Chapter 27
Why are So Many Ion Channels Mechanosensitive?
Chapter Outline
III. Eukaryotic MS Channels – Bilayer Structure, Bilayer Deformation
IV. Channel Mechanosensitivity – Tuning of Channel Behavior
V. VGCS and the Mechanosensitivity of Discrete Transitions
VI. Bilayer Structure in X, Y and Z – One LPP Here, Another LPP There
I Summary
Reports of stretch-sensitive channels started appearing in the mid-1980s. Briefly, the unidentified mechanosensitive (MS) channels were seen as representatives of a new subclass of channels. There were speculations that “mechano-gating motifs” would soon be discovered. Gradually, however, it emerged that channels showing no mechanosensitivity are the outliers and it became clear that gating energy is supplied by bilayer deformations. Over the last decade, what has been particularly helpful in clarifying the role of bilayer mechanics in the mechanosensitive responses of diverse eukaryotic channels have been converging advances on three fronts: (1) high resolution structure/function data for voltage-gated channels (VGCs); (2) the demonstration that VGCs are inherently mechanosensitive; and (3) experimental and computational data showing how mechanosensitivity emerges from the energetics at the interface of dynamically structured bilayers and dynamically structured proteins. An ongoing task is to establish if and where the reversible mechanosensitive responses of ion channels are physiologically relevant; do they, for instance, contribute to cardiac mechano-electric feedback? Also enormously important, in my view, is to learn what the irreversible MS behaviors of channel reveal about the pathological membrane phenomena associated with trauma, ischemia, inflammation and, in fact, any condition where channel-bearing membranes undergo irreversible structural changes. One likely payoff: “smarter” drugs designed to target not simply channel-X, but channel-X in bilayer-structure-Y – say, leaky sodium channel in nodes of Ranvier where lipid packing has become disorderly and where leaflet asymmetry has been compromised by traumatic brain injury.
II Introduction
Even before “channel” entered the physiology lexicon, mechanosensitive (MS) membrane conductances were studied in eukaryotic organisms (Gray and Sato, 1953). Sixty years on, much evidence points to ion channels as the mechanotransducers in vertebrate and invertebrate mechanosensory cells. Transduction channels absorb mechanical gating energy from cytoarchitectural elements arranged to focus and amplify small stimuli; candidates include Peizo1 and Peizo2, proteins with no known homologies (Coste et al., 2010), “TRPs” (transient receptor potentials), multimodal channels with overwhelmingly diverse homologies (Patel et al., 2010) and proteins homologous to “ENaC” (epithelial Na channel) (Cueva et al., 2007). Prokaryotic walled cells express several molecularly unrelated multimeric proteins that, at near-lytic bilayer strain, open as osmolyte channels (Kung et al., 2010); additionally, expanding closed-closed transitions noted for some might serve as membrane spandex, providing tension-relief in high turgor conditions (Boucher et al., 2009). Together, these mechanosensory specialists – unidentified eukaryote mechanotransducers and identified prokayote osmovalves – operate at opposite ends of the mechanostimulus intensity spectrum.
Here I discuss a different phenomenon, that of the many non-specialist ion channels that respond to mechanical stimuli. Non-specialist MS channels, like the prokaryote osmovalve specialists, derive gating energy from bilayer deformation. Eukaryotic mechanotransduction, however, is thought to require stiff vectorial protein assemblies whose gating springs control displacement of a gate (Hudspeth, 2008; Kung et al., 2010) on to a channel that, in stark contrast to the non-specialist MS channels, might be designed to ignore all bilayer “noise”.
III Eukaryotic MS Channels – Bilayer Structure, Bilayer Deformation
Shortly after gigaohm patch-clamp techniques were adopted, reports of stretch-activated cation channels began appearing, first for skeletal muscle, then oocytes (Fig. 27.1A), then everywhere. Early assumptions (Guharay and Sachs, 1984) (1) that these must be MS specialists and (2) that bilayer deformation could not provide sufficient mechano-gating energy to account for the stretch-sensitive activity were dropped (Morris, 1990; Sachs and Morris 1998; Hamill and Martinac, 2001). Interrogated under cell-attached or excised-patch-clamp conditions, it emerged that most channels exhibit mechanosensitivity due, almost certainly, to bilayer deformation (Tabarean and Morris, 2002; Liu et al., 2008). Two reports in 2010 exemplify these points: (1) 20 years after cytic fibrosis transport regulator (CFTR) unitary current was first reported, CFTR was shown to be a “stretch-channel” (Zhang et al., 2010); and (2) a putative cytoplasmic linker protein was shown to be irrelevant, not mandatory, to MS responses of big calcium-activated K channel (BKCa) channels (Wang et al., 2010).
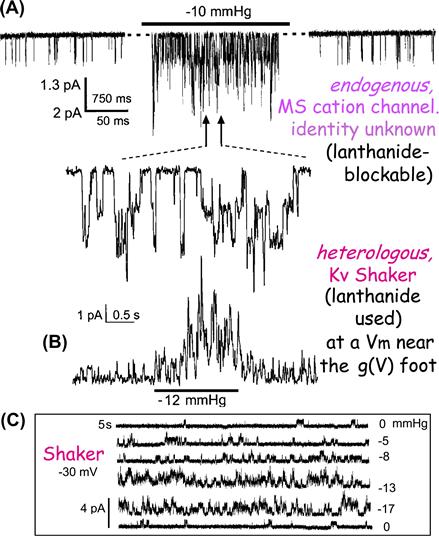
FIGURE 27.1 An unidentified “stretch-activated cation channel” and an equally stretch-sensitive identified “stretch-activated” potassium channel (i.e. an archetypical voltage gated K channel, Kv, Shaker WT, inactivation removed) recorded from oocyte patches, before, during and after stretch due to patch suction (A, B) and Shaker current (C) at a fixed voltage near the foot of the Shaker activation Boltzmann, without (0 mmHg), with (−5,−8,−13,−17 mmHg) and again without stretch (see Gu et al., 2001; Morris and Juranka, 2007a for details).
MS cation channels were soon joined by stretch-activated and stretch-inactivated potassium and anion channels, then NMDA-glutamate channels and L-type Ca current (whole-cell recordings were used as well as patches in some cases). Wherever it is sought – from fibroblasts to fish skin to fungi – MS channel activity is found (Morris, 1990, 2001a; Sachs and Morris, 1998; Hamill and Martinac, 2001; Morris and Juranka, 2007a).
Membrane trauma (Fig. 27.2), which implies plastic (irreversible), as opposed to elastic (reversible), changes in membrane structure in response to a mechanical stimulus, can uncover inherent stretch sensitivity in channels normally mechano-protected by a cortical cytoskeleton meshwork (Morris and Horn, 1991; Wan et al., 1999; Hamill and Martinac, 2001; Morris, 2001b; Morris et al., 2006; Liu et al., 2008). Membrane trauma cannot be completely avoided during patch formation and, certainly, patch excision causes plastic changes. Structurally, the plasma membrane becomes a bleb bilayer. During progressive bleb development (Bailey et al., 2009), adhesion to cortical proteins is lost, lateral order in lipids is diminished, lateral mobililty increases and leaflet asymmetry is lost.
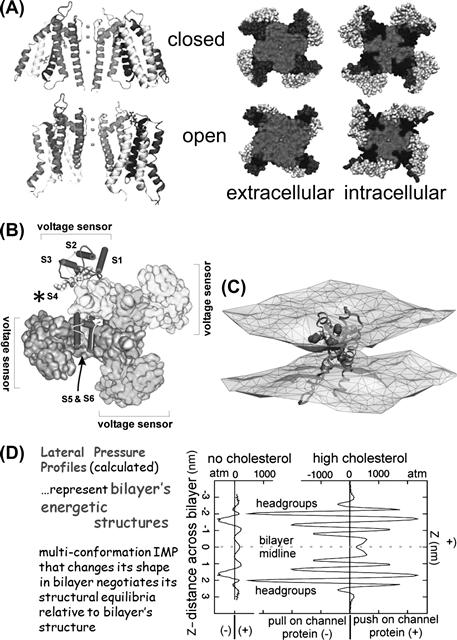
FIGURE 27.2 Protein structure and bilayer structure are interdependent; thence “MS channels”. (A) Kv channel, structural models for two conformations. Voltage sensors occupy periphery; note different protein–lipid interfaces and (B) Kv channel indicating peripheral location of S4-based voltage sensors (A, B modified fromSchmidt and MacKinnon, 2008 and references therein). (C) An activated-state voltage sensor domain locally deforms/thins the bilayer (modified fromKrepkiy et al., 2009 ). (D) Different bilayers have composition-dependent structure and energetics. Lateral pressure profiles calculated for symmetric bilayers without/with cholesterol (see Finol-Urdaneta et al., 2010 for details).
In situ, channels could be strongly to weakly mechano-protected, according to the status of the cortical spectrin–actin–myosin membrane skeleton (Morris, 2001b). Local modifications (cellular or subcellular, chronic or transient) might allow for developmentally or physiologically meaningful mechanical signals from channels. To establish convincingly such phenomena, however, identified MS channels with mutant mechano-phenotypes will likely be needed, as will discrete amphiphilic agents that preferentially inhibit (or activate) the identified channels during bilayer deformation (Fig. 27.3).
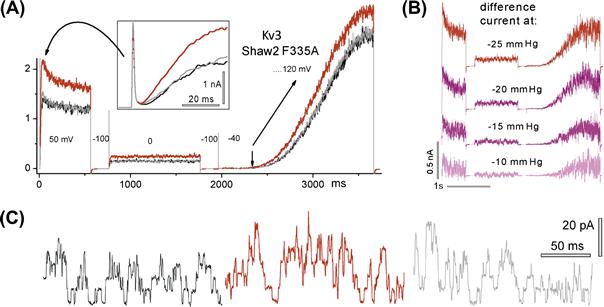
FIGURE 27.3 Kv3 channel activity, before, during and after stretch (black, red and gray traces – same meaning in Figs. 27.6, 27.7, 27.8). (A) Currents during a complex voltage protocol (large then small depolarizing steps followed by a V-ramp); boxed inset, early current); (B) stretch difference currents for different stretch intensities; (C) unitary currents at 0 mV (see Laitko et al., 2006 for details).
Disease mutant MS channels in rhythmically active cells (cardiomyocytes, smooth muscle) might provide fertile ground for in situ studies. Consider the contracting, pumping heart. Cardiac rhythm varies with mechanical load and, where causal links exist between mechanical events and cardiac electrophysiology, there is mechano-electric feedback. Voltage-gated channels (VGCs) shape cardiac rhythms and all classes of VGC have proven to be inherently MS (see Figs. 27.1B,C, 27.4–27.8). To argue a priori that the abundant and ubiquitous VGCs of the myocardium never feel the impacts of shear or stretch forces in the pumping heart would be difficult and yet, evidence showing that any specific MS channel participates in cardiac mechano-electric feedback is lacking. Molecular and genetic tools and modeling approaches now available for VGCs might be usefully exploited to make headway on this question (Morris, 2011a, b).
What can be said, a priori, about ion channel mechanosensitivity? The following: for any structurally dynamic integral membrane protein with >1 bilayer/protein interface conformation, structural deformation of the bilayer will elicit re-equilibration among protein conformations (see Fig. 27.4A). The scope of this simple notion is wide. Consider, for example, voltage-gated sodium channel (Nav) responses in the following circumstances: depletion or addition of membrane cholesterol or fatty acids (Andersen and Koeppe, 2007), traumatic brain injury (Wang et al., 2009), focused ultrasound applied to cortical brain regions (Tufail et al., 2010), stretch of ventricular myocyte sarcolemma (Banderali et al., 2010). Modified Nav channel activity in each scenario is explained most simply in terms of modified channel/bilayer interactions. This applies generally for VGC channels (Schmidt and MacKinnon, 2008) as well as for Nav channels in particular (Morris and Juranka, 2007a; Wang et al., 2009; Banderali et al., 2010; Morris, 2011b).
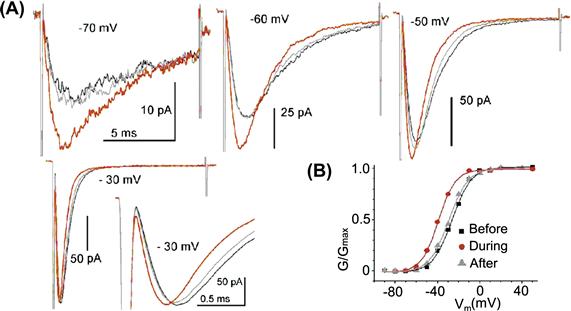
FIGURE 27.4 Nav1.5 currents before during and after stretch. (A) Voltages as labeled; expanded current at −30 mV (which is ≈gmax) reveals the “purely kinetic” effect of stretch, which is to accelerate current onset and inactivation (modified fromMorris and Juranka, 2007b); (B) the Nav1.5 g(V) left-shifts reversibly with stretch (due to pipette suction of −30 mmHg) (modified from Banderali et al., 2010).
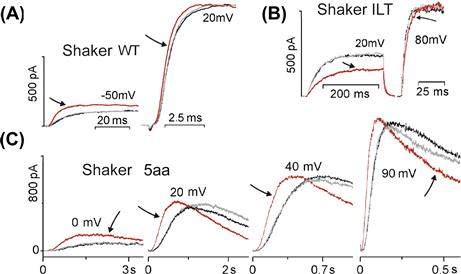
FIGURE 27.5 VGCs are MS channels in which the effects of stretch on particular transitions can be discerned. (A, B, C) Shaker WT and ILT and 5aa as described in the text. Arrows to red traces highlight the during stretch traces. Note that stretch reversibly slows current onset in ILT, while speeding it in WT (as for Nav1.5, the purely kinetic effect is evident at or beyond gmax). Not shown, WT g(V) reversibly left-shifts and ILT g(V) reversibly right-shifts with stretch (see Laitko and Morris, 2004; Laitko et al., 2006 for details).
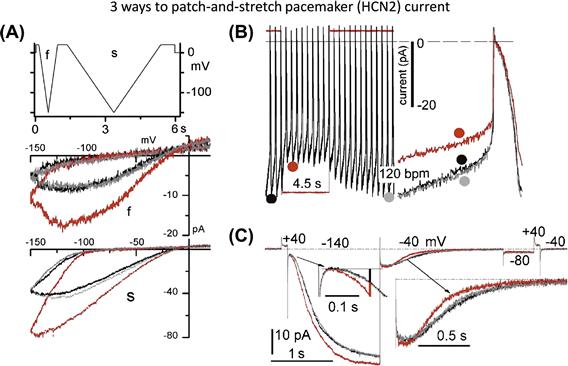
FIGURE 27.6 The pacemaker channel, HCN2, can generate both “SA” and “SI” cation current (Stretch-Augmented, Stretch-Inhibited): (A) V-ramp clamp at two speeds; (B) passive action potential clamp (the net effect under these circumstance: a reversible stretch-inhibition of cation current); and (C) V-step clamp reveals stretch acceleration of both HCN2 current onset and turn-off (the turn-off is “equivalent” to Shaker turn-on vis à vis voltage-sensor movements during depolarization). If cells had a background HCN2 conductance, its stretch-accelerated turn-off could be construed as a “SI cation conductance”. (Consult Lin et al., 2007, Morris, 2011a, b, for details.)
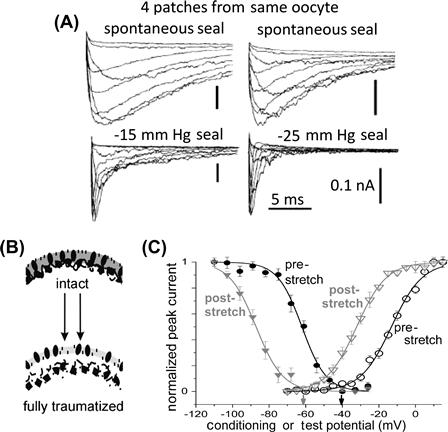
FIGURE 27.7 Membrane trauma can irreversibly alter channel behavior. Trauma here results from gigaohm seal formation accompanied by unintended (A) or intended (C) patch stretch. (A) Four patches, different trauma intensities, same oocyte, Nav1.4 (no beta subunit) current families (see Morris and Juranka, 2007a; Tabarean et al., 1999). (B) Membrane trauma that induces bleb formation irreversibly alters the bilayer environment (▵packing order, ▵thickness, ▵asymmetry) for channels (see Wang et al., 2009). (C) For several VGCs, gating is irreversibly left-shifted by trauma as in this example for Nav1.6 activation and steady-state inactivation (Wang et al., 2009).
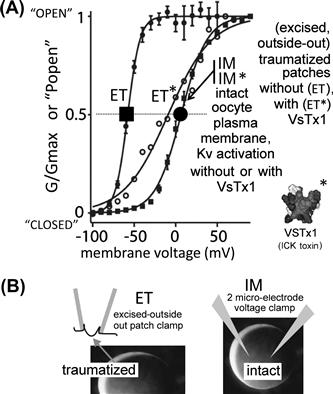
FIGURE 27.8 Bilayer mechanics allows an amphiphilic channel toxin to act like a “silver bullet”, targetting only channels in traumatized membrane. A Kv channel that is irreversibly left-shifted by excision trauma (compare g(V) for IM and ET). Kv is unaffected in intact membrane (IM∗) by the amphiphilic toxin, VsTx1(∗), but is strongly inhibited (right-shifted) by the toxin in traumatized membrane (ET∗). (Modified from Schmidt and MacKinnon, 2008, see further discussion in Morris, 2011a, b).
“Structure” at the angstroms to tens of nanometers scale encompasses shape, dimensional size and local charge density; together this equates closely to “mechanics”. Thus, secondary and tertiary protein structure can be inferred from the set of mechanical forces needed to pull the protein apart from various angles. Applying structure=mechanics thinking to bilayers is fruitful when considering bilayer/membrane protein interfaces (e.g. Butterwick and MacKinnon, 2010). At various depths through the bilayer (“Z”-dimension), bilayer lipids collectively push or pull any embedded protein, with the sign and magnitude of these forces switching dramatically within angstroms (see Fig. 27.4D) and with the Z-dimension integral of push/pull forces in the equilibrated bilayer being zero. For any given bilayer (e.g. Fig. 27.9A,B), this equilibrium “Z-force” summary is its lateral pressure profile (LPP). The prevailing equilibrium (and hence the LPP) can be perturbed by changes in the bilayer’s chemical make-up, by applied physical forces (stretch, hydrostatic pressure, enforced curvature) and by temperature changes (Baumgart et al., 2003, 2011; Morris and Juranka, 2007a; Patel et al., 2010; Morris, 2011b).
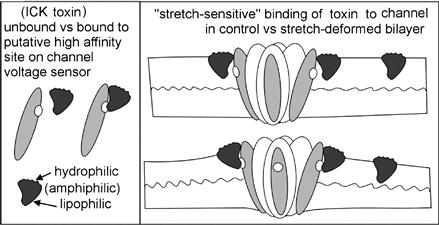
FIGURE 27.9 The efficacy of amphiphilic ICK peptide voltage sensor toxins depends on bilayer mechanics and so some of these toxins might be stretch-sensitive as cartooned here. Other classes of amphiphilic peptide agents with specific binding targets on different classes of channels or non-channel membrane proteins are likely to share this feature (see Morris, 2011a, b ).
LPPs are good mnemonics for these ideas, but emerge from calculations, not direct measurements. Channel activity, of course, is exquisitely accessible to measurement. If at least one of a channel’s several structural states (i.e. conformations, e.g. open, closed, inactivated, desensitized, partially-open… etc.) differs at the protein/bilayer interface then any bilayer deformation at that interface has the potential to modulate the channel’s time-averaged activity and a measurable activity change justifies calling the channel “mechanosensitive”.
The first-cloned VGC, Shaker (Kv1), is thus an MS channel based on its response to patch stretch (see Figs. 27.1B,C and 27.7). More generally than just stretch, one can alter bilayer thickness and orderliness by pressure, temperature and by removal/addition of multitudes of different amphiphiles (e.g. fatty acids, cholesterol, sphingomyelin, alkanols, drugs, soluble gases). For “MS-channel-X”, each of these bilayer mechanical perturbors could alter the probability of being open (Popen) in some fashion. VGCs behave this way (Tilman and Cascio, 2003;Andersen and Koeppe, 2007; Morris and Juranka, 2007a; Finol-Urdaneta et al., 2010), but “MS-channel-X” encompasses the whole alphabet soup of channel families, from ATP-binding cassettes (ABCs) to ligand-gated ion channels (LICs) to VGCs. Moreover, any multiconformation membrane protein with >1 lipid/bilayer interface structure could be mechanosensitive even though “read-outs” might be trickier in, say, Na/K pumps or Na/Ca exchangers than in Nav, Cav, Kv channels. Thanks to precision readouts from rhodopsin, G-protein coupled receptor bilayer mechanics are rather well understood (Soubias et al., 2010). At the brutal extreme of cytomechanics, gross perturbation of bilayers embedding rhodopsin and axonal Nav channels might explain “seeing stars” and “knock-out punches” (Wang et al., 2009).
IV Channel Mechanosensitivity – Tuning of Channel Behavior
Fine-tuning of membrane protein function via bilayer structure can have major implications for development, for physiology and for biomedicine (pathology/pharmacology/anesthesiology). Mass spectrometry-based membrane lipidomics is uncovering enormous diversity among bilayer lipids (Shevchenko and Simons, 2010). Understanding their contributions to general bilayer structure and to particular protein/bilayer interfaces will be a task of many years, but already there are big changes. Until recently, the plasma membrane bilayer was seen mostly as a two-dimensional amphiphilic solvent that reliably orients membrane proteins while simultaneously providing an osmotic barrier to define intra- from extracellular. Lipid imaging, biochemistry (Shevchnko and Simons, 2010; Baumgarten et al., 2011) plus high-resolution channel structures (Schmidt et al., 2009) and computational approaches (Marsh, 2008; Krepkiy et al., 2009; Patel et al., 2010; Baumgarten et al., 2011) have helped bilayers garner respect. No longer are VGCs routinely depicted as cylinders with fixed lateral walls or cartooned with their voltage sensors sequestered from bilayer lipids. Structural models of VGCs now depict protein/lipid interfaces regions for different conformations (e.g. open-like and closed-like for Kv channels (see Fig. 27.4A) with different lateral interfaces.
Bilayers at their most basic are established and maintained by thermally-driven processes; they continually self-organize, minimizing the system’s free energy (Boal, 2001). Channel activity too is thermally-driven, as is directly evident from unitary currents, whose stochastics, quantified for different conditions, yield “channel kinetics”.
e.g. for a one-Nav-channel membrane patch, step 1000X from −100 mV to 0 mV, make a 1000-item histogram of the random-lengthed times-till-first-opening, from plotted histogram obtain the characteristic “first latency” at 0 mV. Repeat for steps to different voltages, plot voltage-dependence of this kinetic parameter. Likewise for open times and closed times (Horn and Vandenberg, 1984).
Taken together, these susceptibilities to thermal energy predict that the rates at which channel conformation changes occur will vary as bilayer structure is varied. Bilayer structure and protein/bilayer interface structure are both subject to the thermal environment; protein and bilayer will therefore accommodate each other structurally (Marsh, 2008). This mutual accommodation has “real-time” consequences – i.e. consequences on the time scales characteristic for activity in any particular channel, whether Popen changes re-equilibrate over hundreds of microseconds or hundreds of milliseconds.
To illustrate this last point, consider how stretch affects fast and ultra-slow variants of a particular VGC channel (Tabarean and Morris, 2002). In both, stretch of a given intensity accelerates activation approximately the same-fold. Not, that is, by the absolute amount, but by the same relative amount (say, 1.6-fold). For both channels, stretch tips the relevant energy landscape the same number of kT units. In a Boltzmann equation for Popen(V), i.e. for ([open]/[open+closed])(V), the affected term is that for the conformational energy difference between the two states, which is “the pre-exponential term” in an Arrhenius equation for thermally-activated exponential behavior. Imagine a generic VGC in a membrane whose voltage corresponds to the activation-Boltzmann midpoint for that channel: by definition, the VGC is equally stable open or closed. Single-channel recordings under such conditions directly reveal thermal energy nudging a Kv channel (say) back and forth between open and closed. Now, change the bilayer structure via stretch (see Fig. 27.1C), for example, or via hyperbaric pressure see Morris and Juranka, 2007a) or via amphiphiles (Fig. 27.10) (Andersen and Koeppe, 2007; Finol-Urdaneta et al., 2010; Patel et al., 2010). Voltage is unchanged but the channel has a new Popen. These considerations apply for elastic (reversible) change and also for plastic change, like the irreversible bilayer restructuring that occurs when membranes bleb due to trauma, ischemia and inflammatory conditions (see Fig. 27.2).
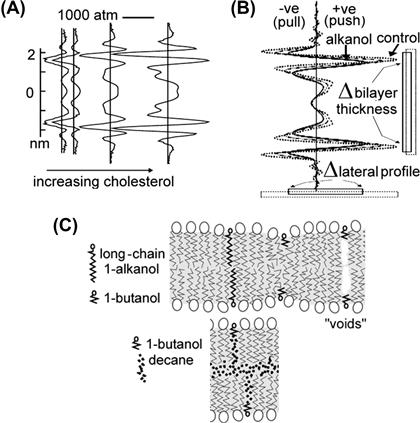
FIGURE 27.10 Calculated LPPs for two different symmetrical bilayers. To each is added increasing abundances of (A) cholesterol and (B) a short chain alkanol. In (C) top, a cartoon depicts impacts of short- and long-chain alkanols in a “bio-bilayer” or any other solvent-free membrane. The planar bilayers from which data in Figure 27.10 were obtained, did have solvent – decane; as cartooned below, a short-chain alkanol can team with decane (which prevents “void collapse”) and thus behave like a long-chain alkanol (see Finol-Urdaneta et al., 2010 for details).
Bilayer mechanics operate when a bilayer/protein interface structure impacts channel function. Outside the purview of bilayer mechanics is a lipid (e.g. an inositol phosphate) behaving as a ligand at a discrete binding site in a channel. Nevertheless, like ligands or covalent modifications, bilayer-structure-changing amphiphiles extend or “tune” the behavioral repertoire of a membrane protein (Tillman and Cascio, 2003). One gene product in three quite different bilayers is like three slightly different gene products in one bilayer. Auditory hair cells use structural tuning on many fronts to optimize mechano-electrical signaling over the auditory spectrum frequencies. In outer hair cells, electromechanical signaling involves prestin, a charge-transferring membrane protein whose in-plane expand/contract conformation changes generate cellular motion. Since cholesterol and fatty acid levels affect the rates of those transitions (Nilsen et al., 2011), it is inevitable to wonder if prestin “resonances” occur along the cochlea’s low-high frequency axis, associated with a gradient of peri-prestin lipid-packing density.
V VGCS and the Mechanosensitivity of Discrete Transitions
Voltage sensor proteins in their activated conformations elicit a local (nanometers) thinning of the bilayer (Krepkiy et al., 2009) so, unsurprisingly, membrane stretch favors activated states of VGCs. This cavalier-sounding sentence could not have been written before late 2009 (see Fig. 27.5C) but it is now evident that applied stretch would relieve a voltage sensor of some of the bilayer-thinning work it must do to go from resting to activated. More than for most other classes of membrane protein, structure/function relations of VGCs, especially for voltage-gated K channels (Kv) (Bezanilla, 2008; Schmidt et al., 2009; Hulse et al., 2010) are getting less mysterious. Fortunately, the message of this section, that protein dynamics and bilayer structure interdependencies are becoming reasonably explicable, extrapolates to other multiconformation integral membrane proteins (Patel et al., 2010).
Voltage-clamped VGC currents monitored for tens of microseconds to hundreds of seconds allow fast and slow VGC conformation changes to be monitored, yielding gating current, i.e. charge movement associated with voltage sensor motions, and ionic current, i.e. ion flow through a channel’s selectivity filter pore or its omega-current pathways. Simultaneously monitored site-directed fluorescence signals provide read-outs about protein movements. Given the resulting database, discrete conformation changes in VGCs can be monitored in conjunction with bilayer mechanical perturbations in cells or artificial bilayers (Schmidt and MacKinnon, 2008; Finol-Urdaneta et al., 2010). Reversible patch stretch is the simplest case to consider. A stretched bilayer will have more disorderly hydrocarbon tails at mid-plane. In a stretched bilayer, the changed propensity for hydrophobic mismatch at any channel/lipid interfaces will have to be compensated.
In Kv1 and in Nav channels, the rate-limiting voltage dependent transition for activation is, it turns out, also a stretch-sensitive transition: in these VGCs, current turn-on accelerates with stretch (Laitko and Morris, 2004; Banderali et al., 2010). In other VGCs, including Cav and Kv3, stretch increases Popen without any change in the speed of current turn-on (Calabrese et al., 2002; Morris, 2011a). Since the rate-limiting (slowest) transition in the voltage activation pathway of these channels is indifferent to stretch, it probably occurs remote from the bilayer interface.
“ILT” is a mutant Kv1 channel useful because its rate-limiting activation transition happens after the one mentioned above in wild-type (WT) Kv1 channels. In WT-Kv1, each of four identical subunits responds independently to a depolarizing step. Those rate-limiting motions make activation in WT Kv1 current a fourth order process (which, as indicated already, accelerates with stretch). In ILT-Kv1, the next step in the activation pathway, a highly cooperative or “concerted” transition (four subunits together) has been rendered so sluggish that it becomes the slowest step. In ILT-Kv1, as a consequence, activation is a first order process and, interestingly, membrane stretch decelerates (see Fig. 27.7B) that single-exponential current onset (Laitko et al., 2006; Morris, 2011a).Thus, for Kv1 in a stretch-deformed bilayer, the independent sensor motions become easier to achieve while the concerted transition becomes harder.
These stretch experiments indicate that in Kv1, concerted and independent transitions both “see” the bilayer interface, while the less-understood rate-limiting activation transitions in Kv3 and Cav do not. What of inactivation transitions? In Nav1.5, the cardiac sodium channel, fast inactivation is a “particle-binding” process strongly coupled to activation but not directly affected by stretch either in WT or in disease mutant-Nav1.5 channels with impaired fast inactivation; in both, stretch accelerates fast inactivation only secondarily, via activation (Banderali et al., 2010). By contrast, for Kv1 slow inactivation (a selectivity filter-occlusion process unrelated to Nav inactivation), stretch is a direct accelerator, distinct from its action on activation (Laitko and Morris, 2004).
Short-chain alkanols (e.g. butanol) are excellent chemical agents for reversibly perturbing bilayer mechanics but, over the years, the qualitatively different, albeit comprehensible, behavior of alkanols and cholesterol in solvent-containing (planar) bilayers versus natural membranes has generated confusion (see Fig. 27.9). Recent work shows that, in KvAP channels, gating kinetics and conductance (see Fig. 27.10) respond simultaneously to these surface active agents (Finol-Urdaneta et al., 2010).
VI Bilayer Structure in X, Y and Z – One LPP Here, Another LPP There
Except in broad biochemical physics terms, plasma membrane bilayer structure in living cells is poorly understood (Shevchenko and Simons, 2010). In the X-Y plane, bilayer structures are non-uniform vis à vis lipid species distribution, leaflet asymmetry, thickness, orderliness and curvature (Baumgart et al., 2010). Sustained high curvature requires protein aggregates, asymmetry requires ATP-flippase activity. Concentrations and arrangements of lipid species of each situation remain to be characterized. Thickness and order differences readily quantified in model systems (Baumgart et al., 2003) are hard to detect in living cells; subdomains in mammalian cells at physiological temperatures are in the 10–200 nm range (Shevchenko and Simons, 2010).
Fluorescent lipid probes like laurdan align parallel to hydrophobic tails of phospholipids in bilayers, emitting at different wavelengths according to nano-environment fluidity/water content. In a fascinating precedent using laurdan, distinctive bilayer heterogeneity is seen in living transparent zebrafish embryos (Owen et al., 2010), poikilotherms for whom lower temperatures are physiological. Gut epithelial cells express transporters and channels differentially along their apical-to-basal axis, so it is fascinating to see, in situ, clear apical-to-basal polarization in the lipid orderliness of living gut cells.
Elevated cholesterol and sphingomyelin typically correlate with thicker more orderly bilayer domains and unsaturated fatty acid with thinner more disorderly bilayers. Large hydrostatic (hyperbaric) pressures increase bilayer thickness by virtue of denser lateral packing (more order), whereas bilayer stretch and elevated temperature both cause thinning and greater disorder. Short-chain alkanols (e.g. butanol) thin native bilayers and increase mid-plain disorder while reducing surface (interfacial) tension (imposed stretch, by contrast, increases the interfacial tension). Lipid molecules differ in shape, size, internal rigidity, headgroup charge and charge dispersion. These molecular features of lipids collectively determine bilayer structure and hence the energetics (Andersen and Koeppe, 2007; Patel et al., 2010) of bilayer/protein interfaces, collectively, modulating the stability of different membrane protein conformations (Morris and Juranka, 2007a).
Channels have some lipid requirements outside the “bilayer mechanics” repertoire. Kv channel activation, for example, has an absolute requirement for phosphatidyl-phosphate/S4-arginine interactions (see Schmidt et al., 2009). Stereo-specific binding of amphiphilic voltage sensor toxins occurs at lipid-embedded voltage sensor sites; this, too, is not bilayer mechanics (see Finol-Urdaneta et al., 2010). Nevertheless, the global efficacy of voltage sensor toxins is strikingly sensitive to bilayer mechanics (see Figs. 27.3 and 27.11) (Schmidt and MacKinnon, 2009; see Morris, 2011b).
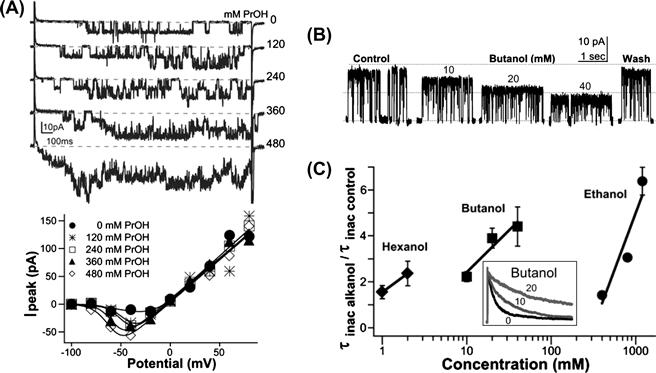
FIGURE 27.11 Surface active agents modulate gating and unitary conductance of a Kv channel in a planar bilayer. (A) KvAP currents traces with increasing abundance of propanol, and below, progressively more left-shifted I/V relations; perhaps short-range bilayer thinning required for voltage sensor activation (see Fig. 27.1C) becomes easier with small alkanols present. (B) Open KvAP in the presence of bilayers of increasing packing order (due to butanol teamed with decane; see Fig. 27.9C) shows a decreasing unitary conductance. (C) Slower entry of KvAP into its slow inactivated state (the state seen in Kv crystal structures) correlates with increased packing order due to alkanols-plus-decane (inset, normalized currents, as labeled) (see Finol-Urdaneta et al., 2010 for details).
Specific binding of inner leaflet cholesterol molecules to discrete locations abutting the transmembrane domains of a Kir channel (Rosenhouse-Dannntsker et al., 2011) appears to be an intriguing instance of a channel using its cytoplasmic domains to regulate the LPP at its own bilayer/protein interface. Could “bilayer auto-mechanics” be the appropriate term here? It seems likely that comparable provisions will turn up in other channels.
The idea that healthy plasma membrane bilayer structure represents a controlled disequilibrium has just begun to be contemplated. Bilayers of living cell plasma membranes have asymmetrical leaflets. The exo-leaflet has little phosphatidyl serine or phosphatidyl ethanolamine and abundant phosphatidyl choline. Vice versa for the cyto-leaflet. ATP-dependent lipid flippases generate asymmetry at endomembrane stages. Properly arrayed adherent proteins help maintain the asymmetry. As has long been recognized for apoptotic blebs, the blebbed membrane of cells in traumatized, ischemic and inflamed tissue loses bilayer asymmetry, but the consequences for channel function have received little attention. In mechanically blebbed membrane, some Kv and Nav channels activate at pathologically hyperpolarized potentials (Schmidt and MacKinnon, 2008; Wang et al., 2009) (see Figs. 27.2C and 27.11A). In ischemic tissue, bilayer disorder due to ATP-starved flippases might achieve the same effect.
Having contemplated bilayer asymmetry, we turn back to symmetric spontaneously-formed bilayers and the lateral pressure profile (LPP) summary of Z-dimension energetics. Extensive formation of weak, thermally-labile bonds (van der Waals, hydrophobic and electrostatic interactions and hydrogen bonds) underlies the free energy minimization that characterizes structural equilibrium. This maximizes the interactions among the lipids’ hydrocarbon tails and maximizes water–water interactions. The outcome is minimal contact between water and lipid-“grease”, while the lipids’ charged-or-polar headgroups are constrained to the two X,Y-plane interfaces separating hydrocarbon from water.
The terms tension, surface tension, interfacial tension, pressure and force are sometimes confusing in bilayer contexts. As in protein structural energetics, forces, in energy units, are what matter. A tension is a force per unit length and pressure is force per unit area. Note that “compression” can take on contradictory context-specific usages. Some speak of a force locally compressing the bilayer where hydrophobic mismatch causes local thinning. Others use it for global bilayer compression under hydrostatic pressure, which causes global bilayer thickening. In an LPP, “lateral pressures” can be in the ±1000 atm range; these are forces acting over some infinitisimally small “area” encircling an imaginary cylinder in the Z-axis. “Living” bilayers with metabolically forced leaflet asymmetries and/or curvatures (Marsh, 2008), sustained in part by peripheral and integral membrane proteins, will be non-equilibrium structures whose LPPs need not integrate to zero. Since membrane proteins are evolutionarily “designed” for asymmetric cholesterol-rich bilayers but crystallized without them, accommodations (Marsh, 2008) to those absences can be expected in even the most elegant of high-resolution structures.
In the LPPs of Figs. 27.4 and 27.9, bilayer thickness is defined by the distance between the two strongly negative lateral pressure regions at the bilayer–water interfaces. Just exterior to that, repulsions between polar headgroups create a smaller positive pressure; positive pressures tend to compress circumferentially any embedded protein. Interacting lipid tails exclude water where pressures go negative at the two bilayer edges. These narrow zones of intense negative pressure will tend to pull circumferentially on any embedded protein (think “suction” from any embedded protein’s point of view) as the lipids fight successfully to stay together. LPPs can be highly structured along Z or flatter. Whereas cholesterol dramatically increases Z-structure in an LLP, addition of short chain alkanols flattens it out. Most eukaryotic membranes are cholesterol-rich in both leaflets, as well as being asymmetrical and so, presumably, most have considerable “Z-structure”. The consequence of adding short alkanols to a membrane is colloquially called “lowering the surface tension” (by analogy to the lowering to air–water interfacial [surface] tension upon addition of alcohol), but in reality, the entire LPP is “lowered” (i.e. flattened). I mention this to emphasize that any “surface active agent”, even if it does not readily cross the bilayer, acts (energetically) across the entire LPP of a bilayer.
LPPs for biological (asymmetrical) bilayers would differ among cell types and among subdomains in a given cell. If membrane protein X occurs where there are different LPPs, both its basic behavior and its responses to amphiphiles will vary whenever the LPP varies. Imagine Channel-X (or GCPR-Z) in diverse neurons with different LPPs. Add amphiphile-A (e.g. propofol) or cocktail-Y (e.g. ethanol + resveratrol + tetrahydrocannabinol + dimethyletcetera). Channel-X (or GCPR-Z) will surely show LPP-dependent “side-effects” when confronted with amphiphile-A or cocktail-Y in quantities sufficient to alter LPPs. Membrane proteins whose conformation changes do not occur at bilayer interfaces could avoid amphiphile modulation but, for VGCs and GCPRs, at least, as well as for all the other MS channels, this is not an option.
VII Physiology? Read with Caution. Proceed with Caution
When it comes to physiology and MS channels, the arena is littered with flawed reports in the highest impact journals and elsewhere. An amazing dynamic range of a few centimeters of water (pipette aspiration pressure) for MS channel activity… surely that indicates exquisitely stretch-sensitive channels? No, it indicates that the so-called “manometer” was corked. Or, readily evident cut-and-paste errors at four points in a report claiming to have cloned an MS calcium-permeant channel – retracted, surely? No, just a “report clarification”. The irreproducible “cloning” of ENaC and of TRPC1 as calcium-permeant MS-channels – retracted? No, but at least the latter effort was “revisited”. A fundamental problem has been lack of proper controls. In our attempt to clone the stretch-activated cation channel of Xenopus oocytes, candidates were tested using blinded stereotyped protocols. After much effort, the dismal conclusion was that none of our clones expressed in mammalian cells yielded more MS current than is seen in controls (Wan et al., 1999; Juranka et al., 2001). Endogenous MS channels tend to activate more readily upon repeated stretch trials; in the absence of blinded stereotyped procedures, this evidently generates the results one expects/wants. Exacerbating problems in this arena have been misuse of gadolinium as a blocker and assertions that GsMTx4 is a specific inhibitor of MS cation channels (see Morris, 2011a, b).
Piezo1 and Piezo2 and CFTR, new entries to the field, and perhaps VGCs, might revitalize the cellular physiology of ion channel mechanosensitivity, though Piezos are probably mechanotransducer specialist proteins. VGCs, the best understood of the identified non-specialist MS channels, generate rhythmic signals in myocardium and smooth muscle. Perhaps arrhythmia-inducing VGC mutants with distinctive mechano-phenotypes, plus the MS actions of voltage sensor toxins (Morris, 2011a, b) will provide leverage for assessing their possible contributions to mechanophysiology.
BIBLIOGRAPHY
1. Andersen OS, Koeppe 2nd RE. Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct. 2007;36:107–130.
2. Bailey RW, Nguyen T, Robertson L, et al. Sequence of physical changes to the cell membrane during glucocorticoid-induced apoptosis in S49 lymphoma cells. Biophys J. 2009;96:2709–2718.
3. Banderali U, Juranka PF, Clark RB, Giles WR, Morris CE. Impaired stretch modulation in potentially lethal cardiac sodium channel mutants. Channels (Austin). 2010;4:12–21.
4. Baumgart T, Capraro BR, Zhu C, Das SL. Thermodynamics and mechanics of membrane curvature generation and sensing by proteins and lipids. Annu Rev Phys Chem. 2011;62:483–506.
5. Baumgart T, Hess ST, Webb WW. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 2003;425:821–824.
6. Bezanilla F. Ion channels: from conductance to structure. Neuron. 2008;60:456–468.
7. Boal D. Mechanics of the Cell. Cambridge University Press 2001.
8. Boucher PA, Morris CE, Joós B. Mechanosensitive closed-closed transitions in large membrane proteins: osmoprotection and tension damping. Biophys J. 2009;97:2761–2770.
9. Butterwick JA, MacKinnon R. Solution structure and phospholipid interactions of the isolated voltage-sensor domain from KvAP. J Mol Biol. 2010;403:591–606.
10. Calabrese B, Tabarean IV, Juranka P, Morris CE. Mechanosensitivity of N-type calcium channel currents. Biophys J. 2002;83:2560–2574.
11. Coste B, Mathur J, Schmidt M, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60.
12. Cueva JG, Mulholland A, Goodman MB. Nanoscale organization of the MEC-4 DEG/ENaC sensory mechanotransduction channel in Caenorhabditis elegans touch receptor neurons. J Neurosci. 2007;27:14089–14098.
13. Finol-Urdaneta RK, McArthur JR, Juranka PF, French RJ, Morris CE. Modulation of KvAP unitary conductance and gating by 1-alkanols and other surface active agents. Biophys J. 2010;98:762–772.
14. Gray JA, Sato M. Properties of the receptor potential in Pacinian corpuscles. J Physiol. 1953;122:610–636.
15. Gu CX, Juranka PF, Morris CE. Stretch-activation and stretch-inactivation of Shaker-IR, a voltage-gated K+ channel. Biophys J. 2001;80:2678–2693.
16. Guharay F, Sachs F. Stretch-activated single ion channel currents in tissue-cultured embryonic chick skeletal muscle. J Physiol. 1984;352:685–701.
17. Hamill OP, Martinac B. Molecular basis of mechanotransduction in living cells. Physiol Rev. 2001;81:685–740.
18. Horn R, Vandenberg CA. Statistical properties of single sodium channels. J Gen Physiol. 1984;84:505–534.
19. Hudspeth AJ. Making an effort to listen: mechanical amplification in the ear. Neuron. 2008;9:530–545.
20. Hulse RE, Li Q, Perozo E. Up a hydrophobic creek with a short paddle. Cell. 2010;142:515–516.
21. Juranka PF, Haghighi AP, Gaertner T, Cooper E, Morris CE. Molecular cloning and functional expression of Xenopus laevis oocyte ATP-activated P2X4 channels. Biochim Biophys Acta. 2001;1512:111–124.
22. Krepkiy D, Mihailescu M, Freites JA, et al. Structure and hydration of membranes embedded with voltage-sensing domains. Nature. 2009;462:473–479.
23. Kung C, Martinac B, Sukharev S. Mechanosensitive channels in microbes. Annu Rev Microbiol. 2010;64:313–329.
24. Laitko U, Morris CE. Membrane tension accelerates rate-limiting voltage-dependent activation and slow inactivation steps in a Shaker channel. J Gen Physiol. 2004;123:135–154.
25. Laitko U, Juranka PF, Morris CE. Membrane stretch slows the concerted step prior to opening in a Kv channel. J Gen Physiol. 2006;127:687–701.
26. Lin W, Laitko U, Juranka PF, Morris CE. Dual stretch responses of mHCN2 pacemaker channels: accelerated activation, accelerated deactivation. Biophys J. 2007;92:1559–1572.
27. Liu X, Huang H, Wang W, Wang J, Sachs F, Niu W. Stretch-activated potassium channels in hypotonically induced blebs of atrial myocytes. J Membr Biol. 2008;226:17–25.
28. Lundbaek JA, Koeppe 2nd RE, Andersen OS. Amphiphile regulation of ion channel function by changes in the bilayer spring constant. Proc Natl Acad Sci USA. 2010;107:15427–15430.
29. Marsh D. Protein modulation of lipids, and vice-versa, in membranes. Biochim Biophys Acta. 2008;1778:1545–1575.
30. Morris CE. Mechanosensitive ion channels. J Membr Biol. 1990;113:93–107.
31. Morris CE. Mechanosensitive ion channels in eukaryotic cells. In: Sperelakis N, ed. Cell Physiology Sourcebook. Academic Press 2001a;:745–760.
32. Morris CE. Mechanoprotection of the plasma membrane in neurons and other non-erythroid cells by the spectrin-based membrane skeleton. Cell Mol Biol Lett. 2001b;6:703–720.
33. Morris CE. Pacemaker, potassium, calcium, sodium: stretch modulation of the voltage-gated channels. In: Kohl P, Sachs F, Franz M, eds. Cardiac Mechano-Electric Coupling and Arrhythmias: from Pipette to Patient. Elsevier Saunders 2011a;:43–49.
34. Morris CE. Voltage gated channel mechanosensitivity. Fact or friction? Front Physiol 2011b; 2:25.
35. Morris CE, Horn R. Failure to elicit neuronal macroscopic mechanosensitive currents anticipated by single-channel studies. Science. 1991;251:1246–1249.
36. Morris CE, Juranka PF. Lipid stress at play: mechanosensitivity of voltage-gated channels. In: Hamill O, Simon S, Benos D, eds. 2007a;:297–337. Mechanosensitive Ion Channels, Part B Curr Top Membr. 59.
37. Morris CE, Juranka PF. Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J. 2007b;93:822–833.
38. Morris CE, Juranka PF, Lin W, Morris TJ, Laitko U. Studying the mechanosensitivity of voltage-gated channels using oocyte patches. Methods Mol Biol. 2006;322:315–329.
39. Nilsen N, Brownell WE, Sun SX, Spector AA. Effect of membrane mechanics on charge transfer by the membrane protein prestin. Biomech Model Mechanobiol 2011; Mar 2.
40. Owen DM, Magenau A, Majumdar A, Gaus K. Imaging membrane lipid order in whole, living vertebrate organisms. Biophys J. 2010;99:L7–9.
41. Patel A, Sharif-Naeini R, Folgering JR, Bichet D, Duprat F, Honoré E. Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflügers Arch. 2010;460:571–581.
42. Phillips R, Ursell T, Wiggins P, Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459:379–385.
43. Rosenhouse-Dantsker A, Logothetis DE, Levitan I. Cholesterol sensitivity of KIR2.1 is controlled by a belt of residues around the cytosolic pore. Biophys J. 2011;100:381–389.
44. Sachs F, Morris CE. Mechanosensitive ion channels in non-specialized cells. Rev Physiol Biochem Pharmacol. 1998;132:1–78.
45. Schmidt D, MacKinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc Natl Acad Sci USA. 2008;105:19276–19281.
46. Schmidt D, Cross SR, MacKinnon R. A gating model for the archeal voltage-dependent K(+) channel KvAP in DPhPC and POPE: POPG decane lipid bilayers. J Mol Biol. 2009;390:902–912.
47. Shevchenko A, Simons K. Lipidomics: coming to grips with lipid diversity. Nat Rev Mol Cell Biol. 2010;11:593–598.
48. Soubias O, Teague Jr WE, Hines KG, Mitchell DC, Gawrisch K. Contribution of membrane elastic energy to rhodopsin function. Biophys J. 2010;99:817–824.
49. Tabarean IV, Morris CE. Membrane stretch accelerates activation and slow inactivation in Shaker channels with S3-S4 linker deletions. Biophys J. 2002;82:2982–2994.
50. Tabarean IV, Juranka P, Morris CE. Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys J. 1999;77:758–774.
51. Tillman TS, Cascio M. Effects of membrane lipids on ion channel structure and function. Cell Biochem Biophys. 2003;38:161–190.
52. Tufail Y, Matyushov A, Baldwin N, et al. Transcranial pulsed ultrasound stimulates intact brain circuits Neuron. 2010;66:681–694.
53. Wan X, Juranka P, Morris CE. Activation of mechanosensitive currents in traumatized membrane. Am J Physiol. 1999;276:C318–C327.
54. Wang JA, Lin W, Morris T, Banderali U, Juranka PF, Morris CE. Membrane trauma and Na+ leak from Nav1.6 channels. Am J Physiol Cell Physiol. 2009;297:C823–C834.
55. Wang W, Huang H, Hou D, et al. Mechanosensitivity of STREX-lacking BKCa channels in the colonic smooth muscle of the mouse. Am J Physiol Gastrointest Liver Physiol. 2010;99:G1231–G1240.
56. Zhang WK, Wang D, Duan Y, Loy MM, Chan HC, Huang P. Mechanosensitive gating of CFTR. Nat Cell Biol. 2010;12:507–512.