Chapter 32
Synaptic Transmission
Chapter Outline
III. Structure and Function of Chemical Synapses: An Overview
IVA. Neurotransmitters and Neurotransmitter Receptors
IVB. Biosynthesis, Storage and Inactivation of Neurotransmitters
IVC1. Quantal-Vesicular Hypothesis of Transmitter Release
IVC2. Essential Role of Ca2+ in Depolarization-Release Coupling
IVD. Generation of Postsynaptic Potentials at Fast Synapses
IVD1. Synaptic Current and Synaptic Equilibrium Potential
IVD2. Relationship Between Synaptic Currents and Postsynaptic Potentials
IVE. Slow Synaptic Transmission Mediated by G-Protein-Coupled Receptors
IVF. Synaptic Integration versus Amplification
I Summary
The transmission of synaptic signals is mediated by chemical neurotransmitter substances. Neurotransmitters are synthesized in presynaptic terminals and stored in synaptic vesicles. Transmitter release is evoked by presynaptic action potentials (APs), which activate influx of Ca2+ into terminals and trigger a Ca2+-dependent exocytosis of transmitter from synaptic vesicles into the synaptic cleft. Once released, neurotransmitters activate specific receptor-gated channels in the postsynaptic cell and elicit a transient change in the membrane permeability to cations or anions. Fast synaptic transmission is mediated by ionophoric receptors. Slow synaptic transmission is mediated by G-protein-coupled receptors. Excitatory postsynaptic potentials (EPSPs) are associated with transmitter-induced increase in Na+ and K+ conductance of the synaptic membrane, resulting in net entry of positive charge carried by Na+ and membrane depolarization. Inhibitory postsynaptic potentials (IPSPs) are associated with transmitter-activated influx of Cl− and membrane hyperpolarization. The EPSPs at the skeletal neuromuscular junction are called end-plate potentials (EPPs). In a healthy neuromuscular junction, the EPPs are always large enough to depolarize the muscle membrane to threshold and trigger muscle APs. The EPSPs generated at any single neuro-neuronal synapse are usually too small to depolarize the postsynaptic neuron to threshold. Synaptic signals converging onto a neuron are normally integrated through summation of EPSPs and IPSPs and an AP is triggered only when the resultant membrane potential reaches or exceeds the threshold. Chemical synaptic transmission is subject to modulation by intrinsic and extrinsic factors, including frequency and pattern of AP firing, which can either facilitate or depress the transmission across any given synapse.
II Introduction
The function of nerve cells is to receive, process and transmit information. Intercellular transfer of signals among neurons or from neurons to effector cells is accomplished at specialized intercellular junctions called synapses and is by and large chemically mediated. A characteristic feature of a chemical synapses is the presence of a 20–100 nm wide extracellular space, the synaptic cleft, which separates the presynaptic (transmitting) and postsynaptic (receiving) elements of the synapse. This physical discontinuity prevents efficient transfer of current between the cells and necessitates intervention of a diffusible chemical transmitter substance. The fundamental aspect of chemically-mediated transmission is the transduction of voltage signal into the release of neurotransmitter from the presynaptic neuron and transduction of transmitter binding to a specific receptor into voltage change in the postsynaptic cell.
The basic principles of chemical neurotransmission have been elaborated in a series of seminal experiments conducted on the vertebrate skeletal neuromuscular junction by B. Katz and his collaborators in the 1950s and 1960s (Katz, 1966). At about the same time, the pioneering work by J.C. Eccles and his coworkers on spinal motor neurons showed that the general principles of chemical transmission established at the vertebrate neuromuscular junction also apply to central synapses (Eccles, l964). Since then, new insights into the mechanism of synaptic transmission have been gained on the molecular level through the application of contemporary techniques of molecular biology and electrophysiology.
This chapter focuses on chemical synaptic transmission; however, the reader should be cognizant of another form of signal transfer, referred to as electrical transmission. Electrical transmission is mediated by the gap junctions (Bennett, 1997). Gap junctions consist of channel aggregates that bridge the closely apposed cell membranes and thus provide electrical continuity between the communicating cells. Unlike the chemical synapses, gap junctions and the electrical coupling they mediate are not unique to the nervous system but are also found in many other tissues. Gap junctions and electrotonic transmission are discussed in greater detail elsewhere in this volume.
III Structure and Function of Chemical Synapses: An Overview
Based on their morphofunctional characteristics, synaptic junctions may be classified as directed, fast-acting or non-directed, slow-acting synapses. Although the configurations of synaptic junctions can vary considerably, all share common features (Fig. 32.1). The presynaptic element, usually an axon terminal bouton or axon varicosity, is characterized by the presence of membrane-bound spherical organelles, the synaptic vesicles, which store the transmitter prior to its release. A subset of synaptic vesicles clusters near the presynaptic active zone, a structural specialization of the presynaptic membrane for synaptic vesicle docking and exocytosis. The juxtaposed postsynaptic membrane contains specific receptors for the transmitter released from the presynaptic terminal. A thickening of postsynaptic membrane, the postsynaptic density, is frequently observed and thought to play a role in localizing the neurotransmitter receptors at the synaptic membrane. The width of the synaptic cleft at highly directed (point-to-point), fast-acting synapses typically varies from about 20–50 nm but can be considerably wider at non-directed, slow-acting synapses. The synaptic cleft contains proteinaceous material, including various cell adhesion molecules (Hall and Sanes, 1993), thought to serve as an adherent for stabilizing the synapse. The junction is enveloped by glial elements, which insulate the synapse as well as perform other functions such as, for example, transmitter removal from the synaptic cleft. For a detailed discussion of synaptic ultrastructure, the student may wish to consult the articles in Pappas and Purpura (l972) and a more recent review by Burns and Augustine (1995).
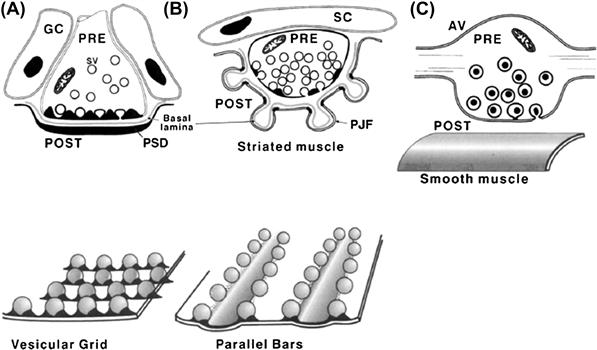
FIGURE 32.1 Schematic representations of neuro-neuronal and neuro-effector synaptic junctions. (A) A directed neuro-neuronal synapse subserving fast synaptic transmission in the CNS. The presynaptic terminal bouton contains mitochondria (M), clear-cored, about 40 nm in diameter, synaptic vesicles (SV), a subset of which is seen docked at the presynaptic vesicular grid. The receptors are localized in the juxtaposed thickening of postsynaptic membrane, the postsynaptic density (PSD). The presynaptic vesicular grid together with the juxtaposed PSD define the active zone of the synapse. The synaptic cleft is typically about 20 nm wide and contains dense material, with filamentous structures seen sometimes to span the cleft from pre-to-postsynaptic membrane. Glial cells (GC) cap and insulate the synapse. (B) The vertebrate skeletal neuromuscular junction (NMJ), a directed synapse subserving fast transmission from motor neuron to striated muscle. These large terminals are filled with many clear-cored, acetylcholine-containing synaptic vesicles. A few dense-cored vesicles can be also seen. A subset of clear-cored synaptic vesicles aggregate at the presynaptic active zone, which is defined by the presence of the presynaptic density. The presynaptic density appears to have the configuration of a bar defined by parallel rows of intramembrane particles, thought to represent the calcium channels, with two rows of vesicles attached on either side of the bar (bottom). The receptors for acetylcholine (AChR) are localized in the crests of the postjunctional folds (PJF) directly opposite the presynaptic active zones. The synaptic cleft at the vertebrate NMJ is typically 50 nm wide. The basal lamina within the synaptic cleft contains AChE, an enzyme that degrades ACh released into the synaptic cleft. The junction is insulated by Schwann cell (SC) processes. (C) A non-directed, slow-acting synapse between a presynaptic axon varicosity (AV) of a sympathetic neuron and smooth muscle. The varicosity contains large (>60 nm) dense-core, norepinephrine-containing vesicles. This synapse is characterized by the absence of distinct active zone structures and a variable but usually wide (>100 nm) separation between the presynaptic and postsynaptic elements of the junction.
Transmission at chemical synapses is unidirectional from the presynaptic to postsynaptic element. The presynaptic terminals are specialized for transmitter biosynthesis, packaging of transmitter into the synaptic vesicles and vesicular transmitter exocytosis. The transmitter receptors in the postsynaptic membrane transduce transmitter binding into ionic current which generates the postsynaptic potential (PSP). Postsynaptic potentials at fast synapses are generated by activation of ionophoric, or channel-forming, receptors. The synaptic potentials are typically fast in onset and last for only a few milliseconds. The generation of the postsynaptic potentials is, however, not instantaneous but rather registers 0.3–0.5 ms after the arrival of the action potential at the presynaptic terminal. This synaptic delay is a characteristic feature of chemical synapses and reflects, for the most part, the time required for the molecular events associated with the transmitter release. At non-directed, slow synapses, channels are not directly transmitter-gated but rather are coupled to the receptor via G protein and second messenger systems. These receptors are referred to as metabotropic. The synaptic potential generated by activation of the metabotropic receptors is slower in onset and longer lasting.
The process of chemical neurotransmission involves (1) synthesis and vesicular uptake of neurotransmitter in presynaptic terminal, (2) exocytotic release of vesicular transmitter into the synaptic cleft, (3) diffusion and binding of transmitter to postsynaptic receptors and generation of synaptic potential and (4) termination of synaptic activity. As indicated above, transmitters are synthesized in the nerve ending of the presynaptic neuron and are concentrated and stored in synaptic vesicles. The intravesicular packet or quantum of transmitter that is stored in the vesicles is released by Ca2+-dependent exocytosis. Exocytosis is triggered when an AP in the presynaptic neurons invades and depolarizes the nerve terminal, thereby opening the presynaptic voltage-gated Ca2+ channels and allowing influx of Ca2+ into the terminal. The resulting transient rise in [Ca2+] i triggers fusion of synaptic vesicles with the plasmalemma and transmitter exocytosis. The requirement for Ca2+ influx is absolute. In absence of Ca2+, the presynaptic AP will fail to trigger release.
Following its release into the synaptic cleft, transmitter combines with specific receptors in the postsynaptic membrane, causing a change in its permeability to specific ions. Change in membrane permeability to ions gives rise to synaptic current which, depending on ions involved, depolarizes or hyperpolarizes the postsynaptic membrane (Fig. 32.2). The depolarizing potential is called an excitatory postsynaptic potential (EPSP) because it tends to bring the cell membrane potential toward the threshold for an AP. EPSPs are associated with a change in the postsynaptic membrane permeability to Na+ and K+ ions and a net influx of positive charges (inward current carried by Na+). The hyperpolarizing potential is called an inhibitory postsynaptic potential (IPSP), because it tends to move or hold the membrane potential away from the threshold, thus decreasing the likelihood of an AP being fired. The IPSPs elicited by activation of ionophoric receptors at fast synapses are associated with an increase in the postsynaptic membrane permeability to Cl−. PSPs associated with activation of metabotropic receptors usually involve change in membrane permeability to K+ ions.
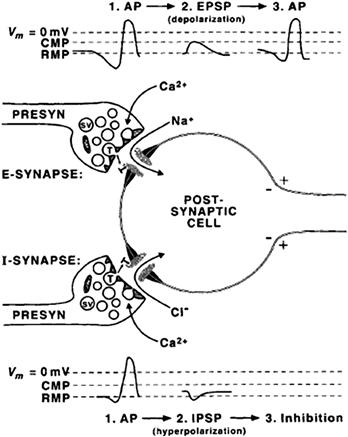
FIGURE 32.2 Fast excitatory versus inhibitory synaptic transmission. At an excitatory (E) synapse, the presynaptic action potential releases transmitters that activate cationic channels, resulting in an inward synaptic current carried by Na+ ions and depolarizing potential, the excitatory postsynaptic potential (EPSP). EPSPs increase the likelihood of an action potential (AP) being fired by the postsynaptic cell. At an inhibitory (I) synapse, the transmitter activates receptor-gated chloride channels and influx of Cl− ions, resulting in hyperpolarizing potential, the inhibitory postsynaptic potential (IPSP). The likelihood of an AP is diminished by activation of I-synapses because IPSPs hold or move the membrane potential of the postsynaptic cell away from the threshold. RMP, resting membrane potential; CMP, critical membrane potential (threshold) at which an AP is triggered.
Transmitter release normally terminates within less than 1 ms. As the presynaptic AP decays and the terminal repolarizes back toward the resting potential, the depolarization-activated calcium channels reclose, Ca2+ influx ceases and [Ca2+]i is rapidly lowered to the prestimulus level. Transmitter action on the receptors in the postsynaptic membrane is terminated by diffusion and enzymatic degradation into an ineffective substance or clearance from the synaptic cleft by reuptake into the nerve terminals and/or glial cells. Reuptake and subsequent catabolism is the primary route of transmitter inactivation for most transmitters. The exception is acetylcholine, in which case inactivation is primarily by means of extracellular degradation of ACh to inactive acetate and choline.
IV Neurotransmission
IVA Neurotransmitters and Neurotransmitter Receptors
Several criteria define a chemical substance as a neurotransmitter. (1) The biosynthetic enzymes for the synthesis of the substance must be present in the identified presynaptic neuron to catalyze the synthesis of transmitter in the nerve terminals. (2) The substance must be released by stimulation of the presynaptic neuron in a Ca2+-dependent manner. (3) Application of the substance to the postsynaptic cell must mimic the actions of the neurally released substance. (4) A specific mechanism for inactivation of the transmitter substance, such as a selective uptake system in the presynaptic terminals or the presence of degradative enzyme(s), must be demonstrated at the synapse investigated.
Amino acid glutamate is the major excitatory neurotransmitter and γ-aminobutyric acid (GABA) and glycine serve as inhibitory transmitters in the CNS. Acetylcholine (ACh), dopamine (DA), norepinephrine (NE), epinephrine, serotonin (5-HT), histamine and ATP are all recognized as neurotransmitters or neurotransmitter candidates. Acetylcholine and norepinephrine are the established transmitters in the peripheral nervous system. In addition, there is good evidence that in the vascular smooth muscle, ATP is co-released and acts as a co-transmitter with NE. Neurons may also co-store and co-release various peptide hormones. These may act as co-transmitters or modulate the synaptic actions of conventional transmitters. Common low-molecular-weight transmitter substances and their chemical structures are shown in Fig. 32.3.
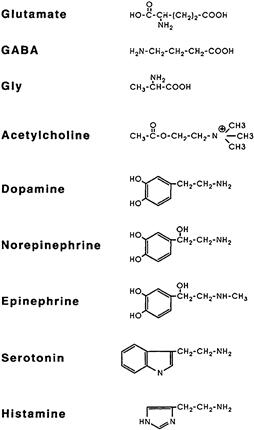
FIGURE 32.3 Structural formulas of common neurotransmitters.
Neurotransmitter receptors (Table 32.1) are integral membrane proteins containing the transmitter recognition site and the transducer site. Ionophoric or channel-forming receptors include the muscle and neural nicotinic acetylcholine receptors; three pharmacologically distinguishable glutamate receptors that are selectively activated by analogs of glutamate – kainate, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) or N-methyl-D-aspartic acid (NMDA); the 5HT3 serotonin receptor; and the P2x purinoceptor for ATP. These relatively non-specific cationic channels permit passage of Na+, K+ and, in some cases, Ca2+ ions, but exclude anions. The anionic (Cl−) channel-forming receptors include the GABAA and GABAC receptors and the glycine receptors (Gly-R).
TABLE 32.1. Common Neurotransmitters and Major Receptor Types
| Transmission type | Neurotransmitter | Receptora |
| Amino acidergic | Glutamic acid | Kainate, AMPA, NMDA, mGluR |
| Gamma-aminobutyric acid (GABA) | GABAA, GABAB, GABAC | |
| Glycine | Gly-R | |
| Cholinergic | Acetylcholine | nAChR, mAChR |
| Aminergic | Dopamine | D1, D2 |
| Norepinephrine | α1, α2, β1, β2 | |
| Serotonin | 5HT1, 5HT2, 5HT3 | |
| Histamine | H1, H2, H3 | |
| Purinergic | ATP, adenosine | P1, P2x, P2y |
a Ionophoric or channel-forming receptors are indicated in bold. Regular lettering indicates metabotropic or G-protein-coupled receptors.
Metabotropic receptors transduce the transmitter binding into a physiological response via G-protein/second messenger-coupled mechanisms. The synaptic actions of monoamines, dopamine, norepinephrine, epinephrine, serotonin and histamine are exerted via the G-protein/second messenger-coupled mechanisms systems. The G-protein-coupled receptors also include the muscarinic ACh receptors, metabotropic glutamate receptor (mGluR), GABAB and several receptors for adenosine and ATP. Ligand-gated and G-protein-coupled channels are discussed elsewhere in this volume.
IVB Biosynthesis, Storage and Inactivation of Neurotransmitters
Conventional, low-molecular-weight neurotransmitters are synthesized locally in the nerve terminals and packaged into synaptic vesicles (SV) prior to release. The local synthesis of transmitter in the nerve terminals assures that transmitter is available for refilling the vesicles after they have released their contents into the synaptic cleft. The vesicular uptake of transmitters is mediated by specific transporters and is driven by an electrochemical gradient generated by electrogenic proton pump in the vesicular membrane (McMahon and Nicholls, 1991). Following release, the synaptic neurotransmitter action is terminated by diffusion, reuptake into presynaptic terminals or glial cells and degradation. Reuptake of transmitters from the extracellular space is energetically coupled to a transmembrane Na+ gradient and is mediated by specific high affinity transporters that are distinct from the vesicular transporters. Two families of plasmalemmal transporters have been identified. The carriers for excitatory amino acids require the presence of extracellular Na+, whereas the carriers for biogenic amines, GABA and glycine require both Na+ and Cl− for uptake (Fig. 32.4). (Amara and Arriza, 1993; Sonders and Amara, 1996.)
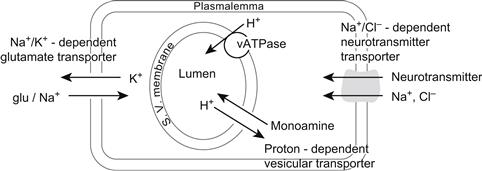
FIGURE 32.4 Neurotransmitter transport systems in vesicular and plasmalemmal membranes. (Based on Amara and Arriza, 1993.)
The key enzyme(s) involved in transmitter synthesis are selectively expressed in the neurons and define their neurotransmitter phenotype. Choline acetyltransferase (ChAT) is the marker enzyme for cholinergic neurons where it catalyzes the synthesis of acetylcholine (ACh) from acetyl coenzyme A (AcCoA) and choline (Ch):
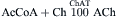
Acetyl coenzyme A derives from the tricarboxylic acid (TCA) cycle whereas choline is transported into nerve terminals from the extracellular medium by the sodium-dependent, high-affinity carrier system that is localized in the presynaptic terminals. Transport of choline and synthesis of ACh are tightly coupled and both the rate of choline uptake and ACh synthesis increase during activity, ensuring adequate supply of the transmitter. The vesicular ACh transporter (VAChT) mediates uptake of ACh from the cytosol into synaptic vesicles. The plasmalemmal Ch uptake is selectively inhibited by a chemical hemicholinium-3 (HC-3) whereas vesamicol blocks vesicular uptake of ACh. Either of these drugs will cause depletion of ACh and failure of cholinergic transmission. Following its release, the synaptic action of ACh is terminated by diffusion and acetylcholinesterase (AChE)-catalyzed hydrolysis to inactive acetate and choline. Much of the choline is recaptured by the nerve terminals and reutilized in new ACh synthesis.
Catecholaminergic neurons and their synaptic connections comprise dopaminergic, noradrenergic (norepinephrine) and adrenergic (epinephrine) systems. The rate-limiting step in catecholamine biosynthesis is the tyrosine hydroxylase (TH)-catalyzed hydroxylation of tyrosine to L-dihydroxyphenylalanine (l-DOPA), which is then decarboxylated by a non-specific aromatic l-amino acid decarboxylase (L-AADC) to dopamine. Dopamine is taken up into synaptic vesicles and serves as a transmitter at dopaminergic synapses. In noradrenergic nerve endings, dopamine is further converted to norepinephrine by intravesicularly localized dopamine-β-hydroxylase (DβH). Methylation of norepinephrine to epinephrine at adrenergic synapses is catalyzed in cytosol by phenylethanolamine-N-methyltransferase (PNMT) in a reaction requiring S-adenosylmethionine as methyl donor:
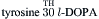
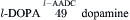
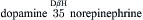
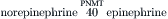
Vesicular monoamine transporters (VMATs) mediate the uptake of catecholamines into synaptic vesicles. Reserpine inhibits the VMATs and thereby depletes vesicular stores of catecholamines. Synaptic actions of catecholamines are terminated by high-affinity neuronal reuptake followed by intracellular degradation or reuptake into synaptic vesicles. The plasmalemmal catecholamine transporters are distinct from VMATs. Plasmalemmal reuptake is insensitive to reserpine but is blocked by psychoactive drugs, such as cocaine and amphetamine, which thereby increase extracellular levels of catecholamines. The major catabolic pathway for catecholamines involves the mitochondrial monoamine oxidase (MAO)-catalyzed oxidative deamination to aldehydes followed by their rapid conversion by hydrogenases and reductases to corresponding acids and alcohols.
Serotonergic neurons utilize serotonin as the transmitter. Serotonin, an indoleamine, is formed by tryptophan hydroxylase-mediated hydroxylation of tryptophan to 5-hydroxytryptophan (5-HTP), followed by l-AADC-mediated decarboxylation of 5-HTP to 5-hydroxytryptamine (5-HT, serotonin):
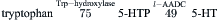
Uptake of serotonin into synaptic vesicles appears to be mediated by the same or a closely related VMAT that mediates vesicular transport of catecholamines. The synaptic action of serotonin is terminated primarily by neuronal sodium-dependent reuptake, mediated by a transporter that is homologous to the plasmalemmal transporters for catecholamines. Following reuptake, serotonin is degraded by MAO-catalyzed oxidative deamination to 5-hydroxyindole acid aldehyde and 5-hydroxyindole acetic acid.
Glutamic acid, GABA and glycine are the major amino acid neurotransmitters in the CNS. Because glutamic acid and glycine are common constituents of amino acid pools found in all cells, their use as neurotransmitters at glutamatergic and glycinergic synapses implies subcompartmentation within the respective nerve terminals. This subcompartment in all likelihood corresponds to synaptic vesicles (SV), which presumably selectively accumulate glutamic acid and glycine from the cytosol and release them during synaptic transmission. The amino acid neurotransmitters are synthesized in nerve terminals from intermediates of the tricarboxylic acid (TCA) cycle. Glutamate is formed by transamination of α-ketoglutaric acid (α-KA) and GABA is formed from glutamate in a reaction catalyzed by glutamic acid dehydrogenase (GAD), a marker enzyme for GABAergic neurons. Inactivation of synaptic actions of glutamate and GABA is through reuptake into nerve terminals as well as the glial cells, where both amino acids are converted to glutamine, which is in turn exported to the nerve terminals and reutilized in the formation of glutamate:

IVC Transmitter Release
Transmitter release is evoked by arrival of an action potential at the presynaptic terminal. Depolarization of the nerve terminal membrane activates (opens) the plasmalemmal voltage-gated Ca2+ channels, allowing influx of extracellular Ca2+ into the terminal. The resultant transient increase of Ca2+ concentration near the plasmalemmal release sites triggers a cascade of biochemical events that culminate in synaptic vesicle–plasmalemma fusion and exocytosis of transmitter into the synaptic cleft. The process terminates upon nerve terminal repolarization when the Ca2+ channels deactivate (close), Ca2+ entry ceases and intracellular Ca2+ returns to prestimulus levels.
IVC1 Quantal-Vesicular Hypothesis of Transmitter Release
The evolution of the current understanding of the mechanism of transmitter release began with the formulation of the quantal-vesicular hypothesis of transmitter release by Bernard Katz and his collaborators (Fatt and Katz, l952; delCastillo and Katz, l954). Katz and colleagues observed that, in the resting neuromuscular junction, i.e. in the absence of stimulation, nerve terminals spontaneously release ACh, giving rise to small depolarizations that occur at random intervals and average about 0.5 mV in amplitude (Fig. 32.5A). These small potentials behaved in all respects as miniature replicas of the end-plate potential (EPP) evoked by presynaptic APs (Fig. 32.5B) and therefore were called miniature end-plate potentials (MEPPs). The crucial insight into the relationship between MEPPs and EPPs was provided by the observation that when evoked release was reduced in low-Ca2+/high-Mg2+ solutions, the size of EPPs fluctuated in a random manner (Fig. 32.5C) such that the EPP amplitudes appeared to be made up of integral multiples of the average MEPP amplitude and could be described by Poisson distribution (Fig. 32.6). Katz concluded that transmitter release is a stochastic process, consisting of random release of multimolecular packets or quanta of ACh each producing a unit response (MEPPs) and that the EPP is a summation of many quantal units released nearly synchronously by a presynaptic AP.
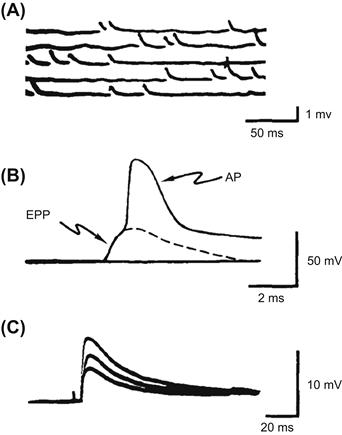
FIGURE 32.5 Intracellular recordings from the frog end-plate. (A) Spontaneous, miniature end-plate potentials (MEPPs) recorded from resting (not stimulated) junction. Note that these small depolarizing potentials are less than 1 mV in amplitude and occur randomly. (B) Postsynaptic response to a presynaptic action potential. The initial hump on the recorded waveform is the end-plate potential (EPP) elicited by ACh released by the presynaptic AP. Note that the EPP is a large depolarization (>40 mV), sufficient to bring the end-plate to threshold and trigger a muscle AP. (C) Fluctuations in EPPs when transmitter output has been reduced by adding 10 mM Mg2+ to the bathing medium. (Adapted from Fatt and Katz, 1952, and delCastillo and Katz, l954.)
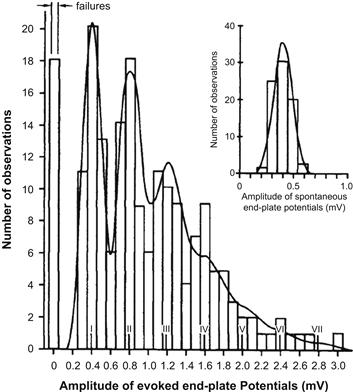
FIGURE 32.6 Distribution of EPP amplitudes recorded from mammalian end-plate under conditions of reduced transmitter release in high (12.5 mM) Mg2+. The inset shows a histogram of spontaneous potentials (MEPPs) recorded from resting junction. Note that EPP amplitudes group around multiples of mean MEPPs amplitude and the number of experimentally observed failures (0 quanta released) and single, double, triple, or more quantal responses, fit the theoretical distribution (solid curve) calculated from the Poisson equation. (From Boyd and Martin, 1956.)
For detailed discussion of the statistics of transmitter release, the interested reader is referred to Martin (l977). For the present purpose, it suffices to state that transmitter release can be described by a simple statistical expression:
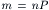
The parameter m is the average number of quanta released per presynaptic impulse when a large number of trials are performed and is called the quantal content of the EPP. The parameter n represents the number of quanta immediately available for release and most likely corresponds to either the population of synaptic vesicles associated with the presynaptic active zones or the number of release sites. P is the probability of any single quantum being released and primarily reflects the probability of a productive Ca2+-dependent vesicle fusion with the plasmalemma as a function of Ca2+ concentration at the release sites. Reducing the availability of Ca2+ to enter terminals would reduce P, thus accounting for a reduction of transmitter release in low-Ca2+/high-Mg2+ media. Conversely, increasing the concentration of Ca2+ at or near the release sites would tend to enhance the probability of exocytosis, i.e. facilitate transmitter release.
IVC2 Essential Role of Ca2+ in Depolarization-Release Coupling
The voltage-gated calcium channels in the presynaptic plasma membrane couple membrane depolarization to transmitter exocytosis. The essential role of Ca2+ influx in transmitter release was demonstrated by Katz and Miledi (l967), who showed that depolarization of presynaptic terminals failed to evoke transmitter release when Ca2+ was absent or its entry into nerve terminals was prevented by high Mg2+ concentration in the extracellular medium (Fig. 32.7). In subsequent experiments carried out at the squid giant synapse, where it is possible to make intracellular recordings from both pre- and postsynaptic cells simultaneously, Miledi (l973) showed that injection of Ca2+ into presynaptic terminals elicited transmitter release, thus providing direct evidence that a rise in intracellular Ca2+ alone is sufficient for activation of the release process. Furthermore, using voltage-clamping to control the membrane potential of the presynaptic terminals at the squid giant synapse, Llinas (l977) showed that the quantity of synaptic transmitter released, monitored as the size of the postsynaptic potential, is related to the size of the presynaptic calcium current which, in turn, depends on the extent of nerve terminal depolarization (Fig. 32.8).
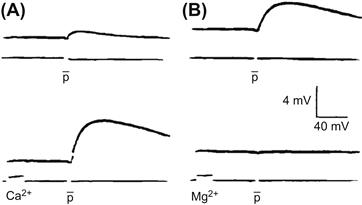
FIGURE 32.7 Calcium is required for transmitter release evoked by depolarization of nerve terminals. In the absence of Ca2+, a depolarizing pulse (p) applied to a presynaptic nerve fails to evoke transmitter release and the postsynaptic response is nearly absent (A, top). Application of a pulse of Ca2+ to the neuromuscular junction shortly before applying stimulus (p) to the presynaptic nerve enables transmitter release as shown by the appearance of a postsynaptic response (A, bottom). The normal postsynaptic response in Ca2+-containing media (B, top) is blocked by applying a pulse of high Mg2+ prior to the depolarizing pulse (p) (B, bottom). The failure to elicit transmitter release in the presence of high Mg2+ is due to block of Ca2+ influx into the terminals. (Adapted from Katz and Miledi, l967.)
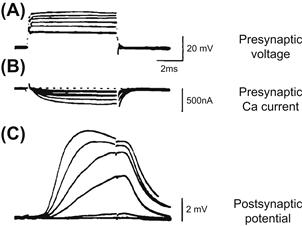
FIGURE 32.8 Experiments in the squid giant synapse illustrating the relationship between magnitude of presynaptic Ca2+ current and transmitter release monitored by recording postsynaptic potentials. Graded depolarizations of presynaptic terminals (A) evoke graded inward Ca2+currents (B) that correlate with graded postsynaptic potentials (C), which reflect the amount of transmitter released. (Adapted from Llinas, l977.)
The presynaptic calcium channels that couple membrane depolarization to transmitter release at fast synapses appear to be strategically localized near the release sites (Robitaille et al., 1990). Opening of these channels results in domains of calcium entry, giving rise to localized subplasmalemmal calcium concentrations that may reach 0.1 mM or higher very rapidly and for a very brief duration (Smith and Augustine, 1988). These subplasmalemmal calcium transients represent more than a thousand-fold increase of [Ca2+] relative to the resting cytosolic calcium level of about 0.1 μM and provide a powerful trigger for focal synaptic vesicle exocytosis. The relationship between intracellular calcium concentration and the rate of exocytosis investigated in the terminals of goldfish retinal bipolar neurons indicates half-saturation at about 200 μM and cooperative interactions involving at least four calcium ions in activation of synaptic vesicle exocytosis (Heidelberger et al., 1994). Transmitter release occurs within about 60 μs following calcium entry into presynaptic terminals, indicating that Ca2+-triggered exocytosis involves activation of a preassembled vesicle–plasmalemma docking/fusion complex (Bruns and Jahn, 1995; Sabatini and Regehr, 1996).
IVC3 Exocytosis and Recycling of Synaptic Vesicles
The active zones in the presynaptic nerve terminals provide plasmalemmal specializations for synaptic vesicle docking and exocytosis. In ingenious experiments, Heuser and coworkers (1979) employed a specially constructed quick-freeze apparatus that enabled them to capture images of vesicle exocytosis at the active zone of the frog neuromuscular junction (Fig. 32.9). The comparison of the number of exocytotic pores captured in the quick-freeze experiment correlated with the estimated number of quanta released under similar conditions of stimulation, providing the most direct, structural evidence that exocytosis of synaptic vesicles at the active zone region is the likely mechanism of quantal transmitter release.
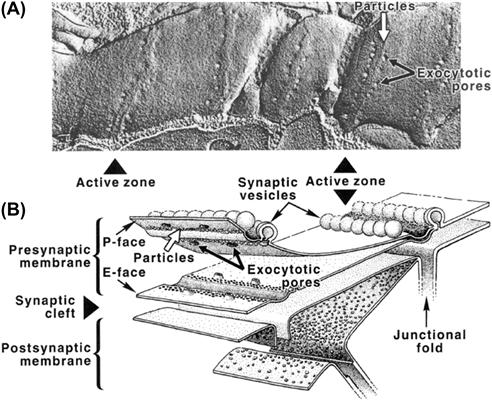
FIGURE 32.9 Synaptic vesicle exocytosis captured by a rapid-freezing technique. (A) P-face of freeze-fractured replica of a stimulated and rapidly frozen nerve terminal showing images of synaptic vesicles caught in the process of exocytosis at the active zones. The junction was activated in presence of 1 mM 4-aminopyridine to enhance the level of transmitter release per single presynaptic impulse. (Adapted fromHeuser et al. (1979). J Cell Biol. 81, 275–300.) (B) Three-dimensional representation of pre- and postsynaptic membranes in relation to the freeze-fracture image above. The freeze-fracture splits membrane bilayers into protoplasmic (P-face) and extracellular (E-face) leaflets of the bilayer. The rows of intramembrane particles seen in P- and E-faces are thought to be the presynaptic Ca2+ channels. The openings in the P-face and their craterlike continuations in the E-face are the exocytotic pores formed when synaptic vesicles fuse with the plasmalemma along the parallel bars of the active zone. (Adapted from Heuser et al. (1979). J Cell Biol. 81, 275–300.)
Synaptic vesicles within the terminal are distributed among the so-called readily available and reserve pools. The readily available pool is thought to correspond to the vesicles docked at the active zone and immediately available for release, whereas the reserve pool comprises the vesicles distributed within the terminal at some distance from the active zone. It is evident that, in order to maintain the availability of quanta for release, exocytosis must be accompanied by mobilization of new vesicles from the reserve pool within the terminal to the plasmalemmal release sites. Mobilization of vesicles is thought to involve alteration in vesicle cytoskeleton interactions that are regulated by phospho-dephosphorylation of synaptic vesicle-associated proteins, synapsins. Dephosphorylated synapsin seems to stabilize vesicle–cytoskeletal interactions and inhibit vesicle mobilization, whereas phosphorylation of synapsin by the Ca2+-calmodulin-dependent protein kinase II is thought to promote vesicle mobilization (Llinas et al., 1991; Greengard et al., 1993).
The mechanism of vesicle docking and initiation of exocytosis by Ca2+ has not been yet completely worked out; however, remarkable progress has been made since the early 1990s. Studies initially conducted in model systems such as mast cells or chromaffin cells and then extended to synaptic preparations indicate that secretion is accompanied by a stepwise increase in cell capacitance, consistent with fusion of secretory granules with the cell membrane. Electrical measurements further suggest that the first event in exocytosis may be the formation of a pore that connects vesicle lumen with the extracellular space and may provide a channel for the release of soluble contents into the synaptic cleft and/or promote collapse and complete fusion of vesicle with the plasma membrane (Lindau and Almers, 1995; Matthews, 1996).
Beginning with the identification and cloning of synaptic proteins in the early 1990s (reviewed in Südhof, 1995), extraordinarily rapid progress has been made during the last few years in defining the molecular machinery of exocytosis. Current evidence indicates that the synaptic exocytotic apparatus makes use of constitutive membrane fusion machinery that has been placed under control of a calcium sensor. The elements of the constitutive docking–fusion apparatus at synapses are the synaptic vesicle membrane-associated protein (VAMP) synaptobrevin and the plasma membrane proteins syntaxin and SNAP-25 (25-kDa, synaptosome-associated protein). The VAMP, syntaxin and SNAP-25 belong to a class of highly conserved membrane-targeting proteins that serve as SNAP (soluble NSF attachment protein) receptors and hence are referred to as SNAREs (Söllner et al., 1993a, b). The SNAREs have been shown to associate into a stable, 7-S core complex that links the apposed vesicle and plasma membranes through formation of parallel bundles of four interacting α-helices, with syntaxin and synaptobrevin each contributing one helix and SNAP-25 contributing two helices (Sutton et al., 1998). The evidence for the crucial role of SNARE complexes in exocytosis has been provided by the observation that selective proteolysis of either VAMP, syntaxin or SNAP-25 by clostridial neurotoxins prevents formation of the complex and inhibits transmitter release (Niemann et al., 1994; Hayashi et al., 1994). Biochemical studies indicate that the core complex is primed by ATP-dependent, SNAP-assisted binding of NSF (N-maleimide-sensitive factor). NSF is an ATPase whose activation is thought to play an important role in fusion by inducing a conformational change in syntaxin and disassembly of the SNARE complex (Whiteheart et al., 1994; Hanson et al., 1997). The synaptic vesicle membrane protein synaptotagmin 1, whose cytosolic domains contain two Ca2+-binding C2 motifs homologous to the C2 regulatory domain of protein kinase C, is believed to serve as the Ca2+-sensor of exocytosis (Brose et al., 1992; Shao et al., 1977). Biochemical experiments indicate that Ca2+ activation of synaptotagmin is associated with simultaneous binding of its C2 domains to the membrane phospholipids and the SNARE complex. The kinetics of synaptotagmin–Ca2+–core complex interactions occur on a microsecond time scale consistent with its postulated function as Ca2+-trigger receptor of exocytosis (Davis et al., 1999). Another protein that has been implicated in exocytosis is neurexin (Petrenko et al., 1991), a plasmalemmal protein that provides a target for α-latrotoxin, a component of black widow spider venom that induces massive Ca2+-independent transmitter exocytosis. Through its capacity to associate with synaptotagmin, neurexin could somehow modulate interactions between synaptotagmin and SNAREs. A simplified model of a vesicle docking–fusion complex is illustrated in Fig. 32.10. Several other synaptic vesicle- and plasma membrane-associated proteins have been identified that are likely to have either accessory or regulatory functions in vesicle exocytosis/endocytosis (Südhof, 1995); however, their precise function and mechanism of action still remain by and large unresolved.
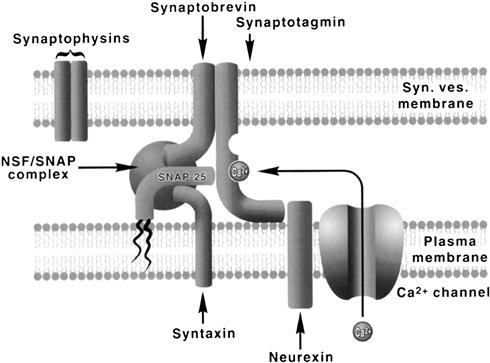
FIGURE 32.10 A simplified model of a vesicle docking–fusion complex. The model incorporates interactions among the proteins of synaptic vesicles (synaptobrevins and synaptotagmins), plasma membrane proteins (syntaxins, SNAP-25, neurexin) and the soluble elements of constitutive fusion machinery (NSF, SNAP). Synaptobrevin, syntaxin and SNAP-25 form a stable, 7-S core complex to which SNAP and NSF (an ATPase) bind to form a 20-S complex. Activation of NSF ATPase leads to disassembly of the complex, perhaps assisting in the final step of membrane fusion. The complex is thought to form in close proximity to the plasmalemmal calcium channels, with synaptotagmin serving as the key Ca2+ sensor. The plasma membrane protein neurexin interacts with synaptotagmin and may modulate its interactions with the core complex.
Following the release of transmitter, exocytosis may be terminated in at least two ways. One is simply through reclosure of the pore and fission of vesicles at the active zone. These postexocytotic vesicles may then refill with locally synthesized transmitter and, by virtue of being already positioned close to or at the active zone, release the newly formed transmitter in preference to the reserve pool. This could explain the well-documented phenomenon of the preferential release of newly synthesized transmitter. A more generally accepted idea is that, following fusion and exocytosis, vesicle membrane collapses into the presynaptic plasmalemma to be retrieved in a series of transformations starting with clathrin-mediated endocytosis followed by endosomal fusion-budding and reformation of functional vesicles (Fig. 32.11). It is likely that both types, the rapid exo-endocytosis as well as the slower, clathrin-mediated endocytosis occur, but the extent to which one predominates over the other depends on the rate of presynaptic stimulation and possibly other factors. The evidence for local recycling of synaptic vesicles in the nerve terminals has been provided by the observation that vesicles become labeled with high-molecular-weight markers such as horseradish peroxidase (HRP) or dextrans when motor nerve terminals are stimulated with these markers in the extracellular medium (Ceccarelli et al., l973). More recent evidence for vesicle recycling has been provided by tracking the movement of fluorescent-labeled synaptic vesicles during stimulation of motor nerve terminals at the frog neuromuscular junction (Betz and Bewick, 1992) and in other synaptic preparations (Ryan et al., 1993; Lagnado et al., 1996).
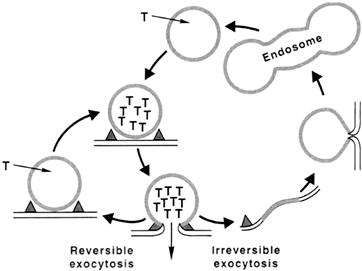
FIGURE 32.11 Synaptic vesicle cycling in nerve endings. Following vesicle docking and fusion, synaptic vesicles can recycle through a short or long pathway. The short pathway (left) involves transient fusion between vesicle and plasma membrane, followed by fission and recharging of the emptied vesicle with new transmitter (T). In the long pathway (right), vesicle membrane collapses into the plasmalemma to be eventually retrieved and reformed via a sequence of steps, then reloaded with new transmitter.
Vesicles undergoing local recycling within the nerve terminals are progressively degraded and replaced by vesicles that are formed de novo in the cell body and transported to the nerve terminals by fast axoplasmic transport. The half-life of vesicles has been estimated at 7–14 days. Evidently, synaptic vesicles can undergo numerous cycles of transmitter release-reloading before being replaced with new ones.
IVD Generation of Postsynaptic Potentials at Fast Synapses
The nature of postsynaptic responses is determined by the type of receptor-gated ionic conductances that are activated in the postsynaptic membrane. At excitatory synapses, the transmitters activate receptor-gated channels that conduct cations, principally Na+ and K+. The net current through the synaptic channels is inward and carried by Na+ ions, causing a depolarization of the postsynaptic membrane, or EPSP. The EPSPs generated at the skeletal neuromuscular junction are called end-plate potentials (EPPs) and those at other peripheral synapses, e.g. at nerve–smooth muscle junctions, are frequently referred to as excitatory junctional potentials, or EJPs. At inhibitory synapses, transmitters activate receptor-gated channels that conduct Cl− ions. Influx of chloride ions through the synaptic channels tends to increase the negativity of the cell interior and hyperpolarizes the postsynaptic membrane. The hyperpolarizing postsynaptic potentials are called inhibitory postsynaptic potentials (IPSPs) because they tend to move the membrane potential away from the threshold.
IVD1 Synaptic Current and Synaptic Equilibrium Potential
The synaptic current (iS) flowing through a single transmitter-activated channel is determined by the channel conductance (γS) and the electrochemical driving force (Vm−Es) acting on the ions moving through the channel:
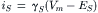 (32.1)
(32.1)
where Vm and ES are membrane potential and the synaptic equilibrium potential, respectively.
In resting synapse, most of the receptor-gated channels are closed and the conductance of the postsynaptic membrane to ions is very low. When transmitter is released by presynaptic impulse, it binds to its receptors in the postsynaptic membrane and opens the associated channels for a short, 1–2 ms duration. The resultant total synaptic current, IS, is a sum of currents through all opened channels:
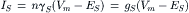 (32.2)
(32.2)
where n is the number of active channels and gS is the total synaptic conductance (nγS).
At excitatory synapses, the transmitter-activated channels permit the passage of both Na+ and K+ ions with about equal ease and the excitatory postsynaptic current IS(E) is the sum of Na+ and K+ currents:
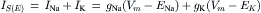 (32.3)
(32.3)
where gNa and gK are Na+ and K+ ion conductances and ENa and EK are the Na+ and K+ equilibrium potentials, respectively. The direction and magnitude of ion flux is determined by the electrochemical driving forces (Vm−Ei). Because gNa (Vm−ENa)>gK(Vm−EK), i.e. INa>IK, the net synaptic current is always inward and carried by Na+ ions, resulting in membrane depolarization. As the membrane potential (Vm) becomes depolarized, the term (Vm−ENa) decreases and (Vm−EK) increases until equilibrium is reached, where the inward Na+ current is exactly equal to the outward K+ current:
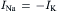 (32.4)
(32.4)
or
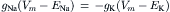 (32.5)
(32.5)
The membrane potential Vm at which this occurs is the synaptic equilibrium potential (ES). By solving for Vm, it is seen that ES is a weighted average of sodium and potassium equilibrium potentials:
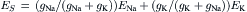 (32.6)
(32.6)
ES is the limiting potential to which the postsynaptic membrane can be depolarized during the transmitter action. Any further depolarization beyond this point would result in IK > INa and reversal of net synaptic current from inward to outward direction. Therefore, the ES is also called a reversal potential (Er).
It is evident that activation of receptor-gated Cl− channels at inhibitory synapses results in the synaptic current given by:
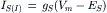 (32.7)
(32.7)
where the equilibrium potential ES is the chloride equilibrium potential, ECl. This is the limiting potential to which a synaptic membrane can be hyperpolarized during the action of transmitter at inhibitory synapse.
IVD2 Relationship Between Synaptic Currents and Postsynaptic Potentials
The relationship between the synaptic current and postsynaptic potential can be analyzed in terms of an equivalent electrical circuit consisting of parallel synaptic and non-synaptic branches, as is illustrated for an excitatory synapse in Fig. 32.12. The synaptic branch represents the synaptic receptor-gated conductance (gS) in series with the synaptic battery of the synaptic equilibrium potential ES. The non-synaptic branch consists of membrane capacitance Cm and leakage channels (gm) in series with the battery of the resting membrane potential, Em.
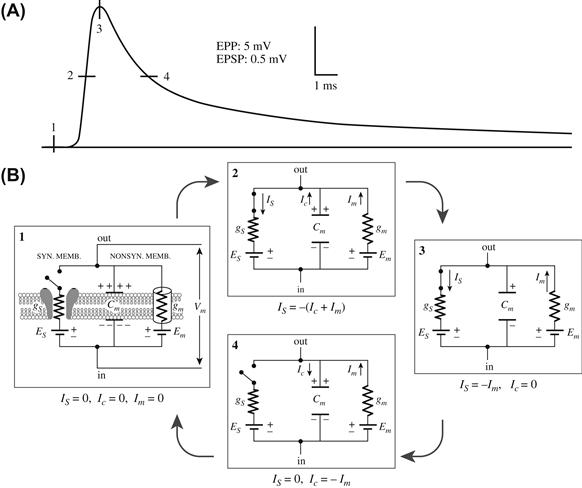
FIGURE 32.12 Equivalent electrical circuit description of excitatory synaptic potential. Four phases of the postsynaptic potential (A) and the corresponding equivalent circuit representations (B) are illustrated. (1) At rest, the transmitter-gated synaptic channels are closed (circuit open; no current flow; dV/dt = 0). (2) Onset of synaptic action (active phase). Synaptic channels are opened by transmitter binding to the receptor (the synaptic switch is closed) and Is flows inward through the synaptic branch (gs) and outward as Ic and Im through the non-synaptic branch. (3) At the peak of synaptic action, capacitance Cm has discharged to its final value, Ic = 0 and Is = −Im (dV/dt = 0). (4) Passive decay phase. Synaptic channels have reclosed (synaptic switch open) and Is = 0. The current through the non-synaptic membrane consists of outward Im and inward Ic, which recharges the membrane capacitance and repolarizes the membrane back to the prestimulus membrane potential. (Based on Kandel et al. (2000). Principles of Neural Science. McGraw-Hill, New York. Reproduced with permission of The McGraw-Hill Companies.)
During the synaptic action of transmitter at the E-synapses, the synaptic current (IS) flows inward through the synaptic branch and outward through the parallel capacitive and resistive elements of the non-synaptic branch as IC and Im. The direction of current flow at the I-synapses is a mirror image of that at excitatory synapses.
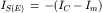 (32.8a)
(32.8a)
and
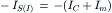 (32.8b)
(32.8b)
where
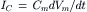 (32.9)
(32.9)
and
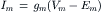 (32.10)
(32.10)
At the onset of synaptic action most of the synaptic current flows through the capacitive branch because the outward driving force (Vm−Em) on current flow through the non-synaptic channels (gm) is small. Once the membrane capacitance is discharged (depolarization) or charged (hyperpolarization) to its final value, all synaptic current exits through the leakage channels (gm). Thus, at the peak of synaptic activation, IC = 0 and
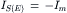 (32.11a)
(32.11a)
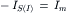 (32.11b)
(32.11b)
at E- and I-synapses, respectively.
Substituting Equations 32.2 and 32.10 into Equation 32.11 and solving for Vm yields the expression for the postsynaptic membrane potential at the peak of synaptic activation:
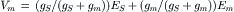 (32.12)
(32.12)
Equation 32.12 shows that the value of membrane potential (Vm) at the peak of synaptic activation is a weighted average of ES and Em, where the weighting factors are the relative magnitudes of synaptic (gS) and non-synaptic (gm) conductances. During peak activation of synaptic channels, gS > gm and the membrane potential will tend toward the ES but the amplitude of PSP (i.e. Vm − Em) will be influenced by gm of the non-synaptic membrane.
IVD3 Time Course of PSPs
The synaptic potentials are electrotonic potentials: they decay passively as a function of time and distance. The time course of the rising phase of the synaptic potential is determined by both active and passive properties of the membrane. As the synaptic channels spontaneously reclose, the PSP decays in an exponential fashion. The decay of the PSP is purely a passive process whose time course is a function of the membrane time constant, τ. The membrane time constant is the time required for an electrotonic potential to decay to 1/e or 37% of its peak value. The time constant of neurons is in the range of 1–20 ms. The amplitude of the synaptic potentials decreases exponentially from the maximum recorded focally at the synapse as a function of the membrane length constant, γ. The length constant is the distance at which electrotonic potentials decay to 1/e or 37% of their amplitude at the point of origin. The length constant of dendrites is typically in the range 0.1 to 1 mm.
IVE Slow Synaptic Transmission Mediated by G-Protein-Coupled Receptors
In contrast to fast synaptic potentials mediated by directly transmitter-gated channels, slow synaptic responses are mediated by a distinct family of proteins that transduce the transmitter binding into cellular responses through activation of GTP-binding regulatory proteins or G proteins. Binding of a transmitter to a specific receptor transforms the associated G protein into an active form that modulates activity of ionic channels at some distance from the receptor either through direct interaction with the channel and/or through second messenger systems. The slow synaptic transmission may involve either increase or decrease in postsynaptic membrane conductance due to a channel opening or channel closure, respectively. For example, activation of muscarinic receptors in heart muscle causes a G-protein-mediated opening of a certain class of voltage-gated K+ channels and relatively long-lasting (seconds) membrane hyperpolarization. Similar, G-protein modulated potassium channels K(G) are present in central neurons, where they mediate slow IPSPs. In contrast, activation of a muscarinic receptor in sympathetic ganglion neurons and certain neurons in the central nervous system is associated with a second-messenger-mediated closure of K+ channels and membrane depolarization. The responses to transmitter activation of G-protein-coupled receptors need not be limited to modulation of ionic channels but also involve modifications of other regulatory proteins resulting in long-term modifications of the postsynaptic cell’s physiology. For a more detailed discussion of G proteins and second messengers in slow synaptic actions of neurotransmitters, the reader is referred to Schulman and Hyman (1999).
IVF Synaptic Integration versus Amplification
Central neurons can receive from several dozens to several thousand excitatory (E) and inhibitory (I) synaptic connections converging on the target neuron from a variety of other neurons in the brain. Whether or not a neuron discharges a propagated AP is determined by the number of E-synapses and I-synapses active at any one time. It should be noted that, even in absence of IPSPs, activation of a single excitatory synapse would be insufficient to discharge an AP, because individual PSPs generated at central synapses are very small, usually in the range of 0.5–2 mV in amplitude. Therefore, summation of several EPSPs is usually necessary to bring the membrane of the postsynaptic neuron to the threshold potential.
The postsynaptic neurons integrate the synaptic potentials by adding the EPSPs and subtracting the IPSPs from the membrane potential at any instant of time. Algebraic summation of two or more topographically separated synapses that are activated nearly simultaneously is called spatial summation. The effectiveness of spatial summation depends on the membrane space constant which, it will be recalled, is the distance at which electrotonic potentials decay to 37% of their amplitude at the point of origin. Clearly, synaptic inputs separated by a distance smaller than the space constant can sum together more effectively than those that are separated by distances larger than the space constant. When a presynaptic neuron fires at a rate such that the interval between successive presynaptic APs is less than the duration of the PSP, each succeeding PSP adds to its predecessor. This process is referred to as temporal summation. Effectiveness of temporal summation depends on the membrane time constant (i.e. the time required for an electrotonic potential to decay to 37% of its peak value). The larger the time constant, the longer is the duration of the PSP and thus the greater the opportunity for summation of successive PSPs to occur. A propagated action potential will be triggered only if the net current is of sufficient magnitude to depolarize the neuronal membrane to the threshold. An action potential is normally triggered at the initial segment of an axon, the region of the neuron with the lowest threshold. Since summation of synaptic currents at the initial segment is the principal determinant of whether or not an AP will be fired, the initial segment is referred to as the integrative zone of the neuron.
In contrast to the integrative activity at central synapses, the function of the neuromuscular junction is to transfer without failure the AP from the presynaptic motor neuron to the postsynaptic muscle fiber. In this case, the excitatory postsynaptic potential referred to as the endplate potential (EPP) is normally always suprathreshold and sufficient to trigger the muscle AP. It may be noted that because the nerve terminal is very small in diameter compared with the muscle fiber it innervates, even if these two membranes were contiguous, the current generated during the invasion of presynaptic terminal by an AP would be insufficient to depolarize the postsynaptic membrane to threshold due to impedance mismatch between the two membranes; i.e. the small nerve terminal cannot provide enough action current to depolarize the large-diameter skeletal muscle fiber much more than about 1 mV. Thus, the function of transmitter at the neuromuscular junction is to amplify the presynaptic signal.
IVG Modulation of Synaptic Transmission
The efficacy of signal transmission at chemical synapses can be modulated by extrinsic and intrinsic factors, including the pattern of ongoing activity as well as history of previous activity. The efficacy of transmission may be altered by mechanisms that affect the dynamics of presynaptic transmitter release and/or modify the postsynaptic receptor-mediated events. This modification may be short lasting or may persist for some time. Thus, synaptic modulation provides for fine-tuning of ongoing synaptic activity as well as for longer lasting changes that are likely to play an important role in learning processes.
IVG1 Depression
Depression or fatigue of synaptic transmission refers to progressive reduction in the amplitudes of postsynaptic potentials in the course of prolonged, relatively high-frequency activation of presynaptic neurons, reflecting progressive depletion of releasable transmitter stores. Recovery from fatigue may take from minutes to hours.
IVG2 Facilitation
Facilitation is a frequency-dependent increase in the amplitude of postsynaptic potentials evoked by closely spaced presynaptic action potentials. The increase in the amplitudes of succeeding PSPs reflects the progressively larger amount of transmitter released with each nerve impulse in the course of stimulation. The mechanism is thought to involve build-up of ionized Ca2+ within the terminals. That is, when presynaptic neuron is stimulated at certain frequency, the diffusion and clearance of Ca2+ from the release sites begins to lag and the residual Ca2+ adds to the Ca2+ transient evoked by the next arriving impulse. Build-up of Ca2+ increases the probability (P) of transmitter quanta being released. Facilitation is a relatively short-lived process that lasts for a few seconds.
IVG3 Post-Tetanic Potentiation
When the nerve is activated with relatively prolonged and/or high-frequency (tetanic) bursts of impulses, the quantity of transmitter is increased upon subsequent stimulation, even after a relatively long intervening rest period. This phenomenon is known as post-tetanic potentiation. In contrast to facilitation, post-tetanic potentiation may last for minutes and sometimes hours, suggesting a long-term modification of presynaptic function secondary to increase in cytosolic Ca2+ levels.
IVG4 Long-Term Potentiation
Long-term potentiation (LTP) refers to a long-lasting increase in EPSP following tetanic stimulation in the presynaptic neurons. It is distinguished from post-tetanic potentiation (PTP) in that the latter is a strictly presynaptic phenomenon, whereas induction and expression of LTP involves both postsynaptic and presynaptic elements. LTP was first described (Bliss and Lomo, 1973) and analyzed most extensively in the hippocampus. In the CA1 region, the LTP involves a special glutamate receptor subtype, the NMDA receptor. The NMDA receptor-gated channel is normally blocked by Mg2+, but can be activated by glutamate when the postsynaptic neuron is sufficiently depolarized so that the Mg2+ blockade of the channel is relieved. The induction of LTP appears to be associated with influx of Ca2+ and activation of Ca2+-dependent protein kinases CaM kinase II and protein kinase C, and possibly other protein kinases in the postsynaptic dendritic spine, which leads to increased efficacy of synaptic transmission that may last for days to weeks. It is thought that while induction of LTP involves the postsynaptic events, the maintenance of LTP may be associated with long-term increase in the probability of transmitter release, a presynaptic event.
IVH Presynaptic Receptors and Transmitter Release
Receptors for neurotransmitters are not confined to postsynaptic sites, but are also found on presynaptic nerve terminals. The modulation of transmitter release by presynaptic receptors that respond to transmitter released by another neuron is referred to as heterosynaptic modulation. Heterosynaptic modulation may involve either inhibition or facilitation of transmitter release. In addition, certain presynaptic autoreceptors recognize the cell’s own neurotransmitter. In this case, the neuron’s transmitter may modulate its own release by interacting with these receptors. This is called automodulation. For example, many cholinergic neuron terminals possess muscarinic autoreceptors and ACh released from these terminals acts on the autoreceptors to inhibit its own release. Although the physiological role of presynaptic receptors has been a subject of debate, it is evident that they provide a potential mechanism for fine-tuning of transmitter release.
BIBLIOGRAPHY
1. Amara SG, Arriza JL. Neurotransmitter transporters: three distinct gene families. Curr Opin Neurobiol. 1993;3:337–344.
2. Bennett MVL. Gap junctions as electrical synapses. J Neurocyt. 1997;26:349–366.
3. Bennett MK, Scheller RH. The molecular machinery for secretion is conserved from yeast to neurons. Proc Natl Acad Sci USA. 1993;90:2559–2563.
4. Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203.
5. Bliss TVP, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356.
6. Boyd IA, Martin AR. The end-plate potential in mammalian muscle. J Physiol. 1956;132:30–38.
7. Brose N, Petrenko AG, Südhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025.
8. Bruns D, Jahn R. Real-time measurement of transmitter release from single synaptic vesicles. Nature. 1995;377:62–65.
9. Burns ME, Augustine GJ. Synaptic structure and function: dynamic organization yields architectural precision. Cell. 1995;83:187–194.
10. Ceccarelli B, Hurlbut WP, Mauro A. Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J Cell Biol. 1973;54:30–38.
11. Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER. Kinetics of synaptotagmin responses to Ca2+and assembly with the core SNARE complex onto membranes. Neuron. 1999;24:363–376.
12. delCastillo J, Katz B. Quantal components of the end-plate potential. J Physiol. 1954;124:560–573.
13. Dreyer F, Peper K, Akert K, Sandri C, Moor H. Ultrastructure of the “active zone” in the frog neuromuscular junction. Brain Res. 1973;62:373–380.
14. Eccles JC. The Physiology of Synapses. New York: Academic Press, Inc; 1964.
15. Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. J Physiol. 1952;117:109–128.
16. Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–784.
17. Hall ZW, Sanes JR. Synaptic structure and development: the neuromuscular junction. Neuron. 1993;72(suppl.):99–121.
18. Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep etch electron microscopy. Cell. 1997;90:523–535.
19. Hayashi T, McMahon H, Yamasaki S, et al. Synaptic vesicle fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 1994;13:5051–5061.
20. Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515.
21. Heuser JE, Reese TS, Dennis MJ, Jan Y, Jan L, Evans L. Synaptic vesicle exocytosis captured by quick freezing and correlated with quantal transmitter release. J Cell Biol. 1979;81:275–300.
22. Kandel ER, Siegelbaum SA. Signalling at the nerve-muscle synapse: directly gated transmission. In: Kandel ER, Schware JH, Jessell TM, eds. Principles of Neural Science. New York: McGraw-Hill; 2000;:187–206.
23. Katz B. Nerve, Muscle, and Synapse. New York: McGraw-Hill Book Co., Inc.; 1966.
24. Katz B, Miledi R. The timing of calcium action during neuromuscular transmission. J Physiol. 1967;189:535–544.
25. Kuffler SW, Nicholls JG. From Neuron to Brain: a Cellular Approach to the Function of the Nervous System. Sunderland: Sinauer Associates, Inc.; 1977.
26. Lagnado L, Gomis A, Job C. Continuous vesicle cycling in the synaptic terminal of retinal bipolar cells. Neuron. 1996;17:957–967.
27. Lindau M, Almers W. Structure and function of fusion pores in exocytosis and ectoplasmic membrane fusion. Curr Opin Cell Biol. 1995;7:509–517.
28. Llinas RR. Calcium and transmitter release in squid synapse. In: Cowan WM, Ferrendelli JA, eds. Approaches to the Cell Biology of Neurons, Society for Neuroscience Symposia, II. Bethesda: Society for Neuroscience; 1977;:139–169.
29. Llinas RR, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca2+-calmod-ulin-dependent protein kinase II of transmitter release in squid giant synapse. J Physiol. 1991;436:257–282.
30. Martin RA. Junctional transmission II Presynaptic mechanisms. In: Bethesda: American Physiological Society; 1977;:329–355. Handbook of Physiology The Nervous System. Vol. 1.
31. Matthews G. Synaptic vesicle exocytosis and endocytosis: capacitance measurements. Curr Opin Neurobiol. 1996;6:358–364.
32. Maycox PR, Hell JW, Jahn R. Amino acid neurotransmission: spotlight on synaptic vesicles. Trends Neurosci. 1990;13:83–87.
33. McMahon HT, Nicholls DG. The bioenergetics of neurotransmitter release. Biochim Biophys Acta. 1991;1059:243–264.
34. Miledi R. Transmitter release induced by injection of calcium ions into nerve terminals. Proc Roy Soc. 1973;183:421–425.
35. Niemann H, Blasi J, Jahn R. Clostridial neurotoxins: new tools for dissecting exocytosis. Trends Cell Biol. 1994;4:179–185.
36. Pappas GD, Purpura DP, eds. Structure and Function of Synapses. New York: Raven Press Publishers; 1972.
37. Petrenko AG, Perin MS, Davletov BA, Ushkaryov YA, Geppert M, Südhof TC. Binding of synaptotagmin to the a-latrotoxin receptor. Nature. 1991;353:65–68.
38. Robitaille R, Adler EM, Charlton MP. Strategic location of calcium channels at transmitter release sites of frog neuro-muscular junction. Neuron. 1990;5:773–779.
39. Ryan TA, Reuters H, Wendland B, Schweizer FE, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724.
40. Sabatini B, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172.
41. Schulman H, Hyman SE. Intracellular signaling. In: Zigmond MJ, Bloom FE, Landis SC, Roberst JL, Squire LR, eds. Fundamental Neuroscience. San Diego: Academic Press; 1999;:269–316.
42. Shao X, Davletov BA, Sutton RB, Südhof TC, Rizo J. Bipartite Ca binding motif in C2 domains of synaptotagmin and protein kinase C. Science. 1997;273:248–251.
43. Smith SJ, Augustine GJ. Calcium ions, active zones and synaptic transmitter release. Trends Neurosci. 1988;11:458–464.
44. Söllner T, Bennett M, Whiteheart S, Scheller R, Rothman J. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation and fusion. Cell. 1993a;75:409–418.
45. Söllner T, Whiteheart SW, Brunner M, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993b;362:318–324.
46. Sonders MS, Amara SG. Channels in transporters. Curr Opin Neurobiol. 1996;6:294–302.
47. Spray DC, Scemes E, Rozental R. Cell-cell communication via gap junctions. In: Zigmond MJ, Bloom FE, Landis SC, Roberts JL, Squire LR, eds. Fundamental Neuroscience. San Diego: Academic Press; 1999;:317–343.
48. Südhof TC. The synaptic vesicle cycle: a cascade of proteinprotein interactions. Nature. 1995;375:645–653.
49. Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353.
50. Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE. N-ethylmaleimide-sen-sitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 1994;126:945–954.