Chapter 22
Biology of Gap Junctions
Chapter Outline
II. Advantages of Electrical Synapses in Excitable Cells
III. Ubiquitous Membrane Permeable Junctions
IV. Structural Candidates for the Permeable Cell Junction
V. Ultrastructural Characterization of Gap Junctions and Correlations with Cell Coupling
VI. Molecular and Structural Studies of Gap Junction Proteins
VII. Two Large Families of Gap Junction Proteins
VIII. Channels within Gap Junctions
IX. Evidence for Charge Selectivity
X. Channel Properties of Different Connexins
XI. Gating by Ions and Second Messengers
XII. Regulation of Functions of Connexin-Based Gap Junctions at Multiple Levels
XIII. Specific Biological Functions of Gap Junctions
XIV. Gap Junctions in Human Disease and in Murine Models of Human Disease
I Introduction
In the late 1950s and early 1960s, physiologists who had been poking fine glass current-injecting and current-recording electrodes into neighboring cells within a variety of tissues made an interesting discovery. They found that while the injection of current into a cell caused a predictable shift in its non-junctional membrane potential, it also caused a similar shift in the non-junctional membrane potential of immediately adjacent cells (Fig. 22.1).
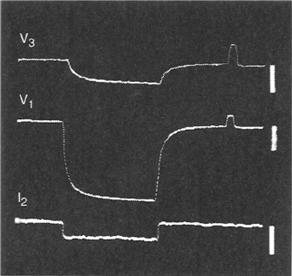
FIGURE 22.1 The injection of current into one cultured WI-26 cell (I2) within a monolayer produces a shift in potential within the injected cell (V1) and in a cell two or three cells removed from the injected cell (V3), indicating that ions may move freely between contacting cells. (From Furshpan, E.J. and Potter, D D. (1968). Low-resistance junctions between cells in embryos and tissue culture. Curr Top Dev Biol. 3, 95, with permission.)
One interpretation of this finding was that the ions emanating from the injection electrode were able to flow freely from the injected cell to the adjacent cell and did so in preference to pathways leading to the extracellular medium or to the intercellular space. Moreover, several studies demonstrated that fluorescent dyes were selectively transferred from cell to cell when injected into the cytoplasm through glass injection pipettes. Many observations such as these led to the hypothesis that some cells were coupled (electrically or with respect to dye transfer) by permeable cell junctions.
Additionally, in the neuronal tissues in which this phenomenon had first been witnessed, it was found that action potentials generated in a presynaptic element could be passed to a postsynaptic element much faster than would occur if the presynaptic and postsynaptic elements were connected by chemical synapses (Fig. 22.2). Ironically, this evidence for the presence of permeable cell junctions that could serve as electrical synapses in the nervous system came to light soon after the common acceptance of Otto Loewi’s findings that supported the idea that neuronal synapses were probably chemical in nature and not electrical as traditionally believed.
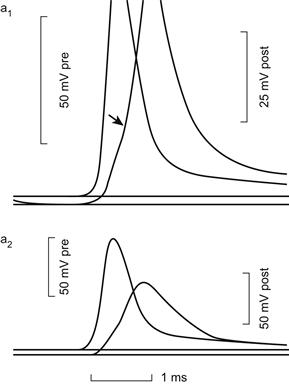
FIGURE 22.2 The upper trace is an AP in the presynaptic element of a crayfish septate axon; the lower trace shows the subsequent AP in the postsynaptic element. The short lag of the latter following the former is typical of electrical synapses. The top and bottom traces were recorded at the same synapse at different amplifications. (From Furshpan, E.J. and Potter, D.D. (1959). Transmission at the giant motor synapses of the crayfish. J. Physiol. 145, 289, with permission.)
II Advantages of Electrical Synapses in Excitable Cells
The utility of electrical synapses in excitable cells is apparent. They may pass action potentials or subthreshold electrical activity more rapidly from one neuronal element to another than can their chemical counterparts. This advantage is especially obvious in neuronal pathways that serve as escape mechanisms, such as those in the tail muscles of crayfish and lobsters and those in the pectoral fins of fishes. In these cases, the rapidity of the animal’s response to imminent danger has selective value. On the other hand, such permeable junctions connecting smooth muscle cells of the uterus or cardiac muscle cells of the heart wall, provide a mechanism for the systematic cell-to-cell spread of depolarization that is required for coordinated and effective contractile activity.
III Ubiquitous Membrane Permeable Junctions
Numerous studies published in the 1960s and 1970s supported the idea that virtually all cells in normal tissues (even those in inexcitable tissues) were coupled by permeable cell junctions. Based on studies with native biological molecules and with tracers of different sizes, it was also suggested that the pores within vertebrate cell-to-cell junctions were approximately 1.2 nm in diameter and that molecules larger than about 1 kDa were excluded. Thus, it was suggested that the biological molecules capable of freely moving from cell to cell in normal tissues would include a wide range of ions and small metabolites including sugars, nucleotides and nucleosides and possible signaling molecules such as cyclic adenosine monophosphate (cAMP).
Since the pores initially appeared to select molecules only with respect to size, it was suggested that one function of permeable cell junctions was to buffer the concentrations of small metabolites throughout the tissue. Direct evidence for the cell contact-mediated exchange of metabolites, for example, was provided by metabolic cooperation experiments in which it was shown that a normal wild-type cell, able to incorporate thymidine into DNA, could transfer DNA precursor molecules (probably the nucleoside triphosphate form of thymidine) to mutant thymidine kinase-deficient cells only if the wild-type and mutant cell were in contact. In these “kiss-of-life” experiments, the mutant cells were then able to incorporate this nucleoside into DNA required for continued proliferation and survival.
IV Structural Candidates for the Permeable Cell Junction
As electron microscopes first came into common use in the late 1950s, several different kinds of cell–cell junctions were discovered. The tight junction, or occludens-type junction, was characterized by the fusion of membranes of adjacent cells and so held some appeal as a potential permeable cell junction. Septate junctions were also good candidates for the permeable cell junction since they were characterized by cell-to-cell bridges or septa covering a large region of cell–cell apposition in many of the invertebrate tissues shown to be well coupled by electrophysiological or dye-tracing techniques. The basic argument against their function as permeable cell junctions, however, was that their distribution among tissues and organisms was significantly more limited than was the coupling phenomenon. The septate junction, for example, could not be identified in coupled vertebrate cells. The apparent fusion of plasmalemma between juxtaposed cells in mammalian smooth and cardiac muscle was termed the nexus in the early 1960s and was demonstrated to be the site of action potential propagation when hypertonic solutions that separated the nexal membranes also blocked action potential propagation.
V Ultrastructural Characterization of Gap Junctions and Correlations with Cell Coupling
In a pioneering study, Revel and Karnovsky (1967) infiltrated the intercellular spaces of heart and liver with an electron opaque dye (lanthanum hydroxide) and then examined these preparations in the electron microscope. They found regions where the membranes of adjacent cells were apposed to one another across a uniform lanthanum-infiltrated intercellular space about 2–4 nm in width (Fig. 22.3B). In addition, in cross-section, they observed small unstained structures bridging the stain-filled gap between adjacent cells in these regions (Fig. 22.3A) and, in en face views, these bridges were packed hexagonally within the intercellular space. Ultimately, freeze-fracture studies provided evidence that these intercellular bridges were in continuity with structures that spanned the lipid bilayers of both cells and thereby could theoretically provide the structural foundation for the cell-to-cell conduit implied by earlier electrophysiological studies.
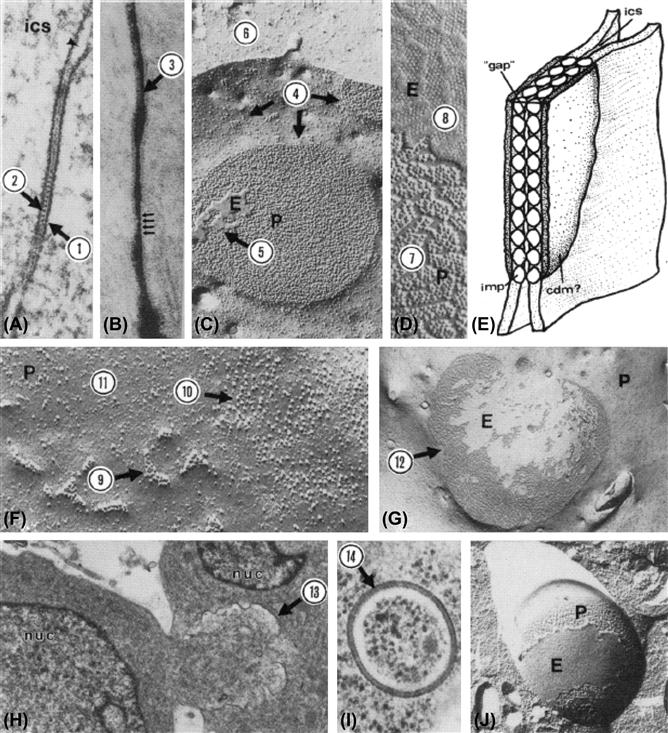
FIGURE 22.3 (A) Thin section of typical gap junction. The entire width of both apposed membranes and the intercellular space is about 18 nm. Dense material is often associated with cytoplasmic surface of gap junction (1). Some gap junctions are characterized by stained periodicites evident at level of “gap” (2). Magnification, 157 500×. (From Larsen, W.J., Skowron-Lomneth, C. and Carron, C. (1988). Gap junction modulation: possible role in tumor cell behavior. In (H.A. Milman and E. Elmore, eds), Biochemical Mechanisms and Regulation of Intercellular Communication, Vol. 14, p. 151. Princeton Scientific Publ. Co., Princeton, NJ, with permission.) (B) Lanthanum infiltrated gap junction. The lanthanum infiltrated intercellular “gap” (3) is about 2–4 nm. Magnification, 212 400× (Reproduced from The Journal of Cell Biology, 1975, vol. 67, p. 801, by copyright permission of the Rockefeller University Press.) (C) Freeze-fracture image of a gap junction. Each junctional membrane contains a set of particles and a set of pits. In vertebrate tissues, the particles adhere to the protoplasmic leaflet of the lipid bilayer (protoplasmic fracture face, P), whereas the pits remain associated with the extracellular leaflet (extracellular fracture face, E). Gap junction particles aggregate within particle-poor zones (4). The fracture may jump between the two membranes, resulting in adhesion of bits of the E-fracture face from the membrane of the adjacent cell (5) on the P-fracture face or vice versa. Occasionally, the fracture plane leaves the membrane and cuts into the cytoplasm (6). Magnification, 64 800×. (From Larsen, W.J. (1977). Structural diversity of gap junctions: a review. Tissue Cell, 9, 373, with permission.) (D) Enlargement of P- and E-fracture faces showing details of gap junction particles (7) and corresponding pits (8). Magnification, 139 000×. (From Larsen, W.J. (1977). Structural diversity of gap junctions: a review. Tissue Cell, 9, 373, with permission.) (E) Simple model of gap junction showing particle-particle (imp) contact within the “gap” at the level of the intercellular space (ics). Cytoplasmic dense material (cdm) is particularly apparent in gap junctions in excitable tissues. (From Larsen, W.J. (1977). Structural diversity of gap junctions: a review. Tissue Cell, 9, with permission.) (F) Gap junctions form by the aggregation (9) of single 11-nm particles (10) in particle-poor regions of the membrane (11). Magnification, 222 300×. (From Larsen, W.J. and Risinger, M.A. (1985). The dynamic life histories of intercellular membrane junctions. In (B. Satir, ed.), Modern Cell Biology, Vol. 4, p. 151. John Wiley & Sons, New York. Copyright 1985 John Wiley & Sons. Reprinted by permission of Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.) (G) Very large gap junctions (12) containing thousands of gap junction particles may represent terminal gap junction gaps. Magnification, 21 600×. (From Larsen, W.J. (1977). Structural diversity of gap junctions: a review. Tissue Cell, 9, 373, with permission.) (H) The large gap junction caps may invaginate into the cell (13) through an endocytotic mechanism. Magnification, 14 400×. (From Larsen, W.J. and Tung, H. (1978). Origin and fate of gap junction vesicles in rabbit granulosa cells. Tissue Cell, 10, 585, with permission.) (I) Bimembranous gap junction vesicles (14) may pinch off from the invaginating junction to fuse with lysosomes before undergoing degradation. Magnification, 52 200×. (From Larsen, W.J. and Tung, H. (1978). Origin and fate of gap junction vesicles in rabbit granulosa cells. Tissue Cell, 10, 585, with permission.) (J) Freeze-fracture planes within the cytoplasm may reveal the P-face particles (P) and E-face pits (E) within cytoplasmic gap junction vesicles. Magnification, 38 700×. (From Larsen, W.J. and Tung, H. (1978). Origin and fate of gap junction vesicles in rabbit granulosa cells. Tissue Cell, 10, 585, with permission.)
It was also very important that this cell–cell junction, which Revel and Karnovsky called the gap junction, was found in many tissues which were ion- or dye-coupled, including both inexcitable and excitable cells. While well-coupled inexcitable cells, such as liver hepatocytes, possessed large numbers of gap junctions, it was especially satisfying to find that these structures were prominent features of the cell–cell contact regions of excitable cells, such as cardiac myocytes, and were prominent in neuronal systems that had been shown to possess electrical synapses. In addition, in the metabolic cooperation experiments described above, DNA precursor molecules were transferred between test cells only if they were capable of forming gap junctions in regions of cell–cell contact. Moreover, a large body of ultrastructural evidence indicated that gap junctions were components of virtually all tissues in multicellular organisms of the animal kingdom (See Fig. 22.3C–J).
VI Molecular and Structural Studies of Gap Junction Proteins
Application of biochemical and molecular approaches to the study of gap junctions proved to be more perplexing than originally envisioned, but tenacious investigators in a number of laboratories were able to overcome initial obstacles. It is now known that proteins that make up gap junctions in vertebrate tissues are basic and contain significant stretches of hydrophobic sequence. This is not surprising given the fact that these are integral membrane proteins. For these reasons, they proved difficult to isolate and purify. Extraction of tissues with boiling sodium dodecyl sulfate (SDS) proved to be the only reliable method for obtaining morphologically pure fractions of gap junction membranes for many years, but the proteins extracted from these fractions ran at variable molecular weights on polyacrylamide gels. It was controversial as to whether some of these bands were breakdown products or other proteins associated with gap junctions in the cell membrane.
It was not until gentler, non-detergent extraction procedures were developed in the early 1980s that a consistent band on polyacrylamide gels with a predicted molecular weight of 26 kDa could be routinely isolated from rodent livers. Once this native protein was obtained and purified, it was possible to produce a polyclonal antibody. This antibody was used to screen an expression cDNA library to isolate and sequence its gene. This first sequence was published in 1986. Hydropathy analysis of the deduced amino acid sequence of this cDNA revealed a 32-kDa protein which could be interpreted as possessing four potential transmembrane regions, two extracellular loops and an internal loop and amino and carboxyl termini that extended into the cytoplasm (Fig. 22.4). The mapping of these proteins with antibodies directed against specific sequences and their cutting with specific proteases have largely confirmed this initial interpretation of the relationships of gap junction protein segments to the membrane. It was also postulated that six gap junction protein molecules, called connexins, constitute each gap junction intramembrane particle (IMP) or connexon, thus providing a molecular basis for models of the permeable membrane junction deduced from earlier physiological and structural studies (Figs. 22.4 and 22.5). Indeed, more recent electron cryomicroscopy studies of frozen, rehydrated 2D crystals of a recombinant C-terminal truncated form of Cx43 has revealed even more detail regarding the relationship of connexins to the connexon and the relationship of their specific transmembrane domains to the plasma membrane. These studies show that each connexin subunit contributes a transmembrane α-helix that lines the aqueous pore and an additional α- helix adjacent to the surrounding membrane lipids. In addition, the images reveal that the apposing connexons that form each channel are staggered by about 30°.
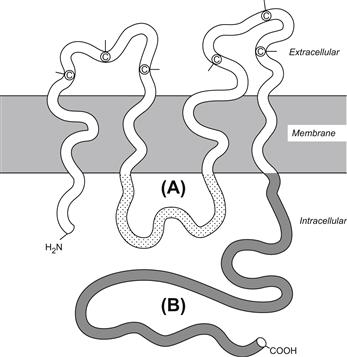
FIGURE 22.4 Structure and topology of a connexin. Both amino and carboxyl termini (B) are located in the cytoplasm along with an intermediate loop (A). Every connexin contains four membrane-spanning regions and two extracellular loops containing highly conserved cyteine residues (C). (From Beyer, E.C., Paul, D.L. and Goodenough, D.L. (1990). Connexin family of gap junction proteins. J. Membr. Biol., 116, 187, with permission.)
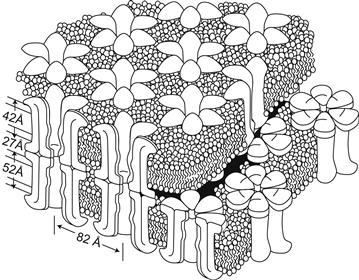
FIGURE 22.5 This diagram of a region of a gap junction isolated from mouse liver is based on electron microscope images and x-ray diffraction data. Each gap junction particle or connexon is composed of six gap junction protein molecules called connexins. Connexons in apposed membranes meet within the intercellular space. (Reproduced from The Journal of Cell Biology, 1977, 74, p. 449, by copyright permission of the The Rockefeller University Press.)
VII Two Large Families of Gap Junction Proteins
Early structural studies, particularly those utilizing the freeze-fracture technique, demonstrated structural diversity among gap junctions distributed throughout the animal kingdom and within a variety of different tissues. The phylogenetic or species variability in some of these structural features was shown to depend upon the tissue in which the gap junction was identified. This variability in the detailed gap junction structure and composition was not initially understood, although it is now known that gap junctions in different tissues, phyla and species are formed by different protein isoforms within two large families of gap junction proteins. The gap junctions in vertebrate tissues are formed by members of the connexin family of gap junction proteins. The gap junctions of invertebrate tissues appear to be formed by members of an analogous innexin family of gap junction proteins.
Members of the connexin family of gap junction proteins range in molecular weight from 23 kDa to 62 kDa (Table 22.1). Each connexin is named for its deduced molecular weight in kilodaltons so that the 26-kDa connexin in humans, for example, is called human connexin 26 (human Cx26), while the 30.3-kDa connexin in mice is called mouse connexin 30.3 (mouse Cx30.3). Sequence analysis supports the possibility that at least two major phylogenetic subfamilies of connexins diverged from each other between 1.3 and 1.9 billion years ago; one group ranging in size from about 26 kDa to 32 kDA (β-group), another from about 33 kDa to 62 kDa (α-group) (Fig. 22.6A). Some lesser divergent connexin subfamilies are also indicated (δ/γ/ε). Twenty different connexin genes have been identified in rodents (e.g. mice), 21 in humans, although transcription of the Cx23 gene has not been demonstrated in humans or mice and the functional expression of some connexins have yet to be described (e.g. Cx25, Cx33).
TABLE 22.1. The Connexin Gene and Protein Family1

1 Adapted from Beyer, E.C. and Berthoud, V.M. (2009). The family of connexin genes. In Connexins: A Guide, p. 8, with permission. Copyright 2009 Humana Press.
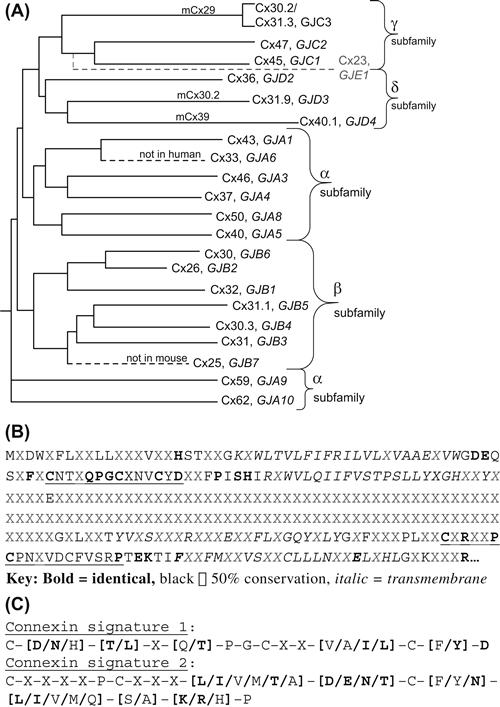
FIGURE 22.6 (A) Dendrogram of the mammalian (human, mouse) connexin family tree constructed by aligning the protein sequences for the known connexins using the nomenclature adapted at the 2007 International Gap Junction Conference (see Table 22.1). (Adapted fromSáez, J.C., Berthoud, V.M., Brañes, M.C., Martínez, A.D. and Beyer, E.C. (2003). Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev., 83, 1363, with permission. Copyright 2003 American Physiological Society.) (B) Consensus sequence for the 20 human connexins (excluding Cx23) illustrating the 50 to 100% conservation of amino acids residues within the amino terminal, first and second extracellular loop and four transmembrane domains with limited sequence homology within the cytoplasmic loop and carboxyl terminal domains. (C) The two connexin signature sequences derived from the consensus sequence underlined in panel B. Cx31 and Cx23 do not match these signature sequences. (Adapted from Beyer, E.C. and Berthoud, V.M. (2009). The family of connexin genes. In Connexins: a Guide, p. 8, with permission. Copyright 2009 Humana Press.)
Typically, it appears that two or more connexins comprise the gap junctions within any given tissue. For example, Cx26 and Cx32 constitute gap junctions of mammalian liver and have been shown to coexist within the same gap junction aggregates. On the other hand, the Cx32 and Cx43 documented in thyroid epithelium, appear to be segregated into separate gap junctions formed in different regions of the lateral cell membrane. Likewise, the distribution of three different connexins (Cx40, Cx43 and Cx45) within the human heart appears to be non-random. For example, Cx45 is the first connexin to appear in the embryonic mouse heart. Ultimately, however, its expression is restricted to the region of the sinoatrial (SA) node, while Cx40 is localized in the atrioventricular (AV) node and in myocytes of the atrium and conductive myocardium. In contrast, Cx43 expression occurs within the crista terminalis and other cardiomyocytes of the heart. As many as six different connexin genes (Cx30.1, Cx31, Cx31.1, Cx40, Cx43 and Cx45) are transcribed and translated in mouse embryos as early as the eight-cell stage. These studies imply differential functions for gap junctions composed of different connexins and consequently stimulate the following question: what part of the connexin molecule encodes functional specificity and how do these specificities differ from connexin to connexin?
In general, the amino termini, transmembrane helicies and extracellular loop cysteine-containing regions of the connexins are highly conserved (see Figs. 22.4 and 22.6B). However, the cytoplasmic loops and cytoplasmic carboxyl termini vary significantly in length and amino acid sequence from one connexin to another. The two triple cysteine-containing segments form the two identified connexin signature sequences less Cx23 which contains only two Cys residues within each extracellular loop (see Fig. 22.6C). Heterotypic gap junctions can be formed only with connexons made of particular connexins, suggesting that sequence specificity within the extracellular domains may be relevant to gap junction assembly. Recent studies, therefore, have focused on the potential functional relevance of specific differences in amino acid sequences within the various domains of the connexin proteins (see Sections IX–XIV).
In invertebrates, proteins of the innexin family are able to form gap junctions based on ultrastructural criteria. In addition, studies with a Xenopus oocyte expression system have shown that some of the innexin proteins are able to form permeable cell-to-cell channels that may be voltage- or pH-sensitive. As many as 24 innexin genes have been identified in the nematode, Caenorhabditis elegans, while six innexin genes have been described in Drosophila. While the innexins bear no sequence homology to the connexin family of proteins, the structural similarities of innexins and connexins are dramatically similar. For example, like the connexins, the innexin proteins have four transmembrane domains, two extracellular loops and cytoplasmic N- and C-termini. Moreover, the functions of the innexin proteins in invertebrates parallel functions of the connexins in vertebrate tissues. For example, the innexin encoded by the shal-B(neural) locus forms electrical synapses of the Drosophila nervous system, while the innexin encoded by the eat-5 locus forms permeable junctions responsible for synchronization of pharyngeal musculature in Drosophila. There is no signature consensus amino acid sequence common to the Arthropoda, Nematoda, Annelida and other invertebrate phylogeny as there is for the connexins. The innexin extracellular loops contain only two cysteine residues each, in contrast to three for the connexins. Three innexin-related genes are found in the vertebrate genome and these are now referred to as the pannexins (panx1, panx2, panx3). Only pannexin1 is capable of forming homologous intercellular channels in Xenopus oocytes, suggesting the possibility that pannexins do not form vertebrate gap junctions. Alternatively, it was hypothesized that pannexins form ATP-releasing hemichannels in complex with the purinergic P2X7 receptor. Evidence for functional panx1/P2X7 hemichannels in astrocytes and other mammalian cells has received considerable support in recent years, suggesting a more patho/physiological role for pannexins in paracrine signaling pathways than conventional gap junctional communication.
VIII Channels within Gap Junctions
Following the identification of the gap junction as the site of cell-to-cell transfer of ions and small hydrophilic molecules, a hypothesis developed that the pathway for this exchange consisted of an array of parallel aqueous channels. The central aqueous pore resides in each IMP particle in the freeze-fracture images or hexagonal bridge in the negatively stained micrographs of a gap junction plaque. An aqueous pore 16 nm in length with a channel diameter of approximately 14 Å should have a unitary conductance of 10−10 Ω (=100 pS). Such a relatively high conductance should produce a detectable electrical signal in support of the channel hypothesis for gap junction permeability. Small stepwise changes in the electrical coupling between paired Xenopus embryonic cells provided evidence for the existence of discrete coupling elements, but the low input resistance owing to the large cell size limited the resolution of the electrical signals. Adaptation of the two-cell voltage clamp to cell pairs of high input resistance achieved precise determination of unitary gap junction channel conductance. Concomitant current records from both voltage-clamped cells always appear as mirror images since each cell resides on opposite sides of the junction (Fig. 22.7). This equal amplitude and opposite polarity is the criterion used to define junctional current signals in the dual whole cell recording configuration. Channel conductance values of 120 and 160 pS were close to the original predicted value and provided supportive evidence for the existence of relatively large non-selective aqueous channels. Conductance values range from 10 to 300 pS for different gap junction channels under essentially physiological salt conditions. For an aqueous diffusion limited pore, the channel conductance will increase in direct proportion to 2πr2. For a long pore of 16 nm in length, a 300-pS gap junction channel requires a 26-Å diameter right cylindrical pore. It follows that higher conductance gap junction channels should exhibit less ionic selectivity and a higher molecular permeability than channels with smaller conductance values.
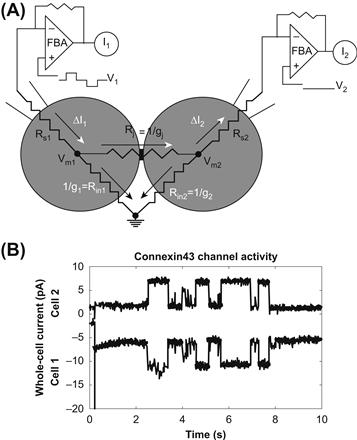
FIGURE 22.7 (A) Dual whole-cell recording configuration. Equivalent resistive circuit for the whole-cell patch-clamp recording configuration from two coupled cells. The voltage command potentials (V1 and V2) are applied via each negative feedback patch-clamp amplifier (FBA) through whole-cell patch electrodes. The difference between the command potentials and the cell membrane potentials (Vm1 and Vm2) is minimized by reducing the series resistance (Rs1 and Rs2) to less than 5% of the cell input resistance (Rin1 and Rin2) and junctional resistance (Rj). Accurate junctional current signals are achieved when a majority of the applied current flows across the junction (white arrowheads) instead of the cell membrane (black arrowheads). This condition is readily obtained by using high input resistance cells (Rin>1 GΩ). Conductances (g) are the reciprocal of the corresponding resistance element. (B) The whole-cell currents for each amplifier (I1 and I2) appear as simultaneous signals of opposite sign. These are unaltered whole-cell currents obtained from a Cx43-transfected N2A cell pair digitized at 1 kHz after low-pass filtering at 100 Hz. The transjunctional voltage was −30 mV.
IX Evidence for Charge Selectivity
The apparent permeability to a variety of water-soluble (hydrophilic) molecules (ions, cAMP, sugars, ATP, nucleosides, etc.) suggests the presence of a large-diameter aqueous pore with minimal selectivity based on electrical charge (equivalent valence at physiological pH). Several permeability studies on mammalian cell and invertebrate gap junctions were instrumental in assigning the commonly accepted molecular permeability limit of 1 kDa or diameter <14 Å. However, two of these same studies used tagged fluorescent tracers with different valences and demonstrated that the molecular permeability limit was lower for molecules with higher negativity. Selective permeability of molecules >600 Da and approximately 10 Å in diameter is not surprising since the size of the permeant molecule is approaching the estimated diameter of the pore. Placing any charged surfaces of the pore and the permeable molecule in close proximity to each other would enhance any electrostatic attractive or repulsive forces that might be present. This suggests that the pore of the gap junction channel contains fixed electronegative sites within the pore which reduce the permeability of large negatively charged molecules relative to their neutral or less negative counterparts.
Evidence for selectivity at the ionic level did not exist until the patch-clamp methodology was adapted to the recording of gap junction channel currents. In two cellular preparations, the junctional membranes of the earthworm septate axon and rat lacrimal gland cells were found to be less permeable to Cl− relative to K+ by ratios of 0.52 and 0.69, respectively. This modest selectivity among ions with nearly identical aqueous mobilities and diameters of less than 4 Å suggests that there are weak interactions between the permeant ion and the wall of the pore. In a simple diffusion-limited pore, ions interact with water molecules but not with other ions. Observations from four different homotypic connexin hemichannels or gap junction channels reported K+ to Cl− permeability ratios of approximately 10:1 with selectivity among monovalent cations in alignment with their relative aqueous mobility sequence. These observations suggest a common theme to the structure of the selectivity filter within the connexin channel pore. This proposed selectivity filter readily permits the passage of hydrated cations while restricting the simultaneous passage of similar aqueous anions by 10-fold. These observations provide direct evidence for cation–anion interactions within the gap junction pore that contradict aqueous diffusion theory. The observed permeability to atomic and large organic cations is consistent with a pore diameter of 10–14 Å for mammalian connexin channels.
The ability to transfer these electrical and chemical signals from cell to cell is important to tissue function. The most direct comparison of connexin-specific molecular permeability differences comes from the use of structurally similar molecules of varying size and valence. Two fluorescein derivatives with varying valences but essentially constant physical dimensions (=10 Å), 2′,7′-dichlorofluorescein (diCl-F) and 6-carboxyfluorescein (6-CF), were readily permeable through rat Cx43 channels. In contrast, both dyes were less permeable through chicken Cx43, human Cx37 and rat Cx40 gap junctions. Chicken Cx45 gap junctions were permeable to diCl-F, but not 6-CF. This indicates that the more anionic 6-CF is restricted in its junctional permeability relative to diCl-F in a connexin-specific manner. Since Cx43 has a lower channel conductance than Cx40 and Cx37, no correlation between the maximum conductance and molecular permeability of the connexin channels exists. This lack of correlation between charge selectivity and channel conductance suggests that pore size and conductance are not directly related. Alternatively, recent channel theory proposes that the pore with the smaller diameter and same electrostatic (fixed structural) charge will exhibit the higher channel conductance due to the increased charge density. This updated charged membrane theory can explain the conductance and selectivity properties of known ion channels, including gap junctions.
X Channel Properties of Different Connexins
The functional expression of different connexins, either by mRNA injection into Xenopus oocytes or stable expression in communication-deficient cell lines derived from mammalian tumors, has yielded tantalizing results. Single-channel unitary conductance values (the ease with which current flows) are connexin-specific. Channel conductance values vary by 30-fold, from 10 to 300 picosiemens (pS = 1/1012 Ω), while observed ionic selectivities vary by approximately 10-fold. The reporting of channel conductance became more complicated due to the presence of multiple conductance states for several of the connexin channels. The ability of some connexins to form heterotypic (two connexons of different connexin composition) or heteromeric (two different connexins within the same connexon) gap junction channels adds another level of complexity to the function of channel conductance. The physiological relevance of these conductance differences is poorly understood, but several observations now indicate that connexin composition can modulate gap junction channel molecular permeability limits. For example, rat hepatocyte gap junctions contain almost exclusively Cx32 while mouse hepatocyte gap junctions are composed of a mixture of Cx26 and Cx32. Isolated mouse liver gap junctions exhibited a lower size limit to uncharged sugar molecules and cAMP than rat liver gap junctions, consistent with a smaller pore size, and they exhibited possible charge selectivity, contributed by Cx26 to otherwise Cx32-containing gap junctions. Connexin composition and mutations can affect gap junction selectivity in detrimental ways (e.g. disease-causing mutations), although the molecular basis for these permeability alterations remain to be determined.
The regulation of gap junction conductance by intracellular pH, intracellular calcium, protein kinases and transjunctional (cell-to-cell) voltage are also connexin-specific. The most significant differences in the amino acid sequences of the connexins occur within the cytoplasmic loop and carboxyl-terminal domains. These two cytoplasmic domains provide the basis for distinct conductance and regulatory properties upon the assembled multimeric connexin channels. Since a specific gene encodes for each connexin, tissue- and developmental-specific patterns of expression exist. Limited comparisons of the channel conductance and regulatory properties of the connexin channels to gap junctions in native cell types demonstrate the presence of at least two distinct connexin channel types. The presence of multiple connexin channels can explain the properties of the gap junctions found in many cell types. In one study, the expression of three embryonic chicken heart connexins, Cx42, Cx43 and Cx45, demonstrated that each connexin exhibited unique conductance properties, which coincided with the observance of multiple conductance states in cultured cardiac myocytes. Furthermore, mixing these three connexins in different proportions modeled the response to transjunctional voltage during different stages of cardiac development. Thus, the observed decline in Cx45 expression could significantly explain the decrease in the transjunctional voltage sensitivity of junctional conductance in the developing chicken heart. Developmental and hormonal changes in connexin expression occur in other tissues, suggesting a possible role for this mechanism in the modulation of gap junction communication in a variety of tissues.
XI Gating by Ions and Second Messengers
Perhaps of greater physiological relevance than transjunctional voltage is the modulation of junctional communication by highly buffered intracellular cations (protons and calcium) and organic second messengers such as cAMP, diacylglycerol and inositoltrisphosphate. Whether cations directly bind to a regulatory site (or sites) on the connexin or act through intermediate accessory proteins is unknown. This issue remains an important topic for investigation since it has been observed that interventions that lower intracellular pH also increase intracellular free calcium. Conversely, there is direct evidence that several of the connexins are substrates for phosphorylation by protein kinases such as protein kinase A (PKA) and PKC. Protein kinase-dependent phosphorylation often increases or decreases junctional communication, depending on the connexins expressed in each tissue. Physiological correlates at the channel level are rare, owing to the difficulty of observing channel activity under distinct ionic or phosphorylation conditions. Transjunctional voltage (Vj) gating and Cx43 phosphorylation by PKC have both been shown to decrease the channel conductance and increase the ionic/molecular selectivity of gap junctions by inducing the formation of a subconductance state. Generally, cAMP increases junctional conductance and PKC has the opposite effect, at least in Cx43-containing cells. PKA and PKC may both phosphorylate the same serine residue (S233) of Cx32. In contrast, tyrosine phosphorylation by pp60v-src produces a rapid and reversible uncoupling of Cx43 gap junctions but has no effect on Cx32 gap junctions. Cx43 contains specific v-src SH2- and SH3-binding domains near a tyrosine phosphorylation site (Y265) by pp60v–src (Fig. 22.8A). Cx32 lacks these binding and tyrosine phosphorylation sites. There is increasing evidence that the diversity in the cytoplasmic loop and carboxyl-tail domains of different connexins confers different conductance and regulatory properties on the connexin-specific gap junctions in response to protein kinases and intracellular ions.
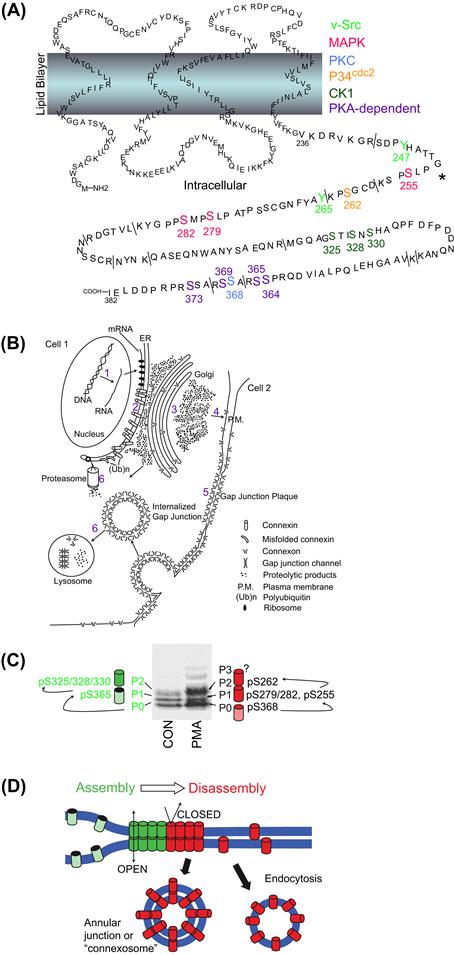
FIGURE 22.8 (A) Graphical illustration of the known protein kinase phosphorylation sites on the carboxyl domain of Cx43 (purple: PKA; blue: PKC; green: CK1; red: MAPK; orange: p34cdc2; lime green: v-src, brackets: trypsin cleavage sites). (Reprinted fromSolan, J.L., and Lampe, P.D. (2005). Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta, 1711, 155 with permission. Copyright 2005 Elsevier Press.) (B) Illustration of the life cycle of the connexins beginning with mRNA transcription (1), translation and protein folding (2), oligomerization and intracellular transport (3), hemichannel insertion into the plasmalemma (4), hemichannel docking and gap junction formation (5), and degradation by the proteasome and lysosome (6). (Reprinted fromBeyer, E.C. and Berthoud, V.M. (2009). The family of connexin genes. In Connexins: a Guide, p. 17, with permission. Copyright 2009 Humana Press.) (C and D) Association of Cx43 phosphorylation-dependent bands on an SDS/PAGE gel with the PO, PI, P2, or P3 isoforms correlated with the protein kinase phosphorylation sites implicated in the assembly (green) or disassembly (red) of gap junctions. Assembled gap junctions are internalized as a double membrane bounded “connexosome” while unassembled hemichannels appear in endocytotic vesicles. (Reprinted from Solan, J.L. and Lampe, P.D. (2007) Connexin43 phosphorylation: structural changes and biological effects. Biochem. J., 419, 262, with permission. Copyright 2007 Biochemical Society.)
XII Regulation of Functions of Connexin-Based Gap Junctions at Multiple Levels
As suggested above, a likely reason for differences in the sequence of the carboxyl tail of connexins may be in the regulation of gap junction-mediated tissue-specific or cell-specific biological activities. Indeed, gap junction regulatory mechanisms described so far appear to fall into one of two general categories. On the one hand, rapid changes in cell coupling that occur within seconds or minutes may reflect regulation of gating of pre-existing pores. On the other hand, long-term changes which occur within minutes or a few hours to days are likely to involve the modulation of synthesis, assembly or degradation of connexins (see Fig. 22.8B). Existence of this latter scheme of modulation is supported by studies that show that the half-lives of several different connexins are relatively short, ranging from 1 to 3 hours, and by more recent direct videomicrography of living cells expressing green fluorescent protein tagged connexins. In these latter studies, connexins have been observed as they concentrate within the developing Golgi; as they move to the surface to form plaques in regions of cell–cell contact; and as large fragments of the connexin-positive plaques are internalized as apparent vesicles (see Figs. 22.3H, I and 22.8B). Moreover, a variety of studies supports the idea that gap junctions may be regulated at virtually any stage of their formation, maturation and degradation.
For example, the possible effect of cAMP on transcription of connexin mRNA and its ultimate translation is supported by studies which show that cAMP-mediated increases in coupling or in junction formation in some cells can be blocked by inhibitors of mRNA or protein synthesis. The potential for control at the level of transcription of connexins is also implied by the characterization of their 5′ flanking sequences. For example, the 5′ flanking sequence of the myometrial Cx43 gene has been shown to possess several consensus activator protein-1 (AP-1) binding sites as well as half-palindromic estrogen response elements. Since the AP-1 proteins, Fos and Jun, are expressed in response to increased estrogen, transcription of Cx43 may be indirectly upregulated through its AP-1, cis-acting elements. In vascular smooth muscle, the transcription of Cx43 may be upregulated by mechanical load induced by stretch, presumably via an upregulation of c-fos, which may act in turn on AP-1 binding sites or upon other sites in the 5′ flanking region of the Cx43 gene. Promoter analysis of other connexin genes has shown that their transcription may also be regulated by transcription factor binding sites. For example, two GC boxes, two GT boxes, a TTAAAA box, a YY1-like binding site and a mammary gland factor binding site have been identified in the proximal promoter region of the human Cx26 gene. Likewise AP-1, AP-2, SP-1, TRE and p53 transcription factor binding sites have been identified in the 5′ flanking sequence of the mouse Cx40 gene, while Cx-B2 and P1 and P2 promoters are implicated in the transcriptional regulation of Cx32.
Yet another level of control of gap junction function is revealed by studies that show the upregulation of coupling or gap junction formation in the absence of transcription or translation, perhaps at a post-translational level of control. The direct application of dibutyryl cAMP (dbcAMP) to mammary tumor cells in culture, for example, increases the number of gap junctions as well as cell–cell coupling in the absence of an increase in Cx43 mRNA or protein. Likewise, connexin 43 trafficking within several cultured cell lines appears to be regulated by post-translational mechanisms. Specifically, the trafficking of Cx43 from the cytoplasm to the plasma membrane and its assembly into functional gap junction aggregates may require phosphorylation of two serine phosphorylation consensus sites in the carboxyl tail of this connexin (see Fig. 22.8C, D). For example, two cell lines were studied that could not form gap junctions or become coupled, but were able to synthesize significant amounts of Cx43, which was stored within vesicles in the cytoplasm. The Cx43 in these cells was phosphorylated at only one of its serine residues. When one of these lines was transfected with a gene for a cell adhesion molecule, L-CAM, the Cx43 in the transfected line was phosphorylated again, translocated to the plasma membrane and assembled into gap junctions. Further evidence for the phosphorylation-dependent trafficking and assembly of Cx43 in cardiomyocytes has been obtained in experiments with monensin, a reagent that inhibits translocation of proteins between Golgi cisternae. These studies suggest that phosphorylation of Cx43 to the mature form occurs in a compartment of the cell between the Golgi apparatus and the plasma membrane. Similar results are obtained in studies of effects of brefeldin A, which blocks both Cx43 phosphorylation and trafficking to the plasma membrane in a mammary tumor cell line. Finally, it has also been suggested that the process of connexon clustering that gives rise to functional gap junction plaques may be facilitated by the action of microfilaments. Thus, the increase in Cx43-positive gap junctions and coupling in the absence of Cx43 mRNA synthesis following treatment of cultured prostatic cells with forskolin may be explained by effects of cAMP on post-translational processing of Cx43, namely their assembly into functional plaques and their translocation from the cytoplasm to the membrane.
Numbers of functional gap junctions within the plasma membrane may also be affected by regulation of their removal and degradation. For example, the treatment of some cells with phorbol esters such as 12-o-tetradecanoylphorbol-13-acetate (TPA) may stimulate the removal and degradation of gap junctions through phosphorylation of connexins by protein kinase C. Similarly, it is thought that the phosphorylation of mitogen-activated protein (MAP) kinase serine phosphorylation sequences in cultured rat liver cells treated with epidermal growth factor, may mediate the loss of functional gap junctions from the cell membrane. In another example, it has been shown that massive endocytosis of gap junctions in granulosa cells of the ovary is stimulated by high titers of follicle stimulating hormone; that endocytosis is facilitated by clathrin and microfilaments; and that the internalized gap junction vesicles fuse with lysosomes prior to degradation (see Fig. 22.8B). Alternatively, it has been suggested that gap junction plaques and their connexins may be disposed of via a ubiquitin-mediated proteasomal proteolytic pathway. This mechanism is supported by studies showing that connexins accumulate in cells treated with proteasomal inhibitors and by immunogold studies that demonstrate the association of ubiquitin with gap junction plaques (Fig. 22.9).

FIGURE 22.9 Image of gap junction in cultured HeLa cell transfected with Cx40 and Cx43. The replica was immunostained with antibodies to Cx40, Cx43, and ubiquitin. The 6-nm gold particles indicate Cx40, the 10-nm gold particles stain for Cx43, and the 15-nm gold particles indicate epitopes for ubiquitin.
In summary, it appears that the amount of functional gap junction membrane at the cell surface may be controlled at virtually any point in the gap junction’s life history; namely through regulation of (1) transcription of connexin mRNA, (2) translation of connexin protein, and then through post-translational modifications, which affect the (3) assembly of functional plaques, (4) gating of junctional channels, (5) channel conductance and (6) degradation of functional gap junctions.
XIII Specific Biological Functions of Gap Junctions
Early speculations regarding the general functional significance of gap junctions, such as the electrical coupling of some neurons and the buffering of metabolites within a tissue mass, have not been seriously challenged for the past three decades (see Sections I, II and IX). Several recent studies, however, have begun to establish even more specific functional relationships between gap junctions and tissue activities and behaviors, most notably with respect to the regulation of smooth muscle contraction and ligand-mediated secretory activity.
Perhaps one of the best-studied cases is that of the myometrial connexin, Cx43. It is known, for example, that the myometrium of the non-pregnant uterus in a variety of species is virtually devoid of gap junctions and expresses very little, if any myometrial Cx43 mRNA or protein. In contrast, however, Cx43 mRNA is rapidly synthesized and translated and then gap junctions appear coincident with rising estrogen and declining progesterone titers just prior to parturition. Experimental manipulations of these hormones also predictably induce or inhibit the appearance of gap junctions in experimental models of preterm labor or tocolysis respectively (effective labor coincides with their induction while myometrial quiescence parallels their inhibition). For example, removal of the ovaries or the injection of RU-486 during the latter part of pregnancy effectively increases the estrogen:progesterone ratio, the expression of Cx43 mRNA and protein, the assembly of myometrial gap junctions and concomitant preterm labor. Conversely, the injection of progesterone several days prior to term inhibits the formation of gap junctions and onset of labor. Thus, it is likely that transcription and translation of Cx43 are required for gap junction formation and coordinated myometrial contractions in humans. Moreover, the evidence for the presence of AP-1 binding sites in 5′ flanking sequences of the myometrial Cx43 gene and the estrogen-mediated expression of Jun and Fos described above (see Section XII) is consistent with this notion. The function of myometrial gap junctions and contractility may also be regulated at the level of post-translational processing. For example, high levels of myometrial Cx43 appear a full day prior to delivery in rats (Fig. 22.10) but most of the Cx43 in these cells is located within perinuclear vesicles which also stain with an antibody against a Golgi-associated protein (Fig. 22.10B). Six to 12 hours prior to onset of delivery, however, Cx43 immunopositive plaques are observed in the plasma membrane (Fig. 22.10C). Conversely, cessation of delivery is accompanied by the loss of gap junctions from the cell surface by a process of endocytosis (Fig. 22.10E).

FIGURE 22.10 Immunofluorescent staining of myometrium sections incubated with site-specific antibodies to the carboxyl terminus of Cx43 from pregnant rat sacrificed on day 19 (A) or day 21 (B), at midnight on day 21 (C), during delivery on day 22 (D), 6 h after delivery (E) or 24 h after delivery (F). Magnification, 500×. (From Hendrix, E.M., Mao, S.J.T., Everson, W. and Larsen, W.J. (1992). Myometrical connexin 43 trafficking and gap junction assembly at term and in preterm labor. Mol. Reprod. Dev., 33, 27, with permission. Copyright 1992 Wiley-Liss, a division of John Wiley & Sons, Inc.)
Gap junctions have also been implicated in the process of secretion and other ligand-mediated functions in many different kinds of cells. For example, gap junctions are well developed in differentiated endocrine and exocrine cells and are typically sparse or absent in proliferating stem cells. Moreover, the induction of secretory activity itself appears to be correlated with increased numbers of gap junctions in luteal cells, adrenal cortical cells, pancreatic exocrine cells, mammary alveolar cells and thyroid cells. Studies of osteoblasts have demonstrated a close correlation between the effects of hormones on the generation of a second messenger, formation of gap junctions and cell function. In addition, gap junctions appear to be more abundant in cultured confluent osteoblasts than proliferating cells and osteoblast cell coupling in culture and cAMP production are enhanced by the application of parathyroid hormone (PTH). Conversely, an analog of PTH which binds to PTH receptors but attenuates cAMP accumulation, results in a decrease in cell coupling. Finally, transfection of osteoblasts with antisense Cx43 mRNA results in significant concomitant reductions in coupling and in cAMP synthesis in response to PTH.
It has been suggested that the development of gap junctions may enhance the sensitivity of hormonally responsive cells to their specific ligand as a consequence of the transfer of cAMP through gap junctions. Thus, cells not receiving direct stimulation by binding of the secretogogue to its receptor may also respond to the ligand-mediated production of cAMP in neighboring cells which possess the appropriate receptor. Such a hypothesis has been invoked to explain the maintenance of meiotic arrest of mammalian primary oocytes. These germ cells initiate the first meiotic division during embryonic life but are then almost immediately arrested at the first meiotic prophase stage. Typically, a primary oocyte does not resume meiotic maturation until it responds to an ovulatory surge of gonadotropic hormones following puberty. However, since meiosis spontaneously resumes when the oocyte is removed from the follicle and cultured in medium-lacking hormones, it has been postulated that the follicular environment is inhibitory to resumption of meiotic maturation. Meiotic arrest may be maintained by follicle cell-generated cAMP which enters the oocyte through a well-developed network of gap junctions. Conversely, it has been suggested that meiotic resumption may be signaled by the LH-mediated disruption of the gap junction pathway within the cumulus mass (following an ovulatory surge of gonadotropins), thus preventing cAMP manufactured within the follicle cells from entering the oocyte. Other hypotheses of both negative and positive regulation of meiotic resumption have also been proposed.
It has recently been proposed that gap junction connexins (specifically Cx43) may function in a role somewhat different than those described above; namely in protein trafficking. It has been shown, for example, that the second PDZ domain (of three) of the tight junction-associated protein ZO-1 binds to the extreme C-terminal end of the Cx43 molecule typically resulting in cell membrane colocalization of Cx43 and ZO-1 in cells that co-express these proteins. In support of the possibility that Cx43 is required for shuttling of ZO-1 to the cell membrane, it was found that both ZO-1 and Cx43 fail to accumulate at the cell membrane in Cx43 knockout astrocytes.
XIV Gap Junctions in Human Disease and in Murine Models of Human Disease
The first hypothesis for a direct link between connexin gap junction function and a human disease was formulated in the 1970s based on the positive correlation between the loss of gap junction function and uncontrolled cell growth as exhibited in carcinogenesis. Over the last two decades, hereditary links to central and peripheral neuropathies, deafness, skin disorders, cataracts, cardiac arrhythmias and multisyndromic craniofacial dysplasias have been identified for 10 different connexins to date. Essentially, all of the known connexin mutations are loss-of-function mutations, either by rendering the resultant gap junction communication-deficient or by preventing the assembly of gap junctions due to connexin misfolding and/or trafficking defects. The precise molecular mechanisms by which a particular connexin mutation produces a communication or trafficking defect are not fully understood but may include alterations in channel permeability, regulatory gating or critical structural components key to correct protein assembly/trafficking and function. A graphical summary of many known connexin-linked human diseases is provided in Fig. 22.11. It should be noted that genetic mutations in tight junction and desmosome intercellular junction protein constituents are also known to occur.
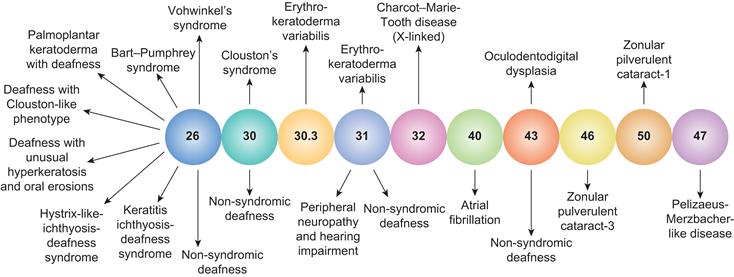
FIGURE 22.11 An illustration of the relationship between inherited connexin mutations and human disease phenotypes. The majority of inherited connexin disorders are autosomal dominant and all involve a loss-of-function due to improper protein folding and trafficking or deficits in gap junction communication. (Adapted from Lai-Chong, J.E., Arita, K. and McGrath, J.A. (2007). Genetic diseases of junctions. J. Invest. Dermatol., 127, 2715, with permission. Copyright 2007 The Society for Investigative Dermatology.)
XIVA Carcinogenesis
Studies that compared the presence or absence of coupling or gap junctions in normal cells and in several tumors and cancer cell lines initially supported this hypothesis. For example, coupling or gap junctions were found to be deficient in certain hepatoma cells and in L-cell derivatives such as the clone-1D cell line in contrast to their normal counterparts. Indeed, more recent studies support these early observations. For example, an extensive study has examined the role of gap junctions in tumor progression in rat liver following initiation by diethylnitrosamine (DEN) and promotion by either phenobarbitol (PB) or 2,3-dichloro-7,8-dibenzo-p-dioxin (TCDD). Transcripts and protein levels of Cx26, Cx32 and Cx43 were measured and reductions in Cx26- and Cx32-positive gap junctions were observed in all resulting neoplasms. However, it was also demonstrated that these decreases were not always associated with reductions in specific m-RNA transcripts for these connexins, suggesting that the loss of junctions could result from the modulation of transcription or translation or from post-translational modification affecting assembly or degradation (see Section XII). Consistent with these observations, the transfection of HeLa cells with cDNA encoding Cx26, but not Cx40 or Cx43, results in inhibition of tumor formation in nude mice, suggesting that Cx26 gap junctions play a pivotal role in growth control. Similarly, the transfection of communication-deficient hepatoma cells (SKHep1) with cDNA encoding Cx32 results in slowing of the growth rate of tumors in nude mice, compared with tumors arising from communication-deficient parental SKHep1 cells.
In conflict with these findings, however, other highly malignant tumor cells have been shown to be well coupled or to possess large numbers of gap junctions. These include Novikoff hepatoma cells, human SW-13 adrenal cortical carcinoma cells and murine B16 melanoma cells. Moreover, other studies have analyzed gap junctions during tumor progression in skin tumors and in hepatocarcinoma and have found that, while gap junctions or coupling may disappear during the transformation of normal cells by cancer-causing agents (such as tumor promoters), their loss seems to follow, rather than lead the changes which transform these normal cells to tumor cells. In response to results such as these, a modified defective communication hypothesis was proposed to include cancer cells which might not necessarily exhibit an absence of, or obvious structural defect in the gap junction itself, but which might be defective with respect to some other part of the gap junction communication mechanism, for example, lack of ability of the cell to synthesize the postulated regulatory message or defects in the receptor which translates the cell’s activity. Other modifications of the hypothesis have also been advanced, proposing, for example, that some cancer cells (e.g. cultured human SW13 cells and diethylstilbestrol-induced tumors of the proximal tubule in the Syrian hamster) are not able to maintain a steady-state gap junction level through a regulated balance of synthesis or degradation of gap junctions. Under some circumstances, these cells possess normal or even excessive gap junction complements and are mitotically quiescent while, under other conditions, the cells lose most or all of their junctions, resulting in cancerous growth. A temporal relationship between gap junction formation and degradation and the rate of proliferation is also exhibited by normal liver cells in partially hepatectomized rats. Normal rat liver cells remaining after partial hepatectomy initially lose their gap junctions and proliferate rapidly. By 40 hours after partial hepatectomy, the liver cells regain their gap junctions coincident with a reduction in the rate of DNA synthesis and cell division.
Another variation of the hypothesis is based upon a series of experiments that demonstrate that normal cells may have the ability to suppress tumor cell behavior in some normal tumor cell co-cultures as long as the tumor cells can also form gap junctions with the co-cultured normal cells. Other tumor cells may not be able to form gap junctions with normal cells but may be able to form gap junctions between themselves. In these tumor cells, contact with normal cells does not suppress the transformed phenotype of the tumor cell. However, the ability of certain tumor cells to make gap junctions with one another may result in more efficient killing by antitumor agents by allowing the cell-to-cell movement of lethal metabolites formed in some, but not all of the tumor cells within the interconnected network. This phenomenon is called the bystander effect. Apoptotic signals may also be propagated by the bystander effect.
In conclusion, despite decades of research, a direct causal link between cancerous growth and deficiencies in gap junction intercellular communication remains elusive. In numerous instances, there is a positive correlation between the development or progression of cancerous tumors and the loss of gap junction communication, but this is not always the case. It is also unlikely that mechanisms of gap junction-mediated growth control will be identical in all cells or tumors where the activity of gap junctions can be demonstrated to play a pivotal role due to the differential expression of connexins.
XIVB Demyelinating Neuropathies of Cx32 and Cx47 Mutations
Spontaneous mutations of Cx32 result in a common peripheral neuropathy in humans, Charcot-Marie-Tooth disease, which affects one of every 2500 individuals. While this demyelinating disease may arise as a consequence of mutations of a peripheral myelin protein PMP22 (on chromosome 17) or of the myelin constituent Po (on chromosome 1), numerous families have been identified with an X-linked dominant form of the disease characterized by as many as 150 different mutations of Cx32 (CMTX). Mutations may involve single-base substitutions, formation of a premature stop codon, frame-shift or elimination of an amino acid residue. They may occur in amino or carboxyl termini, cytoplasmic or extracellular loops, or in transmembrane regions of the connexin. The causal relationship between these diverse mutations of Cx32 and demyelination of peripheral nerves is obviously varied since the mutations are quite diverse and may or may not affect channel properties. In some cases, it seems possible that mutations may directly affect the function of Cx32 gap junctions in peripheral myelin by interfering with their possible function as ATP-sensitive hemichannels, their function in the exchange of nutrients between the perinuclear region of the Schwann cell and the Schmidt–Lantermann incisures and paranodal processes at the node of Ranvier, or their possible function in signaling between internodes. In other cases, it is thought that the mutations may indirectly affect Cx32 function through effects on Cx32 synthesis and trafficking within the cell. Targeted knockout of connexin 32 in mice also results in changes in liver enzyme activities and in glucose mobilization but, most interestingly, these Cx32-deficient mice have an increased susceptibility to hepatocarcinogenesis. Otherwise, they are vital and fertile. There are >300 GJB1 mutations thus far identified. A list of these CMTX mutations can be found at http://www.molgen.ua.ac.be/CMTMutations/Mutations/MutByGene.cfm. Oligodendrocytes in the central nervous system also develop a demyelinating neuropathy, Pelizaeus–Merzbacher–like disease (PLMD1) owing to mutations in Cx47 (GJC2, formerly GJA12). For additional information go to http://www.ncbi.nlm.nih.gov/omim/608804.
XIVC Cataract Formation and Mutations of Cx50 and Cx46
The maintenance of junctional communication between lens fiber cells is necessary for the homeostatic maintenance of normal transparency of the lens since it is an avascular tissue and the cells are devoid of intracellular organelles. Targeted knockout in mice or mutations in Cx50 lead to development of zonular pulverulent cataracts of the lens but also results in microphthalmia. These results thus implicate Cx50 not only in the maintenance of lens transparency but also in the growth of the eye. Cx46 null mice or mutations in Cx46 produce a more severe form of autosomal dominant zonular pulverulent cataracts, but not microphthalmia. Substituting Cx46 for Cx50 prevents the cataract phenotype but does not rescue the microphthalmia, indicative of differential roles for the two lens connexins in the maintenance of lens homeostasis and crystalline solubility, but not developmental growth control. Additional information can be found at http://www.ncbi.nlm.nih.gov/omim/116200 or /601885.
XIVD Deafness and Mutations of Cx26, Cx30, Cx31 and Cx43
A variety of mutations of Cx26 including missense mutations and mutations resulting in frameshifts and formation of premature stop codons have been linked to both autosomal recessive and autosomal dominant hearing loss in humans. It is known that deletion of the Cx26 gene leads to the progressive degeneration of the outer hair cells, supporting cells in the outer and middle organ of Corti and spiral ganglion neurons, presumably by disruption of the gap-mediated ion and endolymph circulation in the cochlea. Greater than 100 total GJB2 mutations have now been identified to cause syndromic (dominant) or non-syndromic (recessive) deafness. Anywhere from two to five non-sydromic deafness mutations have also been identified in the GJA1 (Cx43), GJB3 (Cx31) and GJB6 (Cx30) genes. A database of connexin-linked inherited deafness mutations can be found at http://davinci.crg.es/deafness/index.php?seccion=mut_db&db=synd&synd=cx26synd, or _db&db=nonsynd&nonsynd=cx26mut, …=cx31mut, …=cx30mut, and …=cx43mut. Germline knockout of Cx26 resulted in embryonic lethality as a consequence of placental dysfunction but conditional Cx26 (cCx26) knockout and Cx30 null mice are viable and recapitulate the deafness phenotype.
XIVE Skin Disorders
Several of the Cx26 mutations associated with syndromic deafness also cause ectodermal skin disorders such as palmoplantar keratoderma, hyperkeratosis, ectodermal dysplasia keratitis-ichthyosis and mutilating keratoderma (Vohwinkel’s syndrome). Deafness and non-deafness mutations in Cx31 are known to produce erythrokeratoderma variabilis. Cx30.3 (GJB4) also cause erythrokeratoderma variabilis and non-deafness mutations in Cx30 produce autosomal dominant hidrotic ectodermal dysplasia (Clouston syndrome).
XIVF Oculodentodigital dysplasia (ODDD)
Within the last decade, a multisymptom human disorder including bilateral microphthalmia, microcornea, glaucoma, hypertelorism, narrow nose, microdontia, syndactyly of the fourth and fifth fingers and various neurological and cardiac anomalies were linked to mutations in Cx43. Sixty-two GJA1 mutations are already known and the vast majority are autosomal dominant but exhibit phenotypic variability for unknown reasons (see http://www.ncbi.nlm.nih.gov/omim/164200). Some mutations result in Cx43 trafficking defects while others form non-communicating or communication-deficient gap junctions, the common theme being a loss-of-function. Already, three murine models of Cx43 ODDD mutations (G60S, I130T, G138R) have been demonstrated to exhibit an increased susceptibility to cardiac arrhythmias, whereas clinically cardiac abnormalities are observed in only 3% of human ODDD patients, 10-fold less than the observed rate for neurological disorders and >20 times less frequent than the observed syndactyly. Perhaps the low incidence of cardiac abnormalities observed with Cx43 mutations is that severe defects likely result in embryonic or neonatal lethality.
XIVG Heart Function and Mutations of Cx43 and Cx40
While few specific heart defects related to mutations of Cx43 have been observed in humans, a series of studies with animal models of ischemia support a role for Cx43 gap junctions in recovery from injuries to the myocardium. For example, the number of gap junction plaques within the intercalated disk has been shown to decrease in the region of the border of the healing infarct. In addition, the spatial distribution of gap junctions is also abnormal in these areas. These observations, along with studies that show the critical role of Cx43 in conduction (particularly in the ventricle), have led to suggestions that alterations in Cx43 levels and distributions may play an important role in development of infarct-mediated arrhythmias. Targeted knockouts of Cx43 and mice overexpressing Cx43 are characterized by abnormal development of the pulmonary (right ventricular) outflow tract resulting in their death immediately following birth. Enlargement of the right ventricle with thinning of the wall, attenuation of the ductus arteriosus and formation of one or two abnormal pouches at the base of the pulmonary outflow tract were also observed. It has been suggested that these defects of conotruncal development may result from disruption of the cardiac neural crest. In addition, Cx43 knockout mice of both sexes have small gonads, resulting in part from deficiencies in germ cell development. Since these mice die shortly after birth, the gonads were cultured to determine their capacity for folliculogenesis. It was found that follicles in these ovaries develop only to the primary follicle stage. Targeted knockout of Cx40 results in defects consistent with its localization in the AV node and in conductive myocardium. Electrocardiography of Cx40 knockout animals was characterized by reduced conduction velocities and abnormalities characteristic of first-degree atrioventricular block with associated bundle branch block.
At least half a dozen mutations in Cx40 have been identified in human patients suffering from chronic atrial fibrillation (AF) in recent years and Cx40 polymorphisms have been linked to atrial standstill as well as AF. Conditional Cx43 knockout and Cx43 ODDD mutation mouse models have further revealed that heterogeneous loss of Cx43 from the myocardium increases the susceptibility to atrial or ventricular arrhythmias to a greater extent than uniform reductions in Cx43 expression. Many of these Cx40 or Cx43 mutations again result in a loss-of-function, although the underlying precise molecular mechanisms are poorly understood. Chronic AF and myocardial ischemia or infarction cause cardiac remodeling of gap junctions that sustain AF or increase susceptibility to ventricular arrhythmias due to increased heterogeneity and reductions in the safety margin for conduction disturbances especially during rapid stimulation rates. Our understanding of acute and chronic changes in cardiac gap junction distribution and function will likely evolve through further investigations of Cx40/Cx43 mutations and the molecular mechanisms involved in their redistribution and function.
XIVH Fertility and Targeted Knockout of Cx37
While no human syndrome resulting from mutations of Cx37 (the human ortholog of Xenopus oocyte Cx38 [GJA2]) is known, the targeted knockout of Cx37 in mice has been shown to result in the disruption of folliculogenesis. Follicles develop only to early antral stages, the oocytes fail to acquire meiotic competence and ovulation does not occur. Cx43 is essential to (antral) follicular maturation.
In Memoriam
This chapter is dedicated to the memory of William J. Larsen who previously co-authored this chapter before losing his life to cancer soon after publication of the 3rd edition in 2001.
BIBLIOGRAPHY
1. Alcolea S, Theveniau-Ruissy M, Jarry-Gnichard T, et al. Downregulation of connexin 45 gene products during mouse heart development. Circ Res. 1999;84:1365–1379.
2. Bai S, Schoenfeld A, Pietrangelo A, Burk RD. Basal promoter of the rat connexin 32 gene: identification and characterization of an essential element and its DNA-binding protein. Mol Cell Biol. 1995;15:1439–1445.
3. Barr L, Dewey MM, Berger W. Propagation of action potentials and the structure of the nexus in cardiac muscle. J Gen Physiol. 1965;48:797–823.
4. Bevans CG, Kordel M, Rhee SK, Harris AL. Isoform composition of connexin channels determines selectivity among second messengers and uncharged molecules. J Biol Chem. 1998;272:2808–2816.
5. Beyer EC, Berthoud VM. The family of connexin genes. In: Connexins: A Guide. New York: Humana Press; 2009;:3–26.
6. Beyer EC, Paul DL, Goodenough DL. Connexin family of gap junction proteins. J Membr Biol. 1990;116:187–194.
7. Brink PR, Dewey MM. Evidence for fixed charge in the nexus. Nature. 1980;285:101–102.
8. Brink PR, Fan S-F. Patch clamp recordings from membranes which contain gap junction channels. Biophys J. 1989;56:579–593.
9. Budunova IV, Carbajal S, Viaje A, Slaga TJ. Connexin expression in epidermal cell lines from SENCAR mouse skin tumors. Mol Carcinog. 1996;15:190–201.
10. Bukauskas FF, Bukauskiene A, Verselis VK. Conductance and permeability of the residual state of connexin43 gap junction channels. J Gen Physiol. 2002;119:171–185.
11. Chiba H, Sawada N, Oyamada M, et al. Relationship between the expression of the gap junction protein and osteoblast phenotype in a human osteoblastic cell line during cell proliferation. Cell Struct Func. 1993;19:419–426.
12. Coppen SR, Kodama I, Boyett MR, et al. Connexin45, a major connexin of the rabbit sinoatrial node, is co-expressed with connexin43 in a restricted zone at the nodal-crista terminalis border. J Histochem Cytochem. 1999;47:907–918.
13. Cowan DB, Lye SJ, Langille BL. Regulation of vascular connexin43 gene expression by mechanical loads. Circ Res. 1998;82:786–793.
14. Dahl G, Locovei S. Pannexin: to gap or not to gap, is that a question?. IUBMB Life. 2006;58:409–419.
15. Darrow BJ, Laing JG, Lampe PD, Saffitz JE, Beyer EC. Expression of multiple connexins in cultured neonatal rat ventricular myocytes. Circ Res. 1995;76:381–387.
16. Donahue HJ, McLeod KJ, Rubin CT, et al. Cell-to-cell communication in osteoblastic networks: cell line-dependent hormonal regulation of gap junction function. J Bone Min Res. 1995;10:881–889.
17. Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of connexin43 gap junction channels is regulated through protein kinase C-dependent phosphorylation. Circ Res. 2006;98:1498–1505.
18. Ewart JL, Cohen MF, Meyer RA, et al. Heart and neural tube defects in transgensis mice overexpressing the Cx43 gap junction gene. Development. 1997;124:1281–1292.
19. Filson AJ, Azarnia R, Beyer EC, Loewenstein WR, Brugge JA. Tyrosine phosphorylation of a gap junction protein correlated with inhibition of cell-to-cell communication. Cell Growth Diff. 1990;1:661–668.
20. Flagg-Newton J, Simpson I, Loewenstein WR. Permeability of the cell-to-cell membrane channels in mammalian cell junction. Science. 1979;205:404–407.
21. Forge A, Becker D, Casalotti S, et al. Gap junctions and connexin expression in the inner ear. Novartis Found Symp. 1999;219:134–150.
22. Furshpan EJ, Potter DD. Transmission at the giant motor synapses of the crayfish. J Physiol. 1959;145:289–325.
23. Furshpan EJ, Potter DD. Low-resistance junctions between cells in embryos and tissue culture. Curr Top Dev Biol. 1968;3:95–127.
24. Gerido DA, White TW. Connexin disorders of the ear skin, and lens. Biochim Biophys Acta. 2004;1662:159–170.
25. Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934.
26. Goldberg GS, Martyn KD, Lau AF. A connexin 43 antisense vector reduces the ability of normal cells to inhibit the foci formation of transformed cells. Mol Cytogen. 1994;11:106–114.
27. Gollob DA, Jones DL, Krahn AD, et al. Somatic mutations in the connexin 40 gene (GJA5). N Engl J Med. 2006;354:2677–2688.
28. Goodenough DA, Simon AM, Paul DL. Gap junctional intercellular communication in the mouse ovarian follicle. Novartis Found Symp. 1999;219:226–235.
29. Gutstein DE, Morley GE, Vaidya D, et al. Heterogeneous expression of gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation. 2001;104:1194–1199.
30. Harris AL. Connexin channel permeability to cytoplasmic molecules. Prog Biophys Molec Biol. 2007;94:120–143.
31. Hauer RNW, Groenewegen WA, Firouzi M, Ramanna H, Jongsma HJ. Cx40 polymorphisms in human atrial fibrillation. Adv Cardiol. 2006;42:284–291.
32. Hendrix EM, Mao SJT, Everson W, Larsen WJ. Myometrial connexin 43 trafficking and gap junction assembly at term and in preterm labor. Mol Reprod Develop. 1992;33:27–38.
33. Hendrix EM, Myatt L, Sellers S, Russell PT, Larsen WJ. Steroid hormone regulation of rat myometrial gap junction formation: effects on Cx43 levels and trafficking. Biol Reprod. 1995;53:547–560.
34. Hille B. Ionic Channels of Excitable Membranes. 2nd ed. Sunderland: Sinauer Assoc.; 1992.
35. Huelser DF, Rehkopf B, Traub O. Dispersed and aggregated gap junction channels I identified by immunogold labeling of freeze-fractured membranes. Exp Cell Res. 1997;233:240–251.
36. Iglesias R, Locovei S, Roque A, et al. P2X7 receptor-Panenxin1 complex: pharmacology and signaling. Am J Physiol (Cell Physiol). 2008;295:C752–C760.
37. Jordan K, Solan JL, Dominguez M, et al. Trafficking, assembly, and function of a connexin 43-green fluorescent protein chimera in live mammalian cells. Mol Biol Cell. 1999;10:2033–2050.
38. Juneja SC, Barr KJ, Enders GC, Kidder GM. Defects in the germ line and gonads of mice lacking connexin43. Biol Reprod. 1999;60:1263–1270.
39. Kalcheva N, Qu J, Sandeep N, et al. Gap junction remodeling and cardiac arrhythmogenesis in a murine model of oculodentodigital dysplasia. Proc Natl Acad Sci USA. 2007;104:20512–20516.
40. Kamibayashi Y, Oyamada Y, Mori M, Oyamada M. Aberrant expression of gap junction proteins (connexins) is associated with tumor progression during multistage mouse skin carcinogenesis. Carcinogen. 1995;16:1287–1297.
41. Kanemitsu MY, Loo LWM, Simon S, Lau AF, Eckhart W. Tyrosine phosphorylation of connexin 43 by v-src is mediated by SH2 and SH3 domain interactions. J Biol Chem. 1997;272:22824–22831.
42. Kanter HL, Saffitz JE, Beyer EC. Cardiac myocytes express multiple gap junction proteins. Circ Res. 1992;70:438–444.
43. Kenne K, Fransson-Steen R, Honkasalo S, Warngard L. Two inhibitors of gap junction intercellular communication, TPA and endosulfan: different effects on phosphorylation of connexin 43 in the rat liver epithelial cell line, IAR 20. Carcinogen. 1994;15:1161–1165.
44. Khan-Dawood FS, Yang J, Dawood MY. Expression of gap junction protein connexin-43 in the human and baboon corpus luteum. J Clin Endocrinol Metab. 1996;81:835–842.
45. Kiang DT, Jin N, Tu ZJ, Lin HH. Upstream genomic sequence of the human connexin26 gene. Gene. 1997;199:165–171.
46. Kirchhoff S, Nelles E, Hagendorff A, Kruger O, Traub O, Willecke K. Reduced cardiac conduction velocity and predisposition to arrythmias in connexin40-deficient mice. Curr Biol. 1998;8:299–302.
47. Lai-Chong JE, Arita K, McGrath JA. Genetic diseases of junctions. J Invest Dermatol. 2007;127:2713–2725.
48. Laird DW. Closing the gap on autosomal dominant connexin-26 and connexin-43 mutants linked to human disease. J.Biol Chem. 2007;283:2997–3001.
49. Laird DW, Puranam KL, Revel J-P. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J. 1991;273:67–72.
50. Laird DW, Castillo M, Kasprzak L. Gap junction turnover, intracellular trafficking and phosphorylation of connexin 43 in brefeldin A-treated rat mammary tumor cells. J Cell Biol. 1995;131:1193–1203.
51. Landesman Y, White TW, Starich TA, Shaw JE, Goodenough DA, Paul DL. Innexin-3 forms connexin-like intercellular channels. J Cell Sci. 1999;112:2391–2396.
52. Larsen WJ, Wert SE, Brunner GD. Differential modulation of follicle cell gap junction populations at ovulation. Dev Biol. 1987;122:61–71.
53. Lo CW, Cohen MF, Huang GY, et al. Cx43 gap junction gene expression and gap junctional communication in mouse neural crest cells. Dev Genet. 1997;20:119–132.
54. Loewenstein WR. Junctional intercellular communication and control of growth. Biochim Biophys Acta. 1979;560:1–65.
55. Loewenstein WR. Regulation of cell-to-cell communication by phosphorylation. Biochem Soc Symp. 1985;50:43–58.
56. Loewenstein WR, Kanno Y, Socolar SJ. Quantum leaps of conductance during formation of membrane channels at cell-cell junction. Nature. 1978;274:133–136.
57. Loo LW, Berestecky JM, Kanemitsu MY, Lau AF. pp60src-mediated phosphorylation of connexin 43, a gap junction protein. J Biol Chem. 1995;270:12751–12761.
58. Maestrini E, Korge BP, Ocana-Sierra J, et al. A missense mutation in connexin26, D66H, causes mutiliating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum Mol Genet. 1999;8:1237–1243.
59. Makowski L, Caspar DLD, Phillips WC, Goodenough DA. Gap junction structures II Analysis of the x-ray diffraction data. J Cell Biol. 1977;74:629–645.
60. Mesnil M, Crespin S, Avanzo JL, Dagli ML. Defective gap junctional intercelluar communication in the carcinogenic process. Biochim Biophys Acta. 2005;1719:125–145.
61. Mesnil M, Krutovskikh V, Piccoli C, et al. Negative growth control of HeLa cells by connexin genes: connexin specificity. Cancer Res. 1995;55:629–639.
62. Moennikes O, Buchmann A, Ott T, Willecke K, Schwarz M. The effect of connexin32 null mutation on hepatocarcinogenesis in different mouse strains. Carcinogenesis. 1999;20:1379–1382.
63. Musil LS, Goodenough DA. Gap junctional intercellular communication and the regulation of connexin expression and function. Curr Opin Cell Biol. 1990;2:875–880.
64. Neuhaus IM, Bone L, Wang S, Ionascescu V, Werner R. The human connexin32 gene is transcribed from two tissue-specific promoters. Biosci Rep. 1996;16:239–248.
65. Neveu MJ, Hully JR, Babcock KL, et al. Multiple mechanisms are responsible for altered expression of gap junction genes during oncogenesis in rat liver. J Cell Sci. 1994;107:83–95.
66. Neyton J, Trautmann A. Single-channel currents of an intercellular junction. Nature. 1985;317:331–335.
67. Nonner W, Eisenberg B. Ion permeation and glutamate residues linked by Poisson-Nernst-Planck Theory in L-type calcium channels. Biophys J. 1998;75:1287–1305.
68. Orsino A, Taylor CV, Lye SJ. Connexin-26 and connexin-43 are differentially expressed and regulated in the rat myometrium throughout late pregnancy and the onset of labor. Endocrinology. 1996;137:1545–1553.
69. Pal JD, Berthoud VM, Beyer EC, Mackay D, Shiels A, Ebihara L. Molecular mechanism underlying a Cx50-linked congenital cataract. Am J Physiol. 1999;276:c1443–c1446.
70. Paznekas WA, Karczeski B, Vermeer S, et al. GJA1 mutations, variants, and connexin43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30:724–733.
71. Peters NS. New insights into myocardial arrythmogenesis: distribution of gap junctional coupling in normal ischaemic and hypertrophied human hearts. Clin Sci. 1996;90:447–452.
72. Phelan P. Innexins: members of an evolutionarily conserved family of gap junction proteins. Biochim Biophys Acta. 2005;1711:225–245.
73. Piersanti M, Lye SJ. Increase in messenger ribonucleic acid encoding the myometrial gap junction protein, connexin-43, requires protein synthesis and is associated with increased expression of the activator protein-1, c-fos. Endocrinology. 1995;136:3571–3578.
74. Qu Y, Dahl G. Function of the voltage gate of gap junction channels: selective exclusion of of molecules. Proc Natl Acad Sci USA. 2002;99:697–702.
75. Reaume AG, de Sousa PA, Kulkarni S, et al. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834.
76. Revel J-P, Karnovsky MJ. Hexagonal array of subunits in intercellular junctions in the mouse heart and liver. J Cell Biol. 1967;38:C7–C12.
77. Revel J-P, Hoh JH, John SA, Laird DW, Puranam K, Yancey SB. Aspects of gap junction structure and assembly. Sem Cell Biol. 1992;3:21–28.
78. Sáez JC, Berthoud VM, Brañes MC, Martínez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83 13659–1400.
79. Saez J, Nairn AC, Czernik AJ, et al. Phosphorylation of connexin32, a hepatocyte gap junction protein, by cAMP-dependent protein kinase, protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Eur J Biochem. 1990;192:263–273.
80. Seul KH, Tadros PN, Beyer EC. Mouse connexin40: gene structure and promoter analysis. Genomics. 1997;46:120–126.
81. Simon AM, Goodenough DA, Paul DL. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atriventricular block and bundle branch block. Curr Biol. 1998;8:295–298.
82. Simpson I, Rose B, Loewenstein WR. Size limit of molecules permeating the junctional membrane channels. Science. 1977;195:294–296.
83. Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711:154–163.
84. Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2007;419:261–272.
85. Spray DC, Dermietzel R. X-linked dominant Charcot-Marie-Tooth disease and other potential gap junction diseases of the nervous system. TINS. 1995;18:256–262.
86. Sun Y, Tang W, Chang Q, Wang Y, Kong W, Lin X. Connexin30 null and conditional mice connexin26 null mice display distinct pattern and time course of cellular degeneration in the cochlea. J Comp Neurol. 2009;516:569–579.
87. Swenson KI, Piwnica-Worms H, McNamee H, Paul DL. Tyrosine phosphorylation of the gap junction protein connexin43 is required for the pp60v-src-induced inhibition of communication. Cell Reg. 1990;1:989–1002.
88. Tu ZJ, Kiang DT. Mapping and characterization of the basal promoter of the human connexin26 gene. Biochim Biophys Acta. 1998;1441:169–181.
89. Unger VM, Kumar NM, Gilula NB, Yeager M. Three-dimensional structure of a recombinant gap junction membrane channel. Science. 1999;283:1176–1180.
90. Veenstra RD. Ion permeation through connexin gap junction channels: effects on conductance and selectivity. In: Perrachia C, ed. 2000;:95–129. Current Topics in Membranes. Vol. 49.
91. Veenstra RD, DeHaan RL. Measurement of single channel currents from cardiac gap junctions. Science. 1986;233:972–974.
92. Veenstra RD, Wang H-Z, Beblo DA, et al. Selectivity of connexin-specific gap junctions does not correlate with channel conductance. Circ Res. 1995;77:1156–1165.
93. Veenstra RD, Wang H-Z, Westphale EM, Beyer EC. Multiple connexins confer distinct regulatory and conductance properties of gap junctions in developing heart. Circ Res. 1992;71:1277–1283.
94. Waldo KL, Lo CW, Kirby ML. Connexin43 expression reflects neural crest patterns during cardiovascular development. Dev Biol. 1999;208:307–323.
95. Warn-Cramer BJ, Lampe PD, Kurata WE, et al. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem. 1996;271:3779–3786.
96. White TW, Goodenough DA, Paul PL. Targeted ablation of connexin 50 in mice results in micropthalmia and zonular pulverulent cataracts. J Cell Biol. 1998;143:815–825.
97. Yamaguchi DT, Huang JT, Ma D. Regulation of gap junction intercellular communication by pH in MC3T3-E1 osteoblastic cells. J Bone Min Res. 1995;10:1891–1899.
98. Yamasaki H. Aberrant expression and function of gap junctions during carcinogenesis. Env Health Prosp. 1991;93:191–197.
99. Yang YQ, Liu X, Zhang XL, et al. Novel connexin40 missense mutations in patients with familial atrial fibrillation. Europace. 2010;12:1421–1427.
100. Yu W, Dahl G, Werner R. The connexin43 gene is responsive to oestrogen. Proc Roy Soc (London) Series B Biol Sci. 1994;255:125–132.