CHAPTER 153 The Skin and Disorders of the Kidney and Urinary Tract
Sonja Molin and Thomas Ruzicka
Department of Dermatology and Allergology, Ludwig Maximilian University, Munich, Germany
INTRODUCTION
Skin disorders may be associated with disorders of the kidney and, occasionally, of other parts of the urinary tract. This chapter examines these associations, which may be due to hereditary syndromes or acquired disorders affecting both the skin and kidney, the effects of renal failure on the skin or the effects of skin disease on the kidneys and urinary tract [1–5, 6]. A selection of renocutaneous syndromes is listed in Box 153.1.
HEREDITARY SYNDROMES WITH SKIN AND RENAL INVOLVEMENT
Fabry disease (see Chapter 81). Fabry nephropathy (angiokeratoma corporis diffusum) can develop in early childhood in both sexes, although it may be clinically silent at onset [7]. Symptoms show a wide variation. Affected individuals often may have proteinuria, microalbuminuria, microscopic haematuria and lipiduria. Premature mortality in the condition is often the result of renal failure. Enzyme replacement therapy with α-galactosidase A may not be fully effective if started in the presence of manifest proteinuria or renal impairment. Early diagnosis of Fabry nephropathy is therefore very important.
Neurofibromatosis (see Chapter 80). In neurofibromatosis (von Recklinghausen's disease) urinary outflow obstruction may develop secondary to an impinging neurofibroma [8, 9]. Vascular lesions can result in renal artery thrombosis and subsequent hypertension, which may also develop as a result of an associated phaeochromocytoma or renal artery stenosis.
Nail–patella syndrome (see Chapter 69) Renal involvement is present in up to 40% of patients with nail–patella syndrome (Fong's syndrome or hereditary osteo-onychodysplasia) [10]. Many affected individuals only present with premature loss of renal function but rarely develop symptomatic kidney failure. Only 5–10% of nail–patella syndrome patients develop severe renal impairment progressing to end-stage kidney failure [10].
Tuberous sclerosis (see Chapter 81) Renal involvement in tuberous sclerosis (Bourneville disease) includes angiomyolipomas, renal cysts and malignant tumours such as clear cell carcinoma [11].
Birt–Hogg–Dubé syndrome (see Chapter 80) A seven-fold increase in the incidence of renal tumours is associated with Birt–Hogg–Dubé (BHD) syndrome (Hornstein–Knickenberg syndrome), predominantly various types of renal cell carcinoma. The cutaneous features of BHD include trichofolliculomas, trichodiscomas and skin tags.
Hereditary leiomyomatosis and renal cell carcinoma syndrome (see Chapter 80). Multiple cutaneous leiomyomas, which have an inherited predisposition, are linked to uterine leiomyoma (Reed syndrome) and also appear to be associated with an increased incidence of renal cell carcinoma, usually of the papillary cell type [11].
Von Hippel–Lindau syndrome. Renal lesions in von Hippel–Lindau syndrome (familial cerebelloretinal angiomatosis) are either simple cysts or renal cell carcinoma; usually they are a late manifestation [11].
Multiple hamartoma and neoplasia syndrome (see Chapter 80) Multiple hamartoma and neoplasia syndrome (Cowden's disease) is associated with an increased incidence of renal cell carcinoma or transitional cell carcinoma of the bladder [12].
Familial Mediterranean fever with urticaria, Muckle–Wells syndrome and tumour necrosis receptor associated periodic syndrome (see Chapter 45) These hereditary autoinflammatory syndromes may all be complicated by systemic amyloid A amyloidosis with a significant risk of renal failure [13].
Oro-facial–digital syndrome type 1 (see Chapter 67) Oro-facial–digital syndrome type 1 (OFD1) is characterized by the following abnormalities:
Oral: lobed tongue, hamartomas or lipomas of the tongue, cleft of the hard or soft palate, accessory gingival frenula, hypodontia and other dental abnormalities.
Facial: ocular hypertelorism or telecanthus, hypoplasia of the alae nasi, median cleft or pseudocleft upper lip and micrognathia.
Digital: brachydactyly, syndactyly of varying degrees and clinodactyly of the fifth finger; duplicated hallux (great toe); and preaxial or postaxial polydactyly of the hands.
Brain: intracerebral cysts, corpus callosum agenesis and cerebellar agenesis with or without the Dandy–Walker malformation.
Polycystic disease of the kidneys and liver occurs commonly in this condition. As many as 50% of individuals with OFD1 have some degree of mental retardation, which is usually mild. Almost all affected individuals are female. However, males with OFD1 have been described, mostly as malformed fetuses born to women with OFD1.
SKIN SYMPTOMS AND SIGNS ASSOCIATED WITH RENAL DISORDERS
Metabolic and systemic disorders
Acquired partial lipodystrophy (see Chapter 100) In acquired partial lipodystrophy (Barraquer–Simons syndrome), lipoatrophy of the upper part of the body is associated with lipohypertrophy of the thighs and, in about a quarter of cases, with renal disease, usually a membranoproliferative glomerulonephritis. There is a circulating C3 nephritic factor and reduced levels of complement (C3) [15]. Renal failure at an early age is common.
Amyloid A (AA) amyloidosis [16, 17] Proteinuria leading to the nephrotic syndrome or renal insufficiency is often the first clinical manifestation of AA amyloidosis in patients with chronic inflammatory diseases. Secondary AA amyloidosis is associated with various underlying conditions such as chronic infection, rheumatic diseases (rheumatoid arthritis, ankylosing spondylitis, chronic juvenile arthritis), inflammatory bowel disease and many autoinflammatory syndromes including cryopyrin-associated periodic syndromes (e.g. Muckle–Wells syndrome) and familial Mediterranean fever (see Chapter 45). These conditions frequently have .skin manifestations and may present with specific skin signs. AA amyloidosis is also reported to be a frequent cause of kidney disease in intravenous drug abusers.
Metastatic cutaneous calcification (metastatic calcinosis cutis, calcific panniculitis, benign nodular calcification) (see Chapter 61) Metastatic calcification is a rare phenomenon affecting the dermis and subcutis and affecting predominantly uraemic patients with combined hyperphosphataemia and hypercalcaemia, often in the context of hyperparathyroidism. It typically presents as firm papules, nodules or plaques in the dermis or subcutis, particularly around the large joints or flexural sites. Unlike calciphylaxis it does not lead to tissue necrosis [5].
Calciphylaxis (see Chapter 61) Calcification of small-vessel walls in calciphylaxis (calcific uraemic arteriolopathy) leads to extremely painful cutaneous necrosis and ulceration (Figure 153.1) [18]. It occurs in up to 4% of patients on long-term haemodialysis, but may also be associated with hyperparathyroidism, liver disease, systemic steroid use, malignancy or connective tissue disease in the absence of renal impairment. Prognosis is poor with a mortality rate of up to 80%, mainly due to the risk of sepsis. Careful wound management is particularly important to prevent wound infection; it may include surgical debridement of necrotic tissue, hydrocolloid dressings and systemic antibiotics. Treatment options aiming at a reduction of calciphylaxis risk include a low phosphate diet, low calcium dialysate fluids, non-calcium phosphate binders, parathyroidectomy and, more recently, drugs such as sodium thiosulphate or cinacalcet, which alter calcium metabolism.
Figure 153.1 Early signs of calciphylaxis with superficial skin necrosis and ulceration.
Renal failure and dialysis
Cutaneous signs of renal failure are mainly related to chronicity of disease. Uraemic frosting [15], in which crystalline urea is deposited on the skin, is now very rare due to the widespread use of haemodialysis, but dry, pigmented skin with excoriations is typical.
Xerosis Uraemic patients tend to have a dry skin, sometimes with fine scaling. A reduction in the size of the eccrine sweat glands in uraemia may contribute to this effect [5].
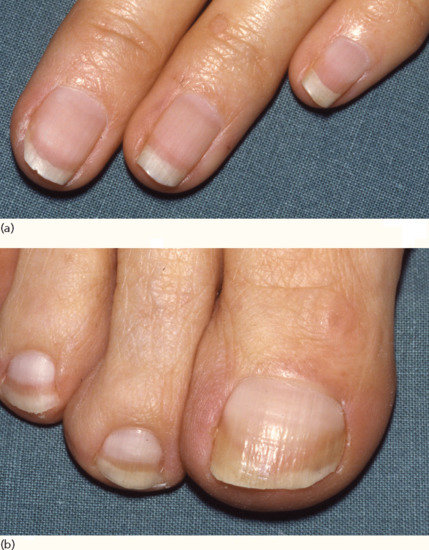
Pigmentation [5] Anaemia presenting as pallor is an early and common sign in renal failure, resulting from reduced erythropoiesis and increased haemolysis (Figure 153.2). Hyperpigmentation is a common clinical sign in patients with end-stage renal disease. The pathomechanisms leading to the development of ‘half-and-half’ nails, a distinctive pattern seen in up to 21% of patients with haemodialysis are not fully understood. They are characterized by a distal brown or reddish colour, combined with a proximal white appearance (Figure 153.3).
Figure 153.2 Lemon yellow pallor in a 27-year-old woman with end-stage renal failure (creatinine 497 μM/L, Hb 77 g/L) resulting from diabetic nephropathy.
Figure 153.3 ‘Half-and-half’ nails readily apparent on (a) the fingers and (b) the toes of a patient with uraemia.
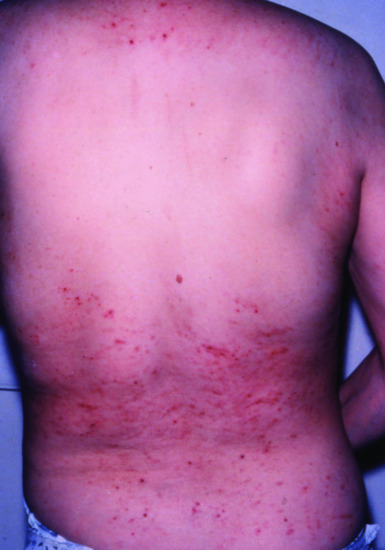
Pruritus [5, 6] Localized (trunk, head) or generalized pruritus occurs in a large proportion of patients with end-stage kidney disease (Figure 153.4). It leads to secondary skin lesions including excoriations, chronic prurigo or acquired perforating dermatosis. The pathogenesis of pruritus in renal failure still remains unclear. Up to 90% of patients on haemodialysis suffer from itching. Other causes of itch should be excluded in every patient. Various treatment modalities have been reported in uraemic pruritus. Among them, topical capsaicin cream has been shown to be effective, as has UVB phototherapy. Parathyroidectomy may be helpful in patients with hyperparathyroidism.
Figure 153.4 Severe uraemic prurigo in a 57-year-old woman with kidney damage following major surgery 1 year earlier. Note the normal skin in areas that cannot be reached to scratch.
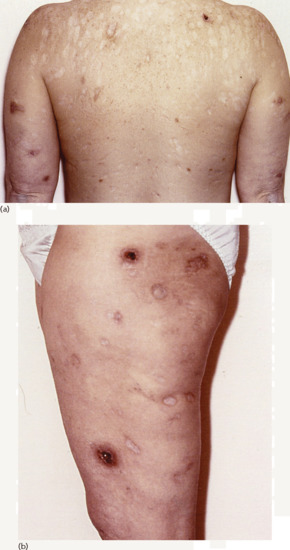
Perforating disorders [5] The acquired perforating dermatoses have been variously labelled reactive perforating collagenosis, perforating folliculitis, Kyrle disease and elastosis perforans serpiginosa depending on the precise clinicopathological presentation (see Chapter 96). The first three of these closely related entities occur principally in patients with end-stage renal disease, particularly in the context of longstanding diabetes (see Chapter 96). They are characterized by the elimination of altered dermal collagen and elastin admixed with degenerate keratin through the follicular wall and/or the epidermis. Pruritus is nearly always present, and up to 11% of dialysis patients may be affected. The cutaneous lesions consist of hyperpigmented papules, plaques and nodules up to 1 cm in diameter with a central keratinous plug. The extensor surfaces of the limbs are more commonly affected but the trunk and face may be involved as well (Figure 153.5).
Figure 153.5 Extensive acquired perforating dermatosis with views of (a) the back and (b) the thigh of a 48-year-old woman with longstanding diabetes complicated by diabetic nephropathy requiring renal transplantation. She had a 12-year history of tender sores that broke down before healing with atrophic scars.
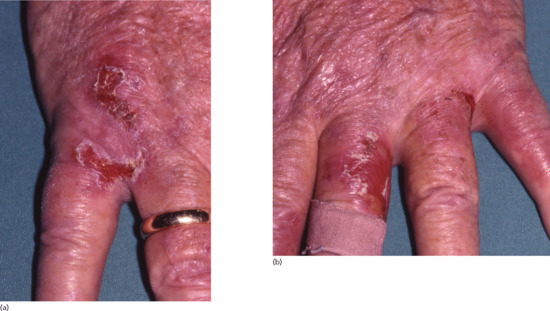
Bullous diseases [5, 6] Pseudoporphyria is a bullous eruption mimicking porphyria cutanea tarda (PCT) that has been reported in patients with end-stage renal disease (chronic renal failure, haemodialysis), with an incidence rate of between 1% and 18% (Figure 153.6). True PCT due to deficiency of uroporphyrinogen decarboxylase is also reported but is uncommon. Distinguishing these two conditions can be difficult as many patients on haemodialysis have elevated blood porphyrin levels slightly above what is regarded as the normal range. However, patients with pseudoporphyria do not present the same porphyrin profile as patients with PCT. Several drugs such as furosemide, oestrogens and non-steroidal anti-inflammatory drugs have been reported as possible triggers of pseudoporphyria and so has UV phototherapy. In all cases, UV protection is important. N-acetylcysteine, which acts as an antioxidant, may be of benefit in patients with pseudoporphyria on haemodialysis. These conditions are discussed in greater detail in Chapter 60.
Figure 153.6 Pseudoporphyria on the backs of the hands of a 64-year-old man receiving renal dialysis for end-stage chronic kidney disease. There was no evidence of elevated porphyrins.
Features related to treatment Premature ageing of the skin and actinic keratoses have been described–a reason for avoiding excessive UV therapy for pruritus. This should be distinguished from the numerous viral, dysplastic and (pre-)malignant skin lesions that may develop in immunosuppressed renal allograft recipients. Cutaneous complications affecting the limb of patients in which their haemodialysis arteriovenous shunt is sited include infection, phlebitis and haematoma. Both irritant and allergic contact eczema may also occur [19], as may pseudo-Kaposi sarcoma. A shunt- associated steal phenomenon can lead to distal ulceration and necrosis.
Nephrogenic systemic fibrosis Nephrogenic systemic fibrosis is linked to renal dysfunction. It was initially described in 2000 as ‘nephrogenic fibrosing dermopathy’ in patients receiving, or with a history of, haemodialysis [5, 20]. It is associated in over 90% of cases with the use of radiocontrast agents containing gadolinium, which are used to enhance magnetic resonance imaging of the blood vessels. Several other risk factors have, however, been suspected to play a role in its development, especially pro-inflammatory conditions including major surgery.
Clinical features include systemic fibrosis, including skin fibrosis with indurated plaques, sometimes with finger-like projections, that may be erythematous, yellowish or skin-coloured. Nodules and contractures occur in more advanced disease. Internal organs that may be involved include the heart, kidney and lungs. A deep skin biopsy down to deep fascia is required for histopathological evaluation. Histological criteria for the diagnosis have been published and include increased spindle cells and compact collagen bundles [20]. As no specific treatment exists, the use of gadolinium should be avoided in patients with significantly impaired renal function.
Renal transplantation
To prevent graft rejection after renal transplantation, immunosuppressive therapy is required. Documentation of the short- and long-term consequences of immunosuppression have been obtained principally from renal transplant recipients. Infections and the development of skin tumours are the most important dermatological consequences.
Infections [21, 22] Fungi, bacteria and viruses all play an important role as causes of cutaneous infections after kidney transplantation. Infections in general are very common in renal transplant patients. They follow a predictable time course after transplantation: within the first month these are mainly donor-derived infections, infections due to surgery-related issues (e.g. wound infection) or nosocomial infections. During the following months, opportunistic infections start to predominate and viral or bacterial infections become more important. By 6 months after transplantation, many patients start to suffer from chronic and progressive infections by, for instance, human papillomaviruses (Figure153.7). Other common viral infections include herpes simplex, herpes zoster and molluscum contagiosum. The predominant bacterial infections in renal transplant recipients are impetigo, folliculitis and erysipelas. Cutaneous fungal infections mainly comprise candidosis, dermatophytosis, including onychomycosis, and pityriasis versicolor [22]. Exotic infections attributable to immunosuppression may occur following kidney transplantation, but these are relatively uncommon.
Figure 153.7 Exuberant viral warts at the oral commissures in a 37-year-old man immunosuppressed following renal transplant.
Skin tumours [23, 24, 25, 26] The incidence of malignancy in renal transplant recipients is particularly high, with skin cancer being the most frequently encountered. This topic is discussed in detail in Chapter 146.
ACQUIRED DISORDERS WITH SKIN AND RENAL INVOLVEMENT
Systemic autoimmune (connective tissue) diseases. Renal involvement is an important feature of many such diseases (see Chapters 54, 55 and 56). Although a majority of patients with systemic lupus erythematosus have renal involvement, this can be clinically inapparent. Levels of complement factors C3 and C4 can help to distinguish between active and inactive lupus nephritis: they are lower in active lupus nephritis [27]. Five percent of patients with systemic sclerosis develop scleroderma renal crisis [27]. It is characterized by severe hypertension, progressive decline of renal function and thrombotic microangiopathy.
Sarcoidosis [28] Nephrolithiasis and nephrocalcinosis can occur in patients with sarcoidosis. Hypercalcaemia may also cause renal impairment and may even precipitate acute renal failure. Renal sarcoidosis can give rise to granulomatous interstitial nephritis due to immune complex deposition. The renal involvement in many cases may, however, remain clinically silent. Rarely, cases of ureteric obstruction, voiding impairment, bladder involvement and sarcoid of the urethra have been reported.
Vasculitis (see Chapter 102) The vasculitides have recently been reclassified due to a better understanding of the different disease entities [29]. The new nomenclature comprises small-, medium-, large- and variable-vessel vasculitis, single-organ vasculitis and vasculitis associated with systemic disease or probable aetiology. Small-vessel vasculitis (SVV) can be further subclassified into antineutrophil cytoplasmic antibody (ANCA) associated vasculitis or immune complex SVV. Granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA) and eosinophilic GPA (formerly Churg–Strauss syndrome) belong to the ANCA-associated SVVs, whereas antiglomerular basement membrane (anti-GBM) disease and IgA vasculitis (formerly Henoch–Schönlein purpura) belong to the group of immune complex SVVs. Behçet disease is considered to be a variable-vessel vasculitis [29].
Renal involvement is common in a variety of vasculitides that present with cutaneous lesions and is the main cause of mortality in many of them. IgA vasculitis is a small-vessel leukocytoclastic vasculitis predominantly affecting the skin, joints, gastrointestinal tract and kidneys, especially in children between 3 and 12 years of age [30]. The renal symptoms include haematuria, proteinuria, nephrotic syndrome, renal impairment and hypertension [31]. Renal involvement (mainly rapidly progressive glomerulonephritis) is common in ANCA-associated SVV, mainly in GPA (50–80%) and MPA (90–100%). In eosinophilic GPA it is less frequent (4–51%) [32]. Renal involvement in Behçet disease may include glomerulonephritis, vascular disease, interstitial nephritis or renal failure [33].
SKIN DISORDERS THAT MAY AFFECT THE KIDNEY AND URINARY TRACT
Bullous pemphigoid Renal disease, including membranous glomerulopathy, diffuse proliferative and mesangioproliferative glomerulonephritis, has been infrequently reported in patients with pemphigoid [34].
Toxic epidermal necrolysis [35] Deteriorating renal function is a poor prognostic factor in this disorder, and is one of the parameters used in the SCORTEN prognostic score.
Epidermolysis bullosa. Renal failure was the second most common cause of death (with a mean age of 35 years) in 12% of adults with generalized severe recessive dystrophic epidermolysis bullosa (Hallopeau–Siemens syndrome) [36]. In epidermolysis bullosa, mucous membrane involvement can be associated with ulceration of the genito-urinary epithelium. Genito-urinary involvement is rare and often asymptomatic, but may clinically present as haematuria, meatal stenosis, sepsis, dysuria and hydronephrosis [37].
Skin infections.Streptococcal impetigo: post-streptococcal glomerulonephritis may occur 1–4 weeks after superficial streptococcal skin infections such as impetigo [38]. Secondary syphilis: this is a rare cause of the nephrotic syndrome [39]. Herpes zoster: if affecting the relevant dermatomes, herpes zoster may cause neurogenic bladder dysfunction leading to acute urinary retention [40].
References
Gupta AK, Gupta MA, Cardella CJ, et al. Cutaneous associations of chronic renal failure and dialysis. Int J Dermatol 1986;25:498–504.
Markova A, Lester J, Wang J, et al. Diagnosis of common dermopathies in dialysis patients: a review and update. Semin Dial 2012;25:408–18.
Kurban MS, Boueiz A, Kibbi AG. Cutaneous manifestations of chronic kidney disease. Clin Dermatol 2008;26:255–64.
Najafian B, Mauer M, Hopkin RJ, et al. Renal complications of Fabry disease in children. Pediatr Nephrol 2013;28:679–87.
Riccardi VM. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis, 2nd edn. Baltimore: Johns Hopkins University Press, 1992.
Han M, Criado E. Renal artery stenosis and aneurysms associated with neurofibromatosis. J Vasc Surg 2005;41:539–43.
Lemley KV. Kidney disease in nail–patella syndrome. Pediatr Nephrol 2009;24:2345–54.
Ferzli PG, Millett CR, Newman MD, et al. The dermatologist's guide to hereditary syndromes with renal tumors. Cutis 2008;81:41–8.
Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Couns 2009;18:13–27.
Obici L, Merlini G. Amyloidosis in autoinflammatory syndromes. Autoimmun Rev 2012;12:14–17.
Thauvin-Robinet C, Cossée M, Cormier-Daire V, et al. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet 2006;43:54–61.
Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore) 2004;83:18–34.
Real de Asúa D, Costa R, Galván JM, et al. Systemic AA amyloidosis: epidemiology, diagnosis, and management. Clin Epidemiol 2014;6:369–77.
Scarpioni R, Rigante D, Cantarini M, et al. Renal involvement in secondary amyloidosis of Muckle-Wells syndrome: marked improvement of renal function and reduction of proteinuria after therapy with human anti-interleukin-1ß monoclonal antibody canakinumab. Clinic Rheumatol 2014, doi 10.1007/s10067-013-2481-2.
Vedvyas C, Winterfield LS, Vleugels RA. Calciphylaxis: a systematic review of existing and emerging therapies. J Am Acad Dermatol 2012;67:e253–60.
Goh CL, Phay KL. Arteriovenous shunt dermatitis in chronic renal failure patients on maintenance haemodialysis. Clin Exp Dermatol 1988;13:379–81.
Weller A, Barber JL, Olsen OE. Gadolinium and nephrogenic systemic fibrosis: an update. Pediatr Nephrol 2014;29:1927–37.
Karuthu S, Blumberg EA. Common infections in kidney transplant recipients. Clin J Am Soc Nephrol 2012;7:2058–70.
Hogewoning AA, Goettsch W, van Loveren H, et al. Skin infections in renal transplant recipients. Clin Transplant 2001;15:32–8.
Bottomley MJ, Harden PN. Update on the long-term complications of renal transplantation. Br Med Bull 2013;106:117–34.
Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part I. Epidemiology of skin cancer in solid organ transplant recipients. J Am Acad Dermatol 2011;65:253–61; quiz 262.
Laing ME, Moloney FJ, Comber H, et al. Malignant melanoma in renal transplant recipients. Br J Dermatol 2006;155:857.
Le Mire L, Hollowood K, Gray D, et al. Melanomas in renal transplant recipients. Br J Dermatol 2006;154:472–7.
Kronbichler A, Mayer G. Renal involvement in autoimmune connective tissue diseases. BMC Med 2013;11:95.
La Rochelle JC, Coogan CL. Urological manifestations of sarcoidosis. J Urol 2012;187:18–24.
Jennette JC. Overview of the 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Clin Exp Nephrol 2013;17:603–6.
Piram M, Mahr A. Epidemiology of immunoglobulin A vasculitis (Henoch-Schönlein): current state of knowledge. Curr Opin Rheumatol 2013;25:171–8.
Tizard EJ, Hamilton-Ayres MJ. Henoch Schonlein purpura. Arch Dis Child Educ Pract Ed 2008;93:1–8.
Millet A, Pederzoli-Ribeil M, Guillevin L, et al. Antineutrophil cytoplasmic antibody-associated vasculitides: is it time to split up the group? Ann Rheum Dis 2013;72:1273–9.
Akpolat T, Dilek M, Aksu K, et al. Renal Behçet's disease: an update. Semin Arthritis Rheum 2008;38:241–8.
Ross EA, Ahmed AR. Bullous pemphigoid-associated nephropathy: report of two cases and review of the literature. Am J Kidney Dis 1989;14:225–9.
Blum L, Chosidow O, Rostoker G, et al. Renal involvement in toxic epidermal necrolysis. J Am Acad Dermatol 1996;34:1088–90.
Fine JD, Johnson LB, Weiner M, et al. National Epidermolysis Bullosa Registry. Inherited epidermolysis bullosa and the risk of death from renal disease: experience of the National Epidermolysis Bullosa Registry. Am J Kidney Dis 2004;44:651–60.
Kajbafzadeh AM, Elmi A, Mazaheri P, et al. Genitourinary involvement in epidermolysis bullosa: clinical presentations and therapeutic challenges. BJU Int 2010;106:1763–6.
Kambham N. Postinfectious glomerulonephritis. Adv Anat Pathol 2012;19:338–47.
Soehardy Z, Hayati SN, Rozita M, et al. Subclinical acquired syphilis masquerading as membranous glomerulonephritis. Med J Malaysia 2006;61:484–6.
Erol B, Avci A, Eken C, et al. Urinary retention, erectile dysfunction and meningitis due to sacral herpes zoster: a case report and review of the literature. Urol Int 2009;82:238–41.