CHAPTER 147
Cutaneous Markers of Internal Malignancy
Lennart Emtestam1 and Karin Sartorius2
1Department of Medicine Huddinge, Karolinska Institutet, Stockholm; Department of Dermatology, Karolinska University Hospital, Stockholm, Sweden
2Department of Clinical Sciences and Education, Karolinska Institutet, Stockholm; and Department of Dermatology, Södersjukhuset, Stockholm, Sweden
INTRODUCTION
The different types of skin change associated with internal malignancy, and the links between systemic neoplasia and associated skin conditions, are so numerous that to report all documented associations here is impracticable. However, the following categories embrace most associations and interactions between skin and internal malignancy:
- Multisystem and haematopoietic tumours that involve the skin.
- Tumour spread.
- Genetically determined syndromes with cutaneous manifestations where there is a recognized predisposition to internal malignancy (also termed ‘genodermatoses with malignant potential’). This category also includes the hamartoneoplastic syndromes.
- Paraneoplastic disorders: cutaneous reaction patterns that have an association with neoplasia involving various internal organ systems.
- Indirect cutaneous markers of internal malignancy (for example, dermatological features of exposure to carcinogens).
Potential cutaneous markers of internal malignancy vary in their reliability for predicting underlying neoplasia. The extent and intensity of the investigations for malignancy should therefore be tempered by a general assessment of the patient. Cutaneous adverse reactions to drugs used to treat malignancy are covered in Chapter 120.
Multisystem and haematopoietic tumours that involve the skin
This category includes a number of tumours in which the skin is involved as part of a multisystem neoplasm. The importance for dermatologists is that the diagnosis may present with skin lesions, or that the skin may be the most accessible site for histological diagnosis. The types of skin involvement that may occur in such disorders range from non-specific signs, such as purpura reflecting thrombocytopenia, to specific features such as cutaneous deposits of the malignancy. This type of involvement of the skin as part of a multisystem tumour is somewhat different from metastases from solid tumours, both in mechanism and in lesion distribution. For example, skin involvement is an important component of haematopoietic malignancies, including lymphoma and Langerhans cell histiocytosis [1–3]; specific cutaneous infiltrations of the skin may occur with myeloproliferative disorders, such as lymphoma and leukaemia (Figure 147.1) [4].
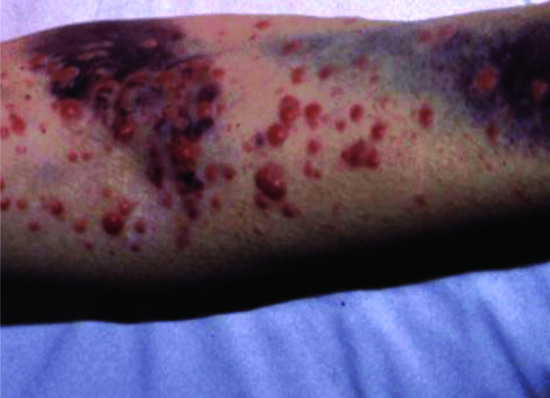
Figure 147.1 Leukaemia cutis (acute myeloid leukaemia) and purpura.
Tumour spread from adjacent and distant tissues
Direct tumour spread and invasion
Other than primary skin neoplasms, the skin may be involved by tumour either through direct invasion, or through local metastasis, or from cutaneous metastases from an internal tumour.
Direct invasion of the skin from a deeper tumour usually causes nodular infiltration, ulceration or inflammation, but may present in less obvious ways: dermal infiltration causing sclerosis (carcinoma en cuirasse), vascular changes (carcinoma telangiectodes), a peau d'orange (orange peel) appearance, and more rarely a carcinoma erysipeloides (inflammatory metastatic carcinoma) pattern (Figures 147.2 and 147.3). Although these patterns may occur as distant metastases, they most commonly occur in the skin in the vicinity of the primary tumour – all being most usually associated with breast cancer – and are therefore usefully considered as a rather different pattern to those tumours that metastasize to distant sites. All of these patterns may be difficult to diagnose unless there is clinical suspicion.
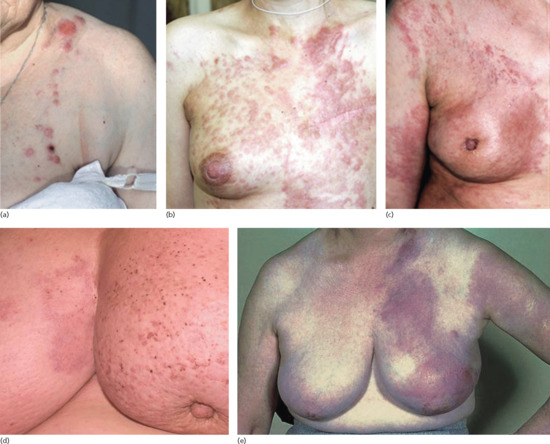
Figure 147.2 Patterns of skin infiltration by carcinoma of the breast. (a, b) Limited (a) and extensive (b) nodular infiltration. (c) Sclerotic infiltration (carcinoma en cuirasse) of breast skin and telangiectatic infiltration (carcinoma telangiectodes) extending beyond the breast. (d) Peau d'orange appearance due to infiltrative carcinoma skin of left breast. (d, e) Erysipelas-like changes (carcinoma erysipeloides) involving the right breast at an early stage (d) and at an advanced stage involving the left upper extremity as well as the breast (e). Note also nipple retraction in parts (c) and (d).
(Parts a–c, courtesy of Dr Olga Mikheeva, Moscow Region Oncological Dispensary, Russia; part d, courtesy of Dr Ken Tsai; part c, courtesy of Dr R. Emmerson, Royal Berkshire Hospital, Reading, UK.)
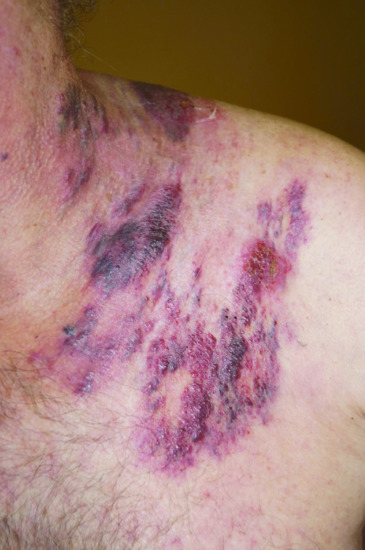
Figure 147.3 Metastatic bronchial carcinoma resembling cutaneous angiosarcoma.
(Courtesy of Dr Olga Mikheeva, Moscow Region Oncological Dispensary, Russia.)
Carcinoma en cuirasse. This may have an early inflammatory stage, and may include some nodularity (Figure 147.2b), but at a later stage is sclerodermoid in appearance (Figure 147.2c). A particular diagnostic problem arises when a breast cancer has been treated with radiotherapy as this may cause post-irradiation morphoea. The latter is relatively well documented but the early inflammatory phase of post-irradiation morphoea that occurs in some patients is much less well recognized and may be alarming clinically. Carcinoma en cuirasse may also occur with lung, gastrointestinal, renal and other malignancies [1].
Carcinoma erysipeloides. This resembles erysipelas, presenting as an extensive, warm, oedematous, tender plaque but without pyrexia or toxaemia (Figure 147.2d). This pattern accounts for nearly a third of cases of cutaneous metastases from breast cancer [2], the malignancy with which it is most commonly associated [3]. Similar presentations have been reported in melanoma, mesothelioma and carcinomas of the lung, prostate, oesophagus, bladder, colon, larynx, rectum, stomach, cutaneous squamous and pancreas [3–23]. Recent studies have demonstrated that the clinical picture is due in the majority of cases to the plugging of dermal lymphatics by tumour cell emboli rather than to intratumoural lymphatic invasion [2, 24]. Histologically, there is plugging of the dermal lymphatics at all levels by aggregates of carcinoma cells (Figure 147.4).
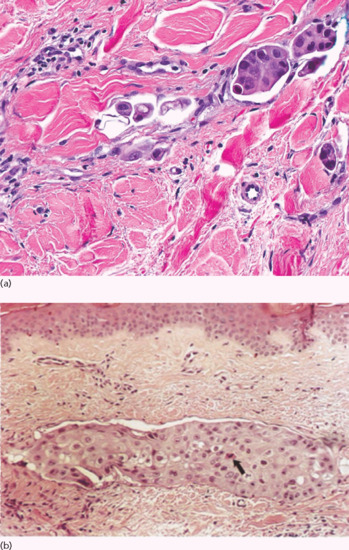
Figure 147.4 Carcinoma erysipeloides. (a) Small aggregates of large epithelioid carcinoma cells within the dilated lymphatics. (b) Dermal lymphatic completely occluded by poorly differentiated epithelioid cells. Note tumour cell necrosis (arrow).
(Part a, courtesy of Dr K. Tsai; part b, from Finkel and Griffiths 1993 [26], with permission of John Wiley.)
Telangiectatic metastatic carcinoma. Telangiectatic metastatic carcinoma is typically associated with breast cancer and may be difficult to diagnose as tumour cells may be quite scanty and the telangiectasia quite subtle. The vascular changes may be more florid and lesional skin can resemble angiosarcoma.
Less common patterns of direct tumour invasion include breast carcinoma presenting as an inframammary intertrigo-like pattern, and lymphatic obstruction by pelvic tumours or lymphadenopathy, which may be accompanied by extensive tumour cells within the lymphatics, presenting as skin nodules. Oral tumours, usually squamous cell carcinomas, may extend to directly involve the skin of the face.
Local and in-transit metastases from primary epidermal tumours are discussed in the relevant chapters in this book; melanoma is the most important tumour in this context. It is a particular feature of malignant lymph node metastases (e.g. breast carcinoma, melanoma), and occasionally arises after diagnostic or therapeutic interventions such as needle aspiration of a tumour, pleural biopsy, drainage of malignant ascites or placement of other drains in the vicinity of a tumour. ‘Tumour spillage’ is the direct contamination of wounds with tumour cells during a laparoscopy or surgical procedure. This was once common, but the rates of laparo- scopic port site metastasis and laparotomy wound metastasis from tumour inoculation are now both in the order of 0.8% [25].
Cutaneous metastasis
Frequency of skin metastases
Metastasis to the skin is not as common as metastasis to liver, lung or bone. Autopsy studies suggest that up to 9% of patients with internal cancer have had skin metastases; a large analysis indicated that 5% is usual [1], and in about 0.5–1% a metastasis is the presenting feature of internal cancer [2, 3]. Of patients with metastatic cancer, 10% have cutaneous metastases [4]. Studies vary in cited frequency of skin metastases in part because some include metastasis from cutaneous melanoma whilst others exclude this and only refer to internally originating tumours. Additionally, it can sometimes be difficult to know whether skin involvement is from close proximity with a primary tumour of a different organ, that is, direct invasion not metastasis [5]. The most common sources of cutaneous metastases are, in generally accepted order of frequency: breast, (melanoma), lung, colon, stomach, upper aerodigestive tract, uterus and kidney. The most common skin metastases from a previously unknown primary tumour originate from the kidney, lung, thyroid or ovary [3, 6, 7].
Appearance of skin metastases
Cutaneous metastases generally present as painless, firm to hard nodules, which may be skin-coloured, blue-brown or reddish purple. The commonest pattern presents with a nodule that is solitary (Figure 147.5), and this presentation is about twice as frequent as multiple nodules (Figure 147.6). Dermal or subcutaneous nodules may be more readily detected by palpation as they may not initially be apparent or symptomatic (Figure 147.7) [6]. The ulceration of nodules may occur but is not usually a feature at initial presentation. Other patterns include morphoea-like sclerotic plaques, scar infiltration, erysipelas-like diffuse skin infiltration, infiltrated areas of alopecia (alopecia neoplastica) and embolic metastasis to digits (Figure 147.8).
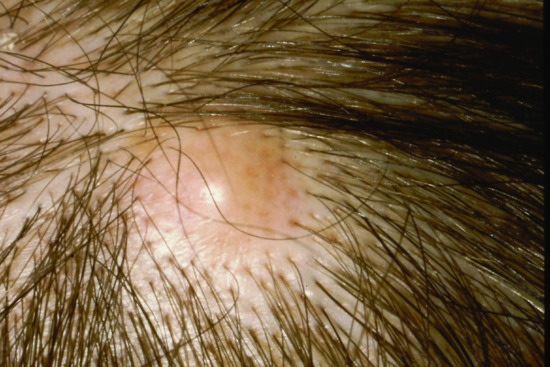
Figure 147.5 Solitary metastasis of squamous carcinoma to the scalp from an unknown primary.
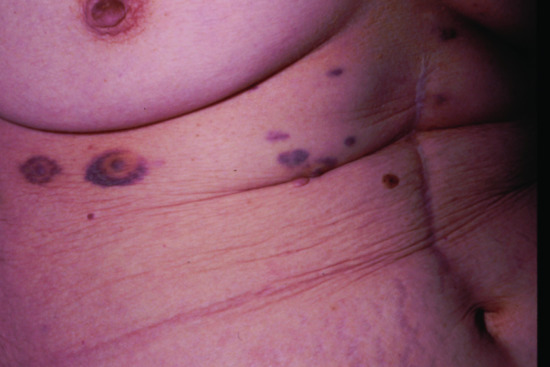
Figure 147.6 Metastatic pancreatic carcinoma manifesting as haemorrhagic nodules in the skin.
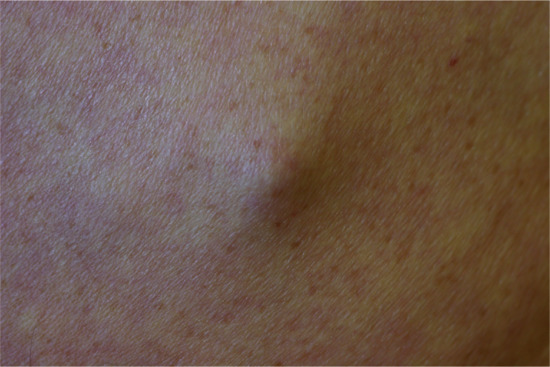
Figure 147.7 Subcutaneous metastasis from lung carcinoma; such metastases are typically multiple and many more firm nodules can usually be palpated if the patient is carefully examined.
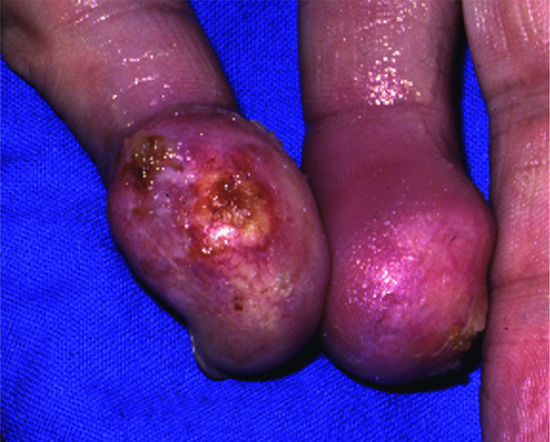
Figure 147.8 Embolic metastasis to the little finger of each hand from carcinoma of oesophagus.
Mechanisms and distribution
Metastasis to the skin occurs as a result of lymphatic or haematogenous dissemination of tumour. The distribution pattern on the skin is not random [3, 5]. Indeed, 75% of metastases are found on the head, neck and upper trunk, which together constitute only 25% of the body surface area [3]. The clinical pattern of metastases may provide clues to the route of metastasis. For example, metastases to extremities suggest intra-arterial embolic spread, widespread skin metastases suggest that tumour cells are present in the general circulation, whilst metastases to the skin in the vicinity of the affected organ is more suggestive of dissemination by lymphatic vessels or by veins (Figure 147.9). Retrograde lymphatic spread may occur from pelvic tumours, causing metastases in the perineal area or on the legs. In general, the head, neck and upper trunk are disproportionately affected by tumours that metastasize to the skin [3, 6–8], possibly because of the high vascularity of this area. Some of the tumour and host factors involved in metastasis, both generally and organ-specific, are reviewed in references [9, 10, 11, 12, 13]; in particular, chemokine receptors on tumour cells may influence the organs involved in metastatic spread [14, 15].
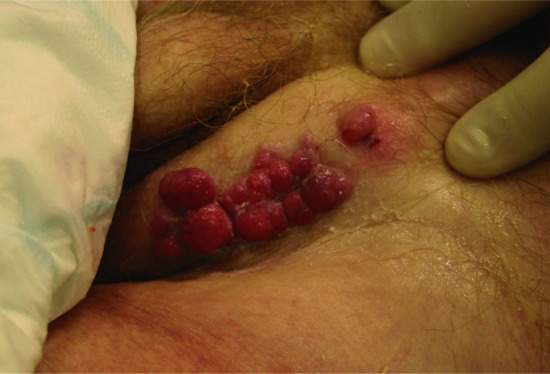
Figure 147.9 Metastasis to the groin in a patient with extensive pelvic prostatic carcinoma.
Other than the scalp, notable sites for skin metastasis include the umbilicus (Sister Mary Joseph nodule, related most commonly to bowel tumours) [7, 16, 17] and recent operative scars. Scar metastasis is most often related to surgery for the primary tumour, and, in view of the low frequency of ‘tumour spillage’, presumably represents spread by local lymphatic or venous drainage. However, recent but distant scars (e.g. skin graft donor sites [18, 19]) may be a site of metastases, suggesting a role for adhesion molecules, metalloproteinases or angiogenesis and tissue repair cytokines. Metastases have also been reported that are localized to a site of irradiation of the skin [20]. An additional, unusual phenomenon is ‘tumour-to-tumour metastasis’, in which metastases localize to another (usually benign) tumour [21]. A postulated mechanism is the trapping effect of fibrin in the vessels of the recipient tumour; most such metastases occur within quite vascular neoplasms, such as thyroid or adrenal adenomas, but metastasis to lipoma and to basal cell carcinoma have both been reported and are relevant to dermatologists [22].
Metastases to the scalp may give rise to focal alopecia, usually from carcinomas of the breast, lung or kidney. Renal and thyroid cancer metastases may be quite vascular in appearance, both clinically and pathologically [23], and are occasionally misdiagnosed as benign haemangiomas or pyogenic granulomas; metastases from renal cell carcinoma may even be pulsatile. Cutaneous metastases may be mistaken for cysts or inflammatory lesions; alopecia over a scalp ‘cyst’ should alert the physician to the possibility of malignancy.
Histopathology
Biopsy of a suspected skin metastasis will usually successfully confirm or exclude malignancy. It is not, however, always useful in determining the organ of origin if the tumour cells are poorly differentiated. Pathological diagnosis of specific metastases and immunohistochemical markers are discussed in references [7, 24]. Occasionally it may be difficult, even with use of immunohistochemical tumour markers, to distinguish reliably between primary skin tumours and metastatic disease. Although the cells of a metastasis usually resemble the cells of the primary tumour, some patterns may prove diagnostically difficult. Spindle cell tumours, and tumours comprising ‘small blue cells’, cause particular difficulty and may need a panel of immunohistochemical markers to aid in diagnosis both between primary skin lesions and metastases, or between different origins of an internal tumour.
In some instances there may be marked oedema or dilated lymphatic vessels that may make diagnosis difficult. In some specific tumours such as renal cell carcinoma, or in some clinical patterns such as carcinoma telangiectodes, the tumour cells may either be scanty or the vascular proliferation may dominate the histopathological appearance. Lymphatic spread of tumour cells may lead to an ‘Indian filing’ appearance in some cases, sometimes with fibrosis.
Prognosis
Cutaneous metastases usually occur in subjects with a known cancer, but may be the first indication of an internal neoplasm, especially in the case of lung cancers. They are usually suggestive of disseminated disease and indicate a correspondingly poor prognosis: survival is typically only about 3 months in patients with disseminated skin metastases [4]. Patients with solitary metastases without other evidence of dissemination may have a better survival rate [25]. Infrequent cases of tumour regression after primary tumour removal have been documented [26]. It is noteworthy that cutaneous metastases do not necessarily relate to a prior, documented tumour; the histological pattern, localization and temporal relationship may occasionally point to a second primary [27]. Treatment options, depending on the primary tumour, may include excision or other destructive therapy (e.g. laser destruction, radiotherapy, photodynamic therapy) for limited numbers of lesions, and chemotherapy or other systemic treatment for disseminated lesions.
Paget disease
Paget disease occurs in mammary and extramammary forms. Paget disease of the breast occurs due to an underlying ductal tumour [1]. Extramammary Paget disease may occur without an underlying tumour, or distant to a tumour, and so does not have the close link with direct tumour spread of the mammary pattern [2].
Paget disease of the breast
This condition presents with scaling and erythema, sometimes with oozing and crusting, on or around the nipple (Figure 147.10). It is generally viewed as a direct epidermal extension of an underlying ductal adenocarcinoma, and is important as the underlying tumour is usually small and superficial so early recognition may allow curative intervention. However, of the 30–50% patients with a palpable underlying lesion, half will have axillary lymph node involvement. The reason for the epidermotropic spread of tumour cells is uncertain but may involve a keratinocyte-derived chemoattractant for Paget cells, termed heregulin-α [2]. The main pathogenetic hypothesis for those cases that do not have an underlying carcinoma is derivation from Toker cells within the epidermis of the nipple and areola [2].
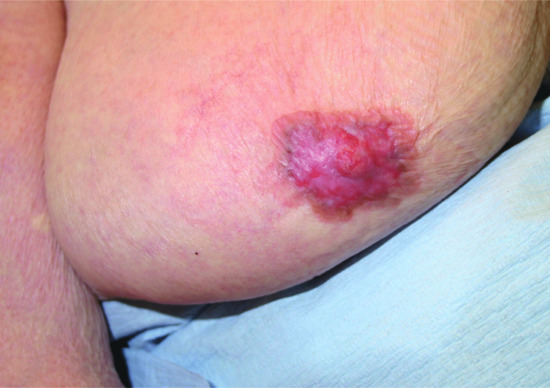
Figure 147.10 Paget disease of the breast causing destruction of the right nipple.
Eczema, psoriasis, hyperkeratosis and erosive adenomatosis are in the clinical differential diagnosis of mammary Paget disease. Histologically, there may be diagnostic problems from other disorders with Pagetoid spread, such as some melanomas or Bowen disease, and also from a benign proliferation of Toker cells termed clear cell papulosis. Useful histopathological markers for mammary Paget disease include epithelial membrane antigen (EMA), carcinoembryonic antigen (CEA), cytokeratins CK7 and CK8/18, as well as mucins such as MUC1 [3, 4]. CD23, a lymphoid and apocrine/eccrine marker, is uniformly positive in mammary or extramammary Paget disease [1, 2].
Extramammary Paget disease
Extramammary Paget disease (EMPD) occurs in apocrine gland-bearing areas such as anogenital and axillary sites (see Chapters 111 and 112). About 60% of cases are vulval, 20% perianal and 15% penile or scrotal; there is thus a female preponderance (although some populations, such as the Japanese [3], have a male preponderance). Its histogenesis is less certain, and only about 25% of patients appear to have an associated invasive malignancy. Of the malignancies associated with vulval Paget disease, a little under half arise locally from apocrine sweat glands or Bartholin's glands; the remainder arise from the vagina, cervix, bladder, ovary, colon or rectum, or occasionally from more distant sites such as the breast or gallbladder [4]. Perianal Paget disease (Figure 147.11a) is associated with an adnexal tumour in about 10% and a distant tumour in about 25% (rectum, stomach, ureter, breast) [4]. Male genital Paget disease (Figure 147.11b) is associated with carcinoma of the prostate, bladder, ureter, kidney or testes in about 10% of cases [4]. In other cases, EMPD arises locally in the epidermis.
Most patients are over 60 years of age. Clinical features include itch, a burning sensation, oedema, bleeding and reddish brown (or sometimes hypopigmented) plaques, often with a prominent margin. The margin may, however, be obscured by secondary infection and may be difficult to define in vaginal mucosa. Genital EMPD is often mistaken for an eczematous process, psoriasis or tinea. Histological and immunohistochemical features are similar to those of mammary Paget disease; the antigen RCAS1 is particularly sensitive [5]. The prognosis is determined by the underlying tumour, if present, and whether or not this is amenable to curative treatment. For associated malignancies of a local epidermal origin, the depth of invasion and lymphatic spread dictate the prognosis, serum CEA being a useful indicator of survival [3].
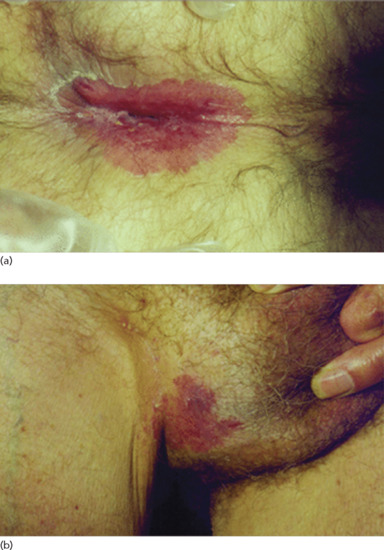
Figure 147.11 Extramammary Paget disease. (a, b) Well-defined erythematous plaques involving perianal skin (a) and the scrotum (b). (From Wagner and Sachse 2011 [6], with permission of John Wiley.)
GENODERMATOSES ASSOCIATED WITH INTERNAL MALIGNANCIES
There are many genodermatoses that are associated with an increased risk of internal malignancy (Table 147.1) [1–10]. A number of mechanisms are involved: these include chromosomal instability, faulty DNA repair mechanisms, abnormal lymphocyte function and immunosurveillance, and, in some cases, a combination of these processes.
Table 147.1 Examples of genodermatoses associated with internal malignancies.
| Main organ affected or usual mode of presentation (many are multisystem disorders) | Genodermatosis | Main neoplasms (may be limited to some families in some of the disorders listed) |
| Gastrointestinal tract | Gardner syndrome | Gastrointestinal polyposis and carcinomas, central nervous system tumours |
| Bannayan–Riley–Ruvalcaba syndrome | ||
| Turcot syndrome (mismatch repair cancer syndrome) | ||
| Peutz–Jeghers syndrome | Gastrointestinal polyposis and carcinomas, pancreatic carcinoma, genital tumours (especially Sertoli cell, sex cord and cervix), breast cancers, lung cancers | |
| Neurological | Ataxia–telangiectasia | Lymphomas, leukaemias |
| Neurofibromatosis | Neurological tumours, sarcomas, phaeochromocytoma | |
| Skin | Xeroderma pigmentosum | Skin cancers, sarcomas, central nervous system tumours, leukaemia, various solid organ tumours |
| Naevoid basal cell carcinoma syndrome (Gorlin) | Basal cell carcinomas of skin, medulloblastoma | |
| Bazex–Dupré–Christol syndrome | Basal cell carcinomas of skin, possible leukaemia | |
| Porphyria cutanea tarda | Hepatocellular carcinoma | |
| Tylosis | Oesophageal carcinoma | |
| Sclerotylosis (Huriez syndrome) | Squamous cell carcinoma of skin; oral and bowel cancers also reported | |
| Muir–Torre syndrome | Colorectal tumours, sebaceous carcinoma | |
| Birt–Hogg–Dubé syndrome and Hornstein–Knickenberg syndrome | Medullary carcinoma of thyroid, renal cell carcinoma | |
| Familial leiomyomas (also uterine) | Renal cell carcinoma, others (see text) | |
| Incontinentia pigmenti | Wilms tumour, rhabdomyosarcomas (renal, paratesticular), retinoblastoma, leukaemias | |
| Familial atypical naevi and melanoma | Pancreatic carcinoma, cutaneous and ocular melanoma | |
| Melanoma–astrocytoma syndrome | Melanomas, astrocytomas and other central nervous system tumours | |
| Supernumerary nipples | Genito-urinary tumours: renal cell carcinoma, Wilms tumour, bladder, testicular, prostate | |
| Ichthyoses (autosomal dominant and X-linked) | Testicular carcinoma | |
| Endocrine | Multiple endocrine neoplasia syndromes | Medullary carcinoma of thyroid, phaeochromocytoma |
| Growth/skeletal | Werner syndrome | Many, especially sarcomas |
| Rothmund–Thomson syndrome | Skin, osteosarcoma | |
| Bloom syndrome | As general population but early onset | |
| Maffucci syndrome | Chondrosarcomas, gliomas, ovarian cancers | |
| Goltz syndrome | Chondrosarcomas, giant cell tumour of bone | |
| Fanconi anaemia (usually presents due to congenital malformations) | Myelodysplastic syndrome, acute myelogenous leukaemia, hepatic carcinoma | |
| Haematopoietic | Dyskeratosis congenita | Mucosal squamous cell carcinoma, haematopoietic malignancy and others |
| Immunological | Wiskott–Aldrich syndrome | Lymphoreticular malignancies |
| Chediak–Higashi syndrome | Lymphoreticular malignancies | |
| Multisystem | Cowden's (multiple hamartoma and neoplasia) syndrome | Breast, thyroid, gastrointestinal, cerebellum, endometrial and renal carcinomas |
| Carney complex | Myxomas, schwannomas, testicular Sertoli cell tumour, pituitary adenomas, thyroid cancer | |
| Von Hippel–Lindau disease | Phaeochromocytoma, renal carcinoma, haemangioblastoma, pancreatic carcinoma | |
| Beckwith–Wiedemann syndrome (exomphalos– macroglossia–gigantism syndrome) | Wilms tumour, adrenal carcinoma, hepatoblastoma, pancreatoblastoma, others (especially in patients with hemihypertrophy) |
Howel–Evans syndrome
This syndrome is the association of autosomal dominantly inherited focal palmoplantar keratoderma (tylosis) with the eventual development of oesophageal carcinoma (see Chapter 65). Oral leukoplakia also frequently occurs in Howel–Evans syndrome [1–8].
Naevoid basal cell carcinoma syndrome
Naevoid basal cell carcinoma syndrome is the association between basal cell carcinomas, mandibular odontogenic keratocysts, skeletal anomalies, abnormal calcification and dyskeratotic pits of palms and soles (see Chapter 141). Patients with this syndrome are at risk of developing medulloblastoma and ovarian tumours. Other rare central nervous system (CNS) tumours have been reported in naevoid basal cell carcinoma syndrome [1–10].
Familial melanoma syndrome
Typically, there is one or more of the following: a family history of melanoma, a personal history of multiple atypical melanocytic naevi, or a family history of multiple atypical melanocytic naevi (see Chapter 143). Large multiple and irregular naevi are the norm, with an early onset of melanoma. Multiple primary melanomas may occur and, in some pedigrees, there is an increase in non-cutaneous malignancies.
The mode of inheritance is probably polygenic. The CDKN2A germline mutation in the 9p21 gene region has been implicated in most cases [1, 2]. Mutations that impair p16 function are linked with increased risk (22-fold) in pancreatic cancer, and specific mutations have been linked with breast cancer in some populations [2]. Cases with pancreatic, gastrointestinal, lung, breast and laryngeal cancers are published.
Melanoma–astrocytoma syndrome
This syndrome comprises an association of melanomas with astrocytomas or other CNS tumours, meningiomas, ependymomas and peripheral nerve tumours, such as malignant schwannoma (see Chapter 143) [1–4]. Deletions of chromosome 9p21, which includes tumour suppressor and cell cycle regulating genes, are implicated. Several genetic loci have been implicated in different families, including all or parts of the p16, p19 and p15 gene cluster (INK4 locus), which includes CDKN2A and CDKN2B (suggesting contiguous suppressor gene deletion) [1–3], and a specific deletion of p14(ARF) [4].
Xeroderma pigmentosum
Xeroderma pigmentosum (XP) characteristically presents at an early age with severe photosensitivity (see Chapter 78). Patients display a marked congenital reduction in threshold for sunburn and present with myriads of lentigines, principally in a sun-exposed distribution. Early onset of photoageing is found in infants, followed by sun-induced dysplasias, basal and squamous cell carcinomas and malignant melanomas, commencing in the first decade of life [1].
The mode of inheritance is autosomal recessive. The mutations causing XP cause abnormal fibroblast sensitivity to ultraviolet radiation, in most cases (complementation groups A–G) resulting from a defective DNA nucleotide excision repair process. The eighth form, termed XP variant, codes for a post-replication repair polymerase. Inactivation of tumour suppressors and activation of oncogenes due to these mutations results in development of multiple tumours [2].
Ocular neoplasms, both melanoma and non-melanoma, occur in 10–20% of individuals with XP. However, there is also a 10–20-fold increased incidence of internal malignancy in XP, including CNS sarcomas, leukaemia and carcinomas of the lung, breast, pancreas, stomach and testes [3].
Von Hippel–Lindau disease
Von Hippel–Lindau disease is an autosomal dominantly inherited condition that carries a high risk of internal malignancy and is associated with non-specific cutaneous manifestations, such as haemangiomas and café-au-lait spots [1, 2]. It is characterized by benign and malignant tumours of various systems, particularly haemangioblastomas of the CNS, angiomatosis of the retinae, phaeochromocytoma (which may be bilateral) [1], renal carcinoma [3], pancreatic adenoma, carcinoma and cysts [4], and epididymal cystadenomas.
The VHL gene has been located on chromosome 3p26-p25, although different mutations have been found to be causative. The gene is a tumour-suppressor gene following the Knudson two-hit model. Families may be characterized by the absence of phaeochromocytoma (type 1, deletions/protein-truncating mutations) or the presence of phaeochromocytoma (type 2, missense mutations) [5, 6].
Neurofibromatosis types 1 and 2
There are two main forms of neurofibromatosis: type 1 (NF1) and type 2 (NF2). Both have an autosomal dominant inheritance; in a proportion of affected individuals the condition is due to a de novo mutation.
The abnormal function of neural crest cells in NF1 leads to the development of multiple peripheral neurofibromas as well as CNS tumours, notably optic glioma (which is one of the diagnostic criteria for NF1) and café-au-lait macules [1–5]. Clinical overlap between NF1 and NF2 occurs particularly in children, who may have café-au-lait macules and peripheral nerve tumours; flexural freckling is indicative of NF1. Café-au-lait macules occur in NF2, but only in half of affected subjects and there are usually fewer than six in number (six or more being one of the diagnostic criteria for NF1). Lisch nodules (pigmented iris hamartomas) are found with slit lamp analysis in 90% of adult patients with NF1 and are fairly specific for NF1. Around 80% of patients with NF2 have posterior subcapsular cataracts, including about a third of affected children.
The associations with benign tumours, malignant tumours and systemic manifestations are varied but patients with NF1 have a 2.5-fold increase in risk of developing a malignancy [4]. NF2 patients develop vestibular and peripheral nerve schwannomas (the vestibular schwannomas characteristically being bilateral), together with CNS tumours such as meningiomas [3, 4]. Spinal schwannomas, astrocytomas or ependymomas eventually occur in about three-quarters of patients with NF2. If meningiomas occur in children, NF2 should be suspected. Perhaps the commonest neoplasm associated with neurofibromatosis is a malignant neurofibrosarcoma [6]. Most superficial neurofibromas have a low malignant potential, change occurring more often in the deep plexiform neurofibromas and those in continuity with peripheral nerves, designated schwannomas. Benign tumours such as acoustic neuromas, dumb-bell tumours and optic gliomas can result in disastrous sequelae when occurring in confined, pressure-sensitive sites. The commonest CNS malignancy is an astrocytoma. Other malignancies include nephroblastoma (Wilms tumour), fibrosarcoma, rhabdomyosarcoma and leukaemia, especially in children [3–8]. There is an association between NF1 and juvenile myelomonocytic leukaemia and juvenile xanthogranulomas [9]. Monosomy 7 myelodysplastic syndrome may also occur [10]. There is an increased frequency of phaeochromocytomas and carcinoid tumours [4], and ocular melanoma has been reported [11].
Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) consists of angiokeratomas, epilepsy and learning difficulties. It may be associated with multisystem tumour involvement, mostly hamartomatous. TSC is an autosomal dominant condition but there is a high spontaneous mutation rate which accounts for over half of cases. There are two known causative genes – TSC1 on chromosome 9q34, encoding hamartin, and TSC2 on chromosome 16p13.3, encoding tuberin; both genes function as tumour suppressor genes. There are large numbers of different mutations, TSC2 mutations being more common, and causing more severe disease, than TSC1 mutations.
Other than skin lesions, the CNS, renal and cardiopulmonary systems are most significantly affected [1–3]. Angiomyolipomas (of vessels, fat and smooth muscle) all show the same loss of heterozygosity at the TSC1 or TSC2 loci. Malignant sarcomatous change can occur, particularly with angiomyolipomas and rhabdomyomas, but is uncommon, and metastases are unusual [4]. Renal cell carcinoma is a recognized, infrequent complication [3–6].
Multiple endocrine neoplasia syndromes
Multiple endocrine neoplasia type 1
This is a familial cancer syndrome, with parathyroid, pancreas and pituitary gland tumours, as well as cutaneous findings. Dermatological features in MEN1 are typically multiple facial angiofibromas (22–88%) and collagenomas (0–72%). Lipomas may occur in more than 33% of patients with MEN1. Café-au-lait macules are also encountered [1–3].
It is caused by mutations in the MEN1 gene, located on chromosome 11q13, which codes for the production of a protein named menin.
MEN1 is characterized by tumours of the parathyroid, anterior pituitary, pancreatic islet cells, cells of neuroendocrine origin, foregut carcinoid and adrenal cortex. It is associated with 20–25% of cases of Zollinger–Ellison syndrome (ZES). In MEN1, 60–100% of cases have gastroenteropancreatic lesions, especially pancreatic islet tumours, also referred to as pancreatic neuroendocrine tumours (NETs). The incidence of tumours is as follows: ZES 54%, insulinoma 21%, glucagonoma 3% and VIPoma (vasoactive intestinal peptide) 1%. In MEN1, associated endocrine disease consists of parathyroid hyperplasia in over 95%, anterior pituitary adenomas in 15–50%, adrenal adenomas in 30% and carcinoid tumours in more than 3% (carcinoid tumours are located in the bronchi, gastrointestinal tract, pancreas or thymus). The pituitary adenomas secrete prolactin in 60%, growth hormone in fewer than 25% and adrenocorticotrophic hormone (ACTH) in 5%. The commonest tumours in MEN1 secrete parathyroid hormone or gastrin.
Metastases from malignant neuroendocrine tumours, in particular malignant pancreatic NET and thymic carcinoid tumours, are one of the commonest causes of mortality in patients with MEN1 [1–6].
Multiple endocrine neoplasia type 2A
Both MEN2A and MEN2B principally involve the thyroid and parathyroid glands, and the adrenal medulla, and are linked with familial medullary thyroid carcinoma [7, 8]. MEN2A lacks the mucosal neuromas and skin lipomas of MEN1 and MEN2B. Café-au-lait macules are only present in those with a combined phenotype of MEN2A with NF1. In MEN2A there may be symmetrical, bilateral, pruritic skin lesions found overlying the scapular area, with hyperpigmentation and hyperkeratosis clinically suggestive of amyloidosis; deposits of keratin-derived amyloid are typically found histologically [4, 6, 9].
MEN2A and MEN2B are caused by mutations of the RET proto-oncogene locus (10q11.2) [10–12]. Both are autosomal dominant, but 50% of cases with MEN2B are due to spontaneous mutations. RET testing has replaced calcitonin screening to diagnose MEN2 carrier status. The specific RET codon mutation will delineate the course of the disease and degree of aggression [4, 5, 11, 12, 13]. Screening of all first-degree relatives should be performed in order to identify RET-mutated gene carriers.
The main internal disorders in MEN2A, described as the triad of cardinal manifestations, are medullary thyroid carcinoma (MTC), phaeochromocytoma and hyperparathyroidism (due to either hyperplasia or adenomas). Other hamartomas and tumours include cerebellar haemangioblastomas, cervical neuroblastoma, pituitary adenomas and pinealomas [9].
Multiple endocrine neoplasia type 2B
This is characterized by mucosal neuromas that are apparent at birth or in the first years of life [14]. Neuromas manifest as asymptomatic, soft, flesh-coloured papules or nodules. They cause a characteristic facial appearance with soft, lumpy (‘blubbery’), protuberant lips; everted, thickened, bumpy eyelids; and prominent eyebrows. Neuromas typically affect the mucosal surfaces, especially the anterior border of the tongue and the buccal mucosa inside the commissures of the lips; gingival, palatal and pharyngeal surfaces may occasionally be affected. Cutaneous nodules or plaques, often linear in shape and hyperpigmented, are occasionally reported, with the histopathological picture of dermal nerve hypertrophy and clinical hyperpigmentation due to chronic scratching and trauma [14]. About 75% of patients have a marfanoid appearance; muscle weakness and musculoskeletal anomalies (especially kyphoscoliosis, pes cavus and bilateral slipped upper femoral epiphysis) may also be present [10].
Hyperparathyroidism due to parathyroid hyperplasia or adenomatosis is much less common than in MEN1 and MEN2A. Intestinal ganglioneuromatosis is more common in type 2B than 2A, occurring in 30% and often presenting early in life due to constipation or abdominal pain.
MEN2B is associated with MTC in 75% of cases, and phaeochromocytoma in almost 50%. MTC in type 2B presents earlier and more aggressively than in type 2A. The MTC is often multicentric and bilateral, occurring in a background of calcitonin-producing cell hyperplasia. Early lymphatic spread may occur, 75% having metastases at presentation [10]. Phaeochromocytomas are often bilateral, but the mortality is greater from MTC than from phaeochromocytoma.
Carney complex
This is a group of disorders in which there are cutaneous pigmented lesions associated with cutaneous, subcutaneous and internal myxomas, and associated endocrinopathy (mainly tumours), including involvement of one or more of the adrenal cortex, thyroid, pituitary and gonads [1]. The NAME syndrome consists of naevi (congenital melanocytic), atrial myxomas, myxoid neurofibromas and ephelides. The LAMB syndrome consists of lentigines, atrial myxomas, mucocutaneous myxomas and blue naevi. The association with myxomas has led to many other names; some prefer to use myxoma syndrome, or the combined term Carney complex/myxoma syndrome [2].
Three of the major diagnostic criteria involve the skin: spotty cutaneous pigmentation (lips, conjunctiva, eyelids, genital mucosa), mucocutaneous myxomas and multiple blue naevi [3]. Cardiac myxomas occur in 61% [4]. The adrenal tumours in Carney complex are typically of the primary pigmented nodular type, an otherwise rare condition. Various other gonadal and endocrine hormone-secreting tumours, including pituitary tumours producing growth hormone, prolactin or ACTH, are found; ovarian tumours are associated, and both benign and malignant thyroid tumours (usually of follicular type) also occur [5, 6]. Testicular tumours, often large-cell calcifying Sertoli cell tumours, occur in about 30% of males, and are often bilateral and multicentric. Psammomatous melanotic schwannoma, usually of the upper gastrointestinal tract or of the paravertebral sympathetic nerves, is very suggestive of this syndrome. Myxoid fibroadenomas of the breast, and mammary ductal adenomas, may be found [2], and myxoid leiomyomas and uterine tumours are described. Lentiginosis occurs in 70–75% and blue naevi in about 50% of patients with Carney complex [2, 3]. Skin myxomas occur in over a third of cases [4]. Multiple mucocutaneous myxomas are the most specific cutaneous marker for Carney complex, however these lesions are difficult to recognize clinically [3].
PTEN hamartoma tumour syndrome
This rare, cancer-associated genodermatosis was first described by Lloyd and Dennis and named after their patient Rachel Cowden [1, 2].
Pathognomonic criteria for the PTEN (phosphatase and tensin homologue) hamartoma tumour syndrome are mucocutaneous lesions, facial trichilemmomas (at least three), acral keratosis (at least six palmar lesions), papillomatous lesions and mucosal lesions. These are found in over 90% of patients. The mucosal lesions comprise a warty, ‘cobblestone’ hyperplasia of the mucosal surfaces, particularly affecting the tongue and buccal mucosa (Figure 147.12). Periorificial facial papules, acral warty keratoses and palmoplantar, semitranslucent, punctate keratosis are characteristic. The lesions, which are grouped especially around the mouth, nose and ears, have a hyperkeratotic, flat-topped, wart-like appearance, as do many lesions elsewhere; these are mostly trichilemmomas or related benign tumours of the follicular infundibulum [3–7]. Multiple hamartomatous lesions of ectodermal, endodermal and mesodermal origin occur. The other cutaneous lesions include ganglioneuromas, lipomas, fibromas, angiomas, angiolipomas, epidermoid cysts and a variety of pigmentary changes. Craniomegaly is common; there may be an adenoid facies, kyphoscoliosis and a high-arched palate.
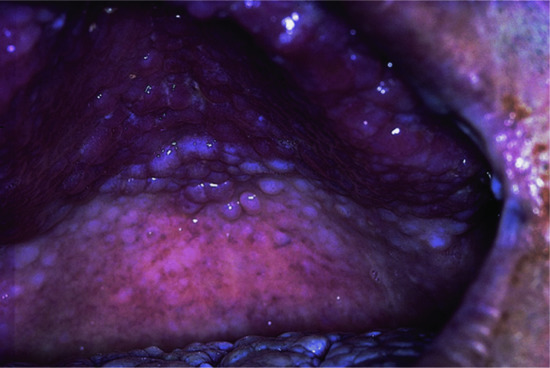
Figure 147.12 Warty papillomatosis of the hard palate in PTEN hamartoma tumour syndrome.
(Courtesy of Dr R. Emmerson, Royal Berkshire Hospital, Reading, UK.)
Seizures and learning difficulties occur (the latter is a minor diagnostic criterion) and there may be an association with meningioma.
Recently, a new entity of neoplasm, called PTEN hamartoma of soft tissue, has been suggested [8]. It is often located in the lower leg and consists of tissue of mesenchymal origin with a histopathologically distinctive picture that differs from other PTEN-associated vascular and connective tissue hamartomas [9].
Benign internal anomalies are numerous, most commonly affecting the breast (severe fibrocystic disease occurs in the majority of women) and the thyroid (mainly multinodular goitres and adenomas). Gastrointestinal polyposis and cysts or polyps of the female genito-urinary system are also frequent.
The cumulative risk of developing cancer in patients with PTEN hamartoma tumour syndrome is elevated (85% at age 70), especially in women [9]. The most frequently reported cancers are female breast cancer (bilateral in almost half of the cases) and thyroid cancer [9]. Fibrocystic breast disease and cancers may have an early onset, and screening of at-risk family members is therefore recommended. Breast, endometrial and thyroid cancers all contribute to a higher mortality in females. Also, renal carcinomas have been linked with this syndrome, and an increased likelihood of melanoma has been suggested [9].
Sebaceous tumours, keratoacanthomas and visceral malignancy
This is a cancer-associated genodermatosis in which there is an association between sebaceous lesions, and to a lesser extent keratoacanthomas, and internal malignancy (see Chapter 138). Inheritance of this syndrome is autosomal dominant with variable expression, males being affected more commonly than females [1–3].
Sebaceous tumours are usually multiple, but occasionally solitary. Although sebaceous adenoma is the commonest, sebaceous carcinoma and epithelioma frequently occur, and within the same patient a variety of different pilosebaceous-derived skin lesions including keratoacanthomas may arise. Most skin tumours occur in middle age, keratoacanthomas occurring in a quarter of affected subjects. Multiple or early onset of keratoacanthomas are suggestive of this diagnosis, as are multiple (especially eyelid) sebaceous tumours.
Up to 60% of affected individuals develop sebaceous neoplasms preceding visceral malignancies [4]. It has been suggested that when a sebaceous neoplasm is identified individuals should be screened for internal malignancies [4]. Criteria for diagnosis (Amsterdam and Bethesda criteria) and recommendations for screening of patients and relatives have been reviewed [5].
The most important internal malignancy is colonic adenocarcinoma, which occurs in almost 50%, often at a relatively young age – around 10 years earlier than in the normal population – and most commonly in the region of the splenic flexure; rectal adenocarcinomas also occur but only in about 5% [6, 7]. Urogenital malignancies are also common, occurring in 25% [6]; bladder, renal, pelvis and endometrial cancers each account for about 5% of cancers in this syndrome. Nearly half of affected patients have two or more internal malignancies [8, 9]. Other notable malignancies are breast cancers, haematological malignancies [6], small intestine adenocarcinoma, head and neck squamous cellcarcinoma and lung carcinoma [7]. Despite the high risk of malignancy, both the malignant cutaneous sebaceous tumours and the colonic tumours tend to have relatively indolent behaviour (the 50% survival time for colonic cancers is about 12 years [2]) and the incidence of metastases is relatively low.
Recently, a clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir–Torre variant of Lynch syndrome was published by Roberts et al. [10].
Hereditary leiomyomatosis and renal cell carcinoma syndrome
Hereditary leiomyomatosis and renal cell cancer (HLRCC) form an autosomal dominant tumour syndrome caused by heterozygous germline mutations in the fumarate hydratase (FH) gene [1, 2].
The syndrome comprises multiple cutaneous leiomyomas with uterine leiomyomas; in some cases there is also an association with aggressive renal cell carcinoma, mainly of the papillary cell type. For cutaneous leiomyomas, the indication for treatment is the alleviation of cosmetic and pain-related complications. For solitary tumours, surgical excision is commonly applied. Multiple painful lesions require management by medical measures [3].
Lifetime risk for renal cancer in FH mutation carriers is estimated to be 15% [4]. Most women with HLRCC syndrome develop uterine leiomyomas (fibroids) with early onset. One study of 35 FH mutation carriers found that cutaneous leiomyomas were present in all subjects older than 40 years. Eleven out of 21 female mutation carriers underwent surgical treatment for symptomatic uterine leiomyomas, at an average of 35 years. Two of the FH mutation carriers had renal cancer [1].
Several other tumours have been reported in FH mutation carriers, including benign ovarian mucinous cystadenomas, renal cysts, adrenal gland adenomas, thyroid lesions, uterine leiomyosarcomas, breast, bladder and brain tumours, lymphoid malignancies and basal cell carcinomas. However, the significance remains unclear as increased risk for these tumours in the FH mutation carriers has not been shown [3, 5].
The main focus of management in HLRCC is prevention of disease and death due to renal cancer. A surveillance protocol has been proposed [4].
Bloom, Rothmund–Thomson and Werner syndromes
Bloom, Rothmund–Thomson and Werner syndromes are rare (≤1/50 000 live births), autosomal recessive diseases. These conditions are considered together as they are all caused by RecQ helicase gene mutations, and they all predispose to abnormal growth, premature ageing and increased incidence of site-specific malignancies (see Chapters 77, 78 and 79) [1, 2].
Bloom syndrome
Affected subjects have small stature and slight build, a sun-sensitive telangiectatic facial rash and café-au-lait macules. Minor congenital anatomical abnormalities commonly occur. Deficient cellular and humoral immunity is common.
Mutations of the gene designated BLM on chromosome 15q26.1 lead to inhibition of the function of the protein product, a DNA helicase enzyme. This loss of function allows genomic instability with the occurrence of significantly increased exchanges between DNA strands during the mitosis, including an increase in sister chromatid exchanges, such that mutations occur throughout the genome.
The occurrence of lymphoproliferative neoplasia (approximately equally divided between leukaemias and lymphomas) and epithelial tissue cancers, particularly of the aerodigestive tract and lower gastrointestinal tumours, is very high; they typically occur at an early age, and the mean age of death is 23 years [1]. Cervical cancer and Wilms’ tumour are additional risks. There is a predisposition to malignancy through mutations in other target genes [2].
Rothmund–Thomson syndrome
This autosomal recessive disease is characterized by developmental abnormalities in the skin and skeletal systems, with photosensitivity, poikiloderma, small stature, premature ageing and juvenile cataracts. There is a predisposition to certain malignancies, including a 30% incidence of osteosarcoma. Fibrosarcoma, myelodysplasia and non-melanoma skin cancer may also occur [1–4].
Werner syndrome
This is a condition of premature ageing, with onset in the second to third decade of life. The key clinical findings include short stature, early greying and loss of hair, bilateral cataracts and scleroderma-like skin changes. Werner syndrome is a chromosome instability syndrome and, as with XP, ataxia-telangiectasia, Bloom syndrome and Fanconi anaemia, is associated with a high incidence of neoplasia [1, 2]. Neoplasms develop in about 10% of cases, although the commonest cause of death is arteriosclerosis. Sarcomas, melanomas, leukaemia, meningiomas and a variety of epithelial-derived carcinomas have been reported [1–7].
IMMUNODEFICIENCY AND NEOPLASIA SYNDROMES (see also Chapter 82)
Wiskott–Aldrich syndrome
Wiskott–Aldrich syndrome is an X-linked recessive immunodeficiency syndrome due to mutations at Xp11.23-p11.22. The Wiskott–Aldrich syndrome protein (WASp) is expressed in all haematopoietic cells, and mutations cause defective T-cell function and thrombocytopenia. In addition to bleeding and susceptibility to infections, severe eczema may occur; older patients also reveal an increased risk of autoimmune disorders and lymphoid malignancies [1]. Non-Hodgkin lymphoma occurs in almost all subjects who survive infections or bleeding due to thrombocytopenia, usually by the age of 30 years. Lymphoma (especially large cell or immunoblastic) and leukaemia also occur; the small intestine is a particular site for lymphomatous involvement. Cerebral tumours such as astrocytoma and various sarcomas have also been reported [2–4]. Recently, haemato-poietic stem cell transplantation treatment with good outcome has been reported [1, 5].
Chediak–Higashi syndrome
This is a fatal, autosomal recessive disorder with features of oculocutaneous albinism, silvery hair, photophobia, neurological abnormalities and severe, recurrent bacterial infections. There is extensive organ infiltration with lymphoid and histiocytic cells. Patients develop fever, jaundice, hepatosplenomegaly, lymphadenopathy, leukaemia-like gingival lesions and sloughing of the oral mucosa, pancytopenia and neurological deterioration. Although strongly suggestive of lymphoma, the infiltrate of affected organs is reported to be of a reactive, diffuse, mononuclear cell type, rather than neoplastic [1]. About 85% of patients develop an accelerated ‘lymphomatous’ phase, which may be triggered by viral infections, especially Epstein–Barr virus [2].
Ataxia-telangiectasia
This is an autosomal recessive condition characterized by progressive cerebellar ataxia and oculocutaneous telangiectasia. Premature ageing of the skin and hair is noted in almost 90% of individuals. Non-infectious cutaneous granulomas, with a tendency to ulcerate, are frequent [2]. The skin findings do not usually occur until 3–6 years of age, and are preceded by ataxia [3]. Peripheral blood lymphocytes are abnormal, and there is a variable but progressive immune deficiency of both the cell-mediated and humoral types [2, 4]. Chronic and recurrent sinopulmonary infections occurs in a majority of patients with ataxia-telangiectasia, and represent the most common cause of death [2].
Most families have one of many different germ-line mutations in the large ATM gene at chromosome 11q22.3. The product, ATM protein, is involved in the handling of chromosome strand breaks and activation of multiple targets, among them the p53 oncogene [2, 3], and has been considered to be a ‘caretaker’ of the genome and tumour suppressor. Mutations are mainly inactivating in type but may be missense [5]; they allow unregulated DNA synthesis, with DNA that is predisposed to instability and hypersensitive to ionizing radiation [3].
There is a high incidence of neoplasia – with an approximately 30% lifetime risk – usually in or before the teenage years, and being the cause of death in 15% of affected individuals [3, 4, 6–9]. The majority (around 80%) of tumours are lymphoproliferative or leukaemic, although carcinomas of various sites also occur, the latter usually in older subjects [7, 8, 9]. Most haematological malignancies are B-cell lymphomas but 25% are leukaemias, notably chronic T-cell leukaemia with chromosome 14 translocations occurring in older patients. However, T-cell leukaemias do occur in younger patients, and both T-cell lymphomas and B-cell leukaemias are also encountered. Most tumours have early onset and may precede diagnosis of ataxia-telangiectasia from the cutaneous features; this is of considerable importance as standard radiotherapy doses are contraindicated. Heterozygotes (carriers) have a 5–10-fold increased risk of tumours, usually not lymphoid, including a fivefold increase in risk of breast malignancy in females [3, 6].
Dyskeratosis congenita
Dyskeratosis congenita (DC) is a multisystem inherited syndrome, with clinical and genetic heterogeneity, characterized by mucocutaneous abnormalities, bone marrow failure and a predisposition to cancer. Most cases have X-linked recessive inheritance but autosomal recessive and autosomal dominant forms also occur [10].
A triad of abnormalities are the most consistent and diagnostic mucocutaneous features, comprising reticulate hyperpigmentation of the skin, nail dystrophy and leukoplakia of the mucous membranes. Dental, skeletal, ocular and gastrointestinal abnormalities are common; learning difficulties, short stature and premature ageing also occur. Aplastic anaemia occurs in 50%, typically in the early teens, and is the main cause of mortality.
Eight DC genes have been characterized, of which a majority are important in telomere maintenance which is defective in DC patients, who usually have very short telomeres. The genetic advances in the last decades have led to the unification with several other severe multisystem disorders, including Hoyeraal–Hreidarsson and Revesz syndromes, as well as a subset of patients with aplastic anaemia, myelodysplasia, leukaemia and idiopathic pulmonary fibrosis [11].
Oro-pharyngeal carcinomas secondary to the mucous membrane lesions are the commonest form of malignancy. There is also increased incidence of internal malignancy, particularly gastrointestinal, including pancreatic adenocarcinoma and other haematological disorders, similar to those found in Fanconi anaemia [12, 13].
Fanconi anaemia
This is a genetically and phenotypically heterogeneous recessive disorder, characterized by diverse congenital malformations, progressive pancytopenia and predisposition to haematological malignancies and solid tumours [14]. The underlying defect in Fanconi anaemia is one of increased DNA cross-linkage (especially radiation induced) and defective DNA repair. The main role of Fanconi anaemia proteins is the repair of DNA interstrand cross-links, and the maintenance of genomic stability. To date, 16 genes have been identified as mutated in these patients and many more interacting genes have been discovered [15, 16].
Dermatologically, Fanconi anaemia is characterized by pigmentary abnormalities that may be diffuse (with accentuation around the neck and over joints), mottled, often with scattered darker macules, and sometimes just exhibiting localized café-au-lait macules. Scattered areas of hypopigmentation are also a common finding.
The main abnormality is progressive pancytopenia, which may lead eventually to the development of leukaemia. Multiple skeletal abnormalities occur, including digital hypoplasias, scoliosis and short stature. The relative risk of cancer in Fanconi anaemia is exceedingly high in comparison with the incidences expected in the general population [16]. Acute myeloid leukaemia and solid tumours are common, especially head and neck or gynaecological squamous cell carcinomas.
PARANEOPLASTIC PHENOMENA INVOLVING THE SKIN
Paraneoplastic dermatoses are skin conditions that have an association with internal malignancy but are not themselves malignant. At least one of the following defining characteristics should be present in order to consider a dermatosis as being related to an underlying malignancy:
- The malignancy and the cutaneous disorder should occur concurrently.
- The two disorders should follow a parallel course.
- There should be a specific tumour site or cell type associated with the cutaneous disease.
- There should be a statistical association between the two processes.
- There should be a genetic association between the two processes.
Paraneoplastic dermatoses may be classified in a variety of ways. Some authors include genodermatoses within the spectrum of paraneoplastic disorders [1] whilst others view these as a separate group [2, 3], or distinguish between paraneoplastic dermatoses [4], hereditary paraneoplastic syndromes [5] and hormonally mediated paraneoplastic syndromes [6]. They may be classified according to strength of association with malignancy, association with certain types of malignancy [7], by the type of eruption that occurs (papulosquamous, vascular, etc.) or by the apparent mechanism (hormone secretion, autoimmune, cytokine/growth factor, etc.).
The likelihood of finding a neoplasm in some of the better known paraneoplastic disorders may be graded as high, intermediate or low (Table 147.2). Nonetheless the evidence supporting paraneoplastic phenomena is highly variable. The majority of the literature is based on case reports or case series: most of the data are hypothesis-generating in nature, and only rarely supported by studies of epidemiology or pathogenetic pathways [7–14].
Table 147.2 Strength of correlation of some potentially paraneoplastic dermatoses with internal malignancy.
| Strength of correlation | Type of reaction pattern | Examples |
| Strong | Papulosquamous and figurate eruptions | Bazex syndrome Erythema gyratum repens Necrolytic migratory erythema |
| Epidermal conditions | Acanthosis palmaris (tripe palms) Florid cutaneous papillomatosis |
|
| Deposition disorders | Primary amyloidosis Scleromyxoedema Necrobiotic xanthogranuloma POEMS syndrome |
|
| Others | Acquired hypertrichosis lanuginosa Paraneoplastic pemphigus Carcinoid syndrome Trousseau syndrome |
|
| Moderate | Papulosquamous and neutrophilic eruptions | Sweet syndrome Pyoderma gangrenosum Dermatomyositis |
| Others | Multicentric reticulohistiocytosis Pityriasis rotunda |
|
| Weak | Epidermal conditions | Acanthosis nigricans in isolation Acquired ichthyosis (unless widespread, deeply fissured, truncal pattern) Eruptive seborrhoeic keratoses (sign of Leser–Trélat) |
| Deposition disorders | Scleredema Calcinosis cutis |
|
| Others | Vasculitis, Raynaud phenomenon, digital ischaemia Erythromelalgia Relapsing polychondritis Erythroderma/exfoliative dermatitis Digital clubbing (unless with hypertrophic osteoarthropathy) Pruritus Erythema annulare centrifugum Cushing syndrome |
POEMS, polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes.
ACANTHOTIC AND ICHTHYOTIC EPIDERMAL DISORDERS
Acanthosis nigricans
Acanthosis nigricans may be divided into two important categories, benign and malignant, although Schwartz [1] described eight types: benign, obesity associated, syndromic, malignant, acral, unilateral, medication induced (especially nicotinic acid) and mixed types.
Benign acanthosis nigricans is often associated with obesity or insulin resistance, and is common and usually mild (see Chapter 87). It has been documented in up to 7% of children, mainly in the teenage years; virtually all childhood cases are of the benign type although malignant acanthosis nigricans (functional adrenocortical tumour) has been reported in a paediatric patient [2].
Malignancy-associated acanthosis nigricans is much less common. It may have a rapid onset and progression to produce symmetrical, hyperpigmented, rugose, velvety plaques (Figure 147.13) [1, 2, 3, 4–6]. The axillae and other flexures are particularly affected, along with the areolar area and nape of the neck and, less commonly, mucosal surfaces. There may be prominent acrochordon-like papillomatosis arising from the plaques; the sign of Leser–Trélat and acanthosis palmaris (tripe palms) may coexist [6]. De novo development of acanthosis nigricans in adults, especially if progressive and associated with weight loss (most patients with insulin resistance have a rather stocky build), should raise suspicions that there is an underlying neoplasm, although cases have been described in which acanthosis nigricans has preceded a malignancy by 10 years or more [3]. If there is also generalized pruritus or the skin changes of tripe palms, then a malignancy is even more likely. Production by tumour cells of either transforming growth factor α or cytokines that activate insulin-like growth factors or their cutaneous receptors have been proposed as the pathogenetic mechanism.
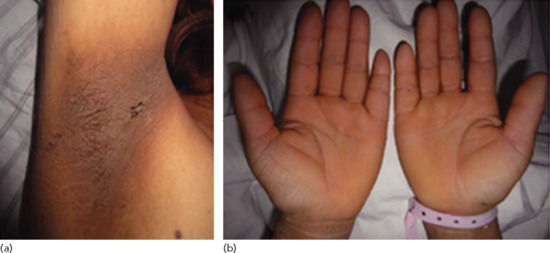
Figure 147.13 (a, b) Paraneoplastic acanthosis nigricans. Gastric adenocarcinoma was found after investigation of this woman with a 1-year history of velvety thickening of the skin of the palms, soles and flexures.
By far the commonest site of underlying neoplasm is the gastrointestinal tract (70–90%); gastric adenocarcinoma is the most frequent [1, 2, 3, 4–7]. A number of different malignancies have been reported, nearly all being adenocarcinomas, at sites including other parts of the intestine, liver or bile duct. Other tumours include lung, breast, endometrium, kidney, bladder, prostate, testis, cervix, thyroid and adrenal. Most are solid organ tumours but lymphoma has been recorded. Sarcomas occur rarely. The prognosis with malignant acanthosis nigricans is related to the survival rate from the neoplasia concerned. However, the skin changes may improve or resolve with eradication of the cancer [7, 8]. Rarely, malignancy-associated acanthosis nigricans has been associated with other paraneoplastic conditions including pachydermoperiostosis, paraneoplastic pemphigus and acquired hypertrichosis lanuginosa.
Acanthosis palmaris
Acanthosis palmaris describes thickened skin of the palms and occasionally the soles with an enhanced dermatoglyphic change, causing a velvety (Figure 147.13) or less commonly a pitted, honeycombed pattern of the hand. It is associated with neoplasia in about 90% of cases; it may be the only paraneoplastic manifestation in 30–40% or it may occur with one or both of malignant acanthosis nigricans or the sign of Leser–Trélat [1–3]. It occurs particularly in men, especially when the underlying tumour is a lung cancer [2]. However, it can occur in isolation without neoplasia, or as a pattern of exfoliative psoriasis or eczema [1, 2], and has been reported with bullous pemphigoid.
As the condition is usually associated with an internal neoplasm, usually of solid organ type, it requires appropriate evaluation and investigation. In the majority of cases, the onset of tripe palms precedes or occurred concurrently with the detection of a previously unsuspected malignancy [3]. Most commonly the underlying tumour is bronchial or gastric, together accounting for over half of the associated malignancies. Many other sites are reported including tumours of the genito-urinary tract (20%), as well as carcinomas in the breast and other organs [2, 3]. Acanthosis palmaris occurring alone is more often associated with bronchial carcinoma compared with combined acanthosis nigricans and acanthosis palmaris, in which gastric carcinoma is more common [3]. If nail clubbing is also present (18% of patients [2]), then bronchial carcinoma is very likely. Thus, tripe palms alone, especially if the patient is male and also has clubbing, very strongly suggests an underlying lung cancer, whilst tripe palms with acanthosis nigricans is more suggestive of an underlying gastric carcinoma. The appearance or exacerbation of tripe palms in a known cancer patient may be a sign of recurrence of the malignancy [3].
Sign of Leser–Trélat
This is the sudden development of numerous seborrhoeic keratoses, in an eruptive fashion, with or without pruritus, as an indicator of internal malignancy. However, the significance of eruptive seborrhoeic keratoses remains unclear, with strong proponents and opponents of its importance [1–4]. Multiple seborrhoeic keratoses are extremely common, especially in elderly people, and may be pruritic or rapidly erupting, without any apparent cause. They may also occur in other situations [4, 5] such as HIV infection, acromegaly and in the resolving phase of erythrodermic dermatoses [6].
Of the cases reported with a neoplasm, half of the tumours are adenocarcinomas, most of which (one-third of the associated tumours) arise in the gastrointestinal tract; this is similar to the distribution of tumours in acanthosis nigricans, which may coexist. Carcinomas of the breast are also frequent, although this may just reflect the incidence of these tumours in an age group who are also likely to have seborrhoeic keratoses. Rare associations have been documented with a variety of other neoplasms, including malignant haemangiopericytoma [7], malignant melanoma [8], renal carcinoma [5] and transitional cell carcinoma of the bladder [9]. Lymphoproliferative disorders, which are rarely associated with acanthosis nigricans, have been more commonly reported with the sign of Leser–Trélat, accounting for about 20% of associated tumours [5]. Eruption of seborrheic keratoses over the upper body occurred in a young patient with relapse of a previously treated pre-B-cell acute lymphocytic leukaemia [10]. Six multiple metachronous cancers in a patient with the sign of Leser-Trélat has been reported [11].
Truly sudden development of multiple seborrhoeic keratosis, especially in younger patients and if associated with pruritus or with acanthosis nigricans, warrants investigation. However, the mere presence of many seborrhoeic keratoses is unlikely to be linked with malignancy and the strength of this sign as a marker of internal malignancy must be viewed as uncertain.
Florid cutaneous papillomatosis
This rare paraneoplastic phenomenon is characterized by a widespread, often pruritic eruption of warty papules associated with an underlying malignancy, particularly gastric adenocarcinoma [1]. The eruption closely resembles disseminated human papilloma virus infection (Figure 147.14) but no evidence of this is found on histological or ultrastructural examination. It may appear concomitantly with other paraneoplastic phenomena, particularly paraneoplastic AN (see above), and the underlying mechanisms are presumed to be similar.
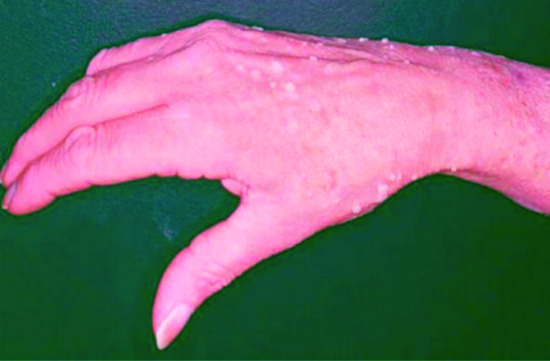
Figure 147.14 Florid cutaneous papillomatosis (From Janniger and Schwartz 2010 [1]. With permission from Wiley.)
Acquired ichthyosis
Many systemic diseases may be associated with acquired ichthyosis including nutritional deficiencies, sarcoidosis, leprosy, HIV infection, hypothyroidism, lupus erythematosus, graft-versus-host disease and drug reactions (see Chapter 87) [1].
However, more sudden onset of ichthyosis similar to the pattern of ichthyosis vulgaris in adult life or with a generalized eczema craquelé appearance suggests the possibility of internal malignancy, particularly if it occurs in a younger age group. Paraneoplastic ichthyosis is typically very extensive, affecting the trunk and having quite prominent fissuring. Other paraneoplastic signs have been reported to be present in conjunction with acquired ichthyosis, including erythema gyratum repens, Bazex syndrome and dermatomyositis [2, 3]. The strongest association is with Hodgkin disease (accounting for over 70% of cases) and other lymphoreticular tumours, including T-cell lymphomas, leukaemias, myelodysplastic syndrome, multiple myeloma and polycythaemia vera [1–3]. Cases linked with solid tumours are also well documented, including cancers of the ovary, kidney, liver and breast, as well as leiomyosarcoma [1–3]. A course paralleling that of an underlying lymphoma (including resolution related to treatment) is usual [4, 5].
OTHER EPIDERMAL DISORDERS
Pityriasis rotunda is a fixed, annular, scaly dermatosis that has been associated with neoplasia, particularly hepatocellular carcinoma. However, it may also be seen in other systemic diseases and in leprosy (see also Chapter 87) [3].
Transient acantholytic dermatosis (Grover disease; Chapter 87) has been linked with internal malignancy, particularly with myelogenous leukaemia [6, 7] and carcinoma of the genito-urinary tract. However, this may be linked in part with therapy, or simply because it may go unrecognized unless it is specifically considered [6].
Acquired seed-like keratoses of the palms and soles are a common normal finding in healthy subjects (36%) over 50 years old, but are apparently more common in individuals with carcinoma of the bladder (87%) or bronchus (71%) [8]. Punctate keratoderma occurring in Cowden's syndrome and seed-like keratoses with arsenic ingestion may also be associated with internal malignancy.
PARANEOPLASTIC PIGMENTATION
The ectopic ACTH syndrome (extracutaneous neuroendocrine melanoderma) occurs due to production of an ACTH-like hormone from tumours; small cell bronchial carcinoma is the cause in over 50% (Table 147.3). Other reports have described the same condition resulting from gastric, pancreatic, oesophageal and ovarian cancers, as well as in thymoma, phaeochromocytoma, carcinoid syndrome and in various APUD (amine precursor uptake and decarboxylation) tumours. The pigmentation in ectopic ACTH syndrome is Addisonian in distribution, diffuse but with photoaccentuation and greater prominence over pressure points and in flexures, genital skin, scars and the oral mucosa [1].
Table 147.3 Pigmentary abnormalities associated with internal malignancy.
| Pigmentary change | Pattern | Examples |
| Hyperpigmentation | Diffuse, or diffuse with localized accentuation (Addisonian pattern of pigmentation) | Melanoma (rarely causes diffuse slate grey pigmentation) Phaeochromocytoma (Addisonian pattern) Ectopic ACTH syndrome (Addisonian pattern) poems syndrome (diffuse or semiconfluent speckled pattern) Hyperpigmentation with scleromyxoedema and gammopathy Diffuse mastocytosis Lymphomas (uncommon) Ependymoma (mild increase in pigmentation) Werner syndrome (localized or diffuse pigmentation) Cachexia due to neoplasia |
| Patchy or reticulated | Fanconi anaemia (various pigmentary changes) Dyskeratosis congenita (reticulate pigmentation) |
|
| Other distributions | Carcinoid syndrome (photodistributed) Pancreatic, gastric and renal tumours (erythema ab igne due to local application of heat) |
|
| Lentigines and freckles | Peutz–Jeghers syndrome (lentigines) Carney complex (lentiginosis is characteristic, freckles also occur) Xeroderma pigmentosum (freckles) Neurofibromatosis (flexural freckle-like macules) (see Chapter 80) Cowden's disease and Bannayan–Riley–Ruvalcaba syndrome (genital lentigines) Gardner syndrome (freckles) Paraneoplastic acral lentiginosis |
|
| Café-au-lait macules | Neurofibromatosis Bloom syndrome Multiple endocrine neoplasia types 1 and 2B Fanconi anaemia Von Hippel–Lindau disease |
|
| With epidermal hyperplasia | Acanthosis nigricans | |
| Melanocytic naevi and melanoma | Associated with pancreatic neoplasia, astrocytomas and other cerebral neoplasms in some families Blue naevi and ordinary naevi occur in Carney complex |
|
| Mixed hyper- and hypopigmentation | Poikiloderma | Dermatomyositis (speckled pigmentation on hypopigmented background) Rothmund–Thomson syndrome (photodistributed poikiloderma) |
| Hypopigmentation | Generalized | Chediak–Higashi syndrome |
| Localized, multiple | Tuberous sclerosis complex (ash leaf macules) Mycosis fungoides (hypopigmented variant) |
|
| Melanoma-associated (other than regression within the primary lesion) | Halo depigmentation around primary tumour or metastases Distant leukoderma, usually with centrifugal spread starting on the trunk |
In Carney complex lentiginosis is typically centrofacial but may be widespread at almost any body site; the buccal mucosa is affected in only 5% of cases. Rarer sites include the conjunctivae and labia minora; the palms, soles and penis are rarely affected. Blue naevi, usually few in number, occur on the face, trunk or limbs but rarely on the extremities [2].
HAIR, NAILS AND SKIN APPENDAGES
Paraneoplastic hypertrichosis lanuginose acquisita
The development of paraneoplastic hypertrichosis lanuginosa acquisita tends to affect the face initially, extending down the body with time. The hair is of fine, downy lanugo type. The mechanism is unclear but prolongation of the anagen growth phase has been proposed. Resolution of hypertrichosis lanuginosa occurs after treatment of the underlying tumour, and regrowth can be related to recurrence of the neoplasm. There may be associated acanthosis nigricans, hypertrophy of papillae of the tongue and glossitis [1]; disturbances of taste or smell also occur [2]. However, the glossitis in at least some patients may be a manifestation of vitamin deficiency rather than a specifically malignancy-related condition [3].
About 70% of cases occur in women, usually aged 40–70 years, and most patients have metastatic tumours at presentation, with correspondingly poor prognosis. The commonest tumour sites in men are lung followed by colorectal, and in women are colorectal followed by lung and breast. Other reported sites or tumour types include endometrium (about 7–8% of cases), ovary, cervix, renal, prostate, bladder, adrenal gland, stomach, gallbladder, skin (including melanoma), parotid gland, sarcoma, lymphoma and leukaemia [2, 4, 5, 6].
Clubbing of nails
In clubbing there is increased transverse and longitudinal nail curvature with hypertrophy of the soft-tissue components of the digit pulp (Chapter 95). Both clubbing and associated hypertrophic osteoarthropathy have been documented with many neoplasms, the commonest being carcinoma of the bronchus. In patients with lung cancer, clubbing has been reported to be present in 29%, especially in females; most lung tumours are squamous cell carcinoma or adenocarcinoma [1]. Resolution of the clubbing after tumour resection has been reported [2]. Clubbing has also been associated with gastrointestinal tumours and tumours metastatic to the lung [3].
A high incidence of hypertrophic osteoarthropathy occurs particularly with mesothelioma, but it may also occur with malignancies of the pulmonary, cardiovascular, gastrointestinal and hepatobiliary systems [4]; it is much less common than clubbing.
Hyperhidrosis
Generalized hyperhidrosis may rarely be associated with malignant disease. It is an almost consistent finding in phaeochromocytoma, in which it may be limited to night-time or may occur at any time. Nocturnal hyperhidrosis (‘night sweats’) may also occur in lymphoma and carcinoid syndrome as well as in non-neoplastic conditions such as thyrotoxicosis, chronic infections and others [1]. Localized hyperhidrosis may occur in POEMS syndrome.
Specific distributions of localized hyperhidrosis may also be important. Hyperhidrosis with autonomic dysreflexia is associated with spinal cord lesions above T6. It is characterized by episodic sweating of the face, neck and upper trunk with vasodilatation in the same distribution, and is accompanied by headache, hypertension and piloerection. Most cases are due to injury or cord compression but intracranial posterior fossa neoplasms can produce similar symptoms [1]. A case of intramedullary thoracic spinal cord ganglioglioma in a 16-year old patient presented with abnormal sweating on the right side of the neck, chest and right arm for 6 years [2].
Paroxysmal unilateral hyperhidrosis of the face and neck, usually severe and unrelated to stimuli such as eating, may be due to an ipsilateral thoracic tumour (adenocarcinoma, squamous cell carcinoma or mesothelioma) compressing or infiltrating the sympathetic trunk. Associated features may include Horner syndrome, facial weakness, sensory disturbance and other features of the primary tumour [1].
DERMATOSES ASSOCIATED WITH INTERNAL MALIGNANCIES
Several dermatoses have a significant association with internal malignancy.
Acrokeratosis paraneoplastica
Acrokeratosis paraneoplastica is a rare paraneoplastic condition, commoner in males than females, that is particularly associated with squamous cell carcinoma of the upper respiratory or gastrointestinal tracts, especially when there are metastases in the cervical lymph nodes.
The cutaneous changes develop gradually, often in several phases, initially with violaceous erythema and scaling on the peripheries, especially the helices of the ears, tip of the nose, hands and feet (particularly the distal portion of digits). The eruption then becomes more hyperkeratotic, with a keratoderma on the hands and feet. Subsequently the eruption may become generalized. Nail dystrophy and paronychia are often present. Changes on the face may appear eczematous or lupus erythematosus-like, whereas acral changes are often psoriasiform. The differential diagnosis can include dermatitis, especially seborrhoeic or contact allergic types, acral psoriasis or reactive arthritis (Reiter syndrome). The histological changes are non-diagnostic, but essentially reflect the clinical appearance with hyperkeratosis, parakeratosis, focal spongiosis and a mixed inflammatory cell infiltrate [1–5].
More than 60% of tumours arise in the oropharynx or larynx or are cervical squamous cell carcinoma metastases with an unknown primary site; lung, oesophageal and other primary or metastatic lesions above the diaphragm make up most of the remainder [5]. Rare associations such as metastatic adenocarcinoma of the prostate and transitional cell carcinoma of the bladder have been reported [6, 7]. The lesions often appear before the cancer is diagnosed [8]. The course mostly parallels the underlying neoplasm. Resolution may occur with successful tumour resection and recurrence may develop on relapse of malignancy. When resection is considered inappropriate or impracticable, systemic retinoids may improve the cutaneous changes [9].
Migratory erythemas
This descriptive term is applied to a variety of annular and figurate eruptions. Two variants, erythema gyratum repens and necrolytic migratory erythema, have a clear association with internal neoplasia. Other migratory erythemas are less clearly associated with neoplasia and erythema annulare centrifugum is usually not associated with neoplasia [1, 2]. If an underlying malignancy is found, a myeloproliferative disorder, specifically lymphoma or leukaemia is most often reported [2]. There are also reports of subacute cutaneous lupus erythematosus-like annular and figurate rashes, linked with myeloproliferative disorders and carcinoma of lung, liver, breast, larynx and oesophagus in individual cases.
Erythema gyratum repens is a rare, bizarre, cutaneous eruption consisting of mobile, concentric, often palpable, erythematous, wave-like bands, which give a ‘wood-grain’ appearance to the skin (Figure 147.15). A peripheral scale or collarette may be present. The complete torso is frequently affected. There is often associated severe pruritus, sometimes ichthyosis, and sometimes bullae within the erythema. The lesions migrate from day to day, usually changing position by about 1 cm daily. It has a strong association with internal malignancy (over 80% of cases), particularly lung cancer [3, 4] which is present in about a third of cases. Other cancer sites include oesophagus, breast, bowel, uterus, cervix, kidney, pancreas and haematological neoplasia [4]. Occasional cases without associated malignancy have been reported [5, 6] but it is important to be aware that 6% are found to have a tumour of unknown primary origin [4]. Identification and resection of the tumour often results in resolution of the eruption.
Figure 147.15 Erythema gyratum repens of the arm secondary to carcinoma of the bronchus.
Necrolytic migratory erythema (NME) is strongly associated with a glucagon-secreting α-cell tumour of the pancreas [7–9]. It presents as a widespread painful migratory rash with repeated eruptions of irregular polycyclic, intensely inflammatory erythematous patches with expanding scaling margins; these blister and break down with superficial epidermal necrolysis and crusting. It may affect any skin site but has a predilection for the anogenital region and trunk. NME is strongly associated with the presence of an underlying glucagon-secreting pancreatic islet cell adenoma, though cases have been described where this was not found. NME is one of the components of the glucagonoma syndrome along with weight loss, diabetes, stomatitis and diarrhoea [9]. If glucagon levels can be restored to normal either by surgery or by the long-acting somatostatin analogue octreotide the rash will usually rapidly remit [10].
CONNECTIVE TISSUE AND RHEUMATOLOGICAL DISORDERS
Malignancies have been reported in association with many disorders that overlap between dermatology and rheumatology. Those that fall into the connective tissue disease group are discussed here.
Dermatomyositis
Dermatomyositis (Figure 147.16) and polymyositis may be associated with internal malignancy in adults (see Chapter 53). The reported likelihood of finding a neoplasm varies widely, but about 25–30% is probably a representative proportion for the various reported studies of dermatomyositis, making this a condition with a significant association with underlying malignancy [1–3, 4, 5, 6]. The association with neoplasia is much stronger for dermatomyositis than for polymyositis or dermatomyositis/autoimmune disease overlap conditions [7]. Conventional teaching has been that malignancy is an uncommon cause of dermatomyositis in subjects less than 40 years of age; however, paediatric cases with neoplasia have been reported [8]. As there is a lower incidence of malignancy in the younger age group, and there are no age-matched comparative studies against a control population in children, it is difficult to judge the strength of the association in this age group [3].
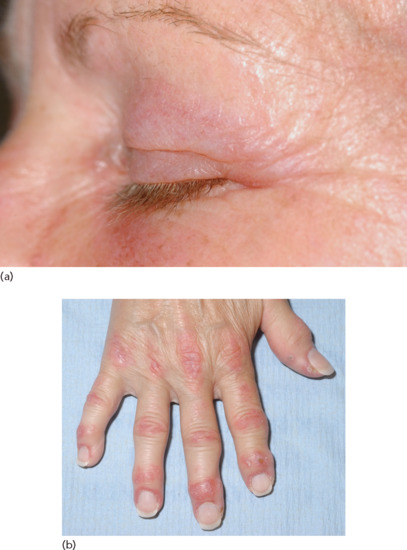
Figure 147.16 Dermatomyositis with typical changes affecting the eyelids (a) and hands (b).
The temporal association of dermatomyositis with neoplasia varies. In one report approximately equal proportions had: (i) a known malignancy at the time that dermatomyositis presented; (ii) a malignancy found due to investigation when dermatomyositis was diagnosed; or (iii) a malignancy found during follow-up (usually in the first 6 months after diagnosis of dermatomyositis) [4].
Accounts of specific malignant associations may be subject to bias by rare case reporting, and in larger series the malignancies identified generally reflect tumour prevalence in the general population: lung cancer in men, breast and gynaecological tumours in women, and colorectal cancers in both sexes. In South-East Asia, there is a higher frequency of naso-pharyngeal carcinoma, that probably also reflects the background risk of this type of neoplasm. The one exception to this generalization is ovarian carcinoma, which appears to be significantly overrepresented and potentially overlooked [2, 4, 5].
The value of extensive screening for neoplasia in dermatomyositis is questionable. Several authors have stressed that the emphasis should be attached to thorough clinical evaluation, simple investigations and then specific investigations as indicated [1, 4]. There should certainly be a low threshold for further or repeated investigations, as indicated at the time of diagnosis or during follow-up if previous neoplasia has been present, when the therapeutic response is poor, or if new symptoms develop. There is also an argument for ongoing screening for ovarian cancers throughout follow-up of female patients [2].
In general, subjects with amyopathic dermatomyositis (dermatomyositis sine myositis) or who have a connective tissue overlap syndrome appear less likely to have an underlying malignancy. However, some patients with amyopathic disease at the outset do eventually develop myositis and tumours have been reported in all such variants, so screening investigations should still be performed [9]. In one study, patients with malignancy were found to have a more rapid onset of dermatomyositis, higher mean creatine kinase and erythrocyte sedimentation rates, and a lower frequency of Raynaud phenomenon compared with patients without an underlying malignancy [10]. Vasculitis or necrosis manifest clinically or in histopathology specimens has also been associated with an increased risk of an associated neoplasm [11, 12]. A recent meta-analysis found several characteristics that may influence cancer development among patients with dermatomyositis and polymyositis: old age at diagnosis, male sex, cutaneous necrosis and dysphagia all increased the risk; arthritis and interstitial lung disease decreased the risk of malignancy [13].
Recent studies have identified new autoantibody specificities in dermatomyositis: melanoma differentiation-associated protein 5 (MDA5), transcription intermediary factor 1 γ (TIF-1γ) and nuclear matrix protein (NXP-2) [14]. It has been demonstrated that anti-NXP-2 and anti-TIF-1 γ antibodies are frequent in dermatomyositis (found in 55% of patients with dermatomyositis) and that either of them is present in most patients (83%) with cancer-associated dermatomyositis, especially in males [14]. Anti-MDA-5 antibodies are found in approximately 10–20% of dermatomyositis patients and are associated with interstitial lung disease [14, 15]. Progress in serotype–phenotype associations may help to determine which patients require more extensive investigation for malignancy [16].
Lupus erythematosus
The possibility of an increased risk of internal malignancy in systemic lupus erythematosus (SLE) [1–3] and subacute cutaneous lupus erythematosus (SCLE) [4, 5] has been debated for many years and still remains controversial. There are individual cases in which a close temporal relationship has been documented [4], and large cohort and population studies have mainly produced results in favour of an increased risk. Studies that have supported an association with malignancy have suggested increases in lymphomas, monoclonal gammopathy and in cervical, lung, hepatobiliary and breast cancer [3, 6, 7, 8], although the latter has been shown recently to be decreased [6]. The majority of large studies suggest an increase in non-Hodgkin lymphoma [3, 6], particularly diffuse large B-cell lymphoma (DLBCL) [7]. The mechanism may be a T-cell defect in SLE or may be a result of immunosuppressive medication used in treatment (especially implicated in the increased risk of haematological malignancies [9]). The evidence for an association between autoimmune conditions and lymphoma is supported by a large study of 3055 patients with non-Hodgkin lymphoma and matched controls; significant associations were found with rheumatoid arthritis, primary Sjögren syndrome, and SLE, again documenting the specific association with DLBCL [10]. The increased tumour risk in patients with Sjögren syndrome has been noted in other reviews [11]. Treatment with antimalarials has been suggested to have a protective effect against the development of cancers in SLE [12].
Rare patterns of lupus erythematosus, such as lupus erythematosus gyratum repens, may carry a higher risk of internal malignancy [13], although this condition is generally reported as isolated cases, so this conclusion is uncertain. Individual reports suggest an association of an SCLE-like eruption with tumours of various organs including myeloproliferative disorders causing a neutrophilic lupus-like figurate erythema and an SCLE-like rash associated with tumours of the lung, liver, larynx, breast and oesophagus [14, 15]. Potential clues to this association are an SCLE-like rash in males, SCLE in an older age group than usual and therapy-resistant SCLE.
Systemic sclerosis and other inflammatory fibrosing conditions of the dermis and subcutis
Like SLE, systemic sclerosis has been linked with the occurrence of internal malignancy [1, 2] (see Chapter 56). A meta-analysis of population-based studies revealed increased risks for lung, liver, haematological and bladder cancers, although absolute risks were relatively low [3]. Likewise, eosinophilic fasciitis has occasionally been linked with contemporaneous diagnosis of a neoplasm [4]. Many of the cases described are not entirely classical of ‘usual’ systemic sclerosis, some cases having a more aggressive course of fibrosis than anticipated, an unusual distribution, extensive fibrotic changes in the subcutaneous fat, or progressive arthritis. To reflect these features, more recent cases have been described under the terms palmar fibrosis/arthritis [5, 6] or cancer-associated fasciitis/arthritis [5, 6] or cancer-associated fasciitis–panniculitis syndrome [7, 8]. The commonest tumour types documented are ovarian and lung; breast, prostate and pancreatic tumours have also been reported.
Panniculitis may occur in patients with acinar cell carcinoma of the pancreas, in whom a syndrome of panniculitis, polyarthritis and eosinophilia can occur. Eosinophilic panniculitis has also been reported in association with other solid tumours or pre-leukaemia [9, 10].
Scleroderma-like skin changes may also be a cutaneous manifestation of carcinoid syndrome, the differential diagnosis from systemic sclerosis being suggested by the presence of flushing and the absence of Raynaud phenomenon.
Post-irradiation morphoea is a phenomenon seen after treatment of breast cancer [11]. Its importance is not as a marker of underlying malignancy, but in the differential diagnosis of carcinoma en cuirasse (or occasionally carcinoma erysipeloides).
BULLOUS DISORDERS ASSOCIATED WITH INTERNAL MALIGNANCY
Paraneoplastic pemphigus
Paraneoplastic pemphigus is a heterogeneous multiorgan autoimmune syndrome in which patients display a spectrum of mucocutaneous manifestations including pemphigus-like, pemphigoid-like, erythema multiforme-like, graft-versus-host disease-like or lichen planus-like patterns, characteristically with oral involvement (see Chapter 50) [1]. In addition, there is an association with small-airways occlusion [2] and the deposition of autoantibody complexes in different organs. The mucosal disease is often severe and progressive.
Associated neoplasms in one large review series were mainly B-cell proliferations and thymoma or thymoma-like neoplasms; specific neoplasms included Hodgkin lymphoma (42%), chronic lymphocytic leukaemia (29%), Castleman tumour (10%), thymoma (6%), spindle cell neoplasms (6%) and Waldenström macroglobulinaemia (6%) [3]. Paraneoplastic pemphigus is distinguished from pemphigus by its clinical features and by the presence of serum autoantibodies to a range of antigens of 250, 230, 210, 190 and 170 kDa (bullous pemphigoid antigen and a range of desmosomal and hemidesmosomal proteins including desmoglein 1, desmoglein 3, envoplakin and periplakin). Direct immunofluorescence of skin biopsies is usually positive for immunoglobulin G (IgG) and C3 but may be negative in some cases. High sensitivity and specificity for this differential diagnosis has more recently been reported by taking account of the association with a lymphoproliferative disorder, by finding antibodies to desmoplakin on indirect immunofluorescence using rat bladder urothelium or envoplakin and/or periplakin bands on immunoblotting [4].
Bullous pemphigoid
Isolated reports have suggested an association between bullous pemphigoid and underlying neoplasia [1–3]. However, larger series do not support a significant association with malignant disease [4, 5]. Despite this, the issue remains controversial and more selective studies have shown there may be a correlation when immunofluorescent findings are negative and mucosal involvement is present [4, 6]. Historical reports are difficult to evaluate with certainty, as some cases may actually have been epidermolysis bullosa acquisita or even bullous pemphigoid-like paraneoplastic pemphigus, which can now be separated from bullous pemphigoid by current immunological techniques. Malignancies have been reported from breast, lung, thyroid, larynx, skin, soft tissue, stomach, colon, lymphoreticular system, prostate, cervix, bladder, kidney and uterus.
Pemphigus
Historically, pemphigus has been linked with various tumours. Some cases, such as those associated with thymoma and Castleman tumour, would probably now be found to have the features of paraneoplastic pemphigus. Pemphigus foliaceus has been associated with acanthosis nigricans-like lesions and hepatocellular carcinoma [7], and pemphigus in Japanese subjects has been associated with lung cancer. The concurrence of internal malignancy and pemphigus may, as with bullous pemphigoid, be a true association [8], although some suggest this to be coincidence [9].
OTHER BLISTERING DISORDERS
Dermatitis herpetiformis Dermatitis herpetiformis (DH) has been documented in some studies to have a link with internal malignancy of various types, especially lymphoma [1–5]. There is logic for an indirect link with lymphoma; DH is always associated with some degree of gluten-sensitive enteropathy and the latter has a well-documented association with small bowel lymphoma. One group of authors who initially found an increased risk of lymphoma in DH [4] later, in a larger study, documented that the overall risk of mortality in DH was lower than in the general population [6] but that the risk of non-Hodgkin lymphoma was increased, with a standardized incidence ratio of 6.0. In this study, one in seven lymphomas in DH was an enteropathy-associated T-cell lymphoma associated with inadequate dietary compliance [6], supporting the earlier documentation that a gluten-free diet reduces the risk of small bowel lymphoma in patients with DH [7]. It therefore appears that there is an increased risk of lymphoma in DH but that the overall risk is small; additionally, the lymphoma risk, particularly of bowel lymphoma, is decreased by adherence to a gluten-free diet.
Linear IgA disease This also appears to have a higher than predicted association with lymphoproliferative malignancy, although this is much less well documented [8].
Epidermolysis bullosa acquisita Epidermolysis bullosa acquisita is commonly linked with autoimmune diseases but has also been reported to occur in association with neoplasia, particularly myeloma and lymphoma [9–11].
Porphyria cutanea tarda and variegate porphyrias These disorders have been associated with hepatocellular carcinoma [12]. Myeloma and visceral carcinoma have also been reported [13].
DEPOSITION DISORDERS
Dermal deposition disorders, such as mucinoses, xanthomas, amyloidosis and calcification, may be linked with internal malignancy [1, 2]. Some of the most important deposition disorders, and their potentially associated internal malignancy, are listed in Table 147.4.
Table 147.4 Some deposition disorders that are linked with internal malignancy.
| Material deposited | Disorder | Associated internal malignancies | Comments |
| Amyloid proteins (see Chapter 58) [1] | Primary and myeloma-associated systemic amyloidosis (AL protein deposition) | Paraproteinaemia, myeloma | Amyloidosis occurs in about 15% of patients withmyelomatosis |
| Secondary amyloidosis (AA protein deposition) | Lymphomas, especially Hodgkin's lymphoma Hypernephroma Other solid tumours |
||
| Mucin/proteoglycans and fibromucinoses (see Chapter 59) | Scleromyxedema/lichen myxoedematosus | Paraproteinaemia, typically a ‘slow gamma region’ | Paraprotein is present in most cases |
| Papular mucinosis | Paraproteinaemia | Uncommon | |
| Scleredema | Paraproteinaemia | More commonly associated with diabetes or streptococcal infection | |
| poems syndrome | Paraproteinaemia | Features are polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes (POEMS) | |
| Lipids (as foamy macrophages) | Necrobiotic xanthogranuloma | Paraproteinaemia, usually monoclonal IgG-κ,present in about 70% of cases | Also associated with cryoglobulinaemia, myeloma, marrow dyscrasias and rarely leukaemia; some cases apparently occur in isolation |
| Normolipaemic plane xanthomatosis | Myeloma | Usually IgG paraprotein | |
| Xanthoma disseminatum | Gammopathy, bone marrow dyscrasias | Usually IgG paraprotein | |
| Calcium | Metastatic calcification | Lung and other squamous carcinomas | Due to ectopic parathyroid-like hormone secretion |
| Due to primary hyperparathyroidism; may be associated with multiple endocrine neoplasia syndromes | |||
| Dystrophic calcification | Pancreatic carcinoma | Calcification of fat |
Calcinoisis cutis is a rare complication of internal carcinoma. However, it should be noted that many cancers cause hypercalcaemia, and that metastatic calcification may occur in other organs, such as the lung or kidney, even if not in the skin. The commonest underlying malignancies are carcinoma of the oesophagus, myeloma, breast cancer, lymphoma or any other tumour responsible for osteolytic metastases [2].
OTHER DERMATOLOGICAL DISORDERS
Lichen planus Lichen planus may rarely be associated with neoplasia (see Chapter 37) [1]. There is also an increased risk, particularly in males, of oral squamous cell carcinoma; this may be due to a combined direct effect and co-factors such as smoking [2]. The strength of the association, however, may have been overestimated, as some of the reports are those that have collected patients presenting with oral squamous cell carcinoma and have then looked for histological evidence of lichen planus as a background factor.
Urticaria With the exception of cold urticaria and peripheral gangrene as a result of circulating cryoglobulins, where there is a possible but uncommon link with myeloma and lymphoma [3], associations of urticaria and neoplasia are difficult to evaluate (see Chapter 42). Certainly it cannot be regarded as an established paraneoplastic phenomenon other than in Schnitzler syndrome (see 45), a distinct autoinflammatory disorder characterized by chronic urticarial wealing, bone pain, hyperostosis, high erythrocyte sedimentation rate, monoclonal IgM gammopathy (usually IgM-κ) and a significant risk of AA-amyloidosis. Although the overall prognosis is reasonable, around 10–15% of patients develop a B-cell lymphoproliferative disorder [4].
Erythroderma and exfoliative dermatitis Exfoliative dermatitis has been linked with malignancy [5, 6]. In most such cases (around 10% in most reported series), the neoplasm is mycosis fungoides or its leukaemic variant, Sézary syndrome. These cases are really a representation of a systemic neoplasm rather than a truly paraneoplastic disorder. However, there are a small number of patients who have neither condition, but present with erythroderma and eventually develop lymphoma or leukaemia [7]. Immunophenotypic studies do not appear to help distinguish benign from malignant cases [8]. There are additional reported cases of erythroderma with cancers of the liver, lung, colon, stomach, pancreas, thyroid, prostate and cervix [5, 6, 9]. Ofuji papuloerythroderma has also been associated with peripheral T-cell non-epidermotrophic cutaneous lymphoma [10].
Granuloma annulare Ganuloma annulare has been reported in association with lymphomas, other haematological malignancies, and uncommonly with solid tumours [11]. However, a causal relationship in such cases is uncertain. It has been suggested that the subcutaneous pattern of granuloma annulare may carry a greater risk of malignancy, but this may reflect the more extensive investigation that is usually performed to establish the diagnosis.
Insect bite-like reactions Florid insect bite reactions are reported in haematological malignancy, usually chronic lymphocytic leukaemia [12].
Cutis verticis gyrata This may occasionally occur as a paraneoplastic phenomenon [13].
Mental neuropathy Mental neuropathy or ‘numb chin syndrome’ has several benign causes, but may occur as a feature of metastatic disease and is considered an indicator of poor prognosis [14]. Relevant tumours include breast, thyroid, renal, lung, prostate, lymphomas and melanoma.
Non-melanoma skin cancer This has been linked with increased risk and poor prognosis of non-Hodgkin lymphoma [15].
Pyoderma gangrenosum and neutrophilic dermatoses Pyoderma gangrenosum, particularly the superficial and bullous forms, has been associated with myeloproliferative diseases, including acute and chronic myeloid leukaemia, acute lymphocytic leukaemia, myeloid metaplasia, polycythaemia rubra vera, multiple myeloma, lymphoma and myelofibrosis (Figure 147.17) (see Chapter 49) [16]. The association of pyoderma gangrenosum with monoclonal gammopathy is uncertain, but it does occur at a frequency higher than expected in the general population and is usually of IgA type, whereas IgG gammopathy is the commonest type overall. Solid tumours reported include carcinoid, colon, bladder, prostate, breast, bronchus, ovary and adrenocortical carcinoma. Sweet syndrome has likewise been associated with several malignancies, especially haematopoietic (see Chapter 148). Chronic recurrent Sweet syndrome appears to have a particularly strong link with myelodysplastic disorders [17].

Figure 147.17 Pyoderma gangrenosum of the lower leg in a patient with myelodysplastic syndrome.
Multicentric reticulohistiocytosis This is a rare condition usually occurring in adult life and characterized by papulonodular lesions of the fingers or other extremities, the face and sometimes mucous membranes. Its synonyms include lipoid dermatoarthritis and reticulocytoma cutis. Papules around the nail fold have been termed the ‘coral bead sign’ (see Chapter 136). A severe, symmetrical polyarthritis, especially affecting the hands, is frequently associated [18]. Approximately 25% of cases are associated with internal neoplasia, virtually always solid tumours, such as those of the breast, lung, colon, pancreas, stomach, ovary or cervix [18–20], although haematological malignancies have also been reported [21].
VASCULAR DISORDERS ASSOCIATED WITH INTERNAL MALIGNANCY
Raynaud phenomenon and digital ischaemia
Persistent, painful digital ischaemia, with an unusual Raynaud syndrome-type appearance but often progressing to gangrene, has been linked to a variety of solid tumours and reticuloendothelial neoplasms [1, 2, 3]. The process may have a vasculitic element [4].
Hyperviscosity syndromes such as polycythaemia vera, leukaemias or myeloma-linked cryoglobulinaemia may give rise to cutaneous ischaemia and phlebitis by microvascular occlusion. Cancer-associated coagulopathy may also cause vascular occlusion [5].
Hypereosinophilic syndrome, for which there is increasing evidence of a malignant clonal proliferation, has also been associated with acrocyanosis, cutaneous microthrombi, digital infarction and gangrene [6–8].
Erythromelalgia
This condition is linked with myeloproliferative disorders, most commonly polycythaemia vera or essential thrombocythaemia in over a third of adult cases (see Chapter 103). In a large series of patients with erythromelalgia linked with haematological malignancy, the feet were most commonly affected, with severe burning pain and erythema; symptoms may occur 2 years or more before the haematological disorder is documented [9]. The mechanism involves microvascular occlusion (typically with platelet aggregates, termed ‘white thrombi’); Raynaud phenomenon may occur.
Palmar erythema
This is rarely linked with malignancy, but an interesting association was observed in a series of patients with cerebral malignancies, in whom nearly 20% had palmar and (to a lesser extent) plantar erythema, either diffuse or mottled. This occurrence seemed to be linked with the vascularity of the tumour, and was particularly seen with high-grade astrocytomas and glioblastomas; a role for vascular endothelial growth factor was proposed [10].
Vasculitis
There appears to be an association of cutaneous vasculitis with neoplasia, particularly in myeloproliferative disorders [11–13] and myeloma [14, 15], although solid tumours have also been reported [4, 16–18] (see Chapter 102). Hairy cell leukaemia appears to be especially associated with vasculitis, which may be of leukocytoclastic or polyarteritis nodosa pattern. Of the solid tumours reported with malignancy, the most common would appear to be those expected in an average population, including breast, lung, colon, oesophageal, renal, prostatic, and head and neck tumours. There is overlap with paraneoplastic digital ischaemia. In a study of 200 patients with antineutrophil cytoplasmic antibody-positive vasculitis (granulomatosis with polyangiitis or microscopic polyangiitis) published in 2004, the relative risk of malignancy preceding or concurrent with vasculitis was sixfold greater than that for the local population. Patients with Henoch–Schönlein purpura (n = 129) had a fivefold relative risk, but only five of 333 patients with SLE had a malignancy [18]. A much lower relative risk (1.54) of approximately 1.5 was found in a recent study of 535 patients. The authors speculated whether this may be due to a reduced reliance on cyclophosphamide in current treatment protocols [19].
The dermatological manifestations include palpable purpura and maculopapular, urticarial and petechial lesions; these presumably reflect a small-vessel vasculitis or even, when ulceration occurs, a necrotizing vasculitis [4, 11]. When linked with a haematological malignancy, vasculitis often antedates bone marrow involvement, as opposed to the more predictable purpura due to thrombocytopenia, which reflects bone marrow infiltration by myeloproliferative disease or carcinoma. It is difficult in some reports to distinguish between microvascular occlusion (for example, by a monoclonal type I cryoglobulin) versus primary vasculitis or therapy-related vessel injury.
Chilblain–like lesions
Lesions resembling perniosis may be a manifestation of leukaemias and myeloproliferative disorders (Figure 147.18) [20, 21]. They are persistent rather than episodic, tend to be refractory to treatment with drugs (such as calcium channel blockers) and on biopsy may show blast cells as well as vascular changes. Metastasis from breast carcinoma has also been documented as resembling chilblains [22].
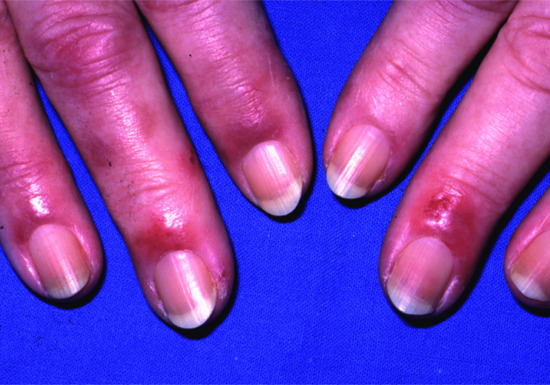
Figure 147.18 Chilblain-like lesions in acute myeloid leukaemia.
Flushing
This is a normal physiological response and may be a problematic menopausal symptom. However, it may be a feature of carcinoid syndrome, mastocytosis, phaeochromocytoma, medullary carcinoma of thyroid, hypogonadism in males, pancreatic tumours producing vasoactive intestinal peptide (VIPomas), basophilic leukaemia, horseshoe kidney (Rovsing syndrome) and renal cell carcinoma [1, 2]. It is also a feature of POEMS syndrome (with polyneuropathy, organomegaly, endocrinopathy, M protein, skin changes), which is associated with myeloma. Plethora, but not flushing, may be apparent in polycythaemia vera.
Carcinoid syndrome This may be difficult to diagnose at an early stage, as flushing is typically provoked by common triggers of physiological flushing such as emotional stress or alcohol ingestion (see Chapter 149). Only about 10% of patients with a carcinoid tumour have a malignant carcinoid syndrome and paroxysmal flushing is present in virtually all cases; episodes typically last a few minutes and may be more widespread than emotional flushing, sometimes involving the whole body. Nearly 75% of carcinoid tumours are gastrointestinal (especially involving the appendix and ileum), 25% are bronchial, and a small number arise at other sites including the larynx, pancreas, gallbladder and ovary [3]. Flushing is described as varying according to the site of the neoplasm. The most common midgut tumours (appendix and ileum) are associated with a gradual development of fixed cyanotic erythema in the flushing distribution, whereas foregut tumours (stomach, pancreas, lung) are associated with a brighter pink flush. This may reflect the production of different mediators; gastric carcinoids typically cause flushing by the production of histamine, whereas midgut carcinoids produce serotonin, bradykinin and prostaglandins. Other symptoms, such as diarrhoea, abdominal pain, dyspnoea, wheezing and occasionally syncope, may occur with progression of the tumour, usually not occurring until liver metastases have developed. Persistent erythema with or without telangiectasia, scleroderma-like change, pigmentary anomalies and a pellagra-like dermatitis eventually develop.
Mastocytosis This is most commonly limited to the skin but may be systemic, in which event it may be classified into four groups: (i) indolent; (ii) associated with a clonal, haematological, non-mast cell lineage disease (myeloproliferative or myelodysplastic disorders); (iii) aggressive (lymphadenopathic with eosinophilia); and (iv) mast cell leukaemia (see Chapter 46) [4]. It may be associated with mutations in the gene for c-kit. The more aggressive forms tend not to be associated with cutaneous mast cell lesions, but flushing, dyspnoea, chest pain, abdominal cramps, palpitations, syncope and other systemic reactions due to mast cell degranulation may occur. Serum tryptase is a useful screening test that may suggest systemic disease. However, the proportion of bone marrow mast cells, eosinophilia (especially in bone marrow) and alkaline phosphatase level are of greater prognostic importance than levels of mast cell mediators [5].
Phaeochromocytomas Phaeochromocytomas may cause pallor with rebound flushing [1, 2], but the more typical dermatological feature is hyperhidrosis.
Medullary carcinoma of thyroid Medullary carcinoma of thyroid, like phaeochromocytomas, may occur as part of a MEN syndrome, but can also occur in isolation. They may produce several other hormones such as ACTH, as well as prostaglandins and amines, which may cause other symptoms such as diarrhoea.
CANCER-ASSOCIATED THROMBOSIS
Three main patterns need to be considered: migratory thrombophlebitis, deep venous thrombosis and Mondor disease (a specific pattern of thrombophlebitis, but sufficiently distinctive that it is described separately). Arterial thrombosis may also occur as a paraneoplastic entity, but is much less common. The aetiology of cancer-associated thrombosis is multifactorial. The interaction of macrophages with cancer cells causes release of tumour necrosis factor and interleukin 6, which damage endothelium, creating a pro-thrombotic situation. The same interaction causes activation of platelets and of clotting factors X and XII [1]. Some tumour proteases are also procoagulant, especially ‘tissue factor’ which activates factor VII, and sialic acid moieties of mucin (released from adenocarcinomas) which activate factor X [1].
Migratory thrombophlebitis
Unlike superficial thrombophlebitis confined to the lower limbs, thrombophlebitis associated with neoplasia is often recurrent and migratory (Trousseau sign). A variety of sites, especially the upper extremities and trunk, can be involved, and lesions are usually multiple. Migratory thrombophlebitis is associated with malignancy in 50% of cases. The mechanism in most cases is an intravascular, low-grade hypercoagulation, which responds poorly to anticoagulant therapy; heparin is usually more effective than warfarin [2]. As well as altered level or function of prothrombotic and anticoagulant proteins, the coagulation and thrombotic process may involve cytokines, angiogenic factors or mucin secretion by tumours. Altered blood viscosity, vascular endothelial changes and production of small tumour emboli may also be contributory. Migratory thrombophlebitis can be associated with any cancer, but particularly occurs with carcinomas of the pancreas, stomach, colon and lung; pancreatic carcinoma accounts for about half of all cases. Leukaemias and lymphomas are less commonly associated with migratory thrombophlebitis. Causative tumours are often highly malignant and metastatic with a poor prognosis.
Deep-vein thrombosis
This is not commonly the presenting feature of malignancy [3, 4], although cancers have a significant risk of causing thrombosis. The likelihood of finding a malignancy is increased by about fourfold in patients with a deep-vein thrombosis (DVT) of the leg compared with expected population rates; the overall risk of such an association is about 10%. By contrast, DVT of the upper limb is more commonly linked with neoplasia (in some cases, the reason is obstruction by an apical lung tumour), and DVT is identified postmortem in half of patients who have died due to cancer. Tumours associated with DVT are usually adenocarcinomas in the gastrointestinal tract, urogenital tract, breast or lung. Mucin secretion, as noted, causes non-enzymatic activation of factor X to factor Xa, initiating the thrombotic cascade. An older age of patients with DVT has been proposed as a factor that should be associated with a lower threshold for malignancy screening.
Mondor disease
This is a rare condition in which a cord-like lesion is palpable in the subcutaneous tissue of the anterior or lateral thorax, or sometimes abdomen. The underlying lesion is a thrombophlebitis of thoracic or epigastric veins, usually unilateral but occasionally bilateral, which may occur as a result of trauma, inflammation or post-surgery. However, 10–15% of patients may have an associated breast carcinoma [3, 4, 5] so careful examination and mammography are indicated unless an alternative cause is obvious.
VENOUS OR LYMPHATIC OBSTRUCTION
In addition to vasculitis and the thrombotic intravascular occlusion described earlier, vessels of different types may become occluded by intravascular tumour cells, or may be compressed by an adjacent tumour. The facial and upper limb suffusion of superior vena caval obstruction is well known. Obstruction of venous flow from the head and neck may also occur due to benign retrosternal thyroid gland enlargement and is typically seen when the arms are elevated (Pemberton sign). Lymphatic obstruction typically occurs at the main lymph node sites, such as in the axilla due to a breast carcinoma, lymphoma or limb melanoma. Apical lung neoplasms may also cause damage to sympathetic nerves, leading to anhidrosis and sometimes hyperhidrosis and flushing of the contralateral, unaffected side. Arterial obstruction by tumours is much less common, although tumour emboli may occur.
Paraneoplastic pruritus
Internal carcinoma is a non-specific, rare but important cause of pruritus (see Chapter 83) [1]. Many mechanisms may be involved, including secondary metabolic effects such as uraemia or cholestasis, or itch related to iron deficiency anaemia, acquired ichthyosis or xerosis. Other mechanisms, such as those that link brain tumours with pruritus, are less well understood. Unfortunately, senile pruritus and asteatosis are not uncommonly encountered in elderly patients, a group who are more at risk from malignancy. It is therefore difficult to dissociate chance from true association, which can lead to difficulties in deciding whether screening is justified [2].
Generalized pruritus
In a 6-year study of 125 patients with generalized pruritus, Paul et al. [2] found no significant increase in malignancy, although of the eight patients with malignancy detected, two had lymphoma – a higher than expected incidence. Other studies support the likelihood of a genuine association of haematological disorders and lymphoma with generalized pruritus; in particular, itch may be a severe problem in patients with Hodgkin disease and may indicate a poorer prognosis [3]. Generalized itching may occur in more than 25% of patients with Hodgkin disease [4]. Other haematological disorders, including Sézary syndrome, mycosis fungoides, myelomatosis and leukaemia, may also cause generalized pruritus [1, 4, 5]; the mechanisms in these conditions are poorly understood. Aquagenic pruritus may be encountered in polycythaemia vera as well as in lymphoproliferative diseases (T-cell lymphoma, myelodysplasia) [1]. Many visceral carcinomas can cause pruritus, including breast and gastrointestinal cancers, and carcinoid syndrome [4]; again, the mechanisms are poorly understood. Generalized itching can also sometimes occur with intra-cranial neoplasia [6].
Treatment of pruritus in malignancy is often difficult; and the number of studies examining the efficacy of treatment are few [1, 4]. In addition to standard topical agents such as moisturizers, current systemic treatment strategies to reduce the itch include blocking of afferent transmission via peripheral and central neural mechanisms [1, 4]. Opioid antagonists, antidepressants and neuroleptics are some of the therapeutic options [1, 4].
Localized pruritus
Nerve damage by a tumour at any site can cause neuropathic pain or pruritus. The patterns that are most likely to present to a dermatologist are brachioradial pruritus and localized facial or nasal pruritus [6]. Cases with localized pruritus in the neck–shoulder–arm region due to a spinal tumour have been reported, with rapid resolution of symptoms after treatment [7]; in one child it was the sole manifestation of an intramedullar tumour [8].
Brain tumours are an uncommon cause of pruritus localized to the face [6]. Variations that have been recorded include unilateral pruritus and pruritus limited to the nostril. The latter is particularly linked with tumours invading the floor of the fourth ventricle but tumours elsewhere in the brain, as well as trigeminal neuralgia, can also produce this distribution of pruritus.
References
Introduction
Multisystem and haematopoietic tumours that involve the skin
- Provost TT, Flynn JA, eds. Cutaneous Medicine: Cutaneous Manifestations of Systemic Disease. Hamilton, Ontario: Marcel Decker, 2001.
- Lebwohl MG. The Skin and Systemic Disease. A Color Atlas and Text, 2nd edn. London: Churchill Livingstone, 2004:185–214.
- Callen JP, Jorizzo JL, Bolognia JL, Piette W, Zone JJ, eds. Dermatological Signs of Internal Disease, 4th edn. Philadelphia: Saunders, 2009.
- Vardy DA, Sion N, Grunswald MH. Specific cutaneous infiltrates in chronic myelogenous leukaemia. Cutis 1989;44:53–5.
Tumour spread from adjacent and distant tissues
Direct tumour spread and invasion
- Rolz-Cruz G, Kim CC. Tumor invasion of the skin. Dermatol Clin 2008;26:89–102.
- Yun SJ, Park HY, Lee JS, Park MH, Lee JB, Won YH. Clinicopathological correlation of cutaneous metastatic breast carcinoma using lymphatic and vascular markers: lymphatics are mainly involved in cutaneous metastasis. Clin Exp Dermatol 2012;37:744–8.
- Hinrichs R, Kirchberg K, Dissemond J, et al. Carcinoma erysipeloides of the facial skin in a patient with metastatic breast cancer. Br J Dermatol 1999;141:940–1.
- Hodge SJ, Mackel S, Owen LG. Zosteriform inflammatory metastatic carcinoma. Int J Dermatol 1979;18:142–5.
- Homler HJ, Goetz CS, Weisenburger DD. Lymphangitic cutaneous metastases from lung cancer mimicking cellulitis. Carcinoma erysipeloides. West J Med 1986;144:610–12.
- Cox SE, Cruz PD Jr. A spectrum of inflammatory metastasis to skin via lymphatics: three cases of carcinoma erysipeloides. J Am Acad Dermatol 1994;30:304–7.
- Yamamura Y, Kodera Y, Kito T. Gastric cancer with carcinoma erysipeloides caused by spontaneous thoracic duct rupture: report of a case. Surg Today 1997;27:166–8.
- Ng CS. Carcinoma erysipeloides from prostate cancer presenting as cellulitis. Cutis 2000;65:215–16.
- Lee SY, Chang SE, Bae GY, et al. Carcinoma erysipeloides associated with anaplastic thyroid carcinoma. Clin Exp Dermatol 2001;26:671–3.
- Ikeda Y, Niimi M, Takami H, Kodaira S. Successfully treated carcinoma erysipeloides from gastric cancer. Ann Oncol 2003;14:1328–9.
- Lee HC, Chu CY, Hsiao CH. Carcinoma erysipeloides from ovarian clear-cell carcinoma. J Clin Oncol 2007;25:5828–30.
- Nikolaou V, Stratigos A, Frangia K, Nikolaidis I, Syrigos K. Carcinoma erysipeloides deriving from a primary cutaneous squamous cell carcinoma. Int J Dermatol 2011;50:754–6.
- Alvarez MA, Casas E, Ruano J, Velez A, Salvatierra J, Moreno JC. Carcinoma erysipeloides of laryngeal origin. Am J Dermatopathol 2012;34:753–4.
- Lee JH, Won CY, Kim EK, Jung JH, Kim GM, Kim SY. Carcinoma erysipeloides from adenocarcinoma of the lung. Ann Dermatol 2013;25:373–5.
- Elston DM, Tuthill RJ, Pierson J, Marden JD, Bergfeld WF. Carcinoma erysipelatoides resulting from genitourinary cancer. J Am Acad Dermatol 1996;35:993–5.
- Webb JM. Carcinoma erysipelatoides from the colon. J Am Acad Dermatol 1996;34:1082–4.
- Han MH, Koh GJ, Choi JH, Sung KJ, Koh JK, Moon KC. Carcinoma erysipelatoides originating from stomach adenocarcinoma. J Dermatol 2000;27:471–4.
- Guberman D, Reinus C. Carcinoma erysipelatoides-cutaneous lymphatic vessel spread of a poorly differentiated naso-pharyngeal carcinoma. Dermatol Online J 2001;7(2):7.
- Kavgaci H, Reis A, Ozdemir F, Bektas O, Arslan M, Aydin F. Carcinoma erysipelatoides resulting from gastric adenocarcinoma: an unusual clinical presentation. Med Princ Pract 2005;14:61–3.
- Choi WJ, Jue MS, Ko JY, Ro YS. An unusual case of carcinoma erysipelatoides originating from gastric adenocarcinoma. Ann Dermatol 2011;23:375–8.
- Tan BB, Marsden JR, Sanders DS. Melanoma erysipeloides: inflammatory metastatic melanoma of the skin. Br J Dermatol 1993;129:327–9.
- Prieto VG, Kenet BJ, Varghese M. Malignant mesothelioma metastatic to the skin, presenting as inflammatory carcinoma. Am J Dermatopathol 1997;19:261–5.
- Yasaka N, Ando I, Kukita A. An acral ‘inflammatory’ cutaneous metastasis of oesophageal carcinoma. Br J Dermatol 1999;141:938–9.
- Marneros AG, Blanco F, Husain S, Silvers DN, Grossman ME. Classification of cutaneous intravascular breast cancer metastases based on immunolabeling for blood and lymph vessels. J Am Acad Dermatol 2009;60:633–8.
- Shoup M, Brennan MF, Karpeh MS, et al. Port site metastasis after diagnostic laparoscopy for upper gastrointestinal tract malignancies: an uncommon entity. Ann Surg Oncol 2002;9:632–6.
- Finkel LJ, Griffiths CE. Inflammatory breast carcinoma (carcinoma erysipeloides): an easily overlooked diagnosis. Br J Dermatol 1993;129:324-6
Cutaneous metastasis
- Reingold IM. Cutaneous metastasis from internal carcinoma. Cancer 1966;19:162–8.
- Provost TT. Cutaneous metastasis. In: Provost TT, Flynn JA, eds. Cutaneous Medicine: Cutaneous Manifestations of Systemic Disease. Hamilton, Ontario: Marcel Decker, 2001:357–66.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol 1995;33:161–82.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol 1993;29:228–36.
- Rolz-Cruz G, Kim CC. Tumor invasion of the skin. Dermatol Clin 2008;26:89–102.
- Marcoval J, Moreno A, Peyri J. Cutaneous infiltration by cancer. J Am Acad Dermatol 2007;57:577–80.
- Weedon D. Skin Pathology, 3rd edn. London: Churchill Livingstone, 2009.
- Brownstein MH, Helwig EB. Patterns of cutaneous metastasis. Arch Dermatol 1972;105:862–8.
- Cao Y, Zhong W. Tumor-derived lymphangiogenic factors and lymphatic metastasis. Biomed Pharmacother 2007;61:534–9.
- Dittmar T, Heyder C, Gloria-Maercker E, et al. Adhesion molecules and chemokines: the navigation system for circulating tumor (stem) cells to metastasize in an organ-specific manner. Clin Exp Metastasis 2008;25:11–32.
- Pedraza-Fariña LG. Mechanisms of oncogenic cooperation in cancer initiation and metastasis. Yale J Biol Med 2006;79:95–103.
- Ramsay AG, Marshall JF, Hart IR. Integrin trafficking and its role in cancer metastasis. Cancer Metastasis Rev 2007;26:567–78.
- Leong SPL, Cady B, Jablons DM, et al. Clinical patterns of metastasis. Cancer Metastasis Rev 2006;25:221–32.
- Dai CY, Haqq CM, Puzas JE. Molecular correlates of site-specific metastasis. Semin Radiat Oncol 2006;16:102–10.
- Kakinuma T, Hwang ST. Chemokines, chemokine receptors, and cancer metastasis. J Leukoc Biol 2006;79:639–51.
- Clements AB. Metastatic carcinoma of the umbilicus. JAMA 1952;150:556–9.
- Duperrat B, Duperrat N. Les métastases ombilicales. A propos de 20 pièces personnelles. Bull Soc Franc Dermatol Syphiligr 1968;75:638–9.
- Taylor CD, Snelling CF, Nickerson D, Trotter MJ. Acute development of invasive squamous cell carcinoma in a split-thickness skin graft donor site. J Burn Care Rehab 1998;19:382–5.
- Serrano OS, Buendia EA, Ortega DRM, Linares SJ. Melanoma metastasis in donor site of full-thickness skin graft. Dermatology (Basel) 2000;201:377–8.
- Diehl LF, Hurwitz MA, Johnson SA, et al. Skin metastases confined to a field of previous irradiation. Report of two cases and review of the literature. Cancer 1984;53:1864–8.
- Richardson JF, Katayama I. Neoplasm to neoplasm metastasis, an acidophil adenoma harbouring metastatic carcinoma: a case report. Arch Pathol 1971;91:135–9.
- Cowley GP, Gallimore A. Malignant melanoma metastasizing to a basal cell carcinoma. Histopathology 1966;29:469–70.
- Rosenthal AL, Lever WF. Involvement of the skin in renal carcinoma: report of two cases with review of the literature. AMA Arch Dermatol 1957;76:96–102.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases. Experience from a cancer center. Arch Dermatol 2007;143:613–20.
- Menter A, Boyd AS, McCaffree DM. Recurrent renal cell carcinoma presenting as skin nodules: two case reports and review of literature. Cutis 1989;44:305–8.
- Braren V, Taylor JN, Pace W. Regression of metastatic renal carcinoma following nephrectomy. Urology 1974;3:777–8.
- Brownstein MH, Helwig EB. Spread of tumours to the skin. Arch Dermatol 1973;107:80–6.
Paget disease
- Paget J. On disease of mammary areola preceding cancer of mammary gland. St Bartholomew's Hosp Rep 1874;10:87–9.
- Kanitakis J. Mammary and extramammary Paget's disease. J Eur Acad Dermatol Venereol 2007;21:581–90.
- Hatta N, Yamada M, Hirano T, et al. Extramammary Paget's disease: treatment, prognostic factors and outcome in 76 patients. Br J Dermatol 2008;158:313–18.
- Chandra JJ. Extramammary Paget's disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol 1985;13:1009–14.
- Liegl B, Liebl S, Gogg-Kamerer M, et al. Mammary and extramammary Paget's disease: an immunohistochemical study of 83 cases. Histopathology 2007;50:439–47.
- Wagner, G. and Sachse, M. M. (2011), Extramammary Paget disease – clinical appearance, pathogenesis, management. JDDG: Journal der Deutschen Dermatologischen Gesellschaft, 9: 448–454
Genodermatoses associated with internal malignancies
- Braverman IM. Skin Signs of Systemic Disease, 3rd edn. Philadelphia: Saunders, 1998.
- Callen JP. Skin signs of internal malignancy. In: Callen JP, Jorizzo JL, eds. Dermatological Signs of Internal Disease, 3rd edn. Philadelphia: Saunders, 2003:95–104.
- Lynch HT, Fusaro RM, eds. Cancer-Associated Genodermatoses. New York: Van Nostrand Reinhold, 1982.
- Callen JP. Skin signs of internal malignancy. Australas J Dermatol 1987;28:106–14.
- Poole S, Fenske NA. Cutaneous markers of internal malignancy, 1: malignant involvement of the skin and the genodermatoses. J Am Acad Dermatol 1993;28:1–13.
- Provost TT, Laman SD, Bell WR. Paraneoplastic dermatoses. In: Provost TT, Flynn JA, eds. Cutaneous Medicine: Cutaneous Manifestations of Systemic Disease. Hamilton, Ontario: Marcel Decker, 2001:367–88.
- Cohen PR, Kurzrock R, eds. Genodermatoses with malignant potential. Dermatol Clin 1995;13:1–243.
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
- Palayoor S, Harper J. Genetic diseases that predispose to malignancy. In: Harper J, Oranje A, Prose N, eds. Textbook of Pediatric Dermatology, 2nd edn. Oxford: Blackwell Publishing, 2006:1596–604.
- Somoano B, Tsao H. Genodermatoses with cutaneous tumors and internal malignancies. Dermatol Clin 2008;26:69–87.
Howel–Evans syndrome
- Harper PS, Harper RM, Howel-Evans AW. Carcinoma of the oesophagus with tylosis. QJM 1970;155:317–33.
- Shine I, Allison PR. Carcinoma of the oesophagus with tylosis. Lancet 1966;i:951–3.
- Zultak M, Blanc D, Merle C, et al. Erytheme annulare centrifuge et leucémie aiguë myeloblastique. Ann Dermatol Vénéréol 1989;116:477–80.
- Bleydon DC, Etheridge SL, Risk JM, et al. RHBDF2 mutations are associated with tylosis, a familiar esophageal cancer syndrome. Am J Hum Genet 2012;90:340-6.
- Bennion SD, Patterson JW. Keratosis punctata palmaris et plantaris and adenocarcinoma of the colon: a possible familial association of punctate keratoderma and gastrointestinal malignancy. J Am Acad Dermatol 1984;10:587–91.
- Blanchet-Bardon C, Nazzaro V, Chevrant-Breton J, et al. Hereditary epidermolytic palmoplantar keratoderma associated with increased breast and ovarian cancer in a large kindred. Br J Dermatol 1987;117:363–70.
- Bianchi L, Orlandi A, Iraci S, et al. Punctate porokeratotic keratoderma: its occurrence with internal neoplasia. Clin Exp Dermatol 1994;19:139–41.
- Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin 2008;26:17–29.
Naevoid basal cell carcinoma syndrome
- Andreev VC, Petkov I. Skin manifestations associated with tumours of the brain. Br J Dermatol 1975;92:675–8.
- Jackson R, Gardere S. Nevoid basal cell carcinoma syndrome. Can Med Assoc J 1971;105:850–9.
- Gorlin SJ. Nevoid basal cell carcinoma syndrome. Dermatol Clin 1995;13:113–25.
- Kimonis VE, Goldstein AM, Pastakia B, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Hum Genet 1997;69:299–308.
- Somoano B, Tsao H. Genodermatoses with cutaneous tumors and internal malignancies. Dermatol Clin 2008;26:69–87.
- Farndon PA. The Gorlin (naevoid basal cell carcinoma) syndrome. In: Harper J, Oranje A, Prose N, eds. Textbook of Pediatric Dermatology, 2nd edn. Oxford: Blackwell Publishing, 2006:1514–31.
- Southwick GT, Schwartz RA. The basal cell nevus syndrome: disasters occurring among a series of 36 patients. Cancer 1979;44:2294–305.
- Motegi S, Nagai Y, Tamura A, Ishikawa O. Multiple skin cysts in nevoid basal cell carcinoma syndrome: a case report and review of the literature. Dermatology 2008;21:159–62.
- Unden AB, Homberg E, Lundh-Rozell B, et al. Mutations in human homolog of Drosophila patched (PTCH) in basal cell carcinomas and the Gorlin syndrome: different in vivo mechinsms of PTCH inactivation. Cancer Res 1996;56:4562–5.
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
Familial melanoma syndrome
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
- Somoano B, Tsao H. Genodermatoses with cutaneous tumors and internal malignancies. Dermatol Clin 2008;26:69–87.
Melanoma–astrocytoma syndrome
- Azizi E, Friedman J, Pavlotsky F, et al. Familial cutaneous malignant melanoma and tumours of the nervous system. A hereditary cancer syndrome. Cancer 1995;76:1571–8.
- Bahuau M, Vidaud D, Kujas M, et al. Familial aggregation of malignant melanoma/dysplastic naevi and tumours of the nervous system: an original syndrome of tumour proneness. Ann Genet 1997;40:78–91.
- Bahuau M, Vidaud D, Jenkins RB, et al. Germline mutation involving the INK4 locus in familial proneness to melanoma and nervous system tumours. Cancer Res 1998;58:2298–303.
- Randerson-Moore JA, Harland M, Williams S, et al. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour family. Hum Mol Genet 2001;1:55–62.
Xeroderma pigmentosum
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum: cutaneous, ocular and neurologic abnormalities in 830 published cases. Arch Dermatol 1987;123:241–50.
- Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, eds. The Genetic Basis of Human Cancer. New York: McGraw-Hill, 1998:211–38.
- Somoano B, Tsao H. Genodermatoses with cutaneous tumors and internal malignancies. Dermatol Clin 2008;26:69–87.
Von Hippel–Lindau disease
- Kiechle-Schwartz M, Neuman HP, Decker HH, et al. Cytogenetic studies on three pheochromocytomas derived from patients with von Hippel–Lindau syndrome. Hum Genet 1989;82:127–30.
- Lynch HT, Fusaro RM, eds. Cancer-Associated Genodermatoses. New York: Van Nostrand Reinhold, 1982.
- Saranya R, Matzkin H, Papo J, Braf Z. Von Hippel–Lindau syndrome with unusual presentations in two brothers. Urology 1989;34:301–4.
- Neumann HP. Basic criteria for clinical diagnosis and genetic counselling in von Hippel–Lindau syndrome. Vasa 1987;16:220–6.
- Maher ER, Kaelin WG Jr. von Hippel–Lindau disease. Medicine (Baltimore) 1997;76:381–91.
- Bar M, Friedman E, Jakobovitz O, et al. Sporadic phaeochromocytomas are rarely associated with germline mutations in the von Hippel–Lindau and RET genes. Clin Endocrinol 1997;47:707–12.
Neurofibromatosis types 1 and 2
- Riccardi VM. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis, 2nd edn. Baltimore: Johns Hopkins University Press, 1992:213–23.
- Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44:81–8.
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
- Somoano B, Tsao H. Genodermatoses with cutaneous tumors and internal malignancies. Dermatol Clin 2008;26:69–87.
- Theos A, Boyd KP, Korf BR. The neurofibromatoses. In: Irvine AD, Hoeger PH, Yan AC, eds. Harper's Textbook of Pediatric Dermatology, 3rd edn. Oxford: Wiley-Blackwell, 2011:128.1.
- Hope DG, Mulvill JJ. Malignancy in neurofibromatosis. Adv Neurol 1981;29:33–56.
- McKeen EA, Bodurtha J, Meadows AT, et al. Rhabdomyosarcoma complicating multiple neurofibromatosis. J Pediatr 1977;93:992–3.
- Stay EJ, Vawter G. The relationship between nephroblastoma and neurofibromatosis (von Recklinghausen's disease). Cancer 1977;39:2550–5.
- Zvulunov A, Barak Y, Metzker A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia. World statistical analysis. Arch Dermatol 1995;131:904–8.
- Maris JM, Wiersma SR, Mahgoub N, et al. Monosomy 7 myelodysplastic syndrome and other second malignant neoplasms in children with neurofibromatosis type 1. Cancer 1997;79:1438–46.
- Wiznia RA, Freeman JK, Mancini AD, et al. Malignant melanoma of the choroid in neurofibromatosis. Am J Ophthalmol 1978;86:684–7.
Tuberous sclerosis complex
- Cowen EW, Callen JP. Skin signs of internal malignancy. In: Callen JP, Jorizzo JL, Bolognia JL, Piette WW, Zone JJ, eds. Dermatological Signs of Internal Disease, 4th edn. Philadelphia: Saunders Elsevier, 2009:107–16.
- Somoano B, Tsao H. Genodermatoses with cutaneous tumors and internal malignancies. Dermatol Clin 2008;26:69–87.
- Krueger DA, Northrup H. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:255–65.
- Lynch HT, Fusaro RM, eds. Cancer-Associated Genodermatoses. New York: Van Nostrand Reinhold, 1982.
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345–56.
- Lynne CM, Carrion HM, Baskshandeh K, et al. Renal angiomyolipoma: polycystic kidney and renal cell carcinoma in a patient with tuberous sclerosis. Urology 1979;14:174–6.
Multiple endocrine neoplasia syndromes
- Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97:2990–3011.
- Guo SS, Sawicki MP. Molecular and genetic mechanism of tumorigenesis in multiple endocrine neoplasia. Mol Endocrinol 2001;15:1653–64.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol 1997;133:853–7.
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
- Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658–71.
- Stratakis CA. Clinical genetics of multiple endocrine neoplasias, Carney's complex and related syndromes. J Endocrinol Invest 2001;24:370–83.
- Sipple JH. The association of phaeochromocytoma with carcinoma of the thyroid gland. Am J Med 1961;31:163–6.
- Cunliffe WJ, Hudgson P, Fulthorpe JJ, et al. A calcitonin secreting medullary thyroid carcinoma, associated with mucosal neuromas, marfanoid features, myopathy and pigmentation. Am J Med 1970;48:120–6.
- Kousseff BG. Multiple endocrine neoplasia 2 (MEN 2)/MEN 2A (Sipple syndrome). Dermatol Clin 1995;13:91–7.
- Holloway KB, Flowers FP. Multiple endocrine neoplasia 2B (MEN 2B)/MEN 3. Dermatol Clin 1995;13:99–103.
- Lee NC, Norton JA. Multiple endocrine neoplasia type 2B: genetic basis and clinical expression. Surg Oncol 2000;9:111–18.
- Huang SC, Torres-Cruz J, Pack SD, et al. Amplification and overexpression of mutant RET in multiple endocrine neoplasia type 2-associated medullary thyroid carcinoma. J Clin Endocrinol Metab 2003;88:459–63.
- Krampitz GW, Norton JA. RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Cancer 2014;120:1920–31.
- Saggini A, Brandi ML. Skin lesions in hereditary endocrine tumor syndromes. Endocr Pract 2011;17(Suppl. 3):47–57.
Carney complex
- Salpea P, Stratakis CA. Carney complex and McCune Albright syndrome: an overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol 2014;386:85–91.
- Cullen MK. Carney complex. In: Nordlund JJ, Boissy RE, Hearing VJ, et al., eds. The Pigmentary System, 2nd edn. Oxford: Blackwell Publishing, 2006:851–62.
- Saggini A, Brandi ML. Skin lesions in hereditary endocrine tumor syndromes. Endocr Pract 2011;17(Suppl. 3):47– 57.
- Carney JA. Carney complex: the complex of myxomas, spotty pigmentation, endocrine overactivity, and schwannomas. Semin Dermatol 1995;14:90–8.
- Stratakis CA. Clinical genetics of multiple endocrine neoplasias, Carney complex and related syndromes. J Endocrinol Invest 2001;24:370–83.
- Lee NC, Norton JA. Multiple endocrine neoplasia type 2B: genetic basis and clinical expression. Surg Oncol 2000;9:111–18.
PTEN hamartoma tumour syndrome
- Lloyd KM, Dennis M. Cowden's disease: a possible new system complex with multiple system involvement. Ann Intern Med 1963;58:136–42.
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
- Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet 2000;37:828–30.
- Mallory S, Mallory SB. Cowden syndrome (multiple hamartoma syndrome). Dermatol Clin 1995;13:27–31.
- Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)?: expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet 2004;41:323–6.
- Uppal S, Mistry D, Coatesworth AP. Cowden disease: a review. Int J Clin Pract 2007;61:645–52.
- Graham RM, Emmerson RW. Multiple hamartoma and neoplasia syndrome. Clin Exp Dermatol 1985;10:262–8.
- Kurek KC, Howard E, Tennant LB, et al. PTEN hamartoma of soft tissue: a distinctive lesion in PTEN syndromes. Am J Surg Pathol 2012;36:671–87.
- Bubien V, Bonnet F, Brouste V, et al. High cumulative risk of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 2013;50:255–63.
Sebaceous tumours, keratoacanthomas and visceral malignancy
- Lynch HT, Fusaro RM, eds. Cancer-Associated Genodermatoses. New York: Van Nostrand Reinhold, 1982.
- Schwarz RA, Torre DP. The Muir–Torre syndrome: a 25-year retrospect. J Am Acad Dermatol 1995;33:90–104.
- Hall NR, Williams AT, Murday VA, et al. Muir–Torre syndrome: a variant of the cancer family syndrome. J Med Genet 1994;31:627–31.
- Ponti G, Pellacani G, Seidenari S, Pollio A, Muscatello U, Tomasi A. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol 2013;85:239–56.
- Jones B, Oh C, Mangold E, Egan CA. Muir–Torre syndrome: diagnostic and screening guidelines. Australas J Dermatol 2006;47:266–9.
- Cohen PR, Kohn SR, Davis DA, Kurzrock R. Muir–Torre syndrome. Dermatol Clin 1995;13:79–89.
- Smoller BR. Muir–Torre syndrome. In: Morgan MB, Smoller BR, Somach SC. Deadly Dermatologic Diseases. Clinicopathologic Atlas and Text. New York: Springer, 2007:53–8.
- Offit K, Kauff ND. Reducing the risk of gynecologic cancer in the Lynch syndrome. N Engl J Med 2006;354:293–5.
- Serleth HJ, Kisken WA. A Muir–Torre syndrome family. Am Surg 1998;64:365–9.
- Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir–Torre variant of Lynch syndrome. Genet Med 2014;16:711–16.
Hereditary leiomyomatosis and renal cell carcinoma syndrome
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 2011;79:49–59.
- Alam NA, Olpin S, Leigh IM. Fumarate hydratase mutations and predisposition to cutaneous leiomyomas, uterine leiomyomas and renal cancer. Br J Dermatol 2005;153:11–17.
- Lehtonen HJ. Hereditary leiomyomatosis and renal cell cancer: update on clinical and molecular characteristics. Fam Cancer 2011;10:397–411.
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer 2014;13:637–44.
- Lehtonen HJ, Kiura M, Ylisaukko-Oja SK, et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet 2006;43:523–6.
Bloom, Rothmund–Thomson and Werner syndromes
- Lindor NM, Furuichi Y, Kitao S, et al. Rothmund–Thomson syndrome due to RecQ helicase gene mutations: report and clinical and molecular comparisons with Bloom syndrome and Werner syndrome. Am J Med Genet 2000;90:223–8.
- Monnat RJ Jr. Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology. Semin Cancer Biol 2010;20:329–39.
Bloom syndrome
- German J. Bloom's syndrome. Dermatol Clin 1995;13:7–18.
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
Rothmund–Thomson syndrome
- Lindor NM, Furuichi Y, Kitao S, et al. Rothmund–Thomson syndrome due to RECQ4 helicase mutations: report and clinical and molecular comparisons with Bloom syndrome and Werner syndrome. Am J Med Genet 2000;90:223–8.
- Narayan S, Fleming C, Trainer AH, et al. Rothmund–Thomson syndrome with myelodysplasia. Pediatr Dermatol 2001;18:210–12.
- Wang LL, Levy ML, Lewis RA, et al. Clinical manifestations in a cohort of 41 Rothmund–Thomson syndrome patients. Am J Med Genet 2001;102:11–17.
- Monnat RJ Jr. Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology. Semin Cancer Biol 2010;20:329–39.
Werner syndrome
- Callen JP. Skin signs of internal malignancy. In: Callen JP, Jorizzo JL, eds. Dermatological Signs of Internal Disease, 3rd edn. Philadelphia: Saunders, 2003:95–104.
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
- Duvic M, Lemak NA. Werner's syndrome. Dermatol Clin 1995;13:163–8.
- Shen J, Loeb LA. Unwinding the molecular basis of the Werner syndrome. Mech Ageing Dev 2001;122:921–44.
- Hrabko RP, Milgrom H, Schwartz RA. Werner's syndrome with associated malignant neoplasms. Arch Dermatol 1982;118:106–8.
- Lindor NM, Furuichi Y, Kitao S, et al. Rothmund–Thomson syndrome due to RECQ4 helicase mutations: report and clinical and molecular comparisons with Bloom syndrome and Werner syndrome. Am J Med Genet 2000;90:223–8.
- Monnat RJ Jr. ‘…Rewritten in the skin’: clues to skin biology and aging from inherited disease. J Invest Dermatol 2015;135:1484–90.
Immunodeficiency and neoplasia syndromes
Wiskott–Aldrich syndrome
- Ariga T. Wiskott–Aldrich syndrome: an X-linked primary immunodeficiency disease with unique and characteristic features. Allergol Int 2013;61:183–9.
- Schaffer, JV, Paller AS. Immunodeficiency syndromes. In: Irvine A, Hoeger P, Yan A, eds. Harper's Textbook of Pediatric Dermatology, 3rd edn. Oxford: Wiley-Blackwell, 2011:2527–60.
- Ormerod AD. The Wiskott–Aldrich syndrome. Int J Dermatol 1985;24:77–81.
- Model LM. Primary reticulum cell sarcoma of the brain in Wiskott–Aldrich syndrome. Report of a case. Arch Neurol 1977;34:633–5.
- Massaad MJ, Ramesh N, Geha RS. Wiskott–Aldrich syndrome: a comprehensive review. Ann NY Acad Sci 2013;1285:26–43.
Chediak-Higashi syndrome, Ataxia-telangiectasia, Dyskeratosis congenital, Fanconi anaemia
- Stolz W, Graubner U, Gerstmeier J, et al. Chediak–Higashi syndrome: approaches in diagnosis and treatment. Curr Probl Dermatol 1989;18:93–100.
- Schaffer, JV, Paller AS. Immunodeficiency syndromes. In: Irvine A, Hoeger P, Yan A, eds. Harper's Textbook of Pediatric Dermatology, 3rd edn. Oxford: Wiley-Blackwell, 2011:2527–60.
- Tsao H. Update on familial cancer syndromes and the skin. J Am Acad Dermatol 2000;42:939–46.
- Spector BD, Fillipivich AH, Perry SS, et al. Epidemiology of cancer in ataxia telangiectasia. In: Bridges BA, Harnden DG, eds. Ataxia Telangiectasia: A Cellular and Molecular Link Between Cancer Neuropathy and Immune Deficiency. New York: Wiley, 1982:103–38.
- Boultwood J. Ataxia telangiectasia gene mutations in leukaemia and lymphoma. J Cutan Pathol 2001;54:512–16.
- Gatti RA. Ataxia-telangiectasia. Dermatol Clin 1995;13:1–6.
- Swift M, Morrell D, Massey RB, et al. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med 1991;325:1831–6.
- Morgan MB. Lethal hereditary vascular disorders: Osler–Weber–Rendu, ataxia telangiectasia, and Fabry's disease. In: Morgan MB, Smoller BR, Somach SC. Deadly Dermatologic Diseases. Clinicopathologic Atlas and Text. New York: Springer, 2007:145–9.
- Callen JP. Skin signs of internal malignancy. In: Callen JP, Jorizzo JL, eds. Dermatological Signs of Internal Disease, 3rd edn. Philadelphia: Saunders, 2003:95–104.
- Drachtman RA, Alter BP. Dyskeratosis congenita. Dermatol Clin 1995;13:33–9.
- Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program 2011:480–6.
- Lambert WC. Genetic diseases associated with DNA and chromosomal instability. Dermatol Clin 1987;5:85–108.
- Connor JM, Teague RH. Dyskeratosis congenita: report of a large kindred. Br J Dermatol 1981;105:321–5.
- Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res 2009;66:4–10.
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013;493:356–63.
- Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol 2014;27:214–21.
Paraneoplastic phenomena involving the skin
- Cowen EW, Callen JP. Skin signs of internal malignancy. In: Callen JP, Jorizzo JL, Bolognia JL, Piette WW, Zone JJ, eds. Dermatological Signs of Internal Disease, 4th edn. Philadelphia: Saunders Elsevier, 2009:107–16.
- Poole S, Fenske NA. Cutaneous markers of internal malignancy, 1: malignant involvement of the skin and the genodermatoses. J Am Acad Dermatol 1993;28:1–13.
- Poole S, Fenske NA. Cutaneous markers of internal malignancy, 2: paraneoplastic dermatoses and environmental carcinogens. J Am Acad Dermatol 1993;28:147–64.
- Provost TT, Laman SD, Bell WR. Paraneoplastic dermatoses. In: Provost TT, Flynn JA, eds. Cutaneous Medicine: Cutaneous Manifestations of Systemic Disease. Hamilton, Ontario: Marcel Decker, 2001:367–88.
- Provost TT, Laman SD, Giardiello FM. Hereditary paraneoplastic syndromes. In: Provost TT, Flynn JA, eds. Cutaneous Medicine: Cutaneous Manifestations of Systemic Disease. Hamilton, Ontario: Marcel Decker, 2001:389–400.
- Provost TT, Gordon AH, Laman SD. Hormonally mediated paraneoplastic syndromes. In: Provost TT, Flynn JA, eds. Cutaneous Medicine: Cutaneous Manifestations of Systemic Disease. Hamilton, Ontario: Marcel Decker, 2001:401–12.
- Cohen PR. Paraneoplastic dermatopathology: cutaneous paraneoplastic syndromes. Adv Dermatol 1996;11:215–52.
- Chung VQ, Moschella SL, Zembowicz A, Liu V. Clinical and pathologic findings of paraneoplastic dermatoses. J Am Acad Dermatol 2006;54:745–62.
- Boyce S, Harper J. Paraneoplastic dermatoses. Dermatol Clin 2002;20:523–32.
- Thomas VD, Thomas CR, eds. Internal malignancy and the skin: paraneoplastic and cancer treatment-related cutaneous disorders. Dermatol Clin 2008;26:l–182.
- Pipkin CA, Lio PA. Cutaneous manifestations of internal malignancies: an overview. Dermatol Clin 2008;26:1–15.
- Stone SP, Buescher LS. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol 2005;23:301–6.
- Thomas I, Schwartz RA. Cutaneous paraneoplastic syndromes: uncommon presentations. Clin Dermatol 2005;23:593–600.
- Zappasodi P, Del Forna C, Corso A, Lazzarino M. Mucocutaneous paraneoplastic syndromes in hematologic malignancies. Int J Dermatol 2006;45:14–22.
Acanthotic, epidermal and ichthyotic disorders
Acanthosis nigricans
- Schwartz RA. Acanthosis nigricans. J Am Acad Dermatol 1994;31:1–19.
- Isaacoff E, Dimitriadi FF, Barrows F, Pawel B, Mattei P, Mostoufi-Moab S. Acanthosis nigricans associated with an adrenocortical tumor in a pediatric patient. Case Rep Endocrinol 2013:174593. doi: 10.1155/2013/174593.
- Thomas I, Schwartz RA. Cutaneous paraneoplastic syndromes: uncommon presentations. Clin Dermatol 2005;23:593–600.
- Stone SP, Buescher LS. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol 2005;23:301–6.
- Curth HO. Classification of acanthosis nigricans. Int J Dermatol 1976;15:592–3.
- Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin 2008;26:17–29.
- Longshore SL, Taylor JS, Kennedy A, Nurko S. Malignant acanthosis nigricans and endometrial carcinoma of the parametrium: the search for malignancy. J Am Acad Dermatol 2003;49:541.
- Moller H, Eriksson S, Holen O, et al. Complete reversibility of paraneoplastic acanthosis nigricans after operation. Acta Med Scand 1978;203:245–6.
Acanthosis palmaris
- Breathnach SM, Wells GC. Acanthosis palmaris: tripe palms – a distinctive pattern of palmar keratoderma frequently associated with internal malignancy. Clin Exp Dermatol 1980;5:181–9.
- Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin 2008;26:17–29.
- Cohen PR, Grossman ME, Silvers DN, et al. Tripe palms and cancer. Clin Dermatol 1993;11:165–73.
Sign of Leser–Trélat
- Holdiness MR. On the classification of the sign of Leser-Trélat. J Am Acad Dermatol 1988;19:754–7.
- Rampen FH, Schwengle LE. The sign of Leser-Trélat: does it exist? J Am Acad Dermatol 1989;21:50–5.
- Turan E, Yesilova Y, Yurt N, Koçarslan S. Leser-Trélat sign: does it really exist? Acta Dermatovenerol Croat 2013;21:123–7.
- Lindelöf B, Sigurgeirsson B, Melander S. Seborrheic keratoses and cancer. J Am Acad Dermatol 1992;26:947–50.
- Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin 2008;26:17–29.
- Flugman SL, McClain SA, Clark RA. Transient eruptive seborrheic keratoses associated with erythrodermic psoriasis and erythrodermic drug eruption: report of two cases. J Am Acad Dermatol 2001;45(6 Suppl.):S212–14.
- Mayou SC, Benn JJ, Sonksen PH, et al. Paraneoplastic rhinophyma and the Leser-Trélat sign. Clin Exp Dermatol 1989;14:253–5.
- Fanti PA, Metri M, Patrizi A. The sign of Leser-Trélat associated with malignant melanoma. Cutis 1989;44:39–41.
- Yaniv R, Servadio Y, Feinstein A, et al. The sign of Leser-Trélat associated with transitional cell carcinoma of the urinary bladder: a case report and short review. Clin Exp Dermatol 1994;19:142–5.
- Fasoldt JJ, Brumwell ER, Lackey JN. Leser-Trélat sign presenting in a patient with recurrent pre-B-cell acute lymphocytic leukaemia. Cutis 2012;89:33–5.
- Ponti G, Luppi G, Losi L, Giannetti A, Seidenari S. Leser-Trélat syndrome in patients affected by six multiple metachronous primitive cancers. J Hematol Oncol 2010;3:2.
Florid cutaneous papillomatosis
- Janniger EJ, Schwartz RA. Florid cutaneous papillomatosis. J Surg Oncol 2010;102:709–12.
Acquired ichthyosis and other epidermal disorders
- Patel N, Spencer LA, English JC III, Zirwas MJ. Acquired ichthyosis. J Am Acad Dermatol 2006;55:647–56.
- Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin 2008;26:17–29.
- Griffin LJ, Massa MC. Acquired ichthyosis and pityriasis rotunda. Clin Dermatol 1993;11:27–32.
- Riesco Martínez MC, Muñoz Martín AJ, Zamberk Majlis P, et al. Acquired ichthyosis as a paraneoplastic syndrome in Hodgkin's disease. Clin Transl Oncol 2009;11:552–3.
- Akpinar TS, Ozkok A, Bakkaloglu OK, Saka B. Acquired ichthyosis as a presenting finding of Hodgkin's lymphoma. Int J Hematol 2012;96:401–2.
- Guana AL, Cohen PR. Transient acantholytic dermatosis in oncology patients. J Clin Oncol 1994;12:1703–9.
- Aldabagh B, Patel RR, Honda K. Leukemia cutis in association with Grover's disease. Am J Dermatopathol 2011;33:e41–3.
- Cuzick J, Harris R, Mortimer PS. Palmar keratoses and cancers of the bladder and lung. Lancet 1984;i:530–3.
Paraneoplastic pigmentation
- Levine N, Burk C. Extracutaneous neuroendocrine melanoderma. In: Nordlund JJ, Boissy RE, Hearing VJ, et al., eds. The Pigmentary System, 2nd edn. Oxford: Blackwell Publishing, 2006:938–9.
- Cullen MK. Carney complex. In: Nordlund JJ, Boissy RE, Hearing VJ, et al., eds. The Pigmentary System, 2nd edn. Oxford: Blackwell Publishing, 2006:851–62.
Hair, nails and skin appendages
Paraneoplastic hypertrichosis lanuginosa acquisita
- Wendolin DS, Pope DN, Mallory SB. Hypertrichosis. J Am Acad Dermatol 2003;48:161–79.
- Stone SP, Buescher LS. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol 2005;23:301–6.
- Cowen EW, Callen JP. Skin signs of internal malignancy. In: Callen JP, Jorizzo JL, Bolognia JL, Piette WW, Zone JJ, eds. Dermatological Signs of Internal Disease, 4th edn. Philadelphia: Saunders Elsevier; 2009:107–16.
- Slee PHTJ, van der Waal RIF, Schagen van Leeuwen JH, et al. Paraneoplastic hypertrichosis lanuginosa acquisita: uncommon or overlooked? Br J Dermatol 2007;157:1087–92.
- Dalcin D, Manser C, Mahler R. Malignant down: hypertrichosis lanuginosa acquisita associated with endometrial adenocarcinoma. J Cutan Med Surg 2015;19:507–10.
- Worsnop F, Campalani E. Paraneoplastic hypertrichosis lanuginosa acquisita: a delayed presentation in quiescent chronic lymphocytic leukaemia. Clin Exp Dermatol 2014;39:669–70.
Clubbing of nails
- Sridhar KS, Lobo CF, Altman RD. Digital clubbing and lung cancer. Chest 1998;114:1535–7.
- Rutherford JD. Digital clubbing. Circulation 2013;127:1997–9.
- Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin 2008;26:59–68.
- Caldwell DS, McCallum RM. Rheumatological manifestations of cancer. Med Clin North Am 1986;70:385–417.
Hyperhidrosis
- Sato K, Kang WH, Saga K, Sato KT. Biology of sweat glands and their disorders. II. Disorders of sweat gland function. J Am Acad Dermatol 1989;20:713–26.
- Murakami T, Koyanagi I, Kaneko T, et al. Intramedullary spinal cord ganglioglioma presenting as hyperhidrosis: unique symptoms and magnetic resonance imaging findings: case report. J Neurosurg Spine 2013;18:184–8.
Dermatoses associated with internal malignancies
Acrokeratosis paraneoplastica
- Bazex A, Griffiths A. Acrokeratosis paraneoplastica: a new cutaneous marker of malignancy. Br J Dermatol 1980;102:301–6.
- Richard M, Giroux JM. Acrokeratosis paraneoplastica. J Am Acad Dermatol 1987;16:178–83.
- Bolognia JL. Bazex syndrome: acrokeratosis paraneoplastica. Semin Dermatol 1995;14:84–9.
- Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin 2008;26:17–29.
- Stone SP, Buescher LS. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol 2005;23:301–6.
- Obasi OE, Garg SK. Bazex paraneoplastic acrokeratosis in prostate carcinoma. Br J Dermatol 1987;117:647–51.
- Arregui MA, Raton JA, Landa NI, et al. Bazex's syndrome (acrokeratosis paraneoplastica): first report of an association with a bladder carcinoma. Clin Exp Dermatol 1993;18:445–8.
- Patel LM, Lambert PJ, Gagna CE, Maghari A, Lambert WC. Cutaneous signs of systemic disease. Clin Dermatol 2011;29:511–22.
- Wishart JM. Bazex paraneoplastic acrokeratosis: a case report and response to Tigason. Br J Dermatol 1986;115:595–9.
Migratory erythemas
- Lazar P. Cancer, erythema annulare centrifugum, autoimmunity. Arch Dermatol 1963;87:247–51.
- Chodkiewicz HM, Cohen PR. Paraneoplastic erythema annulare centrifugum eruption: PEACE. Am J Clin Dermatol 2012;13:239–46.
- De La Torre-Lugo EM, Sanchez JL. Erythema gyratum repens. J Am Acad Dermatol 2011;64:e89–90.
- Stone SP, Buescher LS. Life-threatening paraneoplastic cutaneous syndromes. Clin Dermatol 2005;23:301–6.
- Juhlin L, Lacour JP, Larrouy JC, et al. Episodic erythema gyratum repens with ichthyosis and palmoplantar hyperkeratosis without signs of internal malignancy. Clin Exp Dermatol 1989;14:223–6.
- Campbell L, Freedman JR, O'Donoghue M, Chevalier M, Dy LC, Tharp MD. Erythema gyratum repens without associated malignancy. J Am Acad Dermatol 2011;65:e22–3.
Connective tissue and rheumatological disorders
Dermatomyositis
- Cowen EW, Callen JP. Skin signs of internal malignancy. In: Callen JP, Jorizzo JL, Bolognia JL, Piette WW, Zone JJ, eds. Dermatological Signs of Internal Disease, 4th edn. Philadelphia: Saunders Elsevier, 2009:107–16.
- Provost TT, Flynn JA. Dermatomyositis. In: Provost TT, Flynn JA, eds. Cutaneous Medicine: Cutaneous Manifestations of Systemic Disease. Hamilton, Ontario: Marcel Decker, 2001:82–103.
- Callen JP. Malignancy in polymyositis/dermatomyositis. Clin Dermatol 1988;6:55–63.
- Cox NH, Lawrence CM, Langtry JA, et al. Dermatomyositis: disease associations and evaluation of screening investigations for malignancy. Arch Dermatol 1990;126:61–5.
- Sigurgeiersson B, Lindelöf B, Edhag O, Allander E. Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med 1992;326:363–7.
- Henriksson KG, Sandstedt P. Polymyositis: treatment and prognosis. A study of 107 patients. Acta Neurol Scand 1982;65:280–300.
- Antiochos BB, Brown LA, Li Z, Tosteson TD, Wortmann RL, Rigby WF. Malignancy is associated with dermatomyositis but not polymyositis in Northern New England, USA. J Rheumatol 2009;36:2704–10.
- Kalmanti M, Athanasion A. Neuroblastoma occurring in a child with dermatomyositis. Am J Pediatr Hematol Oncol 1985;7:387–8.
- Callen JP. When and how should the patient with dermatomyositis or amyopathic dermatomyositis be assessed for possible cancer? Arch Dermatol 2002;138:969–71.
- Sparsa A, Liozon E, Herrmann F, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis. A study of 40 patients. Arch Dermatol 2002;138:885–90.
- Hunger RE, Durr D, Brand CU. Cutaneous leukocytoclastic vasculitis in dermatomyositis suggests malignancy. Dermatology 2001;202:123–6.
- Burnouf M, Mahé E, Verpillat P, et al. Cutaneous necrosis is predictive of cancer in adult dermatomyositis. Ann Dermatol Vénéréol 2003;130:313–16.
- Wang J, Guo G, Chen G, Wu B, Lu L, Bao L. Meta-analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol 2013;169:838–47.
- Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum 2013;65:2954–62.
- Hamaguchi Y, Kuwana M, Hoshino K, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol 2011;147:391–8.
- Femia AN, Vleugels RA, Callen JP. Cutaneous dermatomyositis: an updated review of treatment options and internal associations. Am J Clin Dermatol 2013;14:291–313.
Lupus erythematosus
- Cloutier BT, Clarke AE, Ramsey-Goldman R, Gordon C, Hansen JE, Bernatsky S. Systemic lupus erythematosus and malignancies: a review article. Rheum Dis ClinNorth Am 2014;40:497–506.
- Bernatsky S, Boivin JF, Joseph L, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis Rheum 2005;52:1481–90.
- Tarr T, Gyorfy B, Szekanecz E, et al. Occurrence of malignancies in Hungarian patients with lupus erythematosus: results from a single center. Ann NY Acad Sci 2007;1108:76–82.
- Chaudhry SI, Murphy LA, White IR. Subacute cutaneous lupus erythematosus: a paraneoplastic dermatosis? Clin Exp Dermatol 2005;30:655–8.
- Renner R, Sticherling M. Incidental cases of subacute cutaneous lupus erythematosus in association with malignancy. Eur J Dermatol 2008;18:700–4.
- Bernatsky S, Ramsey-Goldman R, Labrecque J, et al. Cancer risk in systemic lupus: an updated international multi-centre cohort study. J Autoimmun 2013;42:130–5.
- Löfström B, Backlin C, Sundström C, et al. A closer look at non-Hodgkin's lymphoma cases in a national Swedish systemic lupus erythematosus cohort: a nested case-control study. Ann Rheum Dis 2007;66:1627–32.
- Ali YM, Urowitz MB, Ibanez D, Gladman DD. Monoclonal gammopathy in systemic lupus erythematosus. Lupus 2007;16:426–9.
- Bernatsky S, Joseph L, Boivin JF, et al. The relationship between cancer and medication exposures in systemic lupus erythematosus: a case-cohort study. Ann Rheum Dis 2008;67:74–9.
- Smedby KE, Hjalgrim H, Askling J, et al. Autoimmune and chronic inflammatory disorders and risk of non-Hodgkin lymphoma by subtype. J Natl Cancer Inst 2006;98:51–60.
- Bernatsky S, Ramsay-Goldman R, Clarke A. Malignancy and autoimmunity. Curr Opin Rheum 2006;18:129–34.
- Ruiz-Irastorza G, Ugarte A, Egurbide MV, et al. Antimalarials may influence the risk of malignancy in systemic lupus erythematosus. Ann Rheum Dis 2007;66:815–17.
- Kreft B, Marsch WC. Lupus erythematosus gyratum repens. Eur J Dermatol 2007;17:79–82.
- Dawn G, Wainwright NJ. Association between subacute cutaneous lupus erythematosus and epidermoid carcinoma of the lung: a paraneoplastic phenomenon? Clin Exp Dermatol 2002;27:714–22.
- Yasim ZF, Walsh MY, Armstrong DKB. Subacute lupus erythematosus-like rash associated with oesophageal carcinoma in situ. Clin Exp Dermatol 2007;32:443–4.
Scleroderma, fasciitis, panniculitis and similar sclerodermoid conditions
- Duncan S, Winkelmann R. Cancer and scleroderma. Arch Dermatol 1979;115:950–5.
- Roumm A, Medsger TJ. Cancer in connective tissue disease. Arthritis Rheum 1982;25:1130–3.
- Onishi A, Sugiyama D, Kumagai S, Morinobu A. Cancer incidence in systemic sclerosis. Meta-analysis of population-based studies. Arthritis Rheum 2013;65:1913–21.
- Chan LS, Hanson CA, Cooper KD. Concurrent eosinophilic fasciitis and cutaneous T-cell lymphoma. Arch Dermatol 1991;127:862–5.
- Naschitz JE, Yeshurun D, Rosner I. Rheumatic manifestations of occult cancer. Cancer 1995;12:2954–8.
- Cohen PR. Paraneoplastic dermatopathology: cutaneous paraneoplastic syndromes. Adv Dermatol 1996;11:215–52.
- Naschitz JE, Yeshuran D, Zuckerman E, et al. Cancer-associated fasciitis–panniculitis. Cancer 1994;73:231–5.
- Cox NH, Ramsay B, Dobson C, Comaish JS. Woody hands in a patient with pancreatic carcinoma: a variant of cancer-associated fasciitisis–panniculitis syndrome. Br J Dermatol 1996;135:995–8.
- Winkelmann RK, Frigas E. Eosinophilic panniculitis: a clinicopathologc study. J Cutan Pathol 1986;13:1–12.
- Marullo S, Dallot A, Carelier-Balloy B, et al. Subcutaneous eosinophilic necrosis associated with refractory anaemia with an excess of myeloblasts. J Am Acad Dermatol 1989;20:320–3.
- Colver GB, Rodger A, Mortimer PS, et al. Post-irradiation morphoea. Br J Dermatol 1989;120:831–5.
Bullous disorders associated with internal malignancy
Paraneoplastic pemphigus
- Nguyen VT, Ndoye A, Bassler KD, et al. Classification, clinical manifestations and immunopathological mechanisms of epithelial variant of paraneoplastic autoimmune multiorgan syndrome. Arch Dermatol 2001;137:193–206.
- Nousari HC, Deterding R, Wojtczack H, et al. The mechanism of respiratory failure in paraneoplastic pemphigus. N Engl J Med 1999;340:1406–10.
- Anhalt GJ. Paraneoplastic pemphigus. Adv Dermatol 1997;12:77–96.
- Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol 2000;43:619–26.
Bullous pemphigoid, Pemphigus
- Hodge L, Marsden RA, Black MM, et al. Bullous pemphigoid: the frequency of mucosal involvement and concurrent malignancy related to indirect immunofluorescence findings. Br J Dermatol 1981;105:65–9.
- Schroeter AL. Pemphigoid and malignancy. Clin Dermatol 1987;5:60–3.
- Tanaka T, Ogino A, Ogura K, et al. A case of bullous pemphigoid and transitional cell carcinoma of the bladder: demonstration of a circulating factor reactive with basement membrane zone of skin and of bladder carcinoma. Arch Dermatol 1983;119:704–5.
- Lindelöf B, Islam N, Eklund G, et al. Pemphigoid and cancer. Arch Dermatol 1990;126:66–8.
- Stone SS, Schroeter AL. Bullous pemphigoid and associated malignant neoplasms. Arch Dermatol 1975;111:991–4.
- Person JR, Rogers RS. Bullous and cicatricial pemphigoid: clinical, histopathologic and immunopathologic correlations. Mayo Clin Proc 1977;52:54–66.
- Muramatsu T, Matsumoto H, Yamashina Y, et al. Pemphigus foliaceus associated with acanthosis nigricans-like lesions and hepatocellular carcinoma. Int J Dermatol 1989;28:462–3.
- Krain LS, Bierman SM. Pemphigus vulgaris and internal malignancy. Cancer 1974;33:1091–9.
- Callen JP. Internal disorders associated with bullous disease of the skin: a critical review. J Am Acad Dermatol 1980;3:107–19.
Other blistering disorders
- Leonard JN, Tucker WF, Fry JS, et al. Increased incidence of malignancy in dermatitis herpetiformis. BMJ 1983;286:16–18.
- Swerdlow AJ, Whittaker S, Carpenter LM, et al. Mortality and cancer incidence in patients with dermatitis herpetiformis: a cohort study. Br J Dermatol 1993;129:140–4.
- Sigurgeirsson B, Agnarsson BA, Lindelöf B. Risk of lymphoma in patients with dermatitis herpetiformis. BMJ 1994;308:13–15.
- Collin P, Pukkala E, Reunala T. Malignancy and survival in dermatitis herpetiformis: a comparison with coeliac disease. Gut 1996;38:528–30.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis: Part I. Epidemiology, pathogenesis and clinical presentation. J Am Acad Dermatol 2011;64:1017–24.
- Viljamaa M, Kaukinen K, Pukkala E, et al. Malignancies and mortality in patients with coeliac disease and dermatitis herpetiformis: 30-year population-based study. Dig Liver Dis 2006;38:374–80.
- Lewis HM, Reunala TL, Garioch JJ, et al. Protective effect of gluten-free diet against development of lymphoma in dermatitis herpetiformis. Br J Dermatol 1996;135:363–7.
- Godfrey KM, Wojnarowska FJ, Leonard J. Disease associations of linear IgA disease. Br J Dermatol 1989;121:48.
- Aractingi S, Bachmeyer C, Prost C, et al. Subepidermal autoimmune bullous skin diseases associated with B-cell lymphoproliferative disorders. Medicine (Baltimore) 1999;78:228–35.
- Baler GR. Epidermolysis bullosa acquisita associated with lymphoma. J Am Acad Dermatol 1987;17:856–9.
- Engineer L, Dow EC, Braverman IM, Ahmed AR. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol 2002;47:943–6.
- Tidman MJ, Higgins EM, Elder GH, et al. Variegate porphyria associated with hepatocellular carcinoma. Br J Dermatol 1989;121:503–5.
- Dandurand M, Guillot B, Guilhou JJ. [Porphyria cutanea tarda and neoplasms. Apropos of 2 cases.] Ann Dermatol Vénéréol 1986;113:679–83.
Deposition disorders
- Touart DM, Sau P. Cutaneous deposition disorders. Part I. J Am Acad Dermatol 1998;39:149–71.
- Weenig RH, Mehrany K. Dermal and pannicular manifestations of internal malignancy. Dermatol Clin 2008;26:31–43.
Other dermatological disorders
- Helm TN, Camisa C, Liu AY, et al. Lichen planus associated with neoplasia: a cell-mediated immune response to tumor antigens? J Am Acad Dermatol 1994;30:219–24.
- Sigurgeirsson B, Lindelöf B. Lichen planus and malignancy. Arch Dermatol 1991;127:1684–8.
- Neittaanmaki H. Cold urticaria: clinical findings in 220 patients. J Am Acad Dermatol 1985;13:636–44.
- Daoud MS, Lust JA, Kyle RA, et al. Monoclonal gammopathies and associated skin disorders. J Am Acad Dermatol 1999;40:507–35.
- Boyce S, Harper J. Paraneoplastic dermatoses. Dermatol Clin 2002;20:523–32.
- Thomas I, Schwartz RA. Cutaneous paraneoplastic syndromes: uncommon presentations. Clin Dermatol 2005;23:593–600.
- Abel EA, Lindae ML, Hoppe RT, et al. Benign and malignant forms of erythroderma: cutaneous immunophenotypic characteristics. J Am Acad Dermatol 1988;19:1089–95.
- Ram-Wolff C, Martin-Garcia N, Bensussan A, Bagot M, Ortonne O. Histopathologic diagnosis of lymphomatous versus inflammatory erythroderma: a morphologic and phenotypic study on 47 skin biopsies. Am J Dermatopathol 2010;32:755–63.
- Nicolis GD, Helwig EB. Exfoliative dermatitis: a clinicopathologic study of 135 cases. Arch Dermatol 1973;108:788–97.
- Grob JJ, Collet-Villete AM, Horchowski N, et al. Ofuji papulo-erythroderma: report of a case with T cell skin lymphoma and discussion of the nature of this disease. J Am Acad Dermatol 1989;20:927–31.
- Cohen PR. Granuloma annulare, relapsing polychondritis, sarcoidosis, and systemic lupus erythematosus: conditions whose dermatologic manifestations may occur as hematologic malignancy-associated mucocutaneous paraneoplastic syndromes. Int J Dermatol 2006;45:70–80.
- Davis MD, Perniciaro C, Dahl PR, et al. Exaggerated arthropod-bite lesions in patients with chronic lymphocytic leukemia: a clinical, histopathologic, and immunopathologic study of eight patients. J Am Acad Dermatol 1998;39:27–35.
- Ross JB, Tompkins MG. Cutis verticis gyrata as a marker of internal malignancy. Arch Dermatol 1989;125:434–5.
- Burt RK, Sharfman WH, Karp B, Wilson WH. Mental neuropathy (numb chin syndrome). A harbinger of tumor progression or relapse. Cancer 1992;70:877–81.
- Hjalgrim H, Frisch M, Storm HH, et al. Non-melanoma skin cancer may be a marker of poor prognosis in patients with non-Hodgkin's lymphoma. Int J Cancer 2000;85:639–42.
- Powell FC, Daniel Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol 1996;34:395–409.
- Vignon-Pennamen MD, Juillard C, Rybojad M, et al. Chronic recurrent lymphocytic Sweet syndrome as a predictive marker of myelodysplasia: a report of 9 cases. Arch Dermatol 2006;142:1170–6.
- Weenig RH, Mehrany K. Dermal and pannicular manifestations of internal malignancy. Dermatol Clin 2008;26:31–43.
- Aldridge RD, Main RA, Daly BM. Multicentric reticulohistiocytosis and cancer. J Am Acad Dermatol 1984;10:296–8.
- Nunnink JC, Krusinski PA, Yates JW. Multicentric reticulohistiocytosis and cancer: a case report and review of the literature. Med Pediatr Oncol 1985;13:273–9.
- Cox NH, West NC, Popple AW. Multicentric reticulohistiocytosis associated with idiopathic myelofibrosis. Br J Dermatol 2001;145:1033–4.
Vascular disorders associated with internal malignancy
Raynaud phenomenon and digital ischaemia, Erythromelalgia, Palmar erythema, Vasculitis, Chilblain-like lesions
- El Tal AK, Tannous Z. Cutaneous vascular disorders associated with internal malignancy. Dermatol Clin 2008;26:45–57.
- Hawley PR, Johnston AW, Rankin JT. Association between digital ischaemia and malignant disease. BMJ 1967;iii:208–12.
- Palmer HM. Digital vascular disease and malignant disease. Br J Dermatol 1974;91:476–7.
- Greer JM, Longley S, Lawrence-Edwards N, et al. Vasculitis associated with malignancy: experience with 13 patients and literature review. Medicine (Baltimore) 1988;67:220–30.
- Boyce S, Harper J. Paraneoplastic dermatoses. Dermatol Clin 2002;20:523–32.
- Jang K-A, Lim Y-S, Choi J-H, et al. Hypereosinophilic syndrome presenting as cutaneous necrotizing eosinophilic vasculitis and Raynaud's phenomenon complicated by digital gangrene. Br J Dermatol 2000;143:641–4.
- Hamada T, Kimura Y, Hayashi S, et al. Hypereosinophilic syndrome with peripheral circulatory insufficiency and cutaneous microthrombi. Arch Dermatol 2007;143:812–13.
- Oppliger R, Gay-Crosier F, Dayer E, Hauser C. Digital necrosis in a patient with hypereosinophilic syndrome in the absence of cutaneous eosinophilic vasculitis. Br J Dermatol 2001;144:1087–90.
- Kurzrock R, Cohen PR. Erythromelalgia and myeloproliferative disorders. Arch Intern Med 1989;149:105–9.
- Noble JB, Boisnic S, Branchet-Gumila MC, Poisson M. Palmar erythema: cutaneous marker of neoplasm. Dermatology 2002;204:209–13.
- Kurzrock R, Cohen PR. Vasculitis and cancer. Clin Dermatol 1993;11:175–87.
- Longley S, Caldwell Pannish RS. Paraneoplastic vasculitis: unique syndrome of cutaneous angiitis and arthritis associated with myeloproliferative disorders. Am J Med 1986;80:1027–30.
- Zappasodi P, Del Forna C, Corso A, Lazzarino M. Mucocutaneous paraneoplastic syndromes in hematologic malignancies. Int J Dermatol 2006;45:14–22.
- Seelen MAJ, de Meijer PHEM, Arnoldus EPJ, et al. A patient with multiple myeloma presenting with severe polyneuropathy caused by necrotizing vasculitis. Am J Med 1997;102:485–6.
- Daoud MS, Lust JA, Kyle RA, et al. Monoclonal gammopathies and associated skin disorders. J Am Acad Dermatol 1999;40:507–35.
- Mita T, Nakanishi Y, Ochiai A, et al. Paraneoplastic vasculitis associated with esophageal carcinoma. Pathol Int 1999;49:643–7.
- Lotti T, Ghersetich I, Comacchi C, Jorizzo JL. Cutaneous small-vessel vasculitis. J Am Acad Dermatol 1998;39:667–87.
- Pankhurst T, Savage COS, Gordon C, Harper L. Malignancy is increased in ANCA-associated vasculitis. Rheumatology (Oxford) 2004;43:1532–5.
- Heijl C, Harper L, Flossmann O, et al.; European Vasculitis Study Group (EUVAS). Incidence of malignancy in patients treated for antineutrophil cytoplasm antibody-associated vasculitis: follow-up data from European Vasculitis Study Group clinical trials. Ann Rheum Dis 2011;70(8):1415–21.
- Yazawa H, Saga K, Omori F, et al. The chilblain-like eruption as a diagnostic clue to the blast crisis of chronic myelocytic leukaemia. J Am Acad Dermatol 2004;50(2 Suppl.):S42–4.
- Affleck AG, Ravenscroft JC, Leach IH. Chilblain-like leukemia cutis. Pediatr Dermatol 2007;24:38–41.
- Tan BB, Lear JT, English JSC. Metastasis from carcinoma of breast masquerading as chilblains. J R Soc Med 1997;90:162.
Flushing
- El Tal AK, Tannous Z. Cutaneous vascular disorders associated with internal malignancy. Dermatol Clin 2008;26:45–57.
- Hemann WR. Flushing, pheochromocytoma, and the dermatologist. J Am Acad Dermatol 2006;55:1075–7.
- Modlin IM, Sandor A. An analysis of 8305 cases of carcinoid tumors. Cancer 1997;79:13–29.
- Tharp MD, Chan IJ. Mastocytosis. Adv Dermatol 2003;19:207–36.
- Pardanani A, Baek J-Y, Li C-Y, et al. Systemic mast cell disease without associated hematologic disorder: a combined retrospective and prospective study. Mayo Clin Proc 2002;77:1169–75.
Cancer-associated thrombosis
- Bick RL. Cancer-associated thrombosis. N Engl J Med 2003;349:109–11.
- Walsh-McMonagle D, Green D. Low molecular-weight heparin in the management of Trousseau's syndrome. Cancer 1997;80:649–55.
- El Tal AK, Tannous Z. Cutaneous vascular disorders associated with internal malignancy. Dermatol Clin 2008;26:45–57.
- Samlaska CP, James WD. Superficial thrombophlebitis. II. Secondary hypercoagulable states. J Am Acad Dermatol 1990;23:1–18.
- Catania S, Zurrida S, Veronesi P, et al. Mondor's disease and breast cancer. Cancer 1992;69:2267–70.
Venous and lymphatic obstruction
- Yosipovitch G. Chronic pruritus: a paraneoplastic sign. Dermatol Ther 2010;23:590–6.
- Paul R, Paul R, Jansen CT. Itch and malignancy prognosis in generalized pruritus: a 6-year follow-up of 125 patients. J Am Acad Dermatol 1987;16:1179–82.
- Feiner AS, Mahmood T, Wallner SF. Prognostic importance of pruritus in Hodgkin's disease. JAMA 1978;240:2738–40.
- Pipkin CA, Lio PA. Cutaneous manifestations of internal malignancies: an overview. Dermatol Clin 2008;26:1–15.
- Thomas I, Schwartz RA. Cutaneous paraneoplastic syndromes: uncommon presentations. Clin Dermatol 2005;23:593–600.
- Andreev VC, Petkov I. Skin manifestations associated with tumours of the brain. Br J Dermatol 1975;92:675–8.
- Johnson RE, Kanigsberg ND, Jimenez CL. Localized pruritus: a presenting symptom of a spinal cord tumor in a child with features of neurofibromatosis. J Am Acad Dermatol 2000;43:958–61.
- Soltani-Arabshahi R, Vanderhooft S, Hansen CD. Intractable localized pruritus as the sole manifestation of intramedullary tumor in a child: case report and review of the literature. JAMA Dermatol 2013;149:446–9.