MN. Alleviation of polyarticular syndrome in multicentric reticulohistiocytosis with intravenous zoledronate. Ann Rheum Dis 2005;64(10):1521–2.
- Codriansky K, Rünger TM, Bhawan J, et al. Multicentric reticulohistiocytosis: a systemic osteoclastic disease? Arthritis Rheum 2008;59 (3):444–8.
Sinus histiocytosis with massive lymphadenopathy
- Juskevicius R, Finley JL. Rosai–Dorfman disease of the parotid gland: cytologic and histopathologic findings with immunohistochemical correlation. Arch Pathol Lab Med 2001;125(10):1348–50.
- Deodhare SS, Ang LC, Bilbao JM. Isolated intracranial involvement in Rosai–Dorfman disease: a report of two cases and review of the literature. Arch Pathol Lab Med 1998;122(2):161–5.
- Lauwers GY, Perez-Atayde A, Dorfman RF, Rosai J. The digestive system manifestations of Rosai–Dorfman disease (sinus histiocytosis with massive lymphadenopathy): review of 11 cases. Hum Pathol 2000;31(3):380–5.
- Grabczynska SA, Toh CT, Francis N, Costello C, Bunker CB. Rosai–Dorfman disease complicated by autoimmune haemolytic anaemia: case report and review of a multisystem disease with cutaneous infiltrates. Br J Dermatol 2001;145(2):323–6.
- Foss HD, Herbst H, Araujo I, et al. Monokine expression in Langerhans' cell histiocytosis and sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease). J Pathol 1996;179(1):60–5.
- Newman B, Hu W, Nigro K, Gilliam AC. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007;56(2):302–16.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): review of the entity. Semin Diagn Pathol 1990;7(1):19–73.
- Perez A, Rodriguez M, Febrer I, Aliaga A. Sinus histiocytosis confined to the skin. Case report and review of the literature. Am J Dermatopathol 1995;17(4):384–8.
- Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai–Dorfman disease isolated to the central nervous system: a report of 11 cases. Mod Pathol 2001;14(3):172–8.
- Goodnight JW, Wang MB, Sercarz JA, Fu YS. Extranodal Rosai–Dorfman disease of the head and neck. Laryngoscope 1996;106(3 Pt 1):253–6.
- Pulsoni A, Anghel G, Falcucci P, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): report of a case and literature review. Am J Hematol 2002;69(1):67–71.
- Lohr HF, Godderz W, Wolfe T, et al. Long-term survival in a patient with Rosai–Dorfman disease treated with interferon-alpha. Eur J Cancer 1995;31A(13–14):2427–8.
- Palomera L, Domingo J, Soria J, Gutierrez M. [Long term survival in a patient with aggressive Rosai–Dorfman disease treated with interferon alpha.] Med Clin (Barc) 2001;116(20):797–8.
- Baildam EM, Ewing CI, D'Souza SW, Stevens RF. Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): response to acyclovir. J R Soc Med 1992;85(3):179–80.
Malignant histiocytosis
- Low SE, Stafford JS. Malignant histiocytosis: a case report of a rare tumour presenting with spontaneous splenic rupture. J Clin Pathol 2006;59(7):770–2.
- Rilke F, Carbone A, Musumeci R, Pilotti S, De Lena M, Bonadonna G. Malignant histiocytosis: a clinicopathologic study of 18 consecutive cases. Tumori 1978;64(2):211–27.
- Ornvold K, Nielsen MH, Clausen N. Malignant histiocytosis in childhood. Clinicopathological study of 4 cases. Acta Pathol Microbiol Immunol Scand A 1986;94(4):291–6.
- Warnke RA, Kim H, Dorfman RF. Malignant histiocytosis (histiocytic medullary reticulosis). I. Clinicopatholigic study of 29 cases. Cancer 1975;35(1):215–30.
- Amsel S, Bijlsma F. Histiocytic medullary reticulosis. Clinical and pathological studies in Uganda. Trop Geogr Med 1974;26(1):31–8.
- Molyneux ME, Tozer RA, Hutt MS. Histiocytic medullary reticulosis in Africa. Lancet 1978;2(8101):1202–3.
- Jain S, Kanarek N, Arceci RJ. Analysis of histiocytosis deaths in the US and recommendations for incidence tracking. J Registry Manag 2010;37(4):156–62.
- Carbone A, Micheau C, Caillaud JM, Carlu C. A cytochemical and immunohistochemical approach to malignant histiocytosis. Cancer 1981;47(12):2862–71.
- Van Heerde P, Feltkamp CA, Hart AA, Somers R, van Unnik JA, Vroom TM. Malignant histiocytosis and related tumors. A clinicopathologic study of 42 cases using cytological, histochemical and ultrastructural parameters. Hematol Oncol 1984;2(1):13–32.
- Nemes Z, Thomazy V. Diagnostic significance of histiocyte-related markers in malignant histiocytosis and true histiocytic lymphoma. Cancer 1988;62(9):1970–80.
- Kamesaki H, Koya M, Miwa H, et al. Malignant histiocytosis with rearrangement of the heavy chain gene and evidence of monocyte–macrophage lineage. Cancer 1988;62(7):1306–9.
- Morgan R, Smith SD, Hecht BK, et al. Lack of involvement of the c-fms and N-myc genes by chromosomal translocation t(2;5)(p23;q35) common to malignancies with features of so-called malignant histiocytosis. Blood 1989;73(8):2155–64.
- Newman B, Hu W, Nigro K, Gilliam AC. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007;56(2):302–16.
- Ducatman BS, Wick MR, Morgan TW, Banks PM, Pierre RV. Malignant histiocytosis: a clinical, histologic, and immunohistochemical study of 20 cases. Hum Pathol 1984;15(4):368–77.
- Bucsky P, Egeler RM. Malignant histiocytic disorders in children. Clinical and therapeutic approaches with a nosologic discussion. Hematol Oncol Clin North Am 1998;12(2):465–71.
- Brugieres L, Caillaud JM, Patte C, et al. Malignant histiocytosis: therapeutic results in 27 children treated with a single polychemotherapy regimen. Med Pediatr Oncol 1989;17(3):193–6.
True histiocytic lymphoma
- Kerl H, Cerroni L. Primary cutaneous B-cell lymphomas: then and now. J Cutan Pathol 2006;33(Suppl. 1):1–5.
- Nemes Z, Thomazy V. Diagnostic significance of histiocyte-related markers in malignant histiocytosis and true histiocytic lymphoma. Cancer 1988;62(9):1970–80.
- Soria C, Orradre JL, Garcia-Almagro D, Martinez B, Algara P, Piris MA. True histiocytic lymphoma (monocytic sarcoma). Am J Dermatopathol 1992;14(6):511–17.
- Alexiev BA, Sailey CJ, McClure SA, Ord RA, Zhao XF, Papadimitriou JC. Primary histiocytic sarcoma arising in the head and neck with predominant spindle cell component. Diagn Pathol 2007;2:7.
- Forestier JY, Schmitt D, Thivolet J. [Cutaneous histiocytosarcoma: clinical, immunohistochemical and ultrastructural aspects; difficulties of study and problems of nosology (author's translation).] Ann Dermatol Venereol 1980;107(1–2):7–19.
- Esteve J, Rozman M, Campo E, Munoz F, Urbano-Ispizua A, Rozman C. Leukemia after true histiocytic lymphoma: another type of acute monocytic leukemia with histiocytic differentiation (AML-M5c)? Leukemia 1995;9(8):1389–91.
Histiocytic sarcoma
- Mainardi C, D'Amore ES, Pillon M, Toffolutti T, Rosolen A. A case of resistant pediatric histiocytic sarcoma successfully treated with chemo-radiotherapy and autologous peripheral blood stem cell transplant. Leuk Lymphoma 2011;52(7):1367–71.
- Black J, Coffin CM, Dehner LP. Fibrohistiocytic tumors and related neoplasms in children and adolescents. Pediatr Dev Pathol 2012;15(Suppl. 1):181–210.
- Pileri SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002;41(1):1–29.
- Castro EC, Blazquez C, Boyd J, et al. Clinicopathologic features of histiocytic lesions following ALL, with a review of the literature. Pediatr Dev Pathol 2010;13(3):225–37.
- Kumar R, Khan SP, Joshi DD, Shaw GR, Ketterling RP, Feldman AL. Pediatric histiocytic sarcoma clonally related to precursor B-cell acute lymphoblastic leukemia with homozygous deletion of CDKN2A encoding p16INK4A. Pediatr Blood Cancer 2011;56(2):307–10.
- Wang E, Hutchinson CB, Huang Q, et al. Histiocytic sarcoma arising in indolent small B-cell lymphoma: report of two cases with molecular/genetic evidence suggestive of a ‘transdifferentiation’ during the clonal evolution. Leuk Lymphoma 2010;51(5):802–12.
- Zeng W, Meck J, Cheson BD, Ozdemirli M. Histiocytic sarcoma transdifferentiated from follicular lymphoma presenting as a cutaneous tumor. J Cutan Pathol 2011;38(12):999–1003.
- Shao H, Xi L, Raffeld M, et al. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol 2011;24(11):1421–32.
- Hure MC, Elco CP, Ward D, et al. Histiocytic sarcoma arising from clonally related mantle cell lymphoma. J Clin Oncol 2012;30(5):e49–53.
- Michonneau D, Kaltenbach S, Derrieux C, et al. BRAFV600E mutation in a histiocytic sarcoma arising from hairy cell leukemia. J Clin Oncol 2014;32:1–5.
- DeMent SH. Association between mediastinal germ cell tumors and hematologic malignancies: an update. Hum Pathol 1990;21(7):699–703.
- Wu W, Tanrivermis Sayit A, Vinters HV, Pope W, Mirsadraei L, Said J. Primary central nervous system histiocytic sarcoma presenting as a postradiation sarcoma: case report and literature review. Hum Pathol 2013;44(6):1177–83.
- Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol 2004;28(9):1133–44.
- Bucsky P, Egeler RM. Malignant histiocytic disorders in children. Clinical and therapeutic approaches with a nosologic discussion. Hematol Oncol Clin North Am 1998;12(2):465–71.
- Gergis U, Dax H, Ritchie E, Marcus R, Wissa U, Orazi A. Autologous hematopoietic stem-cell transplantation in combination with thalidomide as treatment for histiocytic sarcoma: a case report and review of the literature. J Clin Oncol 2011;29(10):e251–3.
- Shukla N, Kobos R, Renaud T, et al. Successful treatment of refractory metastatic histiocytic sarcoma with alemtuzumab. Cancer 2012;118(15):3719–24.
- Bailey KM, Castle VP, Hummel JM, Piert M, Moyer J, McAllister-Lucas LM. Thalidomide therapy for aggressive histiocytic lesions in the pediatric population. J Pediatr Hematol Oncol 2012;34(6):480–3.
- Dalle JH, Leblond P, Decouvelaere A, et al. Efficacy of thalidomide in a child with histiocytic sarcoma following allogeneic bone marrow transplantation for T-ALL. Leukemia 2003;17(10):2056–7.
- Schlick K, Aigelsreiter A, Pichler M, et al. Histiocytic sarcoma – targeted therapy: novel therapeutic options? A series of 4 cases. Onkologie 2012;35(7–8):447–50.
Ontogeny of histiocytes
- Hoeffel G, Wang Y, Greter M, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 2012;209(6):1167–81.
- Ginhoux F, Tacke F, Angeli V, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. 2006 Mar;7(3):265–73.
Function of histiocytes
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature 2003;422(6927):37–44.
- Nikolic T, Leenen P. Histiocyte function and development in the normal immune system. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:40–57.
Classification of histiocytoses
- Chu T, D'Angio GJ, Favara BE, Ladisch S, Nesbit M, Pritchard J. Histiocytosis syndromes in children. Lancet 1987;2(8549):41–2.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997;29(3):157–66.
- Egeler RM, Neglia JP, Arico M, et al. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am 1998;12(2):369–78.
- Weitzman S, Egeler RM. Histiocytic disorders of children and adults: introduction to the problem, overview, historical perspective and epidemiology. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:1–11.
Ontogeny of histiocytes
- Hoeffel G, Wang Y, Greter M, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 2012;209(6):1167–81.
- Ginhoux F, Tacke F, Angeli V, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. 2006 Mar;7(3):265–73.
Function of histiocytes
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature 2003;422(6927):37–44.
- Nikolic T, Leenen P. Histiocyte function and development in the normal immune system. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:40–57.
Classification of histiocytoses
- Chu T, D'Angio GJ, Favara BE, Ladisch S, Nesbit M, Pritchard J. Histiocytosis syndromes in children. Lancet 1987;2(8549):41–2.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997;29(3):157–66.
- Egeler RM, Neglia JP, Arico M, et al. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am 1998;12(2):369–78.
- Weitzman S, Egeler RM. Histiocytic disorders of children and adults: introduction to the problem, overview, historical perspective and epidemiology. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:1–11.
Langerhans cell histiocytosis
- Weitzman S, Egeler RM. Histiocytic disorders of children and adults: introduction to the problem, overview, historical perspective and epidemiology. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:1–11.
- Stalemark H, Laurencikas E, Karis J, et al. Incidence of Langerhans cell histiocytosis: a population-based study. Pediatr Blood Cancer 2008;51(1):76–81.
- Lau LM, Stuurman K, Weitzman S. Skeletal Langerhans cell histiocytosis in children: permanent consequences and health-related quality of life in long-term survivors. Pediatr Blood Cancer 2008;50(3):607–12.
- Arico M, Girschikofsky M, Genereau T, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer 2003;39(16):2341–8.
- Berres ML, Allen CE, Merad M. Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv Immunol 2013;120:127–61.
- Chopin M, Nutt SL. Establishing and maintaining the Langerhans cell network. Semin Cell Dev Biol 2014, http://dx.doi.org/10.1016/j.semcdb.2014.02.001 (last accessed August 2014).
- Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: current concepts and treatments. Cancer Treat Rev 2010;36(4):354–9.
- Merad M, Manz MG, Karsunky H, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol 2002;3(12):1135–41.
- Ginhoux F, Merad M. Ontogeny and homeostasis of Langerhans cells. Immunol Cell Biol 2010;88(4):387–92.
- Hoeffel G, Wang Y, Greter M, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 2012;209(6):1167–81.
- Jaffe R. The diagnostic histopathology of Langerhans cell histiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:14–17.
- Ishii R, Morimoto A, Ikushima S, et al. High serum values of soluble CD154, IL-2 receptor, RANKL and osteoprotegerin in Langerhans cell histiocytosis. Pediatr Blood Cancer 2006;47(2):194–9.
- Senechal B, Elain G, Jeziorski E, et al. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLOS Med 2007;4(8):e253.
- Coury F, Annels N, Rivollier A, et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med 2008;14(1):81–7.
- Yu RC, Chu C, Buluwela L, Chu AC. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 1994;343(8900):767–8.
- Chen W, Wang J, Wang E, et al. Detection of clonal lymphoid receptor gene rearrangements in langerhans cell histiocytosis. Am J Surg Pathol 2010;34(7):1049–57.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116(11):1919–23.
- Badalian-Very G, Vergilio JA, Fleming M, Rollins BJ. Pathogenesis of Langerhans cell histiocytosis. Annu Rev Pathol 2013;8:1–20.
- Donadieu J, Egeler RM, Pritchard J, et al. Langerhans cell histiocytosis: a clinical update. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:95–124.
- Holme SA, Mills CM. Adult primary cutaneous Langerhans' cell histiocytosis mimicking nodular prurigo. Clin Exp Dermatol 2002;27(3):250–1.
- Chang SE, Koh GJ, Choi JH, et al. Widespread skin-limited adult Langerhans cell histiocytosis: long-term follow-up with good response to interferon alpha. Clin Exp Dermatol 2002;27(2):135–7.
- Krafchik B, Pope E, Walsh S, et al. Histiocytosis of skin in children and adults. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:130–47.
- Lau L, Krafchik B, Trebo MM, Weitzman S. Cutaneous Langerhans cell histiocytosis in children under one year. Pediatr Blood Cancer 2006;46(1):66–71.
- Battistella M, Fraitag S, Teillac DH, Brousse N, de Prost Y, Bodemer C. Neonatal and early infantile cutaneous langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol 2010;146(2):149–56.
- Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer 2005;45(6):802–7.
- Stein SL, Paller AS, Haut PR, Mancini AJ. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med 2001;155(7):778–83.
- Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood 2013;121(25):5006–14.
- Kolde G, Schulze P, Sterry W. Mixed response to thalidomide therapy in adults: two cases of multisystem Langerhans' cell histiocytosis. Acta Derm Venereol 2002;82(5):384–6.
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 2013;60(2):175–84.
- Titgemeyer C, Grois N, Minkov M, Flucher-Wolfram B, Gatterer-Menz I, Gadner H. Pattern and course of single-system disease in Langerhans cell histiocytosis data from the DAL-HX 83- and 90-study. Med Pediatr Oncol 2001;37(2):108–14.
- Weitzman S, Egeler RM. Langerhans cell histiocytosis of bone. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:153–70.
- Gadner H, Ladisch S. The treatment of Langerhans cell histiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press, 2005:227–49.
- Minkov M. Multisystem Langerhans cell histiocytosis in children: current treatment and future directions. Paediatr Drugs 2011;13(2):75–86.
- Gadner H, Grois N, Potschger U, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 2008;111(5):2556–62.
- Gadner H, Grois N, Arico M, et al. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr 2001;138(5):728–34.
- Moricke A, Reiter A, Zimmermann M, et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 2008;111(9):4477–89.
- Morimoto A, Ikushima S, Kinugawa N, et al. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer 2006;107(3):613–19.
- Weitzman S, Braier J, Donadieu J, et al. 2'-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). Results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer 2009;53(7):1271–6.
- Steiner M, Matthes-Martin S, Attarbaschi A, et al. Improved outcome of treatment-resistant high-risk Langerhans cell histiocytosis after allogeneic stem cell transplantation with reduced-intensity conditioning. Bone Marrow Transplant 2005;36(3):215–25.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim–Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121(9):1495–500.
- Taverna JA, Stefanato CM, Wax FD, Demierre MF. Adult cutaneous Langerhans cell histiocytosis responsive to topical imiquimod. J Am Acad Dermatol 2006;54(5):911–13.
- Singh A, Prieto VG, Czelusta A, McClain KL, Duvic M. Adult Langerhans cell histiocytosis limited to the skin. Dermatology 2003;207(2):157–61.
- Vogel CA, Aughenbaugh W, Sharata H. Excimer laser as adjuvant therapy for adult cutaneous Langerhans cell histiocytosis. Arch Dermatol 2008;144(10):1287–90.
- Moravvej H, Yousefi M, Barikbin B. An unusual case of adult disseminated cutaneous Langerhans cell histiocytosis. Dermatol Online J 2006;12(6):13.
- McClain K. Histiocyte Society 29th annual meeting abstracts, Washington, DC, USA. Pediatr Blood Cancer 2013;63, doi: 10.1002/pbc.25097.
- Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis 2013;8:72.
- Steen AE, Steen KH, Bauer R, Bieber T. Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br J Dermatol 2001;145(1):137–40.
- Broekaert SM, Metzler G, Burgdorf W, Rocken M, Schaller M. Multisystem Langerhans cell histiocytosis: successful treatment with thalidomide. Am J Clin Dermatol 2007;8(5):311–14.
- Montero AJ, Diaz-Montero CM, Malpica A, Ramirez PT, Kavanagh JJ. Langerhans cell histiocytosis of the female genital tract: a literature review. Int J Gynecol Cancer 2003;13(3):381–8.
Haemophagocytic lymphohistiocytosis
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997;29(3):157–66.
- Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand 1991;80(4):428–35.
- Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002;100(7):2367–73.
- Henter JI, Arico M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12(2):417–33.
- Janka G, Imashuku S, Elinder G, Schneider M, Henter JI. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998;12(2):435–44.
- Takahashi N, Chubachi A, Kume M, et al. A clinical analysis of 52 adult patients with hemophagocytic syndrome: the prognostic significance of the underlying diseases. Int J Hematol 2001;74(2):209–13.
- Jaffe R. The histopathology of hemophagocytic lymphohistiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:321–33.
- Sullivan KE, Delaat CA, Douglas SD, Filipovich AH. Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res 1998;44(4):465–8.
- Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood 2002;99(1):61–6.
- Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: clinical features, pathogenesis and therapy. Expert Rev Clin Immunol 2010;6(1):137–54.
- Ishii E, Ueda I, Shirakawa R, et al. Genetic subtypes of familial hemophagocytic lymphohistiocytosis: correlations with clinical features and cytotoxic T lymphocyte/natural killer cell functions. Blood 2005;105(9):3442–8.
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999;286(5446):1957–9.
- Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003;115(4):461–73.
- Wood SM, Ljunggren HG, Bryceson YT. Insights into NK cell biology from human genetics and disease associations. Cell Mol Life Sci 2011;68(21):3479–93.
- Janka G, Henter J-I, Imashuku S, et al. Clinical aspects and therapy of hemophagocytic lymphohistiocytosis. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:353–73.
- Ambruso DR, Hays T, Zwartjes WJ, Tubergen DG, Favara BE. Successful treatment of lymphohistiocytic reticulosis with phagocytosis with epipodophyllotoxin VP 16-213. Cancer 1980;45(10):2516–20.
- Stephan JL, Donadieu J, Ledeist F, Blanche S, Griscelli C, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood 1993;82(8):2319–23.
- Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics 2007;120(3):e622–8.
- Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer 2013;60(1):101–9.
- Khandelwal P, Lawrence J, Filipovich AH, et al. The successful use of alemtuzumab for treatment of steroid-refractory acute graft-versus-host disease in pediatric patients. Pediatr Transplant 2014;18(1):94–102.
- Weitzman S. Approach to Hemophagocytic Syndromes. Washington, DC: American Society of Hematology Education Program, 2011,178–83.
Juvenile xanthogranuloma
- Dehner LP. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 2003;27(5):579–93.
- Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the Kiel Pediatric Tumor Registry. Am J Surg Pathol 2005;29(1):21–8.
- Isaacs H, Jr. Fetal and neonatal histiocytoses. Pediatr Blood Cancer 2006;47(2):123–9.
- Zelger BW, Cerio R. Xanthogranuloma is the archetype of non-Langerhans cell histiocytoses. Br J Dermatol 2001;145(2):369–71.
- Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-Langerhans cell histiocytoses. Pediatr Blood Cancer 2005;45(3):256–64.
- Newman B, Hu W, Nigro K, Gilliam AC. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007;56(2):302–16.
- Chu AC. The confusing state of the histiocytoses. Br J Dermatol 2000;143(3):475–6.
- Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116(11):1919–23.
- Emile JF, Charlotte F, Amoura Z, Haroche J. BRAF mutations in Erdheim–Chester disease. J Clin Oncol 2013;31(3):398.
- Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012;120(13):2700–3.
- Hoeger PH, Diaz C, Malone M, Pritchard J, Harper JI. Juvenile xanthogranuloma as a sequel to Langerhans cell histiocytosis: a report of three cases. Clin Exp Dermatol 2001;26(5):391–4.
- Tran DT, Wolgamot GM, Olerud J, Hurst S, Argenyi Z. An ‘eruptive’ variant of juvenile xanthogranuloma associated with langerhans cell histiocytosis. J Cutan Pathol 2008;35(Suppl. 1):50–4.
- Bains A, Parham DM. Langerhans cell histiocytosis preceding the development of juvenile xanthogranuloma: a case and review of recent developments. Pediatr Dev Pathol 2011;14(6):480–4.
- Chang MW. Update on juvenile xanthogranuloma: unusual cutaneous and systemic variants. Semin Cutan Med Surg 1999;18(3):195–205.
- Chang MW, Frieden IJ, Good W. The risk intraocular juvenile xanthogranuloma: survey of current practices and assessment of risk. J Am Acad Dermatol 1996;34(3):445–9.
- Krafchik B, Pope E, Walsh S, et al. Histiocytosis of the skin in children and adults In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:130–47.
- Freyer DR, Kennedy R, Bostrom BC, Kohut G, Dehner LP. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr 1996;129(2):227–37.
- Orsey A, Paessler M, Lange BJ, Nichols KE. Central nervous system juvenile xanthogranuloma with malignant transformation. Pediatr Blood Cancer 2008;50(4):927–30.
- Hu WK, Gilliam AC, Wiersma SR, Dahms BB. Fatal congenital systemic juvenile xanthogranuloma with liver failure. Pediatr Dev Pathol 2004;7(1):71–6.
- Weitzman S, Whitlock J. Uncommon histiocytic disorder: the non-Langerhans cell histiocytoses. In: Weitzman S, Egeler RM, eds. Histiocytic Disorders in Children and Adults. Cambridge: Cambridge University Press, 2005:153–170.
- Stover DG, Alapati S, Regueira O, Turner C, Whitlock JA. Treatment of juvenile xanthogranuloma. Pediatr Blood Cancer 2008;51(1):130–3.
- Nakatani T, Morimoto A, Kato R, et al. Successful treatment of congenital systemic juvenile xanthogranuloma with Langerhans cell histiocytosis-based chemotherapy. J Pediatr Hematol Oncol 2004;26(6):371–4.
- Rajendra B, Duncan A, Parslew R, Pizer BL. Successful treatment of central nervous system juvenile xanthogranulomatosis with cladribine. Pediatr Blood Cancer 2009;52(3):413–15.
- Bailey KM, Castle VP, Hummel JM, Piert M, Moyer J, McAllister-Lucas LM. Thalidomide therapy for aggressive histiocytic lesions in the pediatric population. J Pediatr Hematol Oncol 2012;34(6):480–3.
- Simko SJ, Tran HD, Jones J, et al. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai–Dorfman disease. Pediatr Blood Cancer 2014;61(3):479–87.
Benign cephalic histiocytosis
- Zelger BW, Cerio R. Xanthogranuloma is the archetype of non-Langerhans cell histiocytoses. Br J Dermatol 2001;145(2):369–71.
- Sidwell RU, Francis N, Slater DN, Mayou SC. Is disseminated juvenile xanthogranulomatosis benign cephalic histiocytosis? Pediatr Dermatol 2005;22(1):40–3.
- Jih DM, Salcedo SL, Jaworsky C. Benign cephalic histiocytosis: a case report and review. J Am Acad Dermatol 2002;47(6):908–13.
Generalized eruptive histiocytosis
- Jang KA, Ahn SJ, Choi JH, Sung KJ, Moon KC, Koh JK. Histiocytic disorders with spontaneous regression in infancy. Pediatr Dermatol 2000;17(5):364–8.
- Wee SH, Kim HS, Chang SN, Kim DK, Park WH. Generalized eruptive histiocytoma: a pediatric case. Pediatr Dermatol 2000;17(6):453–5.
- Misery L, Kanitakis J, Hermier C, Cambazard F. Generalized eruptive histiocytoma in an infant with healing in summer: long-term follow-up. Br J Dermatol 2001;144(2):435–7.
- Lan Ma H, Metze D, Luger TA, Steinhoff M. Successful treatment of generalized eruptive histiocytoma with PUVA. J Dtsch Dermatol Ges 2007;5(2):131–4.
- Deng YJ, Hao F, Zhou CL, et al. Generalized eruptive histiocytosis: a possible therapeutic cure? Br J Dermatol 2004;150(1):171–3.
Papular xanthoma
- Caputo R, Gianni E, Imondi D, Carminati G, Gianotti R. Papular xanthoma in children. J Am Acad Dermatol 1990;22(6 Pt 1):1052–6.
- Fonseca E, Contreras F, Cuevas J. Papular xanthoma in children: report and immunohistochemical study. Pediatr Dermatol 1993;10(2):139–41.
- Chen CG, Chen CL, Liu HN. Primary papular xanthoma of children: a clinicopathologic, immunohistopathologic and ultrastructural study. Am J Dermatopathol 1997;19(6):596–601.
- Breier F, Zelger B, Reiter H, Gschnait F, Zelger BW. Papular xanthoma: a clinicopathological study of 10 cases. J Cutan Pathol 2002;29(4):200–6.
Progressive nodular histiocytosis
- Zelger BW, Cerio R. Xanthogranuloma is the archetype of non-Langerhans cell histiocytoses. Br J Dermatol 2001;145(2):369–71.
- Beswick SJ, Kirk JM, Bradshaw K, Sanders DS, Moss C. Progressive nodular histiocytosis in a child with a hypothalamic tumor. Br J Dermatol 2002;146(1):138–40.
- Kunimoto K, Uede K, Furukawa F. Progressive nodular histiocytosis. J Dermatol 2010;37(12):1071–3.
- Zelger BW, Staudacher C, Orchard G, Wilson-Jones E, Burgdorf WH. Solitary and generalized variants of spindle cell xanthogranuloma (progressive nodular histiocytosis). Histopathology 1995;27(1):11–19.
- Luftl M, Seybold H, Simon M, Jr, Burgdorf W. [Progressive nodular histiocytosis – rare variant of cutaneous non-Langerhans cell histiocytosis.] J Dtsch Dermatol Ges 2006;4(3):236–8.
- Nofal A, Assaf M, Tawfik A, et al. Progressive nodular histiocytosis: a case report and literature review. Int J Dermatol 2011;50(12):1546–51.
- Chu AC. The confusing state of the histiocytoses. Br J Dermatol 2000;143(3):475–6.
Xanthoma disseminatum
- Calverly DC, Wismer J, Rosenthal D, deSa D, Barr RD. Xanthoma disseminatum in an infant with skeletal and marrow involvement. J Pediatr Hematol Oncol 1995;17(1):61–5.
- Toberer F, Hartschuh W, Witzens-Harig M, Enk AH, Lonsdorf AS. Intertriginous orange-to-brownish papules and plaques: a quiz. Xanthoma disseminatum. Acta Derm Venereol 2013;93(4):493–4.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol 2008;58(Suppl. 2):S47–9.
- Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-Langerhans cell histiocytoses. Pediatr Blood Cancer 2005;45(3):256–64.
- Davies CW, Marren P, Juniper MC, Gray W, Wojnorowska F, Benson MK. Xanthoma disseminatum with respiratory tract involvement and fatal outcome. Thorax 2000;55(2):170–2.
- Ozcelik U, Dogru D, Akcoren Z, Coskun T, Kaya S, Gocmen A. Xanthoma disseminatum: a child with respiratory system involvement and bronchiectasis. Pediatr Pulmonol 2005;39(1):84–7.
- Chepuri NB, Challa VR. Xanthoma disseminatum: a rare intracranial mass. Am J Neuroradiol 2003;24(1):105–8.
- Zak IT, Altinok D, Neilsen SS, Kish KK. Xanthoma disseminatum of the central nervous system and cranium. Am J Neuroradiol 2006;27(4):919–21.
- Carpo BG, Grevelink SV, Brady S, Gellis S, Grevelink JM. Treatment of cutaneous lesions of xanthoma disseminatum with a CO2 laser. Dermatol Surg 1999;25(10):751–4.
- Seaton ED, Pillai GJ, Chu AC. Treatment of xanthoma disseminatum with cyclophosphamide. Br J Dermatol 2004;150(2):346–9.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol 2011;147(4):459–64.
- Lee EH, Kang TW, Kim SC. Successful treatment of xanthoma disseminatum with simvastatin. J Dermatol 2011;38(10):1015–17.
Diffuse plane xanthomatosis
- Hofmann M, Zappel K, Trefzer U, et al. Diffuse normolipemic plane xanthoma in a 9-year-old boy. Pediatr Dermatol 2005;22(2):127–9.
- Cambiaghi S, Boneschi V, Maffeis L, Gelmetti C. Diffuse normolipemic plane xanthoma in a child with common variable immunodeficiency. Pediatr Dermatol 2011;28(1):65–6.
- Marcoval J, Moreno A, Bordas X, Gallardo F, Peyri J. Diffuse plane xanthoma: clinicopathologic study of 8 cases. J Am Acad Dermatol 1998;39(3):439–42.
- Rosmaninho A, Fernandes I, Guimas A, Amorim I, Selores M. Diffuse plane xanthomatosis associated with monoclonal gammopathy. An Bras Dermatol 2011;86(4 Suppl. 1):S50–2.
- Bragg J. Diffuse plane xanthomata. Dermatol Online J 2005;11(4):4.
- Lorenz S, Hohenleutner S, Hohenleutner U, Landthaler M. Treatment of diffuse plane xanthoma of the face with the erbium:YAG laser. Arch Dermatol 2001;137(11):1413–15.
Erdheim–Chester disease
- Zelger B, Burgdorf WH. The cutaneous ‘histiocytoses’. Adv Dermatol 2001;17:77–114.
- Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim–Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996;75(3):157–69.
- Wilejto M, Abla O. Langerhans cell histiocytosis and Erdheim–Chester disease. Curr Opin Rheumatol 2012;24(1):90–6.
- Emile JF, Charlotte F, Amoura Z, Haroche J. BRAF mutations in Erdheim–Chester disease. J Clin Oncol 2013;31(3):398.
- Volpicelli ER, Doyle L, Annes JP, et al. Erdheim–Chester disease presenting with cutaneous involvement: a case report and literature review. J Cutan Pathol 2011;38(3):280–5.
- Arnaud L, Pierre I, Beigelman-Aubry C, et al. Pulmonary involvement in Erdheim–Chester disease: a single-center study of thirty-four patients and a review of the literature. Arthritis Rheum 2010;62(11):3504–12.
- Rush WL, Andriko JA, Galateau-Salle F, et al. Pulmonary pathology of Erdheim–Chester disease. Mod Pathol 2000;13(7):747–54.
- Haroche J, Amoura Z, Dion E, et al. Cardiovascular involvement, an overlooked feature of Erdheim–Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore) 2004;83(6):371–92.
- Lachenal F, Cotton F, Desmurs-Clavel H, et al. Neurological manifestations and neuroradiological presentation of Erdheim–Chester disease: report of 6 cases and systematic review of the literature. J Neurol 2006;253(10):1267–77.
- Arnaud L, Hervier B, Neel A, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim–Chester disease: a multicenter survival analysis of 53 patients. Blood 2011;117(10):2778–82.
- Devouassoux G, Lantuejoul S, Chatelain P, Brambilla E, Brambilla C. Erdheim–Chester disease: a primary macrophage cell disorder. Am J Respir Crit Care Med 1998;157(2):650–3.
- Gupta A, Kelly B, McGuigan JE. Erdheim–Chester disease with prominent pericardial involvement: clinical, radiologic, and histologic findings. Am J Med Sci 2002;324(2):96–100.
- Braiteh F, Boxrud C, Esmaeli B, Kurzrock R. Successful treatment of Erdheim–Chester disease, a non-Langerhans-cell histiocytosis, with interferon-alpha. Blood 2005;106(9):2992–4.
- Haroche J, Amoura Z, Trad SG, et al. Variability in the efficacy of interferon-alpha in Erdheim–Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum 2006;54(10):3330–6.
- Haroche J, Amoura Z, Charlotte F, et al. Imatinib mesylate for platelet-derived growth factor receptor-beta-positive Erdheim–Chester histiocytosis. Blood 2008;111(11):5413–15.
- Janku F, Amin HM, Yang D, Garrido-Laguna I, Trent JC, Kurzrock R. Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol 2010;28(31):e633–6.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim–Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121(9):1495–500.
Reticulohistiocytoma
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol 2006;30(4):521–8.
- Zelger B, Cerio R, Soyer HP, Misch K, Orchard G, Wilson-Jones E. Reticulohistiocytoma and multicentric reticulohistiocytosis. Histopathologic and immunophenotypic distinct entities. Am J Dermatopathol 1994;16(6):577–84.
Familial sea-blue histiocytosis
- Newman B, Hu W, Nigro K, Gilliam AC. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007;56(2):302–16.
- Bigorgne C, Le Tourneau A, Vahedi K, et al. Sea-blue histiocyte syndrome in bone marrow secondary to total parenteral nutrition. Leuk Lymphoma 1998;28(5–6):523–9.
Hereditary progressive mucinous histiocytosis
- Antoni-Bach N, Pfister R, Grosshans E, et al. [Hereditary progressive mucinous histiocytosis.] Ann Dermatol Venereol 2000;127(4):400–4.
- Schroder K, Hettmannsperger U, Schmuth M, Orfanos CE, Goerdt S. Hereditary progressive mucinous histiocytosis. J Am Acad Dermatol 1996;35(2 Pt 2):298–303.
- Wong D, Killingsworth M, Crosland G, Kossard S. Hereditary progressive mucinous histiocytosis. Br J Dermatol 1999;141(6):1101–5.
- Young A, Olivere J, Yoo S, Martins C, Barrett T. Two sporadic cases of adult-onset progressive mucinous histiocytosis. J Cutan Pathol 2006;33(2):166–70.
Malakoplakia
- Hyun KH, Shin HD, Kim DH. Malakoplakia in a healthy young female patient. Korean J Intern Med 2013;28(4):475–80.
- Lee WS, Chen WY, Ou TY, Chen FL, Lin YW. Malakoplakia in a patient with complicated urinary tract infection caused by extended-spectrum beta-lactamase-producing Escherichia coli. J Microbiol Immunol Infect 2014;Feb 13:1–2.
- Long JP, Jr, Althausen AF. Malacoplakia: a 25-year experience with a review of the literature. J Urol 1989;141(6):1328–31.
- Yousef GM, Naghibi B, Hamodat MM. Malakoplakia outside the urinary tract. Arch Pathol Lab Med 2007;131(2):297–300.
- Kohl SK, Hans CP. Cutaneous malakoplakia. Arch Pathol Lab Med 2008;132(1):113–17.
- Afonso JP, Ando PN, Padilha MH, Michalany NS, Porro AM. Cutaneous malakoplakia: case report and review. An Bras Dermatol 2013;88(3):432–7.
- Van der Voort HJ, ten Velden JA, Wassenaar RP, Silberbusch J. Malacoplakia. Two case reports and a comparison of treatment modalities based on a literature review. Arch Intern Med 1996;156(5):577–83.
Necrobiotic xanthogranuloma
- Rose A, Robinson M, Kamino H, Latkowski JA. Necrobiotic xanthogranuloma. Dermatol Online J 2012;18(12):30.
- Hallermann C, Tittelbach J, Norgauer J, Ziemer M. Successful treatment of necrobiotic xanthogranuloma with intravenous immunoglobulin. Arch Dermatol 2010;146(9):957–60.
- Balagula Y, Straus DJ, Pulitzer MP, Lacouture ME. Necrobiotic xanthogranuloma associated with immunoglobulin M paraproteinemia in a patient with Waldenstrom macroglobulinemia. J Clin Oncol 2011;29(11):e305–7.
- Zelger B, Eisendle K, Mensing C. Detection of spirochetal micro-organisms by focus-floating microscopy in necrobiotic xanthogranuloma. J Am Acad Dermatol 2007;57(6):1026–30.
- Inthasotti S, Wanitphakdeedecha R, Manonukul J. A 7-year history of necrobiotic xanthogranuloma following asymptomatic multiple myeloma: a case report. Dermatol Res Pract 2011;2011:1–5.
- Szalat R, Arnulf B, Karlin L, et al. Pathogenesis and treatment of xanthomatosis associated with monoclonal gammopathy. Blood 2011;118(14):3777–84.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol 2009;48(1):1–10.
- Wood AJ, Wagner MV, Abbott JJ, Gibson LE. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol 2009;145(3):279–84.
- Efebera Y, Blanchard E, Allam C, Han A, Lee S, Munshi N. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk 2011;11(3):298–302.
- Kossard S, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinemia. J Am Acad Dermatol 1980;3(3):257–70.
- Shah KC, Poonnoose SI, George R, Jacob M, Rajshekhar V. Necrobiotic xanthogranuloma with cutaneous and cerebral manifestations. Case report and review of the literature. J Neurosurg 2004;100(6):1111–14.
- Stork J, Kodetova D, Vosmik F, Krejca M. Necrobiotic xanthogranuloma presenting as a solitary tumor. Am J Dermatopathol 2000;22(5):453–6.
- Ryan E, Warren LJ, Szabo F. Necrobiotic xanthogranuloma: response to chlorambucil. Australas J Dermatol 2012;53(2):e23–5.
- Vieira V, Del Pozo, Martínez W, et al. Necrobiotic xanthogranuloma associated with lymphoplasmacytic lymphoma. Palliative treatment with carbon dioxide laser. Eur J Dermatol 2005;15(3):182–5.
Multicentric reticulohistiocytosis
- Gorman JD, Danning C, Schumacher HR, Klippel JH, Davis JC, Jr. Multicentric reticuuplohistiocytosis: case report with immunohistochemical analysis and literature review. Arthritis Rheum 2000;43(4):930–8.
- Trotta F, Colina M. Multicentric reticulohistiocytosis and fibroblastic rheumatism. Best Pract Res Clin Rheumatol 2012;26(4):543–57.
- Outland JD, Keiran SJ, Schikler KN, Callen JP. Multicentric reticulohistiocytosis in a 14-year-old girl. Pediatr Dermatol 2002;19(6):527–31.
- Candell Chalom E, Elenitsas R, Rosenstein ED, Kramer N. A case of multicentric reticulohistiocytosis in a 6-year-old child. J Rheumatol 1998;25(4):794–7.
- Havill S, Duffill M, Rademaker M. Multicentric reticulohistiocytosis in a child. Australas J Dermatol 1999;40(1):44–6.
- Pacheco-Tena C, Reyes-Cordero G, Ochoa-Albiztegui R, Rios-Barrera V, Gonzalez-Chavez SA. Treatment of multicentric reticulohistiocytosis with tocilizumab. J Clin Rheumatol 2013;19(5):272–6.
- Fortier-Beaulieu M, Thomine E, Boullie MC, Le Loet X, Lauret P, Hemet J. New electron microscopic findings in a case of multicentric reticulohistiocytosis. Long spacing collagen inclusions. Am J Dermatopathol 1993;15(6):587–9.
- Luz FB, Gaspar AP, Ramos-e-Silva M, et al. Immunohistochemical profile of multicentric reticulohistiocytosis. Skinmed 2005;4(2):71–7.
- Gomez-Mateo Mdel C, Monteagudo C. Nonepithelial skin tumors with multinucleated giant cells. Semin Diagn Pathol 2013;30(1):58–72.
- Campbell DA, Edwards NL. Multicentric reticulohistiocytosis: systemic macrophage disorder. Baillieres Clin Rheumatol 1991;5(2):301–19.
- Kishikawa T, Miyashita T, Fujiwara E, et al. Multicentric reticulohistiocytosis associated with ovarian cancer. Mod Rheumatol 2007;17(5):422–5.
- Malik MK, Regan L, Robinson-Bostom L, Pan TD, McDonald CJ. Proliferating multicentric reticulohistiocytosis associated with papillary serous carcinoma of the endometrium. J Am Acad Dermatol 2005;53(6):1075–9.
- Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and immunophenotypic study. Br J Dermatol 1995;133(1):71–6.
- Yee KC, Bowker CM, Tan CY, Palmer RG. Cardiac and systemic complications in multicentric reticulohistiocytosis. Clin Exp Dermatol 1993;18(6):555–8.
- Lovelace K, Loyd A, Adelson D, Crowson N, Taylor JR, Cornelison R. Etanercept and the treatment of multicentric reticulohistiocytosis. Arch Dermatol 2005;141(9):1167–8.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, Ramos-e-Silva M. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol 2001;15(6):524–31.
- Liang GC, Granston AS. Complete remission of multicentric reticulohistiocytosis with combination therapy of steroid, cyclophosphamide, and low-dose pulse methotrexate. Case report, review of the literature, and proposal for treatment. Arthritis Rheum 1996;39(1):171–4.
- Lonsdale-Eccles AA, Haworth AE, McCrae FC, Young-Min SA. Successful treatment of multicentric reticulohistiocytosis with leflunomide. Br J Dermatol 2009;161(2):470–2.
- Fedler R, Frantzmann Y, Schwarze EW, Frosch PJ. [Multicenter reticulohistiocytosis. Therapy with azathioprine and prednisolone.] Hautarzt 1995;46(2):118–20.
- Satoh M, Oyama N, Yamada H, Nakamura K, Kaneko F. Treatment trial of multicentric reticulohistiocytosis with a combination of predonisolone, methotrexate and alendronate. J Dermatol 2008;35(3):168–71.
- Mavragani CP, Batziou K, Aroni K, Pikazis D, Manoussakis MN. Alleviation of polyarticular syndrome in multicentric reticulohistiocytosis with intravenous zoledronate. Ann Rheum Dis 2005;64(10):1521–2.
- Codriansky K, Rünger TM, Bhawan J, et al. Multicentric reticulohistiocytosis: a systemic osteoclastic disease? Arthritis Rheum 2008;59 (3):444–8.
Sinus histiocytosis with massive lymphadenopathy
- Juskevicius R, Finley JL. Rosai–Dorfman disease of the parotid gland: cytologic and histopathologic findings with immunohistochemical correlation. Arch Pathol Lab Med 2001;125(10):1348–50.
- Deodhare SS, Ang LC, Bilbao JM. Isolated intracranial involvement in Rosai–Dorfman disease: a report of two cases and review of the literature. Arch Pathol Lab Med 1998;122(2):161–5.
- Lauwers GY, Perez-Atayde A, Dorfman RF, Rosai J. The digestive system manifestations of Rosai–Dorfman disease (sinus histiocytosis with massive lymphadenopathy): review of 11 cases. Hum Pathol 2000;31(3):380–5.
- Grabczynska SA, Toh CT, Francis N, Costello C, Bunker CB. Rosai–Dorfman disease complicated by autoimmune haemolytic anaemia: case report and review of a multisystem disease with cutaneous infiltrates. Br J Dermatol 2001;145(2):323–6.
- Foss HD, Herbst H, Araujo I, et al. Monokine expression in Langerhans' cell histiocytosis and sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease). J Pathol 1996;179(1):60–5.
- Newman B, Hu W, Nigro K, Gilliam AC. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007;56(2):302–16.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): review of the entity. Semin Diagn Pathol 1990;7(1):19–73.
- Perez A, Rodriguez M, Febrer I, Aliaga A. Sinus histiocytosis confined to the skin. Case report and review of the literature. Am J Dermatopathol 1995;17(4):384–8.
- Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai–Dorfman disease isolated to the central nervous system: a report of 11 cases. Mod Pathol 2001;14(3):172–8.
- Goodnight JW, Wang MB, Sercarz JA, Fu YS. Extranodal Rosai–Dorfman disease of the head and neck. Laryngoscope 1996;106(3 Pt 1):253–6.
- Pulsoni A, Anghel G, Falcucci P, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): report of a case and literature review. Am J Hematol 2002;69(1):67–71.
- Lohr HF, Godderz W, Wolfe T, et al. Long-term survival in a patient with Rosai–Dorfman disease treated with interferon-alpha. Eur J Cancer 1995;31A(13–14):2427–8.
- Palomera L, Domingo J, Soria J, Gutierrez M. [Long term survival in a patient with aggressive Rosai–Dorfman disease treated with interferon alpha.] Med Clin (Barc) 2001;116(20):797–8.
- Baildam EM, Ewing CI, D'Souza SW, Stevens RF. Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): response to acyclovir. J R Soc Med 1992;85(3):179–80.
Malignant histiocytosis
- Low SE, Stafford JS. Malignant histiocytosis: a case report of a rare tumour presenting with spontaneous splenic rupture. J Clin Pathol 2006;59(7):770–2.
- Rilke F, Carbone A, Musumeci R, Pilotti S, De Lena M, Bonadonna G. Malignant histiocytosis: a clinicopathologic study of 18 consecutive cases. Tumori 1978;64(2):211–27.
- Ornvold K, Nielsen MH, Clausen N. Malignant histiocytosis in childhood. Clinicopathological study of 4 cases. Acta Pathol Microbiol Immunol Scand A 1986;94(4):291–6.
- Warnke RA, Kim H, Dorfman RF. Malignant histiocytosis (histiocytic medullary reticulosis). I. Clinicopatholigic study of 29 cases. Cancer 1975;35(1):215–30.
- Amsel S, Bijlsma F. Histiocytic medullary reticulosis. Clinical and pathological studies in Uganda. Trop Geogr Med 1974;26(1):31–8.
- Molyneux ME, Tozer RA, Hutt MS. Histiocytic medullary reticulosis in Africa. Lancet 1978;2(8101):1202–3.
- Jain S, Kanarek N, Arceci RJ. Analysis of histiocytosis deaths in the US and recommendations for incidence tracking. J Registry Manag 2010;37(4):156–62.
- Carbone A, Micheau C, Caillaud JM, Carlu C. A cytochemical and immunohistochemical approach to malignant histiocytosis. Cancer 1981;47(12):2862–71.
- Van Heerde P, Feltkamp CA, Hart AA, Somers R, van Unnik JA, Vroom TM. Malignant histiocytosis and related tumors. A clinicopathologic study of 42 cases using cytological, histochemical and ultrastructural parameters. Hematol Oncol 1984;2(1):13–32.
- Nemes Z, Thomazy V. Diagnostic significance of histiocyte-related markers in malignant histiocytosis and true histiocytic lymphoma. Cancer 1988;62(9):1970–80.
- Kamesaki H, Koya M, Miwa H, et al. Malignant histiocytosis with rearrangement of the heavy chain gene and evidence of monocyte–macrophage lineage. Cancer 1988;62(7):1306–9.
- Morgan R, Smith SD, Hecht BK, et al. Lack of involvement of the c-fms and N-myc genes by chromosomal translocation t(2;5)(p23;q35) common to malignancies with features of so-called malignant histiocytosis. Blood 1989;73(8):2155–64.
- Newman B, Hu W, Nigro K, Gilliam AC. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol 2007;56(2):302–16.
- Ducatman BS, Wick MR, Morgan TW, Banks PM, Pierre RV. Malignant histiocytosis: a clinical, histologic, and immunohistochemical study of 20 cases. Hum Pathol 1984;15(4):368–77.
- Bucsky P, Egeler RM. Malignant histiocytic disorders in children. Clinical and therapeutic approaches with a nosologic discussion. Hematol Oncol Clin North Am 1998;12(2):465–71.
- Brugieres L, Caillaud JM, Patte C, et al. Malignant histiocytosis: therapeutic results in 27 children treated with a single polychemotherapy regimen. Med Pediatr Oncol 1989;17(3):193–6.
True histiocytic lymphoma
- Kerl H, Cerroni L. Primary cutaneous B-cell lymphomas: then and now. J Cutan Pathol 2006;33(Suppl. 1):1–5.
- Nemes Z, Thomazy V. Diagnostic significance of histiocyte-related markers in malignant histiocytosis and true histiocytic lymphoma. Cancer 1988;62(9):1970–80.
- Soria C, Orradre JL, Garcia-Almagro D, Martinez B, Algara P, Piris MA. True histiocytic lymphoma (monocytic sarcoma). Am J Dermatopathol 1992;14(6):511–17.
- Alexiev BA, Sailey CJ, McClure SA, Ord RA, Zhao XF, Papadimitriou JC. Primary histiocytic sarcoma arising in the head and neck with predominant spindle cell component. Diagn Pathol 2007;2:7.
- Forestier JY, Schmitt D, Thivolet J. [Cutaneous histiocytosarcoma: clinical, immunohistochemical and ultrastructural aspects; difficulties of study and problems of nosology (author's translation).] Ann Dermatol Venereol 1980;107(1–2):7–19.
- Esteve J, Rozman M, Campo E, Munoz F, Urbano-Ispizua A, Rozman C. Leukemia after true histiocytic lymphoma: another type of acute monocytic leukemia with histiocytic differentiation (AML-M5c)? Leukemia 1995;9(8):1389–91.
Histiocytic sarcoma
- Mainardi C, D'Amore ES, Pillon M, Toffolutti T, Rosolen A. A case of resistant pediatric histiocytic sarcoma successfully treated with chemo-radiotherapy and autologous peripheral blood stem cell transplant. Leuk Lymphoma 2011;52(7):1367–71.
- Black J, Coffin CM, Dehner LP. Fibrohistiocytic tumors and related neoplasms in children and adolescents. Pediatr Dev Pathol 2012;15(Suppl. 1):181–210.
- Pileri SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002;41(1):1–29.
- Castro EC, Blazquez C, Boyd J, et al. Clinicopathologic features of histiocytic lesions following ALL, with a review of the literature. Pediatr Dev Pathol 2010;13(3):225–37.
- Kumar R, Khan SP, Joshi DD, Shaw GR, Ketterling RP, Feldman AL. Pediatric histiocytic sarcoma clonally related to precursor B-cell acute lymphoblastic leukemia with homozygous deletion of CDKN2A encoding p16INK4A. Pediatr Blood Cancer 2011;56(2):307–10.
- Wang E, Hutchinson CB, Huang Q, et al. Histiocytic sarcoma arising in indolent small B-cell lymphoma: report of two cases with molecular/genetic evidence suggestive of a ‘transdifferentiation’ during the clonal evolution. Leuk Lymphoma 2010;51(5):802–12.
- Zeng W, Meck J, Cheson BD, Ozdemirli M. Histiocytic sarcoma transdifferentiated from follicular lymphoma presenting as a cutaneous tumor. J Cutan Pathol 2011;38(12):999–1003.
- Shao H, Xi L, Raffeld M, et al. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol 2011;24(11):1421–32.
- Hure MC, Elco CP, Ward D, et al. Histiocytic sarcoma arising from clonally related mantle cell lymphoma. J Clin Oncol 2012;30(5):e49–53.
- Michonneau D, Kaltenbach S, Derrieux C, et al. BRAFV600E mutation in a histiocytic sarcoma arising from hairy cell leukemia. J Clin Oncol 2014;32:1–5.
- DeMent SH. Association between mediastinal germ cell tumors and hematologic malignancies: an update. Hum Pathol 1990;21(7):699–703.
- Wu W, Tanrivermis Sayit A, Vinters HV, Pope W, Mirsadraei L, Said J. Primary central nervous system histiocytic sarcoma presenting as a postradiation sarcoma: case report and literature review. Hum Pathol 2013;44(6):1177–83.
- Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol 2004;28(9):1133–44.
- Bucsky P, Egeler RM. Malignant histiocytic disorders in children. Clinical and therapeutic approaches with a nosologic discussion. Hematol Oncol Clin North Am 1998;12(2):465–71.
- Gergis U, Dax H, Ritchie E, Marcus R, Wissa U, Orazi A. Autologous hematopoietic stem-cell transplantation in combination with thalidomide as treatment for histiocytic sarcoma: a case report and review of the literature. J Clin Oncol 2011;29(10):e251–3.
- Shukla N, Kobos R, Renaud T, et al. Successful treatment of refractory metastatic histiocytic sarcoma with alemtuzumab. Cancer 2012;118(15):3719–24.
- Bailey KM, Castle VP, Hummel JM, Piert M, Moyer J, McAllister-Lucas LM. Thalidomide therapy for aggressive histiocytic lesions in the pediatric population. J Pediatr Hematol Oncol 2012;34(6):480–3.
- Dalle JH, Leblond P, Decouvelaere A, et al. Efficacy of thalidomide in a child with histiocytic sarcoma following allogeneic bone marrow transplantation for T-ALL. Leukemia 2003;17(10):2056–7.
- Schlick K, Aigelsreiter A, Pichler M, et al. Histiocytic sarcoma – targeted therapy: novel therapeutic options? A series of 4 cases. Onkologie 2012;35(7–8):447–50.