CHAPTER 148
The Skin and Disorders of the Haematopoietic and Immune Systems
Robert Gniadecki
Department of Dermatology, Bispebjerg Hospital and University of Copenhagen, Copenhagen, Denmark; and Division of Dermatology, University of Alberta, Alberta, Canada
Introduction
This chapter reviews skin diseases and symptoms which develop secondarily to underlying malignant or benign disease of the haematopoietic system (Table 148.1). Other related diseases are covered in separate chapters: primary cutaneous lymphomas (Chapter 140), mastocytosis (Chapter 46), disorders of coagulation (Chapter 101), antiphospholipid syndrome (Chapter 52), skin manifestations of graft-versus-host disease (Chapter 38) and skin reactions to the drugs used for the treatment of haematological diseases (Chapters 118, 119 and 120).
Table 148.1 Major skin manifestations of diseases of the haematopoietic system.
| Condition | Haematological disease |
| Malignant infiltrations of the skin | Associations |
| Leukaemia cutis |
Acute myeloid leukaemia Myeloproliferative neoplasms Myelodysplastic syndrome Chronic lymphocytic leukaemia Adult T-cell leukaemia/lymphoma T-cell prolymphocytic leukaemia |
| Lymphomatous infiltrates | Mantle cell lymphoma Follicular lymphoma Intravascular lymphoma Lymphomatoid granulomatosis Hodgkin lymphoma |
| Plasma cell infiltrates | Multiple myeloma Waldenström macroglobulinaemia |
| Malignant extramedullary haematopoiesis | Myelodysplastic syndrome |
| Deposition diseases | Associations |
AL amyloidosis Type I cryoglobulinaemia Macroglobulinaemia cutis |
Monoclonal gammopathy of undetermined significance (MGUS) Multiple myeloma Waldenström macroglobulinaemia Plasmacytoma |
| Paraneoplastic conditions | Associations |
Sweet syndrome Pyoderma gangrenosum Neutrophilic eccrine hidradenitis Paraneoplastic pemphigus Exaggerated insect bite-like reactions Scleredema Necrobiotic xanthogranuloma Paraneoplastic pruritus Autoimmune paraneoplastic conditions |
Myeloid neoplasms Lymphoid leukaemias Lymphoma Plasma cell neoplasms MGUS |
| Syndromes with skin manifestations associated with monoclonal gammopathy or haematological malignancies | Associations |
Schnitzler syndrome POEMS syndrome AESOP syndrome TEMPI syndrome |
Monoclonal gammopathy of undetermined significance (MGUS) Multiple myeloma Waldenström macroglobulinaemia Plasmacytoma |
| Leukaemia-predisposing syndrome | Association |
| Neurofibromatosis, juvenile xanthogranuloma and juvenile myeloid leukaemia | Juvenile myelomonocytic leukaemia |
| Idiopathic lymphadenopathies with skin involvement | Associations Kikuchi-Fujimoto disease Kimura disease Rosai-Dorfmann disease IgG4-related disease |
| Skin involvement in primary and acquired immunodeficiencies | Associations |
Infections: bacterial, fungal and viral Eczema Vasculitis Pigmentary changes Autoimmune conditions |
Inherited immunodeficiency syndromes Acquired immunodeficiency |
| Cutaneous and mucosal changes associated with anaemia | Associations |
Iron-deficiency anaemia Megaloblastic anaemia |
|
| Skin ulcers associated with haemoglobinopathies | Associations |
Thalassaemia Sickle-cell anaemia |
|
| Transfusion reactions | Association |
Acute allergic reactions Transfusion-associated graft-versus-host disease Post-transfusion purpura |
Transfusion of blood products |
Skin manifestations of haematological malignancies
Haematological malignancies are neoplasms which originate from cells in the haematopoietic tissue of the bone marrow, lymph nodes or thymus. Lymphoproliferative malignancies originate from the lymphocytes or their precursors whereas the myeloid malignancies are a heterogeneous group of diseases showing differentiation towards one or more non-lymphoid cell types.
There are three broad categories of cutaneous manifestations of haematological neoplasms. In the first, the skin lesions are caused directly by the infiltration of the skin with malignant cells or by their products (e.g. paraproteins). The second category comprises cutaneous manifestations which are indirectly caused by the underlying malignancy, such as is the case of paraneoplastic conditions or dermatological syndromes. Skin manifestations caused by treatment of haematological cancers (e.g. graft-versus-host disease (GVHD) after allogeneic bone marrow transplantation, infections due to immunosuppression or adverse effects of anticancer therapies) belong to the third category.
SKIN DISORDERS CAUSED BY INFILTRATION OF THE SKIN WITH NEOPLASTIC CELLS
This class includes four major groups of diseases: leukaemia cutis, lymphomatous skin infiltrates, malignant plasma cell infiltrates and malignant extramedullary haematopoiesis.
Leukaemia cutis
Definition
Leukaemia cutis is an infiltration of the skin by myeloid or lymphoid neoplastic leukocytes resulting in clinically identifiable cutaneous lesions [1]. When made up of malignant granulocytic precursor cells, leukaemia cutis lesions are also called myeloid sarcoma, granulocytic sarcoma or chloroma.
Epidemiology
Skin lesions mostly develop after or concurrently with the diagnosis of myeloid disorder (60% and 25%, respectively) and only in approximately 5% of cases do they develop before any detectable bone marrow or blood involvement as an aleukaemic leukaemia cutis [2, 3]. In chronic lymphocytic leukaemia (CLL), leukaemia cutis may be the first sign of the disease in 16% of cases [4]. In patients with chronic disease, such as chronic myelomonocytic leukaemia (CMML), skin involvement may be a sign of disease progression and blastic transformation [5]. The likelihood of developing leukaemia cutis depends on the type of underlying haematological neoplasm (Table 148.2). Leukaemia cutis is most common in patients with myeloid neoplasms (10–15%) [5], especially in acute myeloid leukaemia (AML) (2.5–9.1% patients) [6, 7] and in leukaemias with monocytic differentiation, such as CMML [2, 8]. Leukaemia cutis associated with AML accounts for 60–70% of all cases [2]. The risk of leukaemia cutis is higher in AML with neoplastic cells displaying the chromosomal translocation t(8,21) [9]. Within the lymphocytic neoplasms, the risk of leukaemia cutis is relatively high for mature T-cell neoplasms such as adult T-cell leukaemia/lymphoma and T-cell prolymphocytic leukaemia (20–70%) [10, 11, 12] and CLL (4–20%) [3, 12]. In other lymphocytic leukaemias, such as acute B- and T-cell lymphoblastic leukaemias (ALL), the incidence of leukaemia cutis is much lower (below 3%) [3, 12].
Table 148.2 Leukaemia cutis and lymphomatous infiltrates in the setting of a haematological neoplasm.
| Condition | Probability of clinically apparent skin involvementa |
| Myeloid neoplasms | ++ (10–15%) Higher risk in congenital leukaemia andpaediatric leukaemia |
| AML (acute myeloid leukaemia) | ++ (2.5–9.1%) Higher incidence in AML with t(8,21) translocations |
| Other myeloid neoplasmsb | + |
| Lymphoid leukaemias | |
| CLL (chronic lymphocytic leukaemia) | +++ (4–20%) |
| Adult T-cell leukaemia/lymphoma | +++ (20–70%) |
| T-cell prolymphocytic leukaemia | +++ |
| Other lymphocytic leukaemias | + |
| Lymphomas | |
| Hodgkin lymphoma | + (0.5–3.4%) |
| Lymphoblastic lymphoma | + |
| Mantle cell lymphoma | +++ |
| Follicular lymphoma | ++ |
| Diffuse large B-cell lymphoma – intravascular type | +++ |
| Lymphomatoid granulomatosis | +++ (40–50% of patients) |
aStrength of association: +, occasional <4%; ++, frequent 5–14%; +++, very frequent >15%.
bMyeloid neoplasms comprise over 30 well-defined clinicopathological entities in five major groups: myeloproliferative neoplasms (chronic myeloid leukaemia, polycythaemia vera, primary myelofibrosis, essential thrombocythaemia, mastocytosis), myeloid neoplasms with eosinophilia, myelodysplastic neoplasms (e.g. juvenile myelomonocytic leukaemia) and myelodysplastic syndrome (MDS) [21].
The incidence of leukaemia cutis is higher in children with leukaemia and in congenital leukaemias it may be as high as 30% [13]. Isolated leukaemia cutis seems to be more frequent in patients who relapse after allogeneic bone marrow transplant, where frequencies of 8–20% have been reported [14].
Pathophysiology
The molecular basis underlying the migration of leukaemic cells to the skin in leukaemia cutis is not fully understood. Homing of malignant cells to the skin may depend on specific chemokine receptors, such as chemokine receptor 4 (CCR4) interacting with specific intercellular adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) [15].
Clinical features
Leukaemia cutis typically presents as single or multiple monomorphic violaceous, dark-red or haemorrhagic skin nodules, especially on the legs, arms and face (Figures 148.1 and 148.2) [12]. The lesions have a predilection for sites of trauma (catheter or needle insertion) or previous inflammation (e.g. herpes zoster) (Figure 148.3) but may present anywhere on the skin. Other presentations range from small asymptomatic papules or a maculopapular rash to diffuse skin infiltration, ulceration or even erythroderma. It may mimic a variety of benign conditions such as venous leg ulcer, dyshidrotic eczema, cutaneous vasculitis, Jessner lymphocytic infiltration, granuloma annulare or rosacea [2, 12, 16, 17]. The appearance of the skin lesions is not specific for the particular subtype of leukaemia.
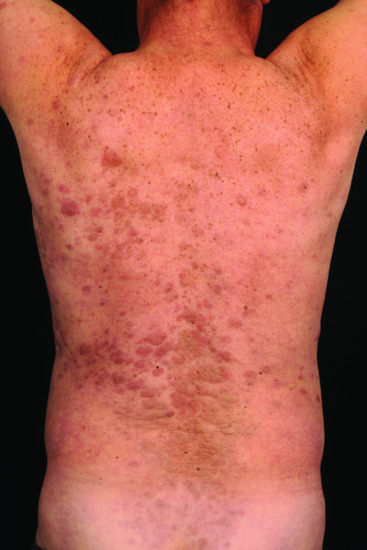
Figure 148.1 Leukaemia cutis in acute myeloid leukaemia (AML): extensive infiltrative, papulonodular lesions over the back.
(Courtesy of Professor Lorenzo Cerroni, University of Graz, Austria.)
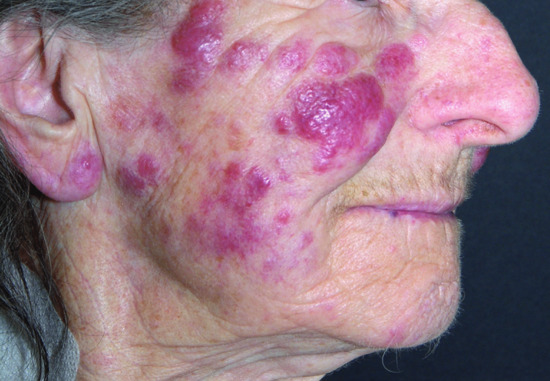
Figure 148.2 Leukaemia cutis in chronic lymphocytic leukaemia (CLL): infiltrated nodules and plaques on the cheek.
(Courtesy of Professor Lorenzo Cerroni, University of Graz, Austria.)
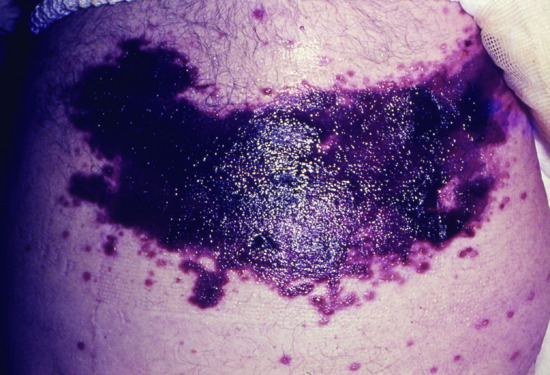
Figure 148.3 Haemorrhagic leukaemic infiltration at the site of thoracic herpes zoster in an elderly man with chronic lymphatic leukaemia (CLL).
Pathology
In leukaemia cutis associated with myeloid neoplasms, skin infiltration is typically both superficial and deep, and consists of proliferating immature myeloid cells (myeloblasts, monoblasts, promonocytes or promyelocytes) [18]. The infiltrate may also be quite subtle and resemble non-specific dermatitis [17], granuloma annulare, histiocytoid Sweet syndrome [2], vasculitis [19] or multicentric reticulohistiocytosis [20]. Immunohistological profiling is therefore an essential part of the diagnostic work-up. The most sensitive markers are CD43, CD68 and lysozyme. Other useful markers are myeloperoxidase, CD33, CD34, CD117 and CD68 [2, 17, 18] but CD117 and myeloperoxidase immunostaining are usually negative in monoblastic skin infiltrates. It is also noteworthy that the immune profile of the circulating leukaemic cells may vary from those in the skin infiltrates, which probably reflects different clonal evolution of the malignant cells in different anatomical compartments [21]. Myeloid leukaemia cutis should be differentiated from blastic plasmacytoid dendritic cell neoplasm which may present in the skin and which is characterized by plasmacytoid CD4+ CD56+ cells thought to derive from dendritic cell precursors (see Chapter 140). Other differential diagnoses comprise some poorly differentiated solid tumours such as metastatic malignant melanoma, Ewing sarcoma or medulloblastoma and histiocytic sarcomas, which may be positive for CD68 and lysozyme but are negative for CD33 and CD13.
The skin infiltrate of CLL comprises small monomorphous lymphocytes with an aberrant marker pattern (CD20+5+43+) in a lichenoid, perivascular or nodular pattern [4]. Analysis of monoclonal immunoglobulin heavy chain (IgH) gene arrangement may be a useful aid to the diagnosis.
Prognosis and management
The prognostic significance of leukaemia cutis has not been firmly established. In some studies, the presence of leukaemia cutis did not appear to affect the 5-year survival of patients with AML [5, 16] but in others it was associated with a significantly poorer survival both in AML (75% 1-year mortality] [18] and in CMML [20]. The presence of leukaemia cutis does not affect the prognosis of CLL except for the large cell transformation of CLL (Richter syndrome) where prognosis is grave in the presence of large blastic cells in the infiltrate [4, 22].
Leukaemia cutis is a cutaneous manifestation of an underlying systemic disease. Hence, treatment should be guided by the underlying leukaemia with supportive care for skin lesions. However, skin lesions may be less responsive to chemotherapy than the disease in the bone marrow or the blood [16]. Radiotherapy with photons or electron beam can be beneficial to treat focal leukaemic skin infiltrates.
Lymphomatous skin infiltrates
In Hodgkin disease, specific skin involvement is uncommon with an estimated prevalence of 0.5–3.4% [23]. Lymphomatous skin involvement in non-Hodgkin lymphomas is more frequent, especially for mantle cell lymphoma, and lymphomatoid granulomatosis (lymphoid dyscrasia caused by Epstein–Barr virus (EBV)) [24, 25, 26].
In Hodgkin lymphoma, the skin infiltrate does not always faithfully replicate the nodal disease. Reed–Sternberg cells in the skin are CD30+ CD45–. In a minority of cases, the neoplastic cells in the skin may be negative for CD15 even in patients with nodal CD15 positivity [27]. The major differential diagnoses are primary cutaneous CD30+ lymphomas and CD30+ transformation of mycosis fungoides.
In Hodgkin lymphoma, the majority of patients die within a few months following the development of skin lesions [23]. Therefore, skin involvement is considered to be an indication of stage IV disease.
Malignant infiltration of the skin in plasma cell disorders
The most common plasma cell malignancies are multiple myeloma and Waldenström macroglobulinaemia. Both conditions are characterized by the presence of >10% malignant plasmacytic cells in the bone marrow and an associated monoclonal gammopathy. It is thought that these malignancies are preceded by a pre-malignant stage in which bone marrow infiltration with abnormal plasma cells is below the threshold of detection (<10%) and the identifiable feature is a monoclonal gammopathy, known as MGUS (monoclonal gammopathy of undetermined significance) MGUS is detected in approximately 3% of the general population aged 50 years or older and is associated with a 1% per year risk of progression to multiple myeloma or Waldenström macroglobulinaemia [28, 29]. According to the type of gammopathy, there are three major classes of MGUS: the immunoglobulin M (IgM) type (progressing to Waldenström macroglobulinaemia), the non-IgM type (IgG or IgA) and the light-chain MGUS which may progress to multiple myeloma [28].
Apart from the bone marrow, neoplastic plasma cells may also develop in extramedullary locations as solitary plasmacytomas, most commonly in the bone or upper respiratory tract. In exceptional situations, a plasmacytoma may be confined to the skin (Figure 148.4). Plasmacytomas may result in MGUS and may progress to multiple myeloma if untreated. Cutaneous plasma cell infiltration in preexisting multiple myeloma and Waldenström macroglobulinaemia is a very rare phenomenon with approximately 100 cases described in the literature [30, 31]. The risk of cutaneous involvement does not depend on the immunoglobulin isotype produced by the neoplastic plasma cells [31]. The lesions are described as firm, violaceous papules and nodules, sometimes with secondary ulceration. The most frequent sites affected are the trunk, followed by the scalp and face.
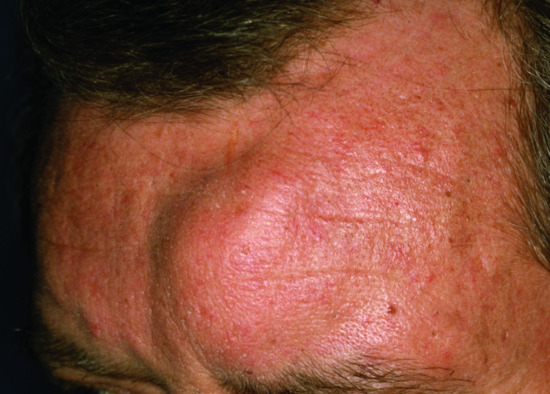
Figure 148.4 Plasmacytoma cutis.
(Courtesy of Professor Lorenzo Cerroni, University of Graz, Austria.)
Histopathologically, the infiltrates are diffuse or nodular aggregations of lymphoplasmacytic cells showing a strong immunoreactivity for CD79a and CD138 and an aberrant expression of CD43 and CD56. In multiple myeloma, the cells are usually negative for pan-B cell markers such as CD19 and CD20. This is in contrast to Waldenström macroglobulinaemia where the neoplastic cells are often CD19+ CD20+. Differential diagnosis includes primary plasmacytoma cutis and EBV-related plasmacytoid hyperplasia which develops in immunosuppressed transplant patients [32]. In only exceptional cases do these conditions present in the skin [33]. In most cases, skin lesions appear relatively late in the course of multiple myeloma or Waldenström macroglobulinaemia and herald an unfavourable outcome.
Extramedullary haematopoiesis
Extramedullary haematopoiesis is the formation of cellular blood components outside the bone marrow. It can be a pathological feature of chronic myeloproliferative syndromes such as primary myelofibrosis [34]; foci commonly occur in the spleen and may in rare instances occur in the skin [35]. The process is probably caused by seeding of abnormal haematopoietic stem cells outside the bone marrow and subsequent aberrant proliferation. Skin manifestations include erythematous or violaceous (angioma-like) papules, nodules, tumours and ulcers, often on the torso, that may appear soon after diagnosis with myelofibrosis (and sometimes following splenectomy). Skin lesions may resemble leukaemia cutis [35] from which they should be differentiated. Histologically, skin lesions are characterized by a polymorphous dermal infiltrate of myeloid and erythroid cell precursors, often with the presence of dysplastic megakaryocytes and occasionally few myeloblasts [36, 37].
Prolonged dermal extramedullary haematopoiesis can also occur in neonates without a haematological neoplasm but as a reactive phenomenon to an infection commencing in utero, such as one of the TORCH group of infections (toxoplasmosis, other (e.g. syphilis, varicella-zoster, parvovirus B19) rubella, cytomegalovirus and herpes infections). The generalized haemorrhagic, purpuric eruption that ensues has led to the term ‘blueberry muffin baby’. In children and adults, reactive extramedullary haematopoiesis may also accompany thalassaemias, sickle cell anaemia or thrombocytopenic purpura but skin involvement in adults is exceptionally rare [36].
IMMUNOGLOBULIN DEPOSITION DISORDERS OF THE SKIN
Circulating abnormal immunoglobulins produced by neoplastic plasma cells in MGUS, multiple myeloma or Waldenström macroglobulinaemia may accumulate in the skin. There are three major manifestations of this phenomenon: macroglobulinaemia cutis, amyloid light-chain (AL) amyloidosis and type I cryoglobulinaemia.
Macroglobulinaemia cutis
Deposition of IgM paraproteins in the skin may produce two distinct patterns of disease: IgM storage papules and immunobullous macroglobulinaemia cutis. Both presentations are very rare. IgM storage papules are translucent flesh-coloured papules, usually localized on the extensor surfaces of the extremities. The deposits are periodic acid–Schiff (PAS) positive and stain positively for IgM on direct immunofluorescence but are negative for amyloid [38, 39, 40]. In rare instances, the IgM paraproteins show affinity for the plasma membrane and result in blistering (immunobullous macroglobulinaemia cutis) [41, 42, 43]. Pathological paraprotein deposition at the dermal–epidermal junction is readily detectable by direct immunofluorescence. Immunobullous macroglobulinaemia cutis has been treated successfully by rituximab or multiagent chemotherapy [41].
Primary systemic or amyloid light–chain amyloidosis
AL amyloidosis is a devastating life-threatening disease which develops as a result of abnormal processing and degradation of the variable portion of monoclonal light chains (see Chapter 58). Over 90% of patients have a monoclonal gammopathy, most commonly MGUS or multiple myeloma; and occasionally Waldenström macroglobulinaemia or solitary plasmacytoma. Median survival without treatment is 12–18 months. AL amyloidosis may involve multiple organs and result in nephrotic syndrome, renal failure, congestive heart failure and peripheral neuropathy. Skin symptoms are common (40% of patients) and are very characteristic with purpura and ecchymoses, typically in the perioral and periorbital areas, waxy papules and plaques with sclerodermatous or bullous change. Macroglossia is another typical manifestation [38, 39], along with patchy alopecia and generalized oedema. Standard treatment approaches by a multidisciplinary team include high-dose melphalan followed by autologous haematopoietic stem cell transplantation or oral melphalan with dexamethasone (MDex) [44].
Type I cryoglobulinaemia
Type I cryoglobulinaemia may develop in the context of any plasma cell disorder with paraproteinaemia. It results from the microvascular deposition of abnormal monoclonal immunoglobulins (usually IgM and less often IgG and IgA) that can precipitate out at low temperatures. This in turn leads to microvascular occlusion in the skin and other tissues. The incidence ranges from 5% for patients with multiple myeloma to 37% for Waldenström macroglobulinaemia [39]. Typical clinical manifestations comprise purpura, cutaneous ulceration, infarction (often on the lower legs or the face), cold urticaria, Raynaud phenomenon, livedo reticularis and inflammatory macules and papules. Horn-like filiform spicules in the follicular ostia of the face, particularly the nose (termed follicular spicules of the nose), have been reported to be a sign of myeloma-associated cryoglobulinaemia [45]. Systemic manifestations include neuropathy, nephropathy and, more rarely, gastrointestinal disturbances [46]. Some patients develop the hyperviscosity syndrome with neurological (headache, confusion), ocular (blurred vision, visual loss), rhino-otological (epistaxis, hearing loss) and renal involvement [46]. It may result in multiorgan failure and thus requires urgent therapy.
The diagnosis of type I cryoglobulinaemia is typically made by direct measurement of cryoglobulins in serum and is supported by typical clinical features and skin biopsy showing intravascular PAS-positive hyaline material [38, 39]. Type I cryoglobulinaemia should be differentiated from other cryoglobulinaemias (see Chapter 125).
PARANEOPLASTIC CONDITIONS STRONGLY ASSOCIATED WITH HAEMATOLOGICAL MALIGNANCIES
Paraneoplastic syndromes (described in detail in Chapter 147) may be associated with haematological malignancies. This section addresses those entities that are most strongly associated with haematological disease. Diagnosis of these paraneoplastic conditions warrants an active search for a possible underlying haematological malignancy. Paraneoplastic conditions may precede the haematological neoplasm by months or years, and the history is therefore an important first step in linking the skin symptoms to an underlying malignancy. Drug intake at the time of disease onset should be determined, as some drug reactions may mimic the paraneoplastic conditions.
Diagnostic screening may include clinical examination of the peripheral lymph nodes, analysis of the peripheral full blood count, blood film and lactose dehydrogenase, detection of monoclonal paraproteins, a bone marrow biopsy (if leukaemia is suspected), and combined 18F fluorodeoxyglucose positron emission and X-ray computed tomography (FDG-PET/CT) scanning. The latter has been advocated to provide a good screening tool, especially for lymphoma and leukaemia [47, 48]. However, not all haematological neoplasms (in particular low-grade lymphomas) will be detected by this technique [49]. Many of these paraneoplastic skin conditions remain refractory to treatment until management of the underlying haematological malignancy is addressed [50].
Sweet syndrome
Sweet syndrome is a neutrophilic dermatosis, also known as acute febrile neutrophilic dermatosis (described in detail in Chapter 49); it may be idiopathic, drug induced or paraneoplastic. The paraneoplastic form comprises approximately 20% of cases [51] and is predominantly associated with haematological neoplasms, particularly myeloid leukaemias and myelodysplasias. Sweet syndrome may precede the development of a haematological malignancy by over a decade [52]. The pathogenesis of paraneoplastic Sweet syndrome has not been elucidated, but a hypersensitivity reaction to tumour antigens or an autoinflammatory, interleukin 1 (IL-1)-mediated process have been suggested as putative mechanisms [53, 54, 55, 56]. Indeed, heterozygous mutations in the gene MEFV known to cause familial Mediterranean fever have been reported in two patients with concurrent Sweet and myelodysplastic syndrome [57].
The clinical features of paraneoplastic Sweet syndrome are very variable. The classical presentation is with acute fever, arthralgia and an eruption of tender, erythematous or violaceous papules and nodules surmounted by pseudovesicles. However, paraneoplastic Sweet syndrome has a tendency to involve extracutaneous sites more often than the idiopathic type. Mucosal lesions, neurological (parkinsonism) and ophthalmological complications (optic neuritis, chorioretinitis) are more common in patients with myeloproliferative diseases than in idiopathic Sweet syndrome [52]. Other clinical variants include acute necrotizing, isomorphic and subcutaneous varieties and the neutrophilic dermatosis of dorsal hands. In the acute necrotizing variant, there is a fulminant onset of oedematous erythematous plaques accompanied by necrosis of the skin and soft tissues [58]. Isomorphic Sweet syndrome develops at the site of trauma such as radiation fields or surgery scars. Subcutaneous Sweet syndrome is a variant overlapping with pyoderma gangrenosum and seems to be particularly associated with myeloproliferative diseases: it presents with tender nodules with a predilection for the buttocks and lower extremities without tissue necrosis and has a tendency to resolve spontaneously [59]. Neutrophilic dermatosis of the dorsal hands is a localized variant of Sweet syndrome which has been described to occur in the context of a variety of malignancies including the haematological neoplasms (Figure 148.5) [60, 61].
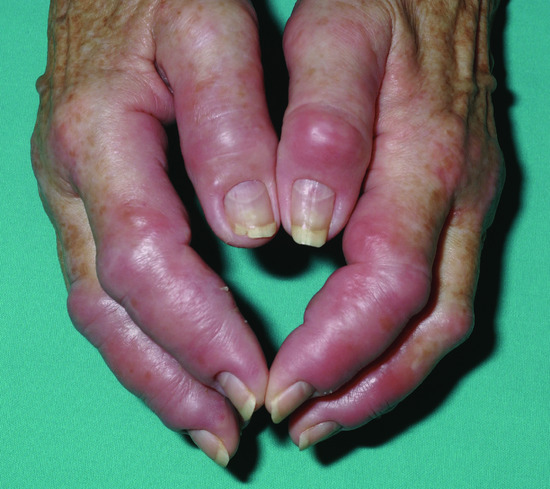
Figure 148.5 Neutrophilic dermatosis of the dorsal hands presenting in an 80-year-old patient subsequently found to have a gastric mucosa-associated lymphoid tissue (MALT) lymphoma.
Paraneoplastic Sweet syndrome should be differentiated from other neutrophilic paraneoplastic conditions (neutrophilic eccrine hidradenitis, pyoderma gangrenosum), vasculitis, erythema elevatum diutinum, infectious neutrophilic panniculitis, panniculitic id reaction, early erythema nodosum and leukaemia cutis. The differential diagnosis may be challenging since paraneoplastic Sweet syndrome has a tendency to coexist with other neutrophilic paraneoplastic conditions in the same patient [52]. Moreover, drug-induced Sweet syndrome should be borne in mind since a number of drugs used in the treatment of haematological malignancies have the potential to precipitate Sweet syndrome (Table 148.3).
Table 148.3 Drugs used in the management of patients with haematological malignancies with known association with Sweet syndrome.
| Cytotoxic anticancer agents | Proteasome inhibitors (bortezomib) Mitoxantrone Cytarabine |
| Other anticancer drugs and biological response modifiers | Tyrosine kinase inhibitors (imatinib mesylate) LenalinomideGranulocyte colony-stimulating factor All-trans-retinoic acidInterferon-α |
| Antibiotics | Clindamycin Tetracyclines Quinolones Trimethoprim with sulfamethoxazole |
| Antivirals | Abacavir Aciclovir |
Pyoderma gangrenosum
Pyoderma gangrenosum (see Chapter 49) is another neutrophilic dermatosis and is classified into four clinical types: the classical ulcerative type, bullous, pustular and the vegetative type. It has been estimated that between 20% and 60% of cases of pyoderma gangrenosum are associated with a haematological malignancy [62, 63]. The non-classical presentations are overrepresented in paraneoplastic pyoderma [63]. The bullous variant is associated with haematological neoplasms in more than 70% of cases (Figure 148.6) [64, 65].
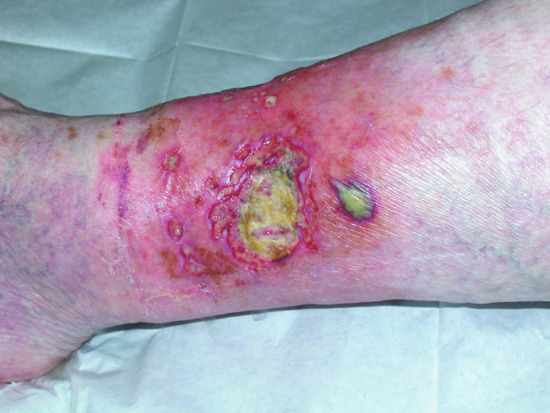
Figure 148.6 Bullous pyoderma gangrenosum in patient with IgA paraproteinaemia.
(Courtesy of Dr Ian Coulson, Burnley General Hospital, UK.)
MGUS, myelodysplastic syndromes and myeloid leukaemias are the most common underlying diseases. Pyoderma gangrenosum and Sweet syndrome may coexist in the same patient with a myeloproliferative disorder. It has been proposed that these entities and neutrophilic eccrine hidradenitis constitute a pathogenic continuum (see earlier) [66].
The development of pyoderma gangrenosum in a patient with a myeloproliferative disease may have a negative prognostic significance. For example, the 1-year mortality for patients with AML and pyoderma gangrenosum is 75% after the appearance of the skin lesions [67].
Neutrophilic eccrine hidradenitis
This condition is the rarest member of a family of paraneoplastic neutrophilic dermatoses that also includes Sweet syndrome and pyoderma gangrenosum. It is characterized by neutrophilic infiltrates around eccrine glands and coils [68]. The condition has a very strong association with an underlying cancer since 90% of patients with neutrophilic eccrine hidradenitis have an underlying haematological malignancy, especially AML. Neutrophilic eccrine hidradenitis has also been reported in association with CLL, Hodgkin lymphoma and a variety of nodal non-Hodgkin lymphomas. It has been debated whether neutrophilic eccrine hidradenitis is a drug-induced condition rather than a paraneoplastic one. However, in 20% of cases, neutrophilic eccrine hidradenitis develops before the onset of therapy and may even precede the presentation of the haematological malignancy, which would indicate that certainly in some cases the condition is paraneoplastic.
In approximately 80% of patients, the occurrence of neutrophilic eccrine hidradenitis can be linked to drug exposure. The offending drugs are similar to those implicated in Sweet syndrome (see Table 148.3). The most common agent is cytarabine followed by decitabine [69], vincristine, imatinib mesylate, topotecan [70], granulocyte–colony-stimulating factor (G-CSF) and antibiotics and antiviral agents [68]. The median time from drug exposure to the onset of symptoms is 9 days [68].
Neutrophilic eccrine hidradenitis can be caused by other factors, especially infections (Staphylococcus aureus, Serratia marcescens, Enterobacter spp.) and has been reported in association with HIV infection (see Chapter 31).
The classical clinical features are infiltrated erythematous papules and plaques with a predilection for the face but there is substantial heterogeneity in its clinical manifestations. The lesions may also involve the upper trunk, arms, thighs and palmoplantar skin (Figure 148.7). They may be annular or linear and may resemble erythema multiforme or Sweet syndrome, from which they should be differentiated. An infectious aetiology should be excluded during differential diagnosis.
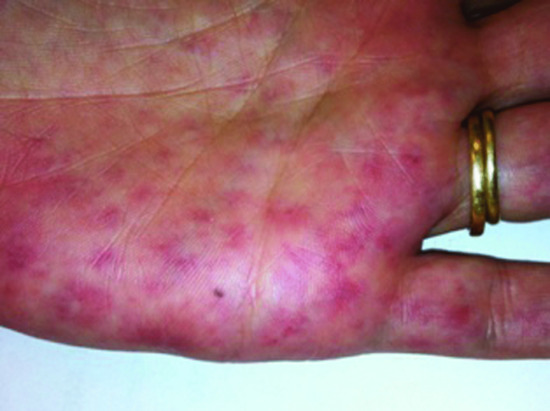
Figure 148.7 Neutrophilic eccrine hidradenitis affecting the palm.
(Courtesy of Professor Lorenzo Cerroni, University of Graz, Austria.)
The optimal treatment is unknown. In some cases, the disease resolves spontaneously, especially after the discontinuation of the offending drug. Therapeutic effects of non-steroidal anti-inflammatory drugs (NSAIDs), systemic steroids or dapsone have been reported in single cases.
Paraneoplastic pemphigus
This disease entity is extensively reviewed in Chapter 50. It is strongly associated with haematological malignancies, particularly of lymphocytic origin. The strongest association is with non-Hodgkin lymphoma (approximately 40%), followed by CLL (20%), Castleman disease and thymoma (20%) [71]. The pathogenesis is unknown, but postulated mechanisms include epitope spreading, antigen mimicry or IL-6 dysregulation by tumour cells [72]. Untreated paraneoplastic pemphigus has a mortality approaching 90% at 2 years following diagnosis. Treatment of the underlying disease may lead to remission; rituximab, the anti-CD20 monoclonal antibody therapy, has produced both partial and complete remissions in a number of cases.
Insect bite–like reaction or exaggerated insect bite reaction
Insect bite-like reaction has been described as a paraneoplastic phenomenon associated with lymphoproliferative neoplasms. This phenomenon is also reported as exaggerated insect bite reaction but the condition does not seem to be precipitated by an actual arthropod bite, but rather occurs spontaneously mimicking an insect bite reaction both clinically and histologically. Pathogenesis is not well understood but the skin reaction may be related to immune dysregulation associated with conditions such as CLL; external factors such as cancer-related immunosuppression and pyogenic infection exacerbate the reaction. In a minority of patients, this reaction precedes the malignancy, but in most cases insect bite-like reactions occur within 2 years after the diagnosis of the underlying malignant disease. Insect bite-like reactions may also occur in immunosuppressed patients (e.g. in the course of HIV infection, congenital agammaglobulinaemia, natural killer lymphocytosis or EBV infection).
The patient develops red infiltrated plaques and, as the condition progresses, vesicles, bullae and skin ulceration (Figure 148.8). CLL is the most commonly associated disease, followed by mantle cell lymphoma, ALL, acute monocytic leukaemia, Burkitt lymphoma and myelodysplastic syndrome [22, 73]. Therapeutic response is often unsatisfactory; oral corticosteroids may produce partial response whereas topical corticosteroids and phototherapy provide only marginal benefit [69]. The prognostic significance is unknown; in CLL patients, the course of the skin eruption is not related to CLL activity or the course of haematological disease [74, 75].
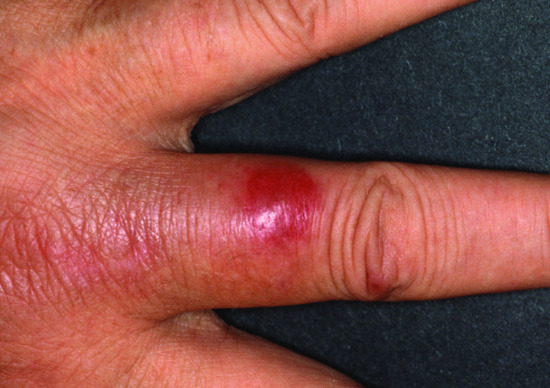
Figure 148.8 Exaggerated insect bite-like reaction in chronic lymphocytic leukaemia (CLL).
(Courtesy of Professor Lorenzo Cerroni, University of Graz, Austria.)
Scleromyxoedema
Scleromyxoedema is a rare generalized cutaneous mucinosis resulting from increased mucin (heavily glycosylated proteins made by fibroblasts) synthesis and deposition following fibroblast proliferation (see Chapter 59). In the absence of thyroid disease, scleromyxoedema develops almost invariably in patients with monoclonal gammopathies, either associated with MGUS, multiple myeloma or plasmacytomas [76, 77]. Over 80% of patients have an IgG- λ paraproteinaemia [39, 78]. The paraneoplastic mechanism of fibroblast activation and mucin synthesis by paraproteins remains unknown. Overexpression of mucin genes, such as MUC1, is associated with many cancers [79].
Clinically, there is a widespread papular and sclerodermatous eruption with a particular predilection for the face, ears and dorsal hands. The papules are firm, slightly translucent and waxy in appearance (Figure 148.9). There are no epidermal changes and inflammation is minimal or absent. Histologically, collagen fibres in the dermis are separated widely by mucin deposits; increased fibroblast numbers and fibrosis are observed.
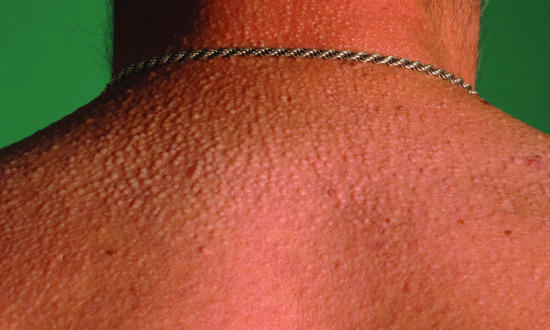
Figure 148.9 Scleromyxoedema in patient with MGUS (monoclonal gammopathy of undetermined significance).
Extracutaneous manifestations are multiple and significantly contribute to the morbidity of scleromyxoedema. The most common are dysphagia, hoarseness and aspiration due to laryngeal involvement, proximal or generalized myopathy, polyarthritis, central nervous system involvement (encephalopathy, seizures, vertigo, psychosis), arrhythmia or myocardial infarction due to myocardial involvement and dyspnoea due to lung involvement.
The course is progressive and mortality is substantial varying between 15% and 50% [78, 80]. The main causes of death are the progression of the haematological disease or central nervous system involvement [80]. Intravenous immunoglobulin provides a rapid response in at least 50% of patients and may be considered first line treatment. Other options are melphalan, systemic corticosteroids, thalidomide and plasmapheresis [76, 80]. Treatment of the underlying paraproteinaemia with agents such as bortezomib and autologous haematopoietic stem cell transplant can be beneficial [81].
Scleredema
Scleredema is a rare skin thickening disorder, characterized by woody induration and non-pitting oedema, typically localized to the face and neck with subsequent spread to the scalp and upper trunk (Figure 148.10). The hands and feet are usually spared (see Chapter 59). Three clinical variants of scleredema are recognized: the classical type (55% of cases) precipitated by a febrile illness, diabetes-associated scleredema and paraneoplastic scleredema (25% of cases). The latter develops in a setting of monoclonal gammopathy or a B-cell lymphoma [82, 83]. In contrast to scleromyxoedema, scleredema may arise in the context of IgG2-κ, IgG3-κ or IgA-κ paraproteins (rather than IgG1-λ) [82, 83]. The paraneoplastic pathogenesis is not clear but there is dermal and subcutaneous thickening due to an increase in glycosaminoglycans separating collagen fibres. No mucin protein deposition is observed in scleredema. Paraneoplastic scleredema does not resolve spontaneously like the classical variant of the disease. Internal organs may be involved, in particular the heart, joints and eye [84, 85]. An association with POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes) has been reported [86]. Treatment is challenging, but psoralens and vitamin A (PUVA) [87], electron beam radiation [83], intravenous immunoglobulins [88], broadband ultraviolet A (UVA1) phototherapy [89] and extracorporeal photopheresis [90] have been used with benefit in single patients. Successful treatment of scleredema with bortezomib-based regimens for the underlying paraprotein is reported [91].

Figure 148.10 Scleredema in a patient with MGUS (monoclonal gammopathy of undetermined significance).
(Courtesy of Dr Ian Coulson, Burnley General Hospital, UK.)
Necrobiotic xanthogranuloma and normolipaemic xanthoma
Diffuse normolipaemic xanthoma and necrobiotic xanthogranuloma may represent paraneoplastic conditions without coexisting hyperlipidaemia (see Chapter 136) [92]. Necrobiotic xanthogranuloma accounts for two thirds of paraneoplastic xanthomas. Normolipaemic xanthomas resemble classical hyperlipaemic xanthomas clinically, whereas necrobiotic xanthogranulomas are firm nodules, papules or plaques in the periorbital area, often with ulceration, crusting or telangiectasia [93]: upper extremities and the upper trunk are affected in less than half of cases. Exceptionally, extracutaneous sites such as oral mucosa, eye, bone, liver or lung can be affected [92]. The histopathological features of normolipaemic xanthogranuloma are similar to the hyperlipaemic variant whereas necrobiotic xanthogranuloma shows a granulomatous inflammation with lymphocytes, foreign body-like giant cells, Touton giant cells and foci of collagen necrobiosis [93].
Necrobiotic xanthogranuloma and normolipaemic xanthoma are associated with monoclonal gammopathy in 80–90% cases. The majority (80%) of these are IgG paraproteinaemias, in particular MGUS (approximately 50%), multiple myeloma (approximately 40%); the remainder are associated with Waldenström macroglobulinaemia, CMML, chronic lymphatic leukaemia or non-Hodgkin lymphoma [92]. In almost all cases, xanthomas are the first sign of the haematological disease.
The differentiation between paraneoplastic necrobiotic xanthogranuloma and other types of xanthoma can be achieved on the basis of clinical manifestations and histology. However, the separation between normolipaemic xanthoma and hyperlipaemic xanthoma may be difficult due to overlapping clinicopathological features and the frequent presence of hyperlipidaemias in patients with monoclonal gammopathies. It has been proposed that low levels of complement C4 and C1 inhibitor are indicative of a paraneoplastic basis [92].
Therapeutic options beyond the treatment of the underlying disease are limited. Surgical excision may provide symptomatic relief in some patients. Systemic steroids and intravenous immunoglobulin have been effective in single patients [93, 94].
Paraneoplastic pruritus
Chronic pruritus, defined as skin itch of longer duration than 6 weeks, has long been associated with haematological neoplasms, in particular malignant lymphomas (see Chapters 85 and 140) [95]. Paraneoplastic pruritus may precede the clinical signs of lymphoma for weeks and months. Its intensity varies from mild to severe and nocturnal exacerbation is typical. The itch may be accompanied by ichthyosiform skin changes on the extremities. The most commonly associated haematological malignancies are Hodgkin lymphoma (30% incidence of paraneoplastic pruritus), non-Hodgkin lymphomas (10–20%), multiple myelomas and lymphatic leukaemia. Aquagenic pruritus is strongly associated with myelodysplastic syndromes (most notably polycythaemia vera) and T-cell lymphomas.
The mechanism of the paraneoplastic itch is largely unknown. Antihistamines and local steroids are largely ineffective. There is recent evidence that paraneoplastic pruritus can be ameliorated by aprepitan [96], nalfurafine [97] and butorphanol [98]. Short courses of oral steroids, UVB phototherapy, thalidomide, gabapentin and serotonin–norepinephrine reuptake inhibitors (SNRI) have been effective in single cases [95].
PARANEOPLASTIC CONDITIONS OCCASIONALLY ASSOCIATED WITH HAEMATOLOGICAL NEOPLASMS
Most known paraneoplastic conditions have been reported in association with haematological neoplasms. Classical paraneoplastic conditions such as erythema gyratum repens or paraneoplastic acanthosis nigricans have only occasionally been linked to haematological neoplasia.
A number of autoimmune conditions have been linked to haematological neoplasia. Association of myelodysplastic syndrome, myeloid leukaemias and chronic lymphatic leukaemia with autoimmune disease may approach 10–15% of cases [99, 100, 101, 102, 103, 104]. Waldenström macroglobulinaemia has been linked to cutaneous vasculitis [105]. In some cases, the autoimmune phenomena may precede the onset of haematological disease. The most common are non-dermatological, in particular pancytopenias, autoimmune haemolytic anaemia and thrombocytopenia. Those that affect the skin are shown in Table 148.4. The development of autoimmune phenomena has been linked to dysfunction of regulatory T cells [106].
Table 148.4 Autoimmune paraneoplastic phenomena associated with haematological neoplasms.
| Haematological neoplasm | Autoimmune condition in the skin | Autoimmune phenomena in other tissues and organs |
| Myeloid neoplasms | Vasculitis Relapsing polychondritis Vitiligo Dermatomyositis Linear IgA dermatosis Acquired angio-oedema Behçet disease |
Chronic inflammatory demyelinating polyneuropathy (CIDP) Haemolytic anaemia (Coombs negative) Autoimmune thrombocytopenia Arthritis Myositis Colitis Episcleritis Glomerulonephritis Cold agglutinin disease |
| Chronic lymphatic leukaemia | Leukocytoclastic vasculitis Paraneoplastic lymphocytic vasculopathy |
Autoimmune cytopenias: autoimmune haemolytic anaemia, autoimmune thrombocytopenia, pure red blood cell aplasia, autoimmune granulocytopenia |
| Waldenström macroglobulinaema | Vasculitis | Myasthenia gravis, haemolytic anaemia, peripheral neuropathy |
Other reported skin manifestations associated with myeloid or lymphoid neoplasms are erythema, erythroderma and exfoliative dermatitis, ichthyosiform skin changes, granuloma annulare, juvenile xanthogranuloma, erythromelalgia, Raynaud phenomenon, cutis laxa, ‘numb chin’ syndrome and mixed cryoglobulinaemia [46].
SYNDROMES ASSOCIATED WITH HAEMATOLOGICAL MALIGNANCIES AND INVOLVING THE SKIN
Schnitzler syndrome [107]
This is an autoinflammatory skin disease occurring in the context of a monoclonal paraproteinaemia, typically IgM and rarely IgG (see Chapter 45). The patients are usually adults of the age of 40–60 and develop a recurrent urticarial rash accompanied by systemic symptoms (fever, arthralgia, myalgia, lymph node enlargement and hepatosplenomegaly) (Figure 148.11). Laboratory tests usually show leucocytosis and elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP).
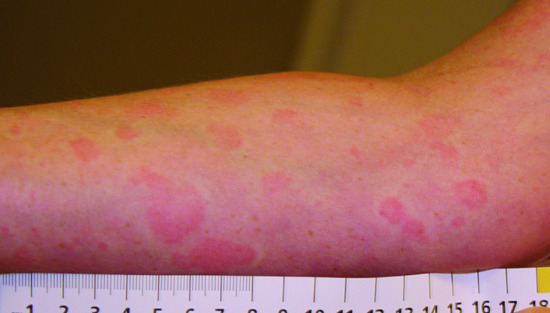
Figure 148.11 Urticaria in Schnitzler syndrome in a patient with MGUS (monoclonal gammopathy of undetermined significance).
The differential diagnoses include urticaria and urticarial vasculitis, systemic adult-onset Still disease and a number of rare autoinflammatory skin diseases, especially Muckle–Wells syndrome and mevalonate kinase deficiency (hyper-IgD syndrome). Mild cases may be treated with colchicine, NSAIDs or hydroxychloroquine whereas treatment with IL-1 receptor antagonists such as anakinra or canakinumab should be considered for non-responders or severely affected patients. The anti-IL6 antibody, tocilizumab, may also be effective in selected cases. Treatment is obligatory since the untreated disease may result in AA amyloidosis, a deposition disease of serum amyloid A (SAA) protein. The patients should be monitored with periodic SAA protein levels and quantification of the monoclonal paraprotein.
POEMS syndrome
POEMS syndrome develops in the context of a monoclonal plasma cell disorder (virtually always of the λ light-chain type). Other common features not included in this acronym include oedema, ascites, pleural effusion, osteosclerotic bone lesions, Castleman disease and thrombocytosis [108]. Skin changes comprise hyperpigmentation, eruptive haemangiomas, hypertrichosis, acrocyanosis, leukonychia, sclerodermoid changes, finger clubbing and facial flushing. The cause of POEMS syndrome is not known but the overproduction of vascular endocrine growth factor (VEGF) and pro-inflammatory cytokines are a major feature [109]. The known symptoms of POEMS syndrome and the diagnostic criteria are summarized in Table 148.5 [110]. The diagnosis of POEMS syndrome is a multidisciplinary task (Table 148.6). The course of the disease is chronic with a reported median survival of nearly 14 years. The total number of POEMS features does not affect survival, but fingernail clubbing, effusions, oedema, ascites and respiratory symptoms have been associated with a significantly shorter overall survival [110]. The treatment of the underlying paraproteinaemia and plasma cell disorder is essential. Isolated lesions of plasmacytoma may be treated by radiation. Other treatment regimen include MDex, bortezomib, lenalinomide and autologous haematopoietic stem cell transplantation. The latter modality has become the first line treatment for younger patients with normal organ function [108, 110].
Table 148.5 Frequency of signs in POEMS syndrome.
| Affected (%) | |
| Polyneuropathy | 100 |
| Monoclonal plasma cell dyscrasia | 100 |
| Organomegaly (hepatomegaly, splenomegaly or other) | 45–85 |
| Lymphadenopathy | 26–74 |
| Castleman disease | 11–25 |
| Endocrinopathy | 67–84 |
| Papilloedema | 29–64 |
| Extravascular volume overload | 29–87 |
| Bone lesions | 27–97 |
| Thrombocytosis | 54–88 |
| Skin changes | 68–89 |
| Hyperpigmentation | 46–93 |
| Acrocyanosis | 19 |
| Hypertrichosis | 26–74 |
| Skin thickening | 5–43 |
From Dispenzieri 2011 [110].
Table 148.6 Diagnostic criteria for POEMS syndrome.
| Major criteria | Minor criteria |
Mandatory
Other
|
• Organomegaly • Extravascular volume overload (oedema, ascites, pleural effusion) • Endocrinopathy • Skin changes • Papilloedema • Thrombocytosis or polycythaemia |
From Dispenzieri 2011 [110].
Diagnosis: two major mandatory criteria and one additional major criterion and one minor criterion.
AESOP [111]
AESOP syndrome is an acronym of adenopathy and extensive skin patch overlying a plasmacytoma. Very few cases have been described. The cardinal features are a slowly enlarging violaceous skin patches or plaques, and enlarged regional lymph nodes (Figure 148.12). Biopsy from the cutaneous patch overlying the plasmacytoma shows increased dermal mucin and vascular hyperplasia. In all described cases, the skin lesion was overlying a solitary plasmacytoma of bone. The patient may share features with POEMS, in particular the polyneuropathy. AESOP syndrome, when recognized at an early stage, may lead to the detection of a curable solitary plasmacytoma of bone.

Figure 148.12 AESOP syndrome (adenopathy and extensive skin patch overlying a plasmacytoma): presternal red-brown patch overlying sternal plasmacytoma.
TEMPI syndrome
This syndrome is associated with monoclonal gammopathy. The acronym describes the following constellation of symptoms: telangiectasia, elevated erythropoietin and erythrocytosis, monoclonal gammopathy, perinephric fluid collections and intrapulmonary shunting [112]. Males and females aged 35–56 years are affected. Skin manifestations are telangiectases on the face, neck and upper trunk. Patients with TEMPI syndrome are at risk of cerebral thrombosis and venous thrombosis. The cause is unknown but untreated, TEMPI syndrome has a progressive course, but responses to bortezomib have been described [112, 113, 114].
Neurofibromatosis type 1, juvenile xanthogranuloma and juvenile chronic myeloid leukaemia
Over 10% of children with juvenile myelomonocytic leukaemia have a clinical diagnosis of neurofibromatosis type 1 (NF1) and the risk of leukaemia in a child with NF1 is 200–500 times above normal [115, 116]. The association with chronic myeloid leukaemia is even more pronounced (additional 20–30-fold) for those children with concurrent finding of NF1 and juvenile xanthogranulomas [117]. The child presents with multiple (>6) café-au-lait patches and at least one juvenile xanthogranuloma (Figure 148.13). This neurocutaneous syndrome is caused by activation of Ras proteins due to defective neurofibromin (the product of the NF1 tumour suppressor gene) causing an increased risk of malignant transformation of haematological and neural crest derived cells [115].

Figure 148.13 Neurofibromatosis, juvenile xanthogranuloma and juvenile myeloid leukaemia syndrome.
(Courtesy of Professor Regina Fölster-Holst, University of Kiel, Germany.)
Other rare syndromes involving skin associated with haematological cancers
A number of rare inherited syndromes with a predisposition to haematological neoplasms have been described. A significant proportion of affected patients may present with skin changes. The genetic defect has been defined in many of these syndromes [118, 119]. The main mechanisms involved are the DNA double-stranded break repair defects (Fanconi anaemia, ataxia telangiectasia, Bloom syndrome, Nijmegen breakage syndrome) or defects in proteins regulating cell signalling and cell differentiation (NF1 and Legius syndrome). It is notable that café-au-lait spots are common to all these syndromes, the main features of which are shown in Table 148.7.
Table 148.7 Rare syndromes associated with haematological malignancies and skin signs [118, 119].
| Condition | Skin signs | Other clinical features | Associated haematological neoplasias | Genetic inheritance and defect |
| Bloom syndromea | Ultraviolet hypersensitivity, skin erythema and maculopapular rash in sun-exposed areas, café-au-lait macules, pigmentary abnormalities | Craniofacial dysmorphism Low birth weight Retarded growth50% risk for cancer |
15% risk for leukaemia (myelodysplastic syndrome, acute myeloid leukaemia, acute lymphoblastic leukaemia); 15% risk for lymphoma | Autosomal recessive. BLM gene encoding helicase |
| Nijmegen breakage syndromeb | Café-au-lait macules | Microcephaly, prominent midface, recurrent infections, marrow failure | 30% risk for lymphoma, 5% risk for leukaemia | Autosomal recessive NBS1 gene |
| Ataxia telangiectasiac | Café-au-lait macules, mucocutaneous telangiectasia, poikiloderma, premature canities and skin ageing | Cerebellar degeneration, immunodeficiency, hypogonadism, insulin resistance, predisposition to cancer (35% incidence by the age of 20) | High risks of leukaemia and non-Hodgkin lymphoma | Autosomal recessive ATM gene |
| Fanconi anaemiac | Pigmentary changes and café-au-lait macules | Short stature, eye malformations, deafness, malformations of the heart, urogenital system, central nervous system, haematopoietic abnormalities | 25% cumulative risk of haematological malignancy by age 45 (acute myeloid leukaemia, myelodysplastic syndrome) | Autosomal recessive (FANC genes: A, D1, D2, E, F, G, I, J, L, M, N, RAD51C) or X-linked (FANCB) |
| Neurofibromatosis type 1d | Neurofibromas, café-au-lait macules, axillary freckling | Scoliosis, epilepsy, glial tumours, learning disabilities, ocular hamartomas | Juvenile myelomonocytic leukaemia | Autosomal dominant (acquired mutation in 50% of cases) NF-1 gene |
| Legius syndromed | Axillary and inguinal freckling, café-au-lait macules, neurofibromas, schwannomas | Brain tumours | Juvenile myelomonocytic leukaemia | Autosomal dominant SPRED1 gene |
aChapter 79
bChapter 82
cChapter 78
dChapter 80.
SKIN INVOLVEMENT IN IDIOPATHIC LYMPHADENOPATHIES
This group of diseases comprises four distinct entities: Kikuchi–Fujimoto disease, Kimura disease, sinus histiocytosis with massive lymphadenopathy and IgG4-related disease (IgG4-RD). They present with lymphadenopathy clinically mimicking lymphoma or histiocytosis, and are associated with pathologies in other tissues and organs including the skin. The underlying aetiologies of these conditions have not been elucidated but some appear to be pre-malignant.
Kikuchi–Fujimoto disease
Also known as Kikuchi disease or Kikuchi histiocytic necrotizing lymphadenitis, this is a rare benign condition of unknown cause that is seen predominantly in East Asia. Whilst originally described in young women, the disorder does occur in male and female adults and young children. The clinical course and histological changes suggest a histiocytic and T-cell response to an infectious agent, such as EBV [120]. Clinical presentation is often with painful cervical lymphadenopathy, fever and leukocytosis in a previously well patient. Skin involvement is present in 25% of patients. The cutaneous manifestations are heterogeneous and include papules, plaques, acneform lesions or erythema multiforme-like changes, often localized on the face and upper trunk (Figure 148.14) [121]. The diagnosis is based on an excisional biopsy of an affected lymph node which, depending on the stage of the disease, may reveal proliferative, necrotic or xanthogranulomatous changes [122]. Kikuchi–Fujimoto resolves spontaneously within 3–4 months [123]. No effective treatment is established but in patients with severe or refractory symptoms high-dose corticosteroids with intravenous immunoglobulin have been used.

Figure 148.14 Kikuchi–Fujimoto syndrome in a 37-year-old South Asian female patient who presented with a 6-day history of malaise, sweating, pyrexia and painful left-sided cervical lymphadenopathy associated with an eruption of ulcerated papules on the face and macules resembling the Janeway lesions of bacterial endocarditis on the toes (inset).
Kimura disease
This is a benign rare chronic inflammatory condition seen predominantly in younger males of East Asian descent. There is a 3–6-fold male preponderance with a median age at onset of 32 years [124]. The classical clinical presentation is with bilateral, cervical lymphadenopathy, occasionally with fever, accompanied by peripheral blood eosinophilia and elevated IgE. Skin involvement is common and ranges from specific infiltration with characteristic histology and presenting as subcutaneous masses, particularly around the head and neck, to a variety of non-specific manifestations including itch, urticaria and chronic eczema [124]. Other sites of involvement are the orbit, oral cavity, and nasal sinuses. Kidney involvement has prognostic significance since 12–16% of patients with Kimura disease develop nephrotic syndrome [125].
Histologically, Kimura disease usually displays an inflammatory infiltrate with eosinophils and follicular hyperplasia, fibrosis and arborizing vascular proliferation of the post-capillary venules [124]. The most important differential diagnoses are other diseases with eosinophilic infiltrates including parasitic infections, drug reactions, angiolymphoid hyperplasia with eosinophilia, Castleman disease and eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome).
Kimura disease has a chronic and relapsing course. There is no effective treatment but the available options are localized radiotherapy (20–45 Gy), local excision of affected tissues, corticosteroid therapy, ciclosporin and low-dose imatinib mesylate [126].
Sinus histiocytosis with massive lymphadenopathy
This disorder, otherwise known as Rosai–Dorfmann disease, affects predominantly children and is more frequently seen in males (see Chapter 136). The characteristic clinical presentation is with significant but painless lymphadenopathy (mainly cervical, mediastinal and axillary), fever, anaemia, neutrophilia and polyclonal hypergammaglobulinaemia [127, 128]. The skin is a common extranodal site of involvement and skin lesions are observed in approximately 10% of cases [126]. Lesions are brownish plaques, papules and nodules, and may resemble xanthomas. In some patients, the lesions are deeper and may mimic morphoea or panniculitis. The key to the diagnosis is the histopathological examination of an affected lymph node which shows dilated sinuses containing neutrophils, lymphocytes, plasma cells and large S100+ CD11c+ CD68+ histiocytes [127]. The main differential diagnoses are lymphoma, Kikuchi–Fujimoto disease and the IgG4-RD.
The course is indolent, but protracted. The disease may disappear spontaneously. Therapies such as systemic corticosteroids, radiotherapy and thalidomide have each been advocated [127, 129, 130].
IgG4–related disease
IgG-RD is an increasingly recognized multiorgan disease affecting predominantly middle-aged and elderly males [131, 132, 133]. The pathogenesis is uncertain and features consistent with both an autoimmune and allergic disorder are found. It remains unclear whether IgG4 itself plays a pathogenic role [131]. The hallmark is sclerosing, autoimmune pancreatitis, lymphadenopathy and elevated IgG4 levels. Skin is involved in less than half of the cases. The patients present with nodules or plaques in the head and neck region. Histologically, the lesions may resemble pseudolymphoma; however, there is stromal fibrosis with dermal and subcutaneous infiltration by lymphocytes, IgG4-positive plasma cells and eosinophils. Other common sites of involvement are the hepatobiliary tract, the orbit and lacrimal and salivary glands. Virtually any organ including the central nervous system may be affected.
The major differential diagnosis is multicentric Castleman disease, which has an abrupt onset and systemic symptoms with a high mortality rate. IgG4 serum and tissue levels may also be elevated in multicentric Castleman disease (which does not itself affect the skin). The other differential diagnosis is cutaneous sinus histiocytosis with massive lymphadenopathy disease (where there may also be an increased proportion of IgG4-positive cells in the dermal infiltrate). The mainstay of treatment for IgG4-RD is corticosteroid therapy. Rituximab therapy has also been reported. The natural history of IgG-RD is not well described. The disorder has been associated with both Hodgkin and non-Hodgkin lymphoma.
SKIN MANIFESTATION IN PRIMARY IMMUNODEFICIENCIES
Primary immunodeficiencies are a heterogeneous group of disorders caused by mutations in the genes encoding functional proteins of the immune cells. They are discussed in detail in Chapter 82. The skin is very often involved and the most common manifestations of this are recalcitrant, relapsing bacterial or fungal infections. Those infections respond poorly to antibiotics and antifungals and have a tendency to recur. Skin infections are accompanied by infections in other organs. Multiple episodes of otitis media, pneumonia, deep abscesses or other recurring serious infections are considered warning signs which may herald a primary immunodeficiency (Table 148.8).
Table 148.8 Warning signs heralding primary immunodeficiency.
| Infectious | Infections with atypical or opportunistic organisms, recurrent or persistent infection |
| >6 episodes of otitis media/year | |
| Resistance to antibiotics (no effect after >2 months of antibiotic treatment) | |
| >2 episodes of pneumonia over a year | |
| Deep infections and abscesses | |
| Other | Family history of primary immunodeficiency |
| Diarrhoea | |
| Failure to thrive |
From Sillevis Smitt et al. [134].
The most common cutaneous infections in immunodeficient patients are caused by Candida, Staphylococcus aureus, Molluscum contagiosum and human papillomavirus (HPV) (Table 148.9).
Table 148.9 Skin diseases and symptoms accompanying primary immunodeficiencies.
| Fungal infections | Candida: oral candidosis, mucocutaneous candidosis, Candida paronychia, granulomatous candidosis Other: disseminated dermatophytosis, aspergillosis, blastomycosis, coccidioidomycosis, cryptococcosis, histoplasmosis, paracoccidioidomycosis, mucormycosis, sporotrichosis |
| Bacterial infections | Staphylococcus aureus: impetigo, persistent folliculitis, furunculosis, cellulitis Ecthyma gangrenosum (Pseudomonas), streptococcal cellulitis and deep cutaneous infections, atypical cellulitis (Helicobacter spp., Campylobacter spp., Haemophilus influenzae) Atypical mycobacterial infections, disseminated Mycobacterium marinum, extensive bacillus Calmette–Guérin (BCG) reaction |
| Viral infections | Molluscum contagiosum, severe infections with varicella-zoster virus (and other herpesviruses) HPV (extensive warts) |
| Eczema | Dermatitis resembling atopic eczema Erythrodermic dermatitis |
| Vasculitis | Small-vessel vasculitis resembling IgA (Henoch–Schönlein) vasculitis |
| Autoimmune conditions | Systemic lupus erythematosus-like disease Vitiligo |
Mucocutaneous candidosis
Chronic mucocutaneous candidosis is the most typical and common phenotype but recalcitrant candidosis may also provide an important clue to the presence of other forms of immunodeficiency (Figure 148.15). Cutaneous Candida species infections are a very sensitive sign of T-cell deficiencies, in particular those affecting Th17 and Th22 cells. Thus, any global T-cell deficiency disorder, such as Di George syndrome, Omenn syndrome or severe combined immunodeficiency is associated with chronic or recurrent candidosis.
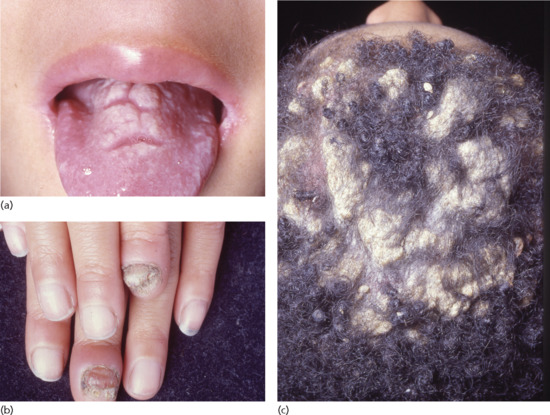
Figure 148.15 Chronic mucocutaneous candidosis in a 6-year-old boy with involvement of (a) the tongue and (b) the fingernails and (c) severe hyperkeratosis of the scalp.
Th17 cells are suppressed in the autosomal dominant form of hyper-IgE syndrome, in which 80% of patients have mucocutaneous candidosis. The cause of hyper-IgE syndrome is a mutation of the transcription factor STAT3. In a physiological situation, STAT3 mediates the expression of RORγt, which in turn causes Th17 cell differentiation. In hyper-IgE syndrome, this differentiation pathway is impaired. These patients have low numbers of circulating Th17 cells (CD4+ CCR6+) and have reduced levels of antimicrobial proteins, such as β-defensin 2 and histatins, the expression of which is stimulated by IL-17 [135].
Deficiency of mannan binding lectin (MBL) has been linked to recurrent vaginal candidosis but whether it is also connected with skin candidosis has not been shown conclusively [136].
Non-syndromic mucocutaneous candidosis may be caused by mutations in the genes coding for IL-17 receptor or in the genes coding the pattern recognition receptors capturing Candida antigens, such as dectin-1 and CARD9 (see Table 148.11). Another interesting mechanism is the gain-of-function STAT1 mutations which cause enhanced responses to Th17 suppressors (interferon (IFN)-γ, IL-27 and IFN-α) with a consequent decrease in Th17 [137, 138].
Table 148.11 Immunodeficiencies caused by mutations in cytokines and signalling proteins.
| Target protein | Symptoms | Remarks |
| IL-17 receptor deficiency [138, 140] | Mucocutaneous candidosis Staphylococcal folliculitis and abscesses |
Mutation of IL-17R and abolition of the responses to IL-17A and IL-17F |
| Dectin-1 deficiency [141] | Mucocutaneous candidosis | Stop codon mutation in the pattern recognition receptor dectin-1, which recognizes Candida antigens. Reduced production of IL-17, TNF-α and IL-6 in response to Candida |
| CARD9 [142, 143] | Mucocutaneous candidosis | CARD9 mediates dectin-1 signalling and the disease is a phenocopy of dectin-1 deficiency |
| IFN-γ receptor 1 deficiency [144] | Mycobacterial skin granulomas (M. bovis, M. avium intracellulare, M. fortuitum, M. ulcerans) Lymphoedema |
Mutations in the IFN-γR1 |
| IL-12p40 and IL-12 receptor β1 deficiencies [145] | Mycobacterial infections including the skin Mucocutaneous candidosis (25% patients) Salmonellosis |
– |
| STAT1 gain of function [137, 146, 147] | Mucocutaneous candidosis, staphylococcal infections, susceptibility to viral infections | Inhibition of Th17 cells due to enhanced STAT1-dependent cellular responses to Th17 repressors (IFN-γ, IL-27 and IFN-α) |
Bacterial infections
Pyogenic skin infections are common in immunodeficiencies. Recalcitrant staphylococcal impetigo, folliculitis and abscesses are the most common manifestations. Bacterial infections are a sensitive marker of immunodeficiencies of the innate immune system targeting granulocytes and cytotoxic T cells (Chediak–Higashi syndrome, Hermansky–Pudlak syndrome, Griscelli syndrome, Wiskott–Aldrich syndrome, chronic granulomatous disease or cyclic haematopoiesis).
Mycobacterial infections are associated with impaired cellular immunity, in particular that mediated by IL-12 and interferons. IL-12 receptor mutations and IFN-γ receptor are strongly associated with atypical mycobacterial skin infections, including M. ulcerans, M. avium and M. marinum.
Persistent HPV infections
Persistent viral warts are associated with suppressed T-cell responses. Examples are congenital CD4 deficiency and WHIM syndrome (warts, hypogammaglobulinaemia, infections and myelokathexis) where persistent viral warts are the dominating symptom (Figure 148.16).
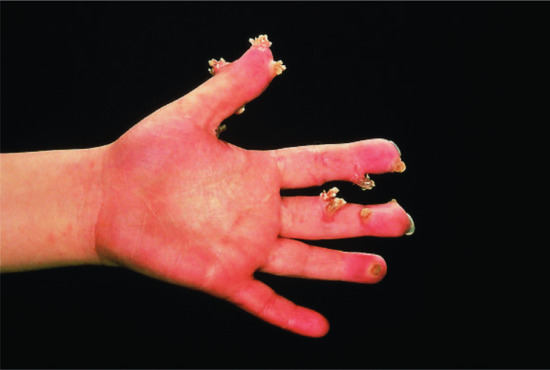
Figure 148.16 Unusually large and rapidly proliferating viral warts in a child with CD4 deficiency.
Other skin conditions associated with immunodeficiencies
Eczematous skin conditions accompany some immunodeficiencies, in particular hyper-IgE syndrome, Omenn syndrome, Wiskott–Aldrich syndrome, common variable immunodeficiency, selective IgA deficiency and X-linked agammaglobulinaemia (Table 148.10). Erythrodermic eczema in a child may be caused by a primary immunodeficiency in up to 30% of cases [139] and is a constant feature of Omenn syndrome.
Table 148.10 Skin manifestations of primary immunodeficiency syndromes.
| Syndrome | Skin manifestations | Other symptoms | Pathogenesis |
| Chediak–Higashi syndrome | Hypopigmented skin, silvery hair (partial albinism) Pyogenic and fungal infections |
Neuropathy Neutropenia |
Mutations in the CHS1 gene (also called LYST) causing defective vesicular transport, melanosome and phagolysosome formation and secretion of cytotoxic granules |
| Hermansky–Pudlak syndrome | Oculocutaneous albinism | Platelet coagulopathy, bleeding Pulmonary fibrosis Granulomatous colitis |
Autosomal recessive Mutations in genes HPS1-7 playing role in lysosome and melanosome biosynthesis |
| Griscelli syndrome | Albinism Pyogenic skin infections |
Hepatosplenomegaly Neutropenia Thrombocytopenia |
Autosomal recessive Mutations in MYO5 (type 1), RAB27A (type 2) and MLPH (type 3) Defective secretion of T-cell cytotoxic granules |
| Wiskott–Aldrich syndrome | Eczema Recurrent staphylococcal skin infections |
Coagulopathy due to thrombocytopenia Leukaemia, lymphomaReduced IgM levels |
X-linked, recessive WAS gene Disorder of actin polymerization, cytoskeletal rearrangement and immunological synapse disruption |
| Omenn syndrome | Graft-versus-host-like symptoms Eczematous lesions Hyperkeratosis and desquamation Erythroderma Persistent bacterial skin infections |
Diarrhoea Hepatosplenomegaly Leukocytosis Lymphadenopathy Elevated IgE |
Autosomal recessive Recombination activated genes RAG1 and RAG2; dysfunction of B and T cells |
| DiGeorge syndrome | Mucocutaneous candidosis Cutaneous calcification |
Thymic hypoplasia Absence of T cells Congenital heart disease Seizures and hypocalcaemic tetany Facial features (short philtrum, low-set ears, hypertelorism) |
22q11.2 deletion syndrome Haploinsufficiency of Tbx1 |
| Hyper-IgE syndrome | Eczematous skin changes Staphylococcal folliculitis and abscesses Mucocutaneous candidosis |
Retained primary teeth Characteristic facial features (prominent forehead, deep-set eyes, broad nasal bridge) Scoliosis Increased IgE and eosinophilia |
Autosomal dominant (STST3 mutation) Autosomal recessive (DOCK8) Defective cytokine signalling |
| Chronic granulomatous disease | Pyogenic and fungal skin infections Perioral dermatitis Granulomatous cheilitis |
Lymphadenopathy | Variable mutations (X-linked recessive gp91phax, autosomal recessive p47phax) resulting in a defect in neutrophil respiratory burst |
| Congenital CD4 deficiency | Persistent viral warts | Increased risk of lymphoma (primary effusion lymphoma, MALT lymphoma, Burkitt lymphoma, diffuse large cell lymphoma) Low numbers of CD4+ cells (<300 cell/μL) on two or more measurements over at least 6 weeks Less than 20% of T lymphocytes are CD4+ |
Unknown |
| WHIM (warts, hypoimmunoglobulinaemia, infections, myelokathexis) | Multiple, large viral warts Recurrent skin bacterial infections |
B-cell dysfunction Neutropenia |
CXCR4 gene |
| Cyclic haematopoiesis | Recurrent aphthous stomatitis Staphylococcal skin infections Periodontitis |
Periodic failure of haematopoietic progenitor cells resulting in dramatic oscillations in neutrophil, monocyte, eosinophil, platelet and reticulocyte counts often in the periods of 20 days Fever |
Autosomal dominant Neutrophil elastase gene ELA2 |
| Common variable immunodeficiency | Recurrent pyogenic skin infections Eczema |
Recurring infections in different organs Bronchiectasis and asthma Viral infections, enlarged lymph nodes and spleen Hypogammaglobulinaemia: low levels of IgG, IgA, IgM |
Variable aetiology: different mutations in six different types of the disease |
| X-linked agammaglobulinaemia | Recurrent pyogenic skin infections | Various infections, as in common variable immunodeficiency | X-linked Mutation in Btk kinase Lack of mature B cells and low levels of immunoglobulins |
Vasculitis associated with primary immunodeficiencies is often of small-vessel type and presents as purpura resembling Henoch–Schönlein disease (IgA vasculitis). It is most typically a feature of Wiskott–Aldrich syndrome.
Paradoxically, autoimmune diseases are overrepresented in people with immunodeficiency disorders. Systemic lupus erythematosus (SLE) is associated with hyper-IgE syndrome and chronic granulomatous disease.
SKIN MANIFESTATIONS OF ACQUIRED IMMUNODEFICIENCIES
HIV infection, diabetes, cancer and iatrogenic immunosuppression are the leading causes of acquired immunodeficiency in adults and their dermatological manifestations have been described in detail elsewhere (see Chapters 31, 64 and 146). Immunodeficiency may also develop in an adult due to the antibodies against cytokines and their receptors (Table 148.12). The clinical symptoms are similar to those seen in children with genetic errors in cytokine receptor genes (Table 148.11). Patients with thymoma may occasionally develop antibodies against IL-12 p35 and p40 subunits, type I interferons and, more rarely, against IL-1α, IL-17A and IL-22 [148]. In 5–10% of patients with thymoma, the immunodeficiency is syndromic (Good syndrome) comprising hypogammaglobulinaemia and variable lymphopenia [149]. The clinical picture is more severe than in those with anticytokine antibodies alone and resembles X-linked agammaglobulinaemia (XLA) and common variable immune deficiency (CVID). Mucocutaneous candidosis is a prominent features of this condition [149].
Table 148.12 Acquired immunodeficiencies due to immunosuppressive antibodies.
| Condition | Antibodies against | Clinical findings |
| Thymoma [148] | IL-12 (p35 and p40) subunits, type I interferons, IL-1α, IL-17A, IL-22 | Mucocutaneous candidosis Mycobacterial infections |
| Good syndrome [149] | IL-12, IL-22, IL-17A | Cutaneous candidosis Recurrent bacterial and fungal infections Hypogammaglobulinaemia Lymphopenia |
| APECED syndrome [150, 151, 152](Figure 148.17) | IL-17A, IL-17F or anti-IL-22 | Autoimmune polyendocrinopathy Cutaneous candidosis Ectodermal dystrophy (APECED) |
| Spontaneous appearance of immunosuppressive antibodies [138, 150, 153, 154] | IFN-γ | Mycobacterial infections Coccidioidomycosis |
| IL-6 | Staphylococcal cellulitis Abscesses |
|
| IL-17, IL-22 | Mucocutaneous candidosis Dermatophytosis |
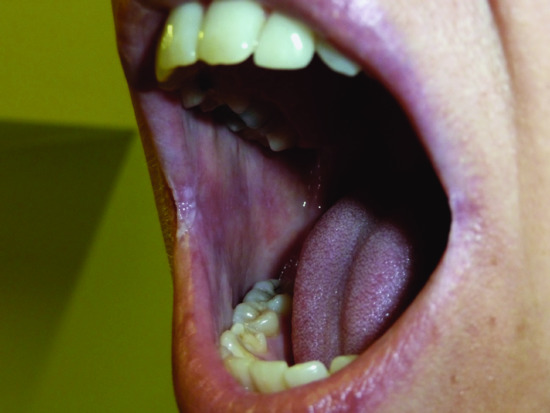
Figure 148.17 Mucocutaneous candidiasis in APECED syndrome (autoimmune polyendocrinopathy, candidiasis and ectodermal dystrophy).
(Courtesy of Dr Nicolas Kluger, University of Helsinki, Finland and Dr Walter Liszewski, University of Minnesota, USA.)
Anti-IL-17A, anti-IL-17F or anti-IL-22 autoantibodies in patients with mucocutaneous candidosis may also develop in the setting of the autoimmune polyendocrinopathy, candidosis and ectodermal dystrophy (APECED) syndrome [150, 151, 152].
Finally, different anticytokine antibodies have been observed in immunodeficient patients [150]. Reported cases include anti-GM-CSF, interferon-γ [155], anti-IL-17 [152], IL-22 [150] and anti-IL-6 [154]. Naturally occurring anti-IFN-γ autoantibodies are associated with cutaneous mycobacterial infections and coccidioidomycosis [156, 157], as could be predicted from the paediatric phenotypes of genetic immunodeficiencies. Anti-IL-6 autoantibodies have been linked to staphylococcal cellulitis and subcutaneous abscesses.
ANAEMIAS AND HAEMOGLOBINOPATHIES
Anaemias
A well-known symptom of anaemia is pallor of the skin and mucosae. It is caused by peripheral vasoconstriction in response to reduced oxygen supply to the skin. Palmar pallor is particularly useful for recognizing anaemia in children under 5 years of age at a primary care level [158, 159]. Pallor of the palmar creases is visible at haemoglobin levels below 70 g/L. Pruritus is reported in 15% of females and 5% of males with iron deficiency anaemia [160]. Glossitis and ulcerative stomatitis are associated with megaloblastic anaemia and vitamin B12 deficiency [159, 161]. The prevalence of oral mucosal symptoms in megaloblastic anaemia is as high as 65% [159]. Glossitis and ulcerative stomatitis are not linked to other types of anaemia, but oral candidosis is more prevalent in iron deficiency anaemia [162].
Fingernail abnormalities such as trachyonychia and koilonychia have been traditionally linked to iron deficiency anaemia but this association has not been confirmed [159].
Haemoglobinopathies
Sickle cell anaemia and thalassaemias are associated with skin ulceration in approximately 5–10% of patients [163]. Skin ulcers resemble venous ulcers and are characteristically localized in the gaiter area of the lower extremities [164]. The pathogenesis of ulceration is probably microvascular obstruction and hypoxia by abnormal erythrocytes, which are dehydrated, dense and inelastic. Treatment with compressive therapy and biomaterial dressings containing the arginine–glycine–aspartate (RGD) peptide seem to be effective [164, 165].
TRANSFUSION REACTIONS
Transfusion reactions with skin manifestations are very rare and fall into one of the following classes: acute allergic reactions, transfusion-associated GVHD and post-transfusion purpura (PTP) (Table 148.13) [166].
Table 148.13 Transfusion reactions with skin involvement.
| Reaction | Skin lesions |
| Acute allergic reaction | Rare. Urticaria, angio-oedema and erythema which occur during or immediately after transfusion |
| Transfusion-associated GVHD | Erythema, maculopapular rash 2–30 days after transfusion |
| Post-transfusion purpura | Thrombocytopenic purpura within 2 weeks after transfusion |
Allergic transfusion reactions
The incidence of allergic reactions to transfused platelets and red blood cells is 3.7% and 0.15%, respectively, and only a minority of those have skin manifestations such as acute urticaria or erythema. Plasma proteins (especially IgA and haptoglobin) are the most frequent allergens and these reactions are more common in patients with IgA deficiency (IgA levels <0.5 mg/L). Haptoglobin deficiency is more prevalent in South-East Asia and may contribute to transfusion reactions in populations from this region. Chemical and food allergens have been reported as causes of allergic transfusion reactions [166]. Examples include methylene blue, used for viral inactivation of fresh plasma, and passive transfer of peanut allergens.
Transfusion–associated graft–versus–host disease
Transfusion-associated GVHD is caused by alloreactive donor leukocytes present in the transfusion product. It presents within 2–30 days after transfusion with an erythematous maculopapular rash, often associated with systemic symptoms such as liver enzyme elevation, jaundice, diarrhoea or vomiting [167, 168]. The diagnosis is supported by skin biopsy and human leukocyte antigen (HLA) typing. Prognosis is grave and the condition is often fatal, especially if gastrointestinal GVHD develops. Immunosuppressed patients and the individuals receiving blood products from related donors are at a higher risk. Various methods for eliminating transfusion-associated GVHD have been adopted in different countries including γ-irradiation, X-ray irradiation and leukoreduction of blood products. GVHD is discussed in greater detail in Chapter 38.
Post–transfusion purpura
PTP is a rare adverse reaction to a transfusion of any platelet-containing product such as packed red blood cells, platelets or granulocyte infusions. It is caused by the production of antibodies to introduced platelet alloantigens, often in a woman previously sensitized by pregnancy [169]. The patient develops antiplatelet antibodies that destroy both the introduced platelets and, surprisingly, their own platelets (the mechanism is unclear) causing sudden onset thrombocytopenia and thrombocytopenic purpura within 4–11 days after transfusion [170]. Treatment with intravenous immunoglobulin infusions and systemic steroids produces satisfactory responses within 2 months after onset [170, 171].
References
- Wagner G, Fenchel K, Back W, Schulz A, Sachse MM. Leukemia cutis – epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges 2012;10:27–36.
- Benet C, Gomez A, Aguilar C, et al. Histologic and immunohistologic characterization of skin localization of myeloid disorders: a study of 173 cases. Am J Clin Pathol 2011;135:278–90.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol 1994;13:223–30.
- Cerroni L, Zenahlik P, Hofler G, Kaddu S, Smolle J, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol 1996;20:1000–10.
- Agis H, Weltermann A, Fonatsch C, et al. A comparative study on demographic, hematological, and cytogenetic findings and prognosis in acute myeloid leukemia with and without leukemia cutis. Ann Hematol 2002;81:90–5.
- Liu PI, Ishimaru T, McGregor DH, Okada H, Steer A. Autopsy study of granulocytic sarcoma (chloroma) in patients with myelogenous leukemia, Hiroshima–Nagasaki 1949–1969. Cancer 1973;31:948–55.
- Wiernik PH, Serpick AA. Granulocytic sarcoma (chloroma). Blood 1970;35:361–9.
- Vitte F, Fabiani B, Benet C, et al. Specific skin lesions in chronic myelomonocytic leukemia: a spectrum of myelomonocytic and dendritic cell proliferations: a study of 42 cases. Am J Surg Pathol 2012;36:1302–16.
- Tallman MS, Hakimian D, Shaw JM, Lissner GS, Russell EJ, Variakojis D. Granulocytic sarcoma is associated with the 8;21 translocation in acute myeloid leukemia. J Clin Oncol 1993;11:690–7.
- Siegel RS, Gartenhaus RB, Kuzel TM. Human T-cell lymphotropic-I-associated leukemia/lymphoma. Curr Treat Options Oncol 2001;2:291–300.
- Magro CM, Morrison CD, Heerema N, Porcu P, Sroa N, Deng AC. T-cell prolymphocytic leukemia: an aggressive T cell malignancy with frequent cutaneous tropism. J Am Acad Dermatol 2006;55:467–77.
- Cho-Vega JH, Medeiros LJ, Prieto VG, Vega F. Leukemia cutis. Am J Clin Pathol 2008;129:130–42.
- Resnik KS, Brod BB. Leukemia cutis in congenital leukemia. Analysis and review of the world literature with report of an additional case. Arch Dermatol 1993;129:1301–6.
- Clark WB, Strickland SA, Barrett AJ, Savani BN. Extramedullary relapses after allogeneic stem cell transplantation for acute myeloid leukemia and myelodysplastic syndrome. Haematologica 2010;95:860–3.
- Petrella T, Meijer CJ, Dalac S, et al. TCL1 and CLA expression in agranular CD4/CD56 hematodermic neoplasms (blastic NK-cell lymphomas) and leukemia cutis. Am J Clin Pathol 2004;122:307–13.
- Bakst RL, Tallman MS, Douer D, Yahalom J. How I treat extramedullary acute myeloid leukemia. Blood 2011;118:3785–93.
- Kaddu S, Zenahlik P, Beham-Schmid C, Kerl H, Cerroni L. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol 1999;40:966–78.
- Klco JM, Welch JS, Nguyen TT, et al. State of the art in myeloid sarcoma. Int J Lab Hematol 2011;33:555–65.
- Paydas S, Zorludemir S, Sahin B. Vasculitis and leukemia. Leuk Lymphoma 2000;40:105–12.
- Mathew RA, Bennett JM, Liu JJ, et al. Cutaneous manifestations in CMML: Indication of disease acceleration or transformation to AML and review of the literature. Leuk Res 2012;36:72–80.
- Cronin DM, George TI, Sundram UN. An updated approach to the diagnosis of myeloid leukemia cutis. Am J Clin Pathol 2009;132:101–10.
- Robak E, Robak T. Skin lesions in chronic lymphocytic leukemia. Leuk Lymphoma 2007;48:855–65.
- Smith JL Jr, Butler JJ. Skin involvement in Hodgkin's disease. Cancer 1980;45:354–61.
- Roglin J, Boer A. Skin manifestations of intravascular lymphoma mimic inflammatory diseases of the skin. Br J Dermatol 2007;157:16–25.
- Beaty MW, Toro J, Sorbara L, et al. Cutaneous lymphomatoid granulomatosis: correlation of clinical and biologic features. Am J Surg Pathol 2001;25:1111–20.
- Sander CA, Flaig MJ, Jaffe ES. Cutaneous manifestations of lymphoma: a clinical guide based on the WHO classification. World Health Organization. Clin Lymphoma 2001;2:86–100; discussion 1–2.
- Cerroni L, Beham-Schmid C, Kerl H. Cutaneous Hodgkin's disease: an immunohistochemical analysis. J Cutan Pathol 1995;22:229–35.
- Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc 2010;85:945–8.
- Kyle RA, Therneau TM, Dispenzieri A, et al. Immunoglobulin m monoclonal gammopathy of undetermined significance and smoldering Waldenstrom macroglobulinemia. Clin Lymphoma Myeloma Leuk 2013;13:184–6.
- Molina-Ruiz AM, Pulpillo A, Lasanta B, Zulueta T, Andrades R, Requena L. A rare case of primary cutaneous plasmacytoma-like lymphoproliferative disorder following renal transplantation. J Cutan Pathol 2012;39:685–9.
- Requena L, Kutzner H, Palmedo G, et al. Cutaneous involvement in multiple myeloma: a clinicopathologic, immunohistochemical, and cytogenetic study of 8 cases. Arch Dermatol 2003;139:475–86.
- Sun X, Peterson LC, Gong Y, Traynor AE, Nelson BP. Post-transplant plasma cell myeloma and polymorphic lymphoproliferative disorder with monoclonal serum protein occurring in solid organ transplant recipients. Mod Pathol 2004;17:389–94.
- Patterson JW, Parsons JM, White RM, Fitzpatrick JE, Kohout-Dutz E. Cutaneous involvement of multiple myeloma and extramedullary plasmacytoma. J Am Acad Dermatol 1988;19:879–90.
- Abdel-Wahab OI, Levine RL. Primary myelofibrosis: update on definition, pathogenesis, and treatment. Annu Rev Med 2009;60:233–45.
- Haniffa MA, Wilkins BS, Blasdale C, Simpson NB. Cutaneous extramedullary hemopoiesis in chronic myeloproliferative and myelodysplastic disorders. J Am Acad Dermatol 2006;55:S28–31.
- O'Malley DP. Benign extramedullary myeloid proliferations. Mod Pathol 2007;20:405–15.
- Nappi O, Boscaino A, Wick MR. Extramedullary hematopoietic proliferations, extraosseous plasmacytomas, and ectopic splenic implants (splenosis). Semin Diagn Pathol 2003;20:338–56.
- Rongioletti F, Patterson JW, Rebora A. The histological and pathogenetic spectrum of cutaneous disease in monoclonal gammopathies. J Cutan Pathol 2008;35:705–21.
- Daoud MS, Lust JA, Kyle RA, Pittelkow MR. Monoclonal gammopathies and associated skin disorders. J Am Acad Dermatol 1999;40:507–35; quiz 36–8.
- Camp BJ, Magro CM. Cutaneous macroglobulinosis: a case series. J Cutan Pathol 2012;39:962–70.
- Chattopadhyay M, Rytina E, Dada M, Bhogal BS, Groves R, Handfield-Jones S. Immunobullous dermatosis associated with Waldenstrom macroglobulinaemia treated with rituximab. Clin Exp Dermatol 2013;38:866–9.
- Whittaker SJ, Bhogal BS, Black MM. Acquired immunobullous disease: a cutaneous manifestation of IgM macroglobulinaemia. Br J Dermatol 1996;135:283–6.
- West NY, Fitzpatrick JE, David-Bajar KM, Bennion SD. Waldenstrom macroglobulinemia-induced bullous dermatosis. Arch Dermatol 1998;134:1127–31.
- Rosenzweig M, Landau H. Light chain (AL) amyloidosis: update on diagnosis and management. J Hematol Oncol 2011;4:47.
- Requena L, Sarasa JL, Ortiz Masllorens F, et al. Follicular spicules of the nose: a peculiar cutaneous manifestation of multiple myeloma with cryoglobulinemia. J Am Acad Dermatol 1995;32:834–9.
- Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet 2012;379:348–60.
- Krug B, Sonet A, Pirson AS, Mahy N, Bosly A, Borght TV. FDG PET utility in paraneoplastic sweet syndrome. Clin Nucl Med 2004;29:91–2.
- Linke R, Voltz R. FDG-PET in paraneoplastic syndromes. Recent Results Cancer Res 2008;170:203–11.
- Wu LM, Chen FY, Jiang XX, Gu HY, Yin Y, Xu JR. 18F-FDG PET, combined FDG-PET/CT and MRI for evaluation of bone marrow infiltration in staging of lymphoma: a systematic review and meta-analysis. Eur J Radiol 2012;81:303–11.
- Fogo A, du Vivier A. The cutaneous manifestations of haematological malignancy. Clin Med 2009;9:366–70.
- Raza S, Kirkland RS, Patel AA, Shortridge JR, Freter C. Insight into Sweet's syndrome and associated-malignancy: a review of the current literature. Int J Oncol 2013;42:1516–22.
- Anzalone CL, Cohen PR. Acute febrile neutrophilic dermatosis (Sweet's syndrome). Curr Opin Hematol 2013;20:26–35.
- Kluger N, Gil-Bistes D, Guillot B, Bessis D. Efficacy of anti-interleukin-1 receptor antagonist anakinra (Kineret(R)) in a case of refractory Sweet's syndrome. Dermatology 2011;222:123–7.
- Delluc A, Limal N, Puechal X, Frances C, Piette JC, Cacoub P. Efficacy of anakinra, an IL1 receptor antagonist, in refractory Sweet syndrome. Ann Rheum Dis 2008;67:278–9.
- Bourke JF, Jones JL, Fletcher A, Graham-Brown RA. An immunohistochemical study of the dermal infiltrate and epidermal staining for interleukin 1 in 12 cases of Sweet's syndrome. Br J Dermatol 1996;134:705–9.
- Oskay T, Anadolu R. Sweet's syndrome in familial Mediterranean fever: possible continuum of the neutrophilic reaction as a new cutaneous feature of FMF. J Cutan Pathol 2009;36:901–5.
- Jo T, Horio K, Migita K. Sweet's syndrome in patients with MDS and MEFV mutations. N Engl J Med 2015;372:686–8.
- Kroshinsky D, Alloo A, Rothschild B, et al. Necrotizing Sweet syndrome: a new variant of neutrophilic dermatosis mimicking necrotizing fasciitis. J Am Acad Dermatol 2012;67:945–54.
- Chan MP, Duncan LM, Nazarian RM. Subcutaneous Sweet syndrome in the setting of myeloid disorders: a case series and review of the literature. J Am Acad Dermatol 2013;68:1006–15.
- Galaria NA, Junkins-Hopkins JM, Kligman D, James WD. Neutrophilic dermatosis of the dorsal hands: pustular vasculitis revisited. J Am Acad Dermatol 2000;43:870–4.
- Willard RJ, Turiansky GW, Genest GP, Davis BJ, Diehl LF. Leukemia cutis in a patient with chronic neutrophilic leukemia. J Am Acad Dermatol 2001;44:365–9.
- Sakiyama M, Kobayashi T, Nagata Y, Fujimoto N, Satoh T, Tajima S. Bullous pyoderma gangrenosum: a case report and review of the published work. J Dermatol 2012;39:1010–15.
- Bennett ML, Jackson JM, Jorizzo JL, Fleischer AB Jr, White WL, Callen JP. Pyoderma gangrenosum. A comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore) 2000;79:37–46.
- Koester G, Tarnower A, Levisohn D, Burgdorf W. Bullous pyoderma gangrenosum. J Am Acad Dermatol 1993;29:875–8.
- Su WP, Davis MD, Weenig RH, Powell FC, Perry HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol 2004;43:790–800.
- Caughman W, Stern R, Haynes H. Neutrophilic dermatosis of myeloproliferative disorders. Atypical forms of pyoderma gangrenosum and Sweet's syndrome associated with myeloproliferative disorders. J Am Acad Dermatol 1983;9:751–8.
- Duguid CM, O'Loughlin S, Otridge B, Powell FC. Paraneoplastic pyoderma gangrenosum. Australas J Dermatol 1993;34:17–22.
- Bachmeyer C, Aractingi S. Neutrophilic eccrine hidradenitis. Clin Dermatol 2000;18:319–30.
- Ng ES, Aw DC, Tan KB, et al. Neutrophilic eccrine hidradenitis associated with decitabine. Leuk Res 2010;34:e130–2.
- Marini M, Wright D, Ropolo M, Abbruzzese M, Casas G. Neutrophilic eccrine hidradenitis secondary to topotecan. J Dermatolog Treat 2002;13:35–7.
- Frew JW, Murrell DF. Paraneoplastic pemphigus (paraneoplastic autoimmune multiorgan syndrome): clinical presentations and pathogenesis. Dermatol Clin 2011;29:419–25, viii.
- Vezzoli P, Berti E, Marzano AV. Rationale and efficacy for the use of rituximab in paraneoplastic pemphigus. Exp Rev Clin Immunol 2008;4:351–64.
- Barzilai A, Shpiro D, Goldberg I, et al. Insect bite-like reaction in patients with hematologic malignant neoplasms. Arch Dermatol 1999;135:1503–7.
- Bairey O, Goldschmidt N, Ruchlemer R, et al. Insect-bite-like reaction in patients with chronic lymphocytic leukemia: a study from the Israeli Chronic Lymphocytic Leukemia Study Group. Eur J Haematol 2012;89:491–6.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol 2005;85:76–7.
- Cokonis Georgakis CD, Falasca G, Georgakis A, Heymann WR. Scleromyxedema. Clin Dermatol 2006;24:493–7.
- Heymann WR. Scleromyxedema. J Am Acad Dermatol 2007;57:890–1.
- Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol 1995;33:37–43.
- Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med 2014;20:332–42.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol 2013;69:66–72.
- Canueto J, Labrador J, Roman C, et al. The combination of bortezomib and dexamethasone is an efficient therapy for relapsed/refractory scleromyxedema: a rare disease with new clinical insights. Eur J Haematol 2012;88:450–4.
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum 2006;35:355–9.
- Angeli-Besson C, Koeppel MC, Jacquet P, Andrac L, Sayag J. Electron-beam therapy in scleredema adultorum with associated monoclonal hypergammaglobulinaemia. Br J Dermatol 1994;130:394–7.
- Rimon D, Lurie M, Storch S, et al. Cardiomyopathy and multiple myeloma. Complications of scleredema adultorum. Arch Intern Med 1988;148:551–3.
- Curtis AC, Shulak BM. Scleredema adultorum. Not always a benign self-limited disease. Arch Dermatol 1965;92:526–41.
- Aarden L, Ruuls SR, Wolbink G. Immunogenicity of anti-tumor necrosis factor antibodies – toward improved methods of anti-antibody measurement. Curr Opin Immunol 2008;20:431–5.
- Hager CM, Sobhi HA, Hunzelmann N, et al. Bath-PUVA therapy in three patients with scleredema adultorum. J Am Acad Dermatol 1998;38:240–2.
- Aichelburg MC, Loewe R, Schicher N, et al. Successful treatment of poststreptococcal scleredema adultorum Buschke with intravenous immunoglobulins. Arch Dermatol 2012;148:1126–8.
- Kroft EB, Berkhof NJ, van de Kerkhof PC, Gerritsen RM, de Jong EM. Ultraviolet A phototherapy for sclerotic skin diseases: a systematic review. J Am Acad Dermatol 2008;59:1017–30.
- Stables GI, Taylor PC, Highet AS. Scleredema associated with paraproteinaemia treated by extracorporeal photopheresis. Br J Dermatol 2000;142:781–3.
- Szturz P, Adam Z, Vasku V, et al. Complete remission of multiple myeloma associated scleredema after bortezomib-based treatment. Leuk Lymphoma 2013;54:1324–6.
- Szalat R, Arnulf B, Karlin L, et al. Pathogenesis and treatment of xanthomatosis associated with monoclonal gammopathy. Blood 2011;118:3777–84.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol 2009;48:1–10.
- Chave TA, Chowdhury MM, Holt PJ. Recalcitrant necrobiotic xanthogranuloma responding to pulsed high-dose oral dexamethasone plus maintenance therapy with oral prednisolone. Br J Dermatol 2001;144:158–61.
- Yosipovitch G. Chronic pruritus: a paraneoplastic sign. Dermatol Ther 2010;23:590–6.
- Duval A, Dubertret L. Aprepitant as an antipruritic agent? N Engl J Med 2009;361:1415–16.
- Inan S, Cowan A. Nalfurafine, a kappa opioid receptor agonist, inhibits scratching behavior secondary to cholestasis induced by chronic ethynylestradiol injections in rats. Pharmacol Biochem Behav 2006;85:39–43.
- Dawn AG, Yosipovitch G. Butorphanol for treatment of intractable pruritus. J Am Acad Dermatol 2006;54:527–31.
- Saif MW, Hopkins JL, Gore SD. Autoimmune phenomena in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk Lymphoma 2002;43:2083–92.
- Fain O, Braun T, Stirnemann J, Fenaux P. [Systemic and autoimmune manifestations in myelodysplastic syndromes.] Rev Med Interne 2011;32:552–9.
- Enright H, Miller W. Autoimmune phenomena in patients with myelodysplastic syndromes. Leuk Lymphoma 1997;24:483–9.
- Zent CS, Kay NE. Autoimmune complications in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol 2010;23:47–59.
- Lulla P, Bandeali S, Baker K. Fatal paraneoplastic systemic leukocytoclastic vasculitis as a presenting feature of chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk 2011;11 Suppl. 1:S14–16.
- Pavlidis NA, Klouvas G, Tsokos M, Bai M, Moutsopoulos HM. Cutaneous lymphocytic vasculopathy in lymphoproliferative disorders – a paraneoplastic lymphocytic vasculitis of the skin. Leuk Lymphoma 1995;16:477–82.
- Stone MJ, Merlini G, Pascual V. Autoantibody activity in Waldenstrom's macroglobulinemia. Clin Lymphoma 2005;5:225–9.
- Jadidi-Niaragh F, Ghalamfarsa G, Yousefi M, Tabrizi MH, Shokri F. Regulatory T cells in chronic lymphocytic leukemia: implication for immunotherapeutic interventions. Tumour Biol 2013;34:2031–9.
- Simon A, Asli B, Braun-Falco M, et al. Schnitzler's syndrome: diagnosis, treatment, and follow-up. Allergy 2013;68:562–8.
- Li J, Zhou DB. New advances in the diagnosis and treatment of POEMS syndrome. Br J Haematol 2013;161:303–15.
- D'Souza A, Hayman SR, Buadi F, et al. The utility of plasma vascular endothelial growth factor levels in the diagnosis and follow-up of patients with POEMS syndrome. Blood 2011;118:4663–5.
- Dispenzieri A. POEMS syndrome: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol 2011;86:591–601.
- Lipsker D, Rondeau M, Massard G, Grosshans E. The AESOP (adenopathy and extensive skin patch overlying a plasmacytoma) syndrome: report of 4 cases of a new syndrome revealing POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes) syndrome at a curable stage. Medicine 2003;82:51–9.
- Sykes DB, Schroyens W, O'Connell C. The TEMPI syndrome – a novel multisystem disease. N Engl J Med 2011;365:475–7.
- Schroyens W, O'Connell C, Sykes DB. Complete and partial responses of the TEMPI syndrome to bortezomib. N Engl J Med 2012;367:778–80.
- Kwok M, Korde N, Landgren O. Bortezomib to treat the TEMPI syndrome. N Engl J Med 2012;366:1843–5.
- Side L, Taylor B, Cayouette M, et al. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N Engl J Med 1997;336:1713–20.
- Side LE, Emanuel PD, Taylor B, et al. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood 1998;92:267–72.
- Zvulunov A, Barak Y, Metzker A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia. World statistical analysis. Arch Dermatol 1995;131:904–8.
- Karalis A, Tischkowitz M, Millington GW. Dermatological manifestations of inherited cancer syndromes in children. Br J Dermatol 2011;164:245–56.
- Seif AE. Pediatric leukemia predisposition syndromes: clues to understanding leukemogenesis. Cancer Genet 2011;204:227–44.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, Chen LM. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. Detection of Epstein–Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol 2000;113:774–81.
- Seno A, Torigoe R, Shimoe K, Tada J, Arata J, Suwaki M. Kikuchi's disease (histiocytic necrotizing lymphadenitis) with cutaneous involvement. J Am Acad Dermatol 1994;30:504–6.
- Kuo TT. Kikuchi's disease (histiocytic necrotizing lymphadenitis). A clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology, and DNA ploidy. Am J Surg Pathol 1995;19:798–809.
- Park HS, Sung MJ, Park SE, Lim YT. Kikuchi–Fujimoto disease of 16 children in a single center of Korea. Pediatr Allergy Immunol 2007;18:174–8.
- Chen H, Thompson LD, Aguilera NS, Abbondanzo SL. Kimura disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 2004;28:505–13.
- Sun QF, Xu DZ, Pan SH, et al. Kimura disease: review of the literature. Intern Med J 2008;38:668–72.
- O'Malley DP, Grimm KE. Reactive lymphadenopathies that mimic lymphoma: entities of unknown etiology. Semin Diagn Pathol 2013;30:137–45.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): review of the entity. Semin Diagn Pathol 1990;7:19–73.
- Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. Current status and future directions. Arch Dermatol 1988;124:1211–14.
- Tjiu JW, Hsiao CH, Tsai TF. Cutaneous Rosai–Dorfman disease: remission with thalidomide treatment. Br J Dermatol 2003;148:1060–1.
- Nasseri E, Belisle A, Funaro D. Rosai–Dorfman disease treated with methotrexate and low-dose prednisone: case report and review of the literature. J Cutan Med Surg 2012;16:281–5.
- Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010;17:303–32.
- Cheuk W, Chan JK. Lymphadenopathy of IgG4-related disease: an underdiagnosed and overdiagnosed entity. Semin Diagn Pathol 2012;29:226–34.
- Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;25:1181–92.
- Sillevis Smitt JH, Wulffraat NM, Kuijpers TW. The skin in primary immunodeficiency disorders. Eur J Dermatol 2005;15:425–32.
- Conti HR, Baker O, Freeman AF, et al. New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol 2011;4:448–55.
- Summers PR. Topical therapy for mucosal yeast infections. Curr Probl Dermatol 2011;40:48–57.
- Boisson-Dupuis S, Kong XF, Okada S, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol 2012;24:364–78.
- Puel A, Cypowyj S, Marodi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol 2012;12:616–22.
- Hoeger PH, Harper JI. Neonatal erythroderma: differential diagnosis and management of the “red baby”. Arch Dis Child 1998;79:186–91.
- Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011;332:65–8.
- Ferwerda B, Ferwerda G, Plantinga TS, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med 2009;361:1760–7.
- Glocker EO, Hennigs A, Nabavi M, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 2009;361:1727–35.
- Lanternier F, Pathan S, Vincent QB, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med 2013;369:1704–14.
- Edeer Karaca N, Boisson-Dupuis S, Aksu G, et al. Granulomatous skin lesions, severe scrotal and lower limb edema due to mycobacterial infections in a child with complete IFN-gamma receptor-1 deficiency. Immunotherapy 2012;4:1121–7.
- de Beaucoudrey L, Samarina A, Bustamante J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) 2010;89:381–402.
- Liu L, Okada S, Kong XF, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011;208:1635–48.
- Aldave JC, Cachay E, Nunez L, et al. A 1-year-old girl with a gain-of-function STAT1 mutation treated with hematopoietic stem cell transplantation. J Clin Immunol 2013;33:1273–5.
- Burbelo PD, Browne SK, Sampaio EP, et al. Anti-cytokine autoantibodies are associated with opportunistic infection in patients with thymic neoplasia. Blood 2010;116:4848–58.
- Kelleher P, Misbah SA. What is Good's syndrome? Immunological abnormalities in patients with thymoma. J Clin Pathol 2003;56:12–16.
- Browne SK, Holland SM. Immunodeficiency secondary to anticytokine autoantibodies. Curr Opin Allergy Clin Immunol 2010;10:534–41.
- Kisand K, Boe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med 2010;207:299–308.
- Puel A, Doffinger R, Natividad A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 2010;207:291–7.
- Browne SK, Burbelo PD, Chetchotisakd P, et al. Adult-onset immunodeficiency in Thailand and Taiwan. N Engl J Med 2012;367:725–34.
- de Beaucoudrey L, Puel A, Filipe-Santos O, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med 2008;205:1543–50.
- Doffinger R, Helbert MR, Barcenas-Morales G, et al. Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clin Infect Dis 2004;38:e10–14.
- Dorman SE, Holland SM. Interferon-gamma and interleukin-12 pathway defects and human disease. Cytokine Growth Factor Rev 2000;11:321–33.
- Rosenzweig SD, Dorman SE, Uzel G, et al. A novel mutation in IFN-gamma receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J Immunol 2004;173:4000–8.
- Sato S. Iron deficiency: structural and microchemical changes in hair, nails, and skin. Semin Dermatol 1991;10:313–19.
- Dawson AA, Ogston D, Fullerton HW. Evaluation of diagnostic significance of certain symptoms and physical signs in anaemic patients. Br Med J 1969;3:436–9.
- Takkunen H. Iron-deficiency pruritus. JAMA 1978;239:1394.
- Piskin S, Sayan C, Durukan N, Senol M. Serum iron, ferritin, folic acid, and vitamin B12 levels in recurrent aphthous stomatitis. J Eur Acad Dermatol Venereol 2002;16:66–7.
- Lu SY, Wu HC. Initial diagnosis of anemia from sore mouth and improved classification of anemias by MCV and RDW in 30 patients. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2004;98:679–85.
- Bartolucci P, Brugnara C, Teixeira-Pinto A, et al. Erythrocyte density in sickle cell syndromes is associated with specific clinical manifestations and hemolysis. Blood 2012;120:3136–41.
- Gniadecka M, Gniadecki R, Serup J, Sondergaard J. Microvascular reactions to postural changes in patients with sickle cell anaemia. Acta Derm Venereol 1994;74:191–3.
- Marti-Carvajal AJ, Knight-Madden JM, Martinez-Zapata MJ. Interventions for treating leg ulcers in people with sickle cell disease. Cochrane Database Syst Rev 2012;11:CD008394.
- Hirayama F. Current understanding of allergic transfusion reactions: incidence, pathogenesis, laboratory tests, prevention and treatment. Br J Haematol 2013;160:434–44.
- Fast LD. Developments in the prevention of transfusion-associated graft-versus-host disease. Br J Haematol 2012;158:563–8.
- Six CK, Haught JM, Safyan EL, Patton T, Stahlfeld K. Transfusion-associated graft-versus-host disease: a case report and review of literature. J Am Acad Dermatol 2012;66:e141–3.
- Lubenow N, Eichler P, Albrecht D, et al. Very low platelet counts in post-transfusion purpura falsely diagnosed as heparin-induced thrombocytopenia. Report of four cases and review of literature. Thromb Res 2000;100:115–25.
- Taaning E, Svejgaard A. Post-transfusion purpura: a survey of 12 Danish cases with special reference to immunoglobulin G subclasses of the platelet antibodies. Transfus Med 1994;4:1–8.
- Waters AH. Post-transfusion purpura. Blood Rev 1989;3:83–7.