Chapter 24
Direct Regulation of Ion Channels by GTP-Binding Proteins
Chapter Outline
II. G-Protein-Coupled Receptors
III. The G-Protein Cyclic Reaction Mediates Receptor-to-Channel Signal Transmission
IV. Electrophysiological Evidence for K+ Channel Activation by G Proteins
V. Electrophysiological Properties of KG Channels
VI. Direct Coupling of KG Channel Subunits to Gβγ
VII. Structural Basis of the Regulation of KG Channel Activity
VIII. RGS Proteins Confer Voltage-Dependent Gating on KG Channel
I Introduction
Various extracellular stimuli can initiate physiological processes by changing membrane potentials. These stimuli regulate ionic flows by controlling the opening and closing of ion channel pores. A cluster of membrane receptors that are coupled to heterotrimeric GTP-binding proteins (G proteins) provide the largest contribution to these signaling pathways. This type of receptor is called a G-protein-coupled receptor (GPCR) and it regulates a relatively small number of different enzymes and ion channels. Because sequential protein interactions mediate GPCR signaling, the signaling pathways cause the temporal range of cellular responses to vary from milliseconds to hours. The responses are obviously slower than those evoked by ionotropic neurotransmitter receptors, which operate within milliseconds. Many ion channels are under direct or indirect control by G proteins. One example of the regulation of an ion channel by the direct action of G proteins is that of activation by acetylcholine (ACh) of the muscarinic K+ channel in the heart (Kurachi, 1995). This action is responsible for the deceleration of the heartbeat upon stimulation of the vagus nerve (Fig. 24.1A) (Loewi, 1921). This K+ channel is also responsible for the formation of the slow inhibitory postsynaptic potential in the central nervous system (Fig. 24.1B) (Lüscher and Slesinger, 2010). Furthermore, it is also known that this system is responsible for inhibition of presynaptic Ca2+ channels (Dolphin, 1998; Currie, 2010). Direct regulation of ion channels by G proteins is therefore an important cell signaling system.
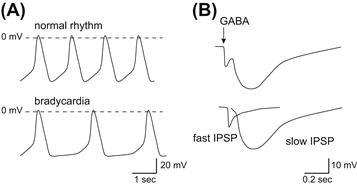
FIGURE 24.1 Physiological implications of the control of cell excitability by KG channels. (A) Control of cell excitability in the sinoatrial nodal cells. Vagus nerve stimulation augments K+ conductance at potentials near the equilibrium potential of K+ and prolongs the interval between action potentials without affecting the action potential configuration. (B) The control of excitability at the inhibitory postsynaptic membranes. γ-Amino butyric acid (GABA) released at the synaptic cleft elicits biphasic changes in the postsynaptic membrane potential of inhibitory synapses (IPSP). While ionotropic GABAA receptors carrying Cl− influx mediate the fast response, metabotropic GABAB receptors increase the K+ efflux via G proteins.
II G-Protein-Coupled Receptors
G-protein-coupled receptors (GPCRs) are encoded by over 900 genes and they constitute the largest family of membrane-embedded proteins in the mammalian genome (Fredriksson et al., 2003; Bjarnadottir et al., 2006). GPCRs are important targets for drugs in all therapeutic areas, and they form the sites for the effects of 25–30% of existing drugs (Overington et al., 2006). GPCRs possess heptahelical transmembrane segments and their N- and C-termini are exposed to the extracellular and intracellular sides of the membrane, respectively (Fig. 24.2A). The GPCRs in the human genome can be divided into five subfamilies that have diverse N-terminal extracellular architectures at the sites where ligands bind and trigger activation of the receptor (Fredriksson et al., 2003). Although the ligands interact with sites differently positioned within GPCRs, they ultimately evoke conformational changes at the cytoplasmic interface to activate G proteins. The most significant conformational change is probably an outward movement of helix VI and subsequent reorientation of the cytoplasmic end of the helix (Fig. 24.2A). This may be linked with the opening of a crevice within the intracellular surface of the receptor and it could allow Gα proteins to interact at their C-termini (Scheerer et al., 2008).
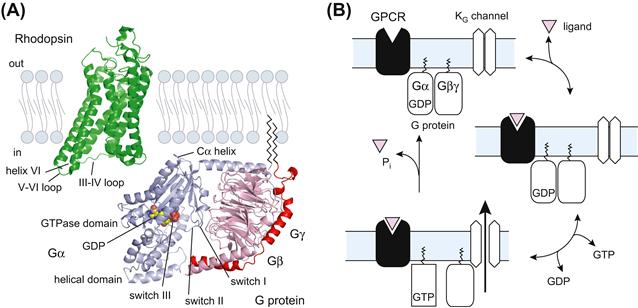
FIGURE 24.2 Molecular architectures of GPCR and G proteins and the G-protein cycle. (A) Molecular architectures of rhodopsin and G protein. The structures of retinal-free rhodopsin (Protein Data Bank identifier 3DQB) is colored in green and the trimeric complexes containing the Gαt/Gαi chimera and the Gβγt subunits (1GOT) are colored in blue-gray and pink, respectively. The bound nucleotide on Gα is represented as a sphere. Palmitoylation and myristoylation are common post-transcriptional modifications at the N-terminus of Gα and the C-terminus of Gγ, respectively. (B) Schematic representation of the G-protein cycle. GPCR: G-protein-coupled receptor; Gα and Gβγ: α and βγ subunits of trimeric G protein, respectively; KG channel: G-protein-gated inwardly rectifying K+ channel.
III The G-Protein Cyclic Reaction Mediates Receptor-to-Channel Signal Transmission
G proteins consist of Gα, Gβ and Gγ subunits (Gilman, 1987; Sprang, 1997; Oldham and Hamm, 2008). To date, at least 16 Gα, 5 Gβ and 12 Gγ genes have been identified. Gα has been subclassified into four groups (Gαs, Gαi, Gαq and Gα12) on the basis of sequence similarity, coupling effectors and sensitivity to toxins. The crystal structures of Gα and Gαβγ have revealed the presence of two preserved domain architectures: the GTPase domain and the helical domain. The former is conserved among all members of G proteins and it hydrolyzes GTP. The latter is specific for Gα, which interacts with the GTPase domain by covering the nucleotide-binding pocket. Under physiological conditions, Gβ and Gγ associate tightly to form a dimer (Gβγ). Gβ is in the form of a seven-bladed propeller, each blade of which is a four-stranded anti-parallel β−sheet. The N-terminus of Gβ possesses an α-helix that forms a coiled-coil with the N-terminus of Gγ. The physiological significance of the possible heterogeneity of Gβγ has been a matter of debate.
In the absence of agonists, Gα exists predominantly in an inactive GDP-bound state (Gα-GDP) (see Fig. 24.2B) (Gilman, 1987; Oldham and Hamm, 2008). Gα-GDP has a high affinity for Gβγ, forming a heterotrimer. Receptor stimulation allows Gα and GDP to dissociate, which results in marked acceleration of the GDP–GTP exchange reaction. Formation of GTP-bound Gα (Gα-GTP) leads to dissociation of Gβγ from Gα-GTP. The separate components of the G protein (Gα-GTP and Gβγ) are subsequently available to transduce signals to the downstream effectors. Gα hydrolyzes the bound GTP to GDP, thereby returning to its GDP-bound state (Gα-GDP), which re-associates with Gβγ. This reaction terminates effector regulation, and the agonist-occupied receptor triggers the heterotrimeric G protein to restart the cycle.
hydrolyzes the bound GTP to GDP, thereby returning to its GDP-bound state (Gα-GDP), which re-associates with Gβγ. This reaction terminates effector regulation, and the agonist-occupied receptor triggers the heterotrimeric G protein to restart the cycle.
The C-terminus of Gα probably binds to the open crevice in a ligand-associated GPCR (Sprang, 1997; Scheerer et al., 2008). Multiple contacts mediate the interaction of Gα with GPCR. Furthermore, Gβ and Gγ stabilize the GPCR-G protein complex. Extensive contact is necessary for a change to occur in the conformation of Gα, leading to the GDP–GTP exchange reaction. The partially overlapped areas are thought to be involved in the specificity of GPCR-coupling on G proteins. Although poor sequence conservation prevents the identification of the source of G-protein-coupling specificity on GPCR, cytoplasmic loops III–IV and the C-terminus are believed to be closely involved.
IV Electrophysiological Evidence for K+ Channel Activation by G Proteins
On stimulation of vagus nerves, ACh released from synaptic terminals decelerates the heartbeat and decreases atrioventricular conduction (see Fig. 24.1A) (Loewi, 1921). ACh causes membrane hyperpolarization in the heart (Del Castillo and Katz, 1955), by increasing K+ efflux across the cell membrane (Hutter and Trautwein, 1956). A specific population of K+ channels, the muscarinic K+ (KACh) channels, is responsible for this efflux (Trautwein and Dudel, 1958; Noma and Trautwein, 1978) and treatment with a toxin from Bordetella pertussis prevents activation of the channel by m2-muscarinic and A1-adenosine receptors (Kurachi et al., 1986a). In cell-free inside-out membrane patches, internal GTP (in the presence of extracellular agonists) and non-hydrolyzable analog of GTP, GTPγS, activate the KACh channel (Kurachi et al., 1986b). These observations led to the proposal that the channel is directly activated by G proteins in a membrane-delimited manner. Subsequently, it was found that Gβγ, but not Gα, was the subunit responsible for activating the KACh channel (Logothetis et al., 1987). Because G-protein-sensitive K+ (KG) channels in neurons and endocrine cells are also regulated by Gβγ like cardiac KACh channels, we refer to these types of channel as “KG channels” henceforth.
V Electrophysiological Properties of KG Channels
The difference between the membrane potential (Vm) and the equilibrium potential of K+ (EK) drives K+ ions through KG channels (Trautwein and Dudel, 1958; Noma and Trautwein, 1978; Kurachi, 1995; Yamada et al., 1998). The manner of K+ conduction through KG channels is unique, involving a large K+ conductance at potentials negative relative to EK, but less outward current flow at potentials positive relative to EK (Fig. 24.3A). Blocking of the outward K+ flow by intracellular Mg2+ and polyamines is responsible for this diode-like conduction and this property is similar to that originally observed as “anomalous” rectifier K+ currents in skeletal muscles of the frog (Katz, 1949). The unidirectional conduction permits a prolongation of the interval of action potential with little effect on the action potential configuration (see Fig. 24.1A). Therefore, the inward rectification property of KG channels is fundamental to their physiological function.
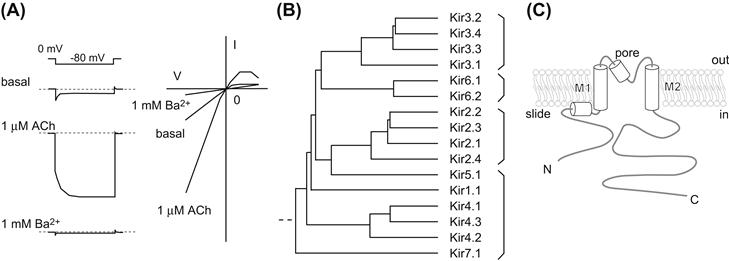
FIGURE 24.3 Kir channel members and their structural description. (A) Representative current response of the KG channel. The reconstituted KG channel with Kir3.1, Kir3.4 and m2-muscarinic receptor shows a rapid change in current followed by a gradual increase on application of hyperpolarizing voltage steps from EK. External Ba2+ practically blocks most Kir channels. (B) Phylogenic tree of Kir channels. The Kir channel members can be classified into four groups: G: protein-gated Kir (KG) channels, Kir3.x; ATP-sensitive Kir channels, Kir6.x; classical Kir channels, Kir2.x; K+ transport-Kir channels, Kir1.1, Kir4.x, Kir5.1 and Kir7.1. (C) Simple representation of Kir channels. Each subunit contains a transmembrane domain that flanks cytoplasmic N- and C-termini.
The activity of the KG channel increases in response to GPCR activation. As discussed below, direct interactions with Gβγ and the KG channel are responsible for this response. How does Gβγ augment the channel activity? The macroscopic current amplitude (I) of the KG channels can be defined as follows:
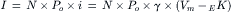 (24.1)
(24.1)
where N is the number of channels in the patch membrane, Po is the probability of the channel being open, i is the single-channel current amplitude and γ is the single-channel conductance. Therefore, the changes in macroscopic current amplitude during continuous recording can be explained in terms of changes in Po and γ. The single-channel conductance of either a native or a recombinant KG channel is dependent on the difference between Vm and EK and is independent of receptor stimulation, thereby showing that the increase in current amplitude caused by Gβγ is the result of an increase in the Po of the channels.
VI Direct Coupling of KG Channel Subunits to Gβγ
Mammalian KG channels form a subgroup of inward rectifier K+ (Kir) channels (see Fig. 24.3B) (Yamada et al., 1998; Hibino et al., 2010). Each channel consists of four Kir3.x subunits: Kir3.1, Kir3.2, Kir3.3 and Kir3.4 (Kubo et al., 2005). Kir3.1/GIRK1 was the first KG channel subunit to be isolated from rat atrium (Kubo et al., 1993). Subsequently, Kir3.2–Kir3.4 were isolated from brain and atrial cDNA libraries (Krapivinsky et al., 1995). These gene transcripts are translated into several splicing variants. Four individual Kir3.x subunits assemble to form functional channels. The primary combination of subunits for the Ikach is Kir3.1 and Kir3.4 (Krapivinsky et al., 1995). In neural tissues, the KG channels consist mainly of a heteromeric Kir3.1/Kir3.2 channel and a homomeric Kir3.2 channel. Kir3.3 also assembles to form neuronal KG channels with Kir3.1 and/or Kir3.2. The primary amino acid sequences of Kir channels show that they possess common transmembrane segments (TM1 and TM2), flanked by a pore-forming region that has a signature motif (T-X-G-Y/F-G) for K+-selective ion channels (see Fig. 24.3C) (Jan and Jan, 1997). Furthermore, it has been speculated that the unique cytoplasmic N- and C-termini to Kir channels play roles in ion conduction and regulation of gating specific to Kir channels.
Proof of the regulation of KG channel activity by direct interactions of Gβγ has been provided by several experiments. First, the co-expression of m2-muscarinic receptor with KG channel subunits in host cells yielded ACh-dependent channel activity (see Fig. 24.3A) (Reuveny et al., 1994). Secondly, further expression of Gβγ resulted in a constitutively active channel, an effect that could be cancelled by additional expression of the Gβγ-binding domain of β-adrenergic receptor kinase 1 (Reuveny et al., 1994). Thirdly, Gβγ directly bound purified cytoplasmic N- and C-termini of KG channel subunits, as well as purified recombinant and native KG channels composed of Kir3.1 and Kir3.4 (Yamada et al., 1998; Bichet et al., 2003). The potential sites for Gβγ binding were thus narrowed down to two distinct regions in the N- and C-termini and several amino acids (the corresponding His57, Leu262, Leu333, and Gly336 in Kir3.1) appear to be critically involved in the Gβγ-dependent activation process.
Because all Kir channels require phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] for activation, the lipid is a prerequisite for opening of the KG channel (Logothetis et al., 2007; Hibino et al., 2010). However, Gβγ and PtdIns(4,5)P2 cannot activate the channel by themselves. How then do Gβγ and PtdIns(4,5)P2 regulate the activity of the KG channel? Electrophysiological and biochemical experiments have shown that the association of Gβγ probably strengthens the channel–PtdIns(4,5)P2 interactions (Hilgemann and Ball, 1996). Differences in the turnover rate of PtdIns(4,5)P2 or in the membrane property of lateral diffusion of PtdIns(4,5)P2 may account for differences in the sensitivity of KG channels to PtdIns(4,5)P2 in different cell lines.
VII Structural Basis of the Regulation of KG Channel Activity
The crystal structures of the isolated cytoplasmic domain of Kir3.1 (Nishida and MacKinnon, 2002) and of the entire bacterial Kir channel homolog KirBac1.1 (Kuo et al., 2003) revealed the structural features of Kir channels (Fig. 24.4A). The Kir channel consists of two functionally and structurally distinct domains: the transmembrane domain and cytoplasmic domain (Bichet et al., 2003; Kuo et al., 2003). The former has a significant degree of similarity with those in other K+ channels and consists primarily of two long helices (TM1 and TM2), two short helices (slide and pore helices) and a selectivity filter (see Figs. 24.3C and 24.4B). The ion-conduction pathway is located at the center of this assembly. The pore starts at the external vestibule and connects to the selectivity filter. In the middle of the membrane, there is a widening in space, called the central cavity. The C-terminal end of the pore helix is oriented toward the midpoint of the cavity and this is presumed to play a role in the positioning of K+. The TM2 helix from each subunit makes a physical constraint at the area closed to the cytoplasm. The slide helix located at the internal surface of membrane is connected to the cytoplasmic N-terminus through a short portion consisting of about 10 amino acids.
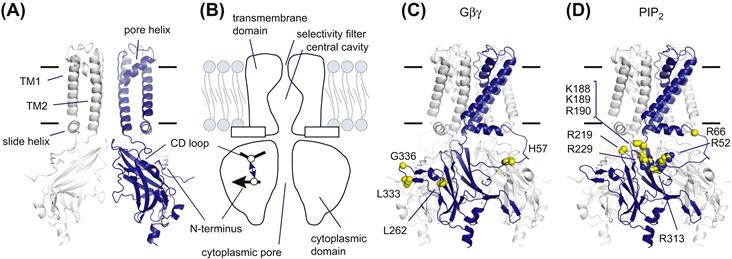
FIGURE 24.4 Structural features of KG channels. (A) Molecular architecture of KG channels. The KG channel has an ion-conduction pathway at the center of a tetrameric assembly. The front and back subunits are omitted for clarity. (B) A schematic representation of KG channels. The channels consist of two structurally distinct domains: a transmembrane domain and a cytoplasmic domain. The inter-subunit ionic bond between the N-terminus and the CD loop lowers the sensitivity to PtdIns(4,5)P2 and stabilizes a closed conformation (Inanobe et al., 2010). (C) Residues involved in Gβγ-interaction. Several residues in the KG channel subunits have been reported to mediate Gβγ-dependent activation. The equivalent residues in Kir3.1 are shown by spheres. (D) Mapping of residues participating in PtdIns(4,5)P2-dependent activation. Many residues in Kir channels have been identified as being involved in PtdIns(4,5)P2-dependent activation. The corresponding residues in Kir3.1 are indicated by spheres.
The cytoplasmic N- and C-termini contribute to a domain folding (see Fig. 24.4A). The domain consists of four individual subunits, each of which has an immunoglobulin-like β-sandwich fold. There is a water-filled pore at the fourfold symmetry axis perpendicular to the membrane. Because the residues involved in the inward rectification are distributed on the internal surface of the pore, the cytoplasmic pore extends the distance that K+ must travel to cross the membrane. The N-terminus of one subunit interacts with the C-terminus of an adjacent subunit.
The residues involved in association with Gβγ and PtdIns(4,5)P2 can be mapped onto the three-dimensional model (see Figs. 24.4C and D). Among the residues involved in Gβγ-dependent channel activation in Kir3.1, His57 is located on the N-terminus, whereas Leu333 and Gly336 are positioned on a loop between the βL and βM strands (see Fig. 24.4C) (Bichet et al., 2003; Logothetis et al., 2007; Hibino et al., 2010). In addition, the regions involved in the binding to Gβγ are clustered at the outer edge of a tetrameric assembly where the sites permit interaction with Gβγ. PtdIns(4,5)P2-sensitive residues are widely distributed in the cytoplasmic domain facing the membrane (see Fig. 24.4D) (Logothetis et al., 2007; Hibino et al., 2010). The cytoplasmic end of TM2 accommodates a cluster of positively charged residues and the linker connecting the cytoplasmic N-terminus to a slide helix and a loop between the βC and βD strands (CD loop) also contains basic amino residues.
What is the mechanism underlying the Gβγ-induced increase in the affinity to PtdIns(4,5)P2? One hypothesis is that the association of Gβγ induces a conformational change that creates a binding pocket for PtdIns(4,5)P2. Another hypothesis is that Gβγ disrupts the inhibitory mechanism that hinders association with PtdIns(4,5)P2. It becomes apparent that, to keep the channel insensitive to PtdIns(4,5)P2, Kir3.2 forms an inter-subunit ionic bond between His69 on the N-terminus and Asp228 on the CD loop (see Fig. 24.4B) (Inanobe et al., 2010); the former residue is equivalent to the residues that are crucial for Gβγ-dependent activation and the latter residue is mandatory for Na+-dependent activation. The crevice between the N-terminus and the CD loop serves as a high-affinity binding site for Cd2+ and the bound Cd2+ tethers the N-terminus and CD loop to inhibit Kir3.2 activity. It seems likely therefore that the mode of action of Gβγ is liberation of the channel from the inactive state to allow the channel to have a high affinity for PtdIns(4,5)P2. These observations also suggest that the cytoplasmic domain of KG channel changes its conformation during gating. How does such conformational change lead to opening of the channel? At the transmembrane domain, the pivoting movement of TM2 and restructuring of the selectivity filter appear to be involved in the control of ionic flow (Jin et al., 2002; Bichet et al., 2003). The rotational and rigid conformational changes of the transmembrane domain against the cytoplasmic domain are also predicted to be coupled with gating (Riven et al., 2003). Furthermore, the widening of the cytoplasmic pore and the rearrangement of the domain interface have also been proposed as potential conformational changes in the bacterial Kir channel (Clarke et al., 2010).
VIII RGS Proteins Confer Voltage-Dependent Gating on KG Channel
Upon hyperpolarization, the KG channel current increases progressively after a current jump, whereas the opposite occurs upon depolarization. The slow time-dependent current change is called “relaxation” and is characteristic for the native KG current (see Fig. 24.3A) (Noma and Trautwein, 1978). The degree of the relaxation varies with the strength of the receptor stimulation, but such relaxation behavior is not observed in cells that express only KG channels and m2-muscarinic receptor.
Regulators of G-protein signaling (RGS) proteins function as GTPase-activating proteins (GAP) specific to Gα to terminate G-protein-mediated signaling (Ross and Wilkie, 2000). To date, more than 20 RGS proteins have been identified. They share a conserved structure of 120 amino acids, designated the “RGS domain”, which is responsible for their GAP activity. Actually, RGS proteins accelerate the time-course of activation and deactivation of KG currents induced by agonists (Saitoh et al., 1997). Subsequently, RGS protein has been recognized as the protein that confers depolarization-induced facilitation on the channel by decreasing the KG channel availability (Fig. 24.5) (Ishii et al., 2001). In the resting state, PtdIns(3,4,5)P3 binds to the RGS domain and the GAP activity of RGS is inhibited. Depolarization facilitates the influx of Ca2+, resulting in the formation of Ca2+/calmodulin (CaM). This complex binds to the RGS domain, thereby ending the PtdIns(3,4,5)P3-mediated inhibition and restoring GAP activity. Therefore, at depolarized potentials, the G-protein cycle is negatively regulated by Ca2+/CaM-bound RGS proteins and the number of active KG channels is decreased by the number of Gβγ moieties available to open the KG channels. This is the mechanism of the voltage-dependent relaxation behavior of KG channels.
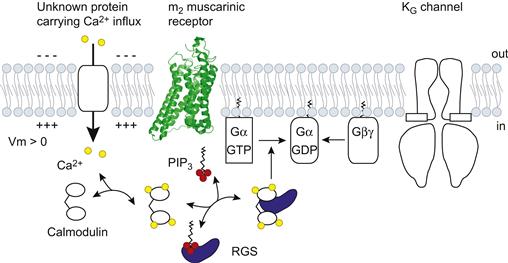
FIGURE 24.5 Regulation of G-protein-mediating channel activation by RGS proteins. Ca2+ influx facilitates the formation of Ca2+/CaM, which binds to RGS proteins and competes with PtdIns(3,4,5)P3. This binding restores the GAP activity and control the G protein cycle negatively.
IX Conclusions
Research in the last decade has provided structural insights into the polypeptides that are involved in G-protein signaling at the atomic level. Since available conformations are limited, it is still difficult to address how proteins change their conformation and transduce the signals. Molecular dynamics simulation is a powerful tool to provide statistical mechanics of the proteins. However, restrictions by computational power and its theory hamper the precise corroboration of the ideas about dynamic features of the proteins and signaling. Therefore, multiple structural models for different functional states are necessary to grasp how the molecule operates and transfers signals. Furthermore, a lipid raft, an organization of specialized membrane microdomains in which cholesterol, sphingolipids and membrane proteins are preferentially associated, has the power to modulate this membrane-delimited signal transduction. Such a system may be linked to the idea that GPCR, G proteins, RGS proteins and KG channels are precoupled, even in the absence of ligands (Lüscher and Slesinger, 2010). In addition, the molecular entity participating in the Ca2+ influx should be clarified to permit understanding of the convergence of two different cellular signaling pathways involving the G protein and the membrane potential, respectively (Ishii et al., 2001). We therefore need to achieve greater clarification regarding the regulatory mechanism of ion channels at the molecular level as well as at the cellular level.
BIBLIOGRAPHY
1. Bichet D, Haass FA, Jan LY. Merging functional studies with structures of inward-rectifier K+ channels. Nat Rev Neurosci. 2003;4:957–967.
2. Bjarnadottir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schioth HB. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics. 2006;88:263–273.
3. Clarke OB, Caputo AT, Hill AP, Vandenberg JI, Smith BJ, Gulbis JM. Domain reorientation and rotation of an intracellular assembly regulate conduction in Kir potassium channels. Cell. 2010;141:1018–1029.
4. Currie KP. G protein modulation of CaV2 voltage-gated calcium channels. Channels (Austin). 2010;4:497–509.
5. Del Castillo J, Katz B. Production of membrane potential changes in the frog’s heart by inhibitory nerve impulses. Nature. 1955;175:1035.
6. Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. J Physiol. 1998;506:3–11.
7. Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272.
8. Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649.
9. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366.
10. Hilgemann DW, Ball R. Regulation of cardiac Na+, Ca2+ exchange and KATP potassium channels by PIP2. Science. 1996;273:956–959.
11. Hutter OF, Trautwein W. Vagal and sympathetic effects on the pacemaker fibers in the sinus venosus of the heart. J Gen Physiol. 1956;39:715–733.
12. Inanobe A, Nakagawa A, Matsuura T, Kurachi Y. A structural determinant for the control of PIP2 sensitivity in G protein-gated inward rectifier K+ channels. J Biol Chem. 2010;285:38517–38523.
13. Ishii M, Inanobe A, Fujita S, Makino Y, Hosoya Y, Kurachi Y. Ca2+ elevation evoked by membrane depolarization regulates G protein cycle via RGS proteins in the heart. Circ Res. 2001;89:1045–1050.
14. Jan LY, Jan YN. Cloned potassium channels from eukaryotes and prokaryotes. Annu Rev Neurosci. 1997;20:91–123.
15. Jin T, Peng L, Mirshahi T, Rohacs T, et al. The βγ subunits of G proteins gate a K+ channel by pivoted bending of a transmembrane segment. Mol Cell. 2002;10:469–481.
16. Katz B. Les constantes electriques de la membrane du muscle. Arch Sci Physiol. 1949;3:285–299.
17. Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374:135–141.
18. Kubo Y, Adelman JP, Clapham DE, et al. International Union of Pharmacology LIV Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. 2005;57:509–526.
19. Kubo Y, Reuveny E, Slesinger PA, Jan YN, Jan LY. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature. 1993;364:802–806.
20. Kuo A, Gulbis JM, Antcliff JF, et al. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926.
21. Kurachi Y. G protein regulation of cardiac muscarinic potassium channel. Am J Physiol. 1995;269:C821–C830.
22. Kurachi Y, Nakajima T, Sugimoto T. Acetylcholine activation of K+ channels in cell-free membrane of atrial cells. Am J Physiol. 1986a;251:H681–H684.
23. Kurachi Y, Nakajima T, Sugimoto T. On the mechanism of activation of muscarinic K+ channels by adenosine in isolated atrial cells: involvement of GTP-binding proteins. Pflügers Arch. 1986b;407:264–274.
24. Loewi O. Über humorale Übertragbarkeit der Herznervenwirkung. Pflügers Arch. 1921;189:239–242.
25. Logothetis DE, Jin T, Lupyan D, Rosenhouse-Dantsker A. Phosphoinositide-mediated gating of inwardly rectifying K+ channels. Pflügers Arch. 2007;455:83–95.
26. Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326.
27. Lüscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci. 2010;11:301–315.
28. Nishida M, MacKinnon R. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 Å resolution. Cell. 2002;111:957–965.
29. Noma A, Trautwein W. Relaxation of the ACh-induced potassium current in the rabbit sinoatrial node cell. Pflügers Arch. 1978;377:193–200.
30. Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71.
31. Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there?. Nat Rev Drug Discov. 2006;5:993–996.
32. Reuveny E, Slesinger PA, Inglese J, et al. Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature. 1994;370:143–146.
33. Riven I, Kalmanzon E, Segev L, Reuveny E. Conformational rearrangements associated with the gating of the G protein-coupled potassium channel revealed by FRET microscopy. Neuron. 2003;38:225–235.
34. Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827.
35. Saitoh O, Kubo Y, Miyatani Y, Asano T, Nakata H. RGS8 accelerates G-protein-mediated modulation of K+ currents. Nature. 1997;390:525–529.
36. Scheerer P, Park JH, Hildebrand PW, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502.
37. Sprang SR. G protein mechanisms: insights from structural analysis. Annu Rev Biochem. 1997;66:639–678.
38. Trautwein W, Dudel J. [Mechanism of membrane effect of acetylcholine on myocardial fibers]. Pflügers Arch. 1958;266:324–334.
39. Yamada M, Inanobe A, Kurachi Y. G protein regulation of potassium ion channels. Pharmacol Rev. 1998;50:723–760.