CHAPTER 137
Soft-tissue Tumours and Tumour-like Conditions
Eduardo Calonje
St John's Institute of Dermatology, Guy's and St Thomas' NHS Foundation Trust, London, UK
Introduction and general description
For many clinical dermatologists, soft-tissue tumours arising in the dermis, subcutis or deeper soft tissues are a confusing group of lesions. This is probably partly explained by the facts that there is a very long list of soft-tissue tumours, and that a large majority of these can arise in the skin or affect it secondarily. Most of these tumours have no characteristic clinical appearance, and present as non-specific, dermal or deep-seated nodules. However, it is necessary for all clinical dermatologists to have an understanding of the range of tumours that may arise in the dermis, and also of the likely biological behaviour of individual lesions. Although cutaneous malignant soft-tissue tumours are rare, many benign lesions may be histologically confused with a malignancy. Furthermore, there is a group of soft-tissue tumours that have low-grade malignant potential (intermediate malignancy) with frequent local recurrences but little or no potential for metastatic spread (e.g. dermatofibrosarcoma protuberans (DFSP)). These tumours may cause important morbidity, and their recognition is therefore essential for the planning of treatment and follow-up. Recognizing a wide range of soft-tissue tumours is also important as a number of these lesions—particularly when multiple—may be markers of genetic syndromes (e.g. multiple neurofibromas and plexiform neurofibroma in neurofibromatosis type 1).
A broad division can be made between tumours according to the morphological lines of differentiation. The latter include fibroblastic, myofibroblastic, neural, vascular, muscular and adipocytic types. In a number of tumours, the line of differentiation is not clear, as a normal cell of origin cannot be identified (e.g. epithelioid sarcoma). In a still larger group of tumours, their origin is descriptively ascribed to fibrohistiocytic cells, but with mounting evidence that many of these lesions have fibroblast and/or myofibroblastic differentiation and almost none display true histiocytic differentiation. The list of tumours discussed in this chapter is not all inclusive. For a full account of the very wide range of these tumours, the reader is referred to the standard major works in this field [1, 2]. True histiocytic tumours (see Chapter 136), and keloids and hypertrophic scars (see Chapter 96) and metastatic malignant tumours (see Chapter 147) are covered elsewhere.
The most useful biological triage is into totally benign lesions; lesions that may recur locally but never or almost never metastasize; and those that are truly malignant and may metastasize. The great majority of dermal or superficial soft-tissue tumours come into the first two categories, whilst truly malignant soft-tissue tumours much more frequently arise below the deep fascia. In the case of these rare malignant tumours, there is a relationship between bulk and prognosis, smaller lesions carrying a better prognosis. More superficially situated lesions tend to carry a better prognosis than those deeply situated. Mitoses (particularly abnormal mitotic figures) and necrosis both tend to be associated with malignant rather than benign lesions.
The usual clinical presentation of many of the tumours described in this chapter is of a non-specific lump or nodule. An incisional biopsy should be arranged, and it must be adequately deep so that the nature of the lesion at its deepest margin can be determined. Once the pathologist has established the nature of the tumour, appropriate definitive surgery can be planned. Prior consultation with the pathologist is strongly recommended, as samples may be needed for cytogenetics, electron microscopy or immunohistochemistry. All of these may be helpful in arriving at an accurate diagnosis.
FIBROUS AND MYOFIBROBLASTIC TUMOURS
Fibrous papule of the face [1]
Definition and nomenclature
A small facial papule with a distinctive fibrovascular component on histological examination.
Epidemiology
Incidence and prevalence
Lesions are very common [2, 3, 4].
Age
Most patients are middle-aged adults.
Sex
Both sexes are equally affected.
Pathophysiology
It has been suggested that the condition may be a variant of a melanocytic naevus [2, 4], but others disagree [3]. S100 protein, is never present in lesional cells giving further support to the theory of a non-melanocytic proliferation.
Pathology [2, 3, 4]
The epidermis appears normal, although there may be an increased number of clear cells overlying the lesion. In the dermis, there is increased collagen with a hyalinized appearance and scattered, somewhat dilated, vascular channels (Figure 137.1). In the background, there is increased cellularity with mono- and multinucleated cells with a histiocyte-like appearance. In some lesions, epithelioid or clear cells and exceptionally granular cells may predominate [5, 6, 7, 8]. There are prominent dilated capillaries.
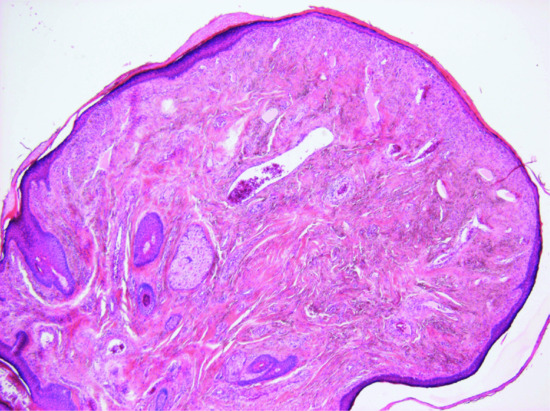
Figure 137.1 Fibrous papule with hyalinized collagen bundles and increased dilated vascular channels.
Clinical features [2, 3, 4]
History and presentation
The lesions usually occur singly on the nose. Occasionally, they may occur on the forehead, cheeks, chin or neck, and there may rarely be multiple. The papule develops slowly as a dome-shaped, skin-coloured or slightly red or pigmented lesion, which is usually sessile. Most are asymptomatic, but about one-third bleed on minor trauma.
Differential diagnosis
The main clinical consideration is that of an intradermal melanocytic naevus and less commonly, a basal cell carcinoma.
Management
The lesion is benign, but it may easily be excised usually by shave biopsy for cosmetic reasons.
Storiform collagenoma [1, 2]
Definition and nomenclature
Storiform collagenoma is a fibrous hypocellular cutaneous lesion which, when multiple, may be associated with Cowden disease or phosphatase and tensin homologue (PTEN) hamartoma syndrome (multiple hamartoma and neoplasia syndrome; see Chapter 147) [3].
Epidemiology
Incidence and prevalence
It is relatively rare.
Age
There is a wide age range with a predilection for adults [2, 3].
Sex
No sex predilection.
Pathophysiology
The aetiology of sporadic cases is unknown. In the setting of Cowden syndrome, the development of multiple lesions is associated with loss-of-function mutations in PTEN, leading to hyperactivity of the mTOR pathway.
Pathology
Storiform collagenoma typically consists of a fairly well-circumscribed dermal nodule with prominent hypocellular hyalinized collagen bundles in a storiform pattern (Figure 137.2). Bland spindle-shaped cells are rare. A similar histological pattern may be seen in the late stages of lesions as diverse as pleomorphic fibroma, fibrous histiocytoma (FH) and myofibroma and it has been proposed that it does not represent a distinctive entity but a reaction pattern [4, 5].
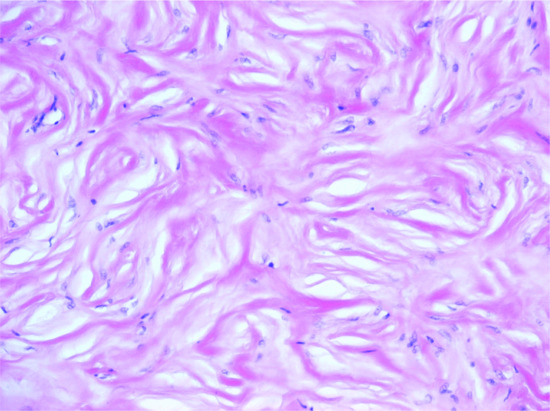
Figure 137.2 Storiform collagenoma. Poorly cellular stroma composed of hyalinized collagen in a storiform pattern.
A more cellular variant containing multinucleated bizarre cells has been described as giant cell collagenoma [6]. The latter is a potential link between pleomorphic fibroma (see later) and sclerotic fibroma as it has been proposed that both entities are part of the same spectrum [7].
Clinical features
History and presentation
Storiform collagenoma usually presents as a small, solitary, asymptomatic papule, with wide anatomical distribution.
Management
Simple excision is curative.
Pleomorphic fibroma
Definition [1]
Pleomorphic fibroma is a relatively rare lesion with features very similar to those of a fibroepithelial polyp (skin tag), but characterized histologically by bizarre mono- or multinucleated stromal cells.
Epidemiology
Incidence and prevalence
Pleomorphic fibroma is relatively rare.
Age
Mainly in adults.
Sex
No sex predilection.
Pathophysiology
The aetiology is unknown.
Pathology
Normal or mildly acanthotic epidermis surrounds a collagenous and vascular stroma containing scattered bizarre mono- or multinucleated cells with hyperchromatic and pleomorphic nuclei. Mitotic figures are rare.
Clinical features
History and presentation
Presentation is in the form of a lesion with clinical findings of a fibroepithelial polyp with wide anatomical distribution with some predilection for perianal skin and the face.
Management
Simple excision is curative, and there is no tendency for local recurrence.
Acquired digital fibrokeratoma [1]
Definition
A benign lesion, possibly a reaction to trauma, which occurs on the fingers and toes [2] (Figure 137.3), although the palms and the soles have occasionally been involved.
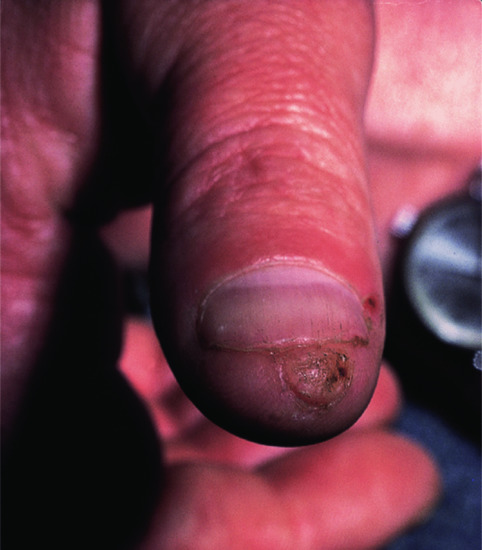
Figure 137.3 Clinical appearance of an acquired digital fibrokeratoma.
Epidemiology
Incidence and prevalence
The incidence is low.
Age
Adults are usually affected.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The histology shows thick collagen bundles, thin elastic fibres and increased vascularity. Occasionally, there is an obvious increase in fibroblasts, and rarely the collagen bundles may be separated by oedema [3]. The epidermis is relatively normal, but acanthosis and hyperkeratosis may occur.
Clinical features
History and presentation
The lesion usually occurs as a solitary dome-shaped lesion, with a collarette of slightly raised skin at its base. Occasionally, it may be elongated or pedunculated. Giant lesions may occasionally occur [4]. The surface may appear to be slightly warty.
Differential diagnosis
There is a wide clinical differential diagnosis, which includes dermatofibroma, viral wart, supernumerary digit and cutaneous horn. Histologically, the lesion is extremely similar to the Koenen tumour [5], the periungual fibrous lesion that arises from the nail fold in tuberous sclerosis.
Management
Simple excision is curative.
Nodular fasciitis [1, 2, 3, 4, 5]
Definition and nomenclature
A rapidly enlarging subcutaneous neoplasm due to a proliferation of myofibroblasts and fibroblasts and that histologically resembles a sarcoma.
Epidemiology
Incidence and prevalence
It is relatively common. The intravascular and cranial variants of fasciitis are rare [6, 7].
Age
It is more common in young adults but can occur at any age. Intravascular fasciitis is more common in young adults and cranial fasciitis tends to occur in children less than 2 years of age [6, 7].
Sex
There is no predilection for either sex except for cranial fasciitis that is more common in males [7].
Pathophysiology
Predisposing factors
There is no clear evidence that trauma initiates the lesions although trauma may play a role in cranial fasciitis [8].
Pathology [1, 2, 3, 4, 5]
These lesions may look extremely worrying in view of the high mitotic rate and rapid growth (see later). The tumour is only focally circumscribed and it is composed of bundles of fairly uniform fibroblasts and myofibroblasts with pink cytoplasm, vesicular nuclei and a single small nucleolus. Myxoid change and mucin deposition is often prominent, resulting in a typical tissue culture-like appearance (Figure 137.4). In the background, there are numerous small delicate blood vessels, extravasated red blood cells and scattered mononuclear inflammatory cells. Multinucleated giant cells may be seen, and they resemble osteoclasts. Mitoses are usually numerous, but there are no abnormal forms. Hyalinized collagen bundles are often present and may display a keloidal appearance. At the periphery, compact bundles of fibroblasts and capillaries probe the fascial planes and may infiltrate fat or skeletal muscle. It is not surprising that this histological picture is relatively often confused with that of a malignant tumour. Variants of nodular fasciitis include those with metaplastic bone (ossifying fasciitis); a variant that involves the periosteum (periosteal fasciitis); a variant that involves the scalp and tends to occur in children (cranial fasciitis) [6]; and a variant within the lumen of a blood vessel (intravascular fasciitis) [7, 9]. A rare variant of intradermal nodular fasciitis has also been described [10, 11]. Intra-articular location may also be seen [12]. Tumour cells are variably positive for smooth muscle actin and calponin [13] and usually negative for smooth muscle markers including desmin and h-caldesmon [14]. The histological diagnosis may be very difficult, especially in small biopsies. Confusion with a sarcoma or with fibromatosis are major pitfalls, with obvious detrimental consequences.
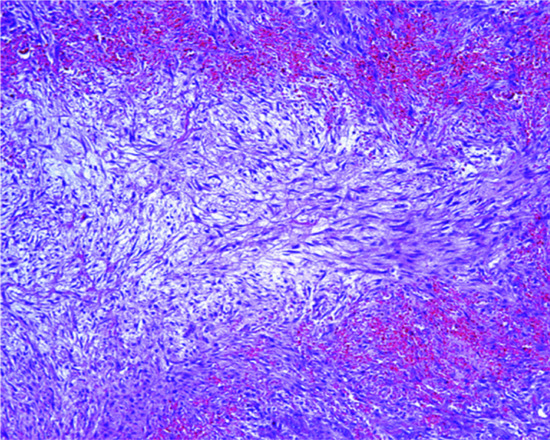
Figure 137.4 Typical tissue culture-like appearance of nodular fasciitis with prominent myxoid background.
Immunohistochemistry may be useful in the distinction between fibromatosis and nodular fasciitis. The former tend to display nuclear β-catenin positivity, while the latter are usually negative or display cytoplasmic positivity only [15]. However, some fibromatoses, especially those superficially located, are negative for this marker and the diagnosis should be based on careful clinicopathological correlation.
Genetics
The MYH9-USP6 fusion gene has consistently been identified in lesions confirming the neoplastic nature of this tumour [16]. Lesions like this with a self-limited life and a distinctive clonal genetic translocation have been referred to as transient neoplasms [16].
Clinical features [1, 2, 3, 4]
History and presentation
The majority of tumours appear as tender rapidly growing masses beneath the skin. The average size is 1–3 cm in diameter. The commonest situation is the upper extremities, particularly the forearm, but the lesion can occur anywhere, including the orbit and the mouth [9]. Lesions on the head and neck often present in children. In nearly half the patients, the tumour has been noticed for only 2 weeks or less when they come for advice.
Differential diagnosis
The rapid growth of the lesion may suggest a clinical diagnosis of malignancy.
Disease course and prognosis
Resolution usually follows incomplete surgical removal. Local recurrence is exceptional.
Management
Simple excision is therefore an adequate treatment.
Fibro-osseous pseudotumour of the digits [1, 2, 3]
Definition
This is a reactive myofibroblastic proliferation with bone formation, which occurs exclusively on the digits.
Epidemiology
Incidence and prevalence
It is rare.
Age
It presents predominantly in young adults although presentation can be at any age.
Sex
Males are more often affected than females.
Pathophysiology
Predisposing factors
Trauma appears to be an important factor in the development of the tumour.
Pathology
The tumour is ill defined and similar to nodular fasciitis, except for the fact that there is formation of osteoid and mature bone. Oedematous stroma, vascular proliferation and bundles of spindle-shaped myofibroblast-like cells are seen intermixed with osteoid and mature bone. Mitotic figures are found and their number depends on the age of the lesion.
Clinical features
History and presentation
The lesion grows rapidly and it is not attached to bone. The fingers are more commonly affected than the toes.
Disease course and prognosis
Local recurrence is rare.
Management
Simple excision is the treatment of choice.
Ischaemic fasciitis [1, 2, 3, 4, 5]
Definition and nomenclature
Ischaemic fasciitis is a reactive pseudosarcomatous fibroblastic/myofibroblastic proliferation that often occurs as a result of alterations in local circulation and sustained pressure in immobilized patients.
Epidemiology
Incidence and prevalence
Ischaemic fasciitis is relatively rare.
Age
Most patients are elderly usually between the seventh and ninth decades of life.
Sex
There is a slight predilection for males.
Pathophysiology
Predisposing factors
Persistent ischaemia and trauma to the affected area in immobilized patients is an important factor in the development of the lesion. However, in many cases there is no association with immobility or debilitation has been found [4].
Pathology
The lesion is poorly circumscribed and contains areas of fibrosis, vascular proliferation, necrosis and focal myxoid change. Thrombosed blood vessels with recanalization and areas of fibrinoid necrosis, focal haemorrhage and mononuclear inflammatory cells are additional features. In the background, there are variable numbers of spindle-shaped myofibroblasts/fibroblasts with vesicular or hyperchromatic nuclei and a prominent nucleolus. Mitotic figures may be seen, but are not prominent.
Clinical features
The lesion presents as an asymptomatic subcutaneous mass, predominantly over bony prominences that may extend to deeper soft tissues and to the overlying dermis.
Management
Excision of the lesion is an adequate treatment.
Fibrous hamartoma of infancy [1-5]
Definition
This is a benign, fibroblastic/myofibroblastic, deep dermal and subcutaneous tumour presenting in children and characterized by three distinctive pathological components, as described below.
Epidemiology
Incidence and prevalence
This is a rare tumour.
Age
The majority of cases present in children under the age of 2. A quarter of the cases present at birth.
Sex
Males are more affected than females.
Pathophysiology
Pathology
The tumour is composed of three components:
- Bundles of interlacing, elongated, bland, wavy spindle-shaped cells in a variable collagenous background.
- Nests of more immature round cells with focal myxoid change.
- Mature adipose tissue.
In a number of cases a focal pseudoangiomatous component is seen [6]. A focal resemblance to a neurofibroma may be seen when the first component predominates, but tumour cells are actin positive and S100 negative [7].
In the dermis overlying the tumour, eccrine glands may show secondary changes including hyperplasia, papillary projections and squamous syringometaplasia [8].
Genetics
Although usually considered to be a hamartoma, it is probably neoplastic in nature. This is further suggested by the presence of complex structural rearrangements demonstrated recently in a single case and involving chromosomes 1, 2, 4 and 17 [9].
Clinical features
History and presentation
Most cases present as an asymptomatic, solitary, skin-coloured plaque/nodule only a few centimetres in diameter. Exceptional tumours are very large and multifocal [10]. Rarely, pigmentary changes and/or hypertrichosis may be seen [11]. The tumour grows rapidly and has a predilection for the axillae, arm and shoulder girdle [1, 2, 3]. Rare cases occur on the head and neck [6]. A familial association has not been reported.
Disease course and prognosis
Local recurrence is exceptional.
Management
Simple excision is the treatment of choice [5]; recurrences are exceptional.
Calcifying fibrous tumour/pseudotumour [1,2,3]
Definition
This is a rare, benign, hypocellular tumour characterized by dense collagen bundles, areas of calcification and a patchy mononuclear cell infiltrate. This lesion has no relation with inflammatory myofibroblastic tumour as was originally suggested [3].
Epidemiology
Incidence and prevalence
This is very rare.
Age
Most lesions occur in children but rare cases may present in young adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The tumour typically consists of haphazardly arranged collagen bundles with scattered bland fibroblasts, focal small calcifications and focal aggregates of lymphocytes and plasma cells. Tumour cells are positive for CD34 and may be focally positive for smooth muscle actin and more rarely for desmin [3].
Clinical features
History and presentation
Lesions present as a fairly large subcutaneous or deeper asymptomatic mass with a wide anatomical distribution. Cases may also occur in internal organs [3].
Disease course and prognosis
Local recurrence is rare.
Management
The treatment of choice is simple excision.
Calcifying aponeurotic fibroma [1, 2]
Definition
This is a rare fibroblastic tumour characterized by a nodular proliferation of bland spindle-shaped cells surrounding nodules at different stages of calcification. Cartilage and, less commonly, bone formation may be seen.
Epidemiology
Incidence and prevalence
Tumours are very rare.
Age
Most cases present in children.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The growth pattern is multinodular. Tumour cells are elongated, with scanty pink cytoplasm, vesicular nuclei and very rare mitotic figures. Tumour nodules frequently contain areas of calcification, which are surrounded by tumour cells in a pattern reminiscent of palisading.
Clinical features
History and presentation
Lesions have a predilection for the hands and, less commonly, the feet. Occurrence at other sites is rare but tumours may present in places as diverse as the knee, back and thigh [1, 2]. Tumours are small, slowly growing and usually asymptomatic. Multiple lesions are exceptional [3].
Disease course and prognosis
Local recurrence is observed in 50% of cases but malignant transformation is exceptional [4].
Management
Simple excision is the treatment of choice.
Dermatomyofibroma
Definition and nomenclature [1-4]
Dermatomyofibroma presents as a benign, dermal and superficial subcutaneous myofibroblastic proliferation microscopically mimicking a fibromatosis. The tumour, however, has no potential for local recurrence and lacks an infiltrative growth pattern.
Epidemiology
Incidence and prevalence
Dermatomyofibroma is relatively rare.
Age
Most patients are young adults with children only exceptionally affected [5, 6, 7].
Sex
There is predilection for females.
Pathophysiology
Pathology
Low-power examination reveals a plaque-like proliferation of fascicles of myofibroblast-like cells with an almost parallel orientation to the epidermis. Tumour cells are bland, and mitotic figures are very rare. The tumour does not destroy adnexal structures, but may extend focally into the subcutaneous tissue. Rare cases with haemorrhage may mimic plaque-stage Kaposi sarcoma (see Chapter 139) [8]. The latter, however, is always positive for human herpes virus 8 (HHV8). Tumour cells are variably positive for smooth muscle actin and calponin. The latter two markers, however, may be negative or minimally positive in some cases. CD34 is focally positive in around 20% of cases [7].
Clinical features
History and presentation
Dermatomyofibroma presents as a solitary, asymptomatic, skin-coloured or hypopigmented plaque measuring less than 4 cm in diameter. Multiple lesions are rarely seen and an exceptional case has presented with a linear pattern [9].
Disease course and prognosis
Local recurrence is almost never seen.
Management
Simple excision is curative.
plaque-like CD34-positive dermal fibroma [1, 2, 3]
Definition and nomenclature
This is a very rare lesion characterized by a superficial dermal plaque-like proliferation of fibroblasts and not of dermal dendrocytes as originally reported [1].
Epidemiology
Incidence and prevalence
Tumours are very rare.
Age
The age range is wide. Earlier reports were mainly in children but tumours also present in adults. Rare lesions are congenital.
Sex
Females are more frequently affected than males.
Pathophysiology
Pathology
The epidermis appears unremarkable or slightly flattend and in the dermis there is a fairly monotonous proliferation of spindle-shaped bland cells in a plaque-like distribution. These cells are positive for CD34 and negative for S100. Only a few scattered cells in the background are positive for factor XIIIa (FXIIIa). The appearance may resemble early DFSP and distinction between the two conditions is very important. Plaque-like CD34-positive dermal fibroma hardly ever extends only focally into the subcutis and does not do it in a lace-like pattern. Furthermore, this tumour does not show the typical t17;22 translocation typically found in DFSP [3].
Clinical features
History and presentation
There is predilection for the trunk and limbs. Lesions are sometimes round or oval and have an atrophic appearance and a yellow-red colour. More often, however, clinical features are non-distinctive.
Disease course and prognosis
Lesions are benign.
Management
Simple excision is the treatment of choice.
Angiomyofibroblastoma [1, 2-4]
Definition
Angiomyofibroblastoma is a distinctive benign neoplasia that occurs almost always in the pelvis and perineum, particularly affecting the vulva. There is some overlap with another tumour that presents in the pelvis and perineum (cellular angiofibroma, see later) and also with aggressive angiomyxoma [5].
Epidemiology
Incidence and prevalence
It is rare.
Age
Young to middle-aged females and very rarely in elderly females.
Sex
Predominantly in females. Exceptional cases in males.
Pathophysiology
Pathology
Lesions are well circumscribed and consist of a mixture of round and spindle-shaped bland cells in a myxoid or oedematous stroma with numerous small dilated blood vessels. There is a tendency for tumour cells to surround the vascular channels. Mitotic activity is not usually present. In a number of cases, there are collections of mature adipocytes [4].
Cytological atypia secondary to degeneration is sometimes seen. Tumour cells are positive for desmin and for oestrogen and progesterone receptors. They are only focally positive for smooth muscle actin and muscle-specific actin. Some tumours are variably positive for CD34.
Clinical features
History and presentation
Tumours present mainly in the vulva and in males usually affect the scrotum. Lesions are subcutaneous, asymptomatic and measure less than 5 cm in diameter. Occasional larger pedunculated lesions have been reported [6].
Disease course and prognosis
Tumours are benign with no tendency for local recurrence. Only one malignant tumour has been reported [7].
Management
The treatment is simple excision.
Cellular angiofibroma [1-4]
Definition and nomenclature
Cellular angiofibroma is a distinctive benign neoplasm that occurs almost exclusively in the vulva and less commonly in the scrotum and inguinal soft tissues of men. Some cases overlap histologically with angiomyofibroblastoma and a relationship with spindle cell lipoma and mammary-type myofibroblastoma has been suggested [2]. The latter is based on histological overlap and also on the presence of a distinctive cytogenetic abnormality (see later).
Epidemiology
Incidence and prevalence
It is relatively rare.
Age
Predominantly in young adults.
Sex
Most tumours occur in females.
Pathophysiology
Pathology
Tumours are sharply circumscribed but not encapsulated and are characterized by short, usually bland, spindle-shaped cells with scanty ill-defined pale pink cytoplasm. These cells are arranged in bundles and the degree of cellularity varies. In the background, there are thin collagen bundles and numerous small to medium-sized blood vessels. Mitotic figures are rare and cytological atypia may be occasionally seen in some cases. Scattered mononuclear inflammatory cells, mainly lymphocytes, and degenerative changes are often identified. The latter consist of haemorrhage, thrombosis, hyalinization and haemosiderin deposition. In myxoid areas, mast cells are present and many tumours contain variable numbers of mature adipocytes. The most consistent immunohistochemical finding is the presence of diffuse positivity for CD34 in many cases. Muscular markers including actin and desmin tend to be negative but positivity has been reported in male tumours. In a few cases, there is focal positivity for oestrogen and progesterone receptors.
Genetics
A monoallelic deletion of RB1 located on chromosome 13q14 is often found [5].
Clinical features
History and presentation
Tumours presenting as a small, well-circumscribed, asymptomatic, subcutaneous nodule. In males, lesions tend to be larger and may be related to a hydrocele or a hernia [2].
Disease course and prognosis
Lesions are benign with little or no tendency for local recurrence. Histologically, exceptional tumours with atypia or sarcomatous transformation have been described but they have not behaved in an aggressive manner, although follow-up was limited [5, 6].
Management
Simple excision is the treatment of choice.
Elastofibroma [1, 2, 3]
Definition and nomenclature
Elastofibroma is a reactive, probably degenerative, process of the elastic fibres of deep soft tissues that occurs almost exclusively around the shoulder. Although the lesion is regarded as degenerative, the finding of chromosomal alterations (see later), and of clonality in some cases, has led to the suggestion that it represents a neoplastic process [4].
Epidemiology
Incidence and prevalence
Unknown. However, computed tomography (CT) scans detected incidental lesions in 2% of persons more than 60 years of age and in 16% of adult autopsies in persons older than 55 [5, 6].
Age
Most lesions occur in old-aged individuals.
Sex
No sex predilection.
Pathophysiology
Predisposing factors
Although elastofibroma has been regarded as the result of a degenerative process involving elastic fibres and in association with trauma, the presence of cytogenetic abnormalities in some tumours suggest that it is more likely to be neoplastic (see later).
Genetics
Comparative genomic hybridization in a series of elastofibromas has found chromosomal alterations in a percentage of cases. The most common alteration consists of gains at chromosome Xq12-q22 [7].
Pathology
The mass is poorly circumscribed, and the appearances are characteristic. Abundant hypocellular hyalinized collagen containing numerous large thick eosinophilic elastic fibres is the most distinctive feature. Sometimes the fibres are beaded and fragmented. Staining for elastic tissue nicely highlights the changes.
Clinical features
History and presentation
It presents as an asymptomatic slowly growing mass on the posterior upper trunk. Pain is very rare. Lesions in other locations, including internal organs, are exceptional. Multiple lesions are usually bilateral and may be symmetrical [8].
Disease course and prognosis
There is no tendency for local recurrence.
Management
Simple excision is the treatment of choice.
Inclusion body (digital) fibromatosis [1-3, 4]
Definition and nomenclature
Inclusion body fibromatosis is a fibro/myofibroblastic proliferation that almost only occurs on the fingers and toes. It is characterized by bright, round, intracytoplasmic, eosinophilic inclusions.
Epidemiology
Incidence and prevalence
Lesions are rare, representing 2% of fibroblastic tumours in childhood [5].
Age
Most lesions present either at birth or during the first year of life. Presentation in adults is exceptional [6].
Sex
Males and females are equally affected.
Pathophysiology
Pathology
Monomorphic bundles of bland myofibroblast-like cells are seen in the dermis (Figure 137.5a) and often the subcutis. Tumour cells have vesicular nuclei, an inconspicuous nucleolus and pink cytoplasm. Some mitotic figures may be seen. A distinctive feature is the presence of variable numbers of small round eosinophilic intracytoplasmic inclusions in tumour cells (Figure 137.5b). These are periodic acid–Schiff (PAS) negative, but stain red with Masson trichrome. They also stain for smooth muscle actin.
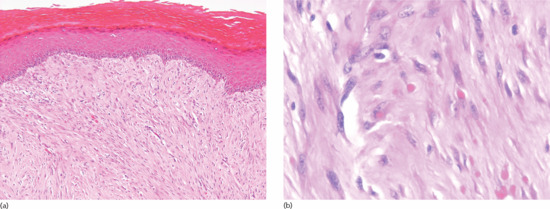
Figure 137.5 (a) Bundles of bland myofibroblast-like cells in the dermis in a case of inclusion body fibromatosis. (b) Numerous typical eosinophilic intracytoplasmic eosinophilic inclusions.
Clinical features
History and presentation
Lesions present as small multiple nodules with a predilection for the dorsal or dorsolateral aspect of the third, fourth and fifth digits. Involvement of the first digits (thumb and hallux) does not occur. Simultaneous involvement of fingers and toes is very rare. New lesions often develop over a long period of time. Only rare cases have been described at other sites including the leg, arm and breast [4, 7].
Disease course and prognosis
Spontaneous regression is sometimes seen [8]. Local recurrence may be seen in up to 25% of cases. Aggressive behaviour has not been described.
Management
Simple excision may be required for lesions that interfere with function, but simple observation of histologically confirmed lesions may be all that is necessary.
Fibroma of tendon sheath [1, 2]
Definition and nomenclature
This is a distinctive well-circumscribed fibroblastic tumour, presenting almost exclusively on the distal extremities.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
Fibroma of tendon sheath presents mainly in young to middle-aged adults and exceptionally in children.
Sex
Males and females are equally affected.
Pathophysiology
Pathology [1, 2]
The neoplasm is multilobular and well circumscribed, and consists of cellular or poorly cellular areas on a background of variably hyalinized stroma. Stromal clefting is usually prominent. Tumour cells are spindle shaped, with scanty cytoplasm and vesicular nuclei. Cytological atypia tends to be absent, and the mitotic count is low. Degenerative changes are seen in some cases and consist of cystic degeneration, myxoid change and bony metaplasia. Rare giant cells are sometimes identified.
Genetics
A translocation at t(2;11)(q31-32;q12) has been demonstrated in a case of fibroma of tendon sheath [3]. This translocation has also been demonstrated in cases of desmoplastic fibroblastoma (p. 137.12).
Clinical features [1, 2]
History and presentation
It is a small slowly growing asymptomatic tumour, with a marked predilection for the distal upper limb, particularly the hand and fingers (1st, 2nd and 3rd). Rare lesions may present with carpal tunnel syndrome [4]. Tumours on the foot are much less common.
Disease course and prognosis
About 20% of cases recur locally but the growth is not destructive.
Management
Simple excision is the treatment of choice.
Desmoplastic fibroblastoma [1, 2]
Definition and nomenclature
Desmoplastic fibroblastoma represents a distinctive subcutaneous fibroblastic tumour consisting of a prominent collagenous stroma.
Epidemiology
Incidence and prevalence
Tumours are relatively common.
Age
Presentation is in middle-aged to old adults.
Sex
Males are twice as frequently affected as females.
Pathophysiology
Pathology
This is a well-circumscribed tumour composed of bland elongated or stellate cells, with a background collagenous stroma and focal myxoid change. Mitotic figures are very rare.
Genetics
A translocation t(2;11)(q31;q12) is characteristically found in this tumour [3]. The rearrangement of the 11q12 chromosome results in the deregulated expression of FOSL1 [3, 4].
Clinical features
History and presentation
Lesions present as an asymptomatic nodule less than 4 cm in diameter, at any body site with a predilection for the back and limbs.
Disease course and prognosis
There is no tendency for local recurrence.
Management
Simple excision is the treatment of choice.
Nuchal–type fibroma [1, 2]
Definition and nomenclature
Nuchal fibroma is a dermal or subcutaneous tumour consisting of hypocellular dense collagen.
Epidemiology
Incidence and prevalence
Occurrence is rare.
Age
Most cases present in adults between the third and fifth decades of life.
Sex
Males are much more commonly affected than females.
Pathophysiology
Predisposing factors
Patients often have type 2 diabetes.
Pathology
Dense aggregates of collagen with very few cells and entrapment of adipose tissue. Inflammation is minimal and consists of a few scattered lymphocytes. In some cases, focal proliferation of nerves is seen mimicking a traumatic neuroma.
Clinical features
History and presentation
The great majority of cases present by far on the nape of the neck. Tumours can also present on the upper back, limbs and face [3]. Coexistence with scleredema is possible, probably reflecting the association with diabetes, and lesions identical to nuchal fibroma are recognized to occur in Gardner syndrome (Chapter 80) and are known as Gardner-associated fibromas [3, 4]. The latter may be multiple, present in various locations and may recur. These lesions may be the first clue as to the existence of Gardner syndrome.
Disease course and prognosis
Local recurrence is common but lesions do not behave aggressively.
Management
Simple excision is the treatment of choice.
Palmar and plantar fibromatosis (superfi cial fibromatoses) [1, 2]
Definition and nomenclature
Palmar and plantar fibromatoses are superficial neoplastic proliferations of fibroblasts and myofibroblasts that have a tendency for local recurrence, but do not metastasize.
Epidemiology
Incidence and prevalence
Palmar fibromatosis is fairly common and more common than plantar fibromatosis. The incidence of both conditions but particularly the former increases with age.
Age
Both conditions affect middle-aged to elderly patients and are uncommon in younger individuals. However, children may rarely be affected, particularly by plantar fibromatosis [3].
Sex
Both lesions are more common in men, but the sex difference is more marked in palmar lesions.
Ethnicity
Affected patients are mainly of northern European origin; non-whites are rarely affected.
Pathophysiology
Predisposing factors
Genetic predisposition, as well as trauma, is thought to play an important role in the pathogenesis of these conditions. Associations with diabetes, alcoholic liver disease and epilepsy have also been described.
Pathology
Early lesions are fairly cellular and consist of bundles of bland fibroblasts with some collagen deposition. The latter increases considerably in older lesions. Interestingly, although superficial fibromatoses are very similar histologically to deep fibromatosis (abdominal, extra-abdominal and mesenteric fibromatosis), the behaviour of superficial fibromatosis is not usually aggressive. This may be due to the fact that deep fibromatosis often display mutations of the APC gene or somatic mutations of the gene encoding β-catenin, while these mutations are absent in superficial fibromatosis. Intriguingly however, although deep fibromatoses often display nuclear expression of β-catenin, this is also seen in a smaller percentage of superficial fibromatoses without gene mutations [4].
Coexistence between the two variants of fibromatoses and desmoid tumours, penile fibromatosis (Peyronie disease) and knuckle pads, may be seen.
Clinical features
History and presentation
Palmar fibromatosis presents as indurated nodules or as an ill-defined area of thickening, bilateral in about 50% of cases that may result in contracture. Plantar fibromatosis usually consists of a single nodule.
Disease course and prognosis
Functional limitation is common. Lesions are prone to local recurrence.
Management
Complete excision is desirable.
Penile fibromatosis [1,2,3]
Definition and nomenclature
Although usually regarded as a variant of superficial fibromatosis, it is more likely that this disease represents a reactive fibrotic disorder of unknown aetiology.
Epidemiology
Incidence and prevalence
The condition is rare.
Age
Most patients are middle aged.
Pathophysiology
Predisposing factors
There is an association with type 2 diabetes.
Pathology
In early lesions, there is a patchy chronic mononuclear inflammatory cell infiltrate and focal vasculitic changes. These changes lead to dense bands of hyalinized collagen in late stages.
Clinical features
History and presentation
It presents as a solitary nodule or multiple nodules close to the corpus cavernosum on the dorsal surface of the shaft, and in most the lesion is small. Pain and curvature of the penis on erection are frequent complaints. The presence of diabetes increases the severity of the disease [4].
Disease course and prognosis
The condition results in penile deformity and sexual dysfunction.
Management
Surgery is the treatment of choice but in recent years less invasive therapies have been attempted. The latter include intralesional injections of interferon α-2b or of collagenase Clostridium histolyticum [5].
Lipofibromatosis [1]
Definition and nomenclature
Lipofibromatosis is a locally aggressive childhood tumour composed of variable amounts of mature adipose and fibroblastic elements.
Epidemiology
Incidence and prevalence
The condition is very rare.
Age
Tumours occur in infants and children; the majority of cases presenting in the first decade of life.
Sex
There is male predominance (around 60% of cases).
Pathophysiology
Pathology
Tumours are infiltrative and consist of lobules of mature adipose tissue intermixed with bundles of fibroblast-like cells with no cytological atypia and low mitotic activity. By immunohistochemistry, tumour cells are focally positive for S100 protein, CD34, bcl-2, actin, epithelial membrane antigen (EMA) and CD99. The lesion closely resembles a fibrous hamartoma of infancy but has more prominent adipose tissue and lacks the third cellular component seen in the latter, which consists of round primitive-looking cells in a myxoid background.
Genetics
A translocation at t(4;9;6) was reported in a single case [2].
Clinical features
History and presentation
Some tumours are congenital. There is a male predominance. The classical presentation is of a slowly growing ill-defined mass. There is a predilection for the hands and feet, but other sites in the limbs, and less commonly on the trunk, may be affected. The rate of local recurrence is high.
Disease course and prognosis
The tumour is locally aggressive with no metastatic potential. There is high tendency for local recurrence.
Management
Complete excision is desirable.
Dermatofibrosarcoma protuberans [1-3]
Definition
Dermatofibrosarcoma protuberans is a locally invasive tumour arising in the dermis and showing fibroblastic differentiation.
Epidemiology [1-3]
Incidence and prevalence
Dermatofibrosarcoma protuberans is uncommon but represents one of the most common dermal sarcomas. The incidence in the USA has been estimated as 4.2 cases per million [4].
Age
Tumours more commonly develop during the third and fifth decades of life. However, presentation during childhood and late life is not particularly rare [5, 6, 7]. Congenital cases have been described [7, 8].
Sex
There is a slight female predilection.
Ethnicity
It is more common in black than in white patients [4].
Pathophysiology
Predisposing factors
Some cases develop at the site of previous trauma and reports have included a burn scar [9] and the site of vaccination. Exceptional cases have been associated with previous radiotherapy to the area [10]. There is an association between DFSP and children with adenosine deaminase deficient severe combined immunodeficiency [11]. Patients affected by the latter have a higher incidence of tumours presenting at early age and often multicentric.
Pathology [1-3]
The tumour is usually a solitary multinodular mass. The dermis and subcutaneous tissue are replaced by bundles of uniform spindle-shaped cells with little cytoplasm and elongated hyperchromatic, but not pleomorphic, nuclei. Usually there is little mitotic activity. Deeper involvement may be seen in some cases. Laterally, the tumour cells infiltrate widely between collagen bundles of the deeper dermis and blend into the normal dermis, forming quite definite bands, which interweave or radiate like the spokes of a wheel; this is described as a ‘storiform’ pattern (Figure 137.6). The interstitial tissue contains collagen fibres, except in the most cellular parts of the tumour. The subcutaneous tissue is extensively infiltrated and replaced in a typical lace-like pattern. Myxoid change may be focal or, rarely, prominent; in the latter setting, the histological diagnosis is difficult [12, 13]. Some tumours are colonized by scattered deeply pigmented melanocytes, a variant known as pigmented DFSP (Bednar tumour) [14, 15]. A further variant consists of myoid nodules and is thought to represent myofibroblastic differentiation [16]. Rare cases show focal granular cell change.

Figure 137.6 Pathological appearance of dermatofibrosarcoma protuberans, showing the storiform or ‘cartwheel’ distribution of the fairly uniform, spindle-shaped tumour cells.
Fibrosarcomatous DFSP [17, 18, 19, 20] is an important variant of this tumour, which is recognized by the focal presence of areas with long sweeping fascicles of tumour cells intersecting at acute angles in a typical ‘herring-bone’ pattern, almost identical to that seen in fibrosarcoma. In these areas, mitoses are increased and there is more nuclear hyperchromatism. P53 expression is increased in fibrosarcomatous areas [20]. Identification of the presence of this pattern, and its quantity, is very important, as it is related to metastatic potential. Fibrosarcomatous areas are more common in recurrent tumours.
Very rare variants of DFSP may show areas of high-grade sarcoma either in the primary tumour or in a recurrence [21].
Dermatofibrosarcoma protuberans may show areas of giant cell fibroblastoma (see later) and either tumour may recur, displaying features of the other tumour [22].
The majority of the lesions are positive on staining with the antibody CD34, although this is not specific for DFSP [23]. Other markers are usually negative but in some cases focal positivity for epithelial membrane antigen may be seen. Fibrosarcomatous areas often show decreased staining with CD34 [19].
Genetics
Cytogenetic studies are helpful, as ring chromosomes indicative of a 17;22 translocation are invariably found [22]. However, it is important to highlight that some cases demonstrate a variant ring chromosome with cryptic rearrangements of chromosomes 17 and 22 [24]. This chromosomal translocation involves the collagen type I α1 (COL1A1) gene on chromosome 17 and the platelet-derived growth factor B (PDGFB) gene on chromosome 22. The abnormal fusion transcripts resulting from this translocation leads to autocrine stimulation of PDGFB and platelet-derived growth factor receptor β (PDGFRB) and cell proliferation. The fusion transcript is found in almost all examples of the tumour by polymerase chain reaction (PCR) and fluorescence in situ hybridization (FISH) [23]. The same cytogenetic abnormality is found in giant cell fibroblastoma, confirming that both tumours are part of the same spectrum.
Clinical features
History and presentation
The tumour is more often situated on the trunk (up to half of the cases), particularly in the flexural regions, than on the extremities or the head [1, 2]. Involvement of the limbs is usually proximal. Presentation on the hands and feet, particularly on the digits, is very rare. It may begin in early adult life with one or more small, firm, painless, flesh-coloured or red dermal nodules (Figure 137.7).
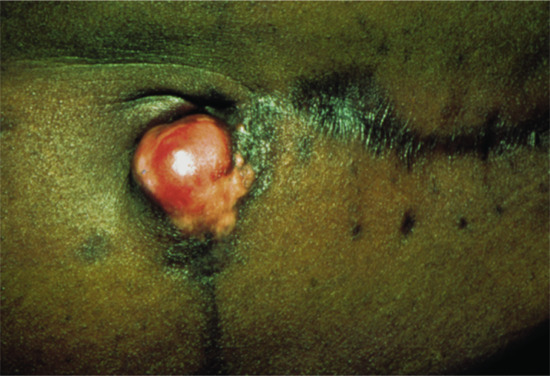
Figure 137.7 Recurrent abdominal dermatofibrosarcoma protuberans.
The tumour starts as a plaque, which may occasionally be atrophic [6, 25]. Progression is usually very slow, and may occur over many years; a significant proportion of tumours only become protuberant after a long period of time [26]. Eventually, nodules develop, coalesce and extend, becoming redder or bluish as they enlarge to form irregular protuberant swellings. At this stage, the base of the lesion is a hard indurated plaque of irregular outline. In the later stages, a proportion of lesions become painful and there may be rapid growth, ulceration and discharge.
Differential diagnosis
In the early stages, it may be impossible to distinguish this tumour from a histiocytoma or a keloid. Some lesions may also be confused with morphoea profunda. The slow progression, deep red or bluish-red colour, and the characteristic irregular contour and extended plaque-like base, are strongly suggestive of DFSP.
Disease course and prognosis
Local recurrence of ordinary DFSP is reported to vary from 15% to up to 60% [3, 27, 28]. The fibrosarcomatous variant has a similar rate of local recurrence but a higher rate of metastatic spread [20, 21, 29, 30, 31]. Metastases to lymph nodes and internal organs tend to be extremely rare in pure DFSP [20, 32, 33] but occur in up to 13% of cases with fibrosarcomatous transformation [20, 21, 22, 30, 31].
Management
The tumour should be excised completely, with a generous margin of healthy tissue [34]. The best chance of achieving a complete cure with no recurrence is early detection of small tumours. Local recurrence invariably follows inadequate removal; the clearance necessary to cure the tumour is often underestimated [35]. A margin of between 2 and 4 cm has been recommended [28, 36]. Mohs micrographic surgery has been reported as effective in reducing the rate of local recurrence and it has become the recommended standard treatment in many large centres [37, 38]. If this type of treatment is used it should be performed using formalin-fixed paraffin-embedded sections rather than frozen sections, and evaluation should be by an experienced pathologist. Although Mohs surgery clearly reduces the rate of local recurrences, the latter still occur and sometimes this happens more than 5 years after surgery [39]. Postsurgical radiotherapy has been advocated to reduce the rate of local recurrence [40] but this type of treatment has not been assessed in large series of patients. In recent years, it has been demonstrated that imatinib mesylate, a potent inhibitor of a number of protein kinases including the PDGFR, results in good clinical response in patients with large unresectable or metastatic tumours [41, 42, 43, 44, 45].
Giant cell fibroblastoma [1-3, 4]
Definition
This is a locally recurrent fibroblastic tumour, closely related to DFSP. It is characterized by spindle-shaped, oval or stellate, mono- or multinucleated cells in a fibromyxoid stroma with irregular pseudovascular spaces lined by tumour cells.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
Most cases present in children. Rare cases are seen in young adults and only exceptionally in older adults.
Sex
About 60% of patients are male.
Pathophysiology
Pathology
Solid fibromyxoid areas with variable collagen deposition contain stellate and spindle-shaped mono- and multinucleated tumour cells with hyperchromatic nuclei. Dilated irregularly branching pseudovascular spaces are commonly seen scattered throughout the lesion. These spaces are lined by tumour cells, which often appear multinucleated (Figure 137.8). Mitotic figures are exceptional. Aggregates of perivascular lymphocytes in an onion-ring pattern and focal haemorrhage are often seen [4]. Focal areas identical to DFSP may be seen and can occupy a substantial part of the tumour. Excised lesions can recur as a pure giant cell fibroblastoma, as a tumour with focal DFSP, or as pure DFSP [4, 5, 6, 7]. All types of tumour cells are positive for CD34.
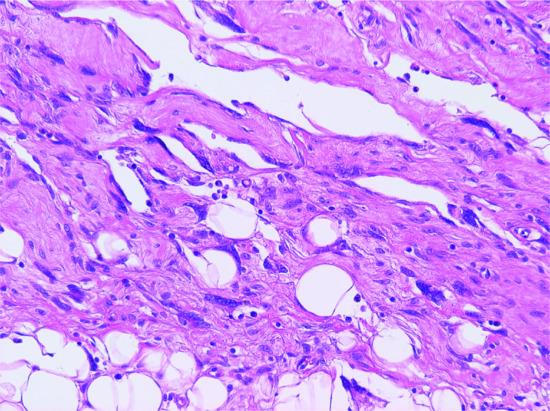
Figure 137.8 Typical pseudovascular spaces focally lined by multinucleated cells in a case of giant cell fibroblastoma.
Genetics
Ring chromosomes with sequences of chromosomes 17 and 22, identical to those found in DFSP, have been described in this tumour, confirming their close histogenetic relationship [8, 9].
Clinical features [1, 2, 3, 4]
History and presentation
The large majority of cases present as a subcutaneous ill-defined mass but rare tumours are polypoid. It is rare in young adults and more exceptional in older adults. The trunk, axilla and groin are much more commonly involved than the proximal limbs. Head and neck tumours are rare. Lesions measure a few centimetres in diameter and tend to be asymptomatic.
Disease course and prognosis
Recurrence may be seen in about half of the cases, but metastasis has not been reported.
Management
Complete surgical excision with adequate margins is the treatment of choice.
Myxoinfl ammatory fibroblastic sarcoma [1-3, 4]
Definition and nomenclature
Myxoinflammatory fibroblastic sarcoma is a distinctive, neoplastic process with marked predilection for acral sites, and with histological features closely mimicking an inflammatory process due to the presence of prominent inflammation and virocyte-like inclusions in the nuclei of tumour cells. The latter features were initially thought to indicate an infectious aetiology. A relationship with haemosiderotic fibrolipomatous tumour is likely (see p. 137.63). Both tumours share a similar translocation (see later) and an example of the latter lesion progressing to a tumour with features of myxoinflammatory fibroblastic sarcoma following recurrence and leading to death of the patient as a result of metastatic disease has been reported [5].
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
Most patients are middle-aged adults. Presentation in children [6] and elderly patients are very rare.
Sex
There is a slight female predilection.
Pathophysiology
Pathology
Lesions are lobulated and poorly circumscribed, and involve the subcutaneous fat and often extend to the dermis and deeper tissues, sparing bone. Low-power examination is misleading and the initial impression is that of an inflammatory process. Lobules of hyalinized and myxoid tissue containing variable numbers of inflammatory cells are seen. The latter include lymphocytes, histiocytes, neutrophils and less commonly eosinophils and plasma cells. Closer examination reveals variable numbers of neoplastic cells that vary from round to spindle shaped. Some of these cells may be multinucleated. Round cells mimic ganglion cells with nucleoli resembling viral inclusions. Less commonly, vacuolated tumour cells resembling lipoblasts are seen. Mitotic activity is very low. Lesions displaying one or more of the following histological features appear to have a higher rate of local recurrence: areas with complex sarcoma-like vasculature, hypercellular areas and increased mitotic activity or the presence of atypical mitotic figures [4]. Immunohistochemistry shows that tumour cells are variably positive for D2-40 (86%), CD34 (50%), keratins (33%), actin (26%), CD68 (27%) and very rarely for desmin, S100 and EMA [4].
Genetics
Very few cases have been subjected to cytogenetic studies [7, 8]. In two cases published, one showed a complex karyotype with a reciprocal translocation t(1;10)(p22;q24) and loss of chromosomes 3 and 13 [7], and the other a translocation t(2;6)(q31;p21.3) [8]. These findings confirm the theory that these lesions are neoplastic.
Clinical features
History and presentation
Characteristically, tumours are longstanding, asymptomatic and slowly growing, multinodular and usually measure no more than 4 cm. The great majority occur on acral sites, particularly the dorsal aspect of the hands and wrists, followed by the feet. However, lesions may rarely present elsewhere on the limbs (the arm, forearm and thigh) [4, 6, 9, 10] and exceptionally elsewhere in the body, including the head and neck [4, 5, 9]. Most cases are clinically diagnosed as a ganglion cyst or as a giant cell tumour of tendon sheath.
Disease course and prognosis
The rate of local recurrence is high, varying from 11 to 67% in different series [1, 2, 4]. The absence of clear surgical margins correlates with higher recurrence rate. Distal metastases are exceptional and are mainly to regional lymph nodes.
Management
The treatment of choice is wide local excision and this often implies amputation.
Malignant fibrous histiocytoma [1-4]
‘Malignant FH’ is an umbrella term encompassing a heterogeneous group of neoplasms that initially included five different clinicopathological subtypes: pleomorphic, myxoid, giant cell, inflammatory and angiomatoid. There is little relation between the different subtypes; the angiomatoid variant has recently been reclassified in the group of fibrohistiocytic tumours and the name changed to ‘angiomatoid FH’. The concept of pleomorphic malignant FH has been challenged as not representing a distinct group of neoplasms but a heterogeneous category, including pleomorphic poorly differentiated sarcomas. If cases classified as such are extensively studied with ancillary studies including immunohistochemistry, electron microscopy and more recently cytogenetics [5, 6], a large percentage may be reclassified as pleomorphic variants of other soft-tissue tumours, including liposarcoma, rhabdomyosarcoma and leiomyosarcoma. The myxoid variant of malignant FH is now known as ‘myxofibrosarcoma’, and it is likely to show fibroblastic differentiation; this tumour often involves the skin because of its frequent origin in the subcutis, and it will therefore be discussed in more detail below. Angiomatoid FH has been described under fibrohistiocytic tumours. The inflammatory and giant cell variants of malignant FH hardly ever involve the skin and will not be discussed further.
Myxofibrosarcoma [1-3,4]
Definition and nomenclature
Myxofibrosarcoma is a neoplasm of the subcutis and deeper soft tissues with variable cellularity, myxoid change and cells with pleomorphic nuclei. The cellular end of the spectrum is identical to a pleomorphic malignant FH, and the diagnosis is made based on the presence of myxoid areas with less cellularity and a lobular pattern. The myxoid change should be seen in 10% or more of the tumour before a lesion is classified as myxofibrosarcoma.
Epidemiology
Incidence and prevalence
Age
Presentation is mainly in middle-aged to old adults.
Sex
There is a slight male predilection.
Pathophysiology
Pathology [4]
These tumours have a lobular growth pattern. They are classified according to the degree of cellularity and pleomorphism into low, medium and high grade. Low-grade tumours are paucicellular and consist of round or elongated bland and pleomorphic cells in a prominent myxoid stroma. The atypical cells have irregular hyperchromatic nuclei, and mitotic figures are relatively frequent. In the background, a fairly prominent number of thin-walled vascular channels with a typical curvilinear pattern are seen. Vacuolated, Alcian blue positive cells, focally mimicking lipoblasts, are relatively frequent. In some tumours, hypocellular areas blend with more cellular areas containing cells with increased pleomorphism; such tumours are classified as intermediate grade. Tumours with high cellularity (high grade) are indistinguishable from the so-called pleomorphic malignant FH and may have necrosis. Grading of lesions is important, because the rate of local recurrence and metastasis varies (see later). Some tumours, particularly high-grade lesions, may have epithelioid morphology [5]. Tumour cells are positive for vimentin and only rarely display very focal positivity for actin.
Clinical features
History and presentation
This tumour mainly presents in the extremities, particularly the lower limbs followed by the upper limbs and less commonly the trunk, head and neck [4]. Typically, an asymptomatic mass, measuring several centimetres in diameter, is found in the subcutis or deeper soft tissues. This is one of the sarcomas that more often involves the dermis as a result of extension from the subcutis or deeper soft tissues, rather than having a dermal origin. About 50% of cases arise in the subcutaneous tissue and involve the overlying dermis [5]. Exceptional cases have been reported in association with a burn scar [6].
Disease course and prognosis
High-grade lesions have a higher tendency for local recurrence and for metastatic spread to regional lymph nodes. The overall 5-year survival is between 60 and 70% [4, 5, 6]. Tumours with epithelioid morphology appear to have a more aggressive behaviour [7].
Management [4]
Excision with clear margins is essential.
Low–grade fibromyxoid sarcoma [1-3]
Definition and nomenclature
This distinctive neoplasm is regarded as a low-grade variant of fibrosarcoma. It is characterized by deceptive, bland, spindle-shaped cells in a stroma with curvilinear blood vessels and either collagenous or myxoid background.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
It is seen mainly in young to middle-aged adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology [1-3]
The tumour consists of a proliferation of wavy, bland, spindle-shaped cells arranged in short fascicles and surrounded by a collagenous or myxoid stroma. Cellularity varies and tumour cells are usually bland with very rare mitotic figures. Frequent, elongated, thin-walled blood vessels are seen throughout the tumour. Only a small number of cases display some degree of cytological atypia. As a result of the deceiving histological appearances, the tumour is often diagnosed as benign. In a proportion of cases there are focal areas with hyalinized collagen surrounded by epithelioid tumour cells forming rosettes. This variant of the tumour was originally described as hyalinizing spindle cell tumour with giant rosettes [5]. The presence of rosettes does not influence the behaviour of the neoplasm. Immunohistochemistry is of limited value as tumour cells are negative for most markers. They may be, however, positive for EMA and this may lead to a misdiagnosis of perineurioma.
Genetics
Tumours show a distinctive translocation, t(7;16)(q33;p11), leading to fusion of the FUS and CREB3L2 genes [4]. This finding is very useful for confirmation of the diagnosis by FISH.
Clinical features
History and presentation
The tumour usually presents as a slowly growing lesion in young to middle-aged adults, with an equal sex incidence; it has a predilection for the proximal extremities, followed by the trunk. Tumours tend to be longstanding and asymptomatic and present as a mass, measuring several centimetres in diameter, and located in the subcutis or deeper soft tissues. Subcutaneous lesions are often clinically diagnosed as a lipoma.
Disease course and prognosis
In the largest series of cases reported so far it has been shown that local recurrence occurs in 9% of cases, metastases in 9% and mortality in 2% [6]. It seems that areas with higher grade morphology do not confer a more aggressive behaviour. However, this needs to be confirmed in further studies. Metastatic spread may occur many years after the original diagnosis and therefore long-term follow-up is indicated.
Management
Excision with clear margins is essential.
FIBROHISTIOCYTIC TUMOURS
Giant cell tumour of tendon sheath [1,2,3,4]
Definition
This is a benign tumour that in its localized variant occurs mainly on the hands, and consists of a nodular proliferation of histiocyte-like cells with scattered multinucleated giant cells and variable numbers of mononuclear inflammatory cells. The diffuse variant of this tumour that involves joints is not discussed further in this chapter.
Epidemiology
Incidence and prevalence
Tumours are relatively rare.
Age
Young to middle-aged adults.
Sex
There is a predilection for females.
Pathophysiology
Pathology
It is a multinodular lesion composed of sheets of histiocyte-like cells with bland vesicular nuclei, intermixed with multinucleated giant cells, foamy cells, siderophages and scattered mononuclear inflammatory cells. Hyalinization, haemosiderin deposition and cholesterol clefts are often seen. No histological features predict lesions that recur locally [3].
Clinical features
History and presentation
Tumours present mainly on the hands with a predilection for the fingers. They are typically between 1 and 3 cm in diameter and asymptomatic, although they may interfere with function. Multiple tumours are very rare [4].
Disease course and prognosis
The rate of local recurrence varies from 5 to 15% [3, 5].
Management
Excision is the treatment of choice.
Fibrous histiocytoma (dermatofi broma) [1-4]
Definition and nomenclature
Fibrous histiocytoma is a benign dermal and often superficial subcutaneous proliferation of oval cells resembling histiocytes, and spindle-shaped cells resembling fibroblasts and myofibroblasts. Their line of differentiation remains uncertain, but these lesions are descriptively classified as fibrohistiocytic tumours because of the microscopic appearance of the tumour cells. The aetiology of FH is unknown, but cytogenetic studies demonstrating clonality favour these lesions being neoplastic [5, 6]. The neoplastic nature of FH is also suggested by their clinical persistence and by the frequency of local recurrence of some variants (cellular, aneurysmal and atypical; see later) as well as the exceptional metastases (rarely leading to death as a result of disseminated disease) of some tumours (cellular, aneurysmal and atypical and more exceptionally, epithelioid and even ordinary types) [7, 8, 9, 10].
Epidemiology
Incidence and prevalence
Ordinary fibrous histiocytoma is probably the most common cutaneous soft-tissue tumour. Important clinicopathological variants (cellular, atypical and aneurysmal) are much more uncommon. Cellular FH represents less than 5% of all FHs [11]. Aneurysmal and atypical FHs are less common than the latter.
Age
Most FHs occur in young to middle-aged adults. Cellular, atypical and epithelioid FHs are more common in young adults.
Sex
Ordinary FH is more common in females. Cellular and atypical FHs are more common in males. The other variants are more common in females.
Pathophysiology
Pathology
The overlying epidermis frequently shows a degree of epidermal hyperplasia [12] (Figure 137.9). The latter displays different patterns including changes mimicking a squamous papilloma, a seborrhoeic keratosis and lichen simplex chronicus. Occasionally, the epidermal proliferation is associated with immature follicular structures, which are often confused with a basal cell carcinoma. In the dermis, there is a localized proliferation of histiocyte-like cells and fibroblast-like cells, associated with variable numbers of mononuclear inflammatory cells. Foamy macrophages, siderophages and multinucleated giant cells are also variably present. A focal storiform pattern is often seen. The tumour blends with the surrounding dermis. Collagen bundles at the periphery of the lesion are surrounded by scattered tumour cells and appear somewhat hyalinized. Focal myofibroblastic differentiation is often suggested, particularly in the cellular variant. Older lesions show focal proliferation of small blood vessels in association with haemosiderin deposition and fibrosis, hence the older name of ‘sclerosing haemangioma’.
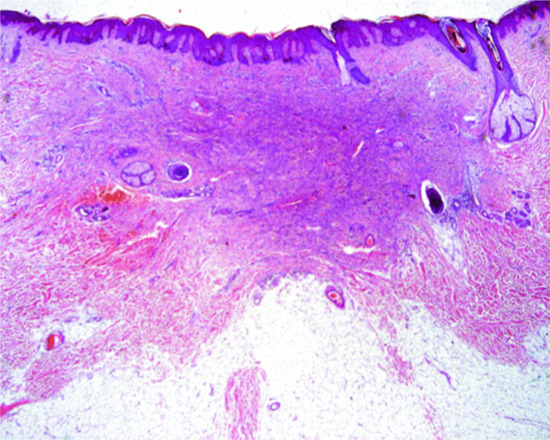
Figure 137.9 Histological appearance of dermatofibroma, showing epidermal hyperplasia overlying the dermal sclerotic component.
Cellular FH [11] also shows epidermal hyperplasia, but the lesions are more cellular, less polymorphic and consist of bundles of spindle-shaped cells with pink cytoplasm and a focal storiform pattern (Figure 137.10). The mitotic rate varies, and necrosis may be found in up to 12% of cases. Extension into the subcutaneous tissue is more prominent than that seen in ordinary FH. However, the pattern of infiltration is mainly along the septae, and only focally into the subcutaneous lobule in a lace-like pattern. The cellularity and growth pattern often make distinction from DFSP difficult, particularly in small biopsies. DFSP is, however, more monomorphic, tends to infiltrate the subcutaneous tissue diffusely and is generally uniformly positive for CD34. Cellular FH may be focally positive for CD34, but this is predominantly seen at the periphery of the tumour. Staining for FXIIIa is positive in FH and negative in DFSP. Furthermore, cellular FH is often focally positive for smooth muscle actin, whereas this marker is negative in DFSP.

Figure 137.10 Cellular fibrous histiocytoma. Note the increased cellularity, fascicular appearance and focal extension into the subcutis.
Aneurysmal FH [13, 14] shows extensive haemorrhage, with prominent cavernous-like pseudovascular spaces (Figure 137.11), which are not lined by endothelial cells. The mitotic rate varies, but may be prominent. The background is that of an ordinary FH.

Figure 137.11 Aneurysmal fibrous histiocytoma. Prominent haemorrhage and cavernous-like spaces obscure the typical background of a fibrous histiocytoma.
Atypical FH [15, 16] shows variable numbers of mono- or multinucleated, pleomorphic, spindle-shaped or histiocyte-like cells on a background of an ordinary FH. These cells may be very prominent, making the histological diagnosis difficult. Mitotic figures, including atypical forms, may be seen. These lesions used to be classified as ‘atypical fibroxanthoma (AFX) occurring in non-sun-exposed skin of young patients’.
Epithelioid FH [17, 18] contains a predominant population of cells with abundant pink cytoplasm and vesicular nuclei, and there is often myxoid change and a prominent vascular component. Distinction from a Spitz naevus may be difficult, but in epithelioid FH there is no junctional component, tumour cells are not nested and they are negative for S100.
Many histological variants of FH have been described; recognizing these variants is important to avoid misdiagnosis. They include lesions with palisading granular cell change [19], abundant lipid (ankle-type) [20], clear cell change [21], balloon cell change [22] and keloidal change [23]. The presence of lipid within lesions of FH is not usually associated with systemic lipid abnormalities [24].
Clinical features
History and presentation
Fibrous histiocytoma is commonest on the limbs and appears as a firm papule which is frequently yellow-brown in colour and slightly scaly (Figure 137.12). If the overlying epidermis is squeezed, the ‘dimple sign’ will be seen, indicating tethering of the overlying epidermis to the underlying lesion. Giant lesions (>5 cm in diameter) are occasionally seen [25] and large tumours are more often encountered in some of the variants (see later). Multiple lesions may develop and eruptive variants have been described. The latter may be familial [26], or may be associated with immunosuppression (e.g. HIV) [27], with systemic disease, including autoimmune diseases such as lupus erythematosus and neoplasia, particularly haematological malignancies [28, 29, 30, 31, 32], and even with highly active antiretroviral therapy (HAART) [33].

Figure 137.12 Clinical appearance of a fibrous histiocytoma or dermatofibroma.
A number of clinicopathological variants of FH mentioned before have been described, which should be recognized by clinicians and pathologists in order to avoid a misdiagnosis of malignancy. These variants include: cellular FH [11], aneurysmal FH [13, 14], atypical FH (pseudosarcomatous FH, dermatofibroma with monster cells) [15, 16] and epithelioid FH [17, 18]. A further variant, described as ‘atrophic’ [34], may mimic a scar and does not usually pose a problem in differential diagnosis. Rare cases may be ulcerated, erosive or lichenoid [35].
Cellular FH, like ordinary FH, has predilection for the limbs. However, the distribution of age and site is wide; cellular FH is not infrequent in children, and on sites such as the head, neck, fingers and toes. The size of these lesions is also larger than that of ordinary FH. Most cellular FHs are less than 2 cm in diameter, but lesions measuring more than 5 cm may occur. Recognition of this variant is important, because it has a local recurrence rate of 25%, and metastases have been reported anecdotally in a small number of cases [7, 9, 10].
Aneurysmal FH is usually rapidly growing and may attain a very large size. They clinically mimic a vascular tumour. Exceptional tumours are multiple [36]. The rate of local recurrence is 19% [20].
Atypical FH presents as a papule, nodule or plaque, usually less than 1.5 cm in diameter. The rate of local recurrence is around 14%, and exceptional metastases have been reported [23].
Epithelioid FH [24, 25] presents on the limbs of young patients, with a predilection for females. The typical clinical appearance is that of a polypoid, often vascular, lesion resembling a non-ulcerated pyogenic granuloma.
Disease course and prognosis
Ordinary and epithelioid FHs hardly ever recur locally. Cellular FH recurs in 25% of cases, Aneurysmal FH recurs in 19% of cases and atypical FH recurs in 14% of cases. Exceptional metastases have been reported in all clinicopathological variants including ordinary and epithelioid FH. This phenomenon, although exceptional, is more common in cellular, aneurysmal and atypical FHs [7, 8, 9, 10]. In one case of cellular FH, transformation to a pleomorphic sarcoma has been reported [10]. Morphological features do no allow prediction of tumours that will behave in a more aggressive manner [9]. Metastatic tumours are more often associated with chromosomal abnormalities as demonstrated by array comparative genomic hybridization [10, 37]. Fatal tumours are associated with the highest number of chromosomal abnormalities [37].
Management
Most FHs are no more than a cosmetic nuisance, and no treatment is necessary. However, cellular, atypical and aneurysmal variants should be completely removed conservatively, because of the risk of local recurrence and the occurrence of occasional distant metastases.
Plexiform fibrohistiocytic tumour [1-4,5]
Definition and nomenclature
Plexiform FH is a distinctive predominantly subcutaneous tumour with two distinctive components:
- A fibro/myofibroblastic fascicular component.
- A nodular histiocytic-like component, which also includes giant cells.
Despite its new name, it does not represent a plexiform variant of an ordinary FH (dermatofibroma). An association with cellular neurothekeoma, a tumour that occurs primarily in the dermis, has been suggested based on morphological similarities [5, 6].
Epidemiology
Incidence and prevalence
Age
It mainly occurs in children and young adults. An exceptional case has been congenital [7].
Sex
It is most common in females.
Pathophysiology
Pathology [1-4,5]
Low-power examination reveals a predominantly subcutaneous tumour with focal involvement of the dermis and a distinctive plexiform growth pattern. Purely dermal lesions are occasionally seen [8]. Two components are usually identified and consist of fascicles of bland spindle-shaped fibro/myofibroblast-like cells and nodules of histiocyte-like cells with scattered giant cells, focal haemorrhage and haemosiderin deposition. In some tumours, one of the components may predominate. The spindle-shaped cells stain focally for smooth muscle actin, and the cells in the nodules are focally positive for CD68.
Clinical features [1-4,5]
History and presentation
Tumours have a predilection for the upper limbs. The tumour is solitary, measures no more than a few centimetres in diameter and is asymptomatic.
Disease course and prognosis
Local recurrences are observed in up to 30% of cases. Metastases to regional lymph nodes or to the lungs have been reported [1, 3, 9].
Management
Complete surgical excision and follow-up are indicated. Histological features do not predict cases with more aggressive behaviour.
Atypical fibroxanthoma [1,2,3,4]
Definition
Atypical fibroxanthoma, by definition, arises in the sun-damaged skin of elderly people. It is a paradoxical tumour with histological features of a highly malignant neoplasm and low-grade clinical behaviour. Tumours with more aggressive histological features should be diagnosed as dermal pleomorphic sarcoma (see later) [5].
Epidemiology
Age
Most patients are elderly in the seventh to eight decades of life. Tumours in younger patients occur very rarely in the setting of xeroderma pigmentosum [6].
Sex
Tumours are much more frequent in males than in females.
Ethnicity
It is a tumour almost exclusively restricted to white people.
Pathophysiology
Predisposing factors
UV radiation-induced p53 mutations have been observed in these lesions, confirming the association with sun-damaged skin [7]. More recently, telomerase reverse transcriptase (TERT) promoter mutations with UV signature have been identified in AFXs giving further support to the relationship to sun exposure [8].
Pathology
The tumours are exophytic, fairly well circumscribed and surrounded by an epidermal collarette. The remarkable and paradoxical feature of AFX is its histological resemblance to a highly malignant soft-tissue sarcoma (Figure 137.13) [9, 10, 11]. It arises in the dermis and may extend very focally into the fat, but the edge is pushing rather than infiltrative. It is composed of large spindle-shaped and histiocyte-like pleomorphic cells, many of which appear multinucleated. The cells are arranged in a haphazard fashion and mitotic figures, including atypical forms, are frequent. The histiocytic cells may contain lipid or haemosiderin [12, 13]. A series of a less pleomorphic spindle cell variant, which may cause considerable problems in differential diagnosis, has been described [14]. Rare cases display prominent sclerosis [15], and partial or total regression may rarely be seen [16]. In some tumours, there is focal or prominent clear or granular cell change, keloid-like areas, prominent myxoid change, osteoclast-like giant cells and pseudoangiomatous areas [17, 18, 19, 20]. Tumours with an infiltrative growth pattern, involvement of deeper tissues, tumour necrosis, lymphovascular invasion and perineural invasion should be classified as dermal pleomorphic sarcomas as they have a more aggressive behaviour than conventional AFXs (see later) [5]. The diagnosis of AFX is a diagnosis of exclusion. An immunohistochemical panel to rule out melanoma (S100), sarcomatoid squamous cell carcinoma (pankeratin, mainly MNF 116 and AE1/AE3, as the low molecular weight keratin Cam 5.2 is usually negative in sarcomatoid squamous cell carcinoma of the skin) and even leiomyosarcoma (desmin, h-caldesmon) should be performed in all cases. The basic panel should be enough to accurately diagnose most cases of AFX. Other markers have been described that are often positive in AFX, and tend to be negative in other tumours that enter the differential diagnosis. These include CD99, CD10 and pro-collagen 1 [21, 22, 23]. CD31 is often focally positive in tumour cells as is EMA and very rare focal positivity for melan-A has been reported [20, 24]. Since the advent of immunohistochemistry, reports of metastatic tumours have been very rare. This suggests that many lesions reported in the past as metastatic AFX, which were diagnosed by examination of H&A stained slides alone, probably represented other tumours, such as spindle cell melanomas or sarcomatoid squamous cell carcinoma. Tumours described in younger patients in non-sun-damaged skin represent examples of atypical FH.
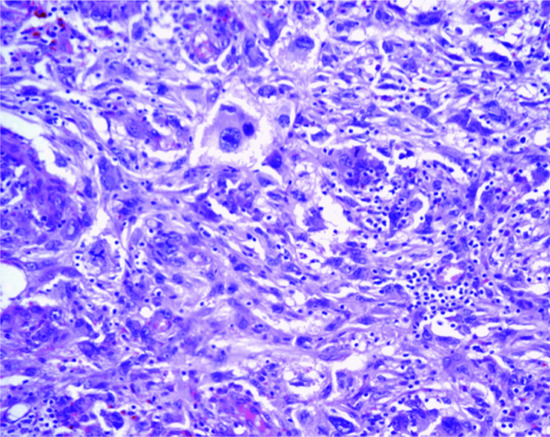
Figure 137.13 Prominent cellular pleomorphism in a case of atypical fibroxanthoma.
Clinical features
History and presentation
The lesions occur most frequently on the ears, bald scalp and cheeks (Figure 137.14). The lesions are often ulcerated and have a red fleshy appearance; they rarely exceed 30 mm in diameter, and are usually of less than 6 months’ duration. Exceptional cases occur as a result of immunosuppression in cardiac transplant [25]. Multiple lesions have been reported [26].
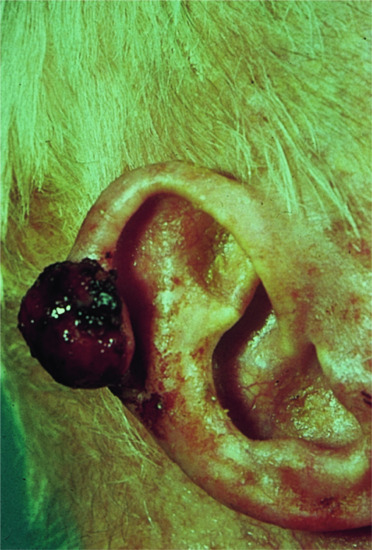
Figure 137.14 Typical clinical appearance of an atypical fibroxanthoma with a polypoid architecture.
Disease course and prognosis
Local recurrence may be seen in about 10% of cases and metastases to lymph nodes and internal organs are occasionally reported [27, 28]. The latter, however, may be examples of tumours that are now classified as dermal pleomorphic sarcomas. The latter have a local recurrence rate of around 28% and metastatic rate of 10% [5].
Management
The benign behaviour of the tumour enables it to be treated by limited local removal. Although rare cases are treated by Mohs micrographic surgery [29], tumours are usually relatively well circumscribed and the former treatment is rarely needed to achieve good clearance. Radiotherapy is not usually recommended.
VASCULAR TUMOURS
Reviews of vascular tumours may be found in [1, 2]. The vascular ectasias (see Chapter 103), verrucous haemangioma, cavernous and capillary haemangiomas and congenital haemangiomas (see Chapter 117) are described elsewhere.
REACTIVE VASCULAR LESIONS
Intravascular papillary endothelial hyperplasia [1,2,3,4]
Definition and nomenclature
Intravascular papillary endothelial hyperplasia is regarded as a form of organizing thrombus in which endothelial cells line hyalinized papillae. It usually presents as a primary phenomenon within a thrombosed blood vessel, usually a vein [2, 3, 4]. The secondary variant is commonly seen as an incidental finding within other vascular tumours, or in lesions such as haemorrhoids. Exceptionally, the same phenomenon is seen within a haematoma [4].
Epidemiology
Incidence and prevalence
A relatively common lesion.
Age
In general, this presents with a wide age range, although the primary form of the disease is more common in young adults.
Sex
The primary form is slightly more common in females.
Pathophysiology
All forms of the condition are the result of reactive proliferation of endothelial cells as a result of an organizing thrombus most often but not always secondary to trauma.
Pathology
The pathology is that of a widely dilated vascular channel in the dermis or subcutis, containing an organizing thrombus and prominent papillary projections with a hyalinized collagenous core. The latter are, usually lined by a single layer of bland endothelial cells. Mitotic figures are rare. The presence of hyalinized collagen lined by endothelial cells produces an appearance similar to the ‘dissection of collagen bundles’ described in angiosarcoma. Distinction from angiosarcoma, however, is easy, as the latter is only exceptionally purely intravascular; it also displays cytological atypia, multilayering and mitotic figures. In secondary forms of Masson tumour, the changes are seen within one or several vascular channels of a vascular tumour, usually a cavernous haemangioma or a vascular malformation.
Clinical features
History and presentation
The primary form presents as a slowly growing solitary asymptomatic or slightly painful bluish nodule less than 20 mm in diameter. The site of predilection is the head and neck, followed by the hand (particularly the fingers). Multiple lesions are exceptional [5].
Disease course and prognosis
Behaviour is benign and there is no tendency for local recurrence.
Differential diagnosis
Clinical presentation is that of a vascular tumour.
Management
Simple excision is usually curative.
Reactive angioendotheliomatosis [1-3]
Definition
A reactive vascular proliferation is usually multifocal and is associated with a number of systemic diseases. In the past, it was divided into a reactive and a malignant form. With the advent of immunohistochemistry, it became apparent that the malignant form is a variant of aggressive intravascular lymphoma (see Chapter 140).
Epidemiology
Incidence and prevalence
The condition is rare.
Age
The age range is wide although most cases occur in adults.
Sex
No sex predilection.
Pathophysiology
Predisposing factors
It has been described in association with systemic diseases, including bacterial endocarditis, peripheral vascular atherosclerotic disease, cryoglobulinaemia [2], liver and renal disease, antiphospholipid syndrome [4], amyloidosis [5] and sarcoidosis [6]. It is not clear how systemic diseases induce the vascular proliferation.
The dermis and, in some cases, the subcutis show a multifocal proliferation of clusters of capillaries lined by plump endothelial cells with little or no cytological atypia. A layer of pericytes surrounds each capillary. In some areas, dilated capillaries appear to contain smaller vascular channels within their lumina. Patients with cryoglobulinaemia show thrombosis of capillaries by hyaline eosinophilic globules.
Clinical features
History and presentation
Most patients present with multiple erythematous and/or haemorrhagic macules, papules and plaques located on the trunk and limbs. Patients with fewer, more localized, lesions may also be seen. In the latter cases, the association with systemic disease is not usually present. In patients with antiphospholipid syndrome or cryoglobulinaemia, ulcerated lesions may be present.
Disease course and prognosis
The condition is self-limited and usually resolves spontaneously within weeks.
Differential diagnosis
Not infrequently patients present with a livedo-like pattern in an unusual location (proximal rather than acral). Distinction from a vasculitic process may be difficult but ulceration is rare except in the very rare cases associated with the antiphospholipid syndrome or with cryoglulinaemia.
Management
There is no treatment available, but the condition is usually self-limited and resolves spontaneously within a few weeks.
Glomeruloid haemangioma [1,2]
Definition
This is a distinctive multifocal vascular proliferation that occurs in association with POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes; see Chapter 148) or with multicentric Castleman disease. This condition is best considered as a form of reactive angioendotheliomatosis in the setting of POEMS syndrome. Exceptional cases with no clinical features of POEMS syndrome have been reported [3, 4].
Epidemiology
Incidence and prevalence
This is a rare disease that presents almost exclusively in the context of POEMS syndrome and multicentric Castleman disease.
Age
It presents in adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The histological appearances in a typical case are striking, consisting of a multifocal dermal proliferation of clusters of closely packed dilated capillaries with a striking similarity to renal glomeruli. A layer of pericytes surrounds each capillary. Vacuolated cells are focally present and, in some cases, there are eosinophilic hyaline globules within the lumina of capillaries. These globules represent deposits of protein.
Clinical features
History and presentation
Patients present with multiple vascular papules on the trunk and limbs. Only a minority of these vascular lesions have the histological appearance of glomeruloid haemangioma; most have the histological appearance of cherry angiomas.
Management
The lesions do not tend to regress spontaneously. Individual lesions can be removed surgically, but because of their numbers this is not generally a practical option.
BENIGN VASCULAR TUMOURS
Tufted angioma
Definition and nomenclature [1-7]
Tufted angioma is a benign vascular tumour which represents a distinctive variant of capillary haemangioma. It has been suggested in recent years that this entity is closely related to kaposiform haemangioendothelioma [8] (see p. 137.33).
Epidemiology [1-5,6,7,8,9,10,11,12,13]
Incidence and prevalence
The incidence is low. More cases have been described in Japan compared to the US [9, 10].
Age
It predominantly occurs in children and young adults. Congenital as well as late-onset cases have been reported.
Sex
There is no sex predilection.
Pathophysiology
Genetics [14,15]
Most cases are sporadic. Familial predisposition has been described.
Pathology (Figure 137.15) [1-5, 6, 7, 9, 10, 11, 12, 13]
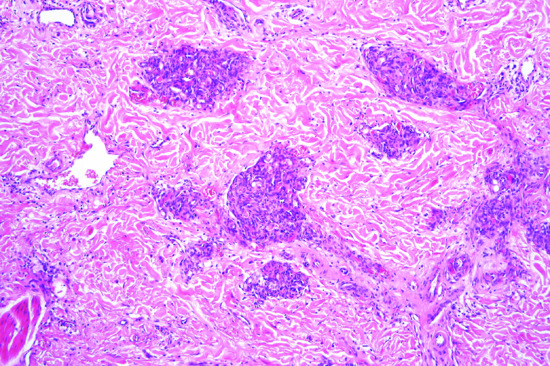
Figure 137.15 Tufted angioma. Multiple circumscribed vascular lobiules in a ‘cannonball’ distribution in the dermis.
Microscopically, multiple circumscribed vascular lobules in a ‘cannonball’ distribution are seen in the dermis and superficial subcutis. Lobules are composed of tightly packed and poorly canalized capillaries lined by endothelial cells and surrounded by pericytes. Some lobules bulge into the walls of dilated lymphatic channels creating a distinctive semilunar appearance at the periphery. In a number of cases, dilated thin-walled lymphatic-like vessels are seen in the background.
Clinical features [1-5, 6, 7, 8, 9, 10, 11, 12, 13, 16-19]
History and presentation
Typically, they present as slowly progressing ill-defined erythematous variably painful macules, papules, plaques or nodules. The lesion may attain a size of up to 10 cm. Multiple lesions are rare. An annular configuration is unusual. Hyperhidrosis and hypertrichosis have been described .A predilection for the neck or trunk is observed although unusual sites such as the oral mucosa have been reported [16]. It may be associated with a vascular malformation or Kasabach–Merritt syndrome [17, 18]. The presence of petechiae and ecchymotic patches are signs of alert.
Prognosis
The tumour is benign. Spontaneous regression may exceptionally occur [19].
Management
Complete excision, although difficult or impossible due to the extent of involvement, is curative.
Papillary haemangioma [1]
Definition
A distinctive dermal tumour characterized by vascular channels with papillary projections.
Epidemiology
Incidence and prevalence
Lesions are very rare.
Age
It presents in adults.
Sex
There is predilection for males.
Pathophysiology
Pathology
Tumours are relatively well circumscribed and consist of multiple dilated thin-walled vascular channels lined by bland endothelial cells and associated with papillary projections containing endothelial cells and pericytes and thick basement membrane-like material. A distinctive feature is the presence of intracytoplasmic eosinophilic globules within the endothelial cells.
Clinical features
History and presentation
Most lesions occur on the head and neck as a single asymptomatic papule.
Disease course and prognosis
Lesions are benign.
Management
Simple excision is the treatment of choice.
Lobular capillary haemangioma (pyogenic granuloma) [1,2]
Definition and nomenclature
A vascular nodule that develops rapidly, often at the site of a recent injury, and which is composed of a lobular proliferation of capillaries in an oedematous stroma.
Epidemiology
Incidence and prevalence
Lesions are very common.
Age
The age range is wide but there is a peak in the second decade of life [3].
Sex
There is predilection for males except lesions presenting in the oral cavity which are more common in females [4].
Pathophysiology
Predisposing factors
In a minority of cases, a minor injury, usually of a penetrating kind, has occurred a few weeks before the nodule appears. Lesions may also occur at the sites of burns [5].
Granuloma gravidarum is a variant of pyogenic granuloma that presents in the oral cavity during pregnancy.
Pathology
There is a lobular proliferation of small blood vessels, which erupt through a breach in the epidermis to produce a globular pedunculated tumour. The epidermis forms a collarette at the base of the lesion and covers part, or all, of the tumour in a thin layer. The proliferating vessels are set in a myxoid stroma, lacking in collagen in the earlier stages and relatively rich in mucin. The endothelial cells are plump, as in new granulation tissue, lining the vessels in a single layer. They are surrounded by a mixed cell population of fibroblasts, mast cells, lymphocytes, plasma cells and, where the surface is eroded, polymorphonuclear leucocytes (Figure 137.16). Mitotic figures may be prominent. Older lesions tend to organize and partly fibrose and may show focal bone formation. Late lesions can display focal degenerative atypia, raising the possibility of malignancy. In rare instances, particularly in children, and sometimes following treatment, satellite lesions, which have a similar pathology to the primary lesion, may develop around a pyogenic granuloma. These respond to simple destructive measures, thus ruling out malignancy [6]. In exceptional cases, extramedullary haemopoiesis may be seen [7]. Bacillary angiomatosis shows an almost identical histology to that of pyogenic granuloma [8]. However, in bacillary angiomatosis, pale epithelioid endothelial cells are prominent, neutrophils and nuclear dust are seen throughout the lesion and violaceous amorphous aggregates of bacilli which are positive with either Giemsa or Warthin–Starry stains are easily identified. The causative organism may also be identified by PCR.

Figure 137.16 Typical lobules of capillaries in a myxoid background in a case of pyogenic granuloma.
Clinical features
History and presentation
The tumour is vascular, bright red to brownish-red or blue-black in colour. It is partially compressible, but cannot be completely blanched and does not show pulsation. The surface of early bright-red lesions is usually thin intact epidermis. Older and darker lesions are frequently eroded and crusted, and may bleed very easily. Occasionally, the surface is raspberry-like or even verrucous. The size is commonly between 5 and 10 mm, but may reach 50 mm. The outline is rounded. The base is often pedunculated and surrounded by a collar of acanthotic epidermis; the lesion may be sessile. The common sites are the hands, especially the fingers (Figure 137.17), the feet, lips, head and upper trunk, and the mucosal surfaces of the mouth and perianal area. The initial evolution is rapid, but growth ceases after a few weeks. Spontaneous disappearance is rare. Lesions are not painful; patients mainly complain of the appearance or of recurrent bleeding. Rarely, pyogenic granulomas may occur within a port-wine stain [9]. There are reported cases of multiple pyogenic granulomas developing after exfoliative dermatitis [10] and, in a periungual location, after HAART [11]. Lesions mimicking pyogenic granuloma may occur during therapy with gefitinib [12], systemic 5-fluorouracil [13] and capecitabine [14]. Eruptive forms of this tumour have rarely been reported [15]. In this setting, distinction from bacillary angiomatosis is crucial, as the latter often presents with multiple lesions that can be clinically and histologically difficult to distinguish from pyogenic granuloma. Multiple lesions closely resembling pyogenic granulomas have been reported after systemic [16] and topical [17] treatment with retinoids. Subcutaneous [18, 19] and intravascular [20] variants are rarely seen and do not have distinctive clinical features. Interestingly, two cases of subcutaneous lesions have been described in patients with antiphospholipid antibodies [21].
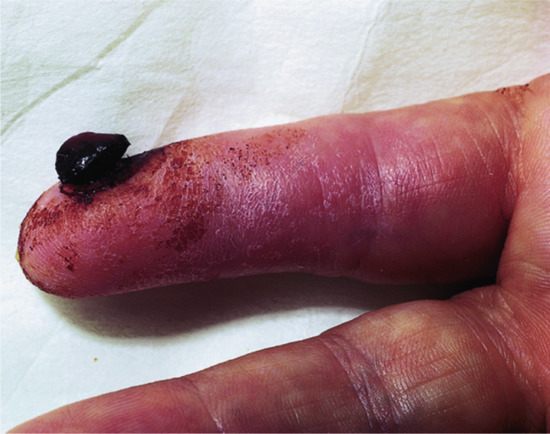
Figure 137.17 Clinical appearance of a pyogenic granuloma on a typical site at the tip of the finger.
Differential diagnosis
In most cases, the history and clinical appearance leave little doubt about the diagnosis, and microscopic confirmation is straightforward. In 38% of one case series, the clinical diagnosis of pyogenic granuloma proved to be wrong [22]. The errors included keratoacanthoma and other epithelial neoplasms, inflamed seborrhoeic keratoses, melanocytic naevi, melanoma and Spitz nevi, viral warts, molluscum contagiosum, angioma, glomus tumour, eccrine poroma, Kaposi sarcoma and metastatic carcinoma.
Disease course and prognosis
Simple excision is the treatment of choice, as lesions do not regress spontaneously. Local recurrence may be seen after incomplete excision.
Management
The pedunculated lesions are easy to treat by curettage with cauterization or diathermy coagulation of the base. A considerable proportion of pyogenic granulomas recur after such treatment, because the proliferating vessels in the base extend in a conical manner into the deeper dermis. In some areas—for instance in the nail fold or on the palmar aspect of a finger—it may be reasonable to carry out curettage and hope for the best. Wherever possible, it is desirable to excise a narrow, but deep, ellipse of skin beneath the lesion and close the wound with sutures. A small number of lesions have been treated with topical imiquimod 5% cream both in children and adults with complete resolution [23, 24]. Other treatment modalities that have been used include Nd:YAG laser [25], cryosurgery [26], intralesional steroids, flash lamp pulsed dye laser, sclerotherapy with sodium tetra decyl sulphate [27] and even injection of absolute ethanol [28].
Cirsoid aneurysm [1,2,3,4]
Definition and nomenclature
Cirsoid aneurysm is a small vascular proliferation characterized by small to medium-sized channels with features of arteries and veins. As opposed to deeper tumours showing similar features, shunting is absent.
Epidemiology
Incidence and prevalence
Lesions are relatively common.
Age
Most patients are young adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The dermis contains a mixture of scattered blood vessels with thick walls and features of veins and arteries (Figure 137.18).
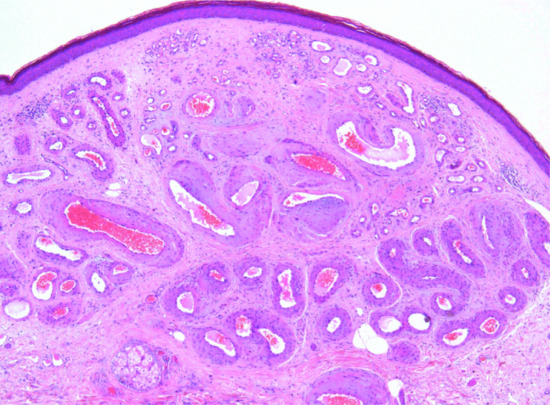
Figure 137.18 A dermal collection of thick- and thin-walled blood vessels in a typical case of cirsoid aneurysm.
Clinical features
History and presentation
Most lesions present on the head and neck region of young adults, with no sex predilection, as a small blue/red asymptomatic papule.
Management
As there is no associated shunting or deep component, simple excision is the treatment of choice.
Epithelioid haemangioma [1,2]
Definition and nomenclature
A benign locally proliferating lesion composed of vascular channels lined by endothelial cells with abundant pink cytoplasm and vesicular nuclei. There has, on occasion, been a difficulty with nomenclature such that the term Kimura disease has been applied but this condition is now viewed as distinct from angiolymphoid hyperplasia with eosinophils. In Kimura disease, the lesions occur in younger patients, are deeper seated, are associated with lymphadenopathy, have no initial overlying skin lesions and do not contain epithelioid endothelial cells [3, 4]. Furthermore, peripheral blood eosinophilia is much more common in Kimura disease. Exceptionally, angiolymphoid hyperplasia with eosinophilia may coexist with Kimura disease [5].
Epidemiology
Incidence and prevalence
These lesions have now been reported from many parts of the world. The cause is unknown, but most studies suggest a reactive process [6].
Age
The age range is wide but lesions most commonly occur in young adults.
Sex
Both sexes are equally affected.
Pathophysiology
Pathology [2,7]
A poorly circumscribed lobular lesion is seen. It is composed of clusters of proliferating capillaries and often thicker blood vessels lined by plump epithelioid endothelial cells (Figure 137.19) with little cytological atypia and rare mitotic figures. Around the blood vessels there is a cellular inflammatory infiltrate composed mainly of lymphocytes and large numbers of eosinophils. However, only less than half of cases contain a prominent infiltrate. Older lesions show sclerosis of the stroma and the epithelioid endothelial cells become more prominent. A frequent finding, particularly in larger lesions, is the involvement of larger blood vessels. Rare cases are entirely intravascular [8, 9]. The endothelial cells stain for vascular markers including CD34, CD31 and ERG (avian v-ets erythroblastosis virus E26 oncogene homologue). In cutaneous cases, the epithelioid endothelial cells are usually negative for keratins.
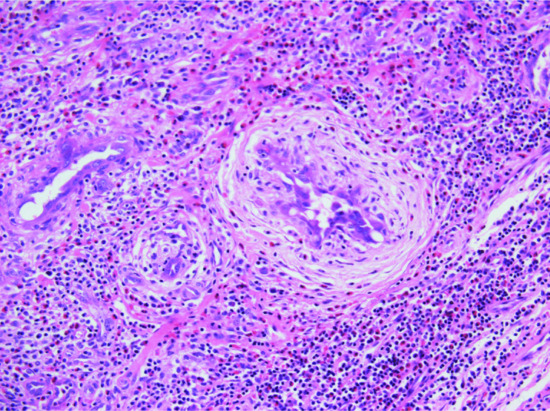
Figure 137.19 Vascular channels lined by epithelioid endothelial cells with abundant pink cytoplasm in a case of epithelioid haemangioma.
Clinical features [10,11,12,13,14] (Figure 137.20)
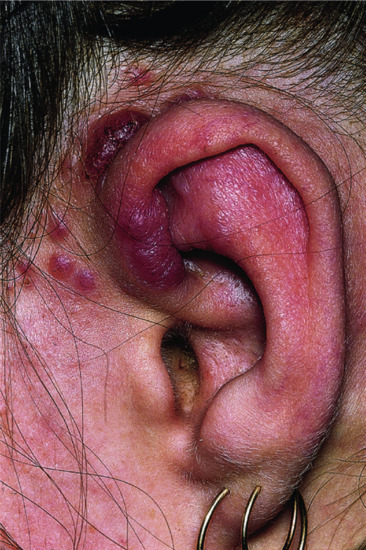
Figure 137.20 Epithelioid haemangioma, or angiolymphoid hyperplasia with eosinophilia.
(Courtesy of and copyright of Dr R. H. Champion, Addenbrooke's Hospital, Cambridge, UK.)
History and presentation
Affected individuals present with a cluster of small translucent nodules on the head and neck, particularly around the ear or the hairline. The lesions may also involve the oral mucosa [15]. Less frequently, lesions can involve the trunk and extremities. Involvement of deeper soft tissues and internal organs, including bone, can be seen. Individual nodules rarely exceed 2–3 cm in diameter, but occasionally deeper extension and larger subcutaneous lesions occur. Peripheral blood eosinophilia may be present but only in less than 10% of patients.
Disease course and prognosis
Spontaneous regression is seen in some cases after a variable period of time.
Management
Both surgery and radiotherapy are effective, but local recurrences are common. Treatment with Nd:YAG laser has been effective [16] and there is an anecdotal report of response to imiquimod cream [17].
Cutaneous epithelioid angiomatous nodule
Definition
This is tumour within the spectrum of vascular lesions characterized by epithelioid endothelial cells [1, 2, 3]. It is regarded by some as part of the spectrum of epithelioid haemangioma.
Epidemiology
Incidence and prevalence
Lesions are rare.
Age
Presentation is usually in adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The majority of lesions are superficial, well circumscribed and surrounded by an epithelial collarette. Occasional deeper tumours may be observed. It is composed of sheets of epithelioid endothelial cells with abundant pink cytoplasm, vesicular nuclei and a single small nucleolus. Cytological atypia is mild or absent and mitotic figures are variable. There is little tendency for formation of vascular channels but individual endothelial cells often contain intracytoplasmic vacuoles. In the background, scattered mononuclear inflammatory cells and eosinophils may be seen.
Clinical features
History and presentation
Lesions consist of a papule or nodule presenting in an adult, with a predilection for the trunk and limbs and, less commonly, involving the face. Multiple lesions are exceptional [1, 4, 5]. A lesion arising in a capillary malformation has been reported [6].
Disease course and prognosis
There is no tendency for recurrence after surgical treatment.
Management
Simple exision is the treatment of choice.
Acquired elastotic haemangioma [1,2]
Definition
This is a distinctive vascular lesion that develops in sun-exposed skin in association with solar elastosis.
Acquired elastotic haemangioma is rare and seems to be aetiologically related to chronic sun exposure.
Epidemiology
Incidence and prevalence
This is a rare lesion.
Age
Middle-aged to elderly people.
Sex
Predilection for females.
Ethnicity
Patients have fair skin.
Pathophysiology
Predisposing factors
The development of the lesion is clearly associated with sun exposure.
Pathology
Lesions are well circumscribed and consist of a superficial band-like proliferation of capillaries in the background of solar elastosis. Each capillary is surrounded by a layer of pericytes.
Clinical features
History and presentation
Lesions present mainly on the forearms or neck as a small red or blue circumscribed and asymptomatic plaque.
Disease course and prognosis
Lesions are benign and there is no tendency for local recurrence after excision.
Management
Simple excision is the treatment of choice and there is no tendency to local recurrence.
Hobnail haemangioma [1,2,3,4]
Definition and nomenclature
This is a benign vascular dermal proliferation characterized by small channels lined by endothelial cells with little cytoplasm and a prominent dark nucleus (hobnail cells). Formation of small papillae is also often seen. The original name proposed for this condition was based on a distinctive targetoid clinical appearance produced by bleeding and haemosiderin deposition. However, only a minority of lesions present with this typical appearance and therefore the alternative name of hobnail haemangioma has, been proposed. More recently, the denomination of superficial haemosiderotic lymphovascular malformation has been proposed [5, 6, 7].
Epidemiology
Incidence and prevalence
It is relatively uncommon.
Age
It has a predilection for young to middle-aged adults. Some lesions occur in children.
Sex
There is a slight predilection for males.
Pathophysiology
Predisposing factors
Trauma may play a part in its pathogenesis [4]. Occasionally, lesions vary according to the timing within the menstrual cycle [8].
Pathology
Pathological examination shows dilated vascular channels in the papillary and high reticular dermis, with a single layer of endothelial cells lining intraluminal papillary projections. These cells have a hobnail (‘matchstick’) appearance. They may occasionally be more numerous and appear to fill the lumen of the vessel. The vascular channels tend to disappear in the mid and lower reticular dermis, and the endothelial cells become less prominent and lose the hobnail appearance. Haemosiderin deposition is prominent and can be highlighted with a Perl stain. The endothelial cells stain for the lymphatic marker podoplanin (D2-40), suggesting that these lesions represent lymphangiomas rather than haemangiomas [9]. This has led to the suggestion that lesions represent a form of lymphatic malformation [5]. This is based on the usually negative staining of the endothelial cells in the proliferation for the endothelial cell marker Wilm tumour 1 gene [6, 7]. The pathological appearance may resemble Kaposi sarcoma, but this differential diagnosis can usually be resolved by clinicopathological correlation, as hobnail haemangioma is a solitary entity whereas Kaposi sarcoma is usually composed of multiple lesions. Histological distinction can be made if attention is paid to the symmetry of the lesion, the presence of hobnail endothelial cells with papillary projections and the absence of inflammation in hobnail haemangioma. Furthermore, hobnail haemangioma is not associated with HHV8 while all cases of Kaposi sarcoma are associated with this virus.
Clinical features
History and presentation
This entity presents as a rapidly developing asymptomatic solitary red or brown lesion, which in some cases has a central raised violaceous papule and is surrounded by a paler brown halo (targetoid appearance) [1]. Lesions with a clinical targetoid appearance are easy to diagnose. Any body site may be affected, but it has a predilection for the lower limbs and trunk. The oral mucosa may also be affected [3].
Disease course and prognosis
Lesions are benign and there is no tendency for local recurrence.
Management
Simple surgical excision is the treatment of choice; there is no tendency for recurrence.
Microvenular haemangioma [1,2,3]
Definition
This is a benign dermal vascular lesion characterized by proliferation of small vascular channels with features suggestive of venules.
Epidemiology
Incidence and prevalence
It is relatively rare.
Age
It presents mainly in young adults. Presentation in children is rare [4].
Sex
There is no sex predilection.
Pathophysiology
Pathology
There is a superficial and deep dermal proliferation of angulated thin-walled vascular channels, all of which are surrounded by a single layer of pericytes. These channels look like venules, are lined by flat bland endothelial cells and are surrounded by somewhat hyalinized collagen. A frequent finding is the infiltration of arrector pili muscles by vascular channels. Inflammation is not usually a feature. These lesions are negative for HHV8.
Clinical features
History and presentation
It presents as a solitary red-brown or bluish papule, nodule or plaque with a predilection for the limbs. Most lesions are less than 10 mm in diameter. Multiple lesions are rarely described [5].
Disease course and prognosis
Lesions are benign and there is no tendency for local recurrence.
Management
Simple surgical excision is the treatment of choice.
Sinusoidal haemangioma [1]
Definition
This is a benign dermal and/or subcutaneous variant of cavernous haemangioma composed of thin-walled dilated vascular spaces in a typical sieve-like distribution.
Epidemiology
Incidence and prevalence
Lesions are rare.
Age
It usually presents in adults.
Sex
There is a slight predilection for females.
Pathophysiology
Pathology
The lesion is usually well circumscribed, but several lobules of subcutaneous tissue may be focally affected by the tumour. A striking feature is the presence of back-to-back, dilated and congested thin-walled vascular channels. These channels are interconnected, and transverse sectioning is, in part, responsible for the distinctive sinusoidal appearance. Pseudopapillary projections are focally present and thrombosis with dystrophic calcification may also be seen. Focal cytological atypia secondary to degenerative changes may be seen. Distinction from angiosarcoma, particularly in tumours presenting in the breast, is based on the fact that the latter occurs in the breast parenchyma and only invades the dermis and subcutis secondarily. Tumour cells in angiosarcoma also display cytological atypia, multilayering and mitotic figures.
Clinical features
History and presentation
Sinusoidal haemangioma presents as a solitary blue asymptomatic nodule, particularly on the trunk or upper limbs. The dermis and subcutaneous tissue overlying the breast is not uncommonly involved and may suggest a diagnosis of angiosarcoma (differentiating features are discussed below).
Disease course and prognosis
Lesions are benign with no tendency for local recurrence.
Management
Simple surgical excision is the treatment of choice.
Spindle cell haemangioma [1-5]
Definition and nomenclature
This is a benign vascular neoplasm. Although initially described as a low-grade malignant lesion with a high tendency for local recurrence and minimal potential for metastasis, further studies demonstrated that it is a benign multifocal process [3, 4, 5]. Confirmation of its benign nature has led to change of the name ‘haemangioendothelioma’ for ‘haemangioma’, as the former implies low-grade (intermediate-grade) malignant potential [6].
Epidemiology
Incidence and prevalence
Spindle cell haemangioma is relatively rare.
Age
The age range is wide but lesions often present in childhood or adulthood.
Sex
Males and females are affected equally.
Pathophysiology
Pathology
Low-power magnification reveals single or multiple fairly well-circumscribed haemorrhagic nodules. Origin from a pre-existing blood vessel is often seen, and individual lesions may be entirely intravascular. Dilated, thin-walled, congested, cavernous-like vascular spaces are intermixed with more cellular areas composed of bland short spindle-shaped cells with the formation of slit-like spaces. Scattered, more epithelioid cells, with pink cytoplasm and prominent vacuolation, are also seen. The spindle-shaped cells are a mixture of endothelial cells, pericytes and fibroblasts. Focal degenerative cytological atypia may be present. Immunohistochemistry reveals staining for CD31 and CD34 and focal staining for smooth muscle actin.
Genetics
R132C IDH1 and IDH2 mutations identical to those found in the enchondromas of Maffucci and Ollier syndrome have been found in this tumour supporting a true neoplasia [7].
Clinical features
History and presentation
The process is often associated with lymphoedema, Maffucci syndrome (multiple enchondromas) [8], early-onset varicose veins or Klippel–Trenaunay syndrome. The majority of cases present in the distal limbs, particularly the hands and feet, as multiple cutaneous or subcutaneous, red or bluish nodules. Deeper tumours are rare. Presentation at other sites including the neck and oral cavity is very rare [9, 10]. Lesions continue to appear over many years, indicating multifocality rather than true recurrences. Most nodules are less than a few centimetres in diameter; they may occasionally be painful.
Disease course and prognosis
Behaviour is benign but new lesions appear over time.
Management
Single lesions are easily treated with simple excision. Treatment is more difficult in the presence of multiple lesions.
Symplastic haemangioma
Definition
This is not a distinctive variant of haemangioma but represents extensive degenerative changes in a pre-existing haemangioma, closely mimicking malignancy [1, 2, 3]. Only a handful of cases have been reported. From personal experience with a small number of cases, the pre-existing vascular lesion is often either not identifiable or it represents a cirsoid aneurysm.
Epidemiology
Incidence and prevalence
Lesions are rare.
Age
It presents in adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
These tumours are often polypoid and well circumscribed, and do not tend to be ulcerated. The typical histological picture consists of dilated and congested, thin- to thick-walled vascular spaces surrounded by a variable cellular stroma with frequent myxoid change and haemorrhage. Stromal cells and smooth muscle cells within the vessel walls show variable cytological atypia, consisting of nuclear enlargement and hyperchromatism. The endothelial cells lining the vascular spaces may be plump but do not display cytological atypia, multilayering or mitotic activity, allowing distinction from an angiosarcoma. Often cells have a bizarre appearance and multinucleated cells are common. Mitotic figures may be found but tend to be rare. Very occasional atypical mitotic figures may also be seen.
Clinical features
History and presentation
These are not distinctive. However, the patient is usually an adult, and the description is usually of a longstanding lesion that has rapidly increased in size.
Disease course and prognosis
Lesions are benign.
Management
Simple excision is the treatment of choice.
VASCULAR TUMOURS OF INTERMEDIATE MALIGNANCY
Kaposiform haemangioendothelioma [1,2,3,4,5]
Definition and nomenclature
Kaposiform haemangioendothelioma is a locally aggressive vascular neoplasm, often associated with Kasabach–Merritt phenomenon (KMP). It occurs in skin and deep soft tissues, the abdominal cavity and within the thorax [1]. It has been suggested that kaposiform haemangioendothelioma and tufted angioma are part of the same spectrum (see p. 137.25) [6, 7].
Epidemiology
Incidence and prevalence
This tumour is rare. The prevalence of kaposiform haemangioendothelioma in Massachusetts, USA, is approximately 0.91 cases per 10 000 children [8].
Age
This presents mainly in young children under the age of 2 years. Up to 60% of patients present during the neonatal period [8]. Presentation in adults in distinctively rare [9].
Sex
There is no sex predilection.
Pathophysiology
Pathology
The growth pattern is lobular and infiltrative. Multiple nodules with haemorrhage and surrounding fibrosis are seen. Tumour lobules are composed of bland spindle-shaped cells with poorly defined pink cytoplasm. Cleft-like spaces are often seen between spindle-shaped cells, and the resemblance to Kaposi sarcoma can be striking. However, numerous capillaries, often associated with microthrombi, are also present in tumour lobules. Epithelioid endothelial cells with focal vacuolation are also present. These features, along with the striking lobular architecture of the tumour and negative staining for HHV8, allow distinction from Kaposi sarcoma. Podoplanin (D2-40), a marker of lymphatic endothelium, is positive in the spindle cells and lymphatic channels around tumour lobules [10]. This finding suggests a lymphatic line of differentiation for this tumour.
Clinical features
History and presentation
In 20% of cases, there is an association with lymphangiomatosis [3]. Although in initial reports the most common presentation was that of a large retroperitoneal infiltrative mass, superficial tumours appear to be more common [8]. Intrathoracic lesions can also occur but they are rare [8]. Involvement of neighbouring organs and the very common association with Kasabach–Merritt syndrome may lead to death. This complication is less common but nonetheless frequent in more superficial tumours, particularly those located in the dermis and subcutaneous tissue [5, 8]. Superficial tumours are associated with KMP in 71% of cases [8]. Tumours involving only bone and those occurring after infancy are not usually associated with KMP [8]. Cutaneous tumours are more common on the limbs mainly in areas overlying the joints [8, 11]. Multifocal lesions are exceptional [12, 13].
Disease course and prognosis
It appears clear that this lesion is truly neoplastic. Although it is not malignant, it causes morbidity and mortality due to its location and the frequent occurrence of consumption coagulopathy (KMP).
Management
Complete excision is desirable as local recurrence is frequent, but this may be difficult to achieve when involvement is extensive. Spontaneous regression does not occur. In cases with large and deep-seated lesions and/or KMP, where surgery is not an option, alternative treatments include embolization, chemotherapy with vincristine, corticosteroids, pirolimus, propranolol, interferon α and even low-dose radiotherapy [7, 14, 15, 16, 17, 18, 19]. Antiaggregant therapy can be added in cases with KMP [20].
Giant cell angioblastoma [1,2]
Definition
This is a very rare entity of which only a handful of cases have been reported. It is not clear whether it represents a true vascular tumour and, until it is more clearly delineated and defined, it has not been included in the latest World Health Organization (WHO) classification of vascular tumours.
Epidemiology
Incidence and prevalence
Tumours are exceptionally rare.
Age
Lesions are congenital.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The tumour is composed of infiltrative vascular channels lined by a single layer of bland endothelial cells and intermixed with solid nodules composed of spindle-shaped cells, histiocyte-like cells and osteoclasts. A plexiform growth pattern is often seen.
Clinical features
History and presentation
Tumours are congenital and have been described on the hand, palate and scalp. The tumour is diffusely infiltrative and slowly growing. Two of the reported cases showed no progression after incomplete excision.
Management
Two cases have been successfully treated with interferon α-2b [3].
Retiform haemangioendothelioma [1,2,3]
Definition and nomenclature
Retiform haemangioendothelioma is a rare variant of low-grade angiosarcoma with a tendency for local aggressive behaviour. It is characterized by arborizing vascular channels lined by endothelial cells with hobnail morphology.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
Presentation is mainly in young adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
Scanning magnification is distinctive and reveals an infiltrative tumour composed of arborizing, thin-walled, narrow, vascular channels with a striking resemblance to the rete testis. Vascular spaces are lined by bland hobnail endothelial cells with prominent nuclei and scanty cytoplasm. Intravascular papillae with collagenous cores, similar to those seen in papillary endolymphatic angioendothelioma, are sometimes seen. The surrounding stroma often appears hyalinized; a prominent mononuclear inflammatory cell infiltrate is common. The endothelial cells stain for vascular markers. The lineage is likely to be lymphatic but positivity for lymphatic endothelial cell markers such as podoplanin is not consistently demonstrated [4]. There is no relationship to HHV8.
Clinical features [1,2,3,5]
History and presentation
Retiform haemangioendothelioma presents as a slowly growing asymptomatic dermal and subcutaneous plaque or nodule. Lesions rarely present as a bruise [6]. Presentation with multiple lesions is exceptional [7]. Rarely, there is an association with lymphoedema or radiotherapy and in one case the tumour arose in the setting of a cystic lymphangioma [8].
Disease course and prognosis
Local recurrence occurs in up to 60% of cases. So far, there has only been one report of a tumour metastasizing to a regional lymph node, and a further lesion has spread locally to soft tissues [9]. No tumour-related deaths have been reported.
Management
Wide local excision is the treatment of choice.
Papillary intralymphatic angioendothelioma [1]
Definition and nomenclature
Defining this entity is difficult because, since its original description in 1969, few further convincing cases have been described [1, 2, 3, 4]. Furthermore, the original series included some examples of what is now known as retiform haemangioendothelioma. Recently, the tumour has been better characterized under the preferred name of ‘papillary endolymphatic angioendothelioma’ [5]. It belongs to the family of tumours with hobnail endothelial cells, and it is characterized by dilated cavernous-like lymphatic spaces with frequent papillary projections.
Epidemiology
Incidence and prevalence
Tumours are very rare.
Age
It presents mainly in infants and children, with 25% of the cases occurring in adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
This tumour is composed of dilated thin-walled channels simulating a cavernous lymphangioma. These channels are lined by bland hobnail endothelial cells with very rare mitotic figures. A striking feature is the formation of intraluminal papillary tufts with hyaline cores. Aggregates of mononuclear inflammatory cells may be seen around the vascular channels.
Clinical features
History and presentation
Presentation is as a slowly growing asymptomatic plaque or nodule with a predilection for the limbs.
Disease course and prognosis
In the original series of six cases, a tendency for local recurrence and metastasis to regional lymph nodes was reported [1], but in a series of 12 cases, none of the eight cases with follow-up recurred locally or metastasized [5]. It therefore seems likely that the behaviour of this tumour is benign. Further studies are needed to confirm whether it deserves to be kept in the group of tumours of intermediate behaviour.
Management
Until the issue regarding the biological behaviour of this tumour is resolved, complete excision is recommended.
Composite haemangioendothelioma [1,2,3]
Definition
This is a tumour defined as a vascular neoplasm made of a mixture of varying proportions of different histological patterns including benign, low grade and/or malignant.
Epidemiology
Incidence and prevalence
Tumours are very rare.
Age
Most tumours present in adults although cases in children have been described exceptionally.
Sex
There is no sex predilection.
Pathophysiology
Pathology
Tumours are infiltrative and occupy the dermis and subutaneous tissue. The proportion of the different components varies and may include spindle cell haemangioma, lymphangioma circumscriptum, retiform haemangioendothelioma, papillary intralymphatic angioendothelioma, epithelioid haemangioendothelioma and conventional angiosarcoma.
Clinical features
History and presentation
The tumours present predominantly on the limbs, with a predilection for the hands and feet, as longstanding nodules or plaques, red or blue in colour, and often haemorrhagic. Most lesions are several centimetres in diameter and lymphoedema is a common occurrence. Rare patients have associated Maffucci syndrome [2].
Disease course and prognosis
It is possibly determined by the tumour with the highest histological grade. There is an increased tendency for local recurrence and lymph node metastases have been documented [3].
Management
Complete excision is the treatment of choice.
Pseudomyogenic haemangioendothelioma [1,2,3,4,5]
Definition and nomenclature
A low-grade malignant vascular rarely metastasizing often multifocal neoplasm lacking histological vasoformative features and displaying features that mimic epithelioid sarcoma or a myogenic tumour [1, 2, 3].
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
It has a predilection for young adults but a smaller number of tumours develop in older individuals.
Sex
There is marked predilection for males.
Pathophysiology
Pathology
Tumours are infiltrative and consist of sheets of cells many of which have abundant pink cytoplasm simulating rhabdomyoblasts. A smaller percentage of cells are spindle shaped or purely epithelioid. Nuclei are vesicular and a small nucleolus is often seen. Cytological atypia that can be pronounced is observed in a small percentage of cases. Mitotic activity is low and up to half of the tumours contain abundant neutrophils [3]. Tumour cells are usually positive for the pankeratin marker AE1/AE3 and also for vascular endothelial cell markers mainly FLI1 and ERG. CD31 is only positive in 50% of cases. Other keratins mainly MNF116 are usually negative.
Genetics
A recurrent translocation t(7;19)(q22;q13) resulting in a SERPINE-FOSB fusion gene has been described [6, 7].
Clinical features
History and presentation
The evolution of the tumours is usually short, not uncommoly less than 2 years. Almost 70% of patients present with multiple nodules that affect the dermis and subcutis, and often deeper soft tissues including skeletal muscle (up to half of patients). Bone involvement is seen in 20% of cases [3]. Most tumours present on lower limbs, less commonly on the trunk and upper limbs and very rarely on the head and neck. Pain is a frequent symptom and tumours are usually less than 3 cm in diameter.
Disease course and prognosis
About 60% of patients develop local recurrence or development of similar lesions in the same area. Regional lymph node metastasis was reported in one case and only two patients have died of the disease [1, 3].
Management
Complete surgical excision is the treatment of choice.
MALIGNANT VASCULAR TUMOURS
Angiosarcoma [1,2-4]
Definition and nomenclature
This is a malignant vascular tumour arising from both vascular and lymphatic endothelium. Except for the pure epithelioid variant of angiosarcoma (see later), cutaneous angiosarcoma almost exclusively occurs in three settings: idiopathic angiosarcoma of the face, scalp and neck [2, 3, 4], angiosarcoma associated with chronic lymphoedema (Stewart–Treves syndrome) [5, 6, 7, 8, 9] and post-irradiation angiosarcoma [10, 11, 12]. In this chapter, the terms ‘angiosarcoma’ and ‘lymphangiosarcoma’ are used interchangeably.
Epidemiology
Incidence and prevalence
Angiosarcoma of the scalp and face of the elderly is very rare.
Stewart–Treves syndrome occurs in 0.5% of patients who survive mastectomy for more than 5 years and the mean interval between mastectomy and the appearance of the tumour is 10.5 years [9]. Not all patients have received radiotherapy in association with the mastectomy, and not all have had axillary nodes removed. Lymph-oedema is not invariably present, or it may be late in appearing and antedate the tumour by only a short time. The incidence and cause of postmastectomy lymphoedema have been reviewed [13]. In the majority of cases, the clinical course and autopsy findings have shown that the treatment of the breast carcinoma was successful and that patients have had less frequent involvement of the axillary nodes than usual [9]. A small number of cases have arisen in lymphoedema of the lower limb, or in the upper limb without breast cancer and mastectomy [14]. Most of these patients were women. Multiple primary malignancies have occurred in 8% of cases of Stewart–Treves syndrome [6] and a systemically acting carcinogen has been suggested [8, 9]. There is no evidence to support this.
Post-irradiation angiosarcoma is rare and most cases arise in the skin after radiotherapy for breast or, less commonly, internal cancer [10, 11, 12].
Angiosarcoma occurring in other settings is very rare. Angiosarcoma may also exceptionally occur in benign vascular tumours including vascular malformations [15], in a large blood vessel [16], in association with a plexiform neurofibroma in neurofibromatosis [17], in a schwannoma [18], in a malignant peripheral nerve sheath tumour [19, 20], in xeroderma pigmentosum [21], in a gouty tophus [22], in association with vinyl chloride exposure [23], in association with immunosuppression in organ transplantation [24] and as the mesenchymal component in a metaplastic carcinoma [25]. An exceptional case of an angiosarcoma producing granulocyte colony-stimulating factor and inducing a leukaemoid reaction has been described [26].
Age
- Angiosarcoma of the scalp and face of the elderly by definition affects patients in the seventh to ninth decades of life and only rarely patients in the sixth decade of life.
- Stewart–Treves syndrome: the mean age at appearance of the angiosarcoma is 62 years.
- Post-irradiation angiosarcoma: occurs mainly in adults, predominantly old-aged patients.
- Angiosarcoma occurring in other settings: mainly adults. Presentation in children is extremely rare [27].
Sex
- Angiosarcoma of the scalp and face of the elderly affects mainly males.
- Stewart–Treves syndrome and post-irradiation angiosarcoma affects mainly women. Two cases of Stewart–Treves syndrome have been reported in men following mastectomy [28].
- Angiosarcoma occurring in other settings: there is no sex predilection.
Pathophysiology
Pathology [4,29-31]
In the well-differentiated tumour, vascular channels infiltrate the normal structures in a disorganized fashion, as if trying to line every available tissue space with a layer of endothelial cells. The collagen is characteristically lined by tumour cells in a pattern that has been described as ‘dissection of collagen’ (Figure 137.21). Tumour cells may be plumper than normal, double-layered in places and form solid intravascular buds. The pattern of growth is more suggestive of lymphatic vessels than blood vessels, but both are probably involved. Haemorrhage is often prominent. Less well-differentiated tumours show more atypical pleomorphic endothelial cells, often with spindle cell morphology, which may be heaped into several layers or become syncytial. Advancing malignancy may be associated with loss of vascular pattern and proliferation of cell masses. Rare cases are composed of granular cells [32]. In exceptional cases, tumours may be extensively infiltrated by lymphocytes or macrophages. In these cases, distinction from a lymphoma may be difficult [33, 34].
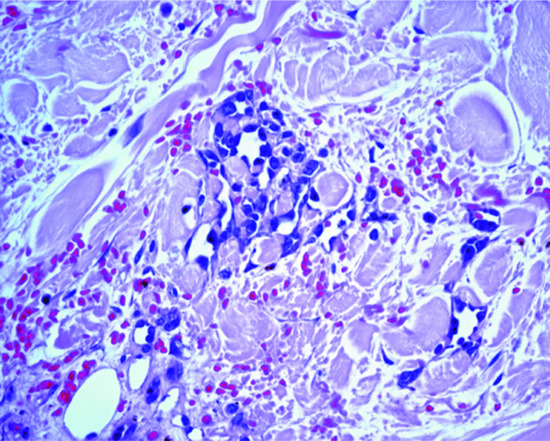
Figure 137.21 Well-differentiated angiosarcoma, with thin-walled irregular vascular channels lined by atypical endothelial cells. Note the dissection of collagen pattern.
Immunohistochemical studies have indicated that antibodies to CD31 are the most reliable markers for routine use, compared with antibodies against von Willebrand factor and CD34 [7]. However, a panel of antibodies including the three markers is recommended in difficult cases as positivity to the various markers varies. Recently, an antibody against the carboxy terminal of the FLI-1 protein, a nuclear transcription factor member of the ETS family of DNA-binding transcription factors, has been shown to be a fairly specific marker of endothelial cells [35]. A more specific and sensitive marker of endothelial cell differentiation is ERG a further member of the ETS family has recently been described [36].
Genetics
Cytogenetics studies of soft-tissue and cutaneous angiosarcoma are limited to a few case reports. Most analysed cases have shown complex cytogenetic aberrations [37, 38]. MYC gene amplification and overexpression by immunohistochemistry has been demonstrated mainly in post-irradiation angiosarcomas [39]. However, it may also occur in other types of cutaneous angiosarcomas [40].
Clinical features
History and presentation [3,4,41,42]
In all types of angiosarcoma, the first sign may be an area of bruising, often thought by the patient to be traumatic (Figure 137.22). Dusky blue or red nodules develop and grow rapidly, and fresh discrete nodules appear nearby. In some cases, haemorrhagic blisters are a prominent feature. As the tumours grow, the oedema may increase and older lesions may ulcerate. Multifocality is a very frequent finding; this makes surgical excision very difficult, particularly in those cases occurring on the face and scalp. Dissemination occurs early, with the first visceral deposits usually being in the lung and pleural cavity.
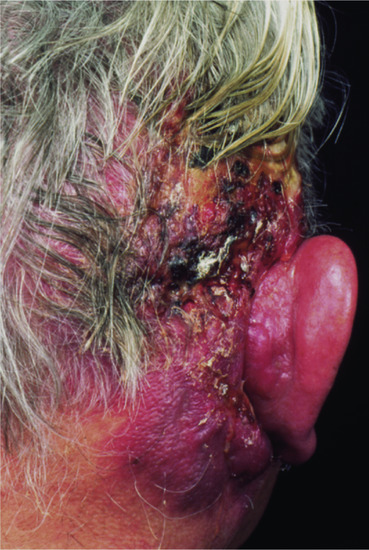
Figure 137.22 Typical haemorrhagic appearance of an angiosarcoma.
Disease course and prognosis
Most studies reporting outcomes have confined their attention to idiopathic angiosarcoma of the face, neck and scalp, in which the reported 5-year survival is low, at between 12% and 33% [3, 43]. Angiosarcomas arising in the setting of chronic lymphoedema and after radiotherapy appear to be equally aggressive. Combined series of idiopathic angiosarcoma of the face and scalp, other cutaneous angiosarcomas and angiosarcomas occurring in internal organs report 5-year survival rates varying between 24 and 34% [1, 44, 45, 46]. Features found to affect prognosis vary between different studies. Tumour size and completeness of excision appear to be more reliable factors to predict outcome [26]. It has been suggested that a high mitotic count correlates with poor prognosis and that a heavy mononuclear inflammatory cell infiltrate correlates with good prognosis [3, 5, 26]. A recent study of cutaneous sporadic angiosarcomas, excluding those associated with radiotherapy or with chronic lymphoedema, has found by univariate analysis that factors associated with higher mortality include older age, lesions on the trunk and limbs compared to those on the head and neck, necrosis and epithelioid cell morphology [47]. Tumours with necrosis and epithelioid morphology were classified as high risk and confirmed by multivariate analysis to be associated with higher mortality. The depth of invasion correlated with higher risk of local recurrence. The caveat with this study is that it combines traditional head and neck angiosarcomas with those showing a more pure epithelioid cell morphology that usually occurs elsewhere and are usually classified under a different category.
Management [3]
All angiosarcomas, regardless of the setting in which they occur, have a bad prognosis. In the less malignant types, wide excision and grafting has controlled some cases. The response to radiotherapy is disappointing and is usually only palliative. In the early stages of angiosarcoma of a limb, radical amputation may offer a hope of cure. In idiopathic angiosarcoma of the head and neck, a very small percentage of patients with smaller lesions (usually less than between 5 and 10 cm in diameter at presentation) can be successfully treated with radical wide-field radiotherapy and surgery [1, 44, 45, 46]. The best chance of survival in these patients resides in wide surgical excision followed by radiotherapy [48]. A combination of radiotherapy and taxanes (paclitaxel and docetaxel), the latter used for induction and maintenance therapy, has been shown to improve the overall survival of patients with cutaneous angiosarcoma compared to those treated with surgery and radiotherapy [49]. Combination of taxanes and anthracyclines has also been described to have some effect in the control of disease [50]. The best approach to the management of these tumours is by individualizing cases at a multidisciplinary setting.
Epithelioid haemangioendothelioma [1,2,3]
Definition
Epithelioid haemangioendothelioma is a distinctive tumour characterized by epithelioid endothelial cells arranged in strands or as individual units, in a myxoid or hyalinized stroma. It was initially described as a low-grade malignant tumour, but it has recently been proposed that it should be classified as a fully malignant neoplasm, in view of the associated morbidity and mortality [3]. However, small primary cutaneous lesions appear to have an indolent behaviour.
Epidemiology
Incidence and prevalence
This tumour may occur in many internal organs, and it is more commonly seen in deeper soft tissues. Involvement of the skin may occur primarily or as a result of direct extension from a deep-seated primary. Less than 10% of cases occur primarily in the skin [4].
Age
There is predilection for middle-aged adults.
Sex
Males are equally affected than females.
Pathophysiology
The neoplasm is infiltrative and is composed of strands, cords and nests of endothelial cells in a hyaline or myxoid stroma. Dermal lesions often consist of a fairly well-defined nodule. The tumour cells have epithelioid morphology and consist of pink cytoplasm, vesicular nuclei and inconspicuous nucleoli. Angiocentricity is commonly seen and tumours often arise from a medium-sized vein or even an artery. Formation of vascular channels is not readily apparent but a common finding is the presence of intracytoplasmic vacuoles with or without red blood cells. A small number of cases display cytological atypia, which may be prominent, and a high mitotic count. There is no clear correlation between cytological grade and behaviour. Occasional tumours overlap with epithelioid angiosarcoma. Staining for endothelial cell markers, including ERG and CD31, is usually positive, and 20–30% of cases are focally positive for keratin [3, 5]. Podoplanin (D2-40) is also frequently positive in tumour cells.
Genetics
Most tumours show a translocation t(1;3) (p36.3;q25) that results in fusion of the WWTR1 and CAMTA1 genes [6, 7, 8, 9]. In a group of tumours, a novel fusion, involving genes YAP1 and TFE3 on chromosomes 11 and X, respectively, has been demonstrated.
Clinical features [1,2,3]
History and presentation
Cutaneous tumours are usually small, but deeper lesions are often several centimetres in diameter. Pain is a frequent complaint, probably due to angiocentricity. Involvement of other organs, including the lung, liver and bone, may be seen in some cases, and it is not clear whether this represents multicentricity or metastatic spread.
Disease course and prognosis
Purely cutaneous tumours appear to have a benign behaviour, but there is some tendency for local recurrence. Deeper tumours have a recurrence rate of up to 15% and a mortality rate of 20% [1, 2, 3].
Management
Complete excision with clear margins is essential.
Epithelioid angiosarcoma [1-3,4]
Definition
A distinctive variant of angiosarcoma composed almost exclusively of endothelial cells with an epithelioid morphology, often mimicking a carcinoma. This tumour represents the malignant end of the spectrum of tumours with epithelioid cell morphology.
Epidemiology
Incidence and prevalence
This is a rare tumour that mainly occurs in deep soft tissue, but that may present primarily in the skin or other organs.
Age
There is predilection for young to middle-aged adults.
Sex
Males are more frequently affected.
Pathophysiology
Pathology [1-3,4]
Sheets of atypical epithelioid cells with abundant pink cytoplasm, vesicular nuclei and a single eosinophilic nucleolus occupy the dermis and/or subcutis. Haemorrhage and haemosiderin deposition is often seen. Formation of vascular channels is not readily apparent, and the main feature is the presence of intracytoplasmic vacuoles with or without red blood cells in variable numbers of tumour cells. Mitotic figures are common. Tumour cells are variably positive for vascular markers including ERG, CD31, CD34, FLI-1 and von Willebrand factor. In 50% of cases, there is positivity for cytokeratin. Focal positivity for epithelial membrane antigen is also seen in 25% of cases [4]. Ordinary angiosarcomas such as those occurring on the head of elderly patients, those associated with radiotherapy and those associated with chronic lymphoedema may display focal areas with epithelioid endothelial cells. These tumours should not be classified as epithelioid angiosarcomas.
Clinical features [1, 2, 3, 4]
History and presentation
Cutaneous tumours present in young to middle-aged adults, with a predilection for the extremities. The typical presentation is that of solitary, or more rarely multiple, asymptomatic papules or nodules which are often haemorrhagic. It is not clear whether multiple lesions represent multifocality or metastatic disease. Occasional cases have been reported in association with a foreign body [5], radiotherapy [1] or an arteriovenous fistula [6]. Epithelioid angiosarcoma arising in another organ may present with cutaneous metastases [7].
Disease course and prognosis
Although it was initially suggested that cutaneous epithelioid angiosarcoma has a relatively good prognosis, this was based on only very few cases with limited follow-up [2]. Overall, the behaviour of these tumours appears to be aggressive with a mortality rate of more than 55% after 3 years [4].
Management
Complete excision and close follow-up are indicated.
LYMPHATIC TUMOURS
Kenneth Y. Tsai
Departments of Dermatology & Immunology, University of Texas MD Anderson Cancer Center, Houston, TX, USA
Cavernous lymphangioma, cystic hygroma and lymphangioma circumscriptum are described in Chapter 105.
Acquired progressive lymphangioma
Definition and nomenclature
This is a benign dermal tumour composed of irregular lymphatic channels dissecting between collagen bundles.
Epidemiology
Incidence and prevalence
Tumours are very rare.
Age
There is a predilection for middle-aged adults but children may be affected.
Sex
Males are slightly more affected than females.
Pathology [1-3,4]
Low-power examination reveals an ill-defined often pan-dermal proliferation of irregular thin-walled lymphatic channels dissecting between collagen bundles. These channels tend to be orientated parallel to the epidermis and are lined by a single layer of bland endothelial cells. Involvement of the subcutaneous tissue is rare. Distinction from the lymphangiomatous variant of Kaposi sarcoma is often very difficult, but in the former there are aggregates of inflammatory cells including plasma cells, and the cells lining the vascular channels are usually positive for HHV8. Distinction from a well-differentiated angiosarcoma is based on the absence of cytological atypia and mitotic figures.
Clinical features [1, 2, 3, 4]
Lesions typically present as slow-growing nondescript solitary vascular or pigmented, 0.3–10 cm macules or papules on the head and neck, and less frequently on the oral mucosa, extremities and trunk [4].
Multiple lesions are exceptional [3].
Management
Excision is all that is required; there is no tendency for local recurrence [1, 4, 5]. Recently, pulsed dye laser has been successfully used [6].
Atypical vascular proliferation after radiotherapy [1-3,4,5,6,7]
Definition and nomenclature
Atypical vascular proliferation after radiotherapy (AVPR) defines a group of vascular lesions that occur months or years after radiotherapy—mainly, but not exclusively, after breast cancer. Lesions may also present after radiotherapy for ovarian and endometrial carcinoma.
Epidemiology
Incidence and prevalence
These lesions are rare.
Age
Most patients are middle-aged to elderly.
Sex
Almost all cases affect females.
Associated diseases
AVPRs have been described almost exclusively in the context of prior radiation therapy for breast cancer, particularly breast-conserving therapy with adjuvant radiation therapy, though cases related to radiation therapy for multiple myeloma, cervical carcinoma, Hodgkin disease, ovarian cancer, endometrial carcinoma and melanoma have been reported [4].
Pathophysiology
MYC gene amplification and overexpression by immunohistochemistry has been demonstrated in post-irradiation angiosarcomas but not in AVPRs and this is an important aid in differential diagnosis [8].
There is a clear aetiological link with radiotherapy [1, 2, 3, 4, 5, 6, 7]. The demonstration of staining of the endothelial cells for lymphatic markers suggests a lymphatic line of differentiation. Although the relationship between AVPR and post-radiation angiosarcoma has been controversial, the more commonly accepted view is that AVPR represents part of a spectrum of lesions with post-radiation angiosarcomas [4]. Most AVPR are banal but occasionally progression from AVPR to angiosarcoma is seen.
Pathology (Figure 137.23a,b)
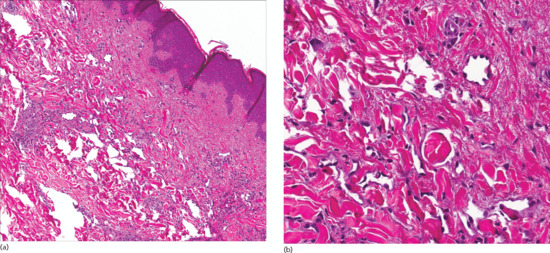
Figure 137.23 (a,b) Histopathology of atypical vascular proliferation after radiotherapy.
Irregular lymphatic-like vascular channels, lined by a single layer of endothelial cells, are seen in the dermis and may be multifocal. The endothelial cells are flat or have a hobnail appearance, and papillary projections can also be found. Careful examination of multiple sections is recommended.
Clinical features
History
AVPR presents a few years or months after radiotherapy for breast cancer (by comparison, post-irradiation angiosarcomas usually but not always tend to present many years after radiotherapy) [1, 2, 3]. As acute and chronic radiation dermatitis typically stabilizes within 3 years, changed noted after this period should prompt suspicion of AVPR. The median latency period following radiation exposure is 5–6 years [4, 6, 7, 9].
Presentation
Characteristic lesions are solitary or grouped erythematous to violaceous macules, papules, nodules or vesicles. These may range in size from 0.3 to 20 cm, with surrounding ecchymoses, induration or telangiectases [1, 4, 6, 9].
Disease course
Although usually benign in clinical course [1, 7], the ultimate outcome is somewhat indeterminate as a result of recurrence, development of new AVPRs, angiosarcoma present upon re-excision (5/11 in one series) and transition from AVPR to angiosarcoma within 2 years in a handful of cases [2, 4, 9, 7].
Prognosis
Most lesions behave in a benign fashion. However, as mentioned before, it is controversial whether these lesions are uniformly benign or whether they overlap with radiation-induced angiosarcoma.
Management
There is no standardized management algorithm and practices differ widely. Given that there have been several reports of AVPR being reclassified as angiosarcoma upon re-excision [6] and transition to angiosarcoma as early as within 2 years [4, 9], wide excision and close clinical follow-up are appropriate, although even in instances where margins were reported to be clear, recurrence was common.
Diffuse lymphangiomatosis
Definition and nomenclature
A rare condition characterized by extensive and variable involvement of the viscera (particularly the lung), bone, soft tissue and occasionally the skin by lymphatic malformations.
Introduction and general description
Diffuse lymphangiomatosis can involve the bone, lungs, spleen, liver, soft tissue and skin, as well as other structures in the thorax [1]. Though histologically benign, the space-occupying nature of the lesions, with the generation of effusions, can severely compromise organ function leading to death. The disease is thought to be congenital, with most clinical presentations in childhood. There is no strong sex predominance or pattern of inheritance [2, 3, 4, 5, 6, 7].
Pathophysiology
Pathology
The histological findings are similar to those of acquired progressive lymphangioma except that they are not confined to the dermis, often penetrating into the subcutis, involving soft tissue, fascia, muscle and organ parenchyma [8, 9]. Involvement of the skin is relatively uncommon in Gorham–Stout syndrome [6, 7, 10], though it can be extensive over affected bone [9]. Haemangiomata are also reported, involving bone, soft tissue and overlying skin [8]. The endothelial cells lining the dilated vascular spaces express CD31, factor VIII, UEA-1, LYVE-1, VEGF-R3, VEGF-C and podoplanin, consistent with lymphatic origin [1, 8, 9, 11].
Clinical features
The clinical features reflect the degree and site of involvement. Pulmonary involvement manifests as cough or dyspnoea, and bone involvement with pain, pathological fracture and osteolytic lesions. Any part of the skeleton may be involved. Diffuse pulmonary disease has a poor prognosis complicated by chylothorax, pleural and pericardial effusions, and infection. Skin involvement is typified by overlying haemangiomata or lymphangiomata [1, 2, 3, 5, 6, 7], and lymphangiomatosis should be considered if accompanied by symptoms of bone or pulmonary involvement.
Management
Generally, treatment is supportive including the draining of effusions, placement of shunts and sclerotherapy. Radiation therapy and surgical correction of skeletal defects can be successful early in the clinical course [1, 10, 12]. Responses to interferon α-2b and bisphosphonates have been reported for bone involvement [3, 13]. Anecdotal successes have been reported with sirolimus (http://clinicaltrials.gov/ct2/show/NCT00975819 last accessed August 2015), bevacizumab [14] and propranolol [15].
Resources
Further information
http://lgdalliance.org (The Lymphangiomatosis & Gorham’s Disease Alliance)
http://www.gorhams.org (Asociacion de Lucha Contra La Enfermedad de Gorham)
All last accessed August 2015.
TUMOURS OF PERIVASCULAR CELLS
Infantile myofi bromatosis and adult myofi broma [1-4,5,6]
Definition and nomenclature
This tumour is composed of cells showing differentiation towards perivascular contractile cells, and has been described in the past as infantile haemangiopericytoma [7]. Infantile myofibromatosis and adult myofibroma/myofibromatosis are best regarded as part of the spectrum of lesions described more recently as myopericytomas (see later) [8, 9].
Epidemiology
Incidence and prevalence
Solitary lesions both in children and adults are relatively rare. Multiple lesions are uncommon.
Age
Most cases of infantile myofibromatosis, present before the age of 2 years, with slight male predominance. Congenital tumours occur in up to a third of the cases. Solitary myofibroma tends to occur in adults, with the same anatomical distribution as that of cutaneous and soft-tissue lesions presenting in infantile myofibromatosis [5, 6]; multiple superficial tumours are rarely seen in adults. Intraoral lesions tend to be more common in young adults [10, 11].
Sex
Tumours are more common in males.
Pathophysiology
Predisposing factors
Rare myopericytomas but not classic myofibromas are associated with Epstein–Barr virus and have been reported in patients with HIV/AIDS [12] (see Chapter 31).
Pathology [1,2,5,6,7,8]
Tumours have a distinctive biphasic growth pattern:
- Areas composed of bundles of mature spindle-shaped myofibroblasts with pink cytoplasm and vesicular nuclei.
- Areas composed of immature round cells, with scanty cytoplasm arranged around small blood vessels, often displaying a haemangiopericytoma-like pattern (‘staghorn-like’).
Protrusion of tumour cells into vascular lumina is frequent, often mimicking vascular invasion. Old lesions often undergo hyalinization of the more mature areas. Mitotic figures and necrosis are relatively common. Tumour cells, particularly in the mature areas, are focally positive for actin.
Genetics
Occasional chromosomal abnormalities have been rarely reported in cases of infantile myofibromatosis [13, 14].
Clinical features [1-3]
History and presentation
Presentation is usually as single or solitary nodules that may be skin coloured or blue/red. Multiple lesions are present in 25% of patients. The preferred sites are the head and neck, followed by the trunk. Familial cases are rare. Involvement of other organs, including the gastrointestinal tract, lungs and bone, is seen in some cases. Multicentric involvement may be associated with mortality. Multiple lesions in the skin and soft tissues behave in a benign fashion and may regress spontaneously. Exceptionally, associated thrombocytopenia has been reported [15].
Disease course and prognosis
Lesions tend to regress spontaneously, but it is important to remember that patients with visceral tumours may die from the disease. Solitary lesions do not tend to recur locally.
Management
Simple excision is the treatment of choice for solitary lesions.
Myopericytoma
Definition
For many years, tumours thought to differentiate towards pericytes (perivascular myoid cells) were divided into two main categories: infantile haemangiopericytoma and adult haemangiopericytoma. Both variants, however, appear to have very little in common except for the histological presence of a pericytomatous pattern characterized by elongated branching thin-walled vascular spaces with a stag-horn pattern. With the combination of immunohistochemistry and electron microscopy, most tumours previously classified as adult haemangiopericytoma on light microscopy represent other tumours, including synovial sarcoma, mesenchymal chondrosarcoma and solitary fibrous tumour [1]. In very few cases the line of differentiation remains obscure, and these represent the ‘true’ adult haemangiopericytomas. They are more likely to arise from an undifferentiated mesenchymal cell than from a pericyte. ‘True’ adult haemangiopericytomas do not usually occur in the skin and will not be discussed further here.
The concept of myopericytoma was introduced to describe a group of lesions characterized by short oval to spindle-shaped myofibroblasts and a characteristic concentric perivascular growth [2]. Tumours tend to be mainly deep dermal and subcutaneous, and include lesions classified in the past as glomangiopericytoma, myopericytoma, myofibroma and myofibromatosis in adults. Infantile haemangiopericytoma and infantile myofibromatosis represent identical conditions and the former term has been abandoned.
Infantile myofibroma/myofibromatosis has already been described in the section on myofibroblastic tumours.
Epidemiology
Incidence and prevalence
Tumours are relatively rare.
Age
Most lesions present in middle-aged adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The histological spectrum of myopericytoma is wide and varies from lesions that are very similar to myofibromatosis, to tumours that closely resemble glomus tumours and even to angioleiomyoma. Tumours in all the latter categories are regarded as belonging within the same spectrum. Lesions are well circumscribed and composed of a mixture of solid cellular areas intermixed with variable numbers of vascular channels. The latter are often elongated and display prominent branching, resulting in a stag-horn appearance (haemangiopericytoma like). The cells in the solid areas are bland, round or short and spindle-shaped with eosinophilic or amphophilic cytoplasm and vesicular nuclei. Mitotic figures are exceptional. A common feature is the presence of concentric layers of tumour cells around vascular channels resulting in a typical onion-ring appearance (Figure 137.24). Myxoid change may be focally prominent. Occasional findings include hyalinization, cystic degeneration and bone formation. Nodules of tumour cells may protrude into the lumina of vascular channels. Rare examples are entirely intravascular [5]. In some cases, tumour cells closely resemble glomus cells and are characterized by round punched-out central nuclei and pale eosinophilic cytoplasm. These cases are referred to as glomangiopericytomas. Tumour cells are positive for smooth muscle actin and in most cases negative for desmin.
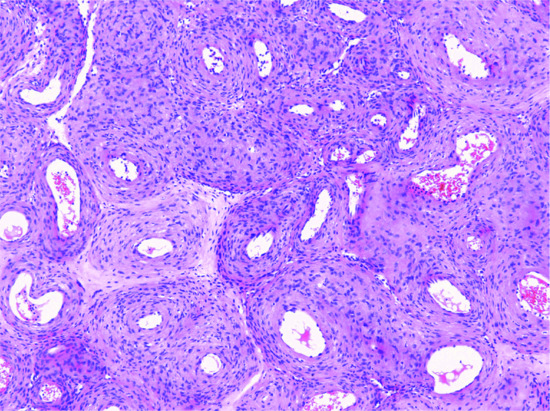
Figure 137.24 Numerous vascular channels surrounded by layers of pericytes in an onion-ring distribution in a case of myopericytoma.
Clinical features
History and presentation
Myopericytoma is relatively rare and presents mainly in middle-aged adults with a predilection for the limbs, especially the distal lower limb. Lesions are small (no more than 2 cm in diameter), longstanding, usually asymptomatic and may be solitary or, rarely, multiple [2, 3, 4]. Exceptionally, they are painful. In the setting of multiple myopericytomas, these often develop simultaneously with a predilection for a single anatomical site.
Disease course and prognosis
Behaviour is benign even in cases with an intravascular location. Very rarely, malignant examples of myopericytoma have been reported [6].
Management
Simple excision is the treatment of choice.
Glomus tumour [1-3]
Definition and nomenclature
A tumour of the myoarterial glomus, composed of vascular channels surrounded by proliferating glomus cells. The tumours have variable quantities of glomus cells, blood vessels and smooth muscle. According to this finding, they are classified as either solid glomus tumour, glomangioma or glomangiomyoma.
Epidemiology
Incidence and prevalence
Glomus tumours are comparatively uncommon.
Age
Some are present at birth; they rarely appear during infancy, but from the age of 7 years onwards the incidence increases gradually. Multiple tumours are 10 times more frequent in children than in adults [3, 4]. Tumours in adults present mainly during the third or fourth decades of life.
Sex
There is no sex predilection.
Pathophysiology
Pathology
The tumour is lobulated, well circumscribed and situated in the dermis. The proportion of glomus cells to vascular spaces varies. The smaller painful lesions tend to be mainly cellular. The larger, multiple and often painless lesions are angiomatous, with only a band of cells around the dilated vascular channels. The glomus cell is cuboidal, with a well-marked cell membrane and a round central nucleus. The cells align themselves in rows around the single layer of endothelial cells of the vascular spaces and in a somewhat less orderly fashion further out. More than 50% of tumours can be classified as glomangiomas, and a minority (less than 15%) are classified as glomangiomyomas (Figure 137.25). Electron microscopy [5, 6, 7] suggests that glomus cells are transversely cut smooth muscle cells and that there are many mast cells around the tumour, but that nerve fibres are not associated with the glomus cells. Tumour cells are universally positive for smooth muscle actin and are usually negative for desmin. Positivity for CD34 may be seen [8]. An oncocytic variant has been described [9], and also variants developing within a cutaneous nerve [10] and within a vein [11]. Malignant glomus tumour (glomangiosarcoma) is exceedingly rare. Even cutaneous tumours that are histologically malignant rarely metastasize, but they have a potential for local recurrence [12, 13].
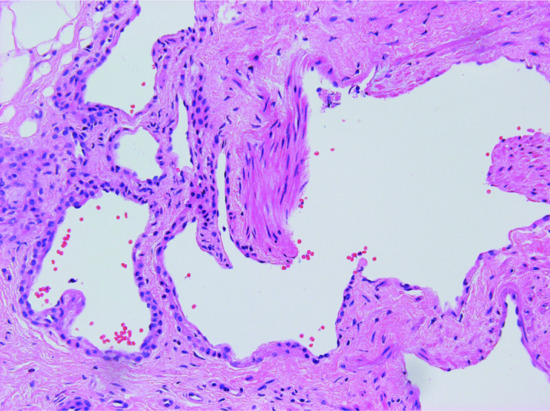
Figure 137.25 A typical case of glomangiomyoma displaying vascular channels, smooth muscle and thin layers of glomus cells.
Genetics
The occurrence of familial cases with autosomal dominant inheritance [1, 14, 15], and the association of multiple tumours with malformation of the same limb, suggests involvement of genetic factors. This theory has been confirmed by the identification of the glomulin gene for multiple inherited glomangiomas (also known as glomuvenous malformations [16]) at chromosome 1p21-22 [17, 18, 19].
Clinical features
History and presentation
A solitary glomus tumour is a pink or purple nodule varying in size from 1 to 20 mm; it is conspicuously painful (Figure 137.26). Pain may be provoked by direct pressure or a change in skin temperature, or may be spontaneous. The commonest site is the hands, particularly the fingers, followed by other sites on the extremities including the head, neck and penis [20]. Tumours beneath the nail are particularly painful, and patients present for treatment while the lesions are still very small. The affected nail has a bluish-red flush. An association between subungual glomus tumour and neurofibromatosis type 1 has been reported [21, 22]. Glomus tumours may also involve internal organs.
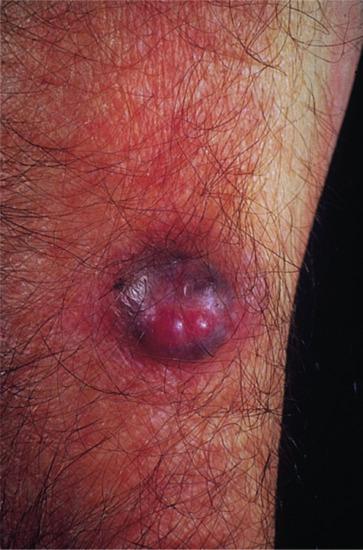
Figure 137.26 Clinical appearance of a glomus tumour.
Multiple glomus tumours are larger and usually dark blue in colour, and are situated deep in the dermis. They are less restricted to the extremities, may be widely scattered and are not usually painful [23, 24, 25, 26]. In some cases, grouped multiple tumours may be painful, and pain, intermittent discoloration and sweating of a limb may precede the development of a palpable tumour.
Differential diagnosis
The solitary tumour is to be distinguished from other painful tumours such as leiomyoma and eccrine spiradenoma. Distinction is usually only possible on histological examination. The multiple glomangioma may be indistinguishable clinically from a cavernous haemangioma, and is possibly identical to ‘blue rubber bleb’ naevus [5].
Disease course and prognosis
Local recurrence is very rare and occurs mainly after incomplete excision. Most recurrences are seen in deeper lesions with an infiltrative growth pattern. These lesions have been described as infiltrating glomus tumours [25].
Management
Surgical excision is usually curative.
PERIPHERAL NEUROECTODERMAL TUMOURS
Reviews of neural tumours may be found in [1, 2].
Neuromuscular hamartoma [1,2]
These lesions appear to be combined hamartomas of both muscular and neural tissue. The clinical appearance is of a subcutaneous mass. Multinodular masses of skeletal muscle are mixed with both myelinated and unmyelinated nerve fibres. Malignant triton tumours, composed of a mixture of schwannoma-like material and rhabdomyosarcoma, are very much commoner than the benign variety of triton tumour. Surgical excision is required.
Multiple mucosal neuromas [1]
In Sipple syndrome, multiple neuromas of the oral mucosa may be associated with phaeochromocytoma, parafollicular thyroid cysts secreting calcitonin, medullary thyroid carcinoma and opaque nerve fibres on the cornea (see Chapter 147).
Amputation stump neuroma [1]
Definition and nomenclature
This is a benign response of nerve tissue to injury.
Pathophysiology
Pathology
Foci of proliferating nerve tissue surrounded by scar tissue are typically seen (Figure 137.27). Accessory digits may show a very similar pattern of tissue involvement.
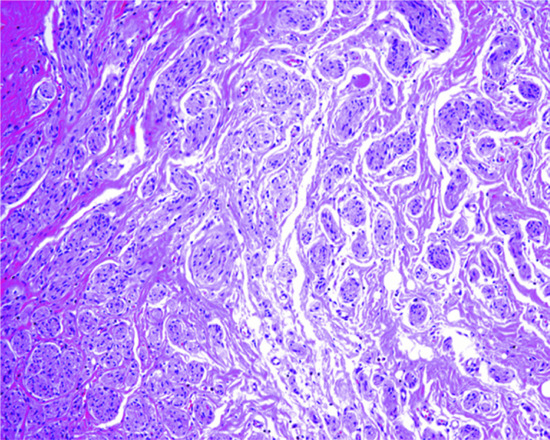
Figure 137.27 Histological appearance of an amputation neuroma. Small nerves proliferate in the dermis in a background of fibrosis.
Clinical features
History and presentation
A small tender nodule is found in a scar site.
Management
Surgical excision is usually required. The problem can be prevented by apposing the ends of nerves at the sites of injury.
Morton neuroma [1,2]
Definition and nomenclature
This is the result of damage to the plantar digital nerve, followed by fibrosis. The condition has been associated with the use of high-heeled footwear.
Epidemiology
Incidence and prevalence
It is rare.
Age
Adults.
Sex
Most common in females.
Pathophysiology
Pathology
On pathological examination, there is very prominent perineurial, endoneurial and epineurial fibrosis. Perivascular fibrosis and intimal thickening are also seen.
Clinical features
History and presentation
Patients complain of severe pain, usually between the third and fourth metatarsals, especially when walking.
Management
Excision is the recommended therapy and is curative.
Solitary circumscribed neuroma
Definition and nomenclature
This is a distinctive variant of cutaneous neuroma composed of variable proportions of the normal components of nerve tissue.
Epidemiology
Incidence and prevalence
Lesions are relatively common.
Age
Most cases present in adults.
Sex
There is equal sex incidence.
Pathophysiology
Pathology [1,2,3]
Examination reveals a well-circumscribed partially encapsulated dermal nodule (Figure 137.28), often associated with a nerve in the deep dermis. It is composed of uniform cells with pink cytoplasm in a collagenous background and with artefactual clefting between bundles. The capsule displays epithelial membrane antigen-positive perineurial cells. Most of the cells within the nodule are S100 positive, and special stains may demonstrate axons.
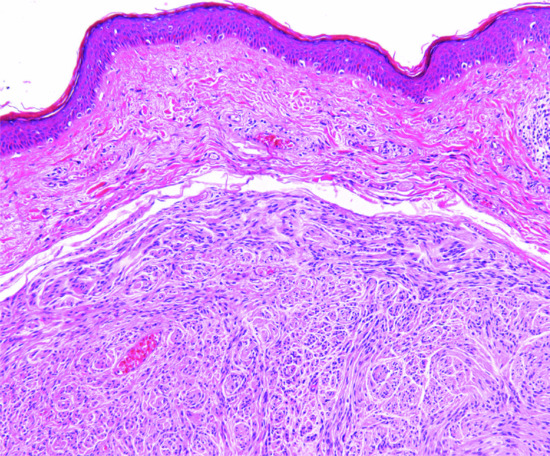
Figure 137.28 Sharply demarcated dermal nodule in a case of solitary circumscribed neuroma.
Clinical features [1,2,3]
History and presentation
It is fairly common and presents mainly on the face of adults as a small asymptomatic papule, which may resemble a naevus.
Disease course and prognosis
Lesions are benign.
Management
Simple excision is curative.
Schwannoma
Definition and nomenclature
A tumour of nerve sheaths composed of Schwann cells.
Epidemiology
Incidence and prevalence
The tumour is relatively rare in the skin and relatively uncommon in other sites including soft tissues.
Age
It may occur at any age, but is most common in the fourth and fifth decades.
Sex
Females are affected more often than males [1].
Pathophysiology
Pathology [2,3]
The tumour is rounded, circumscribed and encapsulated. It is situated in the course of a nerve, usually in the subcutaneous fat. The cells are spindle shaped with poorly defined cytoplasm and elongated wavy basophilic nuclei. Variable amounts of collagen are seen in the background. Cells are arranged in bands, which stream and interweave. The nuclei display palisading and are arranged in parallel rows with intervening eosinophilic cytoplasm in a typical appearance known as Verocay bodies (Figure 137.29). Cellular areas known as Antoni A areas are intermixed with areas showing prominent myxoid change known as Antoni B areas [4]. The latter areas are likely to be the result of degeneration. In some tumours, there is mucous secretion, producing a vacuolated stroma. Scattered mononuclear inflammatory cells are often seen. In some cases, the nerve of origin may be found associated with the capsule. Electron microscopy shows that tumour cells have typical features of Schwann cells [5]. S100 protein staining is strong and uniform [6]. For many years, it was thought that schwannomas lack axons and this was regarded as a useful way of distinguishing these tumours from neurofibromas, which contain variable numbers of axons. The presence of the latter is demonstrated by immunostaining for neurofilaments. This view, however, has been challenged recently as both hereditary and sporadic schwannomas may contain axons, suggesting that both entities may in fact be more closely related than previously thought [7]. This explains the rare occurrence of hybrid lesions combining features of neurofibroma and schwannoma [8].
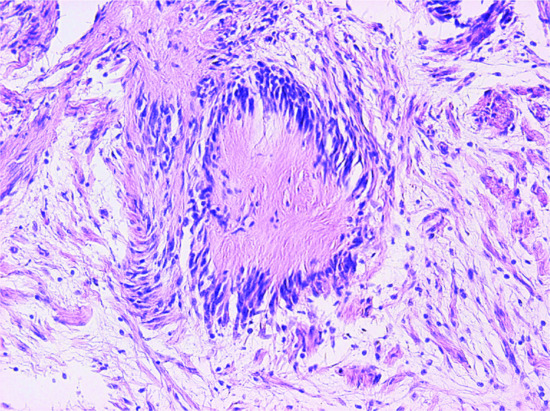
Figure 137.29 Typical Verocay body in a case of schwannoma.
There are several variants of schwannoma, some of which may be confused histologically with other benign or malignant tumours.
Hybrid schwannoma/perineurioma. These are tumours that have two clearly defined populations of cells, Schwann cells and perineural cells positive for S100 protein and EMA, respectively [9].
Ancient schwannoma [10] often occurs in a deep location and is characterized by prominent degenerative changes, which often result in cytological atypia. Ectatic blood vessels, haemorrhage, haemosiderin deposition and focal inflammation consisting of lymphocytes are often seen. There is loss of Antoni A areas, which makes histological diagnosis difficult.
Cellular schwannoma [11] also tends to have a predilection for deep soft tissues. It is characterized by high cellularity, with almost complete absence of Antoni B areas. This, coupled with the presence of mitotic figures, often leads to a misdiagnosis of malignancy [12]. A multinodular plexiform variant may occur in children and some examples are congenital.
Plexiform schwannoma [13, 14] tends to occur in younger patients, may be painful and has a predilection for the dermis. Multiple cellular nodules composed of bland Schwann cells are seen in the dermis. Distinction from plexiform neurofibroma is important, as these tumours are not usually associated with neurofibromatosis type 1. Multiple cutaneous plexiform schwannomas, however, are associated with neurofibromatosis type 2 [1, 15].
Melanotic schwannoma [16] only exceptionally occurs in the skin; it has a predilection for spinal nerve roots. Tumour cells are epithelioid and melanin pigment is prominent. The importance of this variant is that these tumours are regarded as malignant and may be a marker of Carney complex (Chapter 147) [17].
Pacinian schwannoma is a rare variant composed of structures closely resembling the Pacinian corpuscles.
Neuroblastoma-like schwannoma is very rare and characterized histologically by areas composed of round blue small Schwann cells which may form perivascular rosettes or rosettes with collagenous cores [18]. The tumour in other areas has the typical appearance of a schwannoma and the immunohistochemical profile is typical of the latter.
Epithelioid schwannoma is an infrequent type of schwannoma composed predominantly of cells with epithelioid morphology [19]. However, although these tumours are positive for S100 protein, they lack palisading, are composed of uniform tumour cells and contain a population of CD34-positive cells. It has therefore been proposed that these lesions do not represent classical examples of schwannomas [20].
Glandular schwannoma [21, 22] represents in most cases an ordinary schwannoma with entrapment of normal sweat glands.
Clinical features [23]
History and presentation
It arises most frequently from the acoustic nerve. Bilateral acoustic schwannomas are characteristic of neurofibromatosis type 2. A further manifestation of the latter is the occurrence of multiple cutaneous plexiform schwannomas. There is no association with neurofibromatosis type 1. In the peripheral nervous system, it is usually found in association with one of the main nerves of the limbs, usually on the flexor aspect near the elbow, wrist or knee, the hands or the head and neck [23]. It may be seen on the tongue. Other sites include the wall of the gastrointestinal tract and the posterior mediastinum. They are rounded or ovoid circumscribed nodules varying in size up to 5 cm, usually firm (but sometimes soft and cystic) in consistency, and sometimes painful. The colour is pink-grey or yellowish. Small lesions may be intradermal, but larger ones are subcutaneous. They usually grow slowly.
Disease course and prognosis
Most tumours are benign. Malignant transformation of a schwannoma is exceedingly rare and may contain areas of epithelioid angiosarcoma [24, 25, 26].
Management
Simple excision is curative.
Solitary neurofi broma [1, 2, 3, 4]
Definition
An isolated lesion probably arising from the endoneurium and composed of a mixture of Schwann cells, fibroblasts and perineurial fibroblasts. It is not related to neurofibromatosis type 1. Although it was regarded as hamartomatous in nature, the demonstration of clonality suggests a neoplastic origin [5].
Epidemiology
Incidence and prevalence
Lesions are relatively common.
Age
Tumours are more common in adults.
Sex
There is no sex predilection.
Pathophysiology
It has been suggested that cutaneous neurofibromas develop as a result of precursor cells that are plutipotent, probably arise from the hair roots and have a NF1 (+/–) genotype [6].
Genetics
Cytogenetic analysis in a single case of solitary neurofibroma has found a reciprocal translocation t(4;9)(q31;p22) [7].
Pathology
These lesions differ from neurilemmomas in that they do not have a capsule, they are only focally positive for S100 protein and they do not have Antoni A and Antoni B areas. Instead, they are composed of bland spindle-shaped cells with wavy nuclei in a myxoid or collagenous stroma. Mast cells are usually prominent. Degenerative changes are sometimes seen but mitotic activity is absent. Tumours with scattered atypical cells (nuclear enlargement and hyperchromatism) are classified as atypical but they are not associated with aggressive behaviour [8]. Less than 50% of the cells in these lesions are S100 positive. There is also focal positivity for CD34 and EMA.
Several histological variants of neurofibroma have been described, including epithelioid neurofibroma, granular cell neurofibroma, pigmented neurofibroma [9] and a variant with dendritic cells and pseudorosettes [3, 10].
Clinical features
History and presentation
Any body site may be affected. It usually appears as a slow-growing small polypoid lesion. Multiple neurofibromas are rare outside the setting of neurofibromatosis type 1 (Figure 137.30).
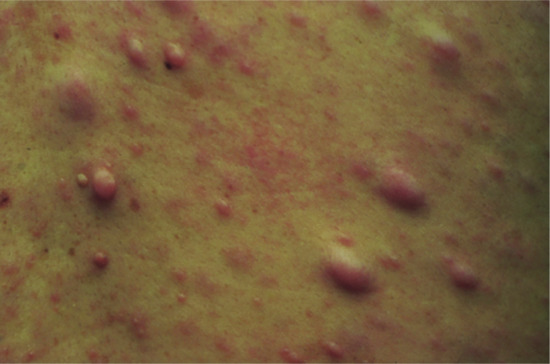
Figure 137.30 Multiple soft papules, typical of neurofibroma in a patient with neurofibromatosis type 1.
Disease course and prognosis
Malignant change is said not to occur outside the setting of neurofibromatosis type 1.
Management
Simple excision is curative.
Plexiform neurofi broma
This tumour is considered to be pathognomonic of neurofibromatosis type 1 (see Chapter 80). However, it has been contested whether the presence of a single plexiform neurofibroma in the absence of other signs of neurofibroma can be regarded as pathognomonic of neurofibromatosis type 1 [1, 2]. Interestingly, cytogenetic analysis of a sporadic plexiform neurofibroma has shown biallelic inactivation of NF1 [3]. It presents in children and young adults of either sex, with a predilection for the lower limbs and the head and neck. Tumours are large and located in the dermis, subcutis and even deeper soft tissues. The overlying skin is folded and hyperpigmented and the lesion is described as having an appearance like a ‘bag of worms’ (Figure 137.31). This reflects the typical histological appearance of nerve trunks of different sizes randomly distributed throughout the involved tissues (Figure 137.32). Careful histological examination of these lesions is necessary because the presence of mitotic activity usually indicates malignant transformation.
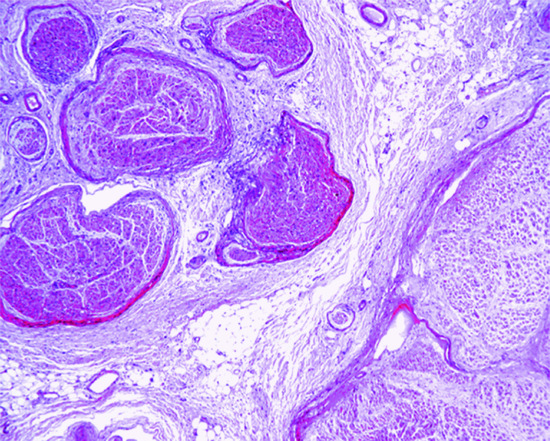
Figure 137.31 Irregular poorly formed nerves in a plexiform neurofibroma.

Figure 137.32 Clinical appearance of a plexiform neurofibroma.
Surgical removal of these lesions is usually very difficult because of the extensive involvement. When planning the surgical removal of these tumours, surgeons should remember that there is a tendency for haemorrhage within the tumour that may lead to morbidity or mortality.
Diffuse neurofibroma
This lesion presents as a diffuse poorly defined induration or plaque-like lesion of the skin and subcutaneous tissue in children or young adults, with a predilection for the trunk and head and neck area. A number of cases are associated with neurofibromatosis type 1. The histological features are identical to those of a solitary neurofibroma except for the fact that there is diffuse replacement of involved tissue by the tumour. Local recurrence is frequent unless the lesion is widely excised. Very rare sporadic tumours recur repeateadly and show a tendency for malignant transformation [1].
Perineurioma [1, 2, 3, 4]
Definition and nomenclature
Perineurioma is a tumour originally described in soft tissues. It is relatively common in the skin and it is composed of cells showing differentiation towards perineural fibroblasts.
Epidemiology
Incidence and prevalence
Lesions are rare.
Age
Most tumours present in young adults.
Sex
There is predilection for females.
Pathophysiology
Pathology
Tumours are well circumscribed and composed of bipolar and slender bland thin spindle-shaped cells with scanty cytoplasm and wavy nuclei. They are often arranged in concentric whorls (Figure 137.33) or in a storiform pattern. Cellularity varies and is low in the sclerosing variant where hyalinized collagen predominates. The architecture may be plexiform in some cases [4]. Tumour cells are distinctively positive for EMA. They are also positive for a tight junction associated protein, claudin-1 [5]. Focal positivity for CD34 may also be seen. Rare cutaneous tumours may be intraneural [6].
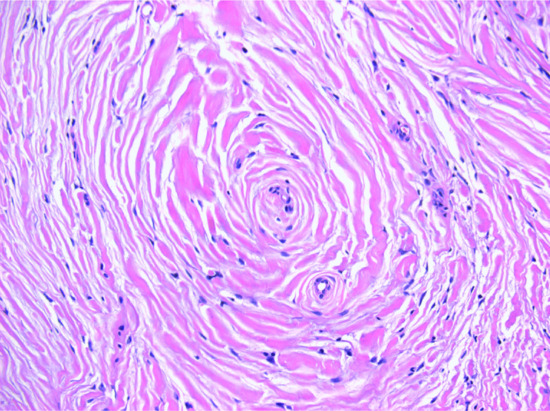
Figure 137.33 Typical whorling appearance and some degree of sclerosis in a case of perineurioma.
Clinical features
History and presentation
The lesion has a predilection for the lower limbs of young females. Tumours are small and asymptomatic. Multiple lesions are exceptional [7]. A distinctive sclerosing variant has been described, affecting most commonly, but not always, the hands [8, 9].
Disease course and prognosis
Lesions are entirely benign.
Management
Simple excision is the treatment of choice.
Dermal nerve sheath myxoma [1,2,3,4]
Definition and nomenclature
This is a myxoid tumour that is thought to display nerve sheath differentiation.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
Young adults.
Sex
There is predilection for males.
Pathophysiology
Pathology [1, 2, 3]
The dermis shows a well-defined tumour composed of lobules that vary in size and shape and are separated by fibrocollagenous stroma. Each lobule is composed of slender stellate or spindle-shaped Schwann cells with bland nuclei and indistinct cytoplasm margins in the background of prominent myxoid change. Mitotic figures are very rare. Tumour cells are uniformly positive for S100. They are also positive for glial fibrillary acid protein and CD57 [4]. EMA-positive cells are seen in the periphery of tumour lobules. These tumours have no relationship with the so-called cellular neurothekeoma [5].
Clinical features [1, 2, 4]
History and presentation
This presents most commonly on the upper limbs (particularly the fingers and hands) and lower limbs (mainly the knees, shins or feet). The trunk and head and neck are rarely affected. Lesions are longstanding, small, usually less than 1 cm, skin coloured and asymptomatic.
Disease course and prognosis
Tumours are benign but there is some tendency for local recurrence [4].
Management
Simple excision is the treatment of choice.
Cellular neurothekeoma [1-4]
Definition
Despite its name, this tumour is not related to dermal nerve sheath myxoma, and its line of differentiation has not been established. Gene expression profiles of this tumour has suggested that they may be related to fibrous histiocytoma [5]. The tumour should not be confused with ordinary nerve sheath myxomas showing focal cellular areas [1, 2, 3].
Epidemiology
Incidence and prevalence
Tumours are relatively common.
Age
The age range is wide but most cases occur in children and young adults.
Sex
It is more common in females.
Pathophysiology
Pathology
In the dermis and frequently extending into the subcutis [6], there is an ill-defined tumour composed of nests and fascicles of epithelioid or spindle-shaped cells (Figure 137.34) with vesicular nuclei and a single small eosinophilic nucleolus. Lesions presenting in the face may extend into the underlying skeletal muscle. Mitotic figures are relatively common and scattered multinucleated cells may be seen. Myxoid change is present in some lesions and it may be extensive [7]. Tumour cells resemble melanocytes, and this often leads to the lesion being confused with a melanoma. However, there is no junctional activity, and cells are invariably negative for S100, HMB45 and melan-A. Rare tumours display perineural extension [7, 8]. Some tumours have larger size, more cytological atypia and increased mitotic count, and these tumours have been classified as atypical cellular neurothekeoma [4, 6, 8]. However, this does not seem to be related to a more aggressive behaviour. Tumour cells are often positive for smooth muscle actin (in about 57% of cases), NKI-C3, neuron-specific enolase, PGP 9.5 and microphthalmia transcription factor [6, 9]. A rare plexiform tumour with hybrid features of perineurioma and cellular neurothekeoma and predilection for the lips has been described [10].
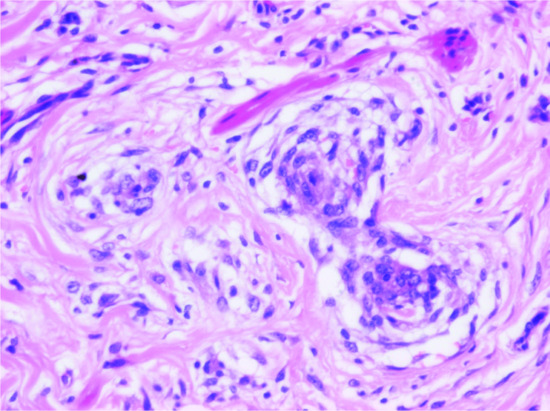
Figure 137.34 Cellular neurothekeoma. Nests of epithelioid cells in the background of a hyalinized stroma. In cases with cytological atypia and mitotic activity, confusion with a melanoma is more likely.
Clinical features
History and presentation
The tumour presents as a small asymptomatic papule with a predilection for the upper limbs and face and neck [4, 6]. Multiple lesions are exceptional [11].
Disease course and prognosis
There is very little tendency for local recurrence even in cases classifed histologically as atypical.
Management
Simple excision is curative.
Granular cell tumour [1, 2, 3]
Definition and nomenclature
A tumour composed of cells with characteristic granular cytoplasm. The histogenesis of the classic granular cell tumour seems to be neural [4, 5]. However, it is worth remembering that many tumours of different histogenesis may show granular cell change, due to the cytoplasmic accumulation of secondary lysosomes.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
It is usually seen between the fourth to sixth decades of life. It can occur in childhood.
Sex
It is more common in males than females.
Pathophysiology
Pathology
Large polyhedral cells arranged in sheets, which infiltrate the dermal connective tissue and subcutaneous fat, form the tumour. The cytoplasm is pale and contains brightly acidophilic granules. Tumour cells often display large eosinophilic granules surrounded by a clear halo and known as pustulo-ovoid bodies of Milian [6]. The nuclei are relatively small and round, and tend to be vesicular. Clear cell change may occasionally be prominent [7]. The epithelium over the area may show pseudoepitheliomatous hyperplasia and in small biopsies this may be confused with a squamous cell carcinoma (Figure 137.35). Perineural extension is often seen. Vascular invasion may be seen occasionally and this does not seem to have any bearing on behaviour [8]. Occasionally, tumour cells involve the epidermis and distinction from melanoma may be difficult [9]. However, although S100 positivity is seen in both tumours, HMB45 is usually negative in granular cell tumour. It is important, however, to take into account that rare granular cell tumours may show focal positivity for melan-A and that microphthalmia transcription factor is positive in a percentage of tumours [10]. The original suggestion that the cells are myoblasts probably arose from the examination of tumours of the tongue in which infiltration between the striated muscle bundles gave the impression of origin from the muscle. The general belief now is that the cells are of neural or nerve sheath origin.
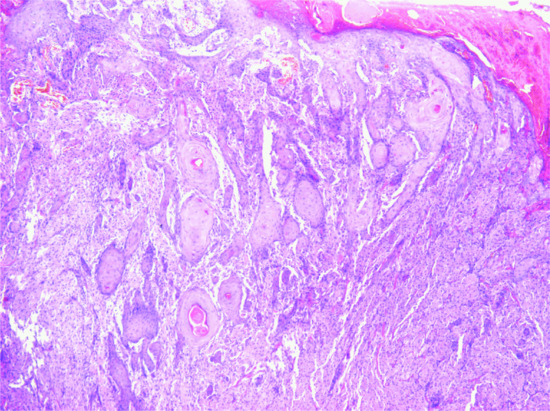
Figure 137.35 Prominent pseudoepitheliomatous hyperplasia mimicking a squamous cell carcinoma in a case of granular cell tumour.
Clinical features
History and presentation
The tumour is usually solitary, situated in the skin, beneath the epithelium of the tongue or in deeper soft tissues or internal organs (mainly the gastrointestinal tract) [2, 3]. It is firm and rounded but with rather indefinite margins, sessile or pedunculated, and between 5 and 20 mm in diameter, although larger tumours may be seen. A warty appearance may be seen due to epidermal hyerplasia. The colour may vary from flesh colour to pink or greyish-brown. Rare tumours are painful. Multiple tumours may occur both in adults and in children [3]. Multiple lesions have also been reported in Noonan syndrome [11]. The tumour grows slowly. Malignant tumours are very rare.
Disease course and prognosis
Local recurrence is sometimes seen after incomplete excision.
Management
Simple excision is the treatment of choice.
Meningothelial heterotopias [1-3,4,5-8]
Definition and nomenclature
Meningeal heterotopias are defined as lesions with meningothelial elements presenting in the skin and soft tissue and can be the result of heterotopias or secondary extension from a primary central nervous system tumour (see later).
Epidemiology
Incidence and prevalence
All types of meningothelial heterotopias are very rare.
Age
See later.
Sex
There is no sex predilection.
Pathophysiology
Pathology [6-8]
Low-power examination often reveals a lesion with a striking resemblance to a lymphangioma. Irregular dilated spaces are seen dissecting between collagen bundles. The spaces are partially lined by plump epithelioid cells, which are also seen in clusters in the surrounding stroma. Focal formation of psammoma bodies may be present. The dermal collagen and blood vessels also appear to be increased. Some lesions contain more solid areas. The presence of meningothelial cells can be demonstrated by positive staining for EMA. These cells are also positive for p63 but negative for keratins and S100 protein [9].
Clinical features
History and presentation
Meningeal heterotopias were divided into three groups by Lopez, et al. [4]. The first two groups of lesions represent meningothelial heterotopias or hamartomas (ectopic meningothelial hamartoma). The main difference between both groups is that affected patients are children in the first group and adults in the second group. The third group consists of intracranial meningiomas that extend secondarily into the skin or soft tissues. This group will not be discussed in more detail here.
A small number of cases of meningothelial heterotopias have been associated with von Recklinghausen disease [1]. The tumour occurs over the scalp or in the paraspinous region of the trunk of children and young adults. Occasionally, it appears to be familial [6]. In the scalp, the area is often bald. The skin is adherent to the mass, which is dermal or subcutaneous, and there may be a central depression with epidermal atrophy or ulceration. A connection with the cranial cavity is not usually demonstrated. The size ranges from 2 to 10 cm.
Management
Simple excision is the treatment of choice.
Glial heterotopic nodules [1,2]
Definition and nomenclature
This represents the presence of heterotopic mature glial tissue in the dermis or subcutis, predominantly on the central face. It may be considered to be a developmental defect in the closure of the neural tube. However, rare cases occur away from the midline, suggesting a different unexplained mechanism for its occurrence [3].
Epidemiology
Incidence and prevalence
Lesions are very rare.
Age
Most lesions present in infants and children. Presentation in adults is exceptional.
Sex
There is no sex predilection.
Pathophysiology
Pathology
Nodules of astrocytes in a neurofibrillar background are characteristic. Less commonly, oligodendrocytes are seen; neuronal elements are exceptional.
Clinical features
History and presentation
Most lesions present as a subcutaneous mass on the bridge of the nose. Communication with the cranial cavity is present in up to 20% of cases.
Management
Excision is curative, but it is very important to make sure that an underlying communication with the cranial cavity is ruled out, as failure to do so may result in complications such as meningitis or cerebrospinal fluid leakage.
Epithelial sheath neuroma [1]
Definition
This is a novel, rare and intriguing dermal lesion that combines a neural and an epithelial component. Although some features suggest that this may be a hamartoma, the fact that it has only been described in adults makes this possibility unlikely. A further theory is that the lesions may represent a variant of nerve hyperplasia [2].
Epidemiology
Incidence and prevalence
Lesions are very rare.
Age
It presents in adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
Histologically there are scattered prominent nerves in the superficial dermis, encased by mature non-dysplastic squamous epithelium with focal keratinization and dyskeratotic cells. Immunohistochemistry displays the normal staining of nerves and epidermis, respectively.
Clinical features
History and presentation
In view of the fact that so few cases have been described, very little can be said about the clinical features. The clinical presentation is not distinctive and lesions appear to have a predilection for the back.
Management
Simple excision is the treatment of choice.
Pigmented neuroectodermal tumour of infancy [1,2]
Definition and nomenclature
A pigmented tumour of childhood, combining neural and melanocytic elements. It is thought to recapitulate the early stages of development of the retinal epithelium although the finding of raised urinary excretion of vanillylmandelic acid in some cases, suggest a neural crest derivation [3, 4, 5, 6].
Epidemiology
Incidence and prevalence
Tumours are very rare.
Age
Most cases present in infants between 3 and 6 months old. Occurrence in adults is exceptional.
Sex
There is no sex predilection.
Pathophysiology [7, 8]
Pathology
A mass of irregular alveolar spaces surrounded by fibrous stroma is seen. Two types of cells are easily recognized: small round blue cells with scanty cytoplasm in a fibrillary matrix, and large epithelioid cells with pink cytoplasm and vesicular nuclei. These cells often contain melanin. Both types of tumour cells stain for synaptophysin and neuron-specific enolase, and are negative for S100. The large cells are positive for cytokeratin and HMB45.
Clinical features
This tumour occurs most frequently in the anterior part of the maxilla, and often presents as a pigmented oral mass [9]. It has been reported also in the anterior fontanelle, shoulder, epididymis, femur, mediastinum and even the foot. It may cause a high urinary excretion of vanillylmandelic acid [3]. This tumour has been mistaken for malignant melanoma, and could also be confused with a cellular blue naevus. The clinical appearance is that of a rapidly expanding nodule in the jaw, which may affect dentition.
Disease course and prognosis
Although classified as benign, the lesions may cause considerable local destruction, and around 5% of cases may metastasize and prove fatal [10].
Management
Complete surgical excision is the treatment of choice.
Malignant peripheral nerve sheath tumour [1,2]
Definition and nomenclature
A malignant tumour arising from the nerve sheath.
Epidemiology
Incidence and prevalence
Tumours are very rare.
Age
Young adults are more commonly affected in the setting of neurofibromatosis type 1. Sporadic cases occur in older individuals.
Sex
Cutaneous tumours usually arise from a plexiform neurofibroma in patients with NF1 [3]. Rare cutaneous lesions may arise within ordinary neurofibromas or de novo [4]. Deep-seated lesions arise de novo or in association with NF1. Patients with this disease develop malignancy in 30–50% of cases.
Pathophysiology
Pathology
The basic pattern is that of fascicles of tumour cells, often with a herringbone pattern and resembling a fibrosarcoma. Tumour cells tend to concentrate around blood vessels and myxoid change is common. The degree of pleomorphism and the number of mitotic figures varies. Immunohistochemical markers are not usually of help in the diagnosis as S100 is only focally positive and often entirely negative. The rare epithelioid variant of the neoplasm consists of sheets of epithelioid cells often mimicking melanoma and tumour cells are usually diffusely positive for S100 but negative for other melanocytic markers.
Clinical features [5]
The diagnosis should be suspected when a previously static tumour in a patient with NF1 begins to enlarge or becomes painful. The pain may become radicular as the lesion progresses but the tumours are not always associated with nerve trunks. The commoner sites are the flexor aspects of the limbs. A minority of cases occur as a complication of radiotherapy.
Disease course and prognosis
Tumours behave in an aggressive manner. Systemic metastases particularly to the lungs are common. The prognosis is worse in cases occurring after previous radiotherapy.
Management
Wide local excision or amputation is necessary.
Peripheral primitive neuroectodermal tumour [1-3,4]
Definition and nomenclature
A small blue round cell tumour displaying a spectrum of neuroectodermal differentiation.
Epidemiology
Incidence and prevalence
Tumours are very rare.
Age
Most tumours present in children but presentation is at a later age than conventional Ewing sarcoma [4].
Sex
There is slight predilection for females.
Pathophysiology
Pathology
Tumours are composed of sheets of small blue round cells that are fairly homogeneous, with scanty pale cytoplasm and fine chromatin. Mitotic figures are common. Tumour cells are diffusely positive for CD99 (in a cytoplasmic membrane pattern) and focally positive for FLI-1. Keratin is postive in up to a third of cases.
Genetics
This tumour usually presents a reciprocal chromosome translocation t(11;22)(q24;q12) that is an important aid in diagnosis and can be demonstrated by FISH.
Clinical features
History and presentation
The tumour presents in children and has no distinctive clinical features, although it is often confused with a vascular tumour.
Disease course and prognosis
Cutaneous tumours appear to have a better prognosis than those presenting in deeper soft tissues [4].
Management
Surgical followed by chemotherapy and in some cases also radiotherapy [4].
TUMOURS OF MUSCLE
Smooth muscle hamartoma
Definition and nomenclature [1]
Smooth muscle hamartoma is a proliferation of smooth muscle within the dermis.
Epidemiology [1,2,3,4]
Incidence and prevalence
It is a rare tumour.
Age
Usually it is congenital. Aquired lesions with onset in puberty or adulthood have been reported.
Sex
There is a slight male predominance.
Pathophysiology
Genetics [5-10]
Familial cases have been reported [5, 6]. An association with Xp microdeletion syndrome [7] or with familial paracentric inversion of chromosome 7q and Michelin tyre syndrome has been suggested [8, 9, 10].
Pathology [1,2,3,4,11-13]
Microscopically, within the dermis or the subcutis, variably orientated bundles of smooth muscle are seen. The overlying epidermis may be acanthotic and hyperpigmented. Follicles may be prominent but are not increased in numbers. Becker naevus may show similar histology; it has been suggested that both lesions may be a part of the same spectrum [11]. Associations with other skin lesions, such as naevus flammeus and blue naevi, have been reported [12, 13].
Clinical features [1,2,3-14]
History and presentation
The typical presentation is that of a solitary macule or plaque of variable size, which may be associated with hyperpigmentation, hypertrichosis or both. The lumbosacral area, trunk and proximal extremities are the sites of predilection. However, unusual sites such as the head and neck, including the oral cavity, scrotum and conjunctiva have been reported. Sometimes lesions may be atrophic or linear. Another unusual presentation is folding of the skin including Michelin tyre syndrome. Pseudo-Darier sign has been described [15].
Prognosis
It is an entirely benign lesion.
Management
Complete excision is curative in almost every case. This is mainly done for cosmetic reasons.
Leiomyoma
Definition
A benign tumour of smooth muscle derived from the arrector pili muscle (pilar leiomyoma), from the media of blood vessels (angioleiomyoma), or from smooth muscle of the scrotum, labia majora or nipples (genital leiomyoma) [1, 2, 3, 4, 5, 6, 7, 8].
Epidemiology [1,2,3,4,5-8]
Incidence and prevalence
Tumours in the three types are relatively uncommon. Pilar leiomyoma is the most common. Genital leiomyoma is the least common followed by angioleiomyoma. The cutaneous variety in general is about six times more frequent than the genital type [2].
Age
Pilar leiomyoma (leiomyoma cutis) can occur at any age from birth onwards, but appears usually in early adult life. Genital leiomyoma (dartoic myoma) can occur at any age. Angioleiomyoma is seen mainly in middle-aged adults.
Sex
Pilar and genital leiomyomas present equally in both sexes. Angioleiomyoma is more frequent in females.
Pathophysiology
Pathology [1,2,3,4,5-8]
The smooth muscle cells proliferate to produce interweaving bundles of spindle-shaped cells, which are strongly eosinophilic (Figure 137.36). The nuclei are long and thin, and the general appearance of the mass in ordinary sections may suggest a hypertrophic fibrous reaction. The smooth muscle cells can be distinguished from collagen by their different reaction with trichrome stains, and by the presence of myofibrils, which stain with phosphotungstic acid haematoxylin, and by their blunt-ended nuclei. Tumour cells are positive for actin and desmin.
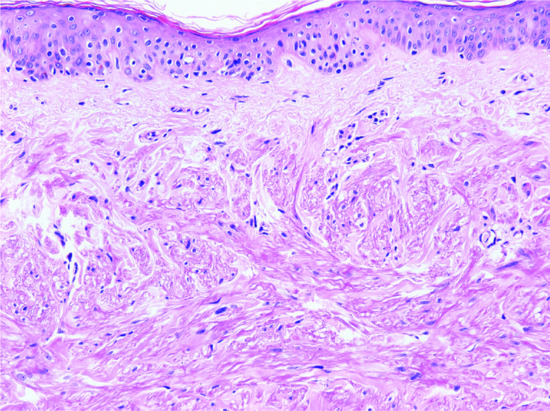
Figure 137.36 Pathology of leiomyoma, showing spindle-shaped cells with eosinophilic cytoplasm arranged in bundles closely resembling the arrector pili muscle.
The tumour of pilomotor origin (leiomyoma cutis, multiple cutaneous leiomyomas) is usually composed of numerous dermal nodules with vague margins where the cells penetrate the surrounding collagen bundles, and an upper border that approaches the papillary body. Associated epidermal hyperplasia is common. Focal nuclear atypia likely to be degenerative in origin and very low mitotic activity (up to one per 10 high-power fields) may be seen without this being indicative of malignant degeneration [1]. Genital leiomyomas are nodular tumours with a similar appearance. Scrotal tumours are less circumscribed and more cellular than those developing in the vulva. The angiomyomas are related to veins in the subcutaneous tissue, and are rounded and well circumscribed [3, 4]. Vessels of variable thickness are intermixed with bundles of mature smooth muscle. Focal degenerative cytological atypia may be seen, but mitotic figures are absent. Calcification, hyalinization and thrombosis of vessels are often seen.
Genetics
The gene that predisposes to multiple pilar leiomyomas has been mapped to chromosome 1q 42.3-q43 [9]. It also predisposes to uterine leiomyomas (multiple cutaneous and uterine leiomyomatosis, Reed syndrome (MCUL)) and to renal cancer (mainly papillary renal cell carcinoma) [10, 11, 12, 13]. This is as a result of mutations in the gene encoding the enzyme fumarate hydratase. In patients with associated renal cancer, the syndrome is known as hereditary leiomyomatosis and renal cancer (HLRCC).
Clinical features [1, 2]
Pilar leiomyoma has been reported in identical twins, in siblings and in several generations of a family. The cases with a familial background have all had multiple tumours and are associated with MCUL and renal cancer syndrome. It generally presents as a collection of pink, red or dusky brown, firm dermal nodules of varying size but usually less than 15 mm diameter (Figure 137.37). The nodules are often subject to episodes of pain and may be tender. The pain can be provoked by touching or chilling the skin, or by emotional disturbance, and is often worse in winter. Some lesions contract and become paler when painful. The condition usually begins with the appearance of one small nodule, which gradually increases in size, and further similar lesions appear nearby or at some other area. Adjacent tumours may coalesce to form a plaque. The areas most commonly affected are the extremities, with the proximal and extensor aspects somewhat favoured. The trunk is involved more often than the head and neck. Multiple lesions may be regional and unilateral, or more than one region can be affected. Solitary lesions may occur, apart from the dartoic type.
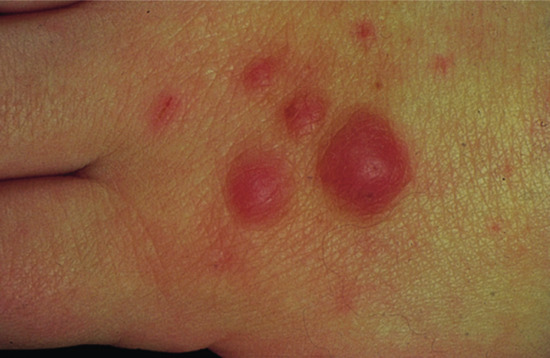
Figure 137.37 Clinical appearance of multiple leiomyomas.
Genital leiomyoma is a solitary dermal nodule occurring most commonly in the scrotum, but also appearing on the penis, labia majora and nipple area [5, 6, 7, 8]. Scrotal tumours are often large. Pain is less frequent than with pilar leiomyoma. Contraction in response to stimulation by touch or cold can occur.
Angioleiomyoma is usually a solitary, flesh-coloured, rounded, subcutaneous or deep dermal tumour up to 40 mm in diameter [3, 4]. It is more frequent on the lower limb than the upper and may appear on the trunk or face. About half the reported cases have been painful. Lesions are longstanding and present between the fourth and sixth decades of life. Pain may be triggered by changes in temperature, pregnancy or menses.
Differential diagnosis
The multiple type should cause little difficulty, and even without pain it is fairly distinctive. The solitary painful lesion may be mistaken for a glomus tumour or an eccrine spiradenoma, and a history of contraction is helpful.
Disease course and prognosis
The behaviour is benign and local recurrences are exceptional.
Management
Surgical excision cures the solitary tumour. The severity of the pain may make the patient demand treatment, and extensive lesions require plastic surgery. Medical treatments that may relieve pain include calcium-channel blockers and gabapentin. However, the effect is not long lasting.
Leiomyosarcoma (dermal and subcutaneous) [1-4]
Definition and nomenclature
A histologically malignant tumour displaying smooth muscle differentiation. Tumours are divided into those occurring in the subcutaneous tissue and those arising in the dermis. Pure dermal lesions have a very different behaviour from those arising in the subcutis and it is therefore important to separate them. Due to the benign behaviour of dermal tumours, it has recently been proposed that they should be renamed as atypical intradermal smooth muscle neoplasms [5].
Epidemiology
Incidence and prevalence
Both variants (dermal and subcutanoeus) are rare.
Age
Atypical intradermal smooth muscle neoplasm (dermal leiomyosarcoma) is more common middle-aged adults. Subcutaneous leiomoysarcoma affects middle-aged to elderly patients.
Sex
Both dermal lesions and subcutaneous leiomyosarcomas have predilection for males.
Pathophysiology
Pathology [3,6,7-10]
The lesion is distinguished from other dermal malignant tumours composed of spindle-shaped cells by the presence of fascicles of eosinophilic spindle-shaped cells with vesicular cigar-shaped nuclei. The degree of differentiation varies and necrosis tends to be present in deeper tumours, and it is rare in those arising primarily in the dermis. Mitotic figures are common in both variants. Most tumours are actin, calponin, desmin and h-caldesmon-positive, but staining may be lost in poorly differentiated variants. Dermal variants tend to be consistently positive for all markers. Rare cases have a desmoplastic stroma making histological diagnosis difficult [11, 12]. About 45% of leiomyosarcomas are immunohistochemically positive for keratin, and this is usually focal.
Clinical features [5,6,13,14]
History and presentation
Both types of tumours are more common in the trunk and lower extremities. Dermal lesions usually present as a skin-coloured or red papule or nodule. The latter is usually larger than a leiomyoma and may be painful. Subcutaneous tumours present as nodular tumours, ulcerated plaques [15] or diffuse swellings [16]. They may invade underlying muscle fascia. Rare subcutaneous tumours may arise from the penis [17] or vulva. An exceptional tumour has been reported arising in a naevus sebaceous [18].
Disease course and progonosis
Atypical intradermal smooth muscle neoplasms have a 5% recurrence rate, but they almost never metastasize [5, 6]. Subcutaneous tumours metastasize in up to 50% of cases and they are associated with a mortality of about 30% [9].
Management
Wide surgical excision is necessary, as local recurrence follows inadequate excision [5].
SKELETAL MUSCLE TUMOURS
Rhabdomyosarcomatous congenital hamartoma [1-8]
Definition and nomenclature [1-3]
Rhabdomyomatous mesenchymal hamartoma is a dermal lesion with a prominent component of skeletal muscle.
Epidemiology [1-4]
Incidence and prevalence
It is a very rare tumour.
Age
Typically it is congenital. Exceptional cases in adults have been reported [4, 5].
Sex
A female predominance is noticed.
Pathophysiology
Genetics [3, 4]
Aberrant embryonic migration of mesodermally derived tissues, or a genetic defect predisposing to the formation of hamartomas, has been suggested as possible aetiological factors.
Pathology [1-7]
Microscopically intersecting bundles of mature skeletal muscle orientated perpendicular to the epidermis are admixed with varying amounts of mature fat, collagen and adnexal structures.
Clinical features [1-8]
History and presentation
The majority of lesions present as papules or polyps, resembling skin tags. The head and neck (especially the central face) is the site of predilection. An association with other congenital abnormalities as well as Delleman syndrome has been reported.
Prognosis
The tumour is benign. Spontaneous regression may rarely occur.
Management
Complete excision is curative.
Rhabdomyoma
Rhabdomyomas are divided into adult, fetal and genital types. They mainly occur in soft tissues, vulva or vagina, upper respiratory tract and internal organs. Presentation in the skin is almost never seen, and they will not be discussed further in this chapter.
Cutaneous rhabdomyosarcoma
Malignant tumours with skeletal-muscle differentiation are classified into two large groups, the embryonal and alveolar types. Although rhabdomyosarcomas represent up to 8% of tumours in children, primary involvement of the skin by this tumour is very rare [1]. Much more common is involvement of the skin by direct extension from deeper soft tissues. Only 16 cases of primary cutaneous rhabdomyosarcoma have been reported in the literature so far, and only five of these have occurred in adults [2, 3]. The most common subtype occurring in the skin is the alveolar variant. The majority of cases have presented on the face. The prognosis is difficult to estimate because of the rarity of these cases and the limited follow-up available.
TUMOURS OF FAT CELLS
Angiolipoma [1-11]
Definition
Angiolipoma is a benign tumour composed of mature white adipose tissue admixed with a variable amount of thin-walled vessels. By definition, some of the vessels should contain fibrin microthrombi.
Epidemiology [1-3]
Incidence and prevalence
It is a relatively common tumour.
Age
Lesions usually occur in young adults. They are rare in children, the middle aged and the elderly.
Sex
A male predominance is observed.
Pathophysiology
Pathology [1-4]
Angiolipomas are encapsulated. They consist of mature adipose tissue intermingled with a prominent vascular component usually more prominent in the periphery of tumour lobules. Some of the blood vessels contain fibrin thrombi. Cases with a prominent vascular component obscuring the adipocytic component have been termed ‘cellular angiolipomas’ [4].
Genetics [5,6]
Cytogenetic studies have consistently shown no karyotypic abnormality. This is a unique finding in adipocytic tumours, which has given rise to arguments regarding its pathogenesis. It has been proposed that the lesion is primarily a vascular tumour and that it should be named lipoangioma.
Clinical features [1-3,7-11]
History and presentation
Typically, angiolipomas are multiple variably painful subcutaneous nodules. They are usually small and well circumscribed. A predilection for the upper limbs is observed, followed by the trunk and lower limbs. Intraosseous cases have been reported [7].
Familial incidence is well documented in a subset of angiolipomas [8, 9, 10, 11].
Prognosis
The tumour is benign. Local recurrence and malignant transformation does not occur.
Management
Complete excision is curative.
Lipoma [1-20]
Definition
Lipoma is a benign tumour composed of variable amounts of mature white adipose tissue.
Epidemiology [1-3]
Incidence and prevalence
It is the most common human mesenchymal neoplasm. It is more frequent in the obese.
Age
Tumours occur in adults (40–60 years old). They are rare in children. Congenital lesions have been reported.
Sex
It is more common in females.
Pathophysiology
Genetics [4-9]
Chromosome aberrations have been found in more than half of the cases examined. Three types of chromosomal abnormalities have been described: tumours with a 12q13-15 rearrangement (the most common), tumours with a 6p21-23 rearrangement and lesions with deletion of 13q.
Pathology [1-3,10-15]
Tumours are usually encapsulated and consist of lobules of mature adipose tissue divided by delicate fibrous septa. Adipocytes are uniform in size and shape. Nuclear atypia is not a feature. There is no mitotic activity. Degenerative changes frequently occur, usually in the form of fat necrosis. Myxoid change is not uncommon.
Some lipomas may contain fibrous tissue in variable amounts. Metaplastic bone, cartilage or muscle tissue may be observed.
Clinical features [1-3,10-20]
History and presentation
The typical presentation is of a slowly growing and painless subcutaneous mass. There is a predilection for the trunk, abdomen and neck. Other sites, such as the proximal extremities, face, scalp and less commonly the hands and feet, may be affected. Spinal cord and pancreatic lipomas have been reported [19, 20].
Lipomas are usually solitary but multiple lesions have been described; some of them in a setting of an autosomal dominant disorder.
Size is variable and large lesions may occur. They are most often well-circumscribed but deep-seated variants, such as intramuscular lipomas, may be ill defined.
Dermal examples may resemble fibroepithelial polyps.
Prognosis
The tumour is benign. Local recurrence is not a frequent feature. Progression to liposarcoma is exceptional.
Management
Complete excision is curative in almost every case.
Hibernoma [1-9]
Definition [1, 2]
Hibernoma is a benign tumour composed of brown granular multivacuolated cells (resembling normal brown fat) admixed with a variable amount of mature white adipose tissue.
Epidemiology [3]
Incidence and prevalence
It is a rare tumour.
Age
It predominantly occurs in young adults (third and fourth decades). Approximately 5% of the tumours occur in children and 7% in elderly individuals.
Sex
A female predominance is noticed.
Pathophysiology
Pathology [3-6]
Hibernomas are usually encapsulated and lobulated. Typical cases consist of adipocytes with an eosinophilic granular multivacuolated cytoplasm and a centrally located nucleous. Mature white adipose tissue is present in a variable proportion. Variants include the lipoma-like, myxoid and spindle cell hibernoma.
By immunohistochemistry, the adipocytes are positive for S100 protein.
Genetics [7, 8]
Cytogenetics studies, in most cases, have shown aberrations of the 11q13-21 region, and in fewer cases of 10q22. Homozygous deletion of the MEN1 suppressor gene, has been found in some cases.
Clinical features [3-5, 9]
History and presentation
Hibernoma presents as painless subcutaneous mass of long duration. The site of predilection is the thigh. This is followed by the trunk, upper limbs and head and neck. Deep-seated lesions can occur. Visceral locations, such as retroperitoneum, have been described.
Prognosis
The tumour is benign. Local recurrence is not a feature.
Management
Local excision is curative.
Lipoblastoma and lipoblastomatosis [1-3]
Definition
Lipoblastoma is a tumour that occurs almost exclusively in infants and children. It is characterized by a proliferation of immature fat cells in a myxoid stroma (that may mimic myxoid liposarcoma) and intermixed with mature adipocytes [1]. Lipoblastoma is a well-circumscribed subcutaneous tumour; lipoblastomatosis refers to a deeper lesion or those that have an infiltrative growth pattern.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
Most cases present during the first few years of life.
Sex
There is predilection for males.
Pathophysiology
Pathology [1, 2, 3]
Tumours have a characteristic lobular appearance. Each tumour lobule is separated by fibrous septae and consists of a mixture of small, univacuolated, signet-ring cells, spindle-shaped or stellate cells and scattered mature adipocytes. In the background, there are prominent myxoid changes and numerous small vessels in a typical ‘crow's feet’ distribution, mimicking a myxoid liposarcoma. Distinction from the latter may be very difficult, especially in small biopsies. The clinical information is therefore crucial, as myxoid liposarcoma is vanishingly rare in children and almost never occurs before the age of 10 years [4]. Furthermore, lipoblastoma tends to be less cellular than myxoid liposarcoma and has a lobular architecture. Over time, maturation occurs, and in some cases most of the tumour is composed of mature fat cells.
Genetics
Cytogenetic studies in lipoblastoma have shown rearrangements on chromosome 8q [5, 6].
Clinical features [1, 2, 3]
The majority of tumours occur on the limbs and trunk as an asymptomatic mass no more than a few centimetres in diameter. Lipoblastoma is much more common than lipoblastomatosis.
Disease course and prognosis
Tumours behave in a benign fashion but deep-seated lesions may recur locally usually as a result of incomplete excision.
Management
The tumour is benign, and simple excision is the treatment of choice.
Spindle cell and pleomorphic lipoma [1-5]
Definition
Spindle cell lipoma is composed of mature adipocytes and variable numbers of short bland spindle-shaped cells with indistinct cytoplasm. Pleomorphic lipoma is composed of mature adipocytes, cells with hyperchromatic nuclei and frequent multinucleation, and collagen bundles. Both types of tumour may overlap, and they are therefore considered to be part of the same spectrum.
Epidemiology
Incidence and prevalence
Lesions are relatively rare.
Age
There is predilection for middle-aged to old patients with a median of 55 years.
Sex
Tumours have a strong predilection for males.
Pathophysiology
Pathology [1, 2, 4]
Spindle cell lipoma presents as a well-circumscribed tumour composed of mature adipocytes intermixed with short spindle-shaped cells with wavy nuclei. Hyalinized collagen bundles and focal myxoid change are prominent. Pseudovascular spaces are prominent in some cases. The spindle-shaped cells stain for CD34 and the adipocytes are positive for S100. Pleomorphic lipoma is also well circumscribed and composed of mature adipocytes intermixed with uninucleated or multinucleated cells with hyperchromatic nuclei. The nuclei in the multinucleated cells are often arranged in a circle (floret cell). The histological diagnosis may be quite difficult in cases with few or no mature fat cells [6]. Rare variants contain real prominent vascular spaces [7].
Genetics
Cytogenetic studies of both tumours have shown variable abnormalities, most commonly in chromosome 16q and rarely in chromosomes 13q and 6p [8, 9].
Clinical features [1, 2, 4, 5]
History and presentation
Spindle cell/pleomorphic lipoma usually presents as a small subcutaneous nodule on the upper back or nape of the neck. Occasional, purely dermal examples may be seen [10]. Multiple lesions, and familial cases, occur rarely [3].
Disease course and prognosis
Local recurrence after excision is very rare.
Management
Simple excision is the treatment of choice.
Atypical lipomatous tumour [1-4]
Definition and nomenclature
This is a lesion composed of lobules of mature adipose cells, with scattered larger cells with variation in nuclear size and hyperchromatism. The term ‘atypical lipomatous tumour’ is usually used for neoplasms occurring in the subcutis or within skeletal muscle. Similar tumours occurring in the abdominal cavity are regarded as well-differentiated liposarcomas, in view of the fact that they have a potential to cause death as a result of extensive growth. Only subcutaneous skin lesions will be discussed here.
Epidemiology
Incidence and prevalence
Tumours are relatively frequent in the subcutaneous tissue and exceptional in the dermis.
Age
Middle-aged to old adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
Typically, lobules of mature adipose tissue, with or without fibrous tissue and myxoid change, are seen. Focal variation in the size and shape of adipocytes is seen and this is associated with nuclear enlargement and hyperchromatism. Vacuolated cells may also be found. Atypical cells are often present in the fibrous tissue. Some tumours are classified as cellular based on the presence of non-lipogenic areas of increased cellularity with low-mitotic activity [5]. Dedifferentiated tumours are lesions that develop a high-grade sarcomatous component that is associated with poor prognosis [6]. This change does not usually develop in tumours that are superficially located.
Genetics
Cytogenetic studies of these neoplasms have found chromosomal abnormalities in most cases. About a third of cases show supernumerary ring chromosomes affecting chromosome 12q.13-15 [4]. This results in amplification of MDM2 and CDK4 genes [7]. Expression of these genes can be detected by FISH, real-time PCR and immunohistochemistry, making it a useful diagnostic aid. Of these methods, FISH and real-time PCR, but particularly the former, are more sensitive than immunohistochemistry for detection of these amplifications [8].
Clinical features [1-3]
History and presentation
Tumours may be large, are asymptomatic and have the same clinical appearance as a lipoma. Dermal tumours may be identical to a fibroepithelial polyp [9].
Disease course and prognosis
There is a tendency for local recurrence, but metastases are not seen unless the tumour undergoes dedifferentiation, which does not tend to happen in superficially located tumours, particularly those in the subcutaneous tissue [6].
Management
Complete surgical excision is indicated.
Liposarcoma [1]
Myxoid and round cell liposarcoma and pleomorphic liposarcoma are vanishingly rare in the skin. Only a few cases of primary cutaneous liposarcoma have been described. Follow-up is limited, but the behaviour seems to be better than that of their deeper counterparts, probably reflecting early detection and treatment and the easy accessibility to the skin. Liposarcoma will not be discussed further in this chapter.
TUMOURS OF UNCERTAIN HISTOGENESIS
Acral fibromyxoma [1-3,4,5]
Definition and nomenclature
Acral fibromyxoma is a distinctive benign dermal and/or subcutaneous, fibroblastic tumour with a strong predilection for digits of both the hands and feet.
Epidemiology
Incidence and prevalence
It is relatively frequent.
Age
The age range is wide but most patients are middle aged.
Sex
There is predilection for males.
Pathophysiology
Pathology
Lesions are circumscribed and consist of bland stellate and spindle-shaped cells in a variably prominent myxoid and collagenous stroma and with small blood vessels in the background. Some lesions are more cellular and have been described in the literature under the rubric of cellular fibroma of the digits [2, 5]. In myxoid areas, mast cells are often seen. Scattered multinucleated cells may be seen in some cases. Mitotic figures are very rare. Tumour cells are diffusely positive for CD34 and may be focally positive for EMA, CD99 and smooth muscle actin.
In the past, it is likely that these tumours were classified as neurofibromas because of the cytomorphology of tumour cells and the myxoid background, with fairly frequent mast cells. However, neurofibroma is rare in acral sites and tumour cells are positive for S100 and only focally positive for CD34.
Clinical features
History and presentation
Most cases present as a longstanding, solitary mass measuring between 1 and 2 cm, on the hands and feet (overwhelmingly involving the digits, often in a subungual location) [1, 2, 3, 4].
Disease course and prognosis
Local recurrence is exceptional and is usually associated with incomplete excision.
Management
Simple local excision is the treatment of choice.
Superficial angiomyxoma [1, 2]
Definition
Superficial angiomyxoma is a dermal or subcutaneous tumour composed of a mixture of small blood vessels and sparse spindle-shaped cells in a prominent myxoid stroma.
Epidemiology
Incidence and prevalence
Lesions are rare.
Age
Most patients are adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
Tumours are multilobulated, with copious myxoid stroma, numerous delicate small blood vessels and spindle-shaped or stellated bland cells, probably representing fibroblasts. Aggregates of inflammatory cells, mainly neutrophils, are frequent. In up to 30% of cases epithelial structures, probably representing hyperplastic trapped adnexal structures (particularly hair follicles), are identified.
Clinical features [1, 2]
Most cases present as an asymptomatic solitary papule or nodule. Lesions are usually less than 3 cm and have a wide anatomical distribution with a predilection for the trunk, head and neck and genital skin. In patients with multiple lesions, the possibility of Carney complex should be considered (see Chapter 147) [3].
Disease course and prognosis
The behaviour is benign. Local recurrence after surgical treatment is seen in up to 30% of cases [1, 2].
Management
Excision is the treatment of choice.
Digital mucous cyst [1]
Digital mucous cyst presents mainly on the fingers as a small solitary painful nodule. Females are much more commonly affected than males and there is a tendency for local recurrence. Lesions are poorly circumscribed and consist of abundant myxoid stroma with only scattered bland spindle-shaped cells (see Chapter 59).
Dermal non–neural granular cell tumour
Definition and nomenclature
This is a distinctive dermal tumour with no specific line of differentiation, initially described as primitive polypoid granular cell tumour in 1991 [1]. Tumours are not related to neural granular cell tumours, which are uniformly S100 positive. They occur both in the dermis and in subcutaneous tissue.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
There is a wide age range but most patients are adults.
Sex
There is no sex predilection.
Pathophysiology
Pathology
Histologically, lesions show rounded or spindle-shaped cells with prominent granular cell change (Figure 137.38). Polypoid tumours have an epithelial collarette. Nuclear pleomorphism varies but does not tend to be prominent. Mitotic figures may be prominent. Multinucleated cells are sometimes seen. There is diffuse positivity for NKI-C3, which is a non-specific marker for lysosomes, and focal positivity for CD68 and neuron-specific enolase (NSE). Tumour cells do not stain for keratin, EMA, actin, desmin, h-caldesmon or S100 [3, 4].
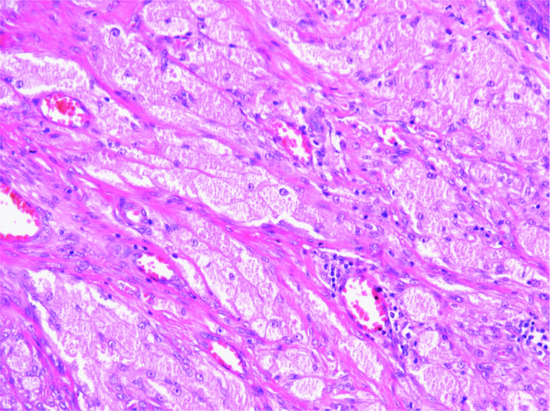
Figure 137.38 Large cells with prominent granular cell change in a case of dermal non-neural granular cell tumour.
Clinical features
History and presentation
This tumour usually presents as an exophytic small lesion with a wide anatomical distribution, wide age range and no sex predilection. Not all tumours are polypoid. Clinical behaviour is usually benign with only rare local recurrences [2, 3, 4]. However, a single case of metastasis to a regional lymph node has been reported [3].
Management
Complete conservative excision is the treatment of choice.
Haemosiderotic fibrolipomatous tumour [1, 2]
Definition and nomenclature
This is a distinctive rare lesion that occurs almost exclusively on the foot, particularly the ankle, and was initially thought to represent a reactive process [1]. A relationship with impaired circulation, particularly stasis, has been suggested [3]. However, it is now believed that lesions are neoplastic. A relation to pleomorphic hyalinizing angiectatic tumour [4] and more recently to myxoinflammatory fibroblastic sarcoma has been suggested [5]. The association with the latter is not only based on the coexistence of both tumours in some cases but also on cytogenetic analysis (see later).
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
There is predilection for middle-aged to elderly adults.
Sex
Females are more frequently affected than males.
Pathophysiology
Pathology
The tumour is fairly circumscribed and it is composed of lobules of mature adipose tissue with scattered areas containing variable numbers of spindle-shaped cells, which may be slightly hyperchromatic and often contain abundant intracytoplasmic haemosiderin. Histiocyte-like cells may also be seen. These cells are bland and only rarely slightly atypical. In the background, there may be a few mononuclear inflammatory cells, mainly lymphocytes. Mitotic figures are exceptional. Spindle cells are positive for calponin and CD34 and may be positive for CD68 but negative for other markers, including desmin and h-caldesmon.
Genetics
As myxoinflammatory fibroblastic sarcoma and confirming their close relationship, these tumours show a t(1;10) translocation with rearrangements of TGFBR3 and MGEA5 [5].
Clinical features
History and presentation
Lesions present as a slowly growing fairly well-defined subcutaneous tumour, which tends to be asymptomatic. Size is variable and may be several centimetres in diameter.
Disease course and prognosis
The behaviour is usually benign with occasional local recurrences. However, recently, a tumour that recurred as a myxoinflammatory fibroblastic sarcoma metastasized resulting in the demise of the patient [6].
Management
The treatment of choice is complete surgical excision.
Perivascular epithelioid cell tumour (‘PEComa’)
Definition
Perivascular epithelioid cell tumour is part of a spectrum of neoplasms that includes clear cell ‘sugar’ tumour of the lung, angiomyolipoma, lymphangioleiomyomatosis and clear cell myomelanocytic tumour of the falciform ligament [1, 2, 3]. Occurrence in the skin and soft tissue is rare but it is likely that the lesion is underrecognized. Although tumour cells are usually positive for melanocytic markers such as HMB45 and Melan-A, the cell of origin has not been identified.
Epidemiology
Incidence and prevalence
Cutaneous tumours are rare [4].
Age
Most patients are young adults.
Sex
There is predilection for females.
Pathophysiology
Pathology [3, 4]
Histology is distinctive and consists of bland epithelioid cells, typically arranged radially around thin-walled vascular channels. A smaller population of spindle cells is often seen. Tumour cells have pale pink cytoplasm and vesicular nuclei. Malignant examples may be seen but have not been reported in the skin. The immunophenotype is distinctive, as the tumour cells stain for melanocytic markers including HMB45, MITF-1, Melan-A and tyrosinase and for muscular markers including SMA, desmin and calponin. They are usually negative for S100 and keratin.
Genetics
Recurrent chromosomal alterations leading to inactivation of the TSC1 or TSC2 genes or rearrangments of TFE3 have been demonstrated in visceral PEComas. However, the latter rearrangement does not seem to occur in cutaneous tumours [5].
Clinical features
History and presentation
Tumours present as a small nondescript lesion, more frequently on the lower extremities.
Disease course and prognosis
Behaviour is benign. Malignant tumours are very rare.
Management
Simple excision is the treatment of choice.
Deep (‘aggressive’) Angiomyxoma [1, 2, 3]
Definition
Deep ‘aggressive’ angiomyxoma is a distinctive tumour occurring in the genital region and pelvis, predominantly in females. It is characterized by bland spindle-shaped cells in the background of a prominent myxoid stroma and frequent thick-walled blood vessels.
Epidemiology
Incidence and prevalence
Tumours are rare.
Age
Most cases present during the reproductive years particularly during the fourth decade of life [3]. Presentation after the menopause and in children is rare [4].
Sex
It occurs almost exclusively in females although rare lesions have been described in males [5].
Pathophysiology
Pathology
The lesion is infiltrative and is composed of spindle- or stellate-shaped bland cells with scanty cytoplasm, surrounded by prominent myxoid stroma. Small to medium-sized thick-walled blood vessels are seen throughout the tumour. Mitotic figures are very rare. Rare cases contain multinucleated giant cells [7]. Interestingly, tumour cells are positive for actin and desmin. Tumour cells are often positive for oestrogen and/or progesterone receptors [8]. Immunohistochemistry for HMGA2 protein is a useful aid in the diagnosis of this neoplasm as it is positive in most aggressive angiomyxomas and negative or minimally positive in other mimics [9].
Genetics
Cytogenetics demonstrate a translocation involving the chromosomal region 12q14-15, involving a rearrangement of HMGA2 (an architectural trancription factor) [6].
Clinical features
History and presentation
Cases in children may involve the spermatic cord [4]. Tumours are slowly growing, and by the time of presentation they are large and ill defined, often measuring 10 cm or more. The most commonly affected sites are the vulva and perineum. Extension into deeper soft tissues is often found.
Disease course and prognosis
Local recurrence is observed in up to a third of cases. Exceptional metastases have been reported [10, 11].
Management
Complete surgical excision is the treatment of choice but it is usually difficult because of the infiltration of surrounding tissue. However, recurrences are not usually destructive, and radical surgical procedures are therefore not indicated [12]. Furthermore, the outcome of patients with resection positive margins does not seem to be very different compared to those with resection negative margins [2]. An alternative to surgical management may be the use of gonadotrophin-releasing hormone agonists, a treatment that appears to be promising in isolated case reports [13, 14].
Angiomatoid fibrous histiocytoma [1,2,3-5]
Definition and nomenclature
Angiomatoid FH was initially described as a variant of malignant FH [1]. It has recently been reclassified as a neoplasm with low-grade malignant behaviour, unrelated to malignant FH. Although it is considered to be ‘fibrohistiocytic’ due to the cytological resemblance of tumour cells to histiocytes, focal positivity of these cells to desmin raises the possibility of muscular differentiation. However, the lesion is now regarded as of uncertain histiogenesis.
Epidemiology
Incidence and prevalence
Represents a rare tumour.
Age
It presents in children and young adults and rarely in older patients.
Sex
There is no sex predilection.
Pathophysiology
Pathology [1, 2, 3, 4]
Low-power examination reveals haemorrhagic, pseudovascular, cavernous-like, cystic spaces filled with red blood cells. Mononuclear inflammatory cells are prominent (including lymphocytes, histiocytes and plasma cells) and germinal centres and sclerosis, are present in some cases [8]. Eosinophils may be seen. Tumour cells are arranged in sheets and consist of short spindle-shaped and round cells with pink cytoplasm and vesicular nuclei. Cytological atypia is sometimes present, and the mitotic count tends to be low. Tumour cells are focally positive to desmin in about half of the cases [3, 4]. Positivity for other markers including EMA, muscle-specific actin, calponin, CD99 and CD68 may be seen.
Genetics
Initial cytogenetic studies demonstrated a translocation between chromosomes 16p11 involving the FUS (TLS) gene and chromosome 12q13 involving the ATF1 gene. The resultant protein (FUS/ATF1) is similar to the protein present in clear cell sarcoma (EWS/ATF1) involving t(12;22)(q13;q12) [6]. However, other cases show a different fusion gene, EWSR1-CREB1, which seems to be the most common fusion gene in these tumours [7].
Clinical features
History and presentation
It presents as an asymptomatic blue or skin-coloured subcutaneous or deeper mass. Primary dermal tumours are exceptional. Most cases occur on the limbs and patients may present with systemic symptoms including fever, anaemia and weight loss. Generalized lymphadenopathy may also be seen.
Disease course and prognosis
Local recurrence is observed in about 15% of patients and, exceptionally, metastasis to neighbouring soft tissues or regional lymph nodes may occur. Complete excision and follow-up are therefore indicated. Local recurrence is more likely with deep tumours, those with an infiltrative growth pattern and those that are incompletely removed [2].
Management
Most cases are cured after adequate excision.
Epithelioid sarcoma [1, 2, 3, 4]
Definition
A distinctive, malignant mesenchymal tumour composed of cells with epithelial differentiation. It is divided into two clinicopathological subtypes: conventional (classic) involving mainly acral sites (granuloma-like) and proximal-type involving the trunk and proximal limbs (solid growth) [5, 6].
Epidemiology
Incidence and prevalence
It is an uncommon tumour representing less than 1% of all soft-tissue sarcomas [7]. The proximal type variant is much less common than the conventional variant.
Age
Conventional tumours are more common in early adult life. The proximal type variant is more common in older patients. Both variants can present in any age group.
Sex
Males are more often affected than females in both variants.
Pathophysiology
The tumour is composed of firm nodules, 5–50 mm or larger in diameter surrounded by fibrous tissue and fat. It is often closely associated with fascia, periosteum, tendon or nerve sheaths. The cut surface is greyish-white and flecked or mottled with yellow or brown, reflecting the presence of areas of necrosis. Microscopically, there are masses of large round polygonal or spindle cells with acidophilic cytoplasm. Spindle cells are often present and may predominate. The larger nodules have necrotic centres and show so-called ‘geographical necrosis’, which may be mistaken on scanning power microscopy for a granuloma. Mitotic figures are common, and binucleate cells occur. Variable cytological atypia is always present and may be prominent. Intercellular hyalinized collagen increases the acidophilia, while calcification, with osteoid or bone formation (in about 20% of cases), may take place in the necrotic areas. The tumour spreads along dense fibrous structures and may ulcerate in areas with little subcutaneous fat. Local recurrence after excision is common, and metastasis, principally to lymph nodes, lung and pleura, may occur. Tumour cells show clear histological, ultrastructural and immunohistochemical evidence of epithelial differentiation.
The proximal type of epithelioid sarcoma shows a similar multinodular growth pattern but tumour cells are larger and with a more rhabdoid appearance consisting of abundant cytoplasm and large nuclei with or without a prominent eosinophilic nucleolus [5, 6].
Immunohistochemically, tumour cells in both variants of epithelioid sarcoma have the same profile. They are positive for vimentin, keratin and EMA, and 50% of cases are positive for CD34 [8]. Most examples of both variants of the tumour show loss of nuclear expression of INI1 [9].
Genetics
Tumours often display complex chromosomal abnormalities involving chromosome 22q11. Some of these changes involve deletions or mutations of the tumour suppressor gene SMARCB1 (INI1, hSNF5). Loss of nuclear expression of the latter can be demonstrated by immunohistochemistry and this is a very useful aid in the differential diagnosis of these tumours [9] (see later).
Clinical features [1, 2, 3, 4, 8, 12]
History and presentation
The presenting sign can be a dermal nodule that grows outwards and may ulcerate early, a nodule or lobular subcutaneous tumour that is painless and grows slowly, or a tumour attached to deeper structures that is rather poorly defined and causes pain, paraesthesiae or muscular wasting when growing along a large trunk nerve. As a result of prominent perineurial and perivascular extension of tumour cells, multiple nodules in a sporotrichoid distribution may be seen. The distal extremities are the usual situation for the tumour, particularly the flexor aspect of the finger and the palm. It may grow at a deceptively slow rate.
The proximal-type epithelioid sarcoma presents as a large often deep-seated mass on the proximal limbs, genitalia, buttocks, trunk (mainly axilla) or head and neck [5, 6].
Disease course and prognosis
Local recurrence and metastasis may occur years after the original diagnosis. The survival rate has been estimated at between 60% and 80% at 5 years and between 42% and 62% at 10 years [8, 14, 15, 16, 17, 18]. The proximal-type variant has a worse prognosis. Features associated with poorer prognosis include male sex, older age at diagnosis, proximal location, deep involvement, rhabdoid phenotype, tumour size, mitotic rate, necrosis, vascular invasion, local recurrence and lymph node metastasis [3, 12, 13, 14, 15, 16, 17, 18].
Management
Complete removal by surgical excision is essential if local recurrence and eventual metastasis are to be avoided, and the earlier this is done the less likely is the process to spread along fascial planes. Surgical excision followed by radiotherapy is often recommended. Involvement of regional lymph nodes is associated with distant metastasis (mainly to the lungs) and death [18].
Clear cell sarcoma [1-4,5]
Definition and nomenclature
Clear cell sarcoma is a distinctive, malignant soft-tissue tumour that displays melanocytic differentiation.
Epidemiology
Incidence and prevalence
Tumours are rare. Primary dermal clear cell sarcomas are exceptional [6, 7].
Age
The tumour is more common in young adults.
Sex
There is predilection for females.
Pathophysiology
Pathology
The lesion has a lobular growth pattern. Tumour cells are fairly homogeneous and contain clear or pale pink cytoplasm and a prominent eosinophilic nucleolus. Mitotic figures are not prominent, but multinucleated giant cells with a wreath-like arrangement of the nuclei are often identified. Loose thin bands of collagen surround tumour cells. Secondary involvement of the dermis is relatively common. Necrosis is sometimes seen. Melanin is sometimes identified, and S100, HMB45 and Melan-A are usually positive. Exceptional cases of clear cell sarcoma may display a junctional component [8]. This may lead to a misdiagnosis of Spitz naevus. Electron-microscopic examination of tumour cells reveals the presence of melanosomes.
Genetics
Cytogenetic analysis often reveals a translocation t(12;22)(q13;q12); this translocation is not found in melanoma, which often has mutations in the BRAF gene (a feature exceptional in clear cell sarcoma) [9, 10, 11, 12]. The clear cell sarcoma translocation results in an EWSR1-ATF1 fusion gene. Rare cases of clear cell sarcoma may also show KIT mutations [12].
Clinical features
Most cases occur on the lower limbs, with a predilection for the foot. The upper limb is affected in about 25% of cases. Tumours tend to grow around tendons, are usually less than 3 cm in diameter and are often painful.
Disease course and prognosis
About 50% of patients develop metastatic disease, often many years after the initial diagnosis. Prognosis is associated with mitotic index, size of the tumour and presence of necrosis [13, 14]. The 5- and 10-year survival rates are 52% and 25%, respectively [14]. Tumour size appears to be associated with worse prognosis [15]. It is not clear whether purely dermal tumours have a better prognosis than those deeply located.
Management [1-4,13,14,15]
Wide excision is the treatment of choice. Chemotherapy does not seem to be effective in the treatment of disseminated disease.
References
Introduction
- Goldblum JR Folpe Al, Weiss SW. Enzinger and Weiss's Soft Tissue Tumors, 6th edn. St Louis: Mosby, 2013.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. WHO Classification of Tumours of Bone and Soft Tissue, 4th edn. Geneva, Switzerland: WHO, 2013.
Fibrous and myofibroblastic tumours
Fibrous and myofibroblastic tumours
- Okun M. Fibrous papules and nevocellular nevi. J Am Acad Dermatol 1984;10:670–1.
- Meigel WN, Ackerman AB. Fibrous papule of face. Am J Dermatopathol 1979;1:329–40.
- Saylan T, Marks R, Wilson Jones E. Fibrous papule of the nose. Br J Dermatol 1971;85:111–18.
- de Cambourg G, Cribier B. Fibrous papules of the face: a retrospective anatomoclinical study of 238 cases. Ann Dermatol Venereol 2013;140:763-70.
- Kucher C, McNiff JM. Epithelioid fibrous papule—a new variant. J Cutan Pathol 2007;34:571–5.
- Lee AN, Stein SL, Cohen LM. Clear cell fibrous papule with NKI/C3 expression: clinical and histologic features in six cases. Am J Dermatopathol 2005;27:296–300.
- Chiang YY, Tsai HH, Lee WR, et al. Clear cell fibrous papule: report of a case mimicking a balloon cell nevus. J Cutan Pathol 2009;36:381–4.
- Jacyk WK, Rütten A, Requena L. Fibrous papule of the face with granular cells. Dermatology 2008;216:56–9.
Storiform collagenoma
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol 1989;20:266–71.
- Metcalf JS, Maize JC, LeBoit PE. Circumscribed storiform collagenoma (sclerosing fibroma). Am J Dermatopathol 1991;13:122–9.
- Starink TM, Meijer CJLM, Brownstein MH. The cutaneous pathology of Cowden's disease: new findings. J Cutan Pathol 1985;12:83–93.
- Chen TM, Purohit SK, Wang AR. Pleomorphic sclerotic fibroma: a case report and literature review. Am J Dermatopathol 2002;24:54–8.
- High WA, Stewart D, Essary LR, et al. Sclerotic fibroma-like change in various neoplastic and inflammatory skin lesions: is sclerotic fibroma a distinct entity? J Cutan Pathol 2004;31:373–8.
- Rudolph P, Schubert C, Harms D, et al. Giant cell collagenoma:a benign dermal tumor with distinctive multinucleate cells. Am J Surg Pathol 1998;22:57–63.
- García Doval I, Casas L, Toribio J. Pleomorphic fibroma of the skin, a form of sclerotic fibroma: an immunohistochemical study. Clin Exp Dermatol 1998;23:22–4.
Pleomorphic fibroma
- Kamino H, Lee JY, Berke A. Pleomorphic fibroma of the skin: a benign neoplasm with cytologic atypia—a clinicopathologic study of eight cases. Am J Surg Pathol 1989;13:107–13.
Acquired digital fibrokeratoma
- Hare PJ, Smith AJ. Acquired digital fibrokeratoma. Br J Dermatol 1969;81:667–70.
- Berger RS, Spielvogel RL. Dermal papule on a distal digit. Arch Dermatol 1988;124:1559.
- Kint A, Baran R, de Keyser H. Acquired (digital) fibrokeratoma. J Am Acad Dermatol 1985;12:816–21.
- Kakurai M, Yamada T, Kiyosawa T, et al. Giant acquired digital fibrokeratoma. J Am Acad Dermatol 2003;48(5 Suppl.):S67–8.
- Kint A, Baran R. Histopathologic study of Koenen tumors: are they different from acquired digital fibrokeratoma? J Am Acad Dermatol 1988;18:369–72.
Nodular fasciitis
- Bernstein KE, Lattes R. Nodular (pseudosarcomatous) fasciitis, a nonrecurrent lesion: clinicopathologic study of 134 cases. Cancer 1982;49:1668–78.
- Konwaler BE, Keasbey L, Kaplan L. Subcutaneous pseudosarcomatous fibromatosis (fasciitis). J Clin Pathol 1955;25:241–52.
- Shimizu S, Hashimoto H, Enjoji M. Nodular fasciitis: an analysis of 250 patients. Pathology 1984;16:161–6.
- Hutter RVP, Stewart FW, Foote FW. Fasciitis: a report of 70 cases with follow-up proving the benignity of the lesion. Cancer 1962;15:992–1003.
- Pandian TK, Zeidan MM, Ibrahim KA, et al. Nodular fasciitis in the pediatric population. J Pediatr Surg 2013;48:1486–9.
- Patchefsky AS, Enzinger FM. Intravascular fasciitis. A report of 17 cases. Am J Surg Pathol 1981;5:29-36.
- Hussein MR. Cranial fasciitis of childhood: a case report and review of the literature. J Cutan Pathol 2008;35:212–14.
- Summers LE, Florez L, Berberian ZJ, et al. Postoperative cranial fasciitis. Report of two cases and review of the literature. J Neurosurg 2007;106:1080–5.
- Dayan D, Nasrallah V, Vered M. Clinicopathologic correlations of myofibroblastic tumors of the oral cavity: 1. Nodular fasciitis. J Oral Pathol Med 2005;34:426–35.
- Price S, Kahn L, Saxe N. Dermal and intravascular fasciitis. Am J Dermatopathol 1993;15:539–43.
- Nishi SP, Brey NV, Sanchez RL. Dermal nodular fasciitis: three case reports of the head and neck and literature review. J Cutan Pathol 2006;33:378–82.
- Hornick JL, Fletcher CD. Intrarticular nodular fasciitis—a rare lesion: clinicopathologic analysis of a series. Am J Surg Pathol 2006;30:237–41.
- Pérez-Montiel MD, Plaza JA, Domínguez-Malagón H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol 2006;28:105–11.
- Ceballos KM, Nielsen GP, Selig MK, et al. Is anti-h-caldesmon useful for distinguishing smooth muscle and myofibroblastic tumors? An immunohistochemical study. Am J Clin Pathol 2000;114:746–53.
- Bhattacharya B, Dilworth HP, Iocabuzio-Donahue C, et al. Nuclear beta-catenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am J Surg Pathol 2005;29:653–9.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest 2011;91:1427–33.
Fibro-osseous pseudotumour of the digits
- Dupree WB, Enzinger FM. Fibro-osseous pseudotumor of the digits. Cancer 1986;58:2103–9.
- de Silva MV, Reid R. Myositis ossificans and fibroosseous pseudotumor of digits: a clinicopathological review of 64 cases with emphasis on diagnostic pitfalls. Int J Surg Pathol 2003;11:187–95.
- Chaudhry IH, Kazakov DV, Michal M, et al. Fibro-osseus pseudotumor of the digit: a clinicopathological study of 17 cases. J Cutan Pathol 2010;37:323–9.
Ischaemic fasciitis
- Montgomery EA, Meis JM, Mitchell MS, et al. Atypical decubital fibroplasia: a distinctive fibroblastic pseudotumor occurring in debilitated patients. Am J Surg Pathol 1992;16:708–15.
- Baldassano MF, Rosenberg AE, Flotte TJ. Atypical decubital fibroplasia: a series of three cases. J Cutan Pathol 1998;25:149–52.
- Perosio PM, Weiss SW. Ischemic fasciitis: a juxta-skeletal fibroblastic proliferation with a predilection for elderly patients. Mod Pathol 1993;6:69–72.
- Liegl B, Fletcher CD. Ischemic fasciitis: analysis of 44 cases indicating an inconsistent association with immobility or debilitation. Am J Surg Pathol 2008; 32: 1546-52.
- Scanlon R, Kelehan P, Flannelly G, et al. Ischemic fasciitis: an unusual vulvovaginal spindle cell lesion. Int J Gynecol Pathol 2004;23:65–7.
Fibrous hamartoma of infancy
- Enzinger FM. Fibrous hamartoma of infancy. Cancer 1965;18:241–8.
- Mitchell ML, di Sant'Agnese PA, Gerber JE. Fibrous hamartoma of infancy. Hum Pathol 1982;13:586–8.
- Paller AS, Gonzalez-Crussi F, Sherman JO. Fibrous hamartoma of infancy: eight additional cases and a review of the literature. Arch Dermatol 1989;125:88–91.
- Sotelo-Avila C, Bale PM. Subdermal fibrous hamartoma of infancy: pathology of 40 cases and differential diagnosis. Pediatr Pathol 1994;14:39–52.
- Carretto E, Dall'Igna P, Alaggio R, et al. Fibrous hamartoma of infancy: an Italian multi-institutional experience. J Am Acad Dermatol 2006;54:800–3.
- Saab ST, McClain CM, Coffin CM. Fibrous hamartoma of infancy: a clinicopathologic analysis of 60 cases. Am J Surg Pathol 2014;38:394–401.
- Groisman G, Lichtig C. Fibrous hamartoma of infancy: an immunohistochemical and ultrastructural study. Hum Pathol 1991;22:914–18.
- Grynspan D, Meir K, Senger C, et al. Cutaneous changes in fibrous hamartoma of infancy. J Cutan Pathol 2007;34:39–43.
- Tassano E, Nozza P, Tavella E, et al. Cytogenetic characterization of a fibrous hamartoma of infancy with complex translocations. Cancer Genet Cytogenet 2010;201:66–9.
- McGowan J 4th, Smith CD, Maize J Jr, et al. Giant fibrous hamartoma of infancy: a report of two cases and review of the literature. J Am Acad Dermatol 2011;64:579-86.
- Yon TY, Kim JW. Fibrous hamartoma of infancy manifesting as multiple nodules with hypertrichosis. J Dermatol 2006;33:427–9.
Calcifying fibrous tumour/pseudotumour
- Rosenthal NS, Abdul-Karim FW. Childhood fibrous tumor with psammoma bodies. Arch Pathol Lab Med 1988;112:565–8.
- Fetsch JF, Montgomery EA, Meis JM. Calcifying fibrous pseudotumour. Am J Surg Pathol 1993;17:502–8.
- Nascimento AF, Ruiz R, Hornick JL, et al. Calcifying fibrous ‘pseudotumor’: clinicopathologic study of 15 cases and analysis of its relationship to inflammatory myofibroblastic tumor. Int J Surg Pathol 2002;10:189–96.
Calcifying aponeurotic fibroma
- Allen PW, Enzinger FM. Juvenile aponeurotic fibroma. Cancer 1970;26:857–67.
- Fetsch JF, Miettinen M. Calcifying aponeurotic fibroma: a clinicopathologic study of 22 cases arising in uncommon sites. Hum Pathol 1998;29:1504–10.
- Hassel B. Calcifying aponeurotic fibroma. A case of multiple primary tumours. Case report. Scand J Plast Reconstr Surg Hand Surg 1992;26:115–16.
- Lafferty KA, Nelson EL, Demuth RJ, et al. Juvenile aponeurotic fibroma with disseminated fibrosarcoma. J Hand Surg (Am) 1986;11:737–40.
Dermatomyofibroma
- Hugol H. Die plaqueformige dermale Fibromatose. Hautarzt 1991;42:223–6.
- Kamino H, Reddy VB, Gero M, et al. Dermatomyofibroma: a benign cutaneous plaque-like proliferation of fibroblasts and myofibroblasts in young adults. J Cutan Pathol 1992;19:85–91.
- Mentzel T, Calonje E, Fletcher CDM. Dermatomyofibroma – additional observations of a distinctive cutaneous myofibroblastic tumour with emphasis on differential diagnosis. Br J Dermatol 1993;129:69–73.
- Colome MI, Sanchez RL. Dermatomyofibroma: report of two cases. J Cutan Pathol 1994;21:371–6.
- Mortimore RJ, Whitehead KJ. Dermatomyofibroma: a report of two cases, one occurring in a child. Australas J Dermatol 2001;42:22–5.
- Tardio JC, Azorin D, Hernandez-Nunez A, et al. Dermatomyofibromas presenting in pediatric patients: clinicopathologic characteristics and differential diagnosis. J Cutan Pathol 2011;38:967–72.
- Mentzel T, Kutzner H. Dermatomyofibroma: clinicopathologic and immunohistochemical analysis of 56 cases and reappraisal of a rare and distinct cutaneous entity. Am J Dermatopathol 2009;31:44–9.
- Mentzel T, Kutzner H. Haemorrhagic dermatomyofibroma (plaque-like dermal fibromatosis): clinicopathological and immunohistochemical analysis of three cases resembling plaque-stage Kaposi's sarcoma. Histopathology 2003;29:426–9.
- Trotter MJ, McGregor GI, O'Connell JX. Linear dermatomyofibroma. Clin Exp Dermatol 1996;21:307–9.
Plaque-like CD34-positive dermal fibroma
- Rodríguez-Jurado R, Palacio C, Durán-McKinster C., et al. Medallion-like dermal dendrocyte hamartoma: a new clinically and histopathologically distinct lesion. J Am Acad Dermatol 2004;51:359–63.
- Shah KN, Anderson E, Junkins-Hopkins J, et al. Medallion-like dermal dendrocyte hamartoma. Pediatr Dermatol 2007;24:632–6.
- Kutzner H, Mentzel T, Palmedo G, et al. Plaque-like CD34-positive dermal fibroma (‘Medallion-like dermal dendrocyte hamartoma’). Clinicopathologic, immunohistochemical, and molecular analysis of 5 cases emphasizing its distinction from superficial, plaque-like dermatofibrosarcoma protuberans. Am J Surg Pathol 2010;34:190–201.
Angiomyofibroblastoma
- Fletcher CD, Tsang WY, Fisher C, et al. Angiomyofibroblastoma of the vulva. A benign neoplasm distinct from aggressive angiomyxoma. Am J Surg Pathol 1992;16:373–82.
- Hisaoka M, Kouho H, Aoki T, et al. Angiomyofibroblastoma of the vulva: a clinicopathologic analysis of 7 cases. Pathol Int 1995;45:487–92.
- Nielsen GP, Rosenberg AE, Young RH, et al. Angiomyofibroblastoma of the vulva and vagina. Mod Pathol 1996;9:284–91.
- Laskin WB, Fetsch JF, Tavasoli FA. Angiomyofibroblastoma of the female genital tract analysis of 17 cases including a lipomatous variant. Hum Pathol 1997;28:1046–55.
- Granter SR, Nucci MR, Fletcher CD. Aggressive angiomyxoma: reappraisal of its relationship to angiomyofibroblastoma in a series of 16 cases. Histopathology 1997;30:3–10.
- Nagai K, Asdachi K, Saito H. Huge pedunculated angiomyofibroblastoma of the vulva. Int J Clin Oncol 2010;15:201–5.
- Nielsen GP, Young RH, Dickersin GR, et al. Angiomyofibroblastoma of the vulva with sarcomatous transformation (‘angiomyofibrosarcoma’). Am J Surg Pathol 1997;21:1104–8.
Cellular angiofibroma
- Nucci MR, Granter SR, Fletcher CD. Cellular angiofibroma: a benign neoplasm distinct from angiomyofibroblastoma and spindle cell lipoma. Am J Surg Pathol 1997;21:636–44
- Laskin WB, Fetsch JF, Mostofi FK. Angiomyofibroblastoma-like tumor of the male genital tract: analysis of 11 cases with comparison to female angiomyofibroblastoma and spindle cell lipoma. Am J Surg Pathol 1998;22:6–16.
- Lane JE, Walker AN, Mullis EN Jr, et al. Cellular angiofibroma of the vulva. Gynecol Oncol 2001;81:326–9.
- Iwasa Y, Fletcher CDM. Cellular angiofibroma: clinicopathologic and immunohistochemical analysis of 51 cases. Am J Surg Pathol 2004;28:1426–35.
- Flucke U, van Krieken JHJM, Mentzel T. Cellular angiofibroma: analysis of 25 cases emphasizing its relationship to spindle cell lipoma and mammary-type myofibroblastoma. Mod Pathol 2011;24:82–9.
- Chen E, Fletcher CD. Cellular angiofibroma with atypia or sarcomatous transformation: clinicopathologic analysis of 13 cases. Am J Surg Pathol 2010;34:707–14.
Elastofibroma
- Jarvi OH, Saxen AE, Hopsu HV, et al. Elastofibroma: a degenerative pseudotumor. Cancer 1969;23:42–63.
- Nagamine N, Nohara Y, Ito E. Elastofibroma in Okinawa: a clinicopathologic study of 170 cases. Cancer 1982;50:1794–805.
- Fukuda Y, Miyake H, Masuda Y, et al. Histogenesis of unique elastinophilic fibers of elastofibroma: ultrastructural and immunohistochemical studies. Hum Pathol 1987;18:424–9.
- Hisaoka M, Hashimoto H. Elastofibroma: clonal fibrous proliferation with predominant CD34-positive cells. Virchows Arch 2006;448:195–9.
- Brandser EA, Goree JC, El-Khoury GY. Elastofibroma dorsi: prevalence in an elderly patient population as revealed by CT. Am J Roentgenol 1998;171:977–80.
- Jarvi OH, Lansimies PH. Subscapular elastofibromas in the scapular region in an autopsy series. Acta Pathol Microbiol Scand A 1975;83:87–108.
- Nishio JN, Iwasaki H, Ohjimi Y, et al. Gain of Xq detected by comparative genomic hybridization in elastofibroma. Int J Mol Med 2002;10:277–80.
- Shimizu S, Yasui C, Tateno M, et al. Multiple elastofibromas. J Am Acad Dermatol 2004;50:126–9.
Inclusion body (digital) fibromatosis
- Reye R. Recurring digital fibrous tumors of childhood. Arch Pathol 1965;80:228–36.
- Beckett JH, Jacobs AH. Recurring digital fibrous tumors of childhood: a review. Pediatrics 1977;59:401–6.
- Choi KC, Hashimoto K, Setoyama M, et al. Infantile digital fibromatosis: immunohistochemical and immunoelectron microscopic studies. J Cutan Pathol 1990;17:225–32.
- Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibromatosis: a clinicopathological and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol 2009;33:1–13.
- Coffin CM, Dehner LP. Fibroblastic-myofibroblastic tumors in children and adolescents: a clinicopathologic study of 108 examples in 103 patients. Pediatr Pathol 1991;11:569–88.
- Viale G, Doglioni C, Iuzzolino P, et al. Infantile digital fibromatosis-like tumor (inclusion body fibromatosis) of adulthood: report of two cases with ultrastructural and immunocytochemical findings. Histopathology 1988;12:415–24.
- Purdy LJ, Colby TV. Infantile digital fibromatosis occurring outside the digit. Am J Surg Pathol 1984;8:787–90.
- Kawaguchi M, Mitsuhashi Y, Hozumi Y, et al. A case of infantile digital fibromatosis with spontaneous regression. J Dermatol 1998;25:523–6.
Fibroma of tendon sheath
- Chung EB, Enzinger FM. Fibroma of tendon sheath. Cancer 1979;19:45–54.
- Pulitzer DR, Martin PC, Reed RJ. Fibroma of tendon sheath: a clinicopathologic study of 32 cases. Am J Surg Pathol 1989;13:472–9.
- Dal Cin P, Sciot R, De Smet L, et al. Translocation 2;11 in a fibroma of tendon sheath. Histopathology 1998;32:433–5.
- Tiong WH, Ismael TS, Regan PJ. Fibroma of tendon sheath: a rare cause of carpal tunnel syndrome. J Hand Surg (Br) 2006;31:579–80.
Desmoplastic fibroblastoma
- Evans HL. Desmoplastic fibroblastoma: a report of seven cases. Am J Surg Pathol 1995;19:1077–81.
- Mietinnen M, Fetsch JF. Collagenous fibroma (desmoplastic fibroblastoma): a clinicopathological analysis of 63 cases of a distinctive soft tissue lesion with stellate-shaped fibroblasts. Hum Pathol 1998;29:676–82.
- Macchia G, Trombetta D, Moller E, et al. FOSL1 as a candidate target gene for 11q12 rearrangements in desmoplastic fibroblastoma. Lab Invest 2012;92:735-42.
- Bernal K, Nelson M, Neff JR, et al. Translocation (2;11)(q31;q12) is recurrent in collagenous fibroma (desmoplastic fibroblastoma). Cancer Genet Cytogenet 2004;149:161–3.
Nuchal-type fibroma
- Balachandran K, Allen RW, McCormac LB. Nuchal fibroma: a clinicopathological analysis of nine cases. Am J Surg Pathol 1995;19:313–17.
- Michal M, Fetsch JF, Hes O, et al. Nuchal-type fibroma: a clinicopathologic study of 52 cases. Cancer 1999;85:156–63.
- Wehrli BM, Weiss SW, Yandow S, et al. Gardner-associated fibromas (GAF) in young patients: a distinct fibrous lesion that identifies unsuspected Gardner syndrome and risk for fibromatosis. Am J Surg Pathol 2001;25:645–61.
- Michal M. Non-nuchal-type fibroma associated with Gardner's syndrome. A hitherto-unreported mesenchymal tumor different from fibromatosis and nuchal-type fibroma. Pathol Res Pract 2000;196:857–60.
Palmar and plantar fibromatosis (superficial fibromatoses)
- Allen PW. The fibromatoses: a clinicopathologic classification based on 140 cases. Am J Surg Pathol 1977;1:255–70.
- Mikkelsen OA. Dupuytren's disease:initial symptoms, age of onset and spontaneous course. Hand 1977;9:11–15.
- Fetsch JF, Laskin WB, Miettinen M. Palmar-plantar fibromatosis in children and pre-adolescents: a clinicopathologic study of 56 cases with newly recognized demographics and extended follow-up information. Am J Surg Pathol 2005;29:1095–105.
- Montgomery E, Lee JH, Abraham SC, et al. Superficial fibromatoses are genetically distinct from deep fibromatosis. Mod Pathol 2001;14:695–701.
Penile fibromatosis
- Smith BH. Peyronie's disease. Am J Clin Pathol 1966;85:670–8.
- McRoberts JW. Peyronie's disease. Surg Gynecol Obstet 1969;129:1291–4.
- Billig R, Baker R, Immergut Maxted W. Peyronie's disease. Urology 1975;6:409–18.
- Kendirci M, Trost L, Sikka SC, et al. Diabetes mellitus is associated with severe Peyronie's disease. BJU Int 2007;99:383–6.
- Jordan GH, Carson CC, Lipshultz LJ. Minimally invasive treatment of Peyronie's disease. BJU Int 2014;114:16–24.
Lipofibromatosis
- Fetsch JF, Miettinen M, Laskin WB, et al. A clinicopathologic study of 45 soft tissue tumors with an admixture of adipose tissue and fibroblastic elements, and a proposal for classification as lipofibromatosis. Am J Surg Pathol 2000;24:1491–500.
- Kenney B, Richkind KE, Friedlaender G, et al. Chromosomal rearrangements in lipofibromatosis. Cancer Genet Cytogenet 2007;179:136–9.
Dermatofibrosarcoma protuberans
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer 1962;15:717–25.
- Burkhardt BR, Soule EH, Winkelmann RK, et al. Dermatofibrosarcoma protuberans: study of 56 cases. Am J Surg 1966;111:638–44.
- Gloster HM. Dermatofibrosarcoma protuberans. J Am Acad Dermatol 1996;35:355–74.
- Criscione VD, Weinstock MA. Descriptive epidemiology of dermatofibrosarcoma protuberans in the United States, 1973 to 2002. J Am Acad Dermatol 2007;56:968–73.
- McKee PH, Fletcher CDM. Dermatofibrosarcoma presenting in infancy and childhood. J Cutan Pathol 1991;18:241–6.
- Martin L, Combemale P, Dupin MJ, et al. The atrophic variant of dermatofibrosarcoma protuberans in childhood: a report of six cases. Br J Dermatol 1998;139:719–25.
- Checketts SR, Hamilton TK, Baughman RD. Congenital and childhood dermatofibrosarcoma protuberans: a case report and review of the literature. J Am Acad Dermatol 2000;42:907–13.
- Maire G, Fraitag S, Galmiche L, et al. A clinical, histologic, and molecular analysis of 9 cases of congenital dermatofibrosarcoma protuberans. Arch Dermatol 2007;143:203–10.
- Tanaka A, Hatoko M, Tada H, et al. Dermatofibrosarcoma protuberans arising from a burn scar of the axilla. Ann Plast Surg 2004;52:423–5.
- Huber GF, Matthews TW, Dort JC. Radiation-induced soft tissue sarcomas of the head and neck. J Otolaryngol 2007;36:93–7.
- Kesserwan C, Sokolic R, Cowen EW, et al. Multicentric dermatofibrosarcoma protuberans in patients with adenosine deaminase-deficient severe combined immune deficiency. J Allergy Clin Immunol 2012;129:762–9.
- Reimann JD, Fletcher CD. Myxoid dermatofibrosarcomas protuberans: a rare variant analyzed in a series of 23 cases. Am J Surg Pathol 2007;31:1371–7.
- Mentzel T, Sharer L, Kazakov DV, et al. Myxoid dermatofibrosarcoma protuberans: clinicopathologic, immunohistochemical, and molecular analysis of eight cases. Am J Dermatopathol 2007;29:443–8.
- Bednar B. Storiform neurofibromas of the skin, pigmented and nonpigmented. Cancer 1957;10:368–75.
- Fletcher CDM, Theaker JM, Flanagan A, et al. Pigmented dermatofibrosarcoma protuberans (Bednar tumour): melanocytic colonization or neuroectodermal differentiation? A clinicopathological and immunohistochemical study. Histopathology1988;13:631–43.
- Calonje E, Fletcher CDM. Myoid differentiation in dermatofibrosarcoma protuberans and its fibrosarcomatous variant: clinicopathologic analysis of 5 cases. J Cutan Pathol 1996;23:30–6.
- Ding J, Hashimoto H, Enjoji M. Dermatofibrosarcoma protuberans with fibrosarcomatous areas: a clinicopathologic study of nine cases and a comparison with allied tumors. Cancer 1989;64:721–9.
- Conelly JH, Evans HL. Dermatofibrosarcoma protuberans: a clinicopathologic review with emphasis on fibrosarcomatous areas. Am J Surg Pathol 1992;16:921–5.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous (‘high grade’) dermatofibrosarcoma protuberans. Clinicopathologic study and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol 1998;22:576–87.
- Abbott JJ, Oliveira AM, Nascimento AG. The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans. Am J Surg Pathol 2006;30:436–43.
- Szollosi Z, Nemes Z. Transformed dermatofibrosarcoma protuberans: a clinicopathological study of eight cases. J Clin Pathol 2005;58:751–6.
- Rubin B, Fletcher J, Fletcher CD. The histologic, genetic and histological relationship between dermatofibrosarcoma protuberans and giant cell fibroblastoma: an unexpected story. Adv Anat Pathol 1997;4:336–41.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol 2008;39:184–93.
- Bigby SM, Oei P, Lambie NK, et al. Dermatofibrosarcoma protuberans: report of a case with a variant ring chromosome and metastases following pregnancy. J Cutan Pathol 2006;33:383–8.
- Zelger BW, Ofner D, Zelger BG. Atrophic variants of dermatofibroma and dermatofibrosarcoma protuberans. Histopathology 1995;26:519–27.
- Martin L, Piette F, Blanc P, et al. Clinical variants of the preprotuberant stage of dermatofibrosarcoma protuberans. Br J Dermatol 2005;153:932–6.
- Fletcher CDM, Evans BJ, Macartney JC, et al. Dermatofibrosarcoma protuberans: a clinicopathological and immunohistochemical study with a review of the literature. Histopathology 1985;9:921–38.
- Rutgers EJ, Kroon BB, Albus-Lutter CE, et al. Dermatofibrosarcoma protuberans: treatment and prognosis. Eur J Surg Oncol 1992;18:241–8.
- McPeak CJ, Cruz T, Nicastri AD. Dermatofibrosarcoma protuberans: an analysis of 86 cases—five with metastasis. Ann Surg 1967;166:803–16.
- Llombart B, Monteagudo C, Sanmartin O, et al. Dermatofibrosarcoma protuberans: a clinicopathological, immunohistochemical, genetic (COL1A1-PDGFB), and therapeutic study of low-grade versus high-grade (fibrosarcomatous) tumors. J Am Acad Dermatol 2011;65:564–75.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation; an evidence-based review of the literature. J Eur Acad Dermatol Venereol 2011;25:1385–91.
- Fisher ER, Helstrom HR. Dermatofibrosarcoma with metastases simulating Hodgkin's disease and reticulum cell sarcoma. Cancer 1966;19:1165–71.
- Brenner W, Schaefler K, Chhabra H, et al. Dermatofibrosarcoma protuberans metastatic to a regional lymph node: report of a case and review. Cancer 1975;36:1897–902.
- Fiore M, Miceli R, Mussi C, et al. Dermatofibrosarcoma protuberans treated at a single institution: a surgical disease with a high cure rate. J Clin Oncol 2005;23:7669–75.
- Smola MG, Soyer HP, Scharnagl E. Surgical treatment of dermatofibrosarcoma protuberans: a retrospective study of 20 cases with review of literature. Eur J Surg Oncol 1991;17:447–53.
- Roses DF, Valensi Q, LaTrenta G, et al. Surgical treatment of dermatofibrosarcoma protuberans. Surg Gynecol Obstet 1986;162:449–52.
- Robinson JK. Dermatofibrosarcoma protuberans resected by Mohs' surgery (chemosurgery): a 5-year prospective study. J Am Acad Dermatol 1985;12:1093–8.
- Thomas CJ, Wood GC, Marks VJ. Mohs micrographic surgery in the treatment of rare aggressive cutaneous tumors: the Geisinger experience. Dermatol Surg 2007;33:333–9.
- Snow SN, Gordon EM, Larson PO, et al. Dermatofibrosarcoma protuberans: a report on 29 cases treated by Mohs micrographic surgery with long-term follow-up and review of the literature. Cancer 2004;101:28–38.
- Dagan R, Morris CG, Zlotecki RA, et al. Radiotherapy in the treatment of dermatofibrosarcoma protuberans. Am J Clin Oncol 2005;28:537–9.
- McArthur GA. Molecular targeting of dermatofibrosarcoma protuberans: a new approach to a surgical disease. J Natl Compr Canc Netw 2007;5:557–62.
- Sjöblom T, Shimizu A, O'Brien KP, et al. Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist ST1571 through induction of apoptosis. Cancer Res 2001;61:5778–83.
- Kondapalli L, Soltani K, Lacouture ME. The promise of molecular targeted therapies: protein kinases inhibitors in the treatment of cutaneous malignancies. J Am Acad Dermatol 2005;53:3291–302.
- Price VE, Fletcher JA, Zielenska M, et al. Imatinib mesylate: an attractive alternative in young children with large, surgically challenging dermatofibrosarcoma protuberans. Pediatr Blood Cancer 2005;44:511–15.
- Sirvent N, Maire G, Pedeutour F. Genetics of dermatofibrosarcoma protuberans family of tumors: from ring chromosomes to tyrosinase inhibitor treatment. Genes Chromosomes Cancer 2003;37:1–19.
Giant cell fibroblastoma
- Dymock RB, Allen PW, Stirling JW, Gilbert EF, Thornbery JM. Giant cell fibroblastoma: a distinctive, recurrent tumour of childhood. Am J Surg Pathol 1987;11:263–71.
- Shmookler BM, Enzinger FM. Giant cell fibroblastoma: a juvenile form of dermatofibrosarcoma protuberans. Cancer 1989;64:2154–61.
- Chou P, Gonzalez-Crussi G, Mangkornikanok M. Giant cell fibroblastoma. Cancer 1989;63:756–62.
- Jha P, Moosavi C, Fanburg-Smith JC. Giant cell fibroblastoma: an update and addition of 86 cases from the Armed Forces Institute of Pathology, in honor of Dr. Franz M. Enzinger. Ann Diagn Pathol 2007;11:81–8.
- Alguacil-García A. Giant cell fibroblastoma recurring as dermatofibrosarcoma protuberans. Am J Surg Pathol 1991;21:184–7.
- Harvell JD, Kilpatrick SE, White WL. Histogenetic relations between giant cell fibroblastoma and dermatofibrosarcoma protuberans. Am J Dermatopathol 1998;20:339–45.
- Beham A, Fletcher CD. Dermatofibrosarcoma protuberans with areas resembling giant cell fibroblastoma: report of two cases. Histopathology 1990;17:165–7.
- Dal Cin P, Sciot R, de Wever I, et al. Cytogenetic and immunohistochemical evidence that giant cell fibroblastoma is related to dermatofibrosarcoma protuberans. Genes Chrom Cancer 1996;15:73–5.
- Terrier-Lacombe MJ, Guillou L, Maire G, et al. Dermatofibrosarcoma protuberans, giant cell fibroblastoma and hybrid lesions in children: clinicopathologic comparative analysis of 28 cases with molecular data—a study of the French Federation of Cancer Centers Sarcoma Group. Am J Surg Pathol 2003;27:27–9.
Myxoinflammatory fibroblastic sarcoma
- Montgomery EA, Devaney KO, Giordano TJ, et al. Inflammatory myxohyaline tumor of distal extremities with virocyte or Reed-Sternberg-like cells: a distinctive lesion with features simulating inflammatory conditions, Hodgkin's disease, and various sarcomas. Mod Pathol 1998;11:384–91.
- Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol 1998;22:911–24.
- Sakaki M, Hirokawa M, Wakatsuki S, et al. Acral myxoinflammatory fibroblastic sarcoma: a report of five cases and review of the literature. Virchows Arch 2003;442:25–30.
- Laskin WB, Fetsch JF, Miettinen M. Myxoinflammatory fibroblastic sarcoma: a clinicopathologic analysis of 104 cases with emphasis on predictors of outcome. Am J Surg Pathol 2014;38:1–12.
- Solomon DA, Antonescu CR, Link TM, et al. Hemosiderotic fibrolipomatous tumor, not an entirely benign entity. Am J Surg Pathol 2013;37:1627–30.
- Weiss VL, Antonescu CR, Alaqqio R, et al. Myxoinflammatory fibroblastic sarcoma in children and adolescents: clinicopathologic aspects of a rare neoplasm. Pediatr Dev Pathol 2013;16:225–31.
- Lambert I, Debiec-Rychter M, Guelinckx P, et al. Acral myxoinflammatory fibroblastic sarcoma with unique clonal chromosomal changes. Virchows Arch 2001;438:509–12.
- Ida CM, Rolig KA, Hulshizer RL, et al. Myxoinflammatory fibroblastic sarcoma showing t(2;6)(q31;p21.3) as a sole cytogenetic abnormality. Cancer Genet Cytogenet 2007;177:139–42.
- Jurcic' V, Zidar A, Montiel MD, et al. Myxoinflammatory fibroblastic sarcoma: a tumor not restricted to acral sites. Ann Diagn Pathol 2002;6:272–80.
- McFarlane R, Meyers AD, Golitz L. Myxoinflammatory fibroblastic sarcoma of the neck. J Cutan Pathol 2005;32:375–8.
Malignant fibrous histiocytoma
- Fletcher CDM, McKee PH. Sarcomas: a clinicopathological guide with particular reference to cutaneous manifestation, 1. Clin Exp Dermatol 1984;9:451–65.
- Lawson CW, Fisher C, Gatter KC. An immunohistochemical study of differentiation in malignant fibrous histiocytoma. Histopathology 1987;11:375–83.
- Weiss SW, Enzinger FM. Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer 1978;41:2250–66.
- Enzinger FM. Malignant fibrous histiocytoma 20 years after Stout. Am J Surg Pathol 1986;10:43–53.
- Fletcher CD. Pleomorphic malignant fibrous histiocytoma: fact or fiction? A critical reappraisal based on 159 tumors diagnosed as pleomorphic sarcoma. Am J Surg Pathol 1992;16:213–28.
- Nakayama R, Nemoto T, Takahashi H, et al. Gene expression analysis of soft tissue sarcomas: characterization and reclassification of malignant fibrous histiocytoma. Mod Pathol 2007;20:749–59.
Myxofibrosarcoma
- Angervall L, Kindblom LG, Merck C. Myxofibrosarcoma: a study of 30 cases. Acta Pathol Microbiol Scand 1977;85:127–40.
- Weiss SW, Enzinger FM. Myxoid variant of malignant fibrous histiocytoma. Cancer 1977;39:1672–85.
- Merck C, Angervall L, Kindblom LG, et al. Myxofibrosarcoma, a malignant soft tissue tumor of fibroblastic-histiocytic origin: a clinicopathologic and prognostic study of 110 cases using multivariate analysis. Acta Pathol Microbiol Immunol Scand 1983;91:3–40.
- Mentzel T, Calonje E, Wadden C, et al. Myxofibrosarcoma: clinicopathologic analysis of 75 cases with emphasis on the low-grade variant. Am J Surg Pathol 1996;20:391–405.
- Sanfilippo R, Miceli R, Grosso F, et al. Myxofibrosarcoma: prognostic factors and survival in a series of patients treated at a single institution. Ann Surg Oncol 2011;18:720–5.
- Kanno S, Asai J, Nakamura N, et al. Myxofibrosarcoma arising from a chronic burn scar. J Dermatol 2014;41:279–80.
- Nascimento AF, Bertoni F, Fletcher CD. Epithelioid variant of myxofibrosarcoma: expanding the clinicomorphologic spectrum of myxofibrosarcoma in a series of 17 cases. Am J Surg Pathol 2007;31:99–105.
Low-grade fibromyxoid sarcoma
- Evans HL. Low-grade fibromyxoid sarcoma: a report of 12 cases. Am J Surg Pathol 1993;17:595–600.
- Goodlad JR, Mentzel T, Fletcher CD. Low-grade fibromyxoid sarcoma: clinicopathological analysis of eleven new cases in support of a distinct entity. Histopathology 1995;26:229–37.
- Zamecnik M, Michal M. Low-grade fibromyxoid sarcoma: a report of 8 cases with histologic, immunohistochemical, and ultrastructural study. Ann Diagn Pathol 2000;4:207–17.
- Folpe AL, Lane KL, Paull G, et al. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol 2000;24:1353–60.
- Lane KL, Shannon RJ, Weiss SW. Hyalinizing spindle cell tumor with giant rosettes: a distinctive tumor closely resembling low-grade fibromyxoid sarcoma. Am J Surg Pathol 1997;21:1481–8.
- Panagopoulos I, Storlazzi CT, Fletcher CD, et al. The chimeric FUS/CREB3l2 gene is specific for low-grade fibromyxoid sarcoma. Genes Chromosomes Cancer 2004;40:218–28.
Fibrohistiocytic tumours
Giant cell tumour of tendon sheath
- Myers BW, Masi AT, Feigenbaum SL. Pigmented villonodular synovitis and tenosynovitis: a clinical epidemiologic study of 166 cases and literature review. Medicine 1980;59:223–38.
- Ushjima M, Hashimoto H, Tsuneyoshi M, et al. Giant cell tumor of the tendon sheath (nodular tenosynovitis): a study of 207 cases to compare the large joint group with the common digit group. Cancer 1986;57:875–84.
- Mohaghan H, Salter DM, Al-Nafussi A. Giant cell tumour of tendon sheath (localised nodular tenosynovitis): clinicopathological features of 71 cases. J Clin Pathol 2001;54:404–7.
- Park JW. Multiple separated giant cell tumors of the tendon sheath in a thumb. J Am Acad Dermatol 2006;54:540–2.
- Di Grazia S, Succi G, Fragatta F, et al. Giant cell tumor of tendon sheath: study of 54 cases and review of the literature. G Chir 2013;34:149–52.
Fibrous histiocytoma (dermatofibroma)
- Niemi KM. The benign fibrohistiocytic tumours of the skin. Acta Dermatol Venereol (Stockh) 1970;50(Suppl. 63):7–42.
- Vilanova JR, Flint A. The morphologic variants of histiocytomas. J Cutan Pathol 1974;1:155–64.
- Gonzáles S, Duarte I. Benign fibrous histiocytoma of the skin: a morphologic study of 290 cases. Pathol Res Pract 1982;174:379–91.
- Calonje E, Fletcher CDM. Cutaneous fibrohistiocytic tumors: an update. Adv Anat Pathol 1994;1:2–15.
- Vanni R, Marras S, Faa G, et al. Cellular fibrous histiocytoma of the skin: evidence of a clonal process with different karyotype from dermatofibrosarcoma. Genes Chromosomes Cancer 1997;18:314–17.
- Calonje E. Dermatofibroma (fibrous histiocytoma): an inflammatory or neoplastic disorder? Histopathology 2001;39:213.
- Colome-Grimmer MI, Evans HL. Metastasizing cellular dermatofibroma: a report of two cases. Am J Surg Pathol 1996;20:1361–7.
- Gu M, Sohn K, Kim D, et al. Metastasizing dermatofibroma in lung. Ann Diagn Pathol 2007;11:64–7.
- Doyle LA, Fletcher CD. Metastasizing “benign” cutaneous fibrous histiocytoma: a clinicopathologic analysis of 16 cases. Am J Surg Pathol 2013;37:484–95.
- Mentzel T, Wiesner T, Cerroni L, et al. Malignant dermatofibroma: clinicopathological, immunohistochmical, and molecular analysis of seven cases. Mod Pathol 2013;26:256–67.
- Calonje E, Mentzel T, Fletcher CDM. Cellular benign fibrous histiocytoma: clinicopathologic analysis of 74 cases of a distinctive variant of cutaneous fibrous histiocytoma with frequent recurrence. Am J Surg Pathol 1994;18:668–76.
- Schoenfeld RJ. Epidermal proliferations overlying histiocytomas. Arch Dermatol 1964;90:266–70.
- Santa Cruz DJ, Kyriakos M. Aneurysmal (‘angiomatoid’) fibrous histiocytoma of the skin. Cancer 1981;47:2053–61.
- Calonje E, Fletcher CDM. Aneurysmal benign cutaneous fibrous histiocytoma: clinicopathologic analysis of a tumor frequently misdiagnosed as a vascular lesion. Histopathology 1995;26:323–31.
- Leyva WH, Santa Cruz DJ. Atypical cutaneous fibrous histiocytoma. Am J Dermatopathol 1986;8:467–71.
- Kaddu S, McMenamin M, Fletcher CD. Atypical fibrous histiocytoma of the skin: clinicopathologic analysis of 59 cases with evidence of infrequent metastasis. Am J Surg Pathol 2002;26:35–46.
- Wilson Jones E, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol 1989;120:185–95.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma: a report of 10 cases including a new cellular variant. Am J Surg Pathol 1994;18:583–90.
- Soyer HP, Metze D, Kerl H. Granular cell dermatofibroma. Am J Dermatopathol 1997;19:168–73.
- Iwata J, Fletcher CDM. Lipidized fibrous histiocytoma: clinicopathologic analysis of 22 cases. Am J Dermatopathol 2000;22:126–34.
- Zelger BW, Steiner H, Kutzner H. Clear cell dermatofibroma: case report of an unusual fibrohistiocytic lesion. Am J Surg Pathol 1996;20:483–91.
- Tran TA, Hayner-Buchan A, Jones DM, et al. Cutaneous balloon cell dermatofibroma (fibrous histiocytoma). Am J Dermatopathol 2007;29:197–200.
- Kuo TT, Hu S, Chan HL. Keloidal dermatofibroma: report of 10 cases of a new variant. Am J Surg Pathol 1998;22:564–8.
- Wagamon K, Somach SC, Bass J, et al. Lipidized dermatofibromas and their relationship to serum lipids. J Am Acad Dermatol 2006;54:494–8.
- Requena L, Farina MC, Fuente C, et al. Giant dermatofibroma. J Am Acad Dermatol 1994;30:714–18.
- Yazici AC, Baz K, Ikizoglu G, et al. Familial eruptive dermatofibromas in atopic dermatitis. J Eur Acad Dermatol Venereol 2006;20:90–2.
- Kanitakis J, Carbonnel E, Delmonte S, et al. Multiple eruptive dermatofibromas in a patient with HIV infection: case report and literature review. J Cutan Pathol 2000;27:54–6.
- Niiyama S, Katsuoka K, Happle R, et al. Multiple eruptive dermatofibromas: a review of the literature. Acta Derm Venereol 2002;82:241–4.
- Yamamoto T, Sumi K, Yokozeki H, et al. Multiple cutaneous fibrous histiocytomas in association with systemic lupus erythematosus. J Dermatol 2005;32:645–9.
- Lee HW, Lee DK, Oh SH, et al. Multiple eruptive dermatofibromas in a patient with primary pulmonary hypertension. Br J Dermatol 2005;153:845–7.
- Alexandrescu DT, Wiernik PH. Multiple eruptive dermatofibromas occurring in a patient with chronic myelogenous leukaemia. Arch Dermatol 2005;141:397–8.
- Chang SE, Choi JH, Sung KJ, et al. Multiple eruptive dermatofibromas occurring in a patient with acute myeloid leukaemia. Br J Dermatol 2000;142:1062–3.
- Bachmeyer C, Cordier F, Blum L, et al. Multiple eruptive dermatofibromas after highly active antiretroviral therapy. Br J Dermatol 2000;143:1336–7.
- Hendi A, Jukic DM, Kress DW, et al. Atrophic dermatofibroma: a case report and review of the literature. Dermatol Surg 2002;28:1085–7.
- Sánchez Yus E, Soria L, de Eusebio E, et al. Lichenoid, erosive and ulcerated dermatofibromas. Three additional clinico-pathologic variants. J Cutan Pathol 2000;27:112–17.
- Ichikawa N, Kobayashi M, Kimoto M, et al. A case of multiple aneurysmal fibrous histiocytomas. Br J Dermatol 2005;153:664–5.
- Charli-Joseph Y, Saggini A, Doyle LA, et al. DNA copy number changes in tumors within the spectrum of cellular, atypical, and metastasizing fibrous histiocytoma. J Am Acad Dermatol 2014;71:253–63.epub ahead of print.
Plexiform fibrohistiocytic tumour
- Enzinger FM, Zhang R. Plexiform fibrohistiocytic tumor presenting in children and young adults: an analysis of 65 cases. Am J Surg Pathol 1988;12:816–26.
- Hollowood K, Holley MP, Fletcher CD. Plexiform fibrohistiocytic tumour: clinicopathological, immunohistochemical and ultrastructural analysis in favour of a myofibroblastic lesion. Histopathology 1991;19:503–13.
- Remstein ED, Arndt CA, Nascimento AG. Plexiform fibrohistiocytic tumor. Clinicopathologic analysis of 22 cases. Am J Surg Pathol 1999;23:662–70.
- Taher A, Pushpanathan C. Plexiform fibrohistiocytic tumor: a brief review. Arch Pathol Lab Med 2007;131:1135–8.
- Moosavi C, Jha P, Fanburg-Smith JC. An update on plexiform fibrohistiocytic tumor and addition of 66 new cases from the Armed Forces Institute of Pathology, in honor of Franz M. Enzinger, MD. Ann Diagn Pathol 2007;11:313–19.
- Jaffer S, Ambrosini-Spaltro A, Mancini AM, et al. Neurothekeoma ands plexiform fibrohistiocytic tumor: mere histologic resemblance or histogenetic relationship? Am J Surg Pathol 2009;33:905–13.
- Leclerc S, Hamel-Teillac D, Oner P, et al. Plexiform fibrohistiocytic tumor: three unusual cases occurring in infancy. J Cutan Pathol 2005;32:572–6.
- Zelger B, Weinlich G, Steiner H, et al. Dermal and subcutaneous variants of plexiform fibrohistiocytic tumor. Am J Surg Pathol 1997;21:235–41.
- Salomao D, Nascimento A. Plexiform fibrohistiocytic tumor with systemic metastases: a case report. Am J Surg Pathol 1997;21:469–76.
Atypical fibroxanthoma
- Beer TW, Drury P, Heenan PJ. Atypical fibroxanthoma: a histological and immunohistochemical review of 171 cases. Am J Dermatopathol 2010;32:533–40.
- \Fretzin DF, Helwig EB. Atypical fibroxanthoma of the skin: a clinicopathological study of 140 cases. Cancer 1973;31:1541–52.
- Kempson RL, McGavran MH. Atypical fibroxanthomas of the skin. Cancer 1964;17:1463–71.
- Mirza B, Weedon D. Atypical fibroxanthoma: a clinicopathological study of 89 cases. Australas J Dermatol 2005;46:235–8.
- Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma: adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am J Surg Pathol 2012;36:1317–26.
- Berk DR, Lind AC, Tapia B, et al. Atypical fibroxanthoma in a child with xeroderma pigmentosum. Pediatr Dermatol 2007;24:450–2.
- dei Tos AP, Maestro R, Doglione C, et al. UV-induced p53 mutations in atypical fibroxanthoma. Am J Pathol 1994;145:11–17.
- Griewank KG, Schilling B, Murali R, et al. TERT promoter mutations are frequent in atypical fibroxanthoma and pleomorphic dermal sarcomas. Mod Pathol 2014;27:502–8
- Dahl I. Atypical fibroxanthoma of the skin: a clinico-pathological study of 57 cases. Acta Pathol Microbiol Scand 1976;84:183–97.
- Kroe OJ, Pitcock JA. Atypical fibroxanthoma of the skin. Am J Clin Pathol 1969;51:487–92.
- Kuwano H, Hashimoto H, Enjoji M. Atypical fibroxanthoma distinguishable from spindle cell carcinoma in sarcoma-like skin lesions. Cancer 1985;55:172–80.
- Leong ASY, Milios J. Atypical fibroxanthoma of the skin: a clinicopathological and immunohistochemical study and a discussion of its histogenesis. Histopathology 1987;11:463–75.
- Reed RJ. Atypical fibroxanthomas and spindle cell carcinomas of the skin. Bull Tulane Univ Med Fac 1967;26:75–89.
- Calonje E, Wadden C, Wilson Jones E, et al. Spindle cell nonpleomorphic atypical fibroxanthoma. Histopathology 1993;22:247–54.
- Bruecks AK, Medlicott SA, Trotter MJ. Atypical fibroxanthoma with prominent sclerosis. J Cutan Pathol 2003;30:336–9.
- Stefanato CM, Robson A, Calonje JE. The histopathologic spectrum of regression in atypical fibroxanthoma. J Cutan Pathol 2010;37:310–15.
- Rudisaile SN, Hurt MA, Santa Cruz DJ. Granular cell atypical fibroxanthoma. J Cutan Pathol 2005;32:314–17.
- Rios-Martín JJ, Delgado MD, Moreno-Ramírez D, et al. Granular cell atypical fibroxanthoma: report of two cases. Am J Dermatopathol 2007;29:84–7.
- Lázaro-Santander R, Andrés-Gozalbo C, Rodríguez-Pereira C, et al. Clear cell atypical fibroxanthoma. Histopathology 1999;35:484–5.
- Luzar B, Calonje E. Morphological and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol 2010;37:301–9.
- Hultgren TL, DiMaio DJ. Immunohistochemical staining of CD10 in atypical fibroxanthomas. J Cutan Pathol 2007;34:415–19.
- Hartel PH, Jackson J, Ducatman BS, et al. CD99 immunoreactivity in atypical fibroxanthoma and pleomorphic malignant fibrous histiocytoma: a useful diagnostic marker. J Cutan Pathol 2006;33(Suppl. 2):24–8.
- Jensen K, Wilkinson B, Wines N, et al. Procollagen 1 expression in atypical fibroxanthoma and other tumors. J Cutan Pathol 2004;31:57–61.
- Thum C, Hollowood K, Birch J, et al. Aberrant Melan-a expression in atypical fibroxanthoma and undifferentiated pleomorphic sarcoma of the skin. J Cutan Pathol 2011;38:954–60.
- Kovach BT, Sams HH, Stasko T. Multiple atypical fibroxanthomas in a cardiac transplant patient. Dermatol Surg 2005;31:467–70.
- Jensen KJ, Peterson SR. Multiple recurrent atypical fibroxanthomas/superficial malignant fibrous histiocytomas of the forehead excised with Mohs micrographic surgery. Dermatol Surg 2006;32:588–91.
- Cooper JZ, Newman SR, Scott GA, et al. Metastasizing atypical fibroxanthoma (cutaneous malignant histiocytoma): report of five cases. Dermatol Surg 2005;31:221–5.
- Lum DJ, King AR. Peritoneal metastases from an atypical fibroxanthoma. Am J Surg Pathol 2006;30:1041–6.
- Seavolt M, McCall M. Atypical fibroxanthoma: review of the literature and summary of 13 patients treated with Mohs micrographic surgery. Dermatol Surg 2006;32:435–41.
Vascular tumours
- Calonje E, Fletcher CDM. Tumors of blood vessels/lymphatics. In: Fletcher CDM, ed. Diagnostic Histopathology of Tumors, 4th edn. London: Churchill Livingstone, 2013:45–86.
- Weiss SW, Goldblum JR, Weiss SW. Enzinger and Weiss's Soft Tissue Tumours, 6th edn. St Louis: Mosby, 2014.
Reactive vascular lesions
Intravascular papillary endothelial hyperplasia
- Masson P. Hémangioendothéliome végétant intra-vasculaire. Bull Soc Anat 1923;93:517–23.
- Kuo T, Sayers CP, Rosai J. Masson's ‘vegetant intravascular hemangioendothelioma’, a lesion often mistaken for angiosarcoma: study of seventeen cases located in the skin and soft tissues. Cancer 1976;38:1227–36.
- Hashimoto H, Daimaru Y, Enjoji M. Intravascular papillary endothelial hyperplasia: a clinicopathologic study of 91 cases. Am J Dermatopathol 1983;5:539–45.
- Pins MR, Rosenthal DI, Springfield DS, et al. Florid extravascular papillary endothelial hyperplasia (Masson's pseudoangiosarcoma) presenting as a soft tissue sarcoma. Arch Pathol Lab Med 1993;117:259–63.
- Reed CN, Cooper PH, Swerlick RA. Intravascular papillary endothelial hyperplasia: multiple lesions simulating Kaposi's sarcoma. J Am Acad Dermatol 1984;10:110–13.
Reactive angioendotheliomatosis
- Wick MR, Rocamora A. Reactive and malignant ‘angioendotheliomatosis’: a discriminant clinicopathologic study. J Cutan Pathol 1988;15:260–71.
- Le Boit PE, Solomon AR, Santa Cruz DJ, et al. Angiomatosis with luminal cryoprotein deposition. J Am Acad Dermatol 1992;27:969–73.
- Krell JM, Sanchez RL, Solomon AR. Diffuse dermal angiomatosis: a variant of reactive cutaneous angioendotheliomatosis. J Cutan Pathol 1994;21:363–70.
- Creamer D, Black MM, Calonje E. Reactive angioendotheliomatosis in association with the antiphospholipid syndrome. J Am Acad Dermatol 2000;42:903–6.
- Ortonne N, Vignon-Pennamen MD, Majdalani G, et al. Reactive angioendotheliomatosis secondary to dermal amyloid angiopathy. Am J Dermatopathol 2001;23:315–19.
- Shyong EQ, Gorevic P, Lebwohl M, et al. Reactive angioendotheliomatosis and sarcoidosis. Int J Dermatol 2002;41:894–7.
- McMenamin ME, Fletcher CD. Reactive angioendotheliomatosis: a study of 15 cases demonstrating a wide clinicopathologic spectrum. Am J Surg Pathol 2002;26:685–97.
Glomeruloid haemangioma
- Chan JKC, Fletcher CDM, Hicklin GA, et al. Glomeruloid hemangioma: a distinctive cutaneous lesion of multicentric Castleman's disease associated with POEMS syndrome. Am J Surg Pathol 1990;14:1036–46.
- Rongioletti F, Gambini C, Lerza R. Glomeruloid hemangioma: a cutaneous marker of POEMS syndrome. Am J Dermatopathol 1994;16:175–8.
- Piña-Oviedo S, López-Patiño S, Ortiz-Hidalgo C. Glomeruloid hemangioma localized to the skin of the trunk with no clinical features of POEMS syndrome. Int J Dermatol 2006;45:1449–50.
- Vélez D, Delgado-Jiménez Y, Fraga J. Solitary glomeruloid hemangioma without POEMS syndrome. J Cutan Pathol 2005;32:449–52.
Benign vascular tumours
Tufted angioma
- Macmillan A, Champion RH. Progressive capillary haemangioma. Br J Dermatol 1971. Nov;85(5):492–3.
- Wilson Jones E. Malignant vascular tumours. Clin Exp Dermatol 1976;1:287–312.
- Kumakiri M, Muramoto F, Tsukinaga I, et al. Crystalline lamellae in the endothelial cells of a type of hemangioma characterized by the proliferation of immature endothelial cells and pericytes–angioblastoma (Nakagawa). J Am Acad Dermatol 1983;8(1):68–75.
- Padilla RS, Orkin M, Rosai J. Acquired ‘tufted’ angioma (progressive capillary hemangioma). Am J Dermatopathol 1987;9:292–300.
- Cho KH, Kim SH, Park KC, et al. Angioblastoma (Nakagawa) – is it the same as tufted angioma? Clin Exp Dermatol 1991;16:110–13.
- Wilson Jones E, Orkin M. Tufted angioma (angioblastoma). A benign progressive angioma, not to be confused with Kaposi's sarcoma or low-grade angiosarcoma. J Am Acad Dermatol 1989;20:214–25.
- Nakagawa K. Case report of angioblastoma of the skin. Jpn J Dermatol 1949;59:92–4.
- Le Huu AR, Jokinen CH, Rubin BP, et al. Expression of prox1, lymphatic endothelial nuclear transcription factor, in kaposiform hemangioendothelioma and tufted angioma. Am J Surg Pathol. 2010;34:1563–73.
- Igarashi M, Oh-i T, Koga M. The relationship between angioblastoma (Nakagawa) and tufted angioma: report of four cases with angioblastoma and a literature-based comparison of the two conditions. J Dermatol Aug 2000;27(8):537–42.
- Ban M, Kamiya H, Kitajima Y. Tufted angioma of adult onset, revealing abundant eccrine glands and central regression. Dermatology 2000;201(1):68–70.
- Osio A, Fraitag S, Hadj-Rabia S, et al. Clinical spectrum of tufted angioma in childhood: a report of 13 cases and a review of the literature. Arch Dermatol 2010;146:758–63.
- Satter EK, Graham BS, Gibbs NF. Congenital tufted angioma. Pediatr Dermatol 2002;19:445–7.
- Lee B, Chiu M, Soriano T, Craft N. Adult-onset tufted angioma: a case report and review of the literature. Cutis 2006;178:341–5.
- Heagerty AH, Rubin A, Robinson TW. Familial tufted angioma. Clin Exp Dermatol 1992;17(5):344–5.
- Tille JC, Morris MA, Brundler MA, et al. Familial predisposition to tufted angioma: identification of blood and lymphatic vascular components. Clin Genet 2003;63:393–9.
- Da Silva AD, Ramos G, Gomes RF, et al. Tufted angioma in children: report of two cases and a review of the literature. Case Rep Dent 2014;2014:942489.
- Maguiness S, Guenther L. Kasabach-Merritt syndrome. J Cutan Med Surg 2002;6:335–9.
- Enjolras O, Mulliken JB, Wassef M, et al. Residual lesions after Kasabach-Merritt phenomenon in 41 patients. J Am Acad Dermatol 2000;42:225–35.
- Ishikawa K, Hatano Y, Ichikawa H, et al. The spontaneous regression of tufted angioma. A case of regression after two recurrences and a review of 27 cases reported in the literature. Dermatology 2005;210:346–8.
Papillary haemangioma
- Suurmeijer AJ, Fletcher CD. Papillary haemangioma. A distinctive cutaneous haemangioma of the head and neck area containing eosinophilic hyaline globules. Histopathology 2007;51:638-48.
Lobular capillary haemangioma (pyogenic granuloma)
- Lee FD. A comparative study of Kaposi's sarcoma and granuloma pyogenicum in Uganda. J Clin Pathol 1968;21:119–28.
- McGeoch AH. Pyogenic granuloma. Aust J Dermatol 1961;6:33–40.
- Harris MN, Desai R, Chuang TY, et al. Lobular capillary hemangioma: an epidemiologic report, with emphasis on cutaneous lesions. J Am Acad Dermatol 2000;42:1012–16.
- Jafarzadeh H, Sanatkhani M, Mohtasham N. Oral pyogenic granuloma: a review. J Oral Sci 2006;48:167–75.
- Bozkurt M, Kulahci Y, Zor F, et al. Multiple giant disseminated pyogenic granuloma in a burn lesion. J Burn Care Res 2006;27:247–9.
- Warner J, Wilson Jones E. Pyogenic granuloma recurring with multiple satellites: a report of 11 cases. Br J Dermatol 1968;80:218–27.
- Vega Harring SM, Niyaz M, Okada S, et al. Extramedullary hematopoiesis in a pyogenic granuloma: a case report and review. J Cutan Pathol 2004;31:555–7.
- LeBoit PE, Berger TG, Egbert BM, et al. Bacillary angiomatosis: the histopathology and differential diagnosis of a pseudoneoplastic infection in patients with human immunodeficiency virus disease. Am J Surg Pathol 1989;13:909–20.
- Sheehan DJ, Lesher JL Jr. Pyogenic granuloma arising within a port-wine stain. Cutis 2004;73:175–80.
- Torres JE, Sánchez JL. Disseminated pyogenic granuloma developing after an exfoliative dermatitis. J Am Acad Dermatol 1995;32:280–2.
- Williams LH, Fleckman P. Painless periungual pyogenic granulomata associated with reverse transcriptase inhibitor therapy in a patient with human immunodeficiency virus infection. Br J Dermatol 2007;156:163–4.
- High WA. Gefitinib: a cause of pyogenic granuloma-like lesions of the nail. Arch Dermatol 2006;142:939.
- Curr N, Saunders H, Murugasu A, et al. Multiple periungual pyogenic granulomas following systemic 5-fluorouracil. Australas J Dermatol 2006;47:130–3.
- Piquet V, Borradori L. Pyogenic granuloma-like lesions during capecitabine therapy. Br J Dermatol 2002;147:1270–2.
- Nappi O, Wick MR. Disseminated lobular capillary hemangioma (pyogenic granuloma): a clinicopathologic study of two cases. Am J Dermatopathol 1986;8:379–85.
- Exner JH, Dahod S, Pochi PE. Pyogenic granuloma-like acne lesions during isotretinoin therapy. Arch Dermatol 1983;119:808–11.
- MacKenzie-Wood AR, Wood G. Pyogenic granuloma-like lesions in a patient using topical tretinoin. Australas J Dermatol 1998;39:248–50.
- Cooper PH, Mills SE. Subcutaneous granuloma pyogenicum: lobular capillary hemangioma. Arch Dermatol 1982;118:30–3.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma). Localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol 2007;29:408–11.
- Cooper PH, McAllister HA, Helwig EB. Intravenous pyogenic granuloma: a study of 18 cases. Am J Surg Pathol 1979;3:221–8.
- Kuroda K, Mizoguchi M. Subcutaneous granuloma pyogenicum in patients with antiphospholipid antibodies. Dermatology 2004;208:331–4.
- Rowe L. Granuloma pyogenicum. AMA Arch Dermatol 1958;78:341–7.
- Fallah H, Fischer G, Zagarella S. Pyogenic granuloma in children: treatment with topical imiquimod. Australas J Dermatol 2007;48:217–20.
- Goldenberg G, Krowchuk DP, Jorizzo JL. Successful treatment of a therapy-resistant pyogenic granuloma with topical imiquimod 5% cream. J Dermatol Treat 2006;17:121–3.
- Bourguignon R, Paquet P, Pierard-Franchimont C, et al. Treatment of pyogenic granulomas with Nd-YAG laser. J Dermatol Treat 2006;17:247–9.
- Mirshams M, Daneshpazhooh M, Mirshekari A, et al. Cryotherapy in the treatment of pyogenic granuloma. J Eur Acad Dermatol Venereol 2006;20:788–90.
- Samatha Y, Reddy TH, Jyothirmai L, et al. Management of oral pyogenic granuloma with sodium tetra decyl sulphate. N Y State Dent J 2013;79:55–7.
- Ichimiya M, Yohikawa Y, Hamamoto Y, et al. Successful treatment of pyogenic granuloma with injection of absolute ethanol. J Dermatol 2004;31:342–4.
Cirsoid aneurysm
- Girard C, Graham JH, Johnson WC. Arteriovenous hemangioma (arteriovenous shunt): a clinicopathological and histochemical study. J Cutan Pathol 1974;1:73–87.
- Connelly MG, Winkelmann RK. Acral arteriovenous tumor: a clinicopathologic review. Am J Surg Pathol 1985;9:15–21.
- Koutlas IG, Jessurun J. Arteriovenous hemangioma: a clinicopathological and immunohistochemical study. J Cutan Pathol 1994;21:343–9.
- Gurbuz Y, Muezzinoglu B, Apaydin R, et al. Acral arteriovenous tumor (cirsoid aneurysm): clinical and histopathological analysis of 6 cases. Adv Clin Pathol 2002;6:25–9.
Epithelioid haemangioma
- Olsen TG, Helwig EB. Angiolymphoid hyperplasia with eosinophilia. J Am Acad Dermatol 1985;12:781–96.
- Wells GC, Whimster IW. Subcutaneous angiolymphoid hyperplasia with eosinophilia. Br J Dermatol 1969;81:1–15.
- Chan JKC, Hui PK, Ng CS, et al. Epithelioid haemangioma (angiolymphoid hyperplasia with eosinophilia) and Kimura's disease in Chinese. Histopathology 1989;15:557–74.
- Fetsch JF, Weiss SW. Observations concerning the pathogenesis of epithelioid haemangioma/angiolymphoid hyperplasia. Mod Pathol 1991;4:449–55.
- Chong WS, Thomas A, Goh CL. Kimura's disease and angiolymphoid hyperplasia with eosinophilia: two entities in the same patient: case report and review of the literature. Int J Dermatol 2006;45:139–45.
- Kuo TT, Shih LY, Chan HL. Kimura's disease: involvement of regional lymph nodes and distinction from angiolymphoid hyperplasia with eosinophilia. Am J Surg Pathol 1988;12:843–54.
- Kung ITM, Gibson JB, Bannatyne PM. Kimura's disease: a clinicopathological study of 21 cases and its distinction from angiolymphoid hyperplasia with eosinophilia. Pathology 1984;16:39–44.
- Rosai J, Ackerman LR. Intravenous atypical vascular proliferation: a cutaneous lesion simulating a malignant blood vessel tumor. Arch Dermatol 1974;109:714–17.
- Koubaa W, Verdier M, Perez M, et al. Intra-arterial angiolymphoid hyperplasia with eosinophilia. J Cutan Pathol 2008;35:495–8.
- Baler GR. Angiolymphoid hyperplasia with eosinophilia: a report of two cases. J Dermatol Surg Oncol 1981;7:229–34.
- Grimwood R, Swinehart JM, Aeling JL. Angiolymphoid hyperplasia with eosinophilia. Arch Dermatol 1979;115:205–7.
- Reed RJ, Terazekis N. Subcutaneous angioblastic lymphoid hyperplasia with eosinophilia (Kimura's disease). Cancer 1972;29:489–97.
- Vazquez-Botet M, Sanchez JL. Angiolymphoid hyperplasia with eosinophilia: report of a case and a review of the literature. J Dermatol Surg Oncol 1978;4:931–6.
- Wilson Jones E. Inflammatory angiomatous nodules with abnormal blood vessels occurring about the ears and scalp (pseudo or atypical pyogenic granuloma). Br J Dermatol 1969;81:804–16.
- Bartralot R, Garcia Patos V, Hueto J, et al. Angiolymphoid hyperplasia with eosinophils affecting the oral mucosa. Br J Dermatol 1996;134:744–8.
- Kadurina MI, Dimitrov BG, Bojinova ST, et al. Angiolymphoid hyperplasia with eosinophilia: successful treatment with Nd:YAG laser. J Cosmet Laser Ther 2007;9:107–11.
- Gencoglan G, Karaca S, Ertekin B. Angiolymphoid hyperplasia with eosinophilia successfully treated with imiquimod. A case report. Dermatology 2007;215:233–5.
Cutaneous epithelioid angiomatous nodule
- Brenn T, Fletcher CDM. Cutaneous epithelioid angiomatous nodule: a distinct lesion in the morphologic spectrum of epithelioid vascular tumors. Am J Dermatopathol 2004;26:14–21.
- Fernández-Flores A, Montero MG, Renedo G. Cutaneous epithelioid angiomatous nodule of the external ear. Am J Dermatopathol 2005;27:175–6.
- Kantrow S, Martin JD, Vnencak-Jones CL, Boyd AS. Cutaneous epithelioid angiomatous nodule: report of a case and absence of microsatellite instability. J Cutan Pathol 2007;34:515–16.
- Sangueza OP, Walsh SN, Sheehan DJ, et al. Cutaneous epithelioid angiomatous nodule: a case series and proposed classification. Am J Dermatopathol 2008;30:16–20.
- Kaushal S, Sharma MC, Raman M, et al. Multifocal cutaneous epithelioid angiomatous nodules of the penis. J Cutan Pathol 2011;38:369–71.
- Shiomi T, Kaddu S, Yoshida Y, et al. Cutaneous epithelioid angiomatous nodule arising in capillary malformation. J Cutan Pathol 2011;38:372–5.
Acquired elastotic haemangioma
- Requena L, Kutzner H, Mentzel T. Acquired elastotic hemangioma: a clinicopathologic variant of hemangioma. J Am Acad Dermatol 2002;47:371–6.
- Martorell-Calatayud A, Balmer N, Sanmartin O, et al. Definition of the features of acquired elastotic hemangioma reporting the clinical and histopathological characteristics of 14 patients. J Cutan Pathol 2010;37:460–4.
Hobnail haemangioma
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol 1988;19:550–8.
- Torrelo A. Hobnail hemangioma. Dermatology 1995;191:154–6.
- Guillou L, Calonje E, Speight P, et al. Hobnail hemangioma: a pseudomalignant vascular lesion with a reappraisal of targetoid hemosiderotic hemangioma. Am J Surg Pathol 1999;23:97–105.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma (‘targetoid hemosiderotic hemangioma’): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol 1999;26:279–86.
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol 2014;31:281–5.
- Trindade F, Kutzner H, Tellechea O, et al. Hobnail hemangioma reclassified as superficial lymphatic malformation: a study of 52 cases. J Am Acad Dermatol 2012;66:112–15.
- Al Dhaybi R, Lam C, Hatami A, et al. Targetoid hemosiderotic hemangiomas (hobnail hemangiomas) are vascular lymphatic malformations: a study of 12 pedoatric cases. J Am Acad Dermatol 2012;66:116–20.
- Ortiz-Rey JA, González-Ruiz A, San Miguel P, et al. Hobnail hemangioma associated with the menstrual cycle. J Eur Acad Dermatol Venereol 2005;19:367–9.
- Franke FE, Steger K, Marks A, et al. Hobnail hemangiomas (targetoid hemosiderotic hemangiomas) are true lymphangiomas. J Cutan Pathol 2004;31:362–7.
Microvenular haemangioma
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol 1991;18:235–40.
- Aloi F, Tomasini C, Pippione M. Microvenular hemangioma. Am J Dermatopathol 1993;15:534–8.
- Fukunaga M, Ushigome S. Microvenular hemangioma. Pathol Int 1998;48:237–9.
- Sànz-Trelles A, Ojeda-Martos A, Jiménez-Fernandéz A, et al. Microvenular haemangioma: a new case in a child. Histopathology 1998;32:89–90.
- Linos K, Csaposs J, Carlson JA. Microvenular hemangioma presenting with numerous bilateral macules, patches, and plaques: a case report and review of the literature. Am J Dermatopathol 2013;35:98–101.
Sinusoidal haemangioma
- Calonje E, Fletcher CDM. Sinusoidal hemangioma. Am J Surg Pathol 1991;15:1130–5.
Spindle cell haemangioma
- Weiss SW, Enzinger FM. Spindle cell hemangioendothelioma: a low grade angiosarcoma resembling a cavernous hemangioma and Kaposi's sarcoma. Am J Surg Pathol 1986;10:521–30.
- Scott GA, Rosai J. Spindle cell hemangioendothelioma: report of seven additional cases of a recently described vascular neoplasm. Am J Dermatopathol 1988;10:281–8.
- Fletcher CDM, Beham A, Schmid C. Spindle cell haemangioendothelioma: a clinicopathological and immunohistochemical study indicative of a nonneoplastic lesion. Histopathology 1991;18:291–301.
- Ding J, Hashimoto H, Imayama S, et al. Spindle cell hemangioendothelioma, probably a benign vascular lesion not a low-grade angiosarcoma: a clinicopathological, ultrastructural and immunohistochemical study. Virchows Arch (A) 1992;420:77–85.
- Imayama S, Murakamai Y, Hashimoto H, et al. Spindle cell hemangioendothelioma exhibits the ultrastructural features of reactive vascular proliferation rather than of angiosarcoma. Am J Clin Pathol 1992;97:279–87.
- Perkins P, Weiss SW. Spindle cell hemangioendothelioma: an analysis of 78 cases with reassessment of its pathogenesis and biologic behavior. Am J Surg Pathol 1996;20:1196–204.
- Kurek KC, Pansuriya TC, van Ruler MA, et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am J Pathol 2013;182:1494–500.
- Fanburg JC, Meis Kindblom JM, Rosenberg AE. Multiple enchondromas associated with spindle cell hemangioendotheliomas: an overlooked variant of Maffucci's syndrome. Am J Surg Pathol 1995;19:1029–38.
- Sheehan M, Roumpf SO, Summerlin DJ, et al. Spindle cell hemangioma: report of a case presenting in the oral cavity. J Cutan Pathol 2007;34:797–800.
- Tosios KI, Gouveris I, Sklavounou A, et al. Spindle cell hemangioma (hemangioendothelioma) of the head and neck: report of an unusual (or underdiagnosed) tumor. Oral Surg Oral Med 2008;105:216–21.
Symplastic haemangioma
- Tsang WYW, Chan JKC, Fletcher CDM, et al. Symplastic hemangioma: a distinctive vascular neoplasm featuring bizarre stromal cells. Int J Surg Pathol 1994;1:202.
- Kutzner H, Winzer M, Mentzel T. [Symplastic hemangioma.] Hautarzt 2000;51:327–31.
- Goh N, Dayrit J, Calonje E. Symplastic hemangioma. Report of two cases. J Cutan Pathol 2006;33:735–40.
Vascular tumours of intermediate malignancy
Kaposiform haemangioendothelioma
- Tsang WY, Chan JK. Kaposi-like infantile hemangioendothelioma: a distinctive vascular neoplasm of the retroperitoneum. Am J Surg Pathol 1991;15:982–9.
- Niedt GW, Alba Greco M, Wieczorek R, et al. Hemangioma with Kaposi's sarcoma-like features: report of two cases. Pediatr Pathol 1989;9:567–75.
- Zukerberg LR, Nickoloff BJ, Weiss SW. Kaposiform hemangioendothelioma of infancy and childhood: an aggressive neoplasm associated with Kasabach–Merritt syndrome and lymphangiomatosis. Am J Surg Pathol 1993;17:321–8.
- Fukunaga M, Ushigome S, Ishikawa E. Kaposiform haemangioendothelioma associated with Kasabach–Merritt syndrome. Histopathology 1996;28:281–4.
- Vin-Christian K, McCalmont TH, Frieden IJ. Kaposiform hemangioendothelioma: an aggressive, locally invasive vascular tumor that can mimic hemangioma of infancy. Arch Dermatol 1997;133:1573–8.
- Chu CY, Hsiao CH, Chiu HC. Transformation between Kaposiform hemangioendothelioma and tufted angioma. Dermatology 2003;206:334–7.
- Drolet BA, Trenor CC, Brandao LR, et al. Consensus-derived practice standards plan for complicated kaposiform hemangioendothelioma. J Pediatr 2013;163:285–91.
- Croteau SE, Liang MG, Kozakewich HP, et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabch-Merritt phenomenon in 107 referrals. J Pediatr 2013;162:142–7.
- Mentzel T, Mazzoleni G, Dei Tos AP, et al. Kaposiform hemangioendothelioma in adults: clinicopathologic and immunohistochemical analysis of three cases. Am J Clin Pathol 1997;108:450–5.
- Debelenko LV, Perez-Atayde AR, Mulliken JB, et al. D2-40 immunohistochemical analysis of pediatric vascular tumors reveals positivity in kaposiform hemangioendothelioma. Mod Pathol 2005;18:1454–60.
- Mac-Moune Lai F, To KF, Choi PC, et al. Kaposiform hemangioendothelioma: five patients with cutaneous lesions and long follow-up. Mod Pathol 2001;14:1087–92.
- Deraedt K, Vander Poorten V, Van Geet C, et al. Multifocal kaposiform haemangioendothelioma. Virchows Arch 2006;448:843–6.
- Gianotti R, Gelmetti C, Alessi E. Congenital cutaneous multifocal kaposiform hemangioendothelioma. Am J Dermatopathol 1999;21:557–61.
- Haisley-Royster C, Enjolras O, Frieden IJ, et al. Kasabach-Merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol 2002;24:459–62.
- Wang Z, Li K, Dong K, et al. Successful treatment of Kasabach-Merritt phenomenon arising from kaposiform hemangioendothelioma. J Pediatr Hematol Oncol 2015;37:72–3.
- Wang Z, Li K, Dong K, et al. Variable response to propranolol treatment of kaposiform hemangioendothelioma, tufted angioma, and Kasabach-Merritt phenomenon. Pediatr Blood Cancer 2014;61:1518–19.
- Kai L, Wang Z, Yao W, et al. Sirolimus, a promising treatment for refractory kaposiform heamngioendothelioma. J Cancer Res Clin Oncol 2014;140:471–6.
- Malhotra Y, Yang CS, McNamara J, et al. Congenital kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon successfully treated with low-dose radiation therapy. Pediatr Dermatol 2013;35:595–8.
- Harper L, Michel JL, Emjolras O, et al. Successful management of a retroperitoneal kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon using alpha-interferon. Eur J Pediatr Surg 2006;16:369–72.
- Fernandez-Pineda I, Lopez-Gutierrez JC, Chocarro G, et al. Long-term outcome of vincristine-aspirin-ticlopidine (VAT) therapy for vascular tumors associated with Kasabach-Merritt phenomenon. Pediatr Blood Cancer 2013;60:1478–81.
Giant cell angioblastoma
- Gonzalez-Crussi F, Choud P, Crawford SE. Congenital infiltrating giant cell angioblastoma, a new entity? Am J Surg Pathol 1991;15:175–83.
- Vargas SO, Perez-Atayde AR, Gonzalez-Crussi F, et al. Giant cell angioblastoma: three additional occurrences of a distinct pathologic entity. Am J Surg Pathol 2001;25:185–96.
- Marler JJ, Rubin JB, Trede NS, et al. Successful antiangiogenic therapy of giant cell angioblastoma with interferon alfa 2b: report of two cases. Pediatrics 2002;109:E37.
Retiform haemangioendothelioma
- Calonje E, Fletcher CD, Wilson Jones E, et al. Retiform hemangioendothelioma: a distinctive form of low-grade angiosarcoma delineated in a series of 15 cases. Am J Surg Pathol 1994;18:115–25.
- Dufau JP, Pierre C, De SaintMaur PP, et al. Hemangioendothelioma retiforme. Ann Pathol 1997;17:47–51.
- Fukunaga M, Endo Y, Masui F, et al. Retiform haemangioendothelioma. Virchows Arch 1996;428:301–4.
- Parsons A, Sheehan DJ, Sangueza OP. Retiform hemangioendotheliomas usually do not express D2-40 and VERFR-3. Am J Dermatopathol 2008;30:31–3.
- Tan D, Kraybill W, Cheney RT, et al. Retiform hemangioendothelioma: a case report and review of the literature. J Cutan Pathol 2005;23:634–7.
- Ioannidou D, Panayiotides J, Krasagakis K, et al. Retiform hemangioendothelioma presenting as bruise-like plaque in an adult woman. Int J Dermatol 2006;45:53–5.
- Duke D, Dvorak AM, Harrist TJ, et al. Multiple retiform hemangioendotheliomas: a low grade angiosarcoma. Am J Dermatopathol 1996;18:606–10.
- Albertini AF, Brousse N, Bodemer C, et al. Retiform hemangioendothelioma developed on the site of an earlier cystic lymphangioma in a six-year-old girl. Am J Dermatopathol 2011;33:e84–7.
- Mentzel T, Stengel B, Katenkamp D. Retiform hemangioendothelioma: clinico-pathologic case report and discussion of the group of low grade malignancy vascular tumors. Pathologe 1997;18:390–4.
Papillary intralymphatic angioendothelioma
- Dabska M. Malignant endovascular angioendothelioma of childhood. Cancer 1969;24:503–9.
- Manivel JC, Wick MR, Swanson PE, et al. Endovascular papillary angio-endothelioma of childhood. Hum Pathol 1986;17:1240–4.
- Morgan J, Robinson NJ, Rosen LB, et al. Malignant endovascular papillary angioendothelioma. Am J Dermatopathol 1989;11:64–8.
- Patterson K, Chandra RS. Malignant endovascular papillary angioendothelioma. Arch Pathol Lab Med 1985;109:671–3.
- Fanburgh-Smith JC, Michal M, et al. Papillary intralymphatic angioendothelioma (PILA): a report of twelve cases of a distinctive vascular tumor with phenotypic features of lymphatic vessels. Am J Surg Pathol 1999;23:1004–10.
Composite haemangioendothelioma
- Nayler SJ, Rubin BP, Calonje E, et al. Composite hemangioendothelioma: a complex, low-grade vascular lesion mimicking angiosarcoma. Am J Surg Pathol 2000;24:352-61.
- Fukunaga M, Suzuki K, Saegusa N, et al. Composite hemangioendothelioma: report of 5 cases including one with associated Maffucci syndrome. Am J Surg Pathol 2007;31:1567–72.
- Requena L, Luis Diaz J, Manzarbeitia F, et al. Cutaneous composite hemangioendothelioma with satellitosis and lymph node metastases. J Cutan Pathol 2008;35:225–30.
Pseudomyogenic haemangioendothelioma
- Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. A fibrohistiocytic/myoid lesion often confused with benign and malignant spindle cell tumors. Cancer 1992;69:1382–95.
- Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol 2003;27:48–57.
- Hornick JL, Fletcher CD. Pseudomyogenic hemangioendothelioma: a distinctive, often nulticentric tumor with indolent behavior. Am J Surg Pathol 2011;35:190–201.
- Requena L, Santonja C, Martinez-Amo JL, et al. Cutaneous epithelioid sarcoma-like (pseudomyogenic) hemangioendothelioma: a little-known low-grade cutaneous vascular neoplasm. JAMA Dermatol 2013;149:459–65.
- Amary MF, O'Donnell P, Berisha F, et al. Pseudomyogenic (epithelioid sarcoma-like) hemangioendothelioma: characterization of five cases. Skeletal Radiol 2013;42:947–57.
- Trombetta D, Magnusson L, von Steyern FV, et al. Translocation t(7;19)(q22;q13) – a recurrent chromosome aberration in pseudomyogenic hemangioendothelioma? Cancer Genet 2011;204:211–15.
- Walther C, Tayebwa J, Lilljebjorn H, et al. A novel SERPINE1-FOSB fusion gene results in a transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma. J Pathol 2014;232:534–40.
Malignant vascular tumours
Angiosarcoma
- Mark RJ, Poen JC, Tran LM, et al. Angiosarcoma: a review of 67 patients and review of the literature. Cancer 1996;77:2400–6.
- Bardwill JM, Mocega EE, Butler JJ, et al. Angiosarcomas of the head and neck region. Am J Surg 1968;11:548–53.
- Holden CA, Spittle MF, Wilson Jones E. Angiosarcoma of the face and scalp: prognosis and treatment. Cancer 1987;48:1907–21.
- Maddox JC, Evans HL. Angiosarcoma of skin and soft tissue: a study of 44 cases. Cancer 1981;48:1907–21.
- Chen KTK, Gilbert EF. Angiosarcoma complicating generalized lymphangiectasia. Arch Pathol Lab Med 1979;103:86–8.
- Eby CS, Brennan MF, Fine G. Lymphangiosarcoma: lethal complication of chronic lymphedema—report of two cases and review of the literature. Arch Surg 1967;94:223–30.
- MacKenzie DH. Lymphangiosarcoma arising in chronic congenital and idiopathic lymphoedema. J Clin Pathol 1971;24:524–9.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer 1948;1:64–81.
- Herrman JB. Lymphangiosarcoma of the chronically edematous extremity. Surg Gynecol Obstet 1965;121:1107–15.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol 1985;12:922–6.
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesions of the skin of the breast after radiation therapy for breast carcinoma. Am J Clin Pathol 1994;102:757–63.
- Karlsson P, Holmberg E, Johansson KA, et al. Soft tissue sarcoma after treatment for breast cancer. Radiother Oncol 1996;38:25–31.
- Treves N. An evaluation of the etiological factors of lymphedema following radical mastectomy: an analysis of 1007 cases. Cancer 1957;10:444–59.
- Scott RB, Nydick I, Conway H. Lymphangiosarcoma arising in lymphedema. Am J Med 1960;28:1008–12.
- Rossi S, Fletcher CD. Angiosarcoma arising in hemangioma/vascular malformation: report of four cases and review of the literature. Am J Surg Pathol 2002;26:1319–29.
- Abratt RP, Williams M, Raff M, et al. Angiosarcoma of the superior vena cava. Cancer 1983;52:740–3.
- Chadhuri B, Ronan SG, Manahgod JR. Angiosarcoma arising in a plexiform neurofibroma. Cancer 1980;46:605–10.
- Trassard M, Le Doussal V, Bui BN, et al. Angiosarcoma arising in a solitary schwannoma (neurilemoma) of the sciatic nerve. Am J Surg Pathol 1996;20:1412–17.
- Mentzel T, Katencamp D. Intraneural angiosarcoma and angiosarcoma arising in benign and malignant peripheral nerve sheath tumours: clinicopathological and immunohistochemical analysis of four cases. Histopathology 1999;35:114–20.
- Morphopoulos GD, Banerjee SS, Ali HH, et al. Malignant peripheral nerve sheath tumour with vascular differentiation: a report of four cases. Histopathology 1996;28:401–10.
- Marcon I, Collini P, Casanova M, et al. Cutaneous angiosarcoma in a patient with xeroderma pigmentosum. Pediatr Hematol Oncol 2004;21:23–6.
- Folpe AL, Johnston CA, Weiss SW. Cutaneous angiosarcoma arising in a gouty tophus: report of a unique case and a review of foreign material-associated angiosarcoma. Am J Dermatopathol 2000;22:418–21.
- Ghandur-Mnaymneh L, Gonzales MS. Angiosarcoma of the penis with hepatic angiomas in a patient with low vinyl chloride exposure. Cancer 1981;47:1318.
- Ahmed I, Hamacher KL. Angiosarcoma in a chronically immunosuppresed renal transplant recipient: report of a case and review of the literature. Am J Dermatopathol 2002;24:330–5.
- Kantrow SM, Boyd AS. Primary cutaneous metaplastic carcinoma: report of a case involving angiosarcoma. Am J Dermatopathol 2007;29:270–3.
- Nake N, Ohsawa M, Tomita Y, et al. Prognostic factors in angiosarcoma: a multivariate analysis of 55 cases. J Surg Oncol 1996;61:170–6.
- Lezana-del Valle P, Gerald WL, Tsai J, et al. Malignant vascular tumors in young patients. Cancer 1998;83:1634–9.
- Oettle AG, van Blerk PJP. Postmastectomy lymphostatic endothelioma of Stewart and Treves in a male. Br J Surg 1963;50:736–43.
- Wilson Jones E. Malignant angioendothelioma of the skin. Br J Dermatol 1964;76:21–39.
- Girard C, Johnson WC, Graham JH. Cutaneous angiosarcoma. Cancer 1970;26:868–83.
- Lydiatt WM, Shaha AR, Sha JP. Angiosarcoma of the head and neck. Am J Surg 1994;168:451–4.
- Hitchcock MG, Hurt MA, Santa Cruz DJ. Cutaneous granular cell angiosarcoma. J Cutan Pathol 1994;21:256–62.
- Brightman LA, Demierre MF, Byers HR. Macrophage-rich epithelioid angiosarcoma mimicking malignant melanoma. J Cutan Pathol 2006;33:38–42.
- Requena L, Santonja C, Stutz N, et al. Pseudolymphomatous cutaneous angiosarcoma: a rare variant of cutaneous angiosarcoma readily mistaken for cutaneous lymphoma. Am J Dermatopathol 2007;29:342–50.
- Folpe AL, Chand EM, Goldblum JR, et al. Expression of Fli-1, a nuclear transcription factor distinguishes vascular neoplasms from potential mimics. Am J Surg Pathol 2001;25:1061–6.
- Miettinen M, Wang ZF, Paetau A, et al. ERG transcription factor as an immunohistochemical marker for vascular endothelial cell tumors and prostatic carcinoma. Am J Surg Pathol 2013;37:1580–5.
- Schuborg C, Mertens F, Rydholm A, et al. Cytogenetic analysis of four angiosarcomas from deep and superficial soft tissue. Cancer Genet Cytogenet 1998;100:52–6.
- Kindblom LG, Stenman G, Angervall L. Morphological and cytogenetic studies of angiosarcoma in Stewart-Treves syndrome. Virchows Arch A Pathol Anat Histopathol 1991;419:439–45.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol 2012;25:75–85.
- Shon W, Sukov WR, Jenkins SM, et al. MYC amplification and overexpression in primary cutaneous angiosarcoma: a fluorescence in-situ hybridization and immunohistochemical study. Mod Pathol 2014;27:509–15.
- Wilson Jones E. Malignant vascular tumours. Clin Exp Dermatol 1976;1:287–312.
- Orchard GE, Zelger B, Wilson Jones E, Russell Jones R. An immunohistochemical assessment of 19 cases of angiosarcoma. Histopathology 1996;28:235–40.
- Hori Y. Malignant hemangioendothelioma of the skin. J Dermatol Surg Oncol 1981;7:130–6.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlations. J Am Acad Dermatol 2004;50:867–74.
- Abraham JA, Hamicek FJ, Kaufman AM, et al. Treatment and outcome of 82 patients with angiosarcoma. Ann Surg Oncol 2007;14:1953–67.
- Mendenhall WM, Mendenhall CM, Werning JW, et al. Cutaneous angiosarcoma. Am J Clin Oncol 2006;29:524–8.
- Deyrup AT, McKenney JK, Tighiouart M, et al. Sporadic cutaneous angiosarcomas: a proposal for risk stratification based on 69 cases. Am J Surg Pathol 2008;32:72–7.
- Pawlik TM, Paulino AF, McGinn C, et al. Cutaneous angiosarcoma of the scalp: a multidisciplinary approach. Cancer 2003;98:1716–26.
- Fujisawa Y, Yoshino K, Kadono T, et al. Chemoradiotherapy with taxane was superior to conventional surgery and radiotherapy in the management of cutaneous angiosarcoma: multi-center retrospective stduy. Br J Dermatol 2014;171:1493–500.
- Letsa I, Benson C, Al-Muderis O, et al. Angiosarcoma of the face and scalp: effective systemic treatment in the older patient. J Geriatr Oncol 2014;5:276–80.
Epithelioid haemangioendothelioma
- Weiss SW, Enzinger FM. Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer 1982;50:970–81.
- Weiss SW, Ishak KG, Dail DH, et al. Epithelioid hemangioendothelioma and related lesions. Semin Diagn Pathol 1986;3:259–87.
- Mentzel T, Beham A, Calonje E, et al. Epithelioid hemangioendothelioma of skin and soft tissues: clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol 1997;21:363–74.
- Quante M, Patel NK, Hill S, et al. Epithelioid hemangioendothelioma presenting in the skin: a clinicopathologic study of eight cases. Am J Dermatopathol 1998;20:541–96.
- Grey MH, Rosenberg AE, Dickersin GR, et al. Cytokeratin expression in epithelioid vascular neoplasms. Hum Pathol 1990;21:212–17.
- Mendlick MR, Nelson M, Pickering D, et al. Translocation t(1;3)(p36.3;q25) is a nonrandom aberration in epithelioid hemangioendothelioma. Am J Surg Pathol 2001;25:684–7.
- Errani C, Zhang L, Sung YS, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 2011;50:644–53.
- Tanas MR, Sboner A, Oliveira AM, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med 2011;3:98.
- Antonescu CR, Le Loarer F, Mosquera JM, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 2013;52:775–84.
Epithelioid angiosarcoma
- Fletcher CDM, Beham A, Bekir S, et al. Epithelioid angiosarcoma of deep soft tissue: a distinctive tumor readily mistaken for an epithelial neoplasm. Am J Surg Pathol 1991;15:915–24.
- Marrogi AJ, Hunt SJ, Santa Cruz DJ. Cutaneous epithelioid angiosarcoma. Am J Dermatopathol 1990;12:350–6.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology 1994;25:421–9.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol 2011;35:60–9.
- Jennings TA, Peterson L, Axiotis CA, et al. Angiosarcoma associated with foreign body material. Cancer 1988;62:2436–44.
- Byers RJ, McMahon RFT, Freemont AJ, et al. Epithelioid angiosarcoma arising in an arteriovenous fistula. Histopathology 1992;21:87–9.
- Val-Bernal JF, Figols J, Arce FP, et al. Cardiac epithelioid angiosarcoma presenting as cutaneous metastases. J Cutan Pathol 2001;28:265–70.
Lymphatic tumours
Acquired progressive lymphangioma
- Jones EW, Winkelmann RK, Zachary CB, et al. Benign lymphangioendothelioma. J Am Acad Dermatol 1990;23:229–35.
- Mehregan DR, Mehregan AH, Mehregan DA. Benign lymphangioendothelioma: report of 2 cases. J Cutan Pathol 1992;19:502–5.
- Watanabe M, Kishiyama K, Ohkawara A. Acquired progressive lymphangioma. J Am Acad Dermatol 1983;8:663–7.
- Guillou L, Flecher CDM. Benign lymphangioendothelioma (acquired progressive lymphangioma), a lesion not to be confused with well-differentiated angiosarcoma and patch stage Kaposi's sarcoma: clinicopathologic analysis of a series. Am J Surg Pathol 2000;24:1047–57.
- Lin SS, Wang KH, Lin YH, Chang SP. Acquired progressive lymphangioma in the groin area successfully treated with surgery. Clin Exp Dermatol 2009. Oct;34(7):e341–2.
- Flores S, Baum C, Tollefson M, Davis D. Pulsed dye laser for the treatment of acquired progressive lymphangioma. Dermatol Surg 2013;40:218–20.
Atypical vascular proliferation after radiotherapy
- Fineberg S, Rosen PP. Cutaneous angiosarcoma and atypical vascular lesion of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol 1994;102:757–63.
- Díaz-Cascajo C, Borghi S, Weyers W, et al. Benign lymphangiomatous papules of the skin after radiotherapy: a report of five new cases and review of the literature. Histopathology 1999;35:319–27.
- Requena L, Kutzner H, Mentzel T, et al. Benign vascular proliferations in irradiated skin. Am J Surg Pathol 2002;26:328–37.
- Brenn T, Fletcher CD. Radiation-induced cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol 2005;29:983–96.
- Brenn T, Fletcher CD. Post-radiation vascular proliferation: an increasing problem. Histopathology 2006;48:106.
- Mattoch IW, Robbins JB, Kempson RL, et al. Post-radiotherapy vascular proliferations in mammary skin: a clinicopathologic study of 11 cases. J Am Acad Dermatol 2007;57:126–33.
- Gengler C, Coindre JM, Leroux A, et al. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favour of a benign process: a study from the French Sarcoma Group. Cancer 2007;109:1584–98.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol 2012;25:75–85.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol 2008 Jun;32(6):943–50.
Diffuse lymphangiomatosis
- Blei F. Lymphangiomatosis: clinical overview. Lymp Res Biol 2011;9(4):185–90.
- Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone); its relation to hemangiomatosis. J Bone Joint Surg Am 1955 Oct;37-A(5):985–1004.
- Venkatramani R, Ma NS, Pitukcheewanont P, Malogolowkin MH, Mascarenhas L. Gorham's disease and diffuse lymphangiomatosis in children and adolescents. Pediatr Blood Cancer 2011 Apr;56(4):667–70.
- Satria MN, Pacheco-Rodriguez G, Moss J. Pulmonary lymphangiomatosis. Lymph Res Biol 2011;9(4):191–3.
- Du MH, Ye RJ, Sun KK, et al. Diffuse pulmonary lymphangiomatosis: a case report with literature review. Chinese Med J 2011 Mar;124(5):797–800.
- Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol 2011 Nov;40(11):1391–7.
- Hu P, Yuan XG, Hu XY, Shen FR, Wang JA. Gorham-Stout syndrome in mainland China: a case series of 67 patients and review of the literature. J Zhejiang Univ Sci B 2013 Aug;14(8):729–35.
- Ramani P, Shah A. Lymphangiomatosis. Histologic and immunohistochemical analysis of four cases. Am J Surg Pathol 1993 Apr;17(4):329–35.
- Leite I, Hernandez-Martin A, Colmenero I, Lopez-Gutierrez JC, Torrelo A. Invasive lymphatic malformation (Gorham-Stout) of the pelvis with prominent skin involvement. Pediatr Dermatol 2013 May–Jun;30(3):374–8.
- Bruch-Gerharz D, Gerharz CD, Stege H, et al. Cutaneous lymphatic malformations in disappearing bone (Gorham-Stout) disease: a novel clue to the pathogenesis of a rare syndrome. J Am Acad Dermatol 2007 Feb;56(2 Suppl.):S21–5.
- Mavrogenis AF, Zambirinis CP, Dimitriadis PA, Tsakanikas A, Papagelopoulos PJ. Gorham-Stout disease. J Surg Orthopaed Adv 2010 Summer;19(2):85–90.
- Heyd R, Micke O, Surholt C, et al. Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys 2011 Nov 1;81(3):e179–85.
- Ozeki M, Funato M, Kanda K, et al. Clinical improvement of diffuse lymphangiomatosis with pegylated interferon alfa-2b therapy: case report and review of the literature. Pediatr Hematol Oncol 2007 Oct–Nov;24(7):513–24.
- Aman J, Thunnissen E, Paul MA, van Nieuw Amerongen GP, Vonk-Noordegraaf A. Successful treatment of diffuse pulmonary lymphangiomatosis with bevacizumab. Ann Internal Med 2012 Jun 5;156(11):839–40.
- Ozeki M, Fukao T, Kondo N. Propranolol for intractable diffuse lymphangiomatosis. N Engl J Med 2011 Apr 7;364(14):1380–2.
Tumours of perivascular cells
Infantile myofibromatosis and adult myofibroma
- Chung EB, Enzinger FM. Infantile myofibromatosis. Cancer 1981;48:1807–18.
- Goldberg NS, Bauer BS, Kraus H, et al. Infantile myofibromatosis: a review of clinicopathology with perspectives on new treatment choices. Pediatr Dermatol 1988;5:37–46.
- Coffin CM, Neilson KA, Ingels S, et al. Congenital generalized myofibromatosis: a disseminated angiocentric myofibromatosis. Pediatr Pathol Lab Med 1995;15:571–87.
- Roggli VL, Kim HS, Hawkins E. Congenital generalized fibromatosis with visceral involvement: a case report. Cancer 1980;45:954–60.
- Beham A, Badve S, Suster S, et al. Solitary myofibroma in adults: clinicopathological analysis of a series. Histopathology 1993;22:335–41.
- Daimaru Y, Hashimoto H, Enjoji M. Myofibromatosis in adults (adult counterpart of infantile myofibromatosis). Am J Surg Pathol 1989;13:859–65.
- Mentzel T, Calonje E, Nascimento AG, et al. Infantile hemangiopericytoma versus infantile myofibromatosis: study of a series suggesting a continuous spectrum of infantile myofibroblastic lesions. Am J Surg Pathol 1994;18:922–30.
- Granter SR, Badizadegan K, Fletcher CDM. Myofibromatosis in adults, glomangiopericytoma and myopericytoma: a spectrum of tumors showing perivascular myoid differentiation. Am J Surg Pathol 1998;22:513–25.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol 2006;30:104–13.
- Vered M, Allon J, Buchner A, et al. Clinico-pathologic correlations of myofibroblastic tumors of the oral cavity. II. Myofibroma and myofibromatosis of the oral soft tissues. J Oral Pathol Med 2007;36:304–14.
- Foss RD, Ellis GL. Myofibromas and myofibromatosis of the oral region: a clinicopathologic analysis of 79 cases. Oral Surg Oral Med 2000;89:57–65.
- Lau PP, Wong OK, Lui PC, et al. Myopericytoma in patients with AIDS: a new class of Epstein-Barr virus-associated tumor. Am J Surg Pathol 2009;33:1666–72.
- Sirvent N, Perrin C, Lacour JP, et al. Monosomy 9q and trisomy 16q in a case of congenital solitary infantile myofiobromatosis. Virchows Arch 2004;445:537–40.
- Stenman G, Nadal N, Persson S, et al. del (6) (q12q15) as the sole cytogenetic abnormality in a case of solitary infantile myofibroma. Oncol Rep 1999;6:1101–4.
- Leon-Villapalos J, Wolfe K, Calonje E, et al. Involuting solitary cutaneous infantile myofibroma and thrombocytopaenia: a previously unreported clinical association. J Plast Reconstr Aesthet Surg 2007;60:1260–2.
Myopericytoma
- Fletcher CDM. Haemangiopericytoma: a dying breed? Reappraisal of an entity and its variants. Curr Diagn Pathol 1994;1:19–25.
- Granter SR, Badizadegan K, Fletcher CD. Myofibromatosis in adults, glomangiopericytoma, and myopericytoma: a spectrum of tumors showing perivascular myoid differentiation. Am J Surg Pathol 1998;22:513–25.
- Dray MS, McCarthy SW, Palmer AA, et al. Myopericytoma: a unifying term for a spectrum of tumors that show overlapping features with myofibroma. A review of 14 cases. J Clin Pathol 2006;59:67–73.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol 2006;30:104–13.
- McMenamin ME, Calonje E. Intravascular myopericytoma. J Cutan Pathol 2002;29:557–61.
- McMenamin ME, Fletcher CDM. Malignant myopericytoma: expanding the spectrum of tumors with myopericytic differentiation. Histopathology 2002;41:450–60.
Glomus tumour
- Carroll RE, Berman AT. Glomus tumors of the hand: review of the literature and report on twenty-eight cases. J Bone Joint Surg 1972;54A:691–703.
- Anagnostou GD, Papademetriou DG, Toumazani MN. Subcutaneous glomus tumors. Surg Gynecol Obstet 1973;136:945–50.
- Kohout E, Stout AP. The glomus tumor in children. Cancer 1961;14:555–66.
- Sluiter JT, Postma C. Multiple glomus tumours of the skin. Acta Derm Venereol (Stockh) 1959;39:98–107.
- Tarnowski WM, Hashimoto K. Multiple glomus tumors: an ultrastructural study. J Invest Dermatol 1969;52:474–8.
- Tsuneyoshi M, Enjoji M. Glomus tumour. Cancer 1982;50:1601–7.
- Venkatachalam MA, Greally JG. Fine structure of glomus tumor: similarity of glomus cells to smooth muscle. Cancer 1969;23:1176–84.
- Mentzel T, Hugel H, Kutzner H. CD34-positive glomus tumor: clinicopathologic and immunohistochemical analysis of six cases with myxoid stromal changes. J Cutan Pathol 2002;29:421–5.
- Slater DN, Cotton DWK, Azzopardi JG. Oncocytic glomus tumour: a new variant? Histopathology 1987;11:523–31.
- Calonje E, Fletcher CDM. Cutaneous intraneural glomus tumour. Am J Dermatopathol 1995;17:395–8.
- Beham A, Fletcher CDM. Intravascular glomus tumour: a previously undescribed phenomenon. Virchows Arch (A) Pathol Anat Histol 1991;418:175–7.
- Gould EW, Manivel JC, Albores-Saavedra J, et al. Locally infiltrative glomus tumors and glomangiosarcoma: a clinical, ultrastructural and immunohistochemical study. Cancer 1990;65:310–18.
- Folpe AL, Fanburgh-Smith JC, Miettinen M, et al. Atypical glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol 2001;25:1–12.
- De Sablet M, Mascaro JM. Tumeurs glomiques multiples et blue rubber bleb naevus. Ann Dermatol Syphiligr 1967;94:35–46.
- Touraine A, Renault P. Tumeurs glomiques multiples du tronc et des membres. Bull Soc Franc Dermatol Syphiligr 1936;43:736–40.
- Henning JS, Kovich OI, Schaffer JV. Glomuvenous malformations. Dermatol Online J 2007;13:17.
- Boon LM, Brouillard P, Irrthum A, et al. A gene for inherited cutaneous venous anomalies (‘glomangiomas’) localizes to chromosome 1p21-22. Am J Hum Genet 1999;65:125–33.
- Calvert JT, Burns S, Riney TJ, et al. Additional glomangioma family link to chromosome 1p: no evidence for genetic heterogeneity. Hum Hered 2001;51:180.
- Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (‘glomangiomas’). Am J Hum Genet 2002;70:866–74.
- Schiefer TK, Parker WL, Anakwenze OA, et al. Extradigital glomus tumors: a 20-year experience. Mayo Clin Proc 2006;81:1337–44.
- Sawada S, Honda M, Kamide R, et al. Three cases of subungual glomus tumors with von Recklinghausen neurofibromatosis. J Am Acad Dermatol 1995;32:277–8.
- Brems H, Park C, Maertens O, et al. Glomus tumors in neurofibromatosis type 1: genetic, functional, and clinical evidence of a novel association. Cancer Res 2009;69:7393-401.
- Chevrant-Breton J, Dunn JE, Laudren A. Multiple glomus tumors associated with multiple neoplasias. Dermatologica 1984;168:290–2.
- Goodman TF, Abele DC. Multiple glomus tumours. Arch Dermatol 1971;103:11–23.
- Gorlin RJ, Fusaro RM, Benton JW. Multiple glomus tumour of the pseudocavernous hemangioma type. Arch Dermatol 1960;8:776–8.
- Rycroft RJG, Menter MA, Sharvill DE, et al. Hereditary multiple glomus tumours. Trans St John's Hosp Dermatol Soc Lond 1975;61:70–81.
Peripheral neuroectodermal tumours
Peripheral neuroectodermal tumours
- Rodriguez-Peralto JL, Riveiro-Falkenbach E, Carrillo R. Bening cutaneous neural tumors. Semin Diagn Pathol 2013;30:45–57.
- Goldblum JR Folpe Al, Weiss SW. Enzinger and Weiss's Soft Tissue Tumors, 6th edn. St Louis: Mosby, 2013.
Neuromuscular hamartoma
- Louhimo I, Rapola J. Intraneural muscular hamartoma: report of two cases in small children. J Pediatr Surg 1972;7:696–9.
- Markel SF, Enzinger FM. Neuromuscular hamartoma: a benign ‘triton tumor’ composed of mature neural and striated muscle elements. Cancer 1982;49:140–4.
Multiple mucosal neuromas
- Gorlin RJ, Sedano HO, Vickers RA, et al. Multiple mucosal neuromas, pheochromocytoma and medullary carcinoma of the thyroid: a syndrome. Cancer 1968;22:293–9.
Amputation stump neuroma
- Cieslak AK, Stout AP. Traumatic and amputation neuromas. Arch Surg 1946;53:646–51.
Morton neuroma
- Lassmann G, Lassmann H, Stockinger L. Morton's metatarsalgia: light and electron microscopic observations and their relation to entrapment neuropathies. Virchows Arch (A) 1976;370:307–21.
- Meachim G, Abberton MJ. Histological findings in Morton's metatarsalgia. J Pathol 1971;103:209–17.
Solitary circumscribed neuroma
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol 1972;106:865–70.
- Fletcher CDM. Solitary circumscribed neuroma of the skin (so-called palisaded, encapsulated neuroma): a clinicopathologic and immunohistochemical study. Am J Surg Pathol 1989;13:574–80.
- Dover JS, From L, Lewis A. Palisaded encapsulated neuromas: a clinicopathologic study. Arch Dermatol 1989;125:386–9.
Schwannoma
- Das Gupta TK, Brasfield RD, Strong EW, et al. Benign solitary schwannomas (neurilemmomas). Cancer 1969;24:355–66.
- Miyakawa T, Kamada N, Kobayashi T, et al. Neurofibromatosis type 2 in an infant with multiple plexiform schwannomas as first symptom. J Dermatol 2007;34:60–4.
- Geschikler CF. Tumours of the peripheral nerves. Am J Cancer 1935;25:377–89.
- Sian CS, Ryan SF. The ultrastructure of neurilemmoma with emphasis on Antoni B tissue. Hum Pathol 1981;12:145–52.
- Waggener JD. Ultrastructure of benign peripheral nerve sheath tumors. Cancer 1966;19:699–709.
- Weiss SW, Langloss JM, Enzinger F. The role of the S100 protein in the diagnosis of soft tissue tumours with particular reference to benign and malignant Schwann cell tumours. Lab Invest 1983;49:299–304.
- Nascimento AF, Fletcher CD. The controversial nosology of benign nerve sheath tumors: neurofilament protein demonstrates intratumoral axons in many sporadic schwannomas. Am J Surg Pathol 2007;31:1363–70.
- Feany MB, Anthony DC, Fletcher CD. Nerve sheath tumours with hybrid features of neurofibroma and schwannoma: a conceptual challenge. Histopathology 1998;32:405–10.
- Hornick JL, Bundock EA, Fletcher CD. Hybrid schwannoma/perineurioma: clinicopathologic analysis of 42 distinctive benign nerve sheath tumors. Am J Surg Pathol 2009;33:1554–61.
- Dahl I. Ancient neurilemmoma (schwannoma). Acta Pathol Microbiol Scand A 1977;85:812–18.
- White WM, Shiu MH, Rosenblum MK, et al. Cellular schwannoma: a clinicopathologic study of 57 patients and 58 tumors. Cancer 1990;66:1266–75.
- Woodruff JM, Scheithauer BW, Kurtkaya-Yapicier O, et al. Congenital and childhood plexiform (multinodular) cellular schwannoma: a troublesome mimic of malignant peripheral nerve sheath tumor. Am J Surg Pathol 2003;27:1321–9.
- Fletcher CDM, Davies SE. Benign plexiform (multinodular) schwannoma: a rare tumour unassociated with neurofibromatosis. Histopathology 1986;10:971–80.
- Kao GF, Laskin WB, Olson TG. Solitary cutaneous plexiform neurilemmoma (schwannoma): a clinicopathologic, immunohistochemical and ultrastructural study of 11 cases. Mod Pathol 1989;2:20–6.
- Lim HS, Jung J, Chung KY. Neurofibromatosis type 2 with multiple plexiform schwannomas. Int J Dermatol 2004;43:336–40.
- Carney JA. Psammomatous melanotic schwannoma: a distinctive heritable tumor with special associations including cardiac myxoma and the Cushing syndrome. Am J Surg Pathol 1990;14:206–22.
- Torrs-Mora J, Dry S, Li X, et al. Malignant melanotic schwannian tumor: a clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification “melanotic schwannoma”. Am J Surg Pathol 2014;38:94–105.
- Goldblum JR, Beals TF, Weiss SW. Neuroblastoma-like neurilemmoma. Am J Surg Pathol 1994;18:266–73.
- Smith K, Mezebish D, Williams JP, et al. Cutaneous epithelioid schwannomas: a rare variant of a benign peripheral nerve sheath tumor. J Cutan Pathol 1998;25:50–5.
- Laskin WB, Fetsch JF, Lasota J, et al. Benign epithelioid peripheral nerve sheath tumors of the soft tissues: clinicopathologic spectrum of 33 cases. Am J Surg Pathol 2005;29:39–51.
- Brooks JJ, Draffen RM. Benign glandular schwannoma. Arch Pathol Lab Med 1992;116:192–5.
- Deng A, Petrali J, Jaffe D, et al. Benign cutaneous pseudoglandular schwannoma: a case report. Am J Dermatopathol 2005;27:432–5.
- Stout AP. The peripheral manifestations of the specific nerve sheath tumour (neurilemmoma). Am J Cancer 1935;24:751–96.
- Woodruff JM, Selig AM, Crowley K, et al. Schwannoma (neurilemmoma) with malignant transformation: a rare, distinctive peripheral nerve tumor. Am J Surg Pathol 1994;18:882–95.
- Trassard M, Le Doussal V, Bui BN, et al. Angiosarcoma arising in a solitary schwannoma (neurilemoma) of the sciatic nerve. Am J Surg Pathol 1996;20:1412–17.
- McMenamin ME, Fletcher CD. Expanding the spectrum of malignant change in schwannomas: epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: a study of 17 cases. Am J Surg Pathol 2001;25:13–25.
Solitary neurofibroma
- Geschikler CF. Tumours of the peripheral nerves. Am J Cancer 1935;25:377–89.
- Reed RJ. Cutaneous manifestations of neural crest disorders (neurocristopathies). Int J Dermatol 1977;16:807–26.
- Megahed M. Histopathological variants of neurofibroma: a study of 114 lesions. Am J Dermatopathol 1994;16:486–95.
- Erlandson RA. Peripheral nerve sheath tumors. Ultrastruct Pathol 1985;9:113–22.
- Colman DS, Williams CA, Wallace MR. Benign neurofibromas in type 1 neurofibromatosis (NF1) show somatic deletions of the NF1 gene. Nature Genet 1995;11:90–2.
- Jouhilahti EM, Peltonen S, Callens T, et al. The development of cutaneous neurofibromas. Am J Pathol 2011;178:500–5.
- Sawyer JR, Parr LG, Gokden N, et al. A reciprocal t(4;9)(q31;p22) in a solitary neurofibroma. Cancer Genet Cytogenet 2005;156:172–4.
- Jokinen CH, Argenyi ZB. Atypical neurofibroma of the skin and subcutaneous tissue: clinicopathologuc analysis of 11 cases. J Cutan Pathol 2010;37:35-42.
- Fetsch JF, Michal M, Mietinnen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol 2000;24:331–43.
- Michal M, Fanburg-Smith JC, Mentzel T, et al. Dendritic cell neurofibroma with pseudorosettes: a report of 18 cases of a distinct and hitherto unrecognized neurofibroma variant. Am J Surg Pathol 2001;25:587–94
Plexiform neurofibroma
- Lin V, Daniel S, Forte V. Is a plexiform neurofibroma pathognomonic of neurofibromatosis type I? Laryngoscope 2004;114:1410–14.
- Fisher DA, Chu P, McCalmont T. Solitary plexiform neurofibroma is not pathognomonic of von Recklinghausen's neurofibromatosis: report of a case. Int J Dermatol 1997;36:439–42.
- Beert E, Brems H, Renard M, et al. Biallelic inactivation of NF1 in a sporadic plexiform neurofibroma. Genes Chromosomes Cancer 2012;51:852–7.
Diffuse neurofibroma
- Evans HL. Sporadic superficial diffuse neurofibromas with repeated local recurrence over many years and a tendency toward malignant change: a report of 3 cases. Am J Surg Pathol 2013;37:987–94.
Perineurioma
- Robson AM, Calonje E. Cutaneous perineurioma: a poorly recognized tumour often misdiagnosed as epithelioid histiocytoma. Histopathology 2000;37:332–9.
- Smith K, Skelton H. Cutaneous fibrous perineuroma. J Cutan Pathol 1998;25:333–7.
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathological study of 15 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol 1997;21:1433–42.
- Zelger B, Weinlich G, Zelger B. Perineuroma. A frequently unrecognized entity with emphasis on a plexiform variant. Adv Clin Pathol 2000;4:25–33.
- Folpe AL, Billings SD, McKenney JK, et al. Expression of claudin-1, a recently described tight junction-associated protein, distinguishes soft tissue perineurioma from potential mimics. Am J Surg Pathol 2002;26:1620–6.
- Santos-Briz A, Gody E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol 2013;35:e45–8.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol 2009;36:60–5.
- Rankine AJ, Filion PR, Platten MA, et al. Perineurioma: a clinicopathologic study of eight cases. Pathology 2004;36:309–15.
- Skelton HG, Williams J, Smith KJ. The clinical and histologic spectrum of cutaneous fibrous perineuriomas. Am J Dermatopathol 2001;23:190–6.
Dermal nerve sheath myxoma
- Gallager RL, Helwig EB. Neurothekeoma: a benign tumor of neural crest origin. Am J Clin Pathol 1980;74:759–64.
- Pulitzer DR, Reed RJ. Nerve-sheath myxoma (perineurial myxoma). Am J Dermatopathol 1985;7:409–21.
- Fletcher CDM, Chen JKC, McKee PH. Dermal nerve sheath myxoma: a study of three cases. Histopathology 1986;10:135–45.
- Fetsch JF, Laskin WB, Mietinen M. Nerve sheath myxoma: a clinicopathologic and immunohistochemical analysis of 57 morphologically distinctive, S-100 protein and GFAP positive myxoid peripheral nerve sheath tumors with a predilection for the extremities and a high local recurrence rate. Am J Surg Pathol 2005;29:1615–24.
- Rosati LA, Fratamico CM, Eusebi V. Cellular neurothekeoma. Appl Pathol 1986;4:186–91.
Cellular neurothekeoma
- Barnhill RL, Mihm MC. Cellular neurothekeoma: a distinctive variant of neurothekeoma mimicking nevomelanocytic tumors. Am J Surg Pathol 1990;14:113–20.
- Calonje E, Wilson-Jones E, Smith NP, et al. Cellular ‘neurothekeoma’: an epithelioid variant of pilar leiomyoma? Morphological and immunohistochemical analysis of a series. Histopathology 1992;20:397–404.
- Barnhill RL, Dickersin GR, Nickeleit V, et al. Studies on the cellular origin of neurothekeoma: clinical, light microscopic, immunohistochemical and ultrastructural observations. J Am Acad Dermatol 1991;25:80–8.
- Busam KJ, Mentzel T, Colpaert C, et al. Atypical or worrisome features in cellular neurothekeoma: a study of 10 cases. Am J Surg Pathol 1998;22:1067–72.
- Sheth S, Li X, Binder S, et al. Differential gene expression profiles of neurothekeomas and nerve sheath myxomas by microarray analysis. Mod Pathol 2011;24:343–54.
- Hornick JL, Fletcher CD. Cellular neurothekeoma: detailed characterization in a series of 133 cases. Am J Surg Pathol 2007;31:329–40.
- Stratton J, Billings SD. Cellular neurothekeoma: analysis of 37 cases emphasizing atypical histologic features. Mod Pathol 2014;27:701–10.
- Cardoso J, Calonje E. Cellular neurothekeoma with perineural extension: a potential diagnostic pitfall. J Cutan Pathol 2012;39:662–4.
- Fox MD, Billings SD, Gleason BC, et al. Expression of MiTF may be helpful in differentiating cellular neurothekeoma from plexiform fibrohistiocytic tumor (histiocytoid predominant) in a partial biopsy specimen. Am J Dermatopathol 2012;34:157–60.
- Requena L, Sitthinamsuwan P, Fried I, et al. A benign cutaneous plexiform hybrid tumor of perineurioma and cellular neurothekeoma. Am J Surg Pathol 2013;37:845–52.
- Mahalingam M, Alter JN, Bhawan J. Multiple cellular neurothekeomas—a case report and review on the role of immunohistochemistry as a histologic adjunct. J Cutan Pathol 2006;33:51–6.
Granular cell tumour
- Garancis JC, Komorowski RA, Kuzma FJ. Granular cell myoblastoma. Cancer 1970;25:542–50.
- Lack EE, Worsham GF, Calliham MD, et al. Granular cell tumor: a clinicopathologic study of 100 patients. J Surg Oncol 1980;13:301–9.
- Seo IS, Azarelli B, Warner TF, et al. Multiple visceral and cutaneous granular cell tumors: ultrastructural and immunocytochemical evidence of Schwann cell origin. Cancer 1984;53:2104–10.
- Bedetti CD, Martinez AJ, Beckford NS, et al. Granular cell tumours arising in myelinated peripheral nerves: light and electron microscopy and immunoperoxidase study. Virchows Arch (Pathol Anat) 1983;402:175–84.
- Miettinen M, Lehtonen E, Lehtola H, et al. Histogenesis of granular cell tumour: an immunological and ultrastructural study. J Pathol 1984;142:221–31.
- Epstein DS, Pashaei S, Hunt Jr E, et al. Pustulo-ovoid bodies of Milian in granular cell tumors. J Cutan Pathol 2007;34:405–9.
- Zedek DC, Murphy BA, Shea CR, et al. Cutaneous clear-cell granular cell tumors: the histologic description of an unusual variant. J Cutan Pathol 2007;34:397–404.
- Battistella M, Cribier B, Feugeas JP, et al. Vascular invasion and other invasive features in granular cell tumours of the skin: a multicentre study of 119 cases. J Clin Pathol 2014;67:19–25.
- Ray S, Jukic DM. Cutaneous granular cell tumor with epidermal involvement: a potential mimic of melanocytic neoplasia. J Cutan Pathol 2007;34:188–94.
- Gleason BC, Nascimento AF. HMB-45 and melan-A are useful in the differential diagnosis between granular cell tumor and malignant melanoma. Am J Dermatopathol 2007;29:22–7.
- Ramaswamy PV, Storm CA, Filiano JJ, et al. Multiple granular cell tumors in a child with Noonan syndrome. Pediatr Dermatol 2010;27:209–11.
Meningothelial heterotopias
- Argenyi ZB, Thieberg MD, Hayes C, et al. Primary cutaneous meningioma associated with von Recklinghausen's disease. J Cutan Pathol 1994;21:549–56.
- Argenyi ZB. Cutaneous neural heterotopias and related tumours relevant for the dermatopathologist. Semin Diagn Pathol 1996;13:60–71.
- Bain GO, Shnitka TK. Cutaneous meningioma (psammoma). AMA Arch Dermatol 1956;74:590–4.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas: a clinico-pathologic study. Cancer 1974;34:728–44.
- Miyamoto T, Mihara M, Hagari Y, et al. Primary cutaneous meningioma of the scalp: report of 2 siblings. J Dermatol 1995;22:611–19.
- Suster S, Rosai J. Hamartoma of the scalp with ectopic meningothelial elements: a distinctive soft tissue lesion that may simulate angiosarcoma. Am J Surg Pathol 1990;14:1–11.
- Bale PM, Hughes L, De Silva M. Sequestrated meningoceles of the scalp: extracranial meningeal hamartoma. Hum Pathol 1990;21:1156–63.
- Theaker JM, Fletcher CDM, Tudway AJ. Cutaneous heterotopic meningeal nodules. Histopathology 1990;16:475–9.
- Fox MD, Billings SD, Gleason BC, et al. Cutaneous meningioma: a potential pitfall in p63 positive cutaneous neoplasms. J Cutan Pathol 2013;40:891–5.
Glial heterotopic nodules
- Fletcher CDM, Carpenter G, McKee PH. Nasal glioma: a rarity. Am J Dermatopathol 1986;8:341–6.
- Theaker JM, Fletcher CDM. Heterotopic glial nodules: a light microscopic and immunohistochemical study. Histopathology 1991;18:255–60.
- McDermott MB, Glasner SD, Nielsen PL, Dehner LP. Soft tissue gliomatosis: morphologic unity and histogenetic diversity. Am J Surg Pathol 1996;20:148–55.
Epithelial sheath neuroma
- Requena L, Grosshans E, Kutzner H, et al. Epithelial sheath neuroma: a new entity. Am J Surg Pathol 2000;24:190–6.
- Maxwell A, Fung MD. Epithelial sheath neuroma: neoplasia or hyperplasia? J Cutan Pathol 2012;39:1052–4.
Pigmented neuroectodermal tumour of infancy
- Koudstaal J, Oldhoff J, Panders AK, et al. Melanotic neuroectodermal tumor of infancy. Cancer 1968;22:151–61.
- Krused-Lösler B, Gaertner C, Bürger H, et al. Melanotic neuroectodermal tumor of infancy: systematic review of the literature and presentation of a case. Oral Surg Oral Pathol 2006;102:204–16.
- Borello ED, Gorlin RJ. Melanotic neuroectodermal tumour of infancy: a neoplasm of neural crest origin. Cancer 1966;19:196–203.
- Cutler LS, Chaudhury AP, Topiazian R. Melanotic neuroectodermal tumour of infancy: an ultrastructural literature review and reevaluation. Cancer 1981;48:257–68.
- Johnson RE, Scheithauer BW, Dahlin DC. Melanotic neuroectodermal tumour of infancy. Cancer 1983;52:661–6.
- Pettinato G, Manivel JC, d'Amore ESG, et al. Melanotic neuroectodermal tumor of infancy: a reexamination of a histogenetic problem based on immunohistochemical, flow cytometric and ultrastructural study of 10 cases. Am J Surg Pathol 1991;15:233–45.
- Johnson RE, Scheithauer BW, Dahlin DC. Melanotic neuroectodermal tumor of infancy: a review of seven cases. Cancer 1983;52:661–6.
- Stirling RW, Powell G, Fletcher CDM. Pigmented neuroectodermal tumour of infancy: an immunohistochemical study. Histopathology 1988;12:425–35.
- Takeda Y, Kuroda M, Suzuki A. Melanocytes in odontoameloblastoma: a case report. Acta Pathol Jpn 1989;39:465–8.
- Dehner LP, Sibley RK, Sauk JJ, et al. Malignant melanotic neuroectodermal tumor of infancy: a clinical, pathologic, ultrastructural and tissue culture study. Cancer 1979;43:1389–410.
Malignant peripheral nerve sheath tumour
- D'Agostino AN, Soule EH, Miller RH. Sarcomas of the peripheral nerves and somatic soft tissues associated with multiple neurofibromatosis (von Recklinghausen's disease). Cancer 1963;16:1015–27.
- George E, Swanson PE, Wick MR. Malignant peripheral nerve sheath tumours of the skin. Am J Dermatopathol 1989;11:213–21.
- Demitsu T, Murata S, Kiyosawa T, et al. Malignant Schwannoma arising in patients with von Recklinghausen's disease. J Dermatol 1995;22:747–54.
- Allison KH, Patel RM, Goldblum JR, et al. Superficial malignant peripheral nerve sheath tumor: a rare and challenging diagnosis. Am J Clin Pathol 2005;124:685–92.
- Giodillo PP, Helson L, Hajdu SI, et al. Malignant schwannoma: clinical characteristics and response to therapy. Cancer 1981;47:2503–9.
Peripheral primitive neuroectodermal tumour
- Banerjee SS, Agbamu DA, Eyden BP, et al. Clinicopathological characteristics of peripheral neuroectodermal tumour of skin and subcutaneous tissue. Histopathology 1997;31:355–66.
- Hasegawa S, Davidson JM, Rutten A, et al. Primary cutaneous Ewing's sarcoma: immunophenotypic and molecular cytogenetic evaluation of five cases. Am J Surg Pathol 1998;22:310–18.
- Shingde MV, Buckland M, Busam KJ, et al. Primary cutaneous Ewing sarcoma/primitive neuroectodermal tumour: a clinicopathological analysis of seven cases highlighting diagnostic pitfalls and the role of FISH testing in diagnosis. J Clin Pathol 2009;62:915–19.
- Delaplace M, Lhommet C, de Pinieux G, et al. Primary cutaneous Ewing sarcoma: a systematic review focused on treatment and outcome. Br J Dermatol 2012;166:721–6.
Tumours of muscle
Smooth muscle hamartoma
- Slifman NR, Harrist TJ, Rhodes AR. Congenital arrector pili hamartoma: a case report and review of the spectrum of Becker's melanosis and pilar smooth muscle hamartoma. Arch Dermatol 1985;12:1034–7.
- Zvulunov A, Rotem A, Merlob P, et al. Congenital smooth muscle hamartoma. Prevalence, clinical findings, and follow-up in 15 patients. Am J Dis Child 1990;144:782–4.
- Darling TN, Kamino H, Murray JC. Acquired cutaneous smooth muscle hamartoma. J Am Acad Dermatol 1993;28:844–5.
- Hsiao GH, Chen JS. Acquired genital smooth-muscle hamartoma. A case report. Am J Dermatopathol 1995;17:67–70.
- Gualandri L, Cambiaghi S, Ermacora E, et al. Multiple familial smooth muscle hamartomas. Pediatr Dermatol 2001;18:17–20.
- García-Gavín J, Pérez-Pérez L, Allegue F, et al. Multiple congenital familial smooth muscle hamartoma in two siblings. Dermatol Online J 2012. May 15;18(5):7.
- Paulger BR, Kraus EW, Pulitzer DR, et al. Xp microdeletion syndrome characterized by pathognomonic linear skin defects on the head and neck. Pediatr Dermatol 1997 Jan–Feb;14(1):26–30.
- Schnur RE, Herzberg AJ, Spinner N, et al. Variability in the Michelin tire syndrome. A child with multiple anomalies, smooth muscle hamartoma, and familial paracentric inversion of chromosome 7q. J Am Acad Dermatol 1993;28:364–70.
- Janicke EC, Nazareth MR, Rothman IL. Generalized smooth muscle hamartoma with multiple congenital anomalies without the “Michelin tire baby” phenotype. Pediatr Dermatol 2014 Nov–Dec;31(6):731–3.
- Rothman IL. Michelin tire baby syndrome: a review of the literature and a proposal for diagnostic criteria with adoption of the name circumferential skin folds syndrome. Pediatr Dermatol 2014 Nov–Dec;31(6):659–63.
- Civatte J, Marinho E, Oliver Santos R. Smooth muscle hamartoma or nevus of Becker? Apropos of 4 cases. Med Cutan Ibero Lat Am 1988;16:145–8.
- Fernandez-Flores A, Saeb-Lima M. Combined cutaneous smooth muscle hamartoma and nevus flammeus. J Cutan Pathol 2014 Jul;41(7):612–16.
- Tzu J, Goldman C, Perry AE, Meehan SA. Combined blue nevus-smooth muscle hamartoma: a series of 12 cases. J Cutan Pathol 2013 Oct;40(10):879–83.
- Kienast A, Maerker J, Hoeger PH. Myokymia as a presenting sign of congenital smooth muscle hamartoma. Pediatr Dermatol 2007;24:628–31.
- Viglizzo G, Nemelka O, Nozza P, et al. Congenital smooth muscle hamartoma presenting with an unusual pseudo-Darier's sign. Clin Exp Dermatol 2006;31:148–9.
Leiomyoma
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol 1997;19:2–9.
- Fisher WC, Helwig EB. Leiomyomas of the skin. Arch Dermatol 1963;88:510–20.
- Duhig JT, Ayer JP. Vascular leiomyoma: a study of sixty-one cases. AMA Arch Pathol 1959;68:424–30.
- Hachisuga T, Hashimoto H, Enjoji M. Angioleiomyoma: a clinical reappraisal of 562 cases. Cancer 1984;54:126–30.
- Nascimento AG, Karas M, Rosen PP, et al. Leiomyoma of the nipple. Am J Surg Pathol 1979;3:151–6.
- Prabhakar BR, Davessar K, Chitkara NL, et al. Leiomyoma of the areolar region of the breast. Int J Cancer 1969;6:260–1.
- Matsubara J, Miura K. Leiomyoma of the scrotum: a case report and review of the literature. Jpn J Cancer Clin 1971;17:151–4.
- Newman PL, Fletcher CDM. Smooth muscle tumours of the external genitalia: clinicopathological analysis of a series. Histopathology 1991;18:523–9.
- Alam NA, Bevan S, Churchman M, et al. Localization of a gene (MCUL1) for multiple cutaneous leiomyomata and uterine fibroids to chromosome 1q,42.3–q43. Am J Hum Genet 2001;68:1264–9.
- Alam NA, Barclay E, Rowan AJ, Tyrer JP, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol 2005;141:199–206.
- Alam NA, Olpin S, Leigh IM. Fumarate hydratase mutations and predisposition to cutaneous leiomyomas, uterine leiomyomas and renal cancer. Br J Dermatol 2005;153:11–17.
- Wei MH, Toure O, Glenn GM, et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with herediatary leiomyomatosis and renal cell cancer. J Med Genet 2006;43:18–27.
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 2011;79:49–59.
Leiomyosarcoma
- Headington JT, Beals TF, Niederhuber JE. Primary leiomyosarcoma of skin. J Cutan Pathol 1977;4:308–17.
- Wang P, Hornstein OP, Schricker KTH. Kutanes Leiomyosarkom und osteomedullaeres Plasmozytom mit Nachweis von IgA kappa-Paraprotein in Serum und Hauttummor. Hautarzt 1976;27:441–8.
- Oliver GF, Reiman HM, Gonchoroff NT, et al. Cutaneous and subcutaneous leiomyosarcoma: a clinicopathological review of 14 cases with reference to antidesmin staining and nuclear DNA patterns studied by flow cytometry. Br J Dermatol 1991;124:252–7.
- Bellezza G, Sidoni A, Cavaliere A, et al. Primary cutaneous leiomyosarcoma: a clinicopathological and immunohistochemical study of 7 cases. Int J Surg Pathol 2004;12:39–44.
- Kraft S, Fletcher CD. Atypical intradermal smooth muscle neoplasms: clinicopathologic analysis of 84 cases and a reappraisal of cutaneous “leiomyosarcoma”. Am J Surg Pathol 2011;35:599–607.
- Kaddu S, Beham A, Cerroni L, et al. Cutaneous leiomyosarcoma. Am J Surg Pathol 1997;21:979–87.
- Akers WA, Prazak G. Leiomyosarcoma metastatic to scalp from primary in retroperitoneal area: report of a case. Arch Dermatol 1960;81:953–7.
- Cháves E, Sa HH, Gadelha N, et al. Leiomyosarcoma in the skin. Acta Dermatol Vénéréol 1972;52:288.
- Dahl I, Angervall L. Cutaneous and subcutaneous leiomyosarcoma: a clinicopathologic study of 47 patients. Pathol Eur 1974;9:307–15.
- Fields JP, Helwig EB. Leiomyosarcoma of the skin and subcutaneous tissue. Cancer 1981;47:156–69.
- Choy C, Cooper A, Kossard S. Primary cutaneous diffuse leiomyosarcoma with desmoplasia. Australas J Dermatol 2006;47:291–5.
- Berzal-Cantalejo F, Sabater-Marco V, Pérez-Vallés A, et al. Desmoplastic cutaneous leiomyosarcoma: case report and review of the literature. J Cutan Pathol 2006;33(Suppl. 2):29–31.
- Haim S, Gellei B. Leiomyosarcoma of the skin: report of two cases. Dermatologica 1970;140:30–5.
- Orellana-Díaz O, Hernández-Pérez E. Leiomyoma cutis and leiomyosarcoma: a 10-year study and a short review. J Dermatol Surg Oncol 1983;9:283–7.
- Karroum KE, Zappi EG, Cockerell CJ. Sclerotic primary cutaneous leiomyosarcoma. Am J Dermatopathol 1995;17:292–6.
- Phelan JT, Sherer W, Mesa P. Malignant smooth-muscle tumors (leiomyosarcomas) of soft tissue origin. N Engl J Med 1962;266:1027–30.
- Greenwood N, Fox H, Edwards EC. Leiomyosarcoma of the penis. Cancer 1972;29:481–3.
- Premalata CS, Kumar RV, Malathi M, et al. Cutaneous leiomyosarcoma, trichoblastoma, and syringocystadenoma papilliferum arising from nevus sebaceous. Int J Dermatol 2007;46:306–8.
Rhabdomyosarcomatous congenital hamartoma
- Mills AE. Rhabdomyomatous mesenchymal hamartoma. Am J Dermatopathol 1989;11:58–63.
- Hendrick SJ, Sanchez RL, Blackwell SJ, et al. Striated muscle hamartoma: description of two cases. Pediatr Dermatol 1986;3:153–7.
- Rosenberg AS, Kirk J, Morgan MB. Rhabdomyomatous mesenchymal hamartoma: un unusual dermal entity with a report of two cases and review of the literature. J Cutan Pathol 2002;29:238-43.
- Sánchez RL, Raimer SS. Clinical and histologic features of striated muscle hamartoma: possible relationship to Delleman's syndrome. J Cutan Pathol 1994;21:40–6.
- Chang CP, Chen GS. Rhabdomyomatous mesenchymal hamartoma: a plaque-type variant in an adult. Kaohsiung J Med Sci 2005;21:185–8.
- Gerosa C, Fanni D, Pilloni L, et al. Rhabdomyomatous mesenchymal hamartoma presenting as a polypoid lesion of the nasal skin in a child: answer. J Pediatr Neonat Individual Med 2014;3.
- Ashfaq R, Timmons CF. Rhabdomyomatous mesenchymal hamartoma of skin. Pediatr Dermatol 1992;12:731–5.
- Williams NP, Shue AC. Rhabdomyomatous mesenchymal hamartoma: clinical overview and report of a case with spontaneous regression. West Indian Med J 2009;58:607–9.
Skeletal muscle tumours
- Brecher AR, Reyes-Mugica M, Kamino H, et al. Congenital primary cutaneous rhabdomyosarcoma in a neonate. Pediatr Dermatol 2003;20:335–8.
- Schmidt D, Fletcher CD, Harms D. Rhabdomyosarcoma with primary presentation in skin. Pathol Res Pract 1993;189:422–7.
- Setterfield J, Sciot R, Debiec-Rychter M, et al. Primary cutaneous epidermotropic alveolar rhabdomyosarcoma with t(2;13) in an elderly woman: case report and review of the literature. Am J Surg Pathol 2002;26:938–44.
Angiolipoma
- Howard WR, Helwig EB. Angiolipoma. Arch Dermatol 1960;82:924–31.
- Räsänen O, Nohteri H, Dammert K. Angiolipoma and lipoma. Acta Chir Scand 1967;133:461–5.
- Dixon AY, McGregor DH, Lee SH. Angiolipomas: an ultrastructural and clinicopathological study. Human Pathol 1981;12:739–47.
- Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol 1990;14:75.
- Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Pathol 1996;148(2):623–30.
- Fletcher CD, Akerman M, Dal Cin P, et al. Correlation between clinicopathological features and karyotype in lipomatous tumors. A report of 178 cases from the Chromosomes and Morphology (CHAMP) Collaborative Study Group. Am J Surg Pathol 1997;21(4):441–4.
- Nguyen L, Zwagerman NT, Grandhi R, et al. Intraosseous angiolipoma of the cranium: case report and review of the literature. Surg Neurol Int 2014;28;5:79.
- Lee SP, Nicholson GI, Hitchcock G. Familial abdominal chemodectomas with associated cutaneous angiolipomas. Pathology 1977;9(2):173–7.
- Namba M, Kohda M, Mimura S, et al. Angiolipoma in brothers. J Dermatol 1977;4(6):255–7.
- Heymann WR, Fiorillo A, Simons J. Eruptive familial angiolipomas occurring during pregnancy. Cutis 1988;42(6):525–6.
- Abbasi NR, Brownell I, Fangman W. Familial multiple angiolipomatosis. Dermatol Online J 2007;27:13(1):3.
Lipoma
- Bick EM. Lipoma of the extremities. Ann Surg 1936;104:139–43.
- Osment LS. Cutaneous lipomas and lipomatosis. Surg Gynecol Obstet 1968;127:129–32.
- Rydholm A, Bery NO. Size, site and clinical incidence of lipoma. Factors in the differential diagnosis of lipoma and sarcoma. Acta Orthop Scand 1983;54:929–34.
- Mandahl N, Heim S, Arheden K, et al. Three major cytogenetic subgroups can be identified among chromosomally abnormal solitary lipomas. Hum Genet 1988;79:203–8.
- Heim S, Mandahl N, Rydholm A, et al. Different karyotypic features characterize different clinicopathologic subgroups of benign lipogenic tumors. Int J Cancer1988;42:863–7.
- Sreekantaiah C, Leong SP, Karakousis CP, et al. Cytogenetic profile of 109 lipomas. Cancer Res 1991;51:422–33.
- Mandahl N, Hoglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer 1994;9:207–15.
- Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: lipoma. Cancer Genet Cytogenet 2004;150:93–115.
- Kubo T, Matsui Y, Naka N, et al. Specificity of fusion genes in adipocytic tumors. Anticancer Res 2010;30:661–4.
- Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of the subcutaneous tissue. J Cutan Pathol 2005;32:379–82.
- Laskin WB, Fetsch JF, Michal M, et al. Sclerotic (fibroma-like) lipoma: a distinctive lipoma variant with a predilection for the distal extremities. Am J Dermatopathol 2006;28:308–16.
- Marshall-Taylor C, Farnburg-Smith JC. Fibrohistiocytic lipoma: twelve cases of a previously undescribed benign fatty tumor. Ann Diagn Pathol 2000;4:354–360.
- Val-Bernal JF, Val D, Garijo MF, et al. Subcutaneous ossifying lipoma: a case report and review of the literature. J Cutan Pathol 2007;34:788–92.
- Meis JM, Enzinger FM. Myolipoma of soft tissue. Am J Surg Pathol 1991;15:121–5.
- Ramdial PK, Madaree A, Singh B. Membranous fat necrosis in lipomas. Am J Surg Pathol 1997;21:841–6.
- Cox NH, Broome G. Piezogenic palmar papules: a novel physical sign of palmar lipoma. Br J Dermatol 2008;159:757–8.
- Richert B, Andre J, Choffray A, et al. Periungual lipoma: about three cases. J Am Acad Dermatol 2004;51(2 suppl.):S91–3.
- Bardazzi F, Savola F, Fanti PA. Subungual lipoma. Br J Dermatol 2003;149:418.
- Srinivasan US, Raghunathan N, Radhi L. Long term outcome of non-dysraphic intramedullary spinal cord lipomas in adults: case series and review. Asian Spine J 2014; 8(4):476–83.
- Gossner J. Pancreatic lipomas – prevalence in patients undergoing abdominal CT. Pol J Radiol 2014;11:79:259-6.
Hibernoma
- Merkel H. On a pseudolipoma of the breast (peculiar fat tumor). Beitr Pathol Anat 1906;39:152–7.
- Gery L. Discussions. Bull Mem Soc Anat (Paris) 1914;89:111–23.
- Furlong MA, Fanburg-Smith JC, Miettinen M. The morphologic spectrum of hibernoma: a clinicopathologic study of 170 cases. Am J Surg Pathol 2001;25:809-14.
- Gaffney EF, Hargreaves HK, Semple E, et al. Hibernoma: distinctive light and electron microscopic features and relationship to brown adipose tissue. Hum Pathol 1983;14:677–87.
- Chirieac LR, Dekmezlan RH, Ayala AG. Characterization of the myxoid variant of hibernoma. Ann Diagn Pathol 2006;10:104–6.
- Moretti VM, Brooks JS, Lackman RD. Spindle-cell hibernoma: a clinicopathologi comparison of this new variant. Orthopedics 2010; 33:52–5.
- Fletcher CDM, Akerman M, Dal Cin P, et al. Correlation between clinicopathological features and karyotype in lipomatous tumors. Am J Pathol 1996;148:623–30.
- Gisselson G, Hoglund M, Mertens F, et al. Hibernomas are characterized by homozygous deletions in the multiple endocrine neoplasia type 1 region. Metaphase fluorescence in situ hybridization reveals complex rearrangements not detected by conventional cytogenetics. Am J Pathol 1999;155:61–6.
- Oñate Celdrán J, Sánchez Rodríguez C, Gonzalez Valverde FM, et al. Retroperitoneal hibernoma. A case report and review of the literature. Gastroenterol Hepatol 2014;37(4):246–7.
Tumours of fat cells
Lipoblastoma and lipoblastomatosis
- Chung EB, Enzinger FM. Benign lipoblastomatosis. Cancer 1973;32:482–92.
- Mentzel T, Calonje E, Fletcher CDM. Lipoblastoma and lipoblastomatosis: a clinicopathological study of 14 cases. Histopathology 1993;23:527–33.
- Collins MH, Chatten J. Lipoblastoma/lipoblastomatosis: a clinicopathologic study of 25 tumors. Am J Surg Pathol 1997;21:1131–7.
- Shmookler BM, Enzinger FM. Liposarcoma occurring in children: an analysis of 17 cases and review of the literature. Cancer 1983;52:567–74.
- Fletcher JA, Kozakewich HP, Schoenberg ML, et al. Cytogenetic findings in pediatric adipose tumors: consistent rearrangement of chromosome 8 in lipoblastoma. Genes Chromosomes Cancer 1993;6:24–9.
- Hicks J, Dilley A, Patel D, et al. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol 2001;25:321–33.
Spindle cell and pleomorphic lipoma
- Enzinger FM, Harvey DA. Spindle cell lipoma. Cancer 1975;36:1852–9.
- Fletcher CDM, Martin-Bates E. Spindle cell lipoma: a clinicopathological study with some original observations. Histopathology 1987;11:803–17.
- Fanburgh-Smith JC, Devaney KO, Miettinen M, Weiss SW. Multiple spindle cell lipomas: a report of 7 familial and 11 nonfamilial cases. Am J Surg Pathol 1998;22:40–8.
- Schmookler BM, Enzinger FM. Pleomorphic lipoma: a benign tumor simulating liposarcoma: a clinicopathologic analysis of 48 cases. Cancer 1981;47:126–33.
- Griffin TD, Goldstein J, Johnson WC. Pleomorphic lipoma: case report and discussion of ‘atypical’ lipomatous tumors. J Cutan Pathol 1992;19:330–3.
- Billings SD, Folpe AL. Diagnostically challenging spindle cell lipomas: a report of 34 ‘low-fat’ and ‘fat-free’ variants. Am J Dermatopathol 2007;29:437–42.
- Zamecnik M, Michal M. Angiomatous spindle cell lipoma: report of three cases with immunohistochemical and ultrastructural study and reappraisal of former ‘pseudoangiomatous’ variant. Pathol Int 2007;57:26–31.
- Fletcher CD, Akerman M, Dal Cin P, et al. Correlation between clinicopathologic features and karyotype in lipomatous tumors. Am J Pathol 1996;148:623–30.
- Rubin BP, Fletcher CD. The cytogenetics of lipomatous tumours. Histopathology 1997;30:507–11.
- Mentzel T. Cutaneous lipomatous neoplasms. Semin Diagn Pathol 2001;18:250–7.
Atypical lipomatous tumour
- Azumi N, Curtis J, Kempson RL, Hendrickson MR. Atypical and malignant neoplasms showing lipomatous differentiation: a study of 111 cases. Am J Surg Pathol 1987;11:161–83.
- Evans HL, Soule EH, Winkelmann RK. Atypical lipoma, atypical intramuscular lipoma, and well-differentiated retroperitoneal liposarcoma: a reappraisal of 30 cases formerly classified as well-differentiated liposarcoma. Cancer 1979;43:574–84.
- Evans HL. Liposarcoma and atypical lipomatous tumors: a study of 66 cases followed for a minimum of 10 years. Surg Pathol 1988;1:41–54.
- Rosai J, Akerman M, Dal Cin P, et al. Combined morphologic and karyotypic study of 59 atypical lipomatous tumors: evaluation of their relationship and differential diagnosis with other adipose tissue tumors (a report of the CHAMP Study Group). Am J Surg Pathol 1996;20:1182–9.
- Evans HL. Atypical lipomatous tumour, its variants, and its combined forms: a study of 61 cases, with a minimum follow-up of 10 years. Am J Surg Pathol 2007;31:1–14.
- Weiss SW, Rao VK. Well-differentiated liposarcoma (atypical lipoma) of deep soft tissue of the extremities, retroperitoneum, and miscellaneous sites: a follow-up study of 92 cases with analysis of the incidence of dedifferentiation. Am J Surg Pathol 1992;16:1051–8.
- Binh MB, Sastre-Garau X, Gillou L, et al. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol 2005;29:1340–7.
- Sirvent N, Coindre JM, Maire G, et al. Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. Am J Surg Pathol 2007;31:1476–89.
- Paredes BE, Mentzel T. Atypical lipomatous tumors/“well-differentiated liposarcoma” of the skin clinically presenting as a skin tag: clinicopathologic, immunohistochemical, and molecular analysis of 2 cases. Am J Dermatopathol 2011;33:603-7.
Liposarcoma
- Dei Tos AP, Mentzel T, Fletcher CD. Primary liposarcoma of the skin: a rare neoplasm with unusual high- grade features. Am J Dermatopathol 1998;20:332–8.
Tumours of uncertain histogenesis
Acral fibromyxoma
- Fetsch JF, Laskin WB, Mietinnen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumour with a predilection for the fingers and toes. Hum Pathol 2001;32:704–14.
- Luzar B, Calonje E. Superficial acral fibromyxoma: clinicopathological study of 14 cases with emphasis on cellular variant. Histopathology 2009;54:375–7.
- Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol 2008;35:1020–6.
- Hollmann TJ, Bovee JVMG, Fletcher CDM. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol 2012;36:789–98.
- McNiff JM, Subtil A, Cowper SE, et al. Cellular digital fibromas: distinctive CD34-positive lesions that may mimic dermatofibrosarcoma protuberans. J Cutan Pathol 2005;32:413–18.
Superficial angiomyxoma
- Allen PW, Dymock RB, MacCormac WB. Superficial angiomyxoma with or without epithelial components: report of 30 tumors in 28 patients. Am J Surg Pathol 1988;12:519–30.
- Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with a tendency for recurrence. Am J Surg Pathol 1999;23:910–17.
- Carney JA, Headington JT, Wu SP. Cutaneous myxomas: a major component of the complex of myxomas, spotty pigmentation and endocrine overactivity. Arch Dermatol 1986;122:790–8.
Digital myxoma
- Johnson WC, Graham JH, Helwig EB. Cutaneous myxoid cyst: a clinicopathologic and histochemical study. JAMA 1965;191:15–20.
Dermal non-neural granular cell tumour
- LeBoit PE, Barr RJ, Burall S, et al. Primitive polypoid granular cell tumor and other cutaneous granular cell neoplasms of apparent non-neural origin. Am J Surg Pathol 1991;15:48–58.
- Lacroix-Triki M, Rochaix P, Marques B, et al. Granular cell tumors of the skin of nonneural origin: report of 8 cases. Ann Pathol 1999;19:94–8.
- Lazar AJ, Fletcher CD. Primitive nonneural granular cell tumors of the skin: clinicopathologic analysis of 13 cases. Am J Surg Pathol 2005;29:927–34.
- Chaudhry IH, Calonje E. Dermal non-neural granular cell tumour (so-called polypoid granular cell tumour): a distinctive entity further delineated in a clinicopathological study of 11 cases. Histopathology 2005;47:179–85.
Haemosiderotic fibrolipomatous tumour
- Marshall-Taylor C, Farnburg-Smith JC. Hemosiderotic fibrohistiocytic lipomatous lesion: ten cases of a previously undescribed fatty lesion of the foot/ankle. Mod Pathol 2000;13:1192–9.
- Browne TJ, Fletcher CD. Haemosiderotic fibrolipomatous tumour (so-called haemosiderotic fibrohistiocytic lipomatous tumour): analysis of 13 new cases in support of a distinct entity. Histopathology 2006;48:453–61.
- Kazakov DV, Sima R, Michal M. Hemosiderotic fibrohistiocytic lipomatous lesion: clinical correlation with venous stasis. Virchows Arch 2005;447:103–6.
- Folpe AL, Weiss SW. Pleomorphic hyalinizing angiectatic tumor: analysis of 41 cases supporting evolution from a distinctive precursor lesion. Am J Surg Pathol 2004;28:1417–25.
- Antonescu CR, Zhang L, Nielsen GP, et al. Consistent t(1;10) with rearrangements of TGFBR3 and MGEA5 in both myxoinflammatory fibroblastic sarcoma and hemosiderotic fibrolipomatous tumor. Genes Chromosomes Cancer 2011;50:757–64.
- Solomon DA, Antonescu CR, Link TM, et al. Hemosiderotic fibrolipomatous tumor, not an enterily benign entity. Am J Surg Pathol 2013;37:1627–30.
Perivascular epithelioid cell tumour
- Folpe AL, Goodman ZD, Ishak KG, et al. Clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres: a novel member of the perivascular epithelioid clear cell family of tumors with predilection for children and young adults. Am J Surg Pathol 2000;24:1239–46.
- Folpe AL, McKenney JK, Li Z, et al. Clear cell myomelanocytic tumor of the thigh: report of a unique case. Am J Surg Pathol 2002;26:809–12.
- Hornick JL, Fletcher CD. PEComa: what do we know so far? Histopathology 2006;48:75–82.
- Mentzel T, Reisshauer S, Rütten A, et al. Cutaneous clear cell myomelanocytic tumour: a new member of the growing family of perivascular epithelioid cell tumours (PEComas). Clinicopathological and immunohistochemical analysis of seven cases. Histopathology 2005;46:498–504.
- Llamas-Velasco M, Mentzel T, Requena L, et al. Cutaneous PEComa does not harbor TFE3 gene fusions: immunohistochemical and molecular study of 17 cases. Histopathology 2013;63:122–9.
Deep (‘aggressive’) angiomyxoma
- Steeper TA, Rosai J. Aggressive angiomyxoma of the female pelvis and peritoneum: report of nine cases of a distinctive type of gynecologic soft tissue neoplasm. Am J Surg Pathol 1983;7:463–75.
- Fetsch JF, Laskin WB, Lefkowitz M, et al. Aggressive angiomyxoma: a clinicopathologic study of 29 female patients. Cancer 1996;78:79–90.
- Sutton BJ, Laudadio J. Aggressive angiomyxoma. Arch Pathol Lab Med 2012;136:217–21.
- Carlinfante G, De Marco L, Mori M, et al. Aggressive angiomyxoma of the spermatic cord. Two unusual cases occurring in childhood. Pathol Res Pract 2001;197:139–44.
- Tsang WY, Chang JK, Lee KC, et al. Aggressive angiomyxoma: a report of four cases occurring in men. Am J Surg Pathol 1992;16:1059–65.
- Kazmierczek B, Wanshura S, Meyer-Bolte K, et al. Cytogenetic and molecular analysis of an aggressive angiomyxoma. Am J Pathol 1995;147:530–5.
- Zamecnik M, Skalova A, Michal M, et al. Aggressive angiomyxoma with multinucleated giant cells: a lesion mimicking liposarcoma. Am J Dermatopathol 2000;22:368–71.
- McCluggage WG, Patterson A, Maxwell P. Aggressive angiomyxoma of pelvic parts exhibits oestrogen and progesterone receptor positivity. J Clin Pathol 2000;53:603–5.
- Dreux N, Marty M, Chibon F, et al. Value and limitation of immunohistochemical expression of HMGA2 in mesenchymal tumors: about a series of 1052 cases. Mod Pathol 2010;23:1657–66.
- Siassi RM, Papadopoulos T, Matzel KE. Metastasizing aggressive angiomyxoma. N Engl J Med 1999;341:1772.
- Blandamura S, Cruz J, Faura Vergara S, et al. Aggressive angiomyxoma: a second case of metastasis with patient's death. Hum Pathol 2003;34:1072–4.
- Chan YM, Hon E, Ngai SW, et al. Aggressive angiomyxoma in females: is radical resection the only option? Acta Obstet Gynecol Scand 2000;79:216–20.
- Fine BA, Munoz AK, Litz CE, et al. Primary medical management of recurrent aggressive angiomyxoma of the vulva with a gonadotropin-releasing agonist. Gynecol Oncol 2001;81:120–2.
- McCluggage WG, Jamieson T, Dobbs SP, et al. Aggressive angiomyxoma of the vulva: dramatic response to gonatropin-releasing agonist therapy. Gynecol Oncol 2006;100:623–5.
Angiomatoid fibrous histiocytoma
- Enzinger FM. Angiomatoid malignant fibrous histiocytoma: a distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer 1979;44:2147–57.
- Costa MJ, Weiss SW. Angiomatoid malignant fibrous histiocytoma: a follow-up study of 108 cases with evaluation of possible predictors of outcome. Am J Surg Pathol 1990;14:1126–32.
- Fletcher CD. Angiomatoid ‘malignant fibrous histiocytoma’: an immunohistochemical study indicative of myoid differentiation. Hum Pathol 1991;22:563–8.
- Fanburgh-Smith JC, Miettinen M. Angiomatoid ‘malignant’ fibrous histiocytoma: a clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum Pathol 1999;30:1336–43.
- Billings SD, Folpe AL. Cutaneous and subcutaneous fibrohistiocytic tumors of intermediate malignancy: an update. Am J Dermatopathol 2004;26:141–55.
- Waters BL, Panagopoulos I, Allen EF. Genetic characterization of angiomatoid fibrous histiocytoma identifies fusion of the FUS and ATF-1 genes induced by a chromosomal translocation involving bands 12q13 and 16p11. Cancer Genet Cytogenet 2000;121:109–16.
- Antonescu CR, Dal Cin P, Nafa K, et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2007;46:1051–60.
- Bohman SL, Goldblum JR, Rubin BP, et al. Angiomatoid fibrous histiocytoma: an expansion of the clinical and histological spectrum. Pathology 2014;46:199–204.
Epithelioid sarcoma
- Enzinger F. Epithelioid sarcoma: a sarcoma simulating granuloma or carcinoma. Cancer 1970;26:1029–41.
- Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med 2009;133:814–19.
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol 2006;13:114–21.
- Santiago H, Feinerman LK, Lattes R. Epithelioid sarcoma: a clinical and pathologic study of 9 cases. Hum Pathol 1972;3:1706–10.
- Guillou L, Wadden C, Coindre JM, et al. Proximal-type epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features: clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol 1997;21:130–46.
- Hasegawa T, Matsuno Y, Shimoda T, et al. Proximal-type epithelioid sarcoma: a clinicopathologic study of 20 cases. Mod Pathol 2001;14:655–63.
- Livi L, Shah N, Paiar F, et al. Treatment of epithelioid sarcoma at the Royal Marsden Hospital. Sarcoma 2003;7:149–52.
- Arber DA, Kandalaft PL, Mehta P, et al. Vimentin-negative epithelioid sarcoma: the value of an immunohistochemical panel that includes CD34. Am J Surg Pathol 1993;17:302–7.
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 2009;33:542–50.
- Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer 2004;41:283–90.
- Lee MW, Jee KJ, Han SS, et al. Comparative genomic hybridization in epithelioid sarcoma. Br J Dermatol 2004;151:1054–9.
- Halling AC, Wollan PC, Pritchard DJ, et al. Epithelioid sarcoma: a clinicopathologic review of 55 cases. Mayo Clin Proc 1996;71:636–42.
- Prat J, Woodruff JM, Marcove RC. Epithelioid sarcoma: an analysis of 22 cases indicating the prognostic significance of vascular invasion and regional lymph node metastases. Cancer 1978;41:1472–87.
- Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol 1993;10:286–91.
- Baratti D, Pennacchioli E, Casali PG, et al. Epithelioid sarcoma: prognostic factors and survival in a series of patients treated at a single institution. Ann Surg Oncol 2007;14:3542–51.
- Rekhi B, Gorad BD, Chinoy RF. Clinicopathological features with outcomes of a series of conventional and proximal-type epithelioid sarcomas, diagnosed over a period of 10 years in a tertiarycancer hospital in India. Virchows Arch 2008;453:141–53.
- Gasparini P, Fachinetti F, Boeri M, et al. Prognostic determinants in epithelioid sarcoma. Eur J Cancer 2011;47:287–95.
- Chase DR, Enzinger FM. Epithelioid sarcoma: diagnosis, prognostic indicators and treatment. Am J Surg Pathol 1985;9:241–63.
Clear cell sarcoma
- Enzinger FM. Clear cell sarcoma of tendons and aponeuroses: an analysis of 21 cases. Cancer 1965;18:1163–76.
- Eckardt JJ, Pritchard DJ, Soule EH. Clear cell sarcoma: a clinicopathologic study of 27 cases. Cancer 1983;52:1482–8.
- Lucas DR, Nascimento AG, Sim FH. Clear cell sarcoma of soft tissues: Mayo Clinic experience with 35 cases. Am J Surg Pathol 1992;16:1197–204.
- Montgomery EA, Meis JM, Ramos AG, et al. Clear cell sarcoma of tendons and aponeurosis: a clinicopathologic study of 58 cases with analysis of prognostic factors. Int J Surg Pathol 1993;1:59–62.
- Meis-Kindblom JM. Clear cell sarcoma of tendon and aponeuroses: a historical perspective and tribute to the man behind the entity. Adv Anat Pathol 2006;13:286–92.
- Hantschke M, Mentzel T, Rutten A, et al. Cutaneous clear cell sarcoma: a clinicopathologic, immunohistochemical, and molecular analysis of 12 cases emphasizing its distincton from dermal melanoma. Am J Surg Pathol 2010;34:216–22.
- Falconieri G, Bacchi CE, Luzar B. Cutaneous clear cell sarcoma: report of three cases of a potentially understimated mimicker of spindle cell melanoma. Am J Dermatopathol 2012;34:619–25.
- Kiuru M, Hameed M, Busam K. Compound clear cell sarcoma misdiagnosed as a Spitz nevus. J Cutan Pathol 2013;40:950–4.
- Reeves BR, Fletcher CD, Gusterson BA. Translocation t(12;22)(q13;q13) is a nonrandom rearrangement in clear cell sarcoma. Cancer Genet Cytogenet 1992;64:101–3.
- Panagopoulos I, Mertens F, Isaksson M, et al. Absence of mutations of the BRAF gene in malignant melanoma of soft parts (clear cell sarcoma of tendons and aponeuroses). Cancer Genet Cytogenet 2005;156:74–6.
- Panagopoulos I, Mertens F, Dëbiec-Rychter M, et al. Molecular genetic characterization of the EWS/ATF1 fusion gene in clear cell sarcoma of tendons and aponeuroses. Int J Cancer 2002;99:560–7.
- Park BM, Jin SA, Choi YD, et al. Two cases of clear cell sarcoma with different clinical and genetic features: cutaneous type BRAF mutation and subcutaneous type with Kit mutation. Br J Dermatol 2013;169:1346–52.
- Clark MA, Johnson MB, Thway K, et al. Clear cell sarcoma (melanoma of soft parts): the Royal Marsden Hospital experience. Eur J Surg Oncol 2008;34:800–4.
- Coindre JM, Hostein I, Terrier P, et al. Diagnosis of clear cell sarcoma by real-time reverse transcriptase-polymerase chain reaction analysis of paraffin embedded tissues: clinicopathological and molecular analysis of 44 patients from the French sarcoma group. Cancer 2005;107:1055–64.
- Hocar O, Le Cesne A, Berissi S, et al. Clear cell sarcoma (malignant melanoma) of soft parts: a clinicopathologic study of 52 cases. Dermatol Res Pract 2012;2012:984096.