CHAPTER 141
Basal Cell Carcinoma
Vishal Madan1 and John T. Lear2
1Dermatology Centre, Salford Royal NHS Foundation Trust, Salford, UK
2Department of Dermatology, Manchester Royal Infi rmary, Manchester, UK
BASAL CELL CARCINOMA
Definition and nomenclature
Basal cell carcinoma (BCC) is the most common human cancer. Although there are distinct clinical and pathogenic differences between BCC and squamous cell carcinomas (SCC) (see Chapter 142), the lower malignant potential of these cancers allows them to be grouped together as non-melanoma skin cancer (NMSC), to distinguish them from ‘melanomas’ the third most common skin cancer; which have a much higher malignant potential. BCC represent approximately 74% of NMSC whilst SCC are less common at 23%.
Introduction and general description
Basal cell carcinomas are nearly as common as all other human cancers combined [1]. Despite difficulties in capturing and registering data leading to underreporting of BCC it is clear that there is a 10% per year rise in the incidence of BCC worldwide. Previous research in European countries estimates the level of underreporting to be 30–50% for BCCs which would indicate that the prevalence of BCC will equal that of all other cancers combined in the near future [2]. Furthermore, it is estimated that 40–50% of patients with a primary BCC will develop at least one or more BCC within 5 years. The estimated incidence of NMSC in the US is over 1000 000 cases per year with approximately 70–80% of these being BCC [3]. Despite their slow growing and indolent nature, BCC have a major impact on Western health economies [4].
Epidemiology
Incidence and prevalence
There is variability between skin cancer registries in terms of data capture, recording and processing of BCC data in the UK and some cancer registries may not record BCC data at all [5]. In 2010, there were 99 549 cases of NMSC registered in the UK with approximately 73% being BCC [6]. Between 2000–2002 and 2008–2010, the recorded incidence of BCC increased by around a third (36% in males and 32% in females) in England, Scotland, Northern Ireland and Ireland combined [7]. This rise may partly be attributed to better coding; however, it is still considered an underestimate as approximately 30–50% of BCC go unrecorded, though this may vary by registry [8].
The incidence of BCC has also been noted to increase in other European countries. An observational study from the Netherlands noted a quadrupling of incidence of BCC in 37 years from 1973 to 2009 [9]. The increase was more marked from 2002 until 2009 with an estimated annual percentage change of 6.8% for men and 7.9% for women. Whilst UV exposure may have a direct role, improved screening measures are also likely to contribute to the increased incidence of BCC [10].
Worldwide, the incidence for BCC varies widely with the highest rates in Australia (>1000/100 000 person-years) and the lowest rates in parts of Africa (<1/100 000 person-years). There are regional differences in the average incidence rates of BCC within countries. In England, the incidence of BCC is 76.21/100 000 person-years with the highest rates in the south-west of England (121.29/100 000 person-years) and the lowest rates in London (0.24/100 000 person-years) [11].
The age shift in the population has contributed to an overall increase in the total number of skin cancers as the incidence of BCC increases with advancing age. Indeed 80% of cases occur in people aged 60 years and over [12]. However, the largest annual increase in the numbers of BCC has been observed among those aged 30–49 years [11].
Compared to women, the incidence of BCC is higher in men. Patients diagnosed with a BCC are at a higher risk of a subsequent one. The 5-year cumulative risk of developing one or more subsequent BCCs is 29.2% [13, 14]. This risk is highest in the first 6 months after first BCC diagnosis. Males are at a 30% higher risk of developing multiple BCCs as compared with females. Patients aged 65–79 years have a more than 80% higher risk of developing subsequent BCC compared with patients younger than 50 years [13].
An individual's risk for the development of BCC depends upon genotypic, phenotypic and environmental factors. This risk is higher in residents of high ambient solar irradiance with markers of UV susceptibility such as fair skin colour, red hair and inability to tan [14].
Markers of chronic photodamage are positively associated with BCC [14]. Having more than 10 actinic keratoses confers a fivefold increase in the risk of BCC. Other factors, including solar elastosis, solar lentigines and telangiectasia have weaker but positive associations with BCC [15].
The geographical variability of NMSC incidence correlates with the amount of ambient sun irradiance and skin type, the reported incidence of NMSC in white populations being about five- to sevenfold higher in the US than in Europe, and about 50- to 100-fold higher in Australia compared to Europe [16]. Proximity to the equator is known to be a strong predictor of skin cancer risk and NMSC incidence within countries correlates with increasing proximity to the equator. There is a strong inverse association between geographical latitude and the risk of BCC [17].
UV radiation is the most important risk factor in the pathogenesis of NMSC; however, there appears to be a significantly greater effect of increasing sun-exposure on the risk of developing SCC than of BCC [18]. Unlike SCC, for which cumulative lifetime sun-exposure shows a strong dose–response relationship, for BCC, intermittent sun-exposure and exposure during childhood may be more important [19, 20].
Pathophysiology
Predisposing factors
Risk factors for the development of BCC are listed in Box 141.1. The primary risk factors for BCC development are UV light exposure and genetic predisposition. Other significant risk factors include Fitzpatrick skin types I and II, immunosuppression, advancing age, male sex, previous BCCs and chronic arsenic exposure [21].
Environmental factors
The mechanism of UV radiation induced mutagenesis has been extensively studied. Both UVB and UVA radiation are mutagenic; however, UVB radiation causes direct DNA and RNA damage by inducing covalent bond formation between adjacent pyrimidines. This leads to the generation of mutagenic photoproducts, e.g. cyclopyrimidine dimers (TT) and pyrimidine–pyrimidine (6-4) adducts [22].
UVA induces indirect DNA damage via formation of reactive oxygen species (ROS) by a ‘photo-oxidative stress’ mediated mechanism. The ROS interact with lipids, proteins and DNA to generate intermediates that combine with DNA to form adducts [23]. Several complex DNA repair systems are needed to prevent the deleterious effects of these premutagenic adducts [24]. UV-induced DNA damage normally results in DNA repair or apoptosis; only very rarely does it lead to tumourigenesis. Patients with xeroderma pigmentosum (XP), where DNA repair mechanisms are impaired, can have an up to 2000-fold increased risk of skin cancer [25].
Basal cell carcinomas have been shown to be the most mutated human cancers with the majority being UV signature mutations. BCC from anatomical regions with chronic UV exposure are associated with higher mutation rates than those with intermittent exposure. The greater mutational burden facilitates an increased antitumour immunological response which results in a less aggressive phenotype [26].
Genetics
Naevoid BCC syndrome (NBCCS; syn. Gorlin syndrome, MIM 109400; see p. 141.18) is an autosomal dominant disorder with distinct clinical features including palmoplantar pits, odontogenic cysts, calcification of the falx cerebri, skeletal abnormalities, medulloblastomas and multiple BCC. Insight into the molecular pathogenesis of BCC derives from the study of patients with NBCCS which results from germline mutations in PTCH1, a segment polarity gene (9q22.3) with tumour suppressor functions [26]. PTCH1 was originally identified in the fruitfly Drosophila melanogaster, and is known to play a critical role in vertebrate development. PTCH1 encodes a 12-pass putative transmembrane protein, Patched-1 (Ptch-1) which acts like the receptor of the diffusible morphogen protein sonic hedgehog (SHH) [27].
Ptch-1 acts as a tumour suppressor, repressing the G-protein-coupled receptor smoothened (Smo). Loss-of-function mutations of PTCH1 result in reduced suppression of Smo which activates the Gli family of transcription factors and promotes their importation into the nucleus resulting in sustained activation of target genes. Gli proteins are bound by Sufu, loss of which produces constitutive activation of Gli (Figure 141.1).
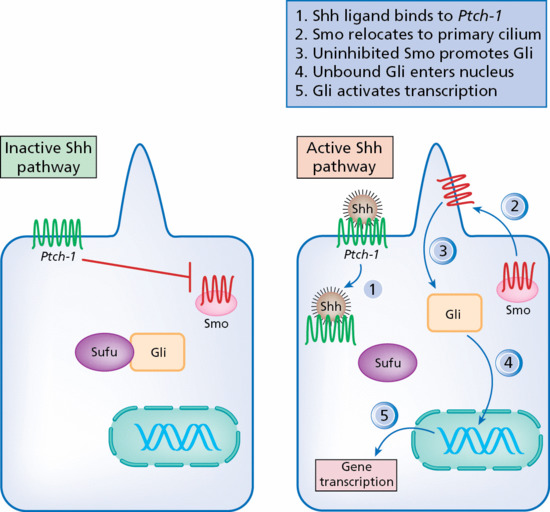
Figure 141.1 The sonic hedgehog (Shh) pathway in the pathogenesis of basal cell carcinoma.
Atypical protein kinase C iota/lambda (aPKC-ι/λ), a novel Gli regulator, and its polarity signalling partners colocalize at the centrosome and form a complex with missing-in-metastasis (MIM), a scaffolding protein that potentiates Hh signalling. Activated aPKC-ι/λ is upregulated in SMO-inhibitor-resistant tumours and targeting aPKC-ι/λ suppresses signalling and growth of resistant BCC cell lines [28].
Somatic PTCH1 mutations have a high frequency in familial BCC [29]. Sporadic BCC tumours also demonstrate loss of function of PTCH1 in 80–90% and SMO in 10–20% of cases [30].
The melanocortin-1 receptor (MC1R) gene variants ASIP and TYR are associated with fair skin, red hair and melanoma risk. Recent evidence suggests that they may also be independent risk factors for BCC [31]. Similarly, the role of the p53 tumour suppressor gene has been examined as 50% of BCC carry a p53 mutation [10, 32].
Both cytochrome P450s (CYP) and glutathione S-transferases (GST) catalyse the detoxication of the products of oxidative stress (e.g. lipid and DNA hydroperoxides). Polymorphism in GST and CYP2D6 (the gene encoding CYP) have been associated with BCC susceptibility (CYP2D6, GSTT1) and some allelic variants of CYP2D6 are associated with a multiple presentation phenotype of BCC [33, 34].
Unlike normal cells, most immortal and tumour cells exhibit significant levels of telomerase activity and show no net loss of telomere length during proliferation, a phenomenon that has also been observed in BCC [35].
The finding that β-catenin and MT1-MMP are increased in high-risk BCC tumour cells, may indicate that they may play an important role in locally invasive and highly destructive growth behaviour of high-risk BCCs [36].
Immunosuppression
In the general population, BCC are three to six times more common than SCC. This ratio reverses in immunosuppressed patients as SCC occur more frequently in transplant recipients [37, 38]. Caucasian transplant recipients have a 10–16-fold higher risk of developing a BCC as compared to the non-transplanted population. HIV-infected patients and those with chronic lymphocytic leukaemia also have more aggressive NMSC [39]. Chronic lymphocytic leukaemia patients are 14 times more likely to suffer recurrences of BCC following Mohs surgery [40].
Other risk factors
Exposure to inorganic arsenic through drinking water has been associated with the development of BCC [41]. Other risk factors for the development of BCC include UVB phototherapy [42] and ionizing radiation [43]. Besides NBCCS, multiple early-onset BCC may be seen in other syndromes [44] (Tables 141.1 and 141.2).
Table 141.1 Genetic syndromes with basal cell carcinoma as a prominent feature
| Condition | Inheritance | Gene(s) | Main additional cutaneous feature(s) | Main additional extracutaneous feature(s) | Main additional neoplasm(s) | Published diagnostic criteria | Available diagnostic test |
| Naevoid BCC syndrome | AD | PTCH1 | Epidermoid cysts, milia, palmoplantar pits | Calcifications of the falx cerebri, coarse face, jaw keratocysts, macrocephaly, spine, limb and oro-facial malformations | Medulloblastomas, rhabdomyosarcomas | Yes | Yes (DNA testing)a |
| Bazex–Dupré–Christol syndrome | XL | Unknown | Flexural hyperpigmentation, follicular atrophoderma, hypohidrosis, hypotrichosis, milia | None | Trichoepitheliomas | No | No |
| Rombo syndrome | AD | Unknown | Atrophoderma vermiculatum, erythematous lesions, hypotrichosis, milia, telangiectasias | None | Trichoepitheliomas | No | No |
| Xeroderma pigmentosum (see Chapter 78) | AR | XPA-XPG, POLH | Actinic keratosis/cheilitis, lentigines, atrophy, photosensitivity, telangiectasias, mottled hypo-/hyperpigmentation | DD/MR, eye anomalies, movement disorder, peripheral neuropathy, photophobia | Melanomas, squamous cell carcinomas, brain and visceral neoplasms | No | Yes |
| Generalized follicular basaloid hamartoma syndrome | AD | Unknown | Comedones, hypohidrosis, hypotrichosis, milia | None | None | No | No |
| Happle–Tinschert syndrome (see Chapter 75) | Sporadic | Unknown | Lesions over Blaschko's lines, atrophy, hyperpigmentation, teeth abnormalities | Body asymmetry, DD/MR, spine and limb malformations | Brain tumours | No | No |
Adapted from Castori et al. 2012 [44] with permission from the copyright holder John Libbey Eurotext.
aIn the presence of developmental delay, craniosynostosis, hydrocephalus, overgrowth and seizures, consider the 9q22.3 microdeletion syndrome.
AD, autosomal dominant; AR, autosomal recessive; DD/MR, developmental delay/mental retardation; XL, X-linked.
Table 141.2 Genetic syndromes with basal cell carcinoma as an ancillary feature
| Condition | Inheritance | Gene(s) | Main additional cutaneous features | Main additional extracutaneous features | Main additional neoplasms | Published diagnostic criteria | Available diagnostic test |
| Genomic instability syndromes | |||||||
| Bloom syndrome (see Chapter 79) | AR | RECQL3 | Café-au-lait spots, photosensitivity, telangiectasias | Diabetes, gastro-oesophageal reflux, growth delay, myelodysplasia, recurrent/chronic infections | Leukaemias, lymphomas, epithelial cancers | No | Yes |
| Werner syndrome (see Chapter 72) | AD (atypical), AR | LMNA (atypical), RECQL2 | Atrophy, calcifications, painful callosities, premature greying, scleroderma | Cataract, diabetes, hypogonadism, peripheral neuropathy, premature atherosclerosis and osteoporosis, typical face | Melanomas, (osteo)sarcomas | No | Yes (DNA testing) |
| Rothmund–Thomson syndrome (see Chapter 77) | AR | RECQL4 | Hypotrichosis, nail dystrophy, palmoplantar keratodema, photosensitivity, poikiloderma, teeth anomalies | Cataract, growth delay, myelodysplasia, osteoporosis, patellar hypoplasia, radial ray defects | Bowen disease, osteosarcomas, squamous cell carcinomas | Yes | Yes (DNA testing) |
| Muir–Torre syndrome (see Chapter 142) | AD | MSH2, MLH1 | None | None | Keratoacanthomas, gastrointestinal, urinary and endometrial neoplasms | No | Yes |
| Disorders of the folliculosebaceous unit | |||||||
| CYLD-associated syndromes (see Chapter 138) | AD | CYLD | None | None | Cylindromas, trichoepitheliomas, spiroadenomas, adenomas of salivary glands | No | Yes (DNA testing) |
| Schöpf–Schulz–Passarge syndrome (see Chapter 138) | AR | WNT10A | Eyelid cysts, hypohidrosis, hypotrichosis, nail dystrophy, milia, palmoplantar keratoderma, teeth anomalies, telangiectasias | None | Apocrine cystoadenomas, eccrine syringofibroadenomas, eccrine poromas, squamous cell carcinomas | No | Yes (DNA testing) |
| Cowden syndrome (see Chapter 138) | AD | PTEN | Acral pseudoverrucoid lesions, acrochordons, fibromas, lipomas, oral and lip papillomas, vascular malformations | Breast fibrocystic disease, Lhermitte–Duclos disease, macrocephaly, thyroid dysfunction | Breast cancer, gastrointestinal polyps, ovarian cancer, thyroid cancer, trichilemmomas, uterine cancer | Yes | Yes (DNA testing) |
| Syndromes with Immunodeficiency | |||||||
| Cartilage–hair hypoplasia (see Chapters 67, 68, 72) | AR | RMRP | Hypotrichosis/alopecia | Disproportionately short stature, metaphyseal dysplasia, peripheral joint laxity, pudgy extremities | Non-Hodgkin lymphomas | No | Yes (DNA testing) |
| Epidermodysplasia verruciformis (see Chapter 25) | AR | EVER1, EVER2 | Polymorphic, erythematous lesions, wart-like/flat papules | None | Bowen disease, squamous cell carcinomas | No | Yes (DNA testing) |
| Disorders of melanin biosynthesis | |||||||
| Oculocutaneous albinism (see Chapters 65, 70) | AR | MATP, P, TYR, TYRP1 | Hair, skin and iris hypopigmentation | Photophobia, reduced colour vision, strabismus, visual loss | Melanomas, squamous cell carcinomas | No | Yes |
| Hermansky–Pudlak syndrome (see Chapter 70) | AR | HPS1-HPS8 | Hair, skin and iris hypopigmentation | Bleeding diathesis, granulomatous colitis, photophobia, pulmonary fibrosis, reduced colour vision, strabismus, visual loss | Melanomas, squamous cell carcinomas | No | Yes |
Adapted from Castori et al. 2012 [44] with permission from the copyright holder John Libbey Eurotext.
AD, autosomal dominant; AR, autosomal recessive.
Pathology
The tumour cells resemble those of the basal layer of the epidermis and the matrix cells of the appendages, in the relatively small amount of cytoplasm they possess and in their ability to interact with the dermis adjacent to them. Their nuclei are compact, rather darkly staining and closely set. Their cytoplasm stains poorly and the cell margins are rather indistinct. Adjacent cells are connected by bridges. The sparsity of keratin fibrils gives these connections a different appearance from the ‘prickles’ of the Malpighian layer, but the presence of desmosomes and tonofibrils has been shown by electron microscopy. The interaction with the dermis, which is one of the principal functions of the normal epidermal basal cell, produces the characteristic marginal palisade of tumour cells and the well-organized stroma that surrounds it. The dependence of the tumour on its stroma has been shown by transplantation experiments [45]. The cells within the palisade usually show little evidence of organization or differentiation. Mitotic figures may be frequent, and it is speculated that the combination of large numbers of mitoses and a slow growth rate result from a high rate of apoptosis. Data on cell kinetics indicate that a considerable proportion of cells in the tumour die fairly rapidly [46]. In some tumours, the cells may become acantholytic and amyloid may be identified [47].
In early lesions, the tumour buds can be seen arising from the epidermis. In very small lesions, multiple buds have been seen. These very soon become confluent, and the three-dimensional examination of superficial BCC shows a coherent margin of tumour with a reticular pattern of growth along the interpapillary ridges and larger, more discrete masses centrally [48]. As the tumour progresses, the masses extend into the dermis, and may separate from each other and from their point of origin. Growth in one area may be accompanied by involution of the tumour in nearby areas leaving an atrophic epidermis. A common site of origin in humans and in the experimental tumours of the rat is the junction between a pilosebaceous duct and the epidermis. From here, the tumour may extend along the epidermis and down the duct. It is difficult to prove a purely adnexal origin for BCC, but some lesions behave as though this were so. In all considerations about the origin of the tumour, one must remember that the tumour can either sever its connection with epithelial structures or establish a secondary connection to structures to which it has grown close.
The variability of the natural history of BCCs is reflected in its pattern of growth. Most tumours are composed of rounded expansile islands. These throw out small buds that grow in the same way to produce multilobular masses with thin strands or septa of fibrous tissue penetrating them [48, 49]. In some regions, a limited capacity to grow around and enclose adjacent connective tissue may be associated with a reticular or cystic pattern of growth. The capacity to invade in thin strands is often accompanied by an excessive and almost exclusive fibroblastic response, in contrast with the lymphocytic response around the expansile masses. Invasive strands may spread for long distances along nerve sheaths. BCC is truly invasive in only a small proportion of cases. In these, the tumours show no tendency to grow as rounded masses, have no palisade or organized stroma, and penetrate the dermis and deeper structures, destroying them as they go. Such tumours are almost always ulcerated, usually from an early stage. In the less invasive tumour, ulceration occurs when the epidermis is replaced by the tumour. An eroded vegetating type of growth is rather uncommon.
Most BCCs provoke a round-cell inflammatory reaction of some degree. It increases in extent with ulceration and is often conspicuous in the papillary body, with superficial patterns of growth. Mast cells are often present in numbers among the fibroblasts of the stroma, and Langerhans cells have been demonstrated within and near the tumour. This infiltrate has recently been correlated with the aggressive nature of the tumour [50].
The diversity of histological patterns of BCC is caused in part by features that have no direct bearing on the clinical course of the tumour. Not infrequently, melanocytes proliferate within the tumour. The melanin they produce causes the tumour to be pigmented, and numerous melanophages collect in the stroma, and sometimes in cystic cavities. Mucin is commonly found in the stroma, particularly at the margin of the tumour, and may be encysted within it. Cystic cavities also form when the centrally placed cells undergo necrosis. There is no evidence that such cavities represent glandular differentiation. Evidence of true sebaceous or sweat gland differentiation has been seen, but is very rare. Within some tumours there are strands of fusiform cells with more abundant eosinophilic cytoplasm, which may form whorls or keratinizing cysts, and which probably represent rudimentary differentiation towards hair roots. Citrulline can be demonstrated as a histochemical confirmation in such cases but does not help in the sometimes difficult separation from trichoepithelioma. Histochemical and electron microscopy investigations show little evidence of differentiation of the tumour cells. However, in vitro culture of tumour cells from nodular tumours produces evidence of keratinization after 30 days, suggesting that the cells possess the biochemical mechanisms for keratinization but that some factor, possibly dermal in origin, inhibits them.
Clinical and histological differentiation between BCC and trichoepitheliomas (see Chapter 138) can be difficult. Whilst the role of immunohistochemistry to differentiate the two tumours is evolving, expression of certain markers such as podoplanin (D2-40, a lymphatic endothelial surface marker) can be useful in in this context. Trichoepitheliomas show weak and focal positivity to D2-40 in 95% of cases as opposed to BCC which show weak and focal positivity in 22% of cases [51]. Other markers which may be useful in differentiating BCC from trichoepitheliomas include CD-10, Bcl-2, cytokeratin 20 and cytokeratin 15 [52].
Histopathological patterns of BCC include superficial, nodular, infiltrative, micronodular and pigmented types (Figure 141.2) [53].
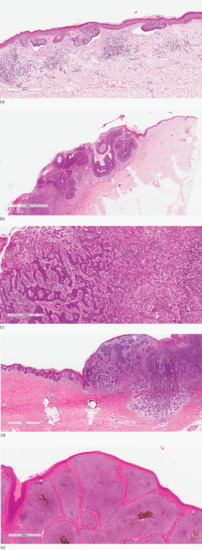
Figure 141.2 Histopathological patterns of basal cell carcinoma. (a) Superficial. Skin with small islands and downgrowths of basaloid cells arising from multiple points from the epidermis with artefactual clefting. Superficial basal cell carcinoma is usually confined to the papillary dermis. (b) Nodular. Ulcerated skin with large solid islands of basaloid cells, some showing central cystic degeneration. Peripheral palisade is present and artefactual clefting focally. (c) Infiltrative. Irregular elongated strands and dermal basaloid islands embedded in a loose desmoplastic stroma with chronic inflammation. (d) Micronodular. A basaloid tumour with numerous small nodules in the deep dermis displaying a degree of infiltration at the edge compared with larger nests in the upper dermis which represent a nodular (solid) pattern. (e) Pigmented. Large solid islands of basaloid cells with central pigmentation. Peripheral palisade is a prominent feature. This lesion may clinically be mistaken for a melanocytic proliferation.
Superficial basal cell carcinoma. Proliferating atypical basaloid cells form an axis parallel to the epidermal surface. Typical features include palisading basal cells which form slit-like spaces containing alcian blue-positive mesenchymal mucoid material – a presumed product of the stromal cells. The atypical basaloid cells rarely show mitoses or apoptotic cells but may colonize the hair follicles and rarely the eccrine adnexal structures.
A band-like lymphoid infiltrate may be present and this should prompt a careful search through multiple levels looking for foci of atypical basaloid cells in skin biopsies from suspected superficial BCC.
Nodular basal cell carcinoma presents histologically as nests of basaloid cells in either the papillary or reticular dermis where solar elastosis may be evident. The nests are separated from the stroma by a slit-like retraction. The surrounding stroma shows myxoid change, and calcification may be seen in discrete islands of tumour or in adjacent stroma. Like in the superficial BCC, which may be coexistent in a third of the cases, mitoses and individual cell necrosis are uncommon. Processing of the specimen may result in drop out of the tumour nests from the stroma which may lead to ‘retraction artefact’.
Micronodular basal cell carcinoma. The tumour nests are smaller than those in nodular BCC and more widely and asymmetrically dispersed in the dermis and/or subcutis. The associated stromal proliferation is similar to that seen in infiltrative BCC. Another distinguishing feature of micronodular BCC is that the retraction spaces are uncommon and the surrounding stroma shows either a myxoid or collagenized morphology.
Morpheaform basal cell carcinoma also known as sclerosing BCC display one to two cells thick columns of basaloid cells enmeshed in a dense collagenous stroma. Such sharply angulated cords of basaloid cells show marked cell necrosis and mitotic activity. Stromal fibroplasia and fibrosis of the tumour cords is commonly seen. Invasion of the deep dermis and subcutis is another feature of morphoeaform BCC.
Infiltrative basal cell carcinoma typically show elongated tumour cell strands, five to eight cells in thickness which present histologically as irregularly sized and shaped nests which are poorly circumscribed and may show invasion of the subcutis and adjacent muscular and other structures. Like the morpheaform variant, these nests show sharp angulation of their peripheral contours with occasional foci of slit-like retraction, and frequent mitotic activity and individual cell necrosis. The stroma is frequently fibrotic with plump proplastic stromal fibroblasts.
Basosquamous or metatypical basal cell carcinoma are tumours that on pathological study appear to have features of both BCC and SCC. The biological significance is that this pathological pattern is associated with a significantly higher incidence of metastatic spread [53, 54]. The pattern in these lesions is of small aggregates of cells lacking classic palisading and embedded in dense and profuse fibrous stroma. The cells are larger with a larger paler nucleus than in the classic BCC and have a more eosinophilic cytoplasm.
Clinical features
Unlike SCC, there is no recognized premalignant stage of BCC. The typical BCC runs a slow progressive course of peripheral extension, which produces the thread-like margin, the nodule with a central depression or the expanding rodent ulcer. Some tumours grow at so slow a rate that they are, for all practical purposes, benign. This is true for many of the superficial lesions and some of the nodular cystic lesions. There may be spontaneous fluctuation in size, and areas of scarring can be found within many superficial tumours. Rapid growth is unusual.
Approximately 80% of all BCC occur on the head and neck, and clinical diagnosis is relatively straightforward [55]. Early BCCs are usually small, translucent or pearly, with raised telangiectatic edges. However, the presentation may be varied and small lesions can be lichenoid or keratotic, excoriated or ulcerated.
More advanced lesions can present as classical rodent ulcer with an indurated edge and an ulcerated centre. Other common subtypes of BCC are nodular or cystic, superficial, morphoeic and pigmented BCC. Pigment, when present, is usually unevenly distributed throughout the tumour.
Certain features will divide BCC into high and low risk which may influence the choice of management (see Table 141.4).
Clinical variants
Nodular basal cell carcinoma (Figure 141.3) is the commonest subtype of BCC and usually presents on the head and neck. The surface contour of this lesion usually becomes more irregular as the lesion grows. The degree of vascularity varies. There may be surface telangiectasia over a flesh-coloured mass or the tumour may be pink or red in colour. Some or all of the component nodules may have cystic centres, which add to the translucent appearance; the cystic parts may be more deeply pigmented than the peripheral parts. Pigmented as well as nodular BCC may cause diagnostic confusion with melanoma (Figure 141.4).
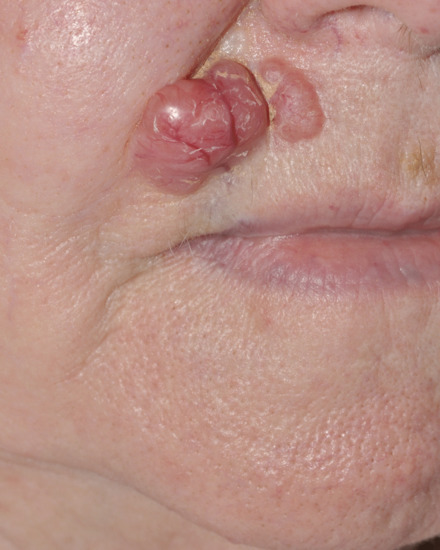
Figure 141.3 Nodular basal cell carcinoma.
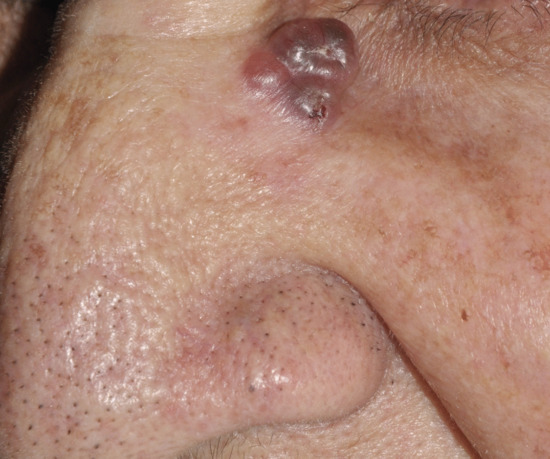
Figure 141.4 Pigmented basal cell carcinoma.
Superficial basal cell carcinomas (Figure 141.5) are less common and predominantly present on the trunk. These are bounded by a slightly raised thread-like margin or a ‘whipcord’ edge, which is irregular in outline and may be deficient at part of the circumference. The epidermis covering the central zone is usually atrophic and may be scaly. This, combined with an increased vascularity, gives a resemblance to Paget disease of the nipple. There may be a series of thickened papular islands of growth within the margin, and these may be crusted or eroded. Patients with initially truncal BCC of superficial histology demonstrate the highest rate of increasing BCC numbers [56]. Superficial BCCs are often pigmented and can sometimes be difficult to differentiate from psoriasis, discoid eczema or Bowen disease (see Chapter 142).
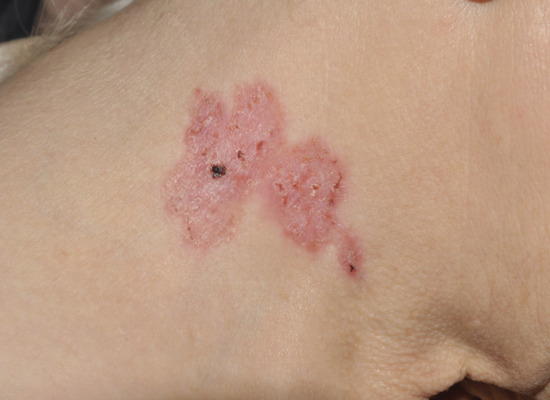
Figure 141.5 Superficial basal cell carcinoma.
Morphoeic basal cell carcinomas (Figure 141.6) are also known as sclerodermiform, so named because dense fibrosis of the stroma produces a thickened plaque rather than a tumour. Morphoeic BCCs account for 5% of all BCC, have ill-defined borders, can be difficult to diagnose clinically and often present late. The exact margin of the lesion is impossible to define, but palpation reveals a firm skin texture that extends irregularly beyond the visible changes. The surface is smooth and may be slightly raised above, or sometimes slightly depressed below, the normal level. The colour is yellowish and has been compared with old ivory. Ulceration is uncommon and only very superficial when it does occur. Many patients, and doctors, may take little notice of this type of BCC until its slow extension produces a sizeable lesion.
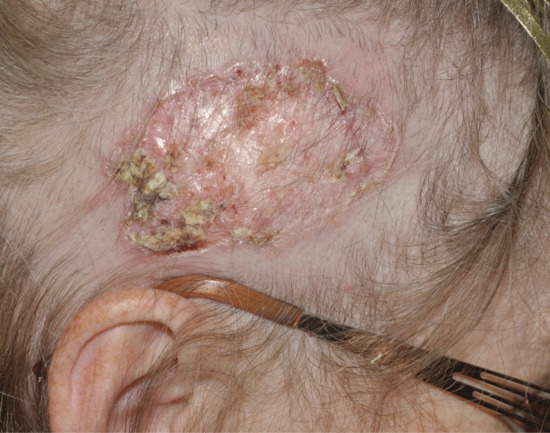
Figure 141.6 Morphoeic basal cell carcinoma.
Ulcerated basal cell carcinomas (Figure 141.7) may start as a small macule or papule but with expansion of the thread-like margins, the attenuated surface ulcerates. The edge is usually indurated and attempts to treat with topical antibiotics are unsuccessful. Atypical ulcerated BCC also has an indurated edge and base but no thread-like margin. The edge is usually raised above the normal level but in some areas, particularly in the naso-labial furrows, it may be flush with the surface. The floor of the ulcer is depressed below the skin surface, fleshy in appearance and not very vascular. Such an ulcerated lesion may have begun as a nodule, but more frequently it is crusted or eroded from an early stage of its evolution. If left, the tumour and its following ulcer may spread deeply and cause great destruction, especially around the eye, nose or ear. There may be wide extension in the periorbital tissues; the bones of the face; the skull; and even the meninges may be invaded, and advanced cases amply justify the title ‘ulcus terebrans’ (penetrating ulcer). Between 10% and 40% of BCCs contain a mixed pattern of two or more of these subtypes, highlighting the need for a clinicopathological diagnosis [57, 58].
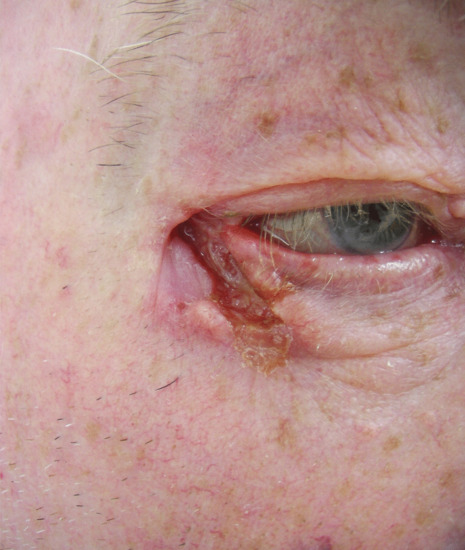
Figure 141.7 Ulcerated basal cell carcinoma.
Fibroepithelial basal cell carcinoma (premalignant fibroepithelial tumour) is now classed as a BCC variant (see later).
Advanced and metastatic basal cell carcinomas (Figure 141.8) are a manifestation of prolonged neglect and are encountered at a rate of 1–2% [59]. Mutilation of the face or scalp, with destruction of the nose or eye and exposure of the paranasal sinuses or the skull, dura or brain may eventually result in death [60]. Progression of advanced BCC to a metastatic form is extremely rare (0.0028–0.55% of BCC) [61]. Authentic cases of bloodstream metastasis are on record in which, for example, deposits in the viscera or spinal column have caused the presenting symptoms of the terminal illness. Other cases have spread via the lymphatics to the regional lymph nodes before disseminating. Unusually, fragments of tumour cells and stroma may be inhaled and become implanted in the lungs [62]. Metastatic BCC usually have a basosquamous (metatypical) histological subtype [54].
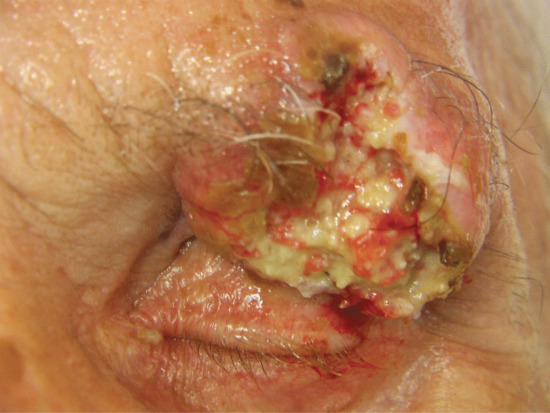
Figure 141.8 Advanced basal cell carcinoma.
Differential diagnosis (Table 141.3)
It may be difficult to differentiate nodular BCC from melanocytic naevus (especially when pigmented), molluscum contagiosum or sebaceous hyperplasia on clinical evaluation alone. Naevi can be distinguished if hairs grow from the surface, and in molluscum contagiosum and sebaceous hyperplasia there is a central keratin-filled pit.
Table 141.3 Differential diagnosis of basal cell carcinoma
| Diagnosis | Distinguishing clinical features | Image |
| Naevi | Lack of rolled telangiectatic edge. May be hairy | 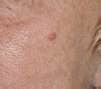 |
| Sebaceous hyperplasia | Central depression, usually multiple on a background of sebaceous quality skin |  |
| Molluscum contagiosum | Central punctum is characteristic | 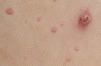 |
| Keratoacanthoma | A central keratin plug is typical, history of evolution followed by involution | 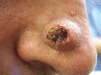 |
| Squamous cell carcinoma | Shorter history and indurated base (better felt on palpation) | 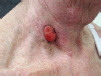 |
| Bowen disease | Scaly edge, lack of thread-like margin | 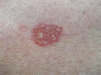 |
| Psoriasis | Silvery scales and response to topical antipsoriatic therapy | 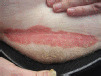 |
| Eczema | History of eczema and response to topical eczema therapy | 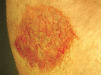 |
| Melanoma | Grey-black discoloration and pigment spill. Shorter history |  |
| Chondrodermatitis nodularis helicis | Usually tender | |
| Viral warts | Keratotic ‘warty’ surface |
Scaling or crusting on the surface of BCC can make differentiation from warts, keratoacanthoma, SCC or molluscum contagiosum difficult. In all cases, the debris should be removed, and this is easily done in BCC. The friable, relatively avascular tissue beneath is characteristic, and if fragments are removed and smeared on a slide the diagnosis can be confirmed by cytology.
Darkly pigmented ulcerated tumours are occasionally confused with malignant melanoma. The margin of BCC is usually rolled, telangiectatic and multinodular, and there is no pigmented halo. The colour tends to be more definitely brown, in contrast with the dusky greyish brown of malignant melanoma. Dermoscopy can be useful here. If diagnostic confusion exists, the lesion is better excised in entirety if possible.
Superficial BCC can be similar morphologically to patches of eczema, psoriasis or Bowen disease (see Table 141.1 in Chapter 142). A thread-like margin on stretching the edge will reveal the true diagnosis. Careful inspection will almost always rule out eczema or psoriasis, which the patient's history will have also made unlikely. There are some cases, however, where distinction from Bowen disease can be made only after biopsy. The consistency of a morphoeic BCC may resemble morphoea; the outline is usually less sharp and the evolution more gradual and relentless.
Chondrodermatitis nodularis helicis (see Chapter 108) is usually tender. Several BCC may not display the typical clinical features. Surface ulceration or excoriation may hinder accurate diagnosis. Clinical diagnostic accuracy in the diagnosis of BCC can be surprisingly poor. One study reported a diagnostic accuracy rate of 70% for academic dermatologists, 65% for dermatologists in private practice and 64% for residents [63].
Investigations
The diagnosis of BCC is primarily clinical. In clinically challenging cases, a biopsy is required. In nodular BCC, this may be a shave biopsy if the aim is to make a clinical diagnosis. A punch biopsy may be more appropriate in identifying morphoeic lesions. The accuracy of punch biopsy in identifying BCC subtypes in mixed tumours is poor [64].
Dermoscopy using cross-polarized light is a useful clinical adjunct as identification of features such as white and grey-brown structureless areas, blue-grey ovoid nests, blue-grey globules, maple-leaf like areas, spoke-wheel areas, concentric structures, ulcerations and blue-grey dots may help differentiate BCC from Bowen disease and melanomas and also help in subtyping the tumour [65] (Figure 141.9). The most frequently observed vasculature included atypical red vessels, arborizing, comma and telangiectactic vessels, short fine telangiectasias and vascular blush.
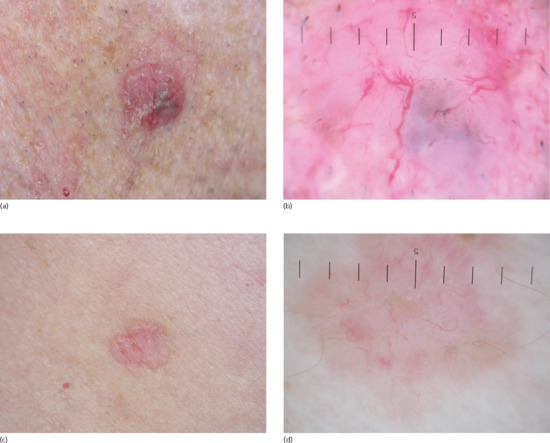
Figure 141.9 Dermoscopic images of basal cell carcinoma. (a) Nodular basal cell carcinoma. (b) Corresponding dermoscopic image showing white and grey-brown structureless areas, blue-grey globules and and telangiectactic vessels. (c) Superficial basal cell carcinoma. (d) Corresponding dermoscopic image: spoke-wheel areas, concentric structures, arborizing, comma and telangiectactic vessels.
Other techniques for the non-invasive diagnosis of BCC include high-frequency ultrasound, optical coherence tomography using infrared light and in vivo confocal microscopy [66].
High-definition optical coherence tomography (HD-OCT) is a high-resolution imaging tool, with micrometre resolution in both transversal and axial directions, enabling visualization of individual cells up to a depth of around 570 μm [67]. Features for four different BCC subtypes were described in both transverse and axial directions. Subepidermal or intradermal aggregations of cells forming islands are surrounded by a less refractile border. This corresponds with palisading and peritumoral mucin production on histology (Figure 141.10). Although still in the experimental stages and not yet widely used in clinics, both optical coherence tomography and confocal microscopy may have a potential role in presurgical margin assessment and non-histological assessment of BCC, respectively [66, 67, 68].
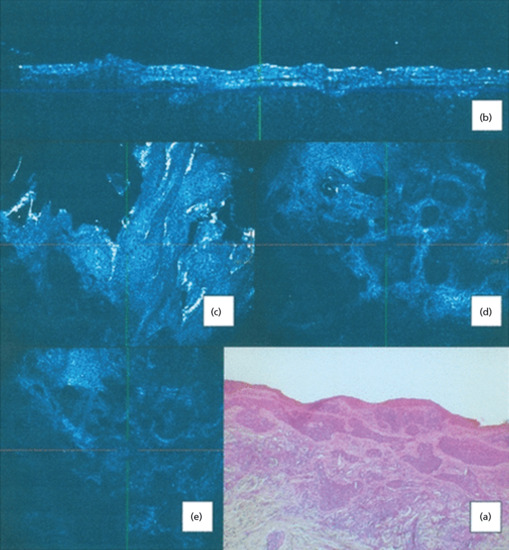
Figure 141.10 Nodular basal cell carcinoma (BCC). (a) H&E stained histological section shows actinic changes in overlying epidermis, islands of basaloid cells with palisading. These islands are surrounded by an abundant mucinous stroma with separation of the tumour cells from the surrounding stroma (clefting). Prominent peritumoral stromal reaction. (b) In high-definition optical coherence tomography (HDOCT) slice mode, lobular patterns of abnormal architecture are seen with dilated blood vessels and high-reflective margins. (c) In HD-OCT en face mode, the overlying epidermis presents an architectural disarray with parakeratosis, pleomorphism and some degree of keratinocyte atypia with variably sized nuclei at the basal cell layer. (d) In en face HDOCT imaging, the nodular BCC islands of tumour cells are noted with intervening areas of low refractility. Inside this low-refractile zone, tumour cells are more refractile. Abundant blood vessels are seen juxtaposed to the islands. (e) En face HDOCT imaging shows a variable refractile stroma with inflammatory cells. (Reproduced from Boone et al. 2012 [67], copyright John Wiley.)
In confocal microscopy, cellular morphology can be observed in real time, in thin optical sections and high-resolution individual images which are stitched together to create mosaics that display low magnification of large areas of tissue as required for Mohs surgery (Figure 141.11) [68]. Contrast agents such as acridine orange in fluorescence and acetic acid in reflectance confocal microscopy have been used in ex vivo imaging to enhance nuclear contrast.
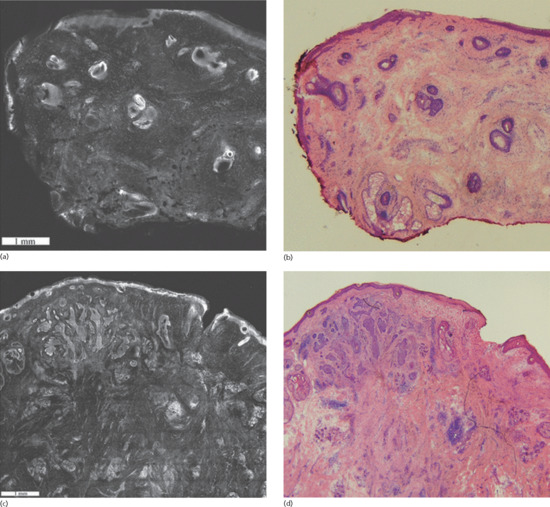
Figure 141.11 Fluorescent confocal submosaics (a,c) and the corresponding H&E stained Mohs frozen sections (b,d) at 4× magnification. The submosaic and frozen section of a negative Mohs stage shows no residual basal cell carcinoma (BCC) (a,b); positive Mohs stage shows residual BCC within the upper dermis (c,d). There is good correlation between the mosaics and the corresponding frozen sections with respect to the overall size, shape, location and morphology of benign and malignant skin structures. (Reproduced from Karen et al. 2009 [98], copyright John Wiley.)
Management
Both tumour (clinical and histological nature, size and site) and patient factors determine the choice of treatment of BCC. Other factors dictating choice of treatment include local experience and availability of treatments, which may indirectly depend upon the cost. Treatment options for BCC have been systematically reviewed and guidelines for the management of BCC have been published by national and international bodies [69, 70, 71]. An approach to the treatment of a patient with a BCC is illustrated in Figure 141.12.
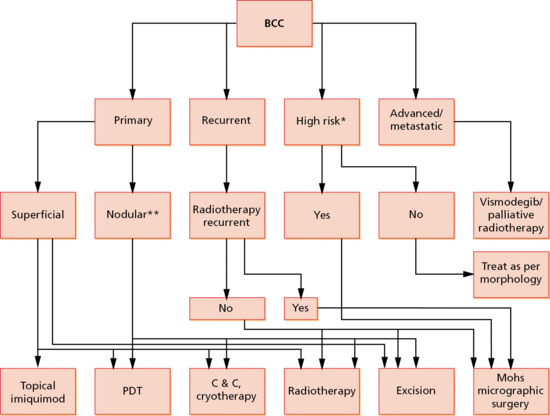
Figure 141.12 An algorithm for the treatment of basal cell carcinomas (BCC). *High-risk BCCs include morphoeic lesions/infiltrative and micronodular growth pattern/greater than 5 cm in size/centrofacial and ear location/perineural and perivascular invasion on histology/host immunosuppression. **Only small nodular BCC should be treated with photodynamic therapy (PDT). C&C, curettage and cautery.
As in any malignancy, the primary aim of treatment is complete removal or destruction of the BCC. Achieving a good and acceptable cosmetic outcome is an important secondary goal. For low-risk superficial and small nodular BCC, non-destructive therapies such as topical treatments and photodynamic therapy have a high success rate and this is usually associated with an excellent cosmetic outcome. Conversely, high-risk and recurrent BCC require histological examination of the excision margins, which is not permitted by all the available surgical techniques.
Treatments for BCC can be classified as medical or surgical [21].
Medical treatments
Imiquimod
Imiquimod stimulates toll-like receptor 7 and 8 expressed on dendritic cells and monocytes, leading to increased production of cytokines and chemokines. These in turn promote both Th1 innate and adaptive cell-mediated immune responses which are crucial in the recognition and destruction of the tumour cells [72].
Topical 5% imiquimod cream, applied five times per week over 6 weeks is effective in clearing 69–100% of superficial BCC and 42–76% of nodular BCC [21, 73]. There is a lower response rate in nodular BCC and thicker superficial BCC [74, 75]. One study suggests that superficial BCC <0.4 mm may be better suited to treatment with imiquimod as such lesions are most likely to respond [75]. When compared to photodynamic therapy, imiquimod may be more effective in treating superficial BCC [76].
Imiquimod is licensed for use in superficial BCC in immunocompetent adults, with a maximum tumour diameter of 2.0 cm.
Patients may experience flu-like symptoms such as fever, chills, body aches and lymphadenopathy. This may be severe enough to warrant discontinuation of treatment.
Erythema, pruritus, erosion, ulceration, dyspigmentation and scabbing are commonly observed side effects of topical imiquimod therapy (Figure 141.13).
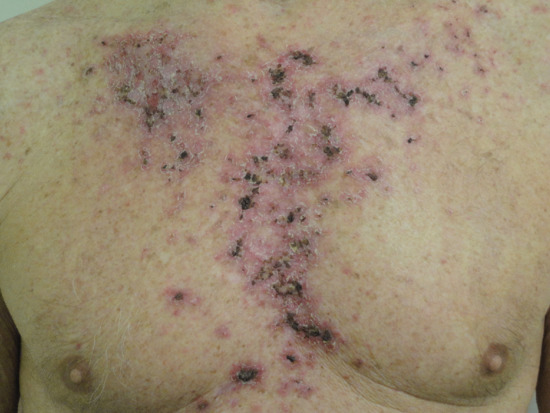
Figure 141.13 Skin reaction to topical imiquimod therapy for superficial basal cell carcinoma.
5-Fluorouracil
Topical 5-fluorouracil (5-FU) disrupts DNA and RNA synthesis by inhibiting the enzyme thymidylate synthetase. This prevents purine and pyrimidine from becoming incorporated into DNA during the S-phase of the cell cycle [77]. Despite high histological cure rates and good cosmesis with topical 5-FU treatment of small and superficial BCC, it is rarely used for this indication [78]. Like imiquimod, 5-FU is not recommended for the treatment of large, nodular and high-risk BCC, and local irritation and skin reaction resulting in erythema, swelling, desquamation and tenderness are common post treatment.
Photodynamic therapy
Photodynamic therapy relies on the production of ROS, which results in necrosis or apoptosis of tumour cells. The role of photodynamic therapy in the treatment of BCC is discussed in Chapter 22.
Intralesional interferon α-2b
The need for treatment to be delivered in multiple sessions and the high associated costs limit the use of peri- and intralesional interferon α-2b treatment of BCC which have otherwise high reported cure rates approaching 98% at 12 years and good cosmetic outcomes [79].
Hedgehog pathway inhibitors
Approximately 90% of human BCCs have loss of function of PTCH1, with the remaining 10% having gained a mutation in SMO. These mutations result in unchecked activation of the hedgehog pathway, resulting in unregulated proliferation of basal cells leading to BCC development (see Figure 141.1) [80].
Vismodegib (GDC-0449) is a small molecular inhibitor of SMO used in the treatment of metastatic and locally advanced BCCs (BCC not considered suitable for existing therapies such as surgery and radiotherapy). Orally administered at a dose of 150 mg daily until disease progression stopped due to side effects or withdrawal from the study, vismodegib was shown to induce a significant response in 43% of patients with advanced BCC and 30% with metastatic BCC [81]. Vismodegib is also a promising treatment in patients with NBCCS [82]. Adverse reactions of vismodegib include fatigue, hyponatraemia, weight loss and dyspnoea, muscle spasm, atrial fibrillation, aspiration, back pain, corneal abrasion, dehydration, keratitis, lymphopenia, pneumonia, urinary tract infection and a prolonged QT interval [80].
Other oral and topical SMO inhibitors are currently being evaluated in trials.
Surgical treatments
Excision with predetermined margins
Surgical excision is one of the most effective treatments for well-defined BCCs as not only does it allow for histological examination of the excised tissue to permit accurate evaluation of the excisional margins, but complete clearance can also be achieved in approximately 95% of the well-defined small BCC with a 4–5 mm surgical margin [83].
Additionally, recurrence rates are low (<2% in 5 years) following histologically complete excision of BCC and cosmetic outcomes are good with surgical excision [21, 84].
Surgical excision may not be appropriate for recurrent, morphoeic or large BCC which require a 3–15 mm margin in order to obtain a similar clearance rate and hence, if available, Mohs micrographic surgery is more appropriate in this setting.
For high-risk, mid-facial, incompletely excised BCC, immediate re-excision with or without frozen section control or with histologically controlled margins is recommended [69]. In certain inoperable cases, or when the patient's situation does not permit re-excision, adjuvant radiotherapy may be an acceptable alternative.
Mohs micrographic surgery
This is discussed in Chapter 20.
Curettage and electrodesiccation (syn. curettage and cautery, C&C)
The friable nature of the BCC lends to a selective removal of abnormal tissue with the traditional Volkmann spoon reusable curettes. This differential effect on tumour tissue is an effective way of delineating the extent of some tumours and can be useful before standard or Mohs excision of some BCCs to define more accurately lateral tumour spread within the epidermis. The disposable loop curettes, have a much sharper edge, so a more gentle initial curettage delineates the tumour. When used therapeutically it is important that the stroma and surrounding dermis are charred with diathermy or cautery to a depth of 1 mm following initial curettage and that this is repeated on two further occasions with curetting of the charred tumour base. As the procedure does not divide the upper dermis connective tissue network, healing is usually predictable and occurs in most cases with mild scarring. However, hypopigmentation or hypertrophic scar formation may occur, the latter particularly when the upper trunk is treated and especially when vigorous diathermy is employed.
Curettage and cautery is widely employed for the treatment of BCC in the UK and can achieve 5-year cure rates of up to 92.3% for selected primary BCCs [85]. Low-risk non-facial lesions are best suited for treatment with curettage and cautery. Curettage and cautery should not be employed for the treatment of recurrent or ill-defined BCC or for high-risk histological subtypes of BCC. Recurrence rates of 27% for histologically aggressive BCCs treated using curettage and cautery alone with median 6.5 years follow-up have been reported [86].
Cryosurgery
The application of liquid nitrogen to skin lesions to cause destruction of the tissue and a margin of surrounding tissue requires the achievement of lesional temperatures of −50 to −60°C. Like curettage and cautery, liquid nitrogen cryosurgery is considered appropriate for the treatment of low-risk primary BCC (Table 141.4). A single 30-s freeze–thaw cycle for superficial truncal BCC is recommended. Compared to surgical excision, for low-risk primary BCC, cryosurgery may achieve a similar recurrence rate at the expense of a worse cosmetic outcome as hypopigmentation is a frequent consequence of cryosurgery [87]. While freezing with liquid nitrogen is a quick and relatively tolerable procedure that does not require local anaesthesia, it can be associated with significant morbidity, particularly when tumours selected for treatment require prolonged freeze times to ensure adequate treatment.
Table 141.4 Features of high-risk basal cell carcinomas
| Features | High-risk basal cell carcinoma |
| Clinical subtype | Morphoeic |
| Tumour size | >5 cm giant BCC |
| Tumour site | Centrofacial, including periocular and ears |
| Histological subtype | Infiltrative and micronodular |
| Histological features of aggression | Perineural and/or perivascular involvement |
| Host characteristic | Immunosuppression |
| Lesion type | Recurrent |
| Lymph node status/other organ involvement | Lymph node involvement or distant metastasis |
Radiotherapy (see Chapter 24)
Superficial and electron beam radiotherapy or brachytherapy are effective therapeutic strategies in primary or surgically recurrent BCC as well as high-risk BCC in patients who are unwilling or unable to tolerate surgery [88, 89]. Radiotherapy may be used as a palliative modality in improving the quality of life in patients with advanced and/or incurable disease [90]. Incomplete clearance of BCC and recurrences after radiotherapy are higher and cosmesis poor when compared with surgical excision [91].
Radiotherapy should not be employed in the treatment of radiotherapy-treated recurrent BCC or for treating patients with NBCCS as they have an increased sensitivity to radiation and a tendency to develop multiple BCC in the irradiated field [92]. Radiotherapy should be used cautiously in young patients (<65 years) in view of the risk of latent tumours in radiotherapy-treated sites [93]. Other situations where radiotherapy is best avoided include BCC on the lower limbs, ear and eyelid, recurrent tumours, lesions previously treated with radiotherapy and tumours with poorly defined clinical margins. Although it is often stated that radiotherapy is best suited for elderly patients, the inconvenience and practical difficulties of frequent out-patient appointments required for a full fractionated course need to be taken into account when it is considered as an alternative to conventional surgical approaches.
Lasers (see Chapter 23)
The tissue heating properties of the carbon dioxide laser can be used to ensure precise ablation of BCC. Whilst high clinical cure rates have been reported, the lack of histological confirmation of cure with carbon dioxide laser ablation means that its use should be restricted to low-risk BCC [94, 95]. Postablative laser recurrent BCC can be more challenging to treat as they have a longer disease interval to diagnosis and a more aggressive histological pattern compared with primary BCC [96].
The pulsed dye laser has also been employed in the treatment of superficial BCC but the high incomplete clearance rate would preclude the use of this technology in the treatment of BCC [97].
FIBROEPITHELIAL BASAL CELL CARCINOMA
Definition and nomenclature
Fibroepithelial BCC is a benign-appearing pedunculated pink premalignant tumour that may resemble an acrochordon or skin tag. It is composed of basaloid cells arranged in a thin honeycomb around a prominent overgrown papillary stroma. It is now considered a variant of BCC.
Introduction and general description
This benign appearing neoplasm has an histological appearance resembling both a reticulated seborrhoeic keratosis and a BCC. It is considered a variant of BCC.
Epidemiology
Incidence and prevalence
This tumour mimics the epidemiology of BCC.
Age
With some exceptions it is seen in fourth to sixth decade of life.
Sex
There is a female preponderance.
Ethnicity
Mostly seen in fair-skinned individuals.
Pathophysiology
Predisposing factors
Probably related to UV exposure.
Pathology
The histological features are of a BCC, with thin anastomosing strands of the basaloid tumour embedded in a loose fibrovascular stroma and some displaying a connection with the epidermis. Small buds of cells may arise from the strands and enlarge to form BCC, replacing part or all of the tumour. The occurrence of Merkel cells in fibroepithelioma of Pinkus is believed to be an indication that the tumour is more closely related to trichoblastoma than to BCC [1].
The thin anastomosing network is composed of PHLDA1 (pleckstrin homology-like domain, family A, member 1), also known as TDAG51 (T-cell death-associated gene 51), positive cells, a follicular bulge marker used to differentiate trichoepithelioma from BCC. The multifocal BCCs that develop from this network are however PHLDA1 negative, supporting the notion of fibroepithelioma of Pinkus as a subtype of BCC [2]. The reticulate pattern is suggestive of seborrhoeic keratosis but absence of horn and pseudohorn pearls is the differentiating feature.
Clinical features
Presentation
A slowly growing, pink or red, fleshy papilloma. Unlike BCC, however, these are seen most commonly on the trunk, especially the lower back and extremities.
Differential diagnosis
- Fibroma.
- Papillomatous naevus.
- Fibroepithelial polyp.
- Amelanotic melanoma.
Investigations
Biopsy is diagnostic. Dermoscopy may be helpful as it shows arborizing vessels or dotted vessels with white streaks [3]. Structureless grey-brown areas of pigmentation and variable numbers of grey-blue dots may also be seen.
Management
Treatment options are as for nodular BCCs.
NAEVOID BASAL CELL CARCINOMA SYNDROME
Definition and nomenclature
An autosomal dominant familial cancer syndrome in which affected individuals are predisposed to the development of multiple BCCs at an early age and a variable combination of other phenotypic abnormalities including a highly characteristic facies (with large forehead), bifid or otherwise misshapen ribs, vertebral and other skeletal anomalies, pits of the skin of the palms and soles, dysgenesis of the corpus callosum, calcification of the falx cerebri (at an earlier age than is seen in non-affected individuals) and macrocephaly.
Epidemiology
Incidence and prevalence
Population-based studies suggest that the prevalence of this disorder in the UK is approximately 1 in 56 000 of the population [1]. In Europe, the approximate prevalence of NBCCS is reported to be 1 in 30 827 (England) to 1 in 256 000 (Italy) [2, 3]. The high rate of new mutations and the variable expressivity of the condition, however, make full ascertainment difficult, particularly in mildly affected individuals where there is no family history of the condition.
Age
Skin lesions including BCCs may develop in infancy but more frequently develop between puberty and 35 years of age with an average age of onset at 25 years [4].
Pathophysiology
Pathology
The histopathological appearance of BCCs from patients with NBCCS are indistinguishable from those seen in sporadic forms. The tumours induce a fibrous stroma as occurs with trichoepithelioma or nodular BCC, and the lesions may become papular or pedunculated. Deeper penetration, ulceration and invasion can occur, with lymphocytic infiltration. There may be pigmentation in and around the masses. The presence of calcification and the general architecture can resemble trichoepithelioma. Palmoplantar pits show focal absence of the stratum corneum with vacuolization of the spinous layer. At an ultrastructural level, pits show evidence of premature desquamation with a reduction in desmosomes and tonofibrils resulting from delay in maturation of the epidermal basal cells [5, 6]. BCCs have developed in palmar pits [5, 6, 7, 8].
Genetics
The NBCCS gene was mapped to chromosome 9q22.3-3.1 and, like other tumour suppressor genes, shows frequent deletion in both sporadic and familial BCCs [9]. The NBCCS gene was identified in 1996 with the identification of mutations in the PATCHED (PTCH1) gene in the germline of NBCCS patients and in sporadic BCC tumour samples [10]. PTCH1 is the human homologue of Protein patched homologue, which was first identified as a key regulator of the evolutionarily conserved Hedgehog (Hh) signalling pathway in elegant genetic studies of embryonic segmentation and imaginal disc specification in Drosophila. This finding was the first reported example of a link between genes important in normal development and cancer, and provided a completely new insight into the molecular pathways important in the development of this common skin cancer. The importance of Hedgehog signalling during normal development explains many of the other phenotypic abnormalities seen in patients with NBCCS and these features are consistent with findings from studies of heterozygote PTCH1 knock-out mice [11]. The PTCH1 mutation rate in NBCCS in published studies appears to range between 40% and 80%. A variety of mutations have been described in these studies including nonsense mutations, in-frame deletion, frame shifts due to deletions and insertions and splice-site mutations [12]. In addition to point mutations, a small number of patients have been described with larger deletions detected by comparative genomic hybridization methodologies. These patients tend to present with additional clinical features which may be caused by codeletion of other genes in addition to PTCH1 [13, 14].
Clinical features
Presentation
The skin manifestations of the syndrome are varied and include BCCs, skin tags, palmoplantar pits (Figure 141.14), milia, epidermoid cysts and lesions that clinically resemble dermal naevi. The number and type of skin lesions is very variable both within and between families and there is a marked difference between white people and African Americans in the number of BCCs [15]. BCCs from NBCCS are often smaller and much more numerous (from a few to thousands) than sporadic BCC [16]. They can present on both sun-exposed and non-exposed areas, and are most commonly located on the face, back and chest. BCC can cause significant tissue destruction leading to disfigurement if treatment is not pursued, particularly on the face. With the exception of the pits that are localized only on the palms and soles, skin lesions can occur in any region. The eyelids, nose, cheeks and forehead are the usual sites, but the neck, trunk and axillae are quite frequently involved. The scalp and limbs are usually spared. Patients with Gorlin syndrome also presumably face psychological challenges especially in terms of previous scarring and disfigurement.
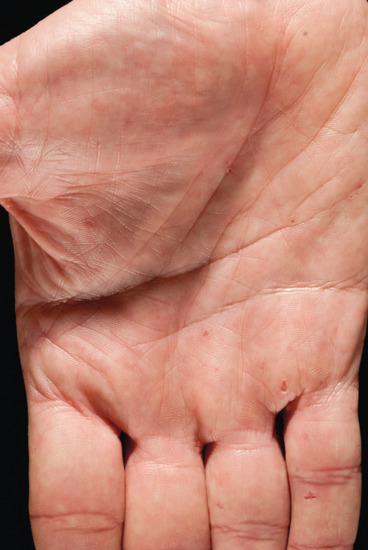
Figure 141.14 Palmar pits in a patient with Gorlin syndrome.
The individual lesions are smooth surfaced rounded elevated papules, flesh coloured or pigmented, varying in size from 1 to 15 mm in diameter. The lesions tend to increase in size and number up to late adolescence. There may be fine telangiectasia and milium-like bodies just below the surface. Tumours of the axillae, neck and eyelids tend to be pedunculated. Most lesions appear to behave in a relatively benign fashion with barely discernible growth and/or evidence of clinical progression. As is the case for patients with sporadic BCCs, some patients with NBCCS develop more aggressive tumours, which can be more difficult to treat and may cause significant morbidity or, rarely, death resulting from extensive invasion and/or recurrence following treatment. The proportion of NBCCS patients who develop very aggressive tumours and the risk factors for this have not been established. Aggressive tumours appear to occur more frequently on the eyelids or nose, and can cause gross destruction. In one study, four of five cases with aggressive BCCs in a series of 36 NBCCS patients received radiotherapy as the initial therapy, which suggests that radiotherapy may be a contributing factor to tumour aggressiveness in some NBCCS patients [17]. A variety of other skin manifestations have also been described, including multiple epidermoid cysts, milia and palmoplantar pits. The pits are a useful diagnostic feature that occur in about 65% of adults with NBCCS but are relatively rare in children. They are characterized by small, more or less circular pits, which may have an erythematous base and are usually 1–2 mm deep. In a recent study, re-examination of the skin phenotype in the context of data implicating Hh signalling in hair follicle biology led to the identification of discrete patches of unusually long, pigmented hair on the skin of three patients with NBCCS from two unrelated families with confirmed heterozygous mutations in the PTCH gene [18].
Differential diagnosis
In many cases, the skin lesions resemble melanocytic naevi, von Recklinghausen neurofibromatosis or skin tags rather than BCC, and their true nature may be suspected only because of associated features or family history. The correlation between the clinical and pathological features of the range of skin lesions seen in NBCCS patients is still poorly understood, which makes it difficult to draw firm conclusions about the natural history of the different skin lesions in these patients.
Complications and co-morbidities
Other diagnostically useful phenotypic abnormalities in NBCCS patients include jaw cysts, a highly characteristic facies (broad nasal root, hypertelorism, frontal bossing), bifid or otherwise misshapen ribs, vertebral and other skeletal anomalies, dysgenesis of the corpus callosum, calcification of the falx cerebri (Figure 141.15) (at an earlier age than is seen in non-NBCCS individuals) and macrocephaly [19]. The dental cysts are usually multiple, occurring in one or both jaws, and are odontogenic keratocysts [20]. Skeletal abnormalities include spina bifida occulta, bifid or splayed ribs, scoliosis or kyphosis, and occur with one-third of the frequency of the cysts or the basal cell naevi [21]. Less commonly associated anomalies include syndactyly, shortened metacarpals, cleft lip and palate, bicornuate uterus, hypogonadism in males, lymphatic cysts of the mesentery, ocular abnormalities including dystopia canthorum, cataracts and congenital blindness, and a variety of neurological disorders [21, 22, 23, 24, 25, 26]. In addition to BCCs, the syndrome is associated with an increased susceptibility to other neoplasms including rhabdomyosarcoma, ovarian and cardiac fibromas and, in particular, medulloblastoma. Approximately 3% of NBCCS patients develop medulloblastomas, and approximately 3% of patients with medulloblastomas have NBCCS [27]. Atypical clinical features such as brachydactyly, pulmonary valve stenosis and mental retardation should prompt consideration of a contiguous gene deletion syndrome.
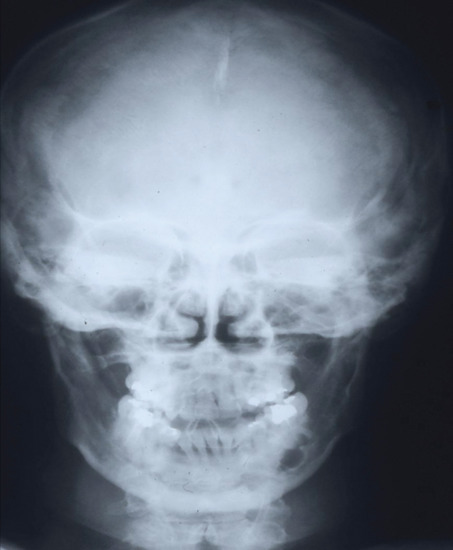
Figure 141.15 Skull X-ray with calcified falx cerebri and jaw cyst.
Investigations
In a patient suspected of NBCCS, skull X-ray and referral for genetic testing for PTCH (Patched), SMO (Smoothened) and SHH (Sonic hedgehog) are required. Other investigations in a confirmed case are listed in Box 141.2.
Management
NBCCS is a complex multisystemic disease and a multidisciplinary approach is crucial in the management of these patients. Such multidisciplinary teams should include input from dental teams (maxillo-facial surgeons, oncologists, radiation oncologists, prosthodontist), dermatologists, geneticist and psychiatrists [28].
The large number of lesions in patients with NBCCS means that primary excision of all lesions is not always practicable. Radiotherapy is contraindicated as many patients show an accelerated rate of development of new BCCs within an irradiated field, and where recurrences do occur they frequently are more aggressive and difficult to manage than the initial primary tumour. Surveillance of NBCCS patients who have developed BCCs can be useful for the early detection and treatment of new lesions. Although no controlled trials have established that reduced sun exposure can reduce the rate of BCC development in NBCCS patients, the reduced prevalence of BCCs in African Americans with this syndrome suggests that advice on reducing sun exposure is important [16].
Treatment of individual lesions should be guided by anatomical location, tumour size, clinical appearance and histology, with primary excision the treatment of choice for BCCs on the central face near critical structures. For superficial BCC on the trunk, approaches such as electrodessication and curettage or cryotherapy are useful. While the recurrence rate is potentially greater with these therapies, they are an effective convenient alternative for small or superficial BCCs distant from critical sites in patients with large numbers of lesions. Where the presence of large numbers of superficial lesions limits the acceptability of conventional therapies, photodynamic therapy with a systemic or topical photosensitizer and an appropriate laser or non-laser light source can be a useful and effective treatment option. Other non-surgical approaches that can be useful in the management of superficial lesions at non-critical sites include topical 5-FU and imiquimod formulations. These approaches have been shown to induce histological clearing of some BCCs and can reduce but not replace the need for more conventional therapies in patients with large numbers of lesions [29, 30, 31, 32]. As Gorlin patients often present with numerous lesions, it is preferable to treat an entire area with field therapy rather than lesional therapy. The value of systemic chemoprevention strategies in the management of patients with NBCCS is still unclear. A few studies suggest that systemic retinoids (isotretinoin >4 mg/kg and etretinate 0.7–1 mg/kg) may reduce the rate of development of new BCCs [32]. The clinical benefits, however, appear small relative to those seen in patients with multiple cutaneous SCCs.
The efficacy of vismodegib in patients with NBCCS was tested in a randomized double-blind placebo-controlled trial [33, 34]. The primary end point was reduction in the incidence of new BCCs that were eligible for surgical resection with vismodegib versus placebo after 3 months; secondary end points included reduction in the size of existing BCCs.
Forty-one patients were treated for a mean of 8 months. This led to a reduction in new BCC and significant reduction in the size of existing BCCs. In some, all BCCs regressed. No tumours progressed whilst on treatment. Patients routinely had grade 1 or 2 adverse events of loss of taste, muscle cramps, hair loss and weight loss. Overall, 54% of patients (14/26) receiving vismodegib discontinued drug treatment owing to adverse events. This study illustrates the dual benefit of this drug in NBCCS patients, both reduction in new lesion development and reduction of existing lesion size and number. However, 50% had to stop therapy due to adverse events. Studies need to assess other treatment regimens to enable more patients to receive the drug for longer and hopefully obtain lasting benefit. In particular, intermittent dosing schedules are being assessed together with other modes of drug delivery. NBCCS patients have also been treated with sonidegib but these trials are yet to be published.
BAZEX–DUPRÉ–CHRISTOL SYNDROME
Definition and nomenclature
A rare genodermatosis that predisposes affected individuals to multiple BCCs. Additional clinical features that allow distinction from NBCCS include follicular atrophoderma, hypotrichosis and hypohidrosis.
Introduction and general description
Bazex–Dupré–Christol syndrome is characterized by hypotrichosis, follicular atrophoderma of the cheeks, milia cysts and BCCs.
Epidemiology
Age
Basal cell carcinomas usually appear after the first decade of life.
Sex
Only females are affected.
Pathophysiology
Genetics
X-linked dominant disorder.
In 1995, Vabres et al. reported a linkage of three families to a 23.3-Mb region on chromosome Xq24-27.1 [1]. These findings were later supported by Parren et al. who reported to have narrowed down the critical region further, to an 11.4-Mb interval on the same chromosome [2].
Clinical features
Presentation
Follicular atrophoderma, hypotrichosis and hypohidrosis are the main presenting features. Follicular atrophoderma is present at birth or in early childhood, and shows as ‘ice-pick marks’, enlarged follicular ostia on the dorsa of the hands, elbows, feet and face. The follicular changes are not caused by injury or inflammation but there may be facial eczema soon after birth. There may be anhidrosis of the face and head, and hypotrichosis. The BCCs appear on the face in the second or third decade and resemble cellular naevi [3]. Pigmented BCC may be seen as early as age 5 years [4]. Milia have been reported as frequently associated features [4].
The association between BCCs and clinical abnormalities of the hair follicle is of interest as BCCs have the same cytokeratin profile as a subpopulation of follicular keratinocytes [5].
Management
As for NBCCS.
References
Basal cell carcinoma
- Levell NJ, Igali L, Wright KA, Greenberg DC. Basal cell carcinoma epidemiology in the UK: the elephant in the room. Clin Exp Dermatol 2013;38:367–9.
- de Vries E, Micallef R, Brewster DH, et al. Population-based estimates of the occurrence of multiple vs first primary basal cell carcinomas in 4 European regions. Arch Dermatol 2012;148:347–54.
- Silverberg E, Boring CC, Squires TS. Cancer statistics 1990. CA Cancer J Clin 1990;40:9–26.
- Cakir BÖ, Adamson P, Cingi C. Epidemiology and economic burden of nonmelanoma skin cancer. Facial Plast Surg Clin North Am 2012;20:419–22.
- Goodwin RG, Holme SA, Roberts DL. Variations in registration of skin cancer in the United Kingdom. Clin Exp Dermatol 2004;29:328–30.
- Cancer Research UK. http://www.cancerresearchuk.org/cancer-info/cancerstats/types/skin/incidence/uk-skin-cancer-incidence-statistics#nmsc (last accessed April 2015).
- National Cancer Intelligence Network (NCIN). Non-melanoma Skin Cancer in England, Scotland, Northern Ireland, and Ireland. London: NCIN, 2013.
- National Cancer Intelligence Network (NCIN) Data Briefing. The Importance of Skin Cancer Registration. London: NCIN, 2010.
- Flohil SC, Seubring I, van Rossum MM, Coebergh JW, de Vries E, Nijsten T. Trends in Basal cell carcinoma incidence rates: a 37-year Dutch observational study. J Invest Dermatol 2013;133:913–18.
- Eisemann N, Waldmann A, Geller AC, et al. Non-melanoma skin cancer incidence and impact of skin cancer screening on incidence. J Invest Dermatol 2014;134:43–50.
- Lomas A, Leonardi-Bee J, Bath-Hextall F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br J Dermatol 2012;166:1069–80.
- Diffey BL, Langtry JA. Skin cancer incidence and the ageing population. Br J Dermatol 2005;153:679–80.
- Flohil SC, Koljenović S, de Haas ER, et al. Cumulative risks and rates of subsequent basal cell carcinomas in the Netherlands. Br J Dermatol 2011;165:874–81.
- Khalesi M, Whiteman DC, Tran B, et al. A meta-analysis of pigmentary characteristics, sun sensitivity, freckling and melanocytic nevi and risk of basal cell carcinoma of the skin. Cancer Epidemiol 2013;37:534–43.
- Flohil SC, van der Leest RJ, Arends LR, de Vries E, Nijsten T. Risk of subsequent cutaneous malignancy in patients with prior keratinocyte carcinoma: a systematic review and meta-analysis. Eur J Cancer 2013;49:2365–75.
- Stern RS. The mysteries of geographic variability in nonmelanoma skin cancer incidence. Arch Dermatol 1999;135:843–4.
- Bauer A, Diepgen TL, Schmitt J. Is occupational solar ultraviolet irradiation a relevant risk factor for basal cell carcinoma? A systematic review and meta-analysis of the epidemiological literature. Br J Dermatol 2011;165:612–25.
- Kricker A, Armstrong BK, English DR, Heenan PJ. A dose–response curve for sun exposure and basal cell carcinoma. Int J Cancer 1995;60:482–8.
- Kricker A, Armstrong BK, English DR, Heenan PJ. Does intermittent sun exposure cause basal cell carcinoma? A case–control study in Western Australia. Int J Cancer 1995;60:489–94.
- Rosso S, Zanetti R, Martinez C, et al. The multicentre south European study ‘Helios’ II: different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer 1996;73:1447–54.
- Madan V, Lear JT, Szeimies RM. Non-melanoma skin cancer. Lancet 2010;375:673–85.
- Rünger TM. How different wavelengths of the ultraviolet spectrum contribute to skin carcinogenesis: the role of cellular damage responses. J Invest Dermatol 2007;127:2103–5.
- Ridley AJ, Whiteside JR, McMillan TJ, Allinson SL. Cellular and sub-cellular responses to UVA in relation to carcinogenesis. Int J Radiat Biol 2009;85:177–95.
- Lear JT, Smith AG, Strange RC, Fryer AA. Detoxifying enzyme genotypes and susceptibility to cutaneous malignancy. Br J Dermatol 2000;142:8–15.
- Lear JT, Hoban P, Strange RC, Fryer AA. Basal cell carcinoma: from host response and polymorphic variants to tumour suppressor genes. Clin Exp Dermatol 2005;30:49–55.
- Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Invest Dermatol 2014;134:213–20.
- Stone DM, Hynes M, Armanini M, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996;384:129–34.
- Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013;494:484–8.
- Soufir N, Gerard B, Portela M, et al. PTCH mutations and deletions in patients with typical nevoid basal cell carcinoma syndrome and in patients with a suspected genetic predisposition to basal cell carcinoma: a French study. Br J Cancer 2006;95:548–53.
- Ali FR, Lear JT. Systemic treatments for basal cell carcinoma (BCC): the advent of dermato-oncology in BCC. Br J Dermatol 2013;169:53–7.
- Gudbjartsson DF, Sulem P, Stacey SN, et al. ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat Genet 2008;40:886–91.
- Brash DE. Roles of the transcription factor p53 in keratinocyte carcinomas. Br J Dermatol 2006;154 Suppl. 1:8–10.
- Lear JT, Smith AG, Strange RC, Fryer AA. Detoxifying enzyme genotypes and susceptibility to cutaneous malignancy. Br J Dermatol 2000;142:8–15.
- Ramachandran S, Lear JT, Ramsay H, et al. Presentation with multiple cutaneous basal cell carcinomas: association of glutathione S-transferase and cytochrome P450 genotypes with clinical phenotype. Cancer Epidemiol Biomarkers Prev 1999;8:61–7.
- Saleh S, King-Yin Lam A, Gertraud BP, et al. Telomerase activity of basal cell carcinoma in patients living in North Queensland, Australia. Hum Pathol 2007;38:1023–9.
- Oh ST, Kim HS, Yoo NJ, Lee WS, Cho BK, Reichrath J. Increased immunoreactivity of membrane type-1 matrix metalloproteinase (MT1-MMP) and β-catenin in high-risk basal cell carcinoma. Br J Dermatol 2011;165:1197–204.
- Moloney FJ, Comber H, Conlon PJ, et al. The role of immunosuppression in the pathogenesis of basal cell carcinoma. Br J Dermatol 2006;154:790–1.
- Moloney FJ, Comber H, O'Lorcain P, O'Kelly P, Conlon PJ, Murphy GM. A population-based study of skin cancer incidence and prevalence in renal transplant recipients. Br J Dermatol 2006;154:498–504.
- Rangwala S, Tsai KY. Roles of the immune system in skin cancer. Br J Dermatol 2011;165:953–65.
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of Basal cell carcinoma after Mohs surgery in patients with chronic lymphocytic leukemia. Arch Dermatol 2004;140:985–8.
- Leonardi G, Vahter M, Clemens F, et al. Inorganic arsenic and basal cell carcinoma in areas of Hungary, Romania, and Slovakia: a case–control study. Environ Health Perspect 2012;120:721–6.
- Man I, Crombie IK, Dawe RS, Ibbotson SH, Ferguson J. The photocarcinogenic risk of narrowband UVB (TL-01) phototherapy: early follow-up data. Br J Dermatol 2005;152:755–7.
- Lichter MD, Karagas MR, Mott LA, et al. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. The New Hampshire Skin Cancer Study Group. Arch Dermatol 2000;136:1007–11.
- Castori M, Morrone A, Kanitakis J, Grammatico P. Genetic skin diseases predisposing to basal cell carcinoma. Eur J Dermatol 2012;22:299–309.
- Van Scott EJ, Reinertson RP. The modulating influence of stromal environment on epithelial cells studied in human autotransplants. J Invest Dermatol 1961;36:109–17.
- Weinstein GD, Frost P. Cell proliferation in human basal cell carcinoma. Cancer Res 1970;30:724–8.
- Weedon D, Shand E. Amyloid in basal cell carcinomas. Br J Dermatol 1979;101:141–6.
- Madsen A. Studies on basal-cell epithelioma of the skin: the architecture, manner of growth, and histogenesis of the tumours – whole tumours examined in serial sections cut parallel to the skin surface. Acta Pathol Microbiol Scand Suppl 1965;117:3–63.
- Sanderson KV. The architecture of basal-cell carcinoma. Br J Dermatol 1961;73:455–74.
- Sherertz EF, Pollack SV, Jegasothy BV. Correlation of basal cell epithelioma aggressiveness with local inhibition of host lymphocyte response. Clin Res 1982;30:266 (Abstract).
- Plaza JA, Ortega PF, Bengana C, Stockman DL, Suster S. Immunolabeling pattern of podoplanin (d2-40) may distinguish basal cell carcinomas from trichoepitheliomas: a clinicopathologic and immunohistochemical study of 49 cases. Am J Dermatopathol 2010;32:683–7.
- Tebcherani AJ, de Andrade HF Jr, Sotto MN. Diagnostic utility of immunohistochemistry in distinguishing trichoepithelioma and basal cell carcinoma: evaluation using tissue microarray samples. Mod Pathol 2012;25:1345–53.
- Crowson AN. Basal cell carcinoma: biology, morphology and clinical implications. Mod Pathol 2006;19 Suppl. 2:S127–47.
- Farmer ER, Helwig EB. Metastatic basal cell carcinoma: a clinicopathologic study of 17 cases. Cancer 1980;46:748–57.
- McCormack CJ, Kelly JW, Dorevitch AP. Differences in age and body site distribution of the histological subtypes of basal cell carcinoma: a possible indicator of differing causes. Arch Dermatol 1997;133:593–6.
- Ramachandran S, Fryer A, Smith A, et al. Cutaneous basal cell carcinomas. Cancer 2001;92:354–8.
- Sexton M, Jones DB, Maloney ME. Histologic pattern analysis of basal cell carcinoma. Study of a series of 1039 consecutive neoplasms. J Am Acad Dermatol 1990;23:1118–26.
- Rippey JJ. Why classify basal cell carcinomas? Histopathology 1998;32:393–8.
- Schwipper V. Invasive basal cell carcinoma of the head and neck (basalioma terebrans). Facial Plastic Surg 2011;27:258–65.
- Gormley LJDE, Hirsch P. Aggressive basal cell carcinoma of the scalp. Arch Dermatol 1978;114:782–3.
- Ting PT, Kasper R, Arlette AP. Metastatic basal cell carcinoma: report of two cases and literature review. J Cutan Med Surg 2005;9:10–15.
- Larson DL, Gillespie JJ, Parsons RW. Metastatic basal cell carcinoma of the lung. South Med J 1980;73:647–9.
- Presser SE, Taylor JR. Clinical diagnostic accuracy of basal cell carcinoma. J Am Acad Dermatol 1987;16:988–90.
- Roozeboom MH, Mosterd K, Winnepenninckx VJ, et al. Agreement between histological subtype on punch biopsy and surgical excision in primary basal cell carcinoma. J Eur Acad Dermatol Venereol 2013;27:894–8.
- Lallas A, Argenziano G, Zendri E, et al. Update on non-melanoma skin cancer and the value of dermoscopy in its diagnosis and treatment monitoring. Exp Rev Anticancer Ther 2013;13:541–58.
- Ulrich M, Stockfleth E, Roewert-Huber J, Astner S. Noninvasive diagnostic tools for nonmelanoma skin cancer. Br J Dermatol 2007;157 S2:56–8.
- Boone MA, Norrenberg S, Jemec GB, Del Marmol V. Imaging of basal cell carcinoma by high-definition optical coherence tomography: histomorphological correlation. A pilot study. Br J Dermatol 2012;167:856–64.
- Bennàssar A, Vilata A, Puig S, Malvehy. Validation of fluorescent confocal microscopy criteria and histopathological correlation for the diagnosis of basal cell carcinoma and most common subtypes. Br J Dermatol 2014;170:360–5.
- Telfer NR, Colver GB, Morton CA; British Association of Dermatologists. Guidelines for the management of basal cell carcinoma. Br J Dermatol 2008;159:35–48.
- Bath-Hextall FJ, Perkins W, Bong J, Williams HC. Interventions for basal cell carcinoma of the skin. Cochrane Database Syst Rev 2007;24;CD003412.
- Bath-Hextall F, Leonardi-Bee J, Somchand N, Webster A, Delitt J, Perkins W. Interventions for preventing non-melanoma skin cancers in high-risk groups. Cochrane Database Syst Rev 2007;4:CD005414.
- Dummer R, Urosevic M, Kempf W, et al. Imiquimod in basal cell carcinoma: how does it work? Br J Dermatol 2003;149 Suppl. 66:57–8.
- Chitwood K, Etzkorn J, Cohen G. Topical and intralesional treatment of nonmelanoma skin cancer: efficacy and cost comparisons. Dermatol Surg 2013;39:1306–16.
- Eigentler TK, Kamin A, Weide BM, et al. A phase III, randomized, open label study to evaluate the safety and efficacy of imiquimod 5% cream applied thrice weekly for 8 and 12 weeks in the treatment of low-risk nodular basal cell carcinoma. J Am Acad Dermatol 2007;57:616–21.
- McKay KM, Sambrano BL, Fox PS, et al. Thickness of superficial basal cell carcinoma (sBCC) predicts imiquimod efficacy: a proposal for a thickness-based definition of sBCC. Br J Dermatol 2013;169:549–54.
- Arits AH, Mosterd K, Essers BA, et al. Photodynamic therapy versus topical imiquimod versus topical fluorouracil for treatment of superficial basal-cell carcinoma: a single blind, non-inferiority, randomised controlled trial. Lancet Oncol 2013;14:647–54.
- Sloan KB, Sherertz EF, McTiernan RG. The effect of 5-fluorouracil on inhibition of epidermal DNA synthesis in vivo: a comparison of the effect of formulations and a prodrug of 5-FU. Arch Dermatol Res 1990;282:484–6.
- Gross K, Kircik L, Kricorian G. 5% 5-Fluorouracil cream for the treatment of small superficial basal cell carcinoma: efficacy, tolerability, cosmetic outcome, and patient satisfaction. Dermatol Surg 2007;33:433–9.
- Tucker SB, Polasek JW, Perri AJ, Goldsmith EA. Long-term follow-up of basal cell carcinomas treated with perilesional interferon alfa 2b as monotherapy. J Am Acad Dermatol 2006;54:1033–8.
- Lear JT. Oral hedgehog pathway inhibitors for basal cell carcinoma. N Engl J Med 2012;366:2225–6.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 2012;366:2171–9.
- Wolfe CM, Green WH, Cognetta AB Jr, et al. Basal cell carcinoma rebound after cessation of vismodegib in a nevoid basal cell carcinoma syndrome patient. Dermatol Surg 2012;38:1863–6.
- Wolf DJ, Zitelli JA. Surgical margins for basal cell carcinoma. Arch Dermatol 1987;123:340–4.
- Walker P, Hill D. Surgical treatment of basal cell carcinomas using standard postoperative histological assessment. Australas J Dermatol 2006;47:1–12.
- Rowe DE, Carroll RJ, Day CL Jr, et al. Long-term recurrence rates in previously untreated (primary) basal cell carcinoma: implications for patient follow-up. J Dermatol Surg Oncol 1989;15:315–28.
- Blixt E, Nelsen D, Stratman E. Recurrence rates of aggressive histologic types of basal cell carcinoma after treatment with electrodesiccation and curettage alone. Dermatol Surg 2013;39:719–25.
- Thissen M, Nieman F, Ideler A, Berretty P, Neumann H. Cosmetic results of cryosurgery versus surgical excision for primary uncomplicated basal cell carcinomas of the head and neck. Dermatol Surg 2000;26:759–64.
- Childers BJ, Goldwyn RM, Ramos D, et al. Long-term results of irradiation for basal cell carcinoma of the skin of the nose. Plast Reconstr Surg 1994;93:1169–73.
- Caccialanza M, Piccinno R, Grammatica A. Radiotherapy of recurrent basal and squamous cell skin carcinomas: a study of 249 re-treated carcinomas in 229 patients. Eur J Dermatol 2001;11:25–8.
- Veness MJ. The important role of radiotherapy in patients with non-melanoma skin cancer and other cutaneous entities. J Med Imaging Radiat Oncol 2008;52:278–86.
- Avril M, Auperin A, Margulis A, et al. Basal cell carcinoma of the face: surgery or radiotherapy? Results of a randomized study. Br J Cancer 1997;76:100–6.
- Kleinerman RA. Radiation-sensitive genetically susceptible pediatric sub-populations. Pediatr Radiol 2009;39:S27–31.
- Landthaler M, Hagspiel HJ, Braun-Falco O. Late irradiation damage to the skin caused by soft X-ray radiation therapy of cutaneous tumors. Arch Dermatol 1995;131:182–6.
- Campolmi P, Brazzini B, Urso C, et al. Superpulsed CO2 laser treatment of basal cell carcinoma with intraoperatory histopathologic and cytologic examination. Dermatol Surg 2002;28:909–11.
- Humphreys TR, Malhotra R, Scharf MJ, et al. Treatment of superficial basal cell carcinoma and squamous cell carcinoma in situ with a high-energy pulsed carbon dioxide laser. Arch Dermatol 1998;134:1247–52.
- Jung DS, Cho HH, Ko HC, et al. Recurrent basal cell carcinoma following ablative laser procedures. J Am Acad Dermatol 2011;64:723–9.
- Ballard CJ, Rivas MP, McLeod MP, et al. The pulsed dye laser for the treatment of basal cell carcinoma. Lasers Med Sci 2011;26:641–4.
- Karen JK, Gareau DS, Dusza SW, et al. Detection of BCCs in Mohs excisions with fluorescence confocal mosaicing microscopy. Br J Dermatol 2009;160:1242–50.
Fibroepithelial basal cell carcinoma
- Bowen AR, LeBoit PE. Fibroepithelioma of Pinkus is a fenestrated trichoblastoma. Am J Dermatopathol 2005;27:149–54.
- Sellheyer K, Nelson P, Kutzner H. Fibroepithelioma of Pinkus is a true basal cell carcinoma developing in association with a newly identified tumour-specific type of epidermal hyperplasia. Br J Dermatol 2012;166:88–97.
- Zalaudek I, Ferrara G, Broganelli P, et al. Dermoscopy patterns of fibroepithelioma of pinkus. Arch Dermatol 2006;142:1318–22.
Naevoid basal cell carcinoma syndrome
- Evans DG, Ladusans EJ, Rimmer S, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population-based study. J Med Genet 1993;30:460–4.
- Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J Rare Dis 2008;3:32.
- Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am J Med Genet A 2010;152:327–32.
- Gorlin RJ. Nevoid basal cell carcinoma syndrome. Dermatol Clin 1995;13(1):113–25.
- Howell JB, Mehregan AH. Pursuit of the pits in the naevoid basal cell carcinoma syndrome. Arch Dermatol 1970;102:586–97.
- Holubar K, Matras H, Smalik AV. Multiple palmar basal cell epitheliomas in basal cell naevus syndrome. Arch Dermatol 1970;10:679–82.
- Howell JB, Freeman RG. Structure and significance of the pits and their tumours in the naevoid basal cell carcinoma syndrome. J Am Acad Dermatol 1980;2:224–38.
- Mason JK, Helwig EB, Graham JH. Pathology of the naevoid basal cell carcinoma syndrome. Arch Pathol 1965;79:401–8.
- Quinn AG. Molecular genetics of human non-melanoma skin cancer. Cancer Surv 1996;26:89–114.
- Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell naevus syndrome. Science 1996;272:1668–71.
- Bale AE, Gailani MR, Leffell DJ. The Gorlin syndrome gene: a tumor suppressor active in basal cell carcinogenesis and embryonic development. Proc Assoc Am Phys 1995;107:253–7.
- Soufir N, Gerard B, Portela M, et al. PTCH mutations and deletions in patients with typical nevoid basal cell carcinoma syndrome and in patients with a suspected genetic predisposition to basal cell carcinoma: a French study. Br J Cancer 2006;95:548–53.
- Nowakowska B, Kutkowska-Kazmierczak A, Stankiewicz P, et al. A girl with deletion 9q22.1-q22.32 including the PTCH and ROR2 genes identified by genome-wide array-CGH. Am J Med Genet A 2007;143:1885–9.
- Olivieri C, Maraschio P, Caselli D, et al. Interstitial deletion of chromosome 9, int del(9)(9q22.31-q31.2), including the genes causing multiple basal cell nevus syndrome and Robinow/brachydactyly 1 syndrome. Eur J Pediatr 2003;162:100–3.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 1997;69:299–308.
- Southwick GJ, Schwartz RA. The basal cell naevus syndrome: disasters occurring among a series of 36 patients. Cancer 1979;44:2294–305.
- Wilson LC, Ajayi-Obe E, Bernhard B, et al. Patched mutations and hairy skin patches: a new sign in Gorlin syndrome. Am J Med Genet A 2006;140:2625–30.
- Gorlin RJ. The naevoid basal cell carcinoma syndrome. Medicine 1987;66:98–113.
- Binkley GW, Johnson HH. Epithelioma adenoides cysticum: basal cell naevi, agenesis of the corpus callosum and dental cysts. Arch Dermatol 1951;63:73–84.
- Anderson DE, Taylor WB, Falls MF, et al. The naevoid basal cell carcinoma syndrome. Am J Hum Genet 1967;19:12–22.
- Berlin NI, van Scott EJ, Clendenning WE, et al. Basal cell naevus syndrome: combined clinical staff conference at the National Institute of Health. Ann Intern Med 1966;64:403–21.
- Clendenning WE, Block JB, Radde IC. Basal cell naevus syndrome. Arch Dermatol 1964;90:38–53.
- Gorlin RJ, Goltz RW. Multiple naevoid basal-cell epithelioma, jaw cysts and bifid rib: a syndrome. N Engl J Med 1960;262:908–12.
- Gorlin RJ, Yunis JJ, Tuna N. Multiple naevoid basal cell carcinoma, odontogenic keratocysts and skeletal anomalies: a syndrome. Acta Derm Venereol (Stockh) 1963;43:39–55.
- Howell JB, Caro MR. The basal-cell naevus: its relationship to multiple cutaneous cancers and associated anomalies of development. AMA Arch Dermatol 1959;79:67–80.
- Van Dijk E, Sanderink JFH. Basal cell naevus syndrome. Dermatologica 1967;134:101–6.
- Evans DG, Farndon PA, Burnell LD, et al. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer 1991;64:959–61.
- Nagy K, Kiss E, Erdei C, et al. Complex care by multiple medical and dental specialists of a patient with aggressive Gorlin–Goltz syndrome. Postgrad Med J 2008;84:330–2.
- Goette DK. Topical chemotherapy with 5-fluorouracil: a review. J Am Acad Dermatol 1981;4:633–49.
- Micali G, De Pasquale R, Caltabiano R, et al. Topical imiquimod treatment of superficial and nodular basal cell carcinomas in patients affected by basal cell naevus syndrome: a preliminary report. J Dermatol Treat 2002;13:123–7.
- Goldberg LH, Rubin HA. Management of basal cell carcinoma: which option is best? Postgrad Med 1989;85:57–8,61–3.
- Cristofolini M, Zumiani G, Scappini P, Piscioli F. Aromatic retinoid in the chemoprevention of the progression of naevoid basal-cell carcinoma syndrome. J Dermatol Surg Oncol 1984;10:778–81.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the Hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med 2012;366:2180–218.
- Lear JT. Oral Hedgehog-pathway inhibitors for basal-cell carcinoma. N Engl J Med 2012;366:2225–6.
- Kiwilsza M, Sporniak-Tutak K. Gorlin–Goltz syndrome – a medical condition requiring a multidisciplinary approach. Med Sci Monit 2012;18:RA145–53.
Bazex–Dupré–Christol syndrome
- Vabres P, Lacombe D, Rabinowitz LG, et al. The gene for Bazex–Dupré–Christol syndrome maps to chromosome Xq. J Invest Dermatol 1995;105:87–91.
- Parren LJ, Abuzahra F, Wagenvoort T, et al. Linkage refinement of Bazex–Dupré–Christol syndrome to an 11·4-Mb interval on chromosome Xq25-27.1. Br J Dermatol 2011;165:201–3.
- Bazex A, Dupré A, Christol B. Atrophodermie folliculaire proliférations baso-cellulaires et hypotrichose. Ann Dermatol Syphiligr 1966;93:241–54.
- Abuzahra F1, Parren LJ, Frank J. Multiple familial and pigmented basal cell carcinomas in early childhood – Bazex–Dupré–Christol syndrome. J Eur Acad Dermatol Venereol 2012;26:117–21.
- Vabres P, Lacombe D, Rabinowitz LG, et al. The gene for Bazex–Dupré–Christol syndrome maps to chromosome Xq. J Invest Dermatol 1995;105:87–91.