NEUROMUSCULAR JUNCTION DISORDERS
Neuromuscular junction (NMJ): Composed of presynaptic, synaptic, & postsynaptic regions. Dzs affect one of these three → failure of transmission across NMJ. Fluctuating symptoms are common in NMJ dz.
Differential Diagnosis of NMJ Dysfunction
Presynaptic: Lambert-Eaton myasthenic syndrome, congenital myasthenic syndromes (CMS), botulism, tick paralysis, some drugs, & venoms.
Synaptic: CMS of end-plate AChE deficiency, cholinesterase-inhibiting drugs, organophosphates.
Postsynaptic: MG, transient neonatal MG, CMS, drugs/venoms.
MYASTHENIA GRAVIS
Epidemiology: Prevalence ~1/5,000 to 1/50,000. Annual incidence ~1/50,000–1/200,000. For age <50 yo, F > M incidence; for age >50 yo, M > F incidence.
Pathophysiology: Autoimmune (AI) d/o of synaptic transmission. Abs interfere w/ fxn, placement, or survival of nicotinic AChRs. Muscarinic receptors not affected → pupillary/autonomic responses spared. Abnormalities at the level of the thymus (intrathymic autosensitization against AChR) &/or peripheral immunoregulatory system (antigenic mimicry) lead to anti-AChR ab, disaggregation of AChR (anti-MuSK/anti-LRP4 ab), & simplification of nl, highly infolded NMJ surface. This loss of functional AChRs → decrease in end-plate potential amplitudes → muscle fiber not depolarized to threshold → NM transmission failure. Muscle-specific tyrosine kinase (MuSK) mediates postsynaptic clustering of AChR, maintains presynaptic structures (Lancet Neurol 2009;8:475–490).
Presentation: Pt history: Variable or fatigable wkns, 20% p/w pharyngeal wkns or hoarse voice, diplopia (blurred vision in subtle cases), orthopnea 2/2 diaphragmatic wkns. Neurologic exam: Nl pupillary reflexes, + ptosis (usu asymmetric & fatigues w/ upgaze), asymmetric ophthalmoparesis, hypercontracted frontalis to maintain eye opening, Cogan lid twitch, “myasthenic snarl,” transverse pucker. Wkns of jaw closure, bulbar muscles, palate, axial & prox limbs, neck flex > ext. Tachypnea & shallow respirations, intact sensation, variably depressed reflexes.
Other subtypes: (1) Anti-MuSK: Young F w/ prominent oculobulbar, neck, shoulder, & ventilatory muscle wkns, frequent crises, & typically not assoc w/ thymoma, +/− triple furrowing of tongue. (2) Ocular: Sx limited to ocular wkns for >2 yr → 90% do not generalize; electrodiagnostic challenge; mono-Rx w/ AChEIs may be sufficient; immunosuppression may have a role. (3) Anti-LRP4 (low-density lipoprotein receptor-related protein): Effects on AChR clustering & agrin-LRP4 interaction could lead to the identification of a new MG subtype. Pts appear to respond to immune suppression. Anti-agrin also under investigation (Arch Neurology 2012;69:445–451; Neuromuscul Disord 2013;23:568–570). (4) Double Ab negative (AChR & MuSK): Classic presentation, neg ab tests.
Ddx: Motor neuron dz (MND), Lambert-Eaton myasthenic syndrome (LEMS), GBS, diphtheria, tick paralysis, thyroid ophthalmopathy, mitochondrial d/os, CMS, botulism, organophosphate & other toxins, inflammatory myopathy, skull-based tumors, cholinergic crisis, oculopharyngeal muscular dystrophy, AIDP.
Dx: (1) Ab testing. (1a) AChR abs: Binding Ab: Se 80%–85% in generalized MG, Se 55% in ocular MG. Blocking Ab: + in isolation in ~1% of MG. Modulating ab: + in 3%–4% MG in isolation, ↑ freq w/ thymoma. At presentation, AChR Ab can be negative and then become detectable later in the course of dz. Ab level does not indicate severity. (1b) Anti-muscle–specific tyrosine kinase abs (Anti-MuSK): Present in 40%–50% of MG pts w/o AChR Abs. (1c) Striated muscle ab: Se 75%–95% for MG w/ thymoma; often present in older MG pts w/o thymoma; utility limited by poor Sp. (1d) Anti-LRP4: 7–33% of double seronegative (MuSK/AChR) pts are +. Not yet commercially available. (2) EMG/NCS: Repetitive nerve stimulation: Low rates of rep stim (2–5 Hz) deplete ACh → decrement >10% (Se 30%–98% for generalized MG & 10%–40% for ocular MG). SFEMG: + blocking &/or increased jitter (variation in contraction time b/n muscle fibers) (Se 80%–99%) (Muscle Nerve 1992;15:720–724). (3) Bedside evaluation. (3a) Tensilon test (edrophonium, an AChEI): IV test dose 2 mg, if no response after 30 s, administer 2 mg at a time up to 10 mg every 10–15 s. Follow clinically observable deficit & only accept unequivocal improvement as (+). Abort if side effects. Onset 30 s, lasts 5–10 min. Side effects: Salivation, sweating, nausea, cramping, fasciculations, bradycardia, ↓ BP, & resp distress. AMBU bag/code cart & atropine at bedside, telemetry. Utility: Se ~90%. (3b) Ice test: Put ice pack on closed eye × 3 min—ptosis improves by at least 2 mm (Se 80%–97% in pts w/ ptosis, use in pts w/ contraindication to Tensilon test) (Neuromuscul Disord 2006;16:459–467; Muscle Nerve 2013;48:902–904).
Additional testing: TFTs, Chest CT, or MRI to eval thymoma.
Prognosis: Prognosis varies & depends on underlying dz distribution & severity. Early in course, exacerbations are often severe, but w/ Rx remissions lasting many months are common. Max dz severity reached in 2 yr in 66% pts. Spontaneous & durable remission in 10%–15%. On avg, active dz lasts 7 yr followed by 10 yr of relative quiescence, then finally “burned-out” phase characterized by some degree of fixed deficit. 15%–40% of pts w/ an isolated ocular presentation will not develop generalized MG. Of those that do, transition to generalized MG is usually within 2 yr (Ann Neurol 1983;14:516–519).
Treatment: Acute Exacerbations/Crisis
Definitions: Myasthenic crisis—Acute flare of wkns generally w/ respiratory failure, often requiring intubation & ICU admission or delay of extubation ≥24 h after surgery.
Causes: (1) Dz progression, (2) Infxn/stressor, (3) withdrawal/↓ in MG Rx, (4) contraindicated medication.
Plasma exchange (PE): Requires large-bore catheter & performed qod. Potential side effects: Line infection, cardiovascular instability, pneumothorax, electrolyte shifts, bleeding.
IVIg: 2 g/kg in divided doses over 2–5 days. Potential side effects: Aseptic meningitis, flulike symptoms (n/v, myalgias, fevers, chills), ARF, CHF exacerbation, allergic Rxn (test IgA levels prior to r/o IgA deficiency), headache common, very rare thrombotic events (MI/stroke).
Pyridostigmine: In intubated pts, might avoid due to increase in secretions. If not intubated, initial adult dose 15–30 mg tid–qid; gradually titrate as necessary (max 360 mg daily). Onset ~30–60 min—pts w/ dysphagia can take 30 min before meal.
Steroids: In intubated pts, can start IV methylprednisolone together w/ PE or IVIg at 500–1,000 mg/day for 5 days followed by 0.8 mg/kg IV daily until able to take PO steroids. If not intubated, initiate prednisone at a dose of 1–1.5 mg/kg/day × 2–4 wk (max 100 mg/day), then taper by 5–10 mg q2–4 wk to lowest tolerated dose. May worsen exacerbation acutely; thus, should use caution & consider hospitalization for monitoring and/or starting PE or IVIg prior to initiating steroids during an exacerbation. Onset of action ≥2 wk.
Supportive care: Aspiration precautions, temporary NPO or NG tube feeding, close resp monitoring w/ NIF & VC, ventilatory support if necessary.
Differential: Cholinergic crisis (salivation, lacrimation, diarrhea, bradycardia). Rx: hold or ↓ AChE meds.
Chronic Treatment
Principles
Time to benefit: (1) Immediate = cholinesterase inhibitors. (2) Very rapid (days) = plasma exchange, IVIg. (3) Short term (2–6 wk) = corticosteroids. (4) Long term (2–12 mo) = mycophenolate mofetil, azathioprine, rituximab, thymectomy.
Prior to initiation of immunosuppression: Negative PPD, baseline CBC w/ diff, CMP, HbA1c, bone density prior to prednisone.
Nonimmune modulatory: Pyridostigmine. Dosing adjusted PRN. Ocular symptoms: Prism glasses.
Immune modulatory: Steroids, steroid-sparing agents, & thymectomy (Amato AA, Russell JA. Neuromuscular Disorders. McGraw Hill, 2008:457–528).
- Steroids: Initiate at a dose of 1–1.5 mg/kg/d × 2–4 wk (max 100 mg/day). Once in remission or improvement plateaus, ↓ dose &/or administration interval (e.g., decrease by 5–10 mg q2–4 wk) to lowest tolerated. Side effects: Cataracts, HTN, osteoporosis, immune suppression, ↑ wt, mood d/o, psychosis, skin sensitivity to sun, glucose intolerance/DM, peptic ulcers, avascular necrosis, steroid myopathy, growth stunting in children.
- Thymectomy: Indicated if thymoma on CT chest (~15% cases). May be done in absence of thymoma in generalized MG, AChR ab+, & onset <50 yo (randomized controlled clinical trial ongoing [MGTX]).
- Steroid-sparing immune therapies.

Medications exacerbating MG: (1) Absolutely contraindicated: Penicillamine, botox telithromycin, IFN-α. (2) Relatively contraindicated: Antibiotics: fluoroquinolones (ciprofloxacin, levofloxacin, etc.), macrolides (erythromycin, azithromycin, etc.), aminoglycosides (gentamicin, tobramycin, etc.), quinine, class IA antiarrhythmics (procainamide, quinidine, lidocaine, etc.), iodinated contrast dye, magnesium, neuromuscular blocking agents. (3) Use w/ caution: Ca channel & β-blockers, Li, statins, steroids.
LAMBERT-EATON MYASTHENIC SYNDROME
Epidemiology: Second most common NMJ d/o. Autoimmune (AI), presynaptic d/o; 2/3 are paraneoplastic (small cell carcinoma ~90% paraneoplastic cases; sx precede tumor by ~10 mo avg). Nonparaneoplastic: often young F w/ other AI dz. ~85% LEMS cases >40 yo.
Pathophysiology: Abs to P/Q-type >> N-type voltage-gated channel on presynaptic membrane → low Ca entry & fewer vesicles released into synapse.
Si/sx: (1) Fatigable wkns; fatigue >> wkns. (2) Proximal LE followed by UE wkns (UE wkns in 80% at some point in dz). (3) Muscle aching/stiffness after exercise. (4) Heat → ↑ wkns. (5) Ocular sx, dysarthria, dysphagia (less common than in MG). (6) Autonomic sx: Xerostomia, xerophthalmia, blurred vision, constipation, urinary hesitancy, impotence.
Exam: Sluggish pupillary rxn. Ptosis may improve w/ upgaze. Wkns worst on initiation of movement, then potentiates, but may fatigue w/ prolonged effort. DTRs: ↓ initially but ↑ following brief sustained effort.
Dx: (1) Serum: Abs for P/Q-type voltage-gated Ca channels in 85%–90%. Abs for N-type Ca channel in 74% of paraneoplastic & 40% of nonparaneoplastic pts. AChR ab in 13%. (2) Electrodiagnostics: CMAP amplitudes diffusely reduced, decrementing CMAP w/ 1–5 Hz RNS, amplitude facilitation w/ 10 s exercise or fast rep stim. EMG: Low amplitude, short motor units that may increase w/ sustained effort. SFEMG: Jitter ± blocking in all/most muscles (not patchy like MG) (N Engl J Med 1995;332:1467–1474).
Rx: (1) Resection of tumor: Can be curative; if no known tumor → close surveillance. (2) Symptomatic Rx: (2a) AChEI (see MG Rx for details); response variable. (2b) Guanidine: ↑s ACh released but side effects limit use—myelosuppression, renal & hepatotoxicity, GI discomfort. (2c) 4-DAP: Use limited by CNS toxicity → sz, confusion. (2d) 3,4 DAP: Not FDA approved; clinical trials ongoing (Jacobus & Catalyst Pharmaceuticals); available on compassionate use basis. (3) Plasmapheresis: ↓s sx w/ transient effect peaking at 2 wk. (4) IVIg: Studied only in small studies suggesting transient effect. (5) Other immune-modulating therapy may be helpful (see MG section).
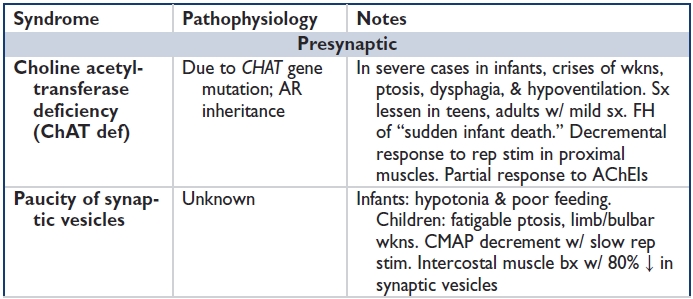
CONGENITAL MYASTHENIC SYNDROMES
Epidemiology: Rare group of syndromes 2/2 genetic defects in NMJ structure & fxn. Typically in neonates-children, but can occur in adolescents/young adults. Transient neonatal myasthenia gravis is not CMS; rather, it is Ab-mediated MG from transfer of mother's abs during pregnancy.
Pathophysiology: Genetic defects → ↓ ACh release (presynaptic), ↓ AChE (synaptic), or ↓ AChR activity (postsynaptic). All → ↓ synaptic transmission or ↓ reserve. Not Ab mediated (immune therapy not indicated).
Clinical features: Neonates-children: Reduced fetal movements, hypotonia, underdeveloped muscles, arthrogryposis, resp crisis, pupillary involvement in congenital AChE deficiency, progressive myopathy, & no response to AChEIs in AChE def & slow channel congenital myasthenic syndrome (SCCMS). Neonates may have high-arched palate, facial dysmorphism, arthrogryposis, scoliosis. Adolescents-adults: Difficult to clinically distinguish from AI MG. Myopathy sometimes present.
Ddx: Infants-young adults w/ MG, seroneg MG, myopathy, peripheral neuropathy, MND.
Dx: Ab testing always neg in CMS; next-generation (whole exome) sequencing can be used to screen for congenital myasthenic syndromes.
Rx: Symptomatic Rx depends on the entity (see table). Some respond to AChEIs, albuterol, ephedrine, 3,4-DAP. No role for immunotherapy; genetic counseling advisable (Curr Opin Neurol 2013;26:561–568).
TOXINS THAT CAUSE NMJ DYSFUNCTION
Presynaptic: Botulinum toxin, corticosteroids, Mg, aminoglycosides, calcium channel blockers, aminopyridines, hemicholinium-3; tetrodotoxin, saxitoxin, ciguatoxin, venoms (Mamba, Australian Tiger snake, Pandinus scorpion, Conus marine snails, multibanded krait, Brazilian rattlesnake, black/brown widow spiders), ticks.
Synaptic: AChEIs (e.g., edrophonium, pyridostigmine, neostigmine), organophosphates.
Postsynaptic: Curare & nondepolarizing agents, succinylcholine & depolarizing agents, tetracyclines, venoms (e.g., banded kraits, Siamese cobra, Conus marine snail).
Botulism
Overview: Rare, toxin-mediated illness 2/2 blockade of NMJ & cholinergic presynaptic neurons. Exotoxin produced by Clostridium botulinum; absorbed via GI tract or wound & spread hematogenously; binds to the presynaptic nerve & internalized via endocytosis.
Si/Sx: Onset over hrs (days for wound botulism); n/v, ± diarrhea, dysphagia, diplopia, dysarthria, wkns of extremities, SOB, xerostomia, blurred vision, constipation. In infants: Variable sx (mild to sudden death); listless, ↓ spontaneous mvmts, ↓ suck, hypotonia (“floppy baby”), ↓ DTRs. Drooling, CN wkns worrisome; ~50% → ventilation. Wks to recovery.
Exam: Ptosis, ↓ gag, dysphagia, dysarthria, facial diplegia, tongue wkns, ± nystagmus. ↓ DTRs, ↓ FVC. Autonomic: Ileus, urinary retention, ↓ pupillary response, ↓ BP, ↓ temp.
Dx: (1) EMG/NCS: ↓ CMAP amplitude; ↑s incrementally to fast rep stim, ↓s decrementally to slow rep stim (most prominent in proximal muscles). EMG variable depending on timing in course: ± pos sharps & fibs, ± MUAPs low amp/short duration, ± early recruitment, & ↓'d interference pattern. (2) Culture: Se ~50%–66% but results take time. (3) Toxin assay: Se ~35%; can be done on gastric contents, stool, serum, wound aspirate, & suspected foods.
Rx: (1) Supportive: ↓ in mortality. (2) Antitoxin: If administered w/in 24 h of sx onset. (3) Human botulinum IgG: ↓s hospitalization duration & ventilation in infantile botulism; no role in adults. (4) GI lavage: Foodborne & infantile botulism (controversial). (5) Wound debridement: For wound botulism. (6) If abx initiated, first give antitoxin to avoid sx worsening as bacteria lyse. (7) Foodborne illness: identify others exposed & test food.
Tick Paralysis
Epid: Children > adults (3:1). M > F. Peak incidence in spring & summer.
Pathophysiology: Varies by species; Ixodes holocyclus (Australia) ↓s ACh release presynaptically. Other tics cause NMJ dysfunction or acute neuropathy.
Si/sx: Ascending wkns over hours-days; ophthalmoplegia, facial wkns, dysarthria, dysphagia, resp failure, sensory sx (pain, numbness, paresthesias, sensory ataxia); ↓ DTRs.
Ddx: MG, GBS, diphtheria, botulism.
Dx: Serology & CSF nl. EMG/NCS: possible mild sensory neuropathy. CMAP amplitudes & motor conduction velocities mildly ↓s in paretic limbs. Pseudofacilitation on rep stim. EMG: abnl recruitment.
Rx: Remove tick; supportive Rx for resp wkns; prognosis favorable if pt supported.