Chapter 26
Regulation of Ion Channel Localization and Activity Through Interactions with the Cytoskeleton
Chapter Outline
III. Mechanisms for Interactions Between the Cytoskeleton and Ion Channels
IIIA. The Spectrin-Based Membrane Skeleton
IIIA1. Characteristics of the Spectrin-Based Membrane Skeleton
IIIA2. The Ankyrins and their Binding Partners
IIIA3. The Spectrin-Based Membrane Skeleton at the Axon Initial Segment and Nodes of Ranvier
IIIA4. The Spectrin-Based Membrane Skeleton in Muscle Physiology
IIIA5. Mutations Affecting Ankyrin Interactions in Human Cardiac Pathology
IIIA6. Regulation of the Association Between Ankyrin and its Integral Partners
IIIB. PDZ Proteins and the Postsynaptic Density
IIIB1. The Postsynaptic Density and the Dendritic Spine: Dynamic Interactions Between Ion Channel Receptors and the Actin Cytoskeleton
IIIB2. PDZ Proteins and the Immobilization of Postsynaptic Receptors
IIIB4. Regulation of Synaptic Strength through Interactions with Scaffolding Proteins
IIIC. Gephyrin and the Inhibitory Synapse
IIID. The Interaction of Epithelial Ion Channels with the Cytoskeleton
I Summary
The physiological function of a variety of cell types is dependent upon their ability to organize and cluster ion channels into specialized areas of the plasma membrane. Studies over the last 20 years have indicated a role for interactions between ion channels and the cellular cytoskeleton in achieving this complex organization. A variety of adaptor/scaffolding proteins that tether ion channels to the cytoskeleton has been identified. In addition to localizing ion channels, interactions with the cytoskeleton may also regulate channel activity. This could be through direct binding to the channel by actin or scaffolding molecules, or by the co-recruitment of signaling molecules that regulate channel activity into a macromolecular complex by scaffolding proteins.
In this chapter, we will review three examples of specialized membrane domains: the axon initial segment and node of Ranvier; the postsynaptic membrane; and the apical domain of the epithelial cell. We will examine the unique adaptor/scaffolding systems found in each of these domains and how they have evolved in response to their particular physiological requirements.
II General Introduction
Ion channels are selective “pores” for the passage of ions across the hydrophobic barrier of the plasma membrane. The physiological activity of a number of cell types is dependent upon their ability to organize integral membrane proteins and, in particular, ion channels, into functionally specialized domains of the plasma membrane. This organization underlies a wide range of cellular functions including the propagation and integration of electrical signals in the nervous and muscular system by “excitable” membranes. A general mechanism whereby ion channels become organized into these membrane domains is shown in Fig. 26.1 and reflects an interaction between the ion channel and the cellular cytoskeleton, particularly the actin microfilament system. This mechanism localizes the lateral mobility of ion channels to a membrane domain through direct tethering of the ion channel to the cytoskeleton utilizing adaptor or scaffold proteins.
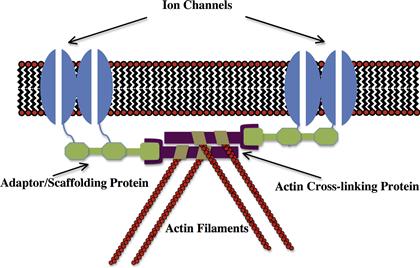
FIGURE 26.1 Conceptual model of a general mechanism for the interaction of ion channels with the cytoskeleton.
As well as restricting the localization of ion channels, interactions with the cytoskeleton and adaptor proteins can also regulate their activity. This could result from direct interactions between actin or adaptor proteins with functional domains of the channel and/or the organization of functional microdomains, whereby ion channels through the activity of scaffold proteins, are brought into close proximity with molecules that can regulate their activity. Conversely, several lines of evidence, particularly from the nervous system, have shown that it is possible for ion channel activity to activate signaling pathways that remodel the actin cytoskeleton.
In this chapter, we will examine a number of different examples of ion channel–cytoskeleton interactions illustrating some of the important concepts in this area. The rapid development of knowledge in this field over the last decade makes it impossible to list all of the known examples of these types of interactions. For example, we have not included discussion of the neuromuscular junction, which is a well-characterized example of the role of the cytoskeleton in the immobilization of postsynaptic receptors or the rapidly growing area of mechanosensory transduction. Apologies are therefore offered to all colleagues where space limitations have prevented mention of their work.
III Mechanisms for Interactions Between the Cytoskeleton and Ion Channels
IIIA The Spectrin-Based Membrane Skeleton
IIIA1 Characteristics of the Spectrin-Based Membrane Skeleton
Originally described in the human erythrocyte, where it plays a major role in maintaining cell shape (reviewed in Bennett and Gilligan, 1993), the spectrin-based membrane skeleton is perhaps the best characterized complex of proteins linking ion channels to the actin cytoskeleton. As shown in Fig. 26.2, at the heart of this complex of proteins is the filamentous protein spectrin, which exists as a 200 nm flexible rod composed of a heterotetramer of α and β subunits. Beta subunits are characterized by the possession of an actin-binding domain at their N-terminus. Heterotetramers and other higher ordered structures form through head to head association of the heterodimer to yield a molecule capable of cross-linking actin through binding sites at opposite ends. In the red cell membrane, short actin oligomers (10–14 monomers) are cross-linked in this fashion to yield a series of pentameric or hexameric structures with actin at the junction (termed the junctional complex) and spectrin filaments organized like spokes of a wheel. Stretched examples of this structure have been visualized by electron microscopy (Byers and Branton, 1985). The molecules protein 4.1 and adducin facilitate the interaction of spectrin with actin at these junctional complexes (Bennett and Gilligan, 1993).
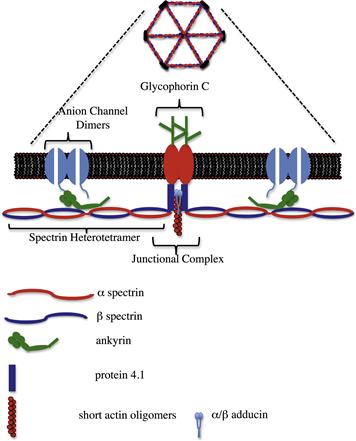
FIGURE 26.2 Cartoon of the major features of the spectrin-based membrane skeleton. Spectrin heterotetramers are arranged around an actin-based junctional complex to generate a hexagonal/pentagonal lattice that can be viewed under the electron microscope when the membrane skeleton is stretched. This lattice is laminated to the bilayer through the interactions of ankyrin with the anion channel and β-spectrin, in addition to interactions between protein 4.1, spectrin and glycophorin C at the junctional complex.
The spectrin-based membrane skeleton forms an electron dense subplasmalemmal undercoating, which can be visualized by electron microscopy and is associated with a number of specialized plasma membrane domains. In red cells, the skeleton is laminated to the bilayer through two major sites of interaction: an association between protein 4.1 which binds both to the β subunit of spectrin and the integral membrane protein glycophorin C and through the molecule ankyrin, which binds both to the β spectrin subunit and to the cytoplasmic domain of the anion channel band 3 (AE1). Mutations that affect the anion channel–ankyrin–spectrin association in red cells result in a loss of membrane lamination, destabilization of the lipid bilayer and a loss of plasma membrane resulting in a shift in cell morphology from discocytes to spherocytes (Palek and Lambert, 1990). In this system, the lateral mobility of the anion channel is restricted in two ways: (1) through the direct interaction of the anion channel with ankyrin and the spectrin bilayer; and (2) through the “corralling” of anion channels within compartments demarcated by the spectrin lattice (Kodippili et al., 2009). However, for the purposes of this review we shall focus on the direct interactions of ankyrins with integral proteins and how this mechanism is used to localize ion channels to specialized plasma membrane domains.
IIIA2 The Ankyrins and their Binding Partners
As shown in Fig. 26.2, the ankyrins are multifunctional adaptors characterized by domains that associate with the β subunit of spectrin and with the cytoplasmic domains of a wide variety of integral membrane proteins, including cell adhesion molecules, transporters and ion channels (Bennett and Healy, 2009). The domain that associates with integral proteins (the membrane-binding domain) consists of 24 copies of the ANK repeat motif now identified in a large number of polypeptides. These repeats are stacked in an array with four basic subdomains such that protein–protein interactions may involve the participation of contact sites from multiple different repeats (Michaely and Bennett, 1993). Binding sites within integral membrane proteins for this domain of ankyrin have arisen independently a number of times during evolution. Hence, it is not easily possible to identify ankyrin-binding sequences within integral proteins from simple analysis of primary sequence. For example, the amino acid sequence associated with ankyrin binding in the family of voltage-gated sodium channels (Garrido et al., 2001; Lemaillet et al., 2003), bears no homology to that identified for members of the L1 family of cell adhesion molecules that also bind ankyrin (Davis and Bennett, 1994). Intriguingly, the same ankyrin-binding sequence that is found in voltage-gated sodium channels is also found in the voltage-gated potassium channels KCNQ2/3 (Pan et al., 2006), responsible for the so-called M-type current. These sequences in KCNQ2/3 appear to have evolved independently and at a later stage in evolution than that of the sodium channels, as one of the first examples of convergent molecular evolution (Hill et al., 2008).
In addition to the membrane and spectrin-binding domains that are highly conserved among different members of the ankyrin family, ankyrins also exhibit an unstructured C-terminal regulatory domain, which is less conserved. The function of this domain is still relatively unclear but it has been implicated in the regulation of the activity of the membrane and spectrin-binding domains (Hall and Bennett, 1997; Abdi et al., 2006). This may well be important as conservation between the various ankyrin membrane-binding domains leads to the question as to how specific ankyrins and their integral partners are associated within specialized membrane domains.
The vertebrate ankyrin gene family consists of three members, ankyrinR (encoded by the ANK1 gene), ankyrinB (ANK2) and ankyrinG (ANK3) reflecting the distribution of these isoforms within tissues (ankyrinR being the isoform initially identified in red cells). Many tissues, such as the nervous system, express variants of all three family members. Diversity in ankyrin structure is also achieved through alternative mRNA processing and the use of alternate promotors. The most prominent of these is the insertion of a large sequence between the ankyrin spectrin-binding and C-terminal regulatory domains to produce the “giant” ankyrin molecules, ankyrinG 480 kDa (Kordeli et al., 1995) and ankyrinB 440 kDa (Otto et al., 1991). These isoforms are almost twice the size of the originally characterized red cell ankyrin (Lambert et al., 1990). The functions of these large inserted sequences remains unknown, although they appear to be mainly associated with isoforms of ankyrin that are targeted to axonal membrane domains (Chan et al., 1993; Kordeli et al., 1995). As the inserted sequences appear to have an extended conformation, they have the potential to link integral proteins in the axonal membrane with structures deep within the axoplasm. “Giant” isoforms of ankyrin have also been reported in Drosophila where the inserted sequences interact with microtubules at the presynaptic neuromuscular junction (Bennett and Healy, 2009).
IIIA3 The Spectrin-Based Membrane Skeleton at the Axon Initial Segment and Nodes of Ranvier
The axon initial segment (AIS) represents a physiologically specialized area of the axon adjacent to the cell body and enriched in cytoplasmic and integral membrane proteins (reviewed in Grubb and Burrone, 2010). It is credited with two major functions in the neuron: (1) the initiation of action potentials; and (2) the formation of a molecular barrier that separates axonal and somatodendritic compartments of the cell to establish and maintain their identity. Key to the organization of this specialized membrane domain is an isoform of ankyrinG (ankyrinG 480 kDa) that has been localized to this area of the axon (Kordeli et al., 1995), although other smaller isoforms of ankyrinG may also be present within this domain. A specialized form of spectrin (β-IV) is also found at the AIS (Grubb and Burrone, 2010). This molecule appears to be important in maintaining or stabilizing integral membrane concentrations at the AIS, as its knockdown does not prevent the initial recruitment of ankyrinG and its membrane-binding partners to the AIS in cultured neurons. The recruitment of ankyrinG to the AIS occurs early in development of the axon, although how this molecule is specifically targeted to the AIS is still unknown. This may reflect the nature of the microtubules in this region of the axon which, in some ways, resemble those of dendrites (Gomis-Ruth et al., 2008). Such microtubules could potentially interact with the ankyrinG 480 kDa inserted region.
AnkyrinG is thought to establish and maintain the localization of a large number of integral proteins from ion channels to cell adhesion molecules at the AIS and it is thought that this high density of integral proteins forms a diffusion barrier that inhibits the migration of non-ankyrin-associated integral proteins between somatodendritic and axonal compartments of the cell (Winckler et al., 1999). It has also been shown that a barrier exists at the AIS for movement within the axonal cytoplasm (Song et al., 2009) and that this selectivity filter is dependent upon the presence of ankyrinG. Knockdown of ankyrinG function in cultured hippocampal neurons not only has been found to dismantle the AIS, but also causes the axon to acquire dendritic characteristics including the formation of spine-like protrusions associated with excitatory synapses (Grubb and Burrone, 2010).
The AIS is the main site of action potential initiation. This is achieved through the establishment of high concentrations of both voltage-gated sodium (vgsc) and potassium channels (vgpc) in the AIS membrane, through interactions with ankyrinG. Vgsc consist of an α subunit composed of 24 transmembrane segments divided into four subdomains that generate an ion-selective pore and an auxillary β subunit that regulates channel activity and membrane expression (Goldin, 1999). There are nine different genes that encode α subunits and proteins encoded by these genes all share an ankyrin-binding sequence within the cytoplasmic domain linking domains II and III (Lemaillet et al., 2003). This sequence is also conserved in the KCNQ2/3 (also referred to as Kv7.2 and Kv7.3) subunits of the M-type potassium channel, responsible for stabilizing the resting membrane potential and preventing repetitive action potentials (Pan et al., 2006). The AIS localization of both vgsc and KCNQ2/3 channels is missing in ankyrinG knockout animals (Zhou et al., 1998; Pan et al., 2006). Consequently, Purkinje neurons from these animals exhibit deficiencies in their ability to initiate action potentials (Zhou et al., 1998).
Ankyrin has also been reported to bind to the accessory β subunits of the voltage-gated sodium channel (Malhotra et al., 2000), although this finding was not supported in alternative studies (Lemaillet et al., 2003). This difference may reflect a requirement for tyrosine phosphorylation within the cytoplasmic domain of the β1 subunit to facilitate ankyrin-binding (Malhotra et al., 2002). Finally, the identification of a di-leucine motif in the C-terminus of the Nav1.2 sodium channel suggests a mechanism whereby voltage-gated sodium channels not bound to ankyrin at the AIS can be selectively removed from the membrane by endocytosis (Garrido et al., 2003) thereby helping to concentrate molecules retained by ankyrinG at the AIS.
Physiologically related to the AIS, the nodes of Ranvier are enriched in ion channels (particularly vgsc) necessary for the propagation of the saltatory action potential (Fig. 26.3). Vgsc densities here are typically 25-fold compared to those observed in the internodal area (Salzer et al., 2008). Nodes are small gaps (1 μM) in the myelin sheath between adjacent myelinating glia. Unlike the AIS whose formation is intrinsic to the neuron, node formation in the PNS requires glial cell contact (Ching et al., 1999). Clustering of sodium channels in the CNS is less understood but at the very least requires soluble factors released from the oligodendrocyte (Kaplan et al., 1997).
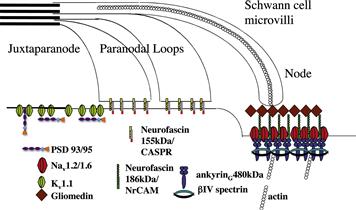
FIGURE 26.3 Mechanisms involved in the localization of ion channels at the nodes of Ranvier. A specialized isoform of ankyrin, ankyrinG 480 kDa binds to voltage-gated sodium channels Nav1.2/1.6 at the nodal membrane along with the cell adhesion molecule neurofascin 186 kDa. This molecule is associated with a specialized isoform of spectrin, β-IV. The release of gliomedin from the myelinating Schwann cell causes neurofascin 186 kDa to cluster, which leads to the recruitment of the ankyrinG 480 kDa and associated Nav1.2/1.6 voltage-gated sodium channels into these clusters. The voltage-gated potassium channel Kv1.1 is able to bind to the PDZ domain containing proteins PSD 93/95 and co-localizes with them in the juxtaparanode.
Key to the formation of the nodes of Ranvier is the binding of ankyrin to members of the L1 family of cell adhesion molecules (CAMs) through a conserved sequence regulated by phosphorylation (Garver et al., 1997; Bennett and Healy, 2009). Both NrCAM and neurofascin 186 kDa from this family are present at both the AIS and nodes of Ranvier and co-localize with ankyrinG 480 kDa and vgsc (Bennett and Healy, 2009). The function of these CAMs at the AIS is unclear, but may be important for the formation of GABAergic synaptic inputs that target the AIS (Grubb and Burrone, 2010). However, at the nodes of Ranvier these CAMs have a clear role in facilitating contacts between the myelinating Schwann cell and the axolemma. Schwann cell microvilli, identified by the presence of ERM (ezrin, radixin, moesin) proteins (Melendez-Vasquez et al., 2001; Gatto et al., 2003) express a unique extracellular matrix that contains multimers of the soluble protein gliomedin complexed with heparin sulphate proteoglycans (HSPGs) (Salzer et al., 2008). Gliomedin binds to the extracellular domain of neurofascin 186 kDa and NrCAM, hence clustering it at the tips of myelinating Schwann cells (Salzer et al., 2008). This results in the recruitment of ankyrinG (Lambert et al., 1997) providing a platform for the immobilization of vgsc and KCNQ2/3 molecules in the nodal membrane. Theoretically, ankyrinG can simultaneously associate with both the CAM and vgsc allowing Schwann cells to regulate directly the clustering of ion channels in the nodal membrane. In addition, the β subunit of the sodium channel has been shown to interact in a cis-like fashion with neurofascin 186 kDa to further promote this clustering (Ratcliffe et al., 2001). As myelination progresses, clusters of nodal complexes associated with the adjoining Schwann cells are brought together to form the mature nodal membrane (Lambert et al., 1997).
Further maintenance of the nodal membrane comes from the formation of the flanking paranodal junctions that act as diffusion barriers to prevent the movement of proteins between nodal and internodal compartments of the axonal membrane (Salzer et al., 2008). Paranodal junctions composed of glial loops anchored to the axolemma through the interactions of the axonal contactin/caspr complex with Schwann cell neurofascin 155 kDa (Salzer et al., 2008). This results in a very close apposition (3–5 nm) between the Schwann cell and the axonal membrane that limits the diffusion of ions between the nodal and internodal areas. The definitive roles of alternatively spliced neurofascin isoforms in nodal development are clearly demonstrated by specific rescues of these isoforms in neurofascin knockout mice (Zonta et al., 2008). Interestingly, the presence of isoforms of neurofascin in the nodal membrane also prevents the invasion of paranodal loops into the nodal membrane domain (Thaxton et al., 2011).
In the juxtaparanodal region of the axon, insulated from the nodal membrane by the paranodal junctions, lie concentrations of vgpc composed of the Kv1 channel subunits Kv1.1, 1.2 and 1.4 (reviewed in Rasband, 2010). As these channels lie under the myelin sheath where they are electrically insulated, their function is somewhat controversial. However, they are thought to modulate action potential propagation and dampen the repetitive firing properties of early myelinating axons. As with ankyrins at the nodal membrane, these proteins exist in a complex that includes both CAMs and adaptors that link them to the cytoskeleton. In this case, an important trans interaction between axon and glial cell is established through an interaction between axonal Caspr2 and glial TAG-1. Caspr2 can also associate with protein 4.1B, a homolog of the molecule characterized in red cells, that links this complex to the spectrin-based membrane skeleton and actin cytoskeleton. Caspr2 also has the ability to link up with multiple PDZ domain containing proteins through its C-terminus. Two of these proteins, PSD-95 and PSD-93 (to be discussed in more detail in the next section), are able to bind to Kv1.1 and 1.2 and have been localized to the juxtaparanodes. However, current observations suggest their activity is not required for vgpc localization (Rasband et al., 2002). Recently, the metalloprotease ADAM22, originally identified as a Kv1 binding partner, has been observed at the juxtaparanode and has been shown to play a role in the clustering of PSD-95 and PSD-93 (Ogawa et al., 2010).
IIIA4 The Spectrin-Based Membrane Skeleton in Muscle Physiology
The wide variety of integral binding partners for ankyrin facilitates its use in the establishment of a range of specialized membranes within a variety of cell types. Studies from the Bennett laboratory in the last decade have shown a range of functions for ankyrin molecules in muscle physiology. As found in other “excitable” cells, ankyrinG is crucial to the localization of vgsc to allow the propagation of action potentials. In cardiomyocytes, this involves the localization and surface expression of Nav1.5 to the intercalated disk, to allow propagation of the action potential between adjoining cells (Mohler et al., 2004).
As well as the intercalated disk, Nav1.5 is also found with ankyrinG in the plasma membrane of the t-tubule (Mohler et al., 2004). T-tubules are extensive invaginations of the plasma membrane that allow it to be brought into close proximity with the sarcoplasmic reticulum at various points along its length. This proximity is crucial for facilitating excitation–contraction coupling in the cardiomyocyte. The t-tubule system consists of a number of specialized microdomains that strictly regulate the movement of calcium between these different membrane compartments to initiate contraction. Key to this activity is a complex between L-type voltage-gated calcium channels in the plasma membrane with ryanodine receptors in the sarcoplasmic reticulum. The localization of Nav1.5 by ankyrinG in microdomains adjacent to this complex is thought to be responsible for activating the voltage-gated calcium channels in this complex. This allows for the passage of small amounts of calcium which, in turn, mobilize calcium stores from the sarcoplasmic reticulum (Bennett and Healy, 2009; Orchard et al., 2009).
A third specialized microdomain within the t-tubule system is enriched in the Na/Ca exchanger (NCX1), which allows for the passage of calcium back across the t-tubule membrane. NCX1 binds to and co-localizes with ankyrinB in these microdomains (Bennett and Healy, 2009). In addition, the Na/K ATPase and the IP3 receptor, which are also known to bind ankyrin, co-localize with ankyrinB in these areas (Mohler et al., 2005). Animals lacking ankyrinB expression in their cardiomyocytes exhibit aberrant localization of these three integral proteins (Mohler et al., 2005) with deficient cardiomyocytes showing increased contractility and increased calcium transients (Mohler et al., 2003, 2005). This suggests an attractive model whereby ankyrinB brings together a functional coupling between the NCX1 and Na/K ATPase with sodium ions entering the cell in exchange for calcium ions. The IP3 receptor in this scenario could be coupled to allow the passage of Ca ions directly from the sarcoplasmic reticulum through NCX1 (Mohler et al., 2005). It would be interesting to determine if ankyrinB is capable of simultaneously associating with both NCX1 and the Na/K ATPase to facilitate formation of a functional macromolecular complex.
Cardimyocytes derived from the ankyrinB knockout mouse also show defects in sodium channel currents suggesting a role for ankyrinB in vgsc activity (Chauhan et al., 2000). These defects include changes in sodium current density detected using whole cell voltage clamp techniques. This suggests a role for ankyrinB in the trafficking and/or stabilization of vgsc on the plasma membrane. Interestingly, changes were also observed in the activation kinetics of the sodium currents suggesting that the binding of ankyrin may also exert functional changes in channel activity.
IIIA5 Mutations Affecting Ankyrin Interactions in Human Cardiac Pathology
Knockout animals have proven invaluable to understanding the role of ankyrins in organizing potentially complex plasma membrane systems such as the t-tubules. Of interest, however, is that some of the same phenotypes can be observed in humans with inherited cardiac disorders and that these phenotypes can be traced to mutations in components of the ankyrin–spectrin based membrane skeleton (reviewed in Ackerman and Mohler, 2010). For example, the E1053K mutation in Nav1.5 is associated with Brugada syndrome, a disorder characterized by cardiac arrhythmia. This mutation falls within the defined ankyrinG binding site (Lemaillet et al., 2003) and abolishes the ability of Nav1.5 to associate with ankyrinG which, in vitro, perturbs the surface expression of the molecule and its localization to intercalated disks.
As well as ankyrinG mutations, mutations within ankyrinB have been associated with cardiac arrhythmias (Ackerman and Mohler, 2010). LQT4 (type 4 long QT syndrome), a disease characterized by prolonged QTc, catecholaminergic polymorphic ventricular arrhythmia and sudden death, was localized to human chromosome 4q25-27 in a large four generation French kindred. This lies within the same region as the ANK2 gene. Further examination of the original kindred showed a missense mutation localized to the regulatory domain of ankyrinB resulting in a loss of function. Subsequent studies have now identified a number of kindreds with similar arrhythmias due to mutations in the ANK2 gene. Phenotypes associated with these individuals range from severe to mild with sinus node dysfunction and atrial fibrillation as hallmarks of this disease. This disease has now been renamed ankyrin-B syndrome (Mohler et al., 2007). These findings illustrate the tip of a potential iceberg in which a number of “channelopathies” might be traced to defects in the adaptor system used to localize them to specific membrane domains or regulate their activity.
IIIA6 Regulation of the Association Between Ankyrin and its Integral Partners
Despite the static nature of the AIS and nodes of Ranvier, evidence has been presented for the dynamic regulation of the ankyrin-integral protein interaction that may be important in the formation or maintenance of these structures. It may also explain one of the conundrums of ankyrin activity. How does a specific ankyrin isoform become targeted to these domains and associate in a non-redundant fashion with its integral partner, given that the ankyrin membrane-binding domains are highly conserved? For example, both ankyrinG and ankyrinB are expressed in axons and both can bind with equal affinitity to ank-CAMs vgsc and vgpc. However, within the axon, ankyrinG is found only at the AIS with neurofascin 186 kDa and vgsc (Bennett and Healy, 2009), whereas ankyrinB is distributed along the length of the axon (Chan et al., 1993). This problem is yet to be fully answered but may lie in the modulation of ankyrin interactions by the divergent regulatory domain (Bennett and Healy, 2009) or control of ankyrin trafficking by the same domain (Mohler et al., 2002). Another possibility is that ankyrin interactions may be regulated by post-translational modifications such as phosphorylation.
Within the cytoplasmic loop between subdomains II and III of the voltage-gated sodium channel are residues phosphorylated by protein kinase CK2 (CK2) (Brechet et al., 2008). Phosphorylation of these residues enhances ankyrin-binding and CK2 is specifically localized to the AIS in neurons through mechanisms currently unknown. Disruption of CK2 activity results in an aberrant localization for voltage-gated sodium channels within the axon (Brechet et al., 2008). This suggests a mechanism whereby the interaction of ankyrinG with vgsc is locally regulated at the AIS.
The ankyrin-binding site in the cytoplasmic C-terminus of neurofascin (the FIGQY motif) contains a tyrosine residue, which when phosphorylated abolishes ankyrin binding (Garver et al., 1997). In axons, neurofascin phosphorylated in this position is excluded from the node of Ranvier and localized to the paranodes (Bennett and Healy, 2009). In contrast, this post-translational event is required for the binding of neurofascin to the microtubule associated protein doublecortin (Bennett and Healy, 2009), suggesting a “switch” mechanism whereby neurofascin is able to associate with different cytoskeletal systems when needed.
IIIB PDZ Proteins and the Postsynaptic Density
IIIB1 The Postsynaptic Density and the Dendritic Spine: Dynamic Interactions Between Ion Channel Receptors and the Actin Cytoskeleton
The postsynaptic compartment of excitatory glutamatergic synapses is localized to small, highly dynamic protruberances that emanate from the dendritic shaft known as dendritic spines. These structures, which are up to a few microns in length, contain all of the signaling molecules and organelles necessary for the receipt, integration and propagation of the synaptic chemical signal. In addition, spines assume a range of dynamic morphologies and mounting evidence has supported the idea that changes in spine morphology alter the propagation of postsynaptic signals. These changes in spine morphology are an important component of synaptic plasticity and hence memory and learning. Dendritic spines themselves are enriched in actin filaments that determine the morphology of the spine and provide a scaffold for the immobilization of postsynaptic receptors and signaling molecules. In turn, these signaling molecules, in response to the activity of synaptic receptors, can regulate the actin cytoskeleton to change the morphology of the spine, thereby regulating synaptic strength. In this section, we will examine the molecules present in the spine, which participate in the immobilization of postsynaptic receptors and signaling molecules regulating dendritic spine morphology.
IIIB2 PDZ Proteins and the Immobilization of Postsynaptic Receptors
The NMDA (N-methyl-D-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoaxazole propionic acid) receptors are the major receptors for glutamate in the postsynaptic membrane. Both are ionotrophic receptors which behave as ion channels for the influx of calcium ions to invoke a membrane depolarization in response to glutamate binding. Quantitative analysis of the numbers of such receptors present in the postsynaptic density indicates that the number of NMDA receptors stays largely fixed at ≈20 molecules/synapse, whereas the AMPA receptor number can vary from ≈5 to 200 (reviewed in Sheng and Hoogenraad, 2007). The variety in AMPA receptor number is believed to correlate with the size of the spine head and the dynamic nature of AMPA receptor trafficking to and from the postsynaptic density to be an important feature of synaptic plasticity (Shepherd and Huganir, 2007). Both of these channels form heterotetrameric pores in the membrane to allow for the passage of calcium. The NMDA receptor consists of two NR1 subunits and two NR2 subunits. There are eight variants of the NR1 (NR1-1a/b, 2a/b, 3a/b and 4a/b) subunit produced from a single gene by alternative mRNA processing and four distinct isoforms of the NR2 variant (NR2A-D) (Hollmann et al., 1993). It is the differential expression of the NR2 subunit across cells that determines the particular electrophysiological characteristics of a receptor. A third subunit (NR3) is also found, which has an inhibitory affect on receptor function. AMPA receptors consist of four subunits (GluR1–4). The majority of subunits found in these receptor types have PDZ-domain interacting motifs in their C-termini, which facilitate their interactions with scaffolding and signaling molecules.
Canonical PDZ domains are ≈90 amino acid sequences consisting of six β-strands and two α-helices arranged in a barrel-like conformation (reviewed in Feng and Zhang, 2009). These domains function in protein–protein recognition and typically recognize a short stretch of amino acids (5–7 residues) at the C-termini of target proteins. There are currently thought to be 335 non-redundant PDZ domains present in the human genome. Many PDZ domain-containing proteins contain more than one PDZ domain, which may or may not have different recognition sequences. This then allows for the clustering of proteins or co-clustering of proteins that may have interrelated functions. Interactions of the PDZ domain lend themselves to analysis using the yeast 2-hybrid technique, which has allowed for the elucidation of a complex network of interacting proteins starting with the C-terminal domains of the NMDA and AMPA receptors and traveling deep within the postsynaptic density and head of the dendritic spine. Details of this network of proteins that form the postsynaptic density is shown in Fig. 26.4.
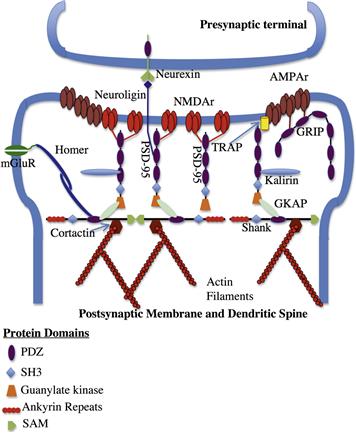
FIGURE 26.4 Interactions of PDZ domain containing proteins at the postsynaptic membrane. NMDA and AMPA receptors at the postsynaptic membrane interact with members of the MAGUK family of scaffolding proteins through their PDZ domains. These scaffolding proteins can also interact with cell adhesion molecules involved in connections with the presynaptic membrane. Interactions with the “higher order” scaffolding proteins (Shanks) link the MAGUKs to the actin cytoskeleton through cortactin. (Adapted from Feng and Zhang, 2009.)
IIIB3 MAGUKS
Studies to identify proteins interacting with the cytoplasmic C-termini of NMDA receptors, in particular the NR2 subunit, led to the identification of a new family of proteins referred to as the MAGUKs (membrane associated guanylate kinases) (Kornau et al., 1995; Niethammer et al., 1996). The possession of three PDZ domains, an SH3 domain involved in protein–protein interactions, and an inactive guanylate kinase domain characterize this family of proteins (reviewed in Dimitratos et al., 1999). MAGUKS have now been identified in a number of specialized membrane domains in cell types where they appear to have a role in the targeting and/or immobilization of integral proteins. The protein found to associate with NMDA receptor subunits in the postsynaptic density was termed PSD-95 (also known as DLG4) and has been found to exist as a family of proteins at the excitatory synapse with PSD-93, SAP-102 and SAP-97. The multimodular nature of these proteins means that they can cluster multiple integral proteins through their multiple PDZ domains or bring together different proteins within functional microdomains. For example, PSD-95 is theoretically able to associate simultaneously with the CAM neuroligin (which associates in a trans fashion with the presynaptic protein neurexin) and the AMPA receptor allowing the presynaptic area to guide development of the postsynaptic density (Feng and Zhang, 2009). Although these proteins are widely expressed throughout the nervous system, their copy number varies according to the neuronal type and developmental stage. The copy number can also vary in accordance with activity-dependent changes underlying synaptic plasticity. It is thought that a typical postsynaptic density contains ≈300–400 copies of MAGUK family members related to PSD-95 (Sheng and Hoogenraad, 2007).
Since the original identification of MAGUKs at the postsynaptic density, a large number of interacting proteins has been identified (see Fig. 26.4). For example AMPA receptors can bind indirectly to PSD-95 family members using the adaptor protein stargazing (Chen et al., 2000) or, alternatively, to the PDZ domain containing protein GRIP (Dong et al., 1997). The development of specific antibodies to these proteins, combined with advances in EM tomography, has allowed a high-resolution anatomical map of the spine head and postsynaptic density to be constructed (Chen et al., 2008). This map divides the postsynaptic density into three layers, the first being at the plasma membrane which encompasses CAMs involved in contact between pre- and postsynaptic areas, ion channels as well as the NMDA and AMPA receptors. Within the spine head the NMDA receptor is observed in the center, whereas AMPA receptors are distributed around the outside of the spine head (Chenet al., 2008). The second layer is enriched in MAGUKs and other scaffolding proteins, arranged such that the N-termini of these molecules are closely associated with the plasma membrane and the C-termini reach down into the density. Finally, the third layer is enriched in large scaffolding proteins termed Shanks (Sh3 domain and Ank repeat domain containing protein).
Shank proteins are large (120–230 kDa) multidomain proteins characterized by the presence of a series of ankyrin repeats, SH3 and PDZ domains, a long proline-rich region and a C-terminal SAM (sterile alpha) domain (reviewed in Kreienkamp, 2008). Three Shank genes have been described which give rise to a diverse range of proteins as a result of alternative mRNA processing and promotor usage. It is estimated that approximately 100 Shank molecules are localized deep within the postsynaptic density (Sheng and Hoogenraad, 2007). Shank proteins are thought to function as higher order scaffolding molecules, organizing the postsynaptic density by connecting different scaffold/receptor complexes through their multiple functional domains. Shank molecules also self-assemble through their C-terminal Sam domain in a head-to-head fashion and connect with the actin cytoskeleton indirectly through an interaction with the actin-binding proteins cortactin and/or fodrin (non-erythroid spectrin) (Kreienkamp, 2008). Receptor scaffolding complexes can anchor to Shank proteins through GKAP (also termed SAP-associated proteins or SAPAP) molecules, which can bind both to the guanylate kinase domain of PSD-95/PSD-93 and with the PDZ domains of Shank (Kreienkamp, 2008). Other ion channels and receptors within the postsynaptic density can also associate with Shank. These include the metabotrophic glutamate receptor (mGluR1), which can associate with the Shank complex through the dimeric linker protein Homer (Tu et al., 1999).
IIIB4 Regulation of Synaptic Strength through Interactions with Scaffolding Proteins
Activity-dependent changes in synaptic strength define synaptic plasticity and hence, memory and learning. At the electrical level, it is possible to regulate synaptic strength either through changing the ionotrophic characteristics of the receptor, or by altering the expression levels of the channel in the plasma membrane. As mentioned previously, the copy number of the NMDA receptor remains fairly fixed in the postsynaptic membrane, whereas that of the AMPA receptor varies widely (Sheng and Hoogenraad, 2007). In the last few years, there has been increasing evidence suggesting that an important component of synaptic strength is the turnover of AMPA receptors in the postsynaptic membrane. At the same time, evidence has accumulated to say that this turnover to some extent is regulated by interactions between the AMPA receptor and scaffolding proteins (reviewed in Elias and Nicoll, 2007).
Evidence of a role for MAGUK proteins in the regulation of AMPA trafficking to the postsynaptic membrane originally came from overexpression studies of PSD-95 in dissociated neurons and slice cultures (Elias and Nicoll, 2007). In these experiments, a large change in AMPA receptor excitatory postsynaptic currents (epscs) was observed when PSD-95 was overexpressed without a concomitant increase in NMDA epscs. This conclusion however, was not supported in studies of knockout animals lacking either PSD-95 or PSD-93, where epscs were unaffected. This finding may reflect functional redundancy between these two similar proteins (Elias and Nicoll, 2007). Indeed, further studies using RNAi to target specific isoforms of MAGUK proteins, when combined with electrophysiology have now shown that the regulation of AMPA receptor surface expression is complex, with multiple MAGUKs within the same neuron playing a role (Elias and Nicoll, 2007).
Perturbing the activity of MAGUKs has also been found to affect the activity of NMDA receptors. The overexpression of PSD-95 has been shown to reduce sensitization of the NMDA receptor in cultured neurons and increases the NMDA receptor opening probability. In heterologous systems, it also results in increased insertion into the plasma membrane (Aoki et al., 2001; Abel et al., 2004). However, these results remain to be reconciled with those of PSD-95 overexpression studies as mentioned above.
IIIB5 Regulation of Dendritic Spine Morphology
Chemical transmission across the glutamergic synapse involving the NMDA and AMPA receptors produces an epsc that is dependent upon the influx of calcium. It is now clear that dendritic spines serve to compartmentalize calcium changes from the dendritic shaft, where the length and diameter of the spine neck influences the strength and nature of the calcium signal in response to glutamate stimulation (Tada and Sheng, 2006). As increasing lines of evidence indicate that spine morphology can be remodeled by synaptic activity, then spine morphology is closely associated with synaptic plasticity. The morphology of dendritic spines reflects the underlying actin cytoskeleton. Hence, the cytoskeleton not only serves to anchor the ion channels necessary for synaptic activity, but is also remodeled in response to that activity. Not surprisingly then, changes in dendritic spine morphology and numbers has been observed in a number of human neurological disorders ranging from autism (Pickett and London, 2005) to Alzheimer’s (Knobloch and Mansuy, 2008).
Although highly varied in their shape, dendritic spines can be roughly classified into three main types: thin, stubby and mushroom, with larger spine heads correlating with increased synaptic strength (Tada and Sheng, 2006). As mentioned earlier, this is thought to reflect increased levels of AMPA receptors in the postsynaptic membrane. Studies using stimuli that induce long-term potentiation (LTP) (Noguchi et al., 2005) and in vivo two-photon microscopy studies of spine turnover (Holtmaat et al., 2005) show that larger spines are more stable and less plastic, whereas smaller spines preferentially undergo LTP. These spines can undergo dramatic changes in their morphology including “splitting” to form two individual postsynaptic areas (Dyson and Jones, 1984). The reason why smaller spines may be susceptible to LTP probably reflects their “neck” geometry, where smaller spines show enhanced elevation of calcium concentrations in the spine head in response to stimuli due to decreased calcium leak through their thinner spine neck (Noguchi et al., 2005).
Dendritic spines are thought to arise from highly dynamic actin-based structures known as filopodia and are induced when these structures contact the nascent presynaptic region (Tada and Sheng, 2006). Finger-like filopodia are associated with a number of motile structures, including the growth cones of neurons and owe their dynamic nature to rapid remodeling of their actin cytoskeleton. Hence, many of the molecules involved in actin remodeling in other cellular structures are also localized in the dendritic spine and play a role in activity-dependent morphological changes. These include molecules that regulate the kinetics of actin polymerization and depolymerization such as profilin, a molecule that associates with actin monomers to limit their availability for the polymerization of actin (Birbach, 2008). Stimulation of NMDA receptors causes the recruitment of profilin to spine heads with the concomitant blocking of actin-related shape changes in the spine (Ackermann and Matus, 2003).
Members of the RhoGTPase family along with proteins that control their activity have also been localized to the spine with their overexpression or inhibition causing major changes in spine morphology (Yoshihara et al., 2009). RhoGTPases act as molecular switches to regulate the actin cytoskeleton (reviewed in Heasman and Ridley, 2008). When associated with GTP, these small G proteins bind to downstream effectors to regulate their activity. A number of proteins are found that cycle RhoGTPases between active (GTP bound) and inactive (GDP bound) states. An example of one of these proteins in dendritic spines is kalirin-7, which is a GEF (guanine-nucleotide exchange factor) that activates the RacGTPase (reviewed in Penzes and Jones, 2008). During synaptogenesis, kalirin-7 activity is induced downstream of signals from the intercellular CAM ephrinB and its receptor. Studies with knockout animals have demonstrated a critical role for kalirin-7 in the regulation of both spine number as well as morphology (Ma, 2010).
Downstream of RacGTPase activity in many cell types is the Lim-kinase cofilin pathway, which is involved in the severing of actin filaments to provide nucleation sites for new actin filament growth. This pathway has also been shown to play a role in synaptic plasticity (Meng et al., 2003). Knockout animals lacking Lim-kinase exhibit dendritic spines with abnormal morphologies and have impaired spatial learning and synaptic plasticity (Meng et al., 2002). As the loss of dendritic spines has been associated with the cognitive defects found in Alzheimer’s disease (Tackenberg et al., 2009), it is not surprising to note that changes in this signaling pathway have also been observed in this disorder (Zhao et al., 2006).
Finally, overexpression or knockdown experiments of scaffold proteins such as Shank or PSD-95 have also been shown to alter the morphology of dendritic spines. This may reflect the functions of these proteins in the recruitment of signaling proteins to these structures (Tada and Sheng, 2006). Given the large number of molecules involved in regulating the actin cytoskeleton that have now been localized to the dendritic spine, the challenge is now to elucidate how the function (or localization) of these molecules is regulated in response to synaptic activity.
IIIC Gephyrin and the Inhibitory Synapse
As described above, the molecules involved in the formation and maintenance of the excitatory synapse, their interactions and localizations have been characterized with a high degree of resolution. Unfortunately, despite its clinical significance, the same cannot be said of the inhibitory synapse. To some extent this may reflect the ease with which the excitatory synapse can be purified compared with its inhibitory counterpart. However, purification of the glycine receptor did yield a binding protein involved in the linkage of these receptor molecules to the microtubule-based cytoskeletal system. This protein has been termed gephyrin (reviewed in Fritschy et al., 2008). Gephyrin has a predicted molecular mass of 83 kDa and consists of three major structural domains, although multiple isoforms are generated by alternative mRNA processing. Two of these domains are termed G and E. This nomenclature comes about because of sequence homologies between gephyrin and the bacterial Moco (molybdenum co-factor) synthesizing enzymes MogA and MoeA. Indeed, gephyrin is highly conserved between species and mediates Moco biosynthesis in addition to serving as a postsynaptic protein (Feng et al., 1998).
Gephyrin’s function in clustering glycine receptors at the inhibitory synapse is based largely on its self-aggregation properties. The protein is thought to form trimers through self-association sites in the G domain and sites in the E domain associate with the large intracellular M3–M4 loops of the pentameric glycine receptor (Fritschy et al., 2008). These trimers are believed to assemble into higher order structures as postulated from aggregation studies of isolated domains in various cell lines (Sola et al., 2001). However, the observation that gephyrin clusters are observed only at the postsynaptic membrane suggests that other factors also regulate this aggregation, including the activation of glycine receptors (Kirsch and Betz, 1998).
As well as binding directly to microtubules, gephyrin associates with the dynein light chain, therefore linking it to motor molecules that utilize the microtubule system for intercellular transport (Fuhrmann et al., 2002). This interaction is supported by live cell imaging studies that show the movement of intracellular complexes of glycine receptor and gephyrin (Maas et al., 2006). In addition, evidence suggests that these complexes may also be able to associate with the actin cytoskeleton as well as providing a second anchorage system for glycine receptor–gephyrin complexes at the inhibitory membrane (Fritschy et al., 2008).
Gephyrin also is found at GABA- (γ-aminobutyric acid) ergic synapses, although interactions between the GABA receptor and gephyrin are currently not well understood (Fritschy et al., 2008). This interaction may be indirect through the protein GABARAP, which has been shown to bind both gephyrin and the GABA receptor (Kanematsu et al., 2007), although it has been shown that this protein is not essential for GABA targeting to the synapse (O’Sullivan et al., 2005). Unlike the glycine receptor, clusters of GABA receptors have been observed independently of gephyrin in neurons. However, gephyrin gene targeting experiments clearly demonstrate a requirement for gephyrin in GABA receptor clustering (Yu et al., 2007), with the recruitment of gephyrin to GABAergic synapses dependent on GABA receptor clustering. This suggests that gephyrin’s role may be in the stabilization of GABA receptor clusters at the postsynaptic membrane, rather than in their formation. The role of gephyrin in the recruitment of GABA receptors remains to be elucidated, along with the question of how different GABA receptors with different subunit compositions are targeted to specific synapses and the role gephyrin plays in that selectivity.
IIID The Interaction of Epithelial Ion Channels with the Cytoskeleton
So far this review has focused on the role of the cytoskeleton in the establishment and maintenance of “excitable” membrane systems such as the AIS and the postsynaptic membrane. In this section, we will now look at how cytoskeleton–ion channel interactions contribute to the polarized nature of epithelial cells. Epithelial cells line cavities and surfaces throughout an organism and form a barrier between the “outside” and the “inside”. The use of an array of junctions including adherens and tight junctions on the lateral surface ensures that the passage of solutes between these two compartments occurs through, rather than around the epithelial cell. In this way epithelial cells control processes such as secretion and selective adsorption, as well as play a role in the detection of sensation. In order to achieve this function, epithelial cells become highly polarized during differentiation into two plasma membrane domains associated with the two physiological compartments that they bridge: an apical domain and a basolateral domain that sits on a specialized extracellular matrix (Fig. 26.5). The restriction of ion channels to these two domains is in part due to their interaction with the actin-based cytoskeleton of these cells. In addition, this interaction can help contribute to the function of these ion channels particularly in the area of sensation detection.
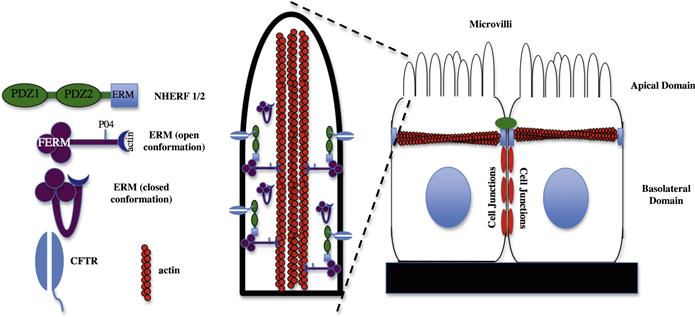
FIGURE 26.5 Interactions that localize CFTR to the apical microvilli of epithelial cells.Epithelial cells are polarized into apical and basolateral domains. The CFTR is localized to the apical compartment of these cells and here is shown localized to apical microvilli. CFTR binds to the PDZ domain of NHERF1/2 which, in turn, binds indirectly to the actin cytoskeleton in the apical domain through an association with ERM (ezrin, radixin, moesin) proteins. As shown, ERM proteins exist in both “open” and “closed” conformations. Binding sites for NHERF1/2 and actin are masked on ERM proteins in the “closed” conformation.
Electrophysiology studies of epithelial ion channel activity first suggested a functional link between these molecules and the actin cytoskeleton. Treatment of epithelial cells with cytochalasins, drugs that cause a depolymerization of the actin cytoskeleton, either activated or inactivated epithelial sodium, chloride and potassium channel activity when examined by patch-clamp electrophysiology (reviewed in Mazzochi et al., 2006). However, from these experiments it was impossible to tell whether affects observed were from binding of the channel to the cytoskeleton or the activation of cytoskeleton-associated second messenger systems that affect channel activity. It is now known that a number of channels associate with the actin cytoskeleton using adaptor and scaffolding systems similar to those described for “excitable” membrane domains in the nervous system.
IIID1 CFTR and PDZ Domain Scaffold Proteins in the Apical Membrane
The cystic fibrosis transmembrane conductance regulator (CFTR) is an ion channel of the ABC-transporter class involved in the movement of chloride and thiocyanate ions across the apical membrane of epithelial cells (Miller, 2010). Essentially, the channel acts as an ATP-gated anion channel allowing anions such as chloride to flow down their electrochemical gradient in response to cAMP activation. This is in contrast to other ABC transporters where ATP is used to drive the passage of ions against the gradient (Miller, 2010). CFTR is found in a number of epithelia but has been well characterized in the lung where the movement of chloride ions from the epithelial cell to the covering mucus results in the movement of water by osmosis and a more fluid mucus (Kreindler, 2010). The importance of this is clearly seen in mutations of the CFTR that give rise to cystic fibrosis, one of the most common genetic disorders among Caucasians. This disease is characterized by the presence of thick mucus that can block airways and glands and acts as a reservoir for bacterial infections (Kreindler, 2010).
The characterization and identification of C-terminal cytoplasmic sequences in CFTR capable of interacting with PDZ-domain containing proteins led to the idea that CFTR maybe anchored to the apical surface through an interaction with a PDZ-domain scaffolding protein. This protein was found to be a previously identified molecule termed NHERF1 (Na+/H+ exchanger regulatory protein) also known as EBP-50 (ezrin-binding protein 50 kDa) (Mazzochi et al., 2006). NHERF proteins are a family of four members that possess either two or four PDZ domains. Family members include the closely related NHERF1 (ebp-50) and NHERF2 (also known as E3KARP) and the four PDZ domain containing proteins NHERF3 (also known as PDZK1, CAP70) and NHERF4 (IKEPP) (Donowitz et al., 2005). As well as their PDZ domains NHERF family members also possess a C-terminal ERM (ezrin-radixin-moesin) binding domain, which therefore indirectly links them to the actin-based cytoskeleton as shown in Fig. 26.5.
NHERF proteins have now been implicated as a major mechanism for the connection of epithelial ion channels to the actin cytoskeleton. These molecules were originally identified as binding partners for the apical membrane Na+/H+ exchanger type 3 (NHE3) (Weinman et al., 1995; Yun et al., 1997; Donowitz et al., 2005). They have been shown to be responsible for the localization of the TRPV5/6 epithelial calcium channels (Embark et al., 2004), TRPC4/5 non-selective cation channels (Tang et al., 2000), the β-adrenergic receptor (Hall et al., 1998), CIC-3 chloride channel (Gentzsch et al., 2003) and ROMK potassium channels (Yoo et al., 2004) in addition to their interactions with CFTR. As with other PDZ domain proteins, the possession of multiple PDZ domains allows NHERF proteins to cluster ion channels and/or establish signaling platforms whereby ion channels are brought into close proximity with proteins that regulate their activity. This is clearly illustrated with CFTR, where the molecule has been shown to bind to both PDZ domains of NHERF1 with physiologically relevant affinity (Raghuram et al., 2001). These interactions have been shown to promote dimerization of the CFTR molecule affecting its activity. In addition, CFTR has been localized in a macromolecular complex with the β-adrenergic receptor bound to NHERF2 (Naren et al., 2003). Given that these two molecules are functionally coupled with activation of the receptor stimulating CFTR activity, interactions with NHERF2 represent an important mechanism for regulating CFTR activity in the cell.
Although interactions between CFTR, NHERFs and ERM proteins are necessary for the trafficking and/or stabilization of CFTR in the apical membrane (Mazzochi et al., 2006), these interactions are found to be fairly dynamic and regulated by cell signaling pathways. Interaction of the CFTR with NHERF1 can be regulated by protein kinase C- (PKC-) mediated phosphorylation of a single residue (S162) within PDZ domain 2 of NHERF1 (Raghuram et al., 2001). Phosphorylation of this residue decreases the binding of CFTR to NHERF1. The binding of NHERFs to ERM proteins is also regulated. ERM proteins are characterized by a globular FERM domain and an extended “tail” region that contains an actin-binding site. Within these two domains are sites of self-association that allow the protein to assume a “closed” conformation, where N- and C-termini interact (reviewed in Fehon et al., 2010). Binding sites for actin and NHERF proteins are blocked in this “closed” conformation (see Fig. 26.5). Movement between “closed” and “open” conformations of the ERM proteins is downstream of cell signaling events. These include an interaction between the FERM domain of ERM proteins with PIP2 (phosphatidylinositol 4,5-bisphosphate) synthesized in the membrane in response to G-protein receptor signaling and phosphorylation of ERM proteins in response to activation of RhoGTPases (Fehon et al., 2010). The dynamic nature of the interaction of CFTR with the actin cytoskeleton through NHERF proteins has been demonstrated using a series of FRAP (fluorescence recovery after photobleaching) studies with GFP-labeled CFTR (Haggie et al., 2004). These studies showed that the diffusion of CFTR in the plane of the membrane is dependent on its ability to bind to PDZ-domain-containing proteins, but that this interaction is highly dynamic with these interactions constantly occurring on a time scale of seconds or faster. In support of the dynamic nature of these interactions, it has been reported that NHERF proteins constitute only a small amount (≈2%) of proteins found in CFTR-containing macromolecular complexes (Li et al., 2004).
IIID2 Interactions of Epithelial Ion Channels with Actin-Binding Proteins
The epithelial sodium channel (ENaC, also known as the amiloride sensitive sodium channel) is present in the apical membrane of epithelial cells (particularly the kidney) where it plays a major role in the reabsorption of sodium. These channels are also closely related to the degenerins family of proteins in Caenorhabditis elegans that are implicated in mechanosensory transduction (Lumpkin et al., 2010). The channel consists of three subunits termed α, β and γ that form a pore with selective permeability for Li+ and Na+ ions (Garty and Palmer, 1997). An interaction between the α subunit of the ENaC and the SH3 domain of α spectrin has been reported (Rotin et al., 1994). This interaction maps to a proline-rich sequence present in the cytoplasmic C-terminal region of the subunit. Microinjection of this domain into rat alveolar epithelial cells causes it to co-localize with α-spectrin in the apical domain. This is in contrast to a domain from the N-terminal region of the ENaC which remains diffuse in the cytoplasm (Rotin et al., 1994).
Other members of the spectrin superfamily of proteins, characterized by the presence of the 106 amino acid spectrin repeat (Hartwig, 1994) are also expressed in epithelial cells. α-Actinin, which consists of four spectrin repeat motifs, is arranged as an anti-parallel homodimer to bind and cross-link actin filaments (Sjoblom et al., 2008). This molecule has been shown to bind to PC2 (polycystin-2 also known as TRPP2) through its spectrin repeat domains (Li et al., 2005), thereby cross-linking PC2 to the actin cytoskeleton. PC2 is a member of the TRP (transient receptor potential) superfamily of ion channels and is a Ca2+-permeable cation channel. Mutations in the PKD2 gene that encodes PC2 are associated with autosomal dominant polycystic kidney disease (ADPKD) (Gonzalez-Perrett et al., 2001). Although PC2 is predominantly localized to the cilia, it is also found in the apical plasma membrane of epithelial cells (Ong and Harris, 2005). Interestingly α-actinin was able to stimulate the activity of PC2 when incorporated into lipid bilayers, suggesting that this interaction may also have a direct regulatory role on PC2 activity as well as regulating its subcellular localization (Li et al., 2005).
IIID3 Direct Interactions Between Epithelial Ion Channels and Actin Filaments
We have so far examined examples of a model in which ion channels couple to the actin cytoskeleton through intermediary adaptor or scaffold proteins. However, increasing lines of evidence support the idea that some ion channels in epithelial cells can directly associate with actin. Actin has been shown to interact directly with the CIC-2 chloride channel using actin overlay and co-sedimentation assays (Ahmed et al., 2000). In addition, treatment of Xenopus laevis oocytes expressing CIC-2 with cytochalasin D or latrunculin that disrupt the actin cytoskeleton significantly increased channel activity. Similarly, changes in conductance of ENaC channels incorporated into lipid bilayers were observed in the presence of short actin filaments (Berdiev et al., 1996) and these channels were found to be activated by protein kinase A (PKA) only in the presence of actin. The concept that ion channel activity may be regulated by the presence of short actin filaments is supported by patch-clamp studies in X. laevis cells showing an increase in ENaC activity within 5 minutes of applying cytochalasin D to the cells (Cantiello et al., 1991). Further experiments utilizing gelsolin, which preferentially stabilizes the polymerization of actin into short oligomers, have demonstrated that it is the interaction of ENaC with short actin filaments, rather than just with actin itself, that regulates ENaC activity (Prat et al., 1993). Mapping studies have localized a putative actin-binding site to the region between E631 and F644 of the α-ENaC molecule (Copeland et al., 2001).
IV General Conclusions
The localization of ion channels to physiologically specialized membrane domains is crucial to the function of a wide variety of metazoan cell types. This localization is dependent upon interactions, either direct or indirect, between ion channels and the actin or microtubule-based cytoskeleton. In this review, we have examined some examples of these interactions and how they contribute to the physiological function of these specialized membrane domains. As is observed, the interaction between ion channels and cytoskeletal elements not only facilitates channel localization, but also may regulate their activity. Regulation of ion channel activity can reflect the affects of direct interactions of the cytoskeleton or cytoskeletal adaptor/scaffolding proteins with components of the channel. An area where this concept becomes crucial, but not covered in this review, is in the area of mechanosensitive transduction. There, the interaction of ion channels with the cytoskeleton and the extracellular matrix serves to regulate channel activity in response to mechanical changes (Lumpkin et al., 2010). Alternatively, ion channel activity can be regulated by the formation of functional microdomains brought about by scaffolding proteins that bring the channel into close proximity with signaling molecules that regulate its activity.
As evidenced in the dendritic spine, the activity of ion channels can also serve to regulate the cytoskeleton to achieve changes in cell shape. These changes can themselves affect how epscs are propagated to the dendritic shaft and are an important component of synaptic plasticity, a crucial concept underlying memory and learning. Dendritic spines themselves contain molecules that have been demonstrated to have a regulatory affect on their morphology. The challenge in upcoming years will be to try to understand how synaptic activity, with its accompanying ion channel involvement, regulates the function of these molecules to change spine morphologies.
A number of insights have occurred over the last 10 years into how these specialized membrane domains are localized and assembled. However, a significant number of important questions remain to be answered. Are ion channels and their associated trafficking proteins trafficked together to the plasma membrane as macromolecular complexes or is assembly localized to these sites? In the case of the excitatory postsynaptic density, we know that at least some of these proteins are already preassembled prior to reaching the postsynaptic membrane (Naisbitt et al., 1999). Similarly, a role for various motor proteins in synapse assembly has been noted (reviewed in Hirokawa et al., 2010), although mechanisms underlying the specific targeting of protein complexes to these sites have yet to be elucidated. The discovery that certain scaffold proteins simultaneously bind to both CAMs and ion channels suggests a mechanism whereby trans interactions between cells could direct ion channel clustering and formation of specialized membrane domains. For example, at the node of Ranvier, gliomedin released from the myelinating Schwann cell clusters the ankyrin-binding molecule neurofascin (Salzer et al., 2008) to direct ankyrin recruitment (Lambert et al., 1997). As ankyrin also binds the vgsc (Lemaillet et al., 2003), a series of molecular interactions allow the Schwann cell to orchestrate directly early assembly of the nodal membrane. However, this model does not explain how these same players are assembled at the AIS, which is an intrinsic feature of the neuron and can be assembled in the absence of cell contacts. Again, the answer to these question lies in understanding mechanisms undergoing the trafficking of these proteins to their specialized membrane domains.
The use of knockout and transgenic animals has been extremely useful in demonstrating the physiological consequences of aberrant interactions between ion channels and the cytoskeleton. However, we have also seen that mutations in these interactions can also be associated with human disease as is seen with ankyrin in the heart (Ackerman and Mohler, 2010) and CFTR in epithelial cells (Kreindler, 2010). In addition, increasing evidence suggests that proteins that affect the dendritic spine cytoskeleton and, hence, its morphology, may be associated with human neurological disorders such as Alzheimer’s disease (Knobloch and Mansuy, 2008; Penzes and Jones, 2008). It may well be that these disorders represent only the tip of the iceberg when it comes to channelopathies that result from aberrant interactions with the cytoskeleton.
BIBLIOGRAPHY
1. Abdi KM, Mohler PJ, Davis JQ, Bennett V. Isoform specificity of ankyrin-B: a site in the divergent C-terminal domain is required for intramolecular association. J Biol Chem. 2006;281:5741–5749.
2. Abel HJ, Lee JC, Callaway JC, Foehring RC. Relationships between intracellular calcium and after hyperpolarizations in neocortical pyramidal neurons. J Neurophysiol. 2004;91:324–335.
3. Ackerman MJ, Mohler PJ. Defining a new paradigm for human arrhythmia syndromes: phenotypic manifestations of gene mutations in ion channel- and transporter-associated proteins. Circ Res. 2010;107:457–465.
4. Ackermann M, Matus A. Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci. 2003;6:1194–1200.
5. Ahmed N, Ramjeesingh M, Wong S, Varga A, Garami E, Bear CE. Chloride channel activity of ClC-2 is modified by the actin cytoskeleton. Biochem J. 2000;352:789–794.
6. Aoki C, Miko I, Oviedo H, et al. Electron microscopic immunocytochemical detection of PSD-95, PSD-93, SAP-102, and SAP-97 at postsynaptic, presynaptic, and nonsynaptic sites of adult and neonatal rat visual cortex. Synapse. 2001;40:239–257.
7. Bennett V, Gilligan DM. The spectrin-based membrane skeleton and micron-scale organization of the plasma membrane. Annu Rev Cell Biol. 1993;9:27–66.
8. Bennett V, Healy J. Membrane domains based on ankyrin and spectrin associated with cell-cell interactions. Cold Spring Harb Perspect Biol. 2009;1:a003012.
9. Berdiev BK, Prat AG, Cantiello HF, et al. Regulation of epithelial sodium channels by short actin filaments. J Biol Chem. 1996;271:17704–17710.
10. Birbach A. Profilin, a multi-modal regulator of neuronal plasticity. Bioessays. 2008;30:994–1002.
11. Brechet A, Fache MP, Brachet A, et al. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J Cell Biol. 2008;183:1101–1114.
12. Byers TJ, Branton D. Visualization of the protein associations in the erythrocyte membrane skeleton. Proc Natl Acad Sci USA. 1985;82:6153–6157.
13. Cantiello HF, Stow JL, Prat AG, Ausiello DA. Actin filaments regulate epithelial Na+ channel activity. Am J Physiol. 1991;261:C882–C888.
14. Chan W, Kordeli E, Bennett V. 440-kD ankyrinB: structure of the major developmentally regulated domain and selective localization in unmyelinated axons. J Cell Biol. 1993;123:1463–1473.
15. Chauhan VS, Tuvia S, Buhusi M, Bennett V, Grant AO. Abnormal cardiac Na(+) channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ Res. 2000;86:441–447.
16. Chen L, Chetkovich DM, Petralia RS, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943.
17. Chen X, Winters C, Azzam R, et al. Organization of the core structure of the postsynaptic density. Proc Natl Acad Sci USA. 2008;105:4453–4458.
18. Ching W, Zanazzi G, Levinson SR, Salzer JL. Clustering of neuronal sodium channels requires contact with myelinating Schwann cells. J Neurocytol. 1999;28:295–301.
19. Copeland SJ, Berdiev BK, Ji HL, et al. Regions in the carboxy terminus of alpha-bENaC involved in gating and functional effects of actin. Am J Physiol Cell Physiol. 2001;281:C231–C240.
20. Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269:27163–27166.
21. Dimitratos SD, Woods DF, Stathakis DG, Bryant PJ. Signaling pathways are focused at specialized regions of the plasma membrane by scaffolding proteins of the MAGUK family. Bioessays. 1999;21:912–921.
22. Dong H, O’Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284.
23. Donowitz M, Cha B, Zachos NC, et al. NHERF family and NHE3 regulation. J Physiol. 2005;567:3–11.
24. Dyson SE, Jones DG. Synaptic remodelling during development and maturation: junction differentiation and splitting as a mechanism for modifying connectivity. Brain Res. 1984;315:125–137.
25. Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352.
26. Embark HM, Setiawan I, Poppendieck S, et al. Regulation of the epithelial Ca2+ channel TRPV5 by the NHE regulating factor NHERF2 and the serum and glucocorticoid inducible kinase isoforms SGK1 and SGK3 expressed in Xenopus oocytes. Cell Physiol Biochem. 2004;14:203–212.
27. Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287.
28. Feng G, Tintrup H, Kirsch J, et al. Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science. 1998;282:1321–1324.
29. Feng W, Zhang M. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat Rev Neurosci. 2009;10:87–99.
30. Fritschy JM, Harvey RJ, Schwarz G. Gephyrin: where do we stand, where do we go?. Trends Neurosci. 2008;31:257–264.
31. Fuhrmann JC, Kins S, Rostaing P, et al. Gephyrin interacts with Dynein light chains 1 and 2, components of motor protein complexes. J Neurosci. 2002;22:5393–5402.
32. Garrido JJ, Fernandes F, Giraud P, Mouret I, et al. Identification of an axonal determinant in the C-terminus of the sodium channel Na(v)1.2. EMBO J. 2001;20:5950–5961.
33. Garrido JJ, Fernandes F, Moussif A, Fache MP, Giraud P, Dargent B. Dynamic compartmentalization of the voltage-gated sodium channels in axons. Biol Cell. 2003;95:437–445.
34. Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396.
35. Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–714.
36. Gatto CL, Walker BJ, Lambert S. Local ERM activation and dynamic growth cones at Schwann cell tips implicated in efficient formation of nodes of Ranvier. J Cell Biol. 2003;162:489–498.
37. Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH, Riordan JR. The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins. J Biol Chem. 2003;278:6440–6449.
38. Goldin AL. Diversity of mammalian voltage-gated sodium channels. Ann NY Acad Sci. 1999;868:38–50.
39. Gomis-Ruth S, Wierenga CJ, Bradke F. Plasticity of polarization: changing dendrites into axons in neurons integrated in neuronal circuits. Curr Biol. 2008;18:992–1000.
40. Gonzalez-Perrett S, Kim K, Ibarra C, et al. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc Natl Acad Sci, USA. 2001;98:1182–1187.
41. Grubb MS, Burrone J. Building and maintaining the axon initial segment. Curr Opin Neurobiol. 2010;20:481–488.
42. Haggie PM, Stanton BA, Verkman AS. Increased diffusional mobility of CFTR at the plasma membrane after deletion of its C-terminal PDZ binding motif. J Biol Chem. 2004;279:5494–5500.
43. Hall RA, Ostedgaard LS, Premont RT, et al. A C-terminal motif found in the beta2-adrenergic receptor, P2Y1 receptor and cystic fibrosis transmembrane conductance regulator determines binding to the Na+/H+ exchanger regulatory factor family of PDZ proteins. Proc Natl Acad Sci, USA. 1998;95:8496–8501.
44. Hall TG, Bennett V. Regulatory domains of erythrocyte ankyrin. J Biol Chem. 1987;262:10537–10545.
45. Hartwig JH. Actin-binding proteins 1: spectrin superfamily. Protein Profile. 1994;1:706–778.
46. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701.
47. Hill AS, Nishino A, Nakajo K, et al. Ion channel clustering at the axon initial segment and node of Ranvier evolved sequentially in early chordates. PLoS Genet. 2008;4:e1000317.
48. Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638.
49. Hollmann M, Boulter J, Maron C, et al. Zinc potentiates agonist-induced currents at certain splice variants of the NMDA receptor. Neuron. 1993;10:943–954.
50. Holtmaat AJ, Trachtenberg JT, Wilbrecht L, et al. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291.
51. Kanematsu T, Mizokami A, Watanabe K, Hirata M. Regulation of GABA(A)-receptor surface expression with special reference to the involvement of GABARAP (GABA(A) receptor-associated protein) and PRIP (phospholipase C-related, but catalytically inactive protein). J Pharmacol Sci. 2007;104:285–292.
52. Kaplan MR, Meyer-Franke A, Lambert S, et al. Induction of sodium channel clustering by oligodendrocytes. Nature. 1997;386:724–728.
53. Kirsch J, Betz H. Glycine-receptor activation is required for receptor clustering in spinal neurons. Nature. 1998;392:717–720.
54. Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer’s disease. Mol Neurobiol. 2008;37:73–82.
55. Kodippili GC, Spector J, Sullivan C, et al. Imaging of the diffusion of single band 3 molecules on normal and mutant erythrocytes. Blood. 2009;113:6237–6245.
56. Kordeli E, Lambert S, Bennett V. AnkyrinG A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem. 1995;270:2352–2359.
57. Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740.
58. Kreienkamp HJ. Scaffolding proteins at the postsynaptic density: shank as the architectural framework. Handb Exp Pharmacol, 2008;:365–380.
59. Kreindler JL. Cystic fibrosis: exploiting its genetic basis in the hunt for new therapies. Pharmacol Ther. 2010;125:219–229.
60. Lambert S, Davis JQ, Bennett V. Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J Neurosci. 1997;17:7025–7036.
61. Lambert S, Yu H, Prchal JT, et al. cDNA sequence for human erythrocyte ankyrin. Proc Natl Acad Sci USA. 1990;87:1730–1734.
62. Lemaillet G, Walker B, Lambert S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem. 2003;278:27333–27339.
63. Li C, Roy K, Dandridge K, Naren AP. Molecular assembly of cystic fibrosis transmembrane conductance regulator in plasma membrane. J Biol Chem. 2004;279:24673–24684.
64. Li Q, Montalbetti N, Shen PY, et al. Alpha-actinin associates with polycystin-2 and regulates its channel activity. Hum Mol Genet. 2005;14:1587–1603.
65. Lumpkin EA, Marshall KL, Nelson AM. The cell biology of touch. J Cell Biol. 2010;191:237–248.
66. Ma XM. Kalirin-7 is a key player in the formation of excitatory synapses in hippocampal neurons. Sci World J. 2010;10:1655–1666.
67. Maas C, Tagnaouti N, Loebrich S, Behrend B, Lappe-Siefke C, Kneussel M. Neuronal cotransport of glycine receptor and the scaffold protein gephyrin. J Cell Biol. 2006;172:441–451.
68. Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL. Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem. 2000;275:11383–11388.
69. Malhotra JD, Koopmann MC, Kazen-Gillespie KA, Fettman N, Hortsch M, Isom LL. Structural requirements for interaction of sodium channel beta 1 subunits with ankyrin. J Biol Chem. 2002;277:26681–26688.
70. Mazzochi C, Benos DJ, Smith PR. Interaction of epithelial ion channels with the actin-based cytoskeleton. Am J Physiol Renal Physiol. 2006;291:F1113–F1122.
71. Melendez-Vasquez CV, Rios JC, Zanazzi G, Lambert S, Bretscher A, Salzer JL. Nodes of Ranvier form in association with ezrin-radixin-moesin (ERM)-positive Schwann cell processes. Proc Natl Acad Sci, SA. 2001;98:1235–1240.
72. Meng Y, Zhang Y, Tregoubov V, et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133.
73. Meng Y, Zhang Y, Tregoubov V, Falls DL, Jia Z. Regulation of spine morphology and synaptic function by LIMK and the actin cytoskeleton. Rev Neurosci. 2003;14:233–240.
74. Michaely P, Bennett V. The membrane-binding domain of ankyrin contains four independently folded subdomains, each comprised of six ankyrin repeats. J Biol Chem. 1993;268:22703–22709.
75. Miller C. CFTR: break a pump, make a channel. Proc Natl Acad Sci USA. 2010;107:959–960.
76. Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3:e423.
77. Mohler PJ, Gramolini AO, Bennett V. The ankyrin-B C-terminal domain determines activity of ankyrin-B/G chimeras in rescue of abnormal inositol 1,4,5-trisphosphate and ryanodine receptor distribution in ankyrin-B (−/−) neonatal cardiomyocytes. J Biol Chem. 2002;277:10599–10607.
78. Mohler PJ, Healy JA, Xue H, et al. Ankyrin-B syndrome: enhanced cardiac function balanced by risk of cardiac death and premature senescence. PLoS One. 2007;2:e1051.
79. Mohler PJ, Rivolta I, Napolitano C, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci USA. 2004;101:17533–17538.
80. Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639.
81. Naisbitt S, Kim E, Tu JC, et al. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582.
82. Naren AP, Cobb B, Li C, et al. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci USA. 2003;100:342–346.
83. Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163.
84. Noguchi J, Matsuzaki M, Ellis-Davies GC, Kasai H. Spine-neck geometry determines NMDA receptor-dependent Ca2+ signaling in dendrites. Neuron. 2005;46:609–622.
85. Ogawa Y, Oses-Prieto J, Kim MY, et al. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J Neurosci. 2010;30:1038–1048.
86. Ong AC, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67:1234–1247.
87. Orchard CH, Pasek M, Brette F. The role of mammalian cardiac t-tubules in excitation–contraction coupling: experimental and computational approaches. Exp Physiol. 2009;94:509–519.
88. O’Sullivan GA, Kneussel M, Elazar Z, Betz H. GABARAP is not essential for GABA receptor targeting to the synapse. Eur J Neurosci. 2005;22:2644–2648.
89. Otto E, Kunimoto M, McLaughlin T, Bennett V. Isolation and characterization of cDNAs encoding human brain ankyrins reveal a family of alternatively spliced genes. J Cell Biol. 1991;114:241–253.
90. Palek J, Lambert S. Genetics of the red cell membrane skeleton. Semin Hematol. 1990;27:290–332.
91. Pan Z, Kao T, Horvath Z, et al. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613.
92. Penzes P, Jones KA. Dendritic spine dynamics – a key role for kalirin-7. Trends Neurosci. 2008;31:419–427.
93. Pickett J, London E. The neuropathology of autism: a review. J Neuropathol Exp Neurol. 2005;64:925–935.
94. Prat AG, Bertorello AM, Ausiello DA, Cantiello HF. Activation of epithelial Na+ channels by protein kinase A requires actin filaments. Am J Physiol. 1993;265:C224–C233.
95. Raghuram V, Mak DO, Foskett JK. Regulation of cystic fibrosis transmembrane conductance regulator single-channel gating by bivalent PDZ-domain-mediated interaction. Proc Natl Acad Sci USA. 2001;98:1300–1305.
96. Rasband MN. Clustered K+ channel complexes in axons. Neurosci Lett. 2010;486:101–106.
97. Rasband MN, Park EW, Zhen D, et al. Clustering of neuronal potassium channels is independent of their interaction with PSD-95. J Cell Biol. 2002;159:663–672.
98. Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA. Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol. 2001;154:427–434.
99. Rotin D, Bar-Sagi D, O’Brodovich H, et al. An SH3 binding region in the epithelial Na+ channel (alpha rENaC) mediates its localization at the apical membrane. EMBO J. 1994;13:4440–4450.
100. Salzer JL, Brophy PJ, Peles E. Molecular domains of myelinated axons in the peripheral nervous system. Glia. 2008;56:1532–1540.
101. Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847.
102. Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643.
103. Sjoblom B, Salmazo A, Djinovic-Carugo K. Alpha-actinin structure and regulation. Cell Mol Life Sci. 2008;65:2688–2701.
104. Sola M, Kneussel M, Heck IS, Betz H, Weissenhorn W. X-ray crystal structure of the trimeric N-terminal domain of gephyrin. J Biol Chem. 2001;276:25294–25301.
105. Song AH, Wang D, Chen G, et al. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136:1148–1160.
106. Tackenberg C, Ghori A, Brandt R. Thin, stubby or mushroom: spine pathology in Alzheimer’s disease. Curr Alzheimer Res. 2009;6:261–268.
107. Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101.
108. Tang Y, Tang J, Chen Z, et al. Association of mammalian trp4 and phospholipase C isozymes with a PDZ domain-containing protein, NHERF. J Biol Chem. 2000;275:37559–37564.
109. Thaxton C, Pillai AM, Pribisko AL, Dupree JL, Bhat MA. Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron. 2011;69:244–257.
110. Tu JC, Xiao B, Naisbitt S, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592.
111. Weinman EJ, Steplock D, Wang Y, Shenolikar S. Characterization of a protein cofactor that mediates protein kinase A regulation of the renal brush border membrane Na(+)-H+ exchanger. J Clin Invest. 1995;95:2143–2149.
112. Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397:698–701.
113. Yoo D, Flagg TP, Olsen O, Raghuram V, Foskett JK, Welling PA. Assembly and trafficking of a multiprotein ROMK (Kir 1.1) channel complex by PDZ interactions. J Biol Chem. 2004;279:6863–6873.
114. Yoshihara Y, De Roo M, Muller D. Dendritic spine formation and stabilization. Curr Opin Neurobiol. 2009;19:146–153.
115. Yu W, Jiang M, Miralles CP, Li RW, Chen G, de Blas AL. Gephyrin clustering is required for the stability of GABAergic synapses. Mol Cell Neurosci. 2007;36:484–500.
116. Yun CH, Oh S, Zizak M, et al. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA. 1997;94:3010–3015.
117. Zhao L, Ma QL, Calon F, et al. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9:234–242.
118. Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–1304.
119. Zonta B, Tait S, Melrose S, et al. Glial and neuronal isoforms of Neurofascin have distinct roles in the assembly of nodes of Ranvier in the central nervous system. J Cell Biol. 2008;181:1169–1177.