Chapter 3
Bonding Weaker Than Covalent
The discussions in Chapters 1 and 2 focused on the structure of molecules as an aggregate of atoms in a distinct 3D arrangement, held together by bonds with energies on the order of 50–100 kcal mol−1 (200–400 kJ mol−1). There are also very weak attractive forces between molecules, on the order of a few tenths of a kilocalorie per mole. These forces, called van der Waals forces,1 are caused by electrostatic attractions, such as those between dipole and dipole, or induced dipole and induced dipole, and are responsible for liquefaction of gases at sufficiently low temperatures. The bonding discussed in this chapter has energies on the order of 2–10 kcal mol−1 (9–40 kJ/mol−1), which is intermediate between the bond orders given above, and produces clusters of molecules. Compounds will also be discussed in which portions of molecules are held together without any attractive forces at all.
3.A. Hydrogen Bonding2
A hydrogen bond is less than a covalent bond, but it is an attractive force between a functional group A–H and an atom or group of atoms B in the same or a different molecule.3 With exceptions to be noted later, hydrogen bonds are assumed to form only when A is oxygen, nitrogen, or fluorine and when B is oxygen, nitrogen, or fluorine.4 The ability of functional groups to act as hydrogen-bond acids and bases can be obtained from either equilibrium constants for 1:1 hydrogen bonding or overall hydrogen-bond constants.4 There are so-called unconventional hydrogen bonds, particularly with organometallic complexes and transition metal or main group hydrides.5 This chapter will largely ignore such compounds. In normal hydrogen bonds, to oxygen or nitrogen, oxygen may be singly or doubly bonded and the nitrogen singly, doubly, or triply bonded. In structures, hydrogen bonds are usually represented by dotted or dashed lines, as shown in the following examples:
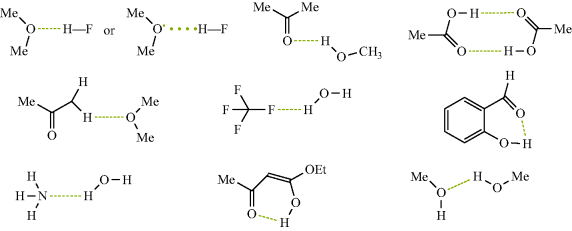
Hydrogen bonds can exist in solid6 and liquid phases, and in solution.7 The efficacy of many organic reactions that will be discussed in later chapters is due to the use of an aqueous media,8 which is due in part, to the hydrogen-bonding nature of such media.9 Even in the gas phase, compounds that form particularly strong hydrogen bonds may remain associated.10 Acetic acid, for example, exists in the gas phase as a dimer, except at very low pressures.11 In the liquid phase and in solution, hydrogen bonds rapidly form and break. The mean lifetime of the NH3 H2O bond is 2 × 10-12 s, for example.12 Except for a few very strong hydrogen bonds13 (e.g., the FH
H2O bond is 2 × 10-12 s, for example.12 Except for a few very strong hydrogen bonds13 (e.g., the FH F− bond) which has an energy of ~50 kcal mol-1 or 210 kJ mol-1, the strongest hydrogen bonds are those connecting one carboxylic acid with another. The energies of these bonds are in the range of 6–8 kcal mol-1 or 25–30 kJ mol-1 (for carboxylic acids, this refers to the energy of each bond). In general, short contact hydrogen bonds between fluorine and HO or NH are rare.14 Other OH
F− bond) which has an energy of ~50 kcal mol-1 or 210 kJ mol-1, the strongest hydrogen bonds are those connecting one carboxylic acid with another. The energies of these bonds are in the range of 6–8 kcal mol-1 or 25–30 kJ mol-1 (for carboxylic acids, this refers to the energy of each bond). In general, short contact hydrogen bonds between fluorine and HO or NH are rare.14 Other OH O and NH
O and NH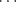 N bonds15 have energies of 3–6 kcal mol-1 (12–25 kJ mol-1). The intramolecular O–H
N bonds15 have energies of 3–6 kcal mol-1 (12–25 kJ mol-1). The intramolecular O–H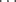 N hydrogen bond in hydroxy-amines is also rather strong.16
N hydrogen bond in hydroxy-amines is also rather strong.16
To a first approximation, the strength of a hydrogen bond will increase with increasing acidity of A–H17 and basicity of B, but the parallel is far from exact.18 A quantitative measure of the strengths of hydrogen bonds has been established, involving the use of an α scale to represent hydrogen-bond donor acidities and a β scale for hydrogen-bond acceptor basicities.19 The use of the β scale, along with another parameter (ξ), allows hydrogen-bond basicities to be related to proton-transfer basicities (pK values).20 A database has been developed to locate all possible occurrences of bimolecular cyclic hydrogen-bond motifs in the Cambridge Structural Database,21 and donor–acceptor, as well as polarity parameters, have been calculated for hydrogen-bonding solvents.22 Bickelhaupt and co-workers22 has stated that hydrogen bonds (X–H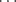 Y) have significant covalent interactions that stem from donor–acceptor orbital interactions between the lone-pair electrons of Y and the empty σ∗ acceptor orbital of X–H, so they are not predominantly electrostatic phenomena.
Y) have significant covalent interactions that stem from donor–acceptor orbital interactions between the lone-pair electrons of Y and the empty σ∗ acceptor orbital of X–H, so they are not predominantly electrostatic phenomena.
When two compounds whose molecules form hydrogen bonds with each other are both dissolved in water, the hydrogen bond between the two molecules is usually greatly weakened or completely removed.23 This finding means that the molecules generally form hydrogen bonds with the water molecules (intermolecular) rather than with each other (intramolecular), presumably because the water molecules are present in such great numbers. In amides, the oxygen atom is the preferred site of protonation or complexation with water.24 In the case of dicarboxylic acids, arguments have been presented that there is little or no evidence for strong hydrogen bonding in aqueous solution,25 although other studies concluded that strong, intramolecular hydrogen bonding can exist in aqueous acetone solutions (0.31 mole-fraction water) of hydrogen maleate and hydrogen cis-cyclohexane-1,2-dicarboxylate.26
Many studies have been made of the geometry of hydrogen bonds,27 and in most (though not all) cases the evidence shows that the hydrogen is on or near the straight line formed by A and B.28 This is true both in the solid state (where X-ray crystallography and neutron diffraction have been used to determine structures),29 and in solution.30 It is significant that the vast majority of intramolecular hydrogen bonding occurs where six-membered rings (counting the hydrogen as one of the six) can be formed, in which linearity of the hydrogen bond is geometrically favorable. Intramolecular hydrogen bonding is much rarer in five-membered rings, where linearity is usually not favored (although it is known). A novel nine-membered intramolecular hydrogen bond has been reported.31
In certain cases X-ray crystallography has shown that a single H–A can form simultaneous hydrogen bonds with two B atoms (bifurcated or three-center hydrogen bonds). An example is an adduct (1) formed from pentane-2,4-dione in its enol form (Sec. 2.N.i) and diethylamine. In 1, the O–H hydrogen simultaneously bonds32 to an O and an N, where the N–H hydrogen forms a hydrogen bond with the O of another pentane-2,4-dione molecule.33 On the other hand, the B atom (in this case oxygen) forms simultaneous hydrogen bonds with two A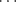 H hydrogens in the adduct (2) formed from 1,8-biphenylenediol and hexamethylphosphoramide (HMPA),34 Another such case is found in methyl hydrazine carboxylate (3).35 Except for the special case of FH
H hydrogens in the adduct (2) formed from 1,8-biphenylenediol and hexamethylphosphoramide (HMPA),34 Another such case is found in methyl hydrazine carboxylate (3).35 Except for the special case of FH F− bonds (see above), the hydrogen is not equidistant between A and B. For example, the O–H distance in ice is 0.97 Å, while the H
F− bonds (see above), the hydrogen is not equidistant between A and B. For example, the O–H distance in ice is 0.97 Å, while the H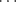 O distance is 1.79 Å.36 A theoretical study of the vinyl alcohol–vinyl alcoholate system (i.e., an enol–enolate anion system) concluded the hydrogen bonding is strong but asymmetric.37 The hydrogen bond in the enol of malonaldehyde, in organic solvents, is asymmetric with the hydrogen atom closer to the basic oxygen atom.38 There is evidence, however, that symmetrical hydrogen bonds to carboxylates should be regarded as two-center rather than three-center hydrogen bonds since the criteria traditionally used to infer three-center hydrogen bonding are inadequate for carboxylates.39 There is also an example of cooperative hydrogen bonding [O–H
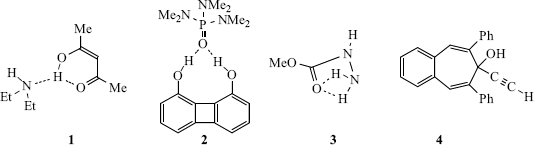
O distance is 1.79 Å.36 A theoretical study of the vinyl alcohol–vinyl alcoholate system (i.e., an enol–enolate anion system) concluded the hydrogen bonding is strong but asymmetric.37 The hydrogen bond in the enol of malonaldehyde, in organic solvents, is asymmetric with the hydrogen atom closer to the basic oxygen atom.38 There is evidence, however, that symmetrical hydrogen bonds to carboxylates should be regarded as two-center rather than three-center hydrogen bonds since the criteria traditionally used to infer three-center hydrogen bonding are inadequate for carboxylates.39 There is also an example of cooperative hydrogen bonding [O–H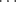 C≡C–H
C≡C–H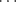 Ph] in crystalline 2-ethynyl-6,8-diphenyl-7H-benzocyclohepten-7-ol (4).40 Related to this discussion is the work that showed the hydrogen bond radii for OH, NH, and acidic CH groups to be 0.60 ± 0.15, 0.76 ± 0.15, and 1.10 ± 0.20 Å, respectively.41
Ph] in crystalline 2-ethynyl-6,8-diphenyl-7H-benzocyclohepten-7-ol (4).40 Related to this discussion is the work that showed the hydrogen bond radii for OH, NH, and acidic CH groups to be 0.60 ± 0.15, 0.76 ± 0.15, and 1.10 ± 0.20 Å, respectively.41
Hydrogen bonding has been detected in many ways, including measurements of dipole moments, solubility behavior, freezing-point lowering, and heats of mixing, but one important way is by the effect of the hydrogen bond on IR spectroscopy.42 The IR frequencies of functional groups (e.g., O–H or C=O) are shifted when the group is hydrogen bonded. Hydrogen bonding always moves the peak toward lower frequencies, for both the A–H and the B groups, though the shift is greater for the former. For example, a free OH group of an alcohol or phenol absorbs at ~ 3590–3650 cm−1, and a hydrogen-bonded OH group is found ~ 50–100 cm-1 lower.43 In many cases, in dilute solution, there is partial hydrogen bonding, that is, some OH groups are free and some are hydrogen bonded. In such cases, two peaks appear.
Infrared spectroscopy can also distinguish between inter- and intramolecular hydrogen bonding, since intermolecular peaks are intensified by an increase in concentration while intramolecular peaks are unaffected. Other types of spectra that have been used for the detection of hydrogen bonding include Raman, electronic,44 and NMR.45 Since hydrogen bonding involves a rapid movement of protons from one atom to another, NMR records an average value that is often broadened. Hydrogen bonding can be detected because it usually produces a chemical shift to a lower field. For example, carboxylic acid–carboxylate systems arising from either mono- or diacids generally exhibit a downfield resonance (16–22 ppm), which indicates “strong” hydrogen bonding in anhydrous, aprotic solvents.46 Hydrogen bonding changes with temperature and concentration, and comparison of spectra taken under different conditions also serves to detect and measure it. As with IR spectra, intramolecular hydrogen bonding in the NMR can be distinguished from intermolecular by its constancy when the concentration is varied. The spin–spin coupling constant across a hydrogen bond, obtained by NMR studies, has been shown to provide a “fingerprint” for hydrogen-bond type.47 Indeed, the determination of 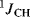 correlates with the strength of the hydrogen bonds formed by an alcohol.48
correlates with the strength of the hydrogen bonds formed by an alcohol.48
Hydrogen bonds are important because of the effects they have on the properties of compounds, among them:
Besides oxygen, nitrogen, and fluorine, there is evidence that weaker hydrogen bonding exists in other systems.50 Although many searches have been made for hydrogen bonding where A is carbon,51 only three types of C–H bonds have been found that are acidic enough to form weak hydrogen bonds.52 These are found in terminal alkynes (RC CH),53 chloroform and some other halogenated alkanes, and HCN. Sterically unhindered C–H groups (CHCl3, CH2Cl2, RCCH) form short contact hydrogen bonds with carbonyl acceptors, where there is a significant preference for coordination with the conventional carbonyl lone-pair direction.54 Weak hydrogen bonds are formed by compounds containing S–H bonds.55 There has been much speculation regarding other possibilities for B. There is evidence that Cl can form weak hydrogen bonds,56 but Br and I form very weak bonds if at all.57 However, the ions Cl−, Br−, and I− form hydrogen bonds that are much stronger than those of the covalently bonded atoms.58 As noted above, the FH
CH),53 chloroform and some other halogenated alkanes, and HCN. Sterically unhindered C–H groups (CHCl3, CH2Cl2, RCCH) form short contact hydrogen bonds with carbonyl acceptors, where there is a significant preference for coordination with the conventional carbonyl lone-pair direction.54 Weak hydrogen bonds are formed by compounds containing S–H bonds.55 There has been much speculation regarding other possibilities for B. There is evidence that Cl can form weak hydrogen bonds,56 but Br and I form very weak bonds if at all.57 However, the ions Cl−, Br−, and I− form hydrogen bonds that are much stronger than those of the covalently bonded atoms.58 As noted above, the FH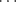 F− bond is especially strong. In this case, the hydrogen is equidistant from the fluorine atoms.59 Similarly, a sulfur atom55 can be the B component (A
F− bond is especially strong. In this case, the hydrogen is equidistant from the fluorine atoms.59 Similarly, a sulfur atom55 can be the B component (A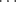 B) in weak hydrogen bonds,60 but the −SH ion forms much stronger bonds.61 There are theoretical studies of weak hydrogen bonding.62 Hydrogen bonding has been directly observed (by NMR and IR) between a negatively charged carbon (see carbanions in Chapter 5) and an OH group in the same molecule.63 Isocyanides (R–+NC−) constitute another type of molecule in which carbon is the B component that forms a rather strong hydrogen bond.64 There is evidence that double and triple bonds, aromatic rings,65 and even cyclopropane rings66 may be the B component of hydrogen bonds, but these bonds are very weak. An interesting case is that of the in-bicyclo[4.4.4]-1-tetradecyl cation 5 (see out–in isomerism, Sec. 4.L The NMR and IR spectra show that the actual structure of this ion is 6, in which both the A and the B component of the hydrogen bond is a carbon.67 These are sometimes called 3-center-2-electron C–H–C bonds.68 A technique called generalized population analysis has been developed to study this type of multicenter bonding.69
B) in weak hydrogen bonds,60 but the −SH ion forms much stronger bonds.61 There are theoretical studies of weak hydrogen bonding.62 Hydrogen bonding has been directly observed (by NMR and IR) between a negatively charged carbon (see carbanions in Chapter 5) and an OH group in the same molecule.63 Isocyanides (R–+NC−) constitute another type of molecule in which carbon is the B component that forms a rather strong hydrogen bond.64 There is evidence that double and triple bonds, aromatic rings,65 and even cyclopropane rings66 may be the B component of hydrogen bonds, but these bonds are very weak. An interesting case is that of the in-bicyclo[4.4.4]-1-tetradecyl cation 5 (see out–in isomerism, Sec. 4.L The NMR and IR spectra show that the actual structure of this ion is 6, in which both the A and the B component of the hydrogen bond is a carbon.67 These are sometimes called 3-center-2-electron C–H–C bonds.68 A technique called generalized population analysis has been developed to study this type of multicenter bonding.69

A weak (~1.5 kcal mol-1, 6.3 kJ mol-1) and rare C–H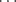 O=C hydrogen bond has been reported in a class of compounds known as a [6]semirubin (a dipyrrinone).70 There is also evidence for a C–H
O=C hydrogen bond has been reported in a class of compounds known as a [6]semirubin (a dipyrrinone).70 There is also evidence for a C–H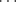 N/CH
N/CH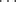 OH bond in the crystal structures of α,β-unsaturated ketones carrying a terminal pyridine subunit,71 and for R3N+–C–H
OH bond in the crystal structures of α,β-unsaturated ketones carrying a terminal pyridine subunit,71 and for R3N+–C–H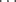 O=C hydrogen bonding.72
O=C hydrogen bonding.72
Deuterium also forms hydrogen bonds; in some systems these seem to be stronger than the corresponding hydrogen bonds; in others, weaker.73 Weak hydrogen bonds can be formed between a π bond (from both alkenes and aromatic compounds) and an appropriate hydrogen. For example, IR data in dilute dichloromethane suggests that the predominant conformation for bis-(amide) (7) contains an N–H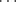 π hydrogen bond involving the C=C unit.74 The strength of an intramolecular π-facial hydrogen bond between an NH group and an aromatic ring in chloroform has been estimated to have a lower limit of −4.5 ± 0.5 kcal mol-1 (−18.8 kJ mol−1).75 A neutron diffraction study of crystalline 2-ethynyladamantan-2-ol (8) shows the presence of an unusual O–H
π hydrogen bond involving the C=C unit.74 The strength of an intramolecular π-facial hydrogen bond between an NH group and an aromatic ring in chloroform has been estimated to have a lower limit of −4.5 ± 0.5 kcal mol-1 (−18.8 kJ mol−1).75 A neutron diffraction study of crystalline 2-ethynyladamantan-2-ol (8) shows the presence of an unusual O–H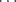 π hydrogen bond, which is short and linear, as well as the more common O–H
π hydrogen bond, which is short and linear, as well as the more common O–H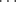 O and C–H
O and C–H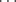 O hydrogen bonds.76
O hydrogen bonds.76
3.B. π–π Interactions
Many theoretical and experimental studies clearly show the importance of π–π interactions,77 which are fundamental to many supramolecular organization and recognition processes.78 Perhaps the simplest prototype of aromatic π–π interactions is the benzene dimer.79 Within dimeric aryl systems such as this, possible π–π interactions are the sandwich and T-shaped interactions shown. It has been shown that all substituted sandwich dimers bind more strongly than a benzene dimer, whereas the T-shaped configurations bind more or less favorably depending on the substituent.80 Electrostatic, dispersion, induction, and exchange-repulsion contributions are all significant to the overall binding energies.80
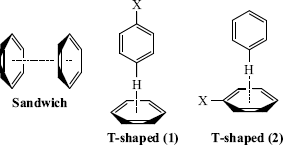
The π electrons of aromatic rings can interact with charged species, yielding strong cation–π interactions that are dominated by electrostatic and polarization effects.81 An interaction with CH units is also possible. For CH–π interactions in both alkyl- and aryl-based model systems, dispersion effects dominate the interaction, but the electrostatics term is also relevant for aryl CH–π interactions.82
Detection of π–π interactions has largely relied on NMR based techniques (e.g., chemical shifts variations),83 and Nuclear Overhauser Effect Spectroscopy (NOESY) or Rotating-Frame NOE Spectroscopy (ROESY).84 Diffusion-ordered NMR spectroscopy (DOSY) has also been used to detect π–π stacked complexes.85
3.C. Addition Compounds
When the reaction of two compounds results in a product that contains all the mass of the two compounds, the product is called an addition compound. There are several kinds. The remainder of this chapter will discuss addition compounds in which the molecules of the starting materials remain more or less intact and weak bonds hold two or more molecules together. There are four broad classes: electron donor–acceptor complexes, complexes formed by crown ethers and similar compounds, inclusion compounds, and catenanes.
3.C.i. Electron Donor–Acceptor Complexes86
In electron donor–acceptor (EDA) complexes,87 there is always a donor and an acceptor molecule. The donor may donate an unshared pair (an n donor) or a pair of electrons in a π orbital of a double bond or aromatic system (a π donor). The electronic spectrum constitutes a good test for the presence of an EDA complex. These complexes generally exhibit a spectrum (called a charge-transfer spectrum) that is not the same as the sum of the spectra of the two individual molecules.88 Because the first excited state of the complex is relatively close in energy to the ground state, there is usually a peak in the visible or near-UV region, and EDA complexes are often colored. Many EDA complexes are unstable and exist only in solutions in equilibrium with their components, but others are stable solids. In most EDA complexes, the donor and acceptor molecules are present in an integral ratio, most often 1:1, but complexes with nonintegral ratios are also known. There are several types of acceptor molecules; complexes formed by two of them will be discussed.
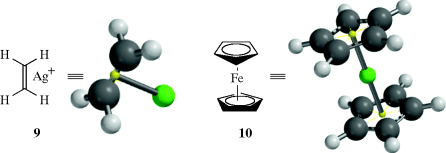
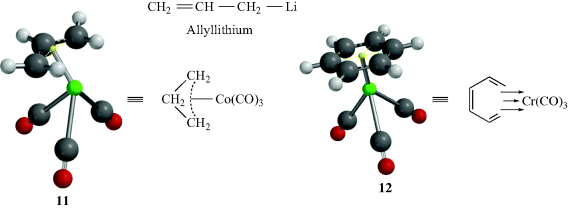
3.C.ii Crown Ether Complexes and Cryptates106
Crown ethers are large-ring compounds that contain several oxygen atoms, usually in a regular pattern. Examples are 12-crown-4 (14; where 12 is the size of the ring and 4 represents the number of coordinating atoms, here oxygen),107 dicyclohexano-18-crown-6 (15), and 15-crown-5 (16). These compounds have the property108 of forming complexes with positive ions, generally metallic ions (though not usually ions of transition metals) or ammonium and substituted ammonium ions.109 The crown ether is called the host and the ion is the guest. In most cases, the ions are held tightly in the center of the cavity.110 Each crown ether binds different ions, depending on the size of the cavity. For example, 14 binds Li+111, but not K+,112 while 15 binds K+ but not Li+.113 Similarly, 15 binds Hg2+, but not Cd2+ or Zn2+, and Sr2+ but not Ca2+.114 18-Crown-5 binds alkali and ammonium cations >1000 times weaker than 18-crown-6, presumably because the larger 18-crown-6 cavity involves more hydrogen bonds.115 The complexes can frequently be prepared as well-defined sharp-melting solids.
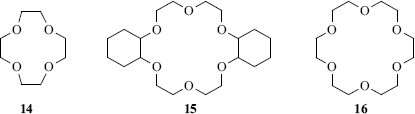
Apart from their obvious utility in separating mixtures of cations,116 crown ethers are widely used in organic synthesis (see the discussion on Sec. 10.G.v). Chiral crown ethers have been used for the resolution of racemic mixtures (Sec. 4.A). Although crown ethers are most frequently used to complex cations, amines, phenols, and other neutral molecules have also been complexed117 (see Sec. 4.L for the complexing of anions).118 Macrocycles containing nitrogen (azacrown ethers) or sulfur atoms (thiacrown ethers)119 (e.g., 17 and 18),120 have complexing properties similar to other crown ethers, as do mixed-heteroatom crown ethers (e.g., 19,12120,122 or 21123).
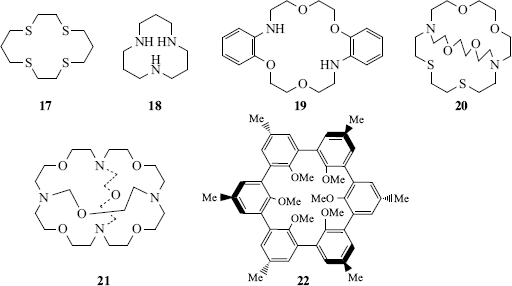
Bicyclic molecules (e.g., 20) can surround the enclosed ion in 3D, binding it even more tightly than the monocyclic crown ethers. Bicyclics and cycles of higher order124 are called cryptands and the complexes formed are called cryptates (monocyclic compounds are sometimes called cryptands). When the molecule contains a cavity that can accommodate a guest molecule, usually through hydrogen-bonding interactions, it is sometimes called a cavitand.125 The tricyclic cryptand 21 has 10 binding sites and a spherical cavity.98 Another molecule with a spherical cavity (though not a cryptand) is 22, which complexes Li+ and Na+ (preferentially Na+), but not K+, Mg2+, or Ca2+.126 Molecules such as these, whose cavities can be occupied only by spherical entities, have been called spherands.83 Other types are calixarenes127 (e.g., 23).128 Spherand-type calixarenes are known.129 There is significant hydrogen bonding involving the phenolic OH units in [4]calixarenes, but this diminishes as the size of the cavity increases in larger ring calixarenes.130 There are also calix[6]arenes131 that have been shown to have conformational isomers (Sec. 4.O) in equilibrium (cone vs alternate) that can sometimes be isolated:132 calix[8]arenes,133 azacalixarenes,134 homooxacalixarenes,135 and calix[9–20]arenes.136 Note that substitution of the unoccupied “meta” positions immobilizes calix[4]arenes and the conformational mobility (Sec. 4.O.iv) in calix[8]arenes is substantially diminished.137 Amide-bridged calix[4]arenes138 calix[4]azulene,139 and quinone-bridged calix[6]arenes.140 are known, and diammoniumcalix[4]arene has been prepared.141 Enantiopure calix[4]resorcinarene derivatives are known,142 and water-soluble calix[4]arenes have been prepared.143 There are also a variety of calix[n]crown ethers,144 some of which are cryptands145 and there is evidence for formation of a calix[4]arene-proton complex.146
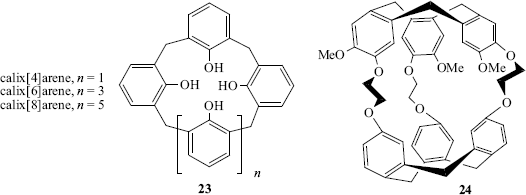
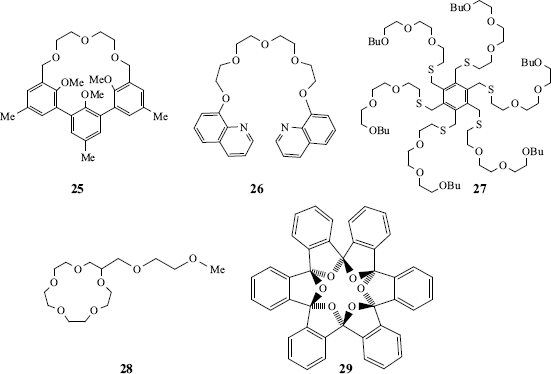
Other molecules include cryptophanes (e.g., 24),147 hemispherands (an example is 25148), and podands.149 The last-named are host compounds in which two or more arms come out of a central structure. Examples are 26150 and 27151 and the latter molecule binds simple cations (e.g., Na+, K+, and Ca2+. Lariat ethers152 are compounds containing a crown ether ring with one or more side chains that can also serve as ligands, (e.g., 28).153 There is also a class of ortho cyclophanes that are crown ethers (see 29) and have been given the name starands.154
The bonding in these complexes is the result of ion-dipole attractions between the heteroatoms and the positive ions. The parameters of the host–guest interactions can sometimes be measured by NMR.155
It has been implied that the ability of these host molecules to bind guests is often very specific, often linked to the hydrogen-bonding ability of the host,156 enabling the host to pull just one molecule or ion out of a mixture. This is called molecular recognition.157 In general, cryptands, with their well-defined 3D cavities, are better for this than monocyclic crown ethers or ether derivatives. An example is the host (30), which selectively binds the dication 31 (n = 5) rather than 31 (n = 4), and 31 (n = 6) rather than 31 (n = 7).158 The host 32, which is water-soluble, forms 1:1 complexes with neutral aromatic hydrocarbons (e.g., pyrene and fluoranthene), and even (though more weakly) with biphenyl and naphthalene, I is also capable of transporting them through an aqueous phase.159
Of course, it has long been known that molecular recognition is very important in biochemistry. The action of enzymes and various other biological molecules is extremely specific because these molecules also have host cavities that are able to recognize only one or a few particular types of guest molecules. Now, organic chemists can synthesize non-natural hosts that can also perform crude (compared to biological molecules) molecular recognition. The macrocycle 33 has been used as a catalyst, for the hydrolysis of acetyl phosphate and the synthesis of pyrophosphate.160

No matter what type of host, the strongest attractions occur when combination with the guest causes the smallest amount of distortion of the host.161 That is, a fully preorganized host will bind better than a host whose molecular shape must change in order to accommodate the guest.

3.C.iii Inclusion Compounds
This type of addition compound is different from either the EDA complexes or the crown ether type of complexes previously discussed. Here, the host forms a crystal lattice that has spaces large enough for the guest to fit into. van der Waals forces constitute the only bonding between the host and the guest. There are two main types, depending on the shape of the space.162 The spaces in inclusion compounds, are in the shape of long tunnels or channels, while the other type, often called clathrate,163 or cage compounds, have spaces that are completely enclosed. In both types, the guest molecule must fit into the space and potential guests that are too large or too small will not go into the lattice, so that the addition compound will not form. Such structures need not be restricted to large molecules. Indeed, the structure and stability of the hydrogen clathrate hydrate with cyclohexanone is known.164
Several important host molecules are known, and inclusion compounds include small molecules (e.g., urea).165 Hydrogen sulfide forms hexagonal clathrate hydrate cages, and a guest molecule (e.g., pinacolone), may be present, as shown in Fig. 3.1.166 Commonly, van der Waals forces between the host and the guest, while small, are essential to the stability of the structure. Which molecules can be a guest is usually dependent on their shapes and sizes and not necessarily on any electronic or chemical effects. For example, octane and 1-bromooctane are suitable guests for urea, but 2-bromooctane, 2-methylheptane, and 2-methyloctane are not. Also, both dibutyl maleate and dibutyl fumarate are guests; neither diethyl maleate nor diethyl fumarate is a guest, but dipropyl fumarate is a guest and dipropyl maleate is not.167 In these complexes, there is usually no integral molar ratio (though by chance there may be). For example, the octane/urea ratio is 1:6.73.168 A deuterium quadrupole echo spectroscopy study of a urea complex showed that the urea molecules do not remain rigid, but undergo 180° flips about the C=O axis at the rate of >106 s–1 at 30 °C.169
Fig. 3.1 X-ray strucutre of pinacolone in a H2S hexagonal clathrate hydrate cage molecule at 100 K.166 [Reprinted with permission from Alavi, S.; Udachin, K.; Ripmeester, J.A. Chem. Eur. J. 2010, 16, 1017, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. Copyright © 2010 by Wiley–VCH Verlag.]
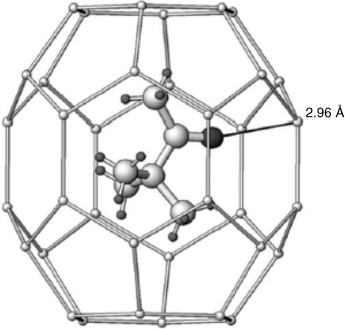
The complexes are solids, but are not useful as derivatives, since they melt, with decomposition of the complex at the melting point of urea. They are useful, however, in separating isomers that would be quite difficult to separate otherwise. Thiourea also forms inclusion compounds though with channels of larger diameter, so that n-alkanes cannot be guests but, (e.g., 2-bromooctane, cyclohexane, and chloroform readily fit).
Hydroquinone is a useful host for clathrates.170 Three molecules, held together by hydrogen bonding, make a cage in which fits one molecule of guest. Typical guests are methanol (but not ethanol), sulfur dioxide, carbon dioxide, and argon (but not neon). One important use is the isolation of anhydrous hydrazine as complex.171 Anhydrous hydrazine is highly explosive, so preparation by distillation of aq hydrazine solutions is both difficult and dangerous. The inclusion complex can be readily isolated and reactions done in the solid state (e.g., the reaction with esters to give hydrazides, Reaction 16–75).172 In contrast to the inclusion compounds, the crystal lattices here can exist partially empty. Another host is water. Usually six molecules of water form the cage and many guest molecules, among them Cl2, propane, and methyl iodide, can fit. The water clathrates (see Fig. 3.1), which are solids, can normally be kept only at low temperatures; at room temperature, they decompose.171 Methane hydrate, which is a promising energy source that exists is vast quantities at in the seabed of various oceans,173 is an example of this type of clathrate. Another inorganic host is sodium chloride (and some other alkali halides), which can encapsulate organic molecules (e.g., benzene, naphthalene, and diphenylmethane).174
Among other hosts175 for inclusion and/or clathrate compounds are deoxycholic acid,176 cholic acid,177 anthracene compounds, (e.g., 34),178 dibenzo-24-crown-8,179 and the compound 35, which has been called a carcerand.180 When carcerand-type molecules trap ions or other molecules (called guests), the resulting complex is called a carciplex.181 It has been shown that in some cases, the motion of the guest within the carciplex is restricted.182
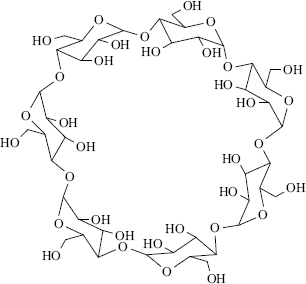
3.C.iv Cyclodextrins
There is one type of host that can form both channel and cage complexes. This type is called cyclodextrins or cycloamyloses.183 The host molecules are made up of six, seven, or eight glucose units connected in a large ring, called, respectively, α-, β-, or γ-cyclodextrin (Fig. 3.2 shows the β or seven-membered ring compound). The three molecules are in the shape of hollow truncated cones (Fig. 3.3) with primary OH groups projecting from the narrow side of the cones and secondary OH group from the wide side. As expected for carbohydrate molecules, all of them are soluble in water and the cavities normally fill with water molecules held in place by hydrogen bonds (6, 12, and 17 H2O molecules for the α, β, and γ forms, respectively), but the insides of the cones are less polar than the outsides, so that nonpolar organic molecules readily displace the water. The polarity of such cavities has been probed by a chemical reaction: the solvolysis of benzoyl halides Reaction (16–57).184 Thus the cyclodextrins form 1:1 cage complexes with many guests, ranging in size from the noble gases to large organic molecules. A guest molecule must not be too large or it will not fit, though many stable complexes are known in which one end of the guest molecule protrudes from the cavity (Fig. 3.4). On the other hand, if the guest is too small, it may go through the bottom hole (though some small polar molecules, e.g., methanol, do form complexes in which the cavity also contains some water molecules). Since the cavities of the three cyclodextrins are of different sizes (Fig. 3.3), a large variety of guests can be accommodated. Since cyclodextrins are nontoxic (they are actually small starch molecules), they are now used industrially to encapsulate foods and drugs.187
Fig. 3.2 β-Cyclodextrin.
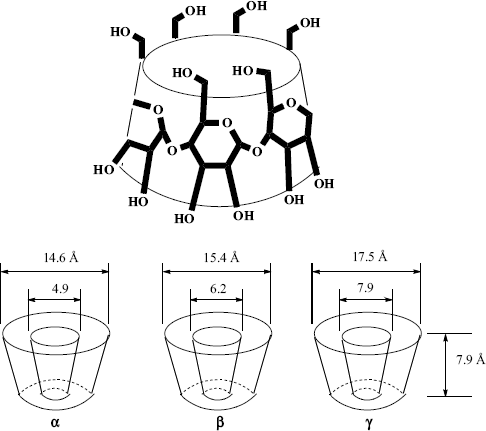
The cyclodextrins also form channel-type complexes, in which the host molecules are stacked on top of each other, like coins in a row.188 For example, α-cyclodextrin (cyclohexaamylose) forms cage complexes with acetic, propionic, and butyric acids, but channel complexes with valeric and higher acids. Capped cyclodextrins are known.189
3.D. Catenanes and Rotaxanes190
These compounds contain two or more independent portions that are not bonded to each other by any valence forces, but nevertheless must remain linked. [n]-Catenanes (where n corresponds to the number of
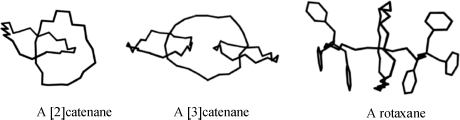
linked rings) are made up of two or more rings held together as links in a chain, while in rotaxanes a linear portion is threaded through a ring and cannot get away because of bulky end groups. Among several types of bulky molecular units, porphyrin units have been used to cap rotaxanes191 as have C60 fullerenes.192 [2]Rotaxanes and [2]catenanes are quite common, and [3]catenanes are known having rather robust amide linkages.193 More intricate variants [e.g., oligocatenanes,194 molecular necklaces (a cyclic oligorotaxane in which a number of small rings are threaded onto a large ring),195 and cyclic daisy chains (an interwoven chain in which each monomer unit acts as a donor and an acceptor for a threading interaction)]196 are known. Ring-in-ring complexes have also been reported.197 Molecular thread, ribbon, and belt assemblies have been synthesized.198 Rotaxanes have been used as the basis for molecular switches,199 and a rotaxane eciplex has been generated that may have applications to molecular-scale photonic devices.200
Transitional isomers are possible in [2]rotaxanes.201 Catenanes and rotaxanes can be prepared by statistical methods or directed syntheses.202 Catenanes can contain heteroatoms and heterocyclic units. In some cases, the catenane exists in equilibrium with the cyclic-non-catenane structures and in some cases this exchange is thought to proceed by ligand exchange and a Möbius strip mechanism.203 An example of a statistical synthesis of a rotaxane is a reaction where a compound A is bonded at two positions to another compound B in the presence of a large ring C. It is hoped that some A molecules would by chance be threaded through C before combining with the two B molecules, so that some rotaxane (D) would be formed along with the normal product E.204 In a directed synthesis,205 the separate parts of the molecule are held together by other bonds that are later cleaved.
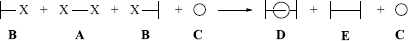
Rotation of one unit through the other catenanes is complex, often driven by making and breaking key hydrogen bonds or π–π interactions. In the case of the isophthaloyl [2]catenane (36), the rate-determining steps do not necessarily correspond to the passage of the bulkiest groups.206

Singly and doubly interlocked [2]catenanes207 can exist as topological stereoisomers208 (see Sec. 4.G for a discussion of diastereomers). Catenanes 37 and 38 are such stereoisomers, and would be expected to have identical mass spectra. Analysis showed that 37 is more constrained and cannot readily accommodate an excess of energy during the mass spectrometry ionization process and, hence, breaks more easily.
Catenanes, molecular knots, and other molecules in these structural categories can exist as enantiomers. In other words, stereoisomers can be generated in some cases. This phenomenon was first predicted by Frisch and Wassermann,209 and the first stereoisomeric catenanes and molecular knots were synthesized by Sauvage and Dietrich-Buchecker.210 Enantiomeric resolution has been achieved.211 A chiral [3]rotaxane containing two achiral wheels, mechanically bonded has been reported,212 generating a cyclodiastereomeric compound, and the enantiomers were separated using chiral HPLC. The terms cycloenantiomerism and cyclodiastereomerism were introduced by Prelog et. al.213 This type of stereoisomerism occurs in cyclic arrangements of several centrally chiral elements in combination with an orientation of the macrocycle.212
A rotaxane can also be an inclusion compound.214 The molecule contains bulky end groups (or “stoppers,” e.g., triisopropylsilyl groups, iPr3Si–) and a chain that consists of a series of –O–CH2CH2–O– groups, but also contains two benzene rings. Cyclodextrins have been threaded onto axle molecules.215 The ring (or bead) around the chain is a macrocycle containing two benzene rings and four pyridine rings. It is preferentially attracted to one of the benzene rings in the chain. The benzene moiety serves as a “station” for the “bead.” However, symmetry of the chain can make the two “stations” equivalent, so that the “bead” is equally attracted to them, and the “bead” actually moves back and forth rapidly between the two “stations,” as shown by the temperature dependence of the NMR spectrum.216 This molecule has been called a molecular shuttle. A copper(I) complexed rotaxane has been prepared with two fullerene (see Sec. 2.L) stoppers.217
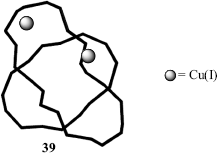
Another variation of these molecules is called molecular knots (e.g., 39), where the represents a metal [in this case, Cu(I)].218 This is particularly interesting since knotted forms of deoxyribonucleic acid (DNA) have been reported.219 There are mechanically interlocked molecules, and one example is known as suit[2]ane.220
3.E. Cucurbit[n]uril-Based Gyroscane
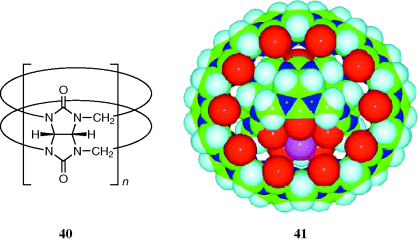
A new molecule known as gyroscane has been prepared, and proposed as a new supramolecular form.221 The class of compounds known as cucurbit[n]urils, abbreviated Qn (40),222 are condensation products of glycoluril and formaldehyde. These macrocycles can act as molecular hosts. The new “supramolecular form is one in which a smaller macrocycle (Q5) is located inside a larger macrocycle (Q10), with facile rotation of one relative to the other in solution (see 41).221 The image of a ring rotating independently inside another ring, which resembles a gyroscope, suggests the name gyroscane for this new class of supramolecular system.”222
[Reprinted with permission from Day, A.I, Blanch, R.J.; Arnold, A.P.; Lorenzo, S.; Lewis, G.R.; Dance, I. Angew Chem. Int. Ed. 2002, 41, 275, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Copyright © 2002 by Wiley-VCH Verlag.]
Notes
1. For a theoretical treatment, see Becke, A.A.A.; Kannemann, F.O. Can. J. Chem. 2010, 88, 1057.
2. For a discussion of hydrogen bonding in organic synthesis, Hydrogen Bonding in Organic Synthesis, see Pihko, M. (Ed.), Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim, 2009.
3. See Schuster, P.; Zundel, G.; Sandorfy, C. The Hydrogen Bond, 3 Vols., North Holland Publishing Co., Amsterdam, The Netherlands, 1976. For a monograph, see Joesten, M.D.; Schaad, L.J. Hydrogen Bonding, Marcel Dekker, NY, 1974. For reviews, see Meot-Ner, M. Mol. Struct. Energ. 1987, 4, 71; Joesten, M.D. J. Chem. Educ. 1982, 59, 362; Gur'yanova, E.N.; Gol'dshtein, I.P.; Perepelkova, T.I. Russ. Chem. Rev. 1976, 45, 792; Kollman, P.A.; Allen, L.C. Chem. Rev. 1972, 72, 283; Huggins, M.L. Angew. Chem. Int. Ed. 1971, 10, 147; Rochester, C.H. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, pp. 327–392. See also, Hamilton, W.C.; Ibers, J.A. Hydrogen Bonding in Solids, W.A. Benjamin, NY, 1968. Also see, Chen, J.; McAllister, M.A.; Lee, J.K.; Houk, K.N. J. Org. Chem. 1998, 63, 4611.
4. See Abraham, M.H.; Platts, J.A. J. Org. Chem. 2001, 66, 3484.
5. Belkova, N.V.; Shubina, E.S.; Epstein, L.M. Acc. Chem. Res. 2005, 38, 624. For a review of hydrogen bonding in cluster ions, see Meot-Ner (Mautner), M. Chem. Rev. 2005, 105, 213.
6. Steiner, T. Angew. Chem. Int. Ed. 2002, 41, 48. See also, Damodharan, L.; Pattabhi, V. Tetrahedron Lett. 2004, 45, 9427.
7. See Nakahara, M.; Wakai, C. Chem. Lett. 1992, 809.
8. Li, C.-J.; Chen, T.-H. Organic Reactions in Aqueous Media, Wiley, NY, 1997.
9. Li, C.-J. Chem. Rev. 1993, 93, 2023.
10. See Curtiss, L.A.; Blander, M. Chem. Rev. 1988, 88, 827.
11. For a review of hydrogen bonding in carboxylic acids and acid derivatives, see Hadž i, D.; Detoni, S. in Patai, S. The Chemistry of Acid Derivatives, pt. 1, Wiley, NY, 1979, pp. 213–266.
12. Emerson, M.T.; Grunwald, E.; Kaplan, M.L.; Kromhout, R.A. J. Am. Chem. Soc. 1960, 82, 6307.
13. For a review of very strong hydrogen bonding, see Emsley, J. Chem. Soc. Rev. 1980, 9, 91.
14. Howard, J.A.K.; Hoy, V.J.; O'Hagan, D.; Smith, G.T. Tetrahedron 1996, 52, 12613. For a discussion of the strength of such hydrogen bonds, see Perrin, C.L. Acc. Chem. Res. 2010, 43, 1550.
15. See Sorensen, J.B.; Lewin, A.H.; Bowen, J.P. J. Org. Chem. 2001, 66, 4105. Also see Ohshima, Y.; Sato, K.; Sumiyoshi, Y.; Endo, Y. J. Am. Chem. Soc. 2005, 127, 1108.
16. Grech, E.; Nowicka-Scheibe, J.; Olejnik, Z.; Lis, T.; Pawêka, Z.; Malarski, Z.; Sobczyk, L. J. Chem. Soc., Perkin Trans. 2 1996, 343. See Steiner, T. J. Chem. Soc., Perkin Trans. 2 1995, 1315.
17. For a comparison of the relative strengths of OH Cl versus OH
Cl versus OH F hydrogen bonds, see Caminati, W.; Melandri, S.; Maris, A.; Paolo Ottaviani, P. Angew. Chem. Int. Ed. 2006, 45, 2438.
F hydrogen bonds, see Caminati, W.; Melandri, S.; Maris, A.; Paolo Ottaviani, P. Angew. Chem. Int. Ed. 2006, 45, 2438.
18. For reviews of the relationship between hydrogen bond strength and acid-base properties, see Pogorelyi, V.K.; Vishnyakova, T.B. Russ. Chem. Rev. 1984, 53, 1154; Epshtein, L.M. Russ. Chem. Rev. 1979, 48, 854.
19. See Abraham, M.H.; Doherty, R.M.; Kamlet, M.J.; Taft, R.W. Chem. Br. 1986, 551; Kamlet, M.J.; Abboud, J.M.; Abraham, M.H.; Taft, R.W. J. Org. Chem. 1983, 48, 2877. For a criticism of the β scale, see Laurence, C.; Nicolet, P.; Helbert, M. J. Chem. Soc., Perkin Trans. 2 1986, 1081. See also, Roussel, C.; Gentric, E.; Sraidi, K.; Lauransan, J.; Guihéneuf, G.; Kamlet, M.J.; Taft, R.W. J. Org. Chem. 1988, 53, 1545; Abraham, M.H.; Grellier, P.L.; Prior, D.V.; Morris, J.J.; Taylor, P.J. J. Chem. Soc., Perkin Trans. 2 1990, 521. Deuterium exchange has been used as an indicator of hydrogen-bond donors and acceptors: see Strobel, T.A.; Hester, K.C.; Sloan Jr., E.D.; Koh, C.A. J. Am. Chem. Soc. 2007, 129, 9544.
20. Kamlet, M.J.; Gal, J.; Maria, P.; Taft, R.W. J. Chem. Soc., Perkin Trans. 2 1985, 1583.
21. Allen, F.H.; Raithby, P.R.; Shields, G.P.; Taylor, R. Chem. Commun. 1998, 1043.
22. Joerg, S.; Drago, R.S.; Adams, J. J. Chem. Soc., Perkin Trans. 2 1997, 2431. See Guerra, C.F.; van der Wijst, T.; Bickelhaupt, F.M. Chem. Eur. J. 2006, 12, 3032; Guerra, C.F.; Zijlstra, H.; Paragi, G.T.; Bickelhaupt, F.M. Chem. Eur. J. 2011, 17, 12612.
23. Stahl, N.; Jencks, W.P. J. Am. Chem. Soc. 1986, 108, 4196.
24. Scheiner, S.; Wang, L. J. Am. Chem. Soc. 1993, 115, 1958.
25. Perrin, C. L. Annu. Rev. Phys. Org. Chem. 1997, 48, 511.
26. Lin, J.; Frey, P. A. J. Am. Chem. Soc. 2000, 122, 11258.
27. Etter, M.C. Acc. Chem. Res. 1990, 23, 120; Taylor, R.; Kennard, O. Acc. Chem. Res. 1984, 17, 320.
28. Stewart, R. The Proton: Applications to Organic Chemistry, Academic Press, NY, 1985, pp. 148–153.
29. A statisical analysis of X-ray crystallographic data has shown that most hydrogen bonds in crystals are nonlinear by ~10–15°: Kroon, J.; Kanters, J.A.; van Duijneveldt-van de Rijdt, J.G.C.M.; van Duijneveldt, F.B.; Vliegenthart, J.A. J. Mol. Struct. 1975, 24, 109. See also, Taylor, R.; Kennard, O.; Versichel, W. J. Am. Chem. Soc. 1983, 105, 5761; 1984, 106, 244.
30. For a discussion of the symmetry of hydrogen bonds in solution, see Perrin, C.L. Pure Appl. Chem. 2009, 81, 571. For reviews of a different aspect of hydrogen bond geometry, see Legon, A.C.; Millen, D.J. Chem. Soc. Rev. 1987, 16, 467, Acc. Chem. Res. 1987, 20, 39.
31. Yoshimi, Y.; Maeda, H.; Sugimoto, A.; Mizuno, K. Tetrahedron Lett. 2001, 42, 2341.
32. Emsley, J.; Freeman, N.J.; Parker, R.J.; Dawes, H.M.; Hursthouse, M.B. J. Chem. Soc., Perkin Trans. 1 1986, 471.
33. For some other three-center hydrogen bonds, see Taylor, R.; Kennard, O.; Versichel, W. J. Am. Chem. Soc. 1984, 106, 244; Jeffrey, G.A.; Mitra, J. J. Am. Chem. Soc. 1984, 106, 5546; Staab, H.A.; Elbl, K.; Krieger, C. Tetrahedron Lett. 1986, 27, 5719.
34. Hine, J.; Hahn, S.; Miles, D.E. J. Org. Chem. 1986, 51, 577.
35. Caminati, W.; Fantoni, A.C.; Schäfer, L.; Siam, K.; Van Alsenoy, C. J. Am. Chem. Soc. 1986, 108, 4364.
36. Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond, W.H. Freeman, San Francisco, 1960, p. 260.
37. Chandra, A.K.; Zeegers-Huyskens, T. J. Org. Chem. 2003, 68, 3618.
38. Perrin, C.L.; Kim, Y.-J. J. Am. Chem. Soc. 1998, 120, 12641.
39. Görbitz, C.H.; Etter, M.C. J. Chem. Soc., Perkin Trans. 2 1992, 131.
40. Steiner, T.; Tamm, M.; Lutz, B.; van der Maas, J. Chem. Commun. 1996, 1127.
41. Lakshmi, B.; Samuelson, A.G.; Jovan Jose, K.V.; Gadre, S.R.; Arunan, E. New J. Chem. 2005, 29, 371.
42. See Symons, M.C.R. Chem. Soc. Rev. 1983, 12, 1; Egorochkin, A.N.; Skobeleva, S.E. Russ. Chem. Rev. 1979, 48, 1198; Aaron, H.S. Top. Stereochem. 1979, 11, 1. For a review of the use of rotational spectra to study hydrogen bonding, see Legon, A.C. Chem. Soc. Rev. 1990, 19, 197.
43. Tichy, M. Adv. Org. Chem. 1965, 5, 115 contains a lengthy table of free and intramolecularly hydrogen-bonding peaks. For a discussion of the role of methyl groups in the formation of hydrogen bonds in dimithyl sulfide(DMS)–methanol mixtures, see Li, Q.; Wu, G.; Yu, Z. J. Am. Chem. Soc. 2006, 128, 1438.
44. See Lees, W.A.; Burawoy, A. Tetrahedron 1963, 19, 419.
45. See Davis, Jr., J.C.; Deb, K.K. Adv. Magn. Reson. 1970, 4, 201. Also see, Kumar, G.A.; McAllister, M.A. J. Org. Chem. 1998, 63, 6968.
46. Bruck, A.; McCoy, L.L.; Kilway, K.V. Org. Lett. 2000, 2, 2007. For a discussion of the effect of solvents on hydrogen bonding, see Cook, J.L.; Hunter, C.A.; Low, C.M.R.; Perez-Velasco, A.; Vinter, J.G. Angew. Chem. Int. Ed. 2007, 46, 3706.
47. Del Bene, J.E.; Perera, S.A.; Bartlett, R.J. J. Am. Chem. Soc. 2000, 122, 3560.
48. Maiti, N.C.; Zhu, Y.; Carmichael, I.; Serianni, A.S.; Anderson, V.E. J. Org. Chem. 2006, 71, 2878.
49. For reviews of the effect of hydrogen bonding on reactivity, see Hibbert, F.; Emsley, J. Adv. Phys. Org. Chem. 1990, 26, 255; Sadekov, I.D.; Minkin, V.I.; Lutskii, A.E. Russ. Chem. Rev. 1970, 39, 179.
50. For a review, see Pogorelyi, V.K. Russ. Chem. Rev. 1977, 46, 316.
51. See Green, R.D. Hydrogen Bonding by C–H Groups, Wiley, NY, 1974. See also, Nakai, Y.; Inoue, K.; Yamamoto, G.; Öki, M. Bull. Chem. Soc. Jpn. 1989, 62, 2923; Seiler, P.; Dunitz, J.D. Helv. Chim. Acta 1989, 72, 1125.
52. For a theoretical study of weak hydrogen-bonds, see Calhorda, M.J. Chem. Commun. 2000, 801.
53. For a review, see Hopkinson, A.C., in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, pt. 1, Wiley, NY, 1978, pp. 75–136. See also, DeLaat, A.M.; Ault, B.S. J. Am. Chem. Soc. 1987, 109, 4232.
54. Streiner, T.; Kanters, J.A.; Kroon, J. Chem. Commun. 1996, 1277.
55. See Zuika, I.V.; Bankovskii, Yu.A. Russ. Chem. Rev. 1973, 42, 22; Crampton, M.R. in Patai, S. The Chemistry of the Thiol Group, pt. 1, Wiley, NY, 1974, pp. 379–396; Pogorelyi, V.K. Russ. Chem. Rev. 1977, 46, 316.
56. See Smith, J.W. in Patai, S. The Chemistry of the Carbon–Halogen Bond, pt. 1; Wiley, NY, 1973, pp. 265–300. See also, Bastiansen, O.; Fernholt, L.; Hedberg, K.; Seip, R. J. Am. Chem. Soc. 1985, 107, 7836.
57. Fujimoto, E.; Takeoka, Y.; Kozima, K. Bull. Chem. Soc. Jpn. 1970, 43, 991; Azrak, R.G.; Wilson, E.B. J. Chem. Phys. 1970, 52, 5299.
58. Fujiwara, F.Y.; Martin, J.S. J. Am. Chem. Soc. 1974, 96, 7625; French, M.A.; Ikuta, S.; Kebarle, P. Can. J. Chem. 1982, 60, 1907.
59. In a few cases, the presence of an unsymmetrical cation causes the hydrogen to be closer to one fluorine than to the other: Williams, J.M.; Schneemeyer, L.F. J. Am. Chem. Soc. 1973, 95, 5780.
60. Schaefer, T.; McKinnon, D.M.; Sebastian, R.; Peeling, J.; Penner, G.H.; Veregin, R.P. Can. J. Chem. 1987, 65, 908; Marstokk, K.; Møllendal, H.; Uggerud, E. Acta Chem. Scand. 1989, 43, 26.
61. McDaniel, D.H.; Evans, W.G. Inorg. Chem, 1966, 5, 2180; Sabin, J.R. J. Chem. Phys. 1971, 54, 4675.
62. Calhorda, M.J. Chem. Commun. 2000, 801.
63. Ahlberg, P.; Davidsson, O.; Johnsson, B.; McEwen, I.; Rönnqvist, M. Bull. Soc. Chim. Fr. 1988, 177.
64. Allerhand, A.; Schleyer, P.v.R. J. Am. Chem. Soc. 1963, 85, 866.
65. For example, see Bakke, J.M.; Chadwick, D.J. Acta Chem. Scand. Ser. B 1988, 42, 223: Atwood, J.L.; Hamada, F.; Robinson, K.D.; Orr, G.W.; Vincent, R.L. Nature 1991, 349, 683.
66. Yoshida, Z.; Ishibe, N.; Kusumoto, H. J. Am. Chem. Soc. 1969, 91, 2279.
67. McMurry, J.E.; Lectka, T.; Hodge, C.N. J. Am. Chem. Soc. 1989, 111, 8867. See also, Sorensen, T.S.; Whitworth, S.M. J. Am. Chem. Soc. 1990, 112, 8135.
68. McMurry, J.E.; Lectka, T. Accts. Chem. Res. 1992, 25, 47.
69. Ponec, R.; Yuzhakov, G.; Tantillo, D. J. J. Org. Chem. 2004, 69, 2992.
70. Huggins, M.T.; Lightner, D.A. J. Org. Chem. 2001, 66, 8402.
71. Mazik, M.; Bläser, D.; Boese, R. Tetrahedron 2001, 57, 5791.
72. Cannizzaro, C.E.; Houk, K.N. J. Am. Chem. Soc. 2002, 124, 7163.
73. Cummings, D.L.; Wood, J.L. J. Mol. Struct. 1974, 23, 103.
74. Gallo, E.A.; Gelman, S.H. Tetrahedron Lett. 1992, 33, 7485.
75. Adams, H.; Harris, K.D.M.; Hembury, G.A.; Hunter, C.A.; Livingstone, D.; McCabe, J.F. Chem. Commun. 1996, 2531. See Steiner, T.; Starikov, E.B.; Tamm, M. J. Chem. Soc., Perkin Trans. 2 1996.
76. Allen, F.H.; Howard, J.A.K.; Hoy, V.J.; Desiraju, G.R.; Reddy, D.S.; Wilson, C.C. J. Am. Chem. Soc. 1996, 118, 4081.
77. Tsuzuki, S.; Lüthi, H.P. J. Chem. Phys. 2001, 114, 3949; Arunan, E.; Gutowsky, H S. J. Chem. Phys. 1993, 98, 4294; Felker, P.M.; Maxton, P.M.; Schaeffer, M.W. Chem. Rev. 1994, 94, 1787; Venturo, V.A.; Felker, P.M. J. Chem. Phys. 1993, 99, 748; Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. J. Am. Chem. Soc. 2002, 124, 104; Hobza, P.; Jure ka, P. J. Am. Chem. Soc. 2003, 125, 15608.
ka, P. J. Am. Chem. Soc. 2003, 125, 15608.
78. Meyer, E.A.; Castellano, R.K.; Diederich, F. Angew. Chem. Int. Ed. 2003, 42, 1210.
79. Sinnokrot, M.O.; Valeev, E.F.; Sherrill, C.D. J. Am. Chem. Soc. 2002, 124, 10887.
80. Sinnokrot, M.O.; Sherrill, C.D. J. Am. Chem. Soc. 2004, 126, 7690.
81. Lindeman, S.V.; Kosynkin, D.; Kochi, J.K. J. Am. Chem. Soc. 1998, 120, 13268; Ma, J.C.; Dougherty, D.A. Chem. Rev. 1997, 97, 1303; Dougherty, D.A. Science 1996, 271, 163; Cubero, E.; Luque, F.J.; Orozco, M. Proc. Natl. Acad. Sci. USA 1998, 95, 5976.
82. Ribas, J.; Cubero, E.; Luque, F.J.; Orozco, M. J. Org. Chem. 2002, 67, 7057.
83. Petersen, S.B.; Led, J.J.; Johnston, E.R.; Grant, D.M. J. Am. Chem. Soc. 1982, 104, 5007.
84. Wakita, M.; Kuroda, Y.; Fujiwara, Y.; Nakagawa, T. Chem. Phys. Lipids 1992, 62, 45.
85. Viel, S.; Mannina, L.; Segre, A. Tetrahedron Lett. 2002, 43, 2515. See also, Ribas, J.; Cubero, E.; Luque, F.J.; Orozco, M. J. Org. Chem. 2002, 67, 7057. For a discussion of substituent effects on aromatic stacking interactions, see Cockroft, S.L.; Perkins, J.; Zonta, C.; Adams, H.; Spey, S.E.; Low, C.M.R.; Vinter, J.G.; Lawson, K.R.; Urch, C.J.; Hunter, C.A. Org. Biomol. Chem.2007, 5, 1062.
86. Foster, R. Organic Charge-Transfer Complexes, Academic Press, NY, 1969; Mulliken, R.S.; Person, W.B. Molecular Complexes, Wiley, NY, 1969; Rose, J. Molecular Complexes, Pergamon, Elmsford, NY, 1967; Poleshchuk, O.Kh.; Maksyutin, Yu.K. Russ. Chem. Rev. 1976, 45, 1077; Banthorpe, D.V. Chem. Rev. 1970, 70, 295; Kosower, E.M. Prog. Phys. Org. Chem. 1965, 3, 81; Foster, R. Chem. Br. 1976, 12, 18.
87. These have often been called charge-transfer complexes, but this term implies that the bonding involves charge transfer, which is not always the case, so that the more neutral name EDA complex is preferable. See Mulliken, R.S.; Person, W.B. J. Am. Chem. Soc. 1969, 91, 3409.
88. Also see Bentley, M.D.; Dewar, M.J.S. Tetrahedron Lett. 1967, 5043.
89. See Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry, 2nd ed, University Science Books, Mill Valley, CA, 1987; Alper, H. Transition Metal Organometallics in Organic Synthesis, 2 Vols., Academic Press, NY, 1976, 1978. For general reviews, see Churchill, M.R.; Mason, R. Adv. Organomet. Chem. 1967, 5, 93; Cais, M. in Patai, S. The Chemistry of Alkenes, Vol. 1, Wiley, NY, 1964, pp. 335–385; Nakamura, A. J. Organomet. Chem. 1990, 400, 35; Bennett, M.A.; Schwemlein, H.P. Angew. Chem. Int. Ed. 1989, 28, 1296; metals-pentadienyl ions, Powell, P. Adv. Organomet. Chem. 1986, 26, 125; complexes of main group metals. For a list of review articles on this subject, see Bruce, M.I. Adv. Organomet. Chem. 1972, 10, 273, pp. 317–321.
90. For a discussion of how this system originated, see Cotton, F.A. J. Organomet. Chem. 1975, 100, 29.
91. Another prefix used for complexes is μ (mu), which indicates that the ligand bridges two metal atoms.
92. See Pearson, A.J. Metallo-organic Chemistry Wiley, NY, 1985; Ittel, S.D.; Ibers, J.A. Adv. Organomet. Chem. 1976, 14, 33; Hartley, F.R. Chem. Rev. 1973, 73, 163; Angew. Chem. Int. Ed. 1972, 11, 596.
93. Dewar, M.J.S. Bull. Soc. Chim. Fr. 1951, 18, C79.
94. Hu, J.; Gokel, G.W.; Barbour, L.J. Chem. Commun. 2001, 1858.
95. See Zeiss, H.; Wheatley, P.J.; Winkler, H.J.S. BenzenoidMetal Complexes, Ronald Press, NY, 1966.
96. Nicholls, B.; Whiting, M.C. J. Chem. Soc. 1959, 551. For reviews of arene–transition metal complexes, see Uemura, M. Adv. Met.-Org. Chem. 1991, 2, 195; Silverthorn, W.E. Adv. Organomet. Chem. 1975, 13, 47.
97. Landesberg, J.M.; Sieczkowski, J. J. Am. Chem. Soc. 1971, 93, 972.
98. See Parini, V.P. Russ. Chem. Rev. 1962, 31, 408; for a review of complexes in which the acceptor is an organic cation, see Kampar, V.E. Russ. Chem. Rev. 1982, 51, 107; also see, Ref. 86.
99. For a review of quinone complexes, see Foster, R.; Foreman, M.I. in Patai, S. The Chemistry of the Quinonoid Compounds, pt. 1, Wiley, NY, 1974, pp. 257–333.
100. See Blackstock, S.C.; Lorand, J.P.; Kochi, J.K. J. Org. Chem. 1987, 52, 1451.
101. See Foster, R. in Patai, S. The Chemistry of Acid Derivatives, pt. 1, Wiley, NY, 1979, pp. 175–212.
102. See Melby, L.R. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 639–669. See also, Fatiadi, A.J. Synthesis 1987, 959.
103. For reviews, see Bender, C.J. Chem. Soc. Rev. 1986, 15, 475; Kampar, E.; Neilands, O. Russ. Chem. Rev. 1986, 55, 334; Bent, H.A. Chem. Rev. 1968, 68, 587.
104. See, for example, Le Fevre, R.J.W.; Radford, D.V.; Stiles, P.J. J. Chem. Soc. B 1968, 1297.
105. Mulliken, R.S.; Person, W.B. J. Am. Chem. Soc. 1969, 91, 3409.
106. See Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, 3 Vols., Academic Press, NY, 1984; Vögtle, F. Host Guest Complex Chemistry I, II, and III (Top. Curr. Chem.1998, 101, 121), Springer, Berlin, 1981, 1982, 1984; Vögtle, F.; Weber, E. Host Guest Complex Chemistry/Macrocycles, Springer, Berlin, 1985; Izatt, R.M.; Christensen, J.J. Synthetic Multidentate Macrocyclic Compounds, Academic Press, NY, 1978. For reviews, see McDaniel, C.W.; Bradshaw, J.S.; Izatt, R.M. Heterocycles, 1990, 30, 665; Sutherland, I.O. Chem. Soc. Rev. 1986, 15, 63; Franke, J.; Vögtle, F. Top. Curr. Chem. 1986, 132, 135; Cram, D.J. Angew. Chem. Int. Ed. 1986, 25, 1039; Gutsche, C.D. Acc. Chem. Res. 1983, 16, 161; Tabushi, I.; Yamamura, K. Top. Curr. Chem. 1983, 113, 145; Stoddart, J.F. Prog. Macrocyclic Chem. 1981, 2, 173; Cram, D.J.; Cram, J.M. Acc. Chem. Res. 1978, 11, 8; Science, 1974, 183, 803; Gokel, G.W.; Durst, H.D. Synthesis 1976, 168; Aldrichim. Acta 1976, 9, 3; Lehn, J.M. Struct. Bonding (Berlin) 1973, 16, 1; Christensen, J.J.; Eatough, D.J.; Izatt, R.M. Chem. Rev. 1974, 74, 351; Pedersen, C.J.; Frensdorff, H.K. Angew. Chem. Int. Ed. 1972, 11, 16. For reviews of acyclic molecules with similar properties, see Vögtle, E. Chimia 1979, 33, 239; Vögtle, E.; Weber, E. Angew. Chem. Int. Ed. 1979, 18, 753. See Angew. Chem. Int. Ed. 1988, 27, pp. 1021, 1009, 89; and Chem. Scr., 1988, 28, pp. 229, 263, 237. See also, the series Advances in Supramolecular Chemistry.
107. Cook, F.L.; Caruso, T.C.; Byrne, M.P.; Bowers, C.W.; Speck, D.H.; Liotta, C. Tetrahedron Lett. 1974, 4029.
108. Discovered by Pedersen, C.J. J. Am. Chem. Soc. 1967, 89, 2495, 7017. For an account of the discovery, see Schroeder, H.E.; Petersen, C.J. Pure Appl. Chem. 1988, 60, 445.
109. See Inoue, Y.; Gokel, G.W. Cation Binding by Macrocycles, Marcel Dekker, NY, 1990.
110. See Izatt, R.M.; Bradshaw, J.S.; Nielsen, S.A.; Lamb, J.D.; Christensen, J.J.; Sen, D. Chem. Rev. 1985, 85, 271; Parsonage, N.G.; Staveley, L.A.K. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 3, Academic Press, NY, 1984, pp. 1–36.
111. Anet, F.A.L.; Krane, J.; Dale, J.; Daasvatn, K.; Kristiansen, P.O. Acta Chem. Scand. 1973, 27, 3395.
112. See Dale, J.; Eggestad, J.; Fredriksen, S.B.; Groth, P. J. Chem. Soc., Chem. Commun. 1987, 1391; Dale, J.; Fredriksen, S.B. Pure Appl. Chem. 1989, 61, 1587.
113. Izatt, R.M.; Nelson, D.P.; Rytting, J.H.; Haymore, B.L.; Christensen, J.J. J. Am. Chem. Soc. 1971, 93, 1619.
114. Kimura, Y.; Iwashima, K.; Ishimori, T.; Hamaguchi, H. Chem. Lett. 1977, 563.
115. Raevsky, O.A.; Solov'ev, V.P.; Solotnov, A.F.; Schneider, H.-J.; Rüdiger, V. J. Org. Chem. 1996, 61, 8113.
116. Crown ethers have been used to separate isotopes of cations, (e.g., 44Ca from 40Ca). For a review, see Heumann, K.G. Top. Curr. Chem. 1985, 127, 77.
117. For reviews, see Vögtle, F.; Müller, W.M.; Watson, W.H. Top. Curr. Chem. 1984, 125, 131; Weber, E. Prog. Macrocycl. Chem. 1987, 3, 337; Diederich, F. Angew. Chem. Int. Ed. 1988, 27, 362.
118. See van Staveren, C.J.; van Eerden, J.; van Veggel, F.C.J.M.; Harkema, S.; Reinhoudt, D.N. J. Am. Chem. Soc. 1988, 110, 4994. See also, Rodrigue, A.; Bovenkamp, J.W.; Murchie, M.P.; Buchanan, G.W.; Fortier, S. Can. J. Chem. 1987, 65, 2551; Fraser, M.E.; Fortier, S.; Markiewicz, M.K.; Rodrigue, A.; Bovenkamp, J.W. Can. J. Chem. 1987, 65, 2558.
119. Voronkov, M.G.; Knutov, V.I. Sulfur Rep. 1986, 6, 137, Russ. Chem. Rev. 1982, 51, 856; Reid, G.; Schröder, M. Chem. Soc. Rev. 1990, 19, 239.
120. For a review of 17 and its derivatives, see Chaudhuri, P.; Wieghardt, K. Prog. Inorg. Chem. 1987, 35, 329. N-Aryl-azacrown ethers are known, see Zhang, X.-X.; Buchwald, S.L. J. Org. Chem. 2000, 65, 8027.
121. Gersch, B.; Lehn, J.-M.; Grell, E. Tetrahedron Lett. 1996, 37, 2213.
122. Newcomb, M.; Gokel, G.W.; Cram, D.J. J. Am. Chem. Soc. 1974, 96, 6810.
123. Ragunathan, K.G.; Shukla, R.; Mishra, S.; Bharadwaj, P.K. Tetrahedron Lett. 1993, 34, 5631.
124. See Potvin, P.G.; Lehn, J.M. Prog. Macrocycl. Chem. 1987, 3, 167; Kiggen, W.; Vögtle, F. Prog. Macrocycl. Chem. 1987, 3, 309; Dietrich, B. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 2, Academic Press, NY, 1984, pp. 337–405; Parker, D. Adv. Inorg. Radichem. 1983, 27, 1; Lehn, J.M. Acc. Chem. Res. 1978, 11, 49, Pure Appl. Chem. 1977, 49, 857.
125. Shivanyuk, A.; Spaniol, T.P.; Rissanen, K.; Kolehmainen, E.; Böhmer, V. Angew. Chem. Int. Ed. 2000, 39, 3497.
126. Bryany, J.A.; Ho, S.P.; Knobler, C.B.; Cram, D.J. J. Am. Chem. Soc. 1990, 112, 5837.
127. Shinkai, S. Tetrahedron 1993, 49, 8933.
128. See Vicens, J.; Böhmer, V. Calixarenes: A Versatile Class of Macrocyclic Compounds, Kluver: Dordrecht, 1991; Gutsche, C.D. Calixarenes; Royal Society of Chemistry, Cambridge, 1989; Gutsche, C.D. Prog. Macrocycl. Chem. 1987, 3, 93. Also see, Geraci, C.; Piattelli, M.; Neri, P. Tetrahedron Lett. 1995, 36, 5429; Zhong, Z.-L.; Chen, Y.-Y.; Lu, X.-R. Tetrahedron Lett. 1995, 36, 6735; No, K.; Kim, J.E.; Kwon, K.M. Tetrahedron Lett. 1995,36, 8453.
129. Agbaria, K.; Aleksiuk, O.; Biali, S.E.; Böhmer, V.; Frings, M.; Thondorf, I. J. Org. Chem. 2001, 66, 2891. See Agbaria, K.; Biali, S.E.; Böhmer, V.; Brenn, J.; Cohen, S.; Frings, M.; Grynszpan, F.; Harrowfield, J.Mc B.; Sobolev, A.N.; Thondorf, I. J. Org. Chem. 2001, 66, 2900.
130. Cerioni, G.; Biali, S.E.; Rappoport, Z. Tetrahedron Lett. 1996, 37, 5797; Molard, Y.; Bureau, C.; Parrot-Lopez, H.; Lamartine, R.; Regnourf-de-Vains, J.-B. Tetrahedron Lett. 1999, 40, 6383.
131. Otsuka, H.; Araki, K.; Matsumoto, H.; Harada, T.; Shinkai, S. J. Org. Chem. 1995, 60, 4862.
132. Kanamathareddy, S.; Gutsche, C.D. J. Org. Chem. 1994, 59, 3871.
133. Cunsolo, F.; Consoli, G.M.L.; Piattelli, M.; Neri, P. Tetrahedron Lett. 1996, 37, 715.
134. Miyazaki, Y.; Kanbara, T.; Yamamoto, T. Tetrahedron Lett. 2002, 43, 7945; Khan, I.U.; Takemura, H.; Suenaga, M.; Shinmyozu, T.; Inazu, T. J. Org. Chem. 1993, 58, 3158.
135. Masci, B. J. Org. Chem. 2001, 66, 1497; Seri, N.; Thondorf, I.; Biali, S.E. J. Org. Chem. 2004, 69, 4774; Tsubaki, K.; Morimoto, T.; Otsubo, T.; Kinoshita, T.; Fuji, K. J. Org. Chem. 2001, 66, 4083.
136. Stewart, D.R.; Gutsche, C.D. J. Am. Chem. Soc. 1999, 121, 4136.
137. Mascal, M.; Naven, R.T.; Warmuth, R. Tetrahedron Lett. 1995, 36, 9361.
138. Wu, Y.; Shen, X.-P.; Duan, C.-y.; Liu, Y.-i.; Xu, Z. Tetrahedron Lett. 1999, 40, 5749.
139. Colby, D.A.; Lash, T.D. J. Org. Chem.2002, 67, 1031.
140. Akine, S.; Goto, K.; Kawashima, T. Tetrahedron Lett. 2000, 41, 897.
141. Aeungmaitrepirom, W.; Hagège, A.; Asfari, Z.; Bennouna, L.; Vicens, J.; Leroy, M. Tetrahedron Lett. 1999, 40, 6389.
142. Shirakawa, S.; Moriyama, A.; Shimizu, S. Eur. J. Org. Chem. 2008, 5957.
143. Shimizu, S.; Shirakawa, S.; Sasaki, Y.; Hirai, C. Angew. Chem. Int. Ed. 2000, 39, 1256.
144. Stephan, H.; Gloe, K.; Paulus, E.F.; Saadioui, M.; Böhmer, V. Org. Lett. 2000, 2, 839; Asfari, Z.; Thuéry, P.; Nierlich, M.; Vicens, J. Tetrahedron Lett. 1999, 40, 499; Geraci, C.; Piattelli, M.; Neri, P. Tetrahedron Lett. 1996, 37, 3899; Pappalardo, S.; Petringa, A.; Parisi, M.F.; Ferguson, G. Tetrahedron Lett. 1996, 37, 3907.
145. Pulpoka, B.; Asfari, Z.; Vicens, J. Tetrahedron Lett. 1996, 37, 6315.
146. Makrlík, E.; Va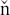 ura, P. Monat. Chemie 2006, 137, 1185-.
ura, P. Monat. Chemie 2006, 137, 1185-.
147. See Collet, A. Tetrahedron 1987, 43, 5725, in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 1, Academic Press, NY, 1984, pp. 97–121.
148. Lein, G.M.; Cram, D.J. J. Am. Chem. Soc. 1985, 107, 448.
149. Fo Kron, T.E.; Tsvetkov, E.N. Russ. Chem. Rev. 1990, 59, 283; Menger, F.M. Top. Curr. Chem. 1986, 136, 1.
150. Tümmler, B.; Maass, G.; Weber, E.; Wehner, W.; Vögtle, F. J. Am. Chem. Soc. 1977, 99, 4683.
151. Vögtle, F.; Weber, E. Angew. Chem. Int. Ed. 1974, 13, 814.
152. For the synthesis of N-pivot lariat ethers, see Elwahy, A.H.M.; Abbas, A.A. J. Het. Chem. 2008, 45, 1.
153. Gatto, V.J.; Gokel, G.W. J. Am. Chem. Soc. 1984, 106, 8240; Nakatsuji, Y.; Nakamura, T.; Yonetani, M.; Yuya, H.; Okahara, M. J. Am. Chem. Soc. 1988, 110, 531.
154. Lee, W.Y.; Park, C.H. J. Org. Chem. 1993, 58, 7149.
155. Wang, T.; Bradshaw, J.S.; Izatt, R.M. J. Heterocylic Chem. 1994, 31, 1097.
156. Fujimoto, T.; Yanagihara, R.; Koboyashi, K.; Aoyama, Y. Bull. Chem. Soc. Jpn. 1995, 68, 2113.
157. For reviews, see Rebek, Jr., J. Angew. Chem. Int. Ed. 1990, 29, 245; Acc. Chem. Res. 1990, 23, 399; Top. Curr. Chem. 1988, 149, 189; Diederich, F. J. Chem. Educ. 1990, 67, 813; Hamilton, A.D. J. Chem. Educ. 1990, 67, 821; Raevskii, O.A. Russ. Chem. Rev. 1990, 59, 219.
158. Mageswaran, R.; Mageswaran, S.; Sutherland, I.O. J. Chem. Soc., Chem. Commun. 1979, 722.
159. Diederich, F.; Dick, K. J. Am. Chem. Soc. 1984, 106, 8024; Diederich, F.; Griebe, D. J. Am. Chem. Soc. 1984, 106, 8037. See also, Vögtle, F.; Müller, W.M.; Werner, U.; Losensky, H. Angew. Chem. Int. Ed. 1987, 26, 901.
160. Hosseini, M.W.; Lehn, J.M. J. Am. Chem. Soc. 1987, 109, 7047. For a discussion, see Mertes, M.P.; Mertes, K.B. Acc. Chem. Res. 1990, 23, 413.
161. See Cram, D.J. Angew. Chem. Int. Ed. 1986, 25, 1039.
162. See Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vols. 1–3, Academic Press, NY, 1984; Weber, E. Top. Curr. Chem. 1987, 140, 1; Gerdil, R. Top. Curr. Chem. 1987, 140, 71; Mak, T.C.W.; Wong, H.N.C. Top. Curr. Chem. 1987, 140, 141; Bishop, R.; Dance, I.G. Top. Curr. Chem. 1988, 149, 137.
163. For reviews, see Goldberg, I. Top. Curr. Chem. 1988, 149, 1; Weber, E.; Czugler, M. Top. Curr. Chem. 1988, 149, 45; MacNicol, D.D.; McKendrick, J.J.; Wilson, D.R. Chem. Soc. Rev. 1978, 7, 65.
164. Strobel, T.A.; Hester, K.C.; Sloan Jr., E.D.; Koh, C.A. J. Am. Chem. Soc. 2007, 129, 9544.
165. For a review of urea and thiourea inclusion compounds, see Takemoto, K.; Sonoda, N. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 2, Academic Press, NY, 1984, pp. 47–67.
166. Taken from Alavi, S.; Udachin, K.; Ripmeester, J.A. Chem. Eur. J. 2010, 16, 1017.
167. Radell, J.; Connolly, J.W.; Cosgrove, Jr., W.R. J. Org. Chem. 1961, 26, 2960.
168. Redlich, O.; Gable, C.M.; Dunlop, A.K.; Millar, R.W. J. Am. Chem. Soc. 1950, 72, 4153.
169. Heaton, N.J.; Vold, R.L.; Vold, R.R. J. Am. Chem. Soc. 1989, 111, 3211.
170. For a review, see MacNicol, D.D. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 2, Academic Press, NY, 1984, pp. 1–45.
171. Toda, F.; Hyoda, S.; Okada, K.; Hirotsu, K. J. Chem. Soc., Chem. Commun. 1995, 1531.
172. For a monograph on water clathrates, see Berecz, E.; Balla-Achs, M. Gas Hydrates; Elsevier, NY, 1983. For reviews, see Jeffrey, G.A. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 1, Academic Press, NY, 1984, pp. 135–190; Cady, G.H. J. Chem. Educ. 1983, 60, 915.
173. Sloan, E.D. Clathrate Hydrate of Natural Gases, Marcel Dekker, Inc., 1998.
174. Kirkor, E.; Gebicki, J.; Phillips, D.R.; Michl, J. J. Am. Chem. Soc. 1986, 108, 7106.
175. See also, Toda, F. Pure App. Chem. 1990, 62, 417, Top. Curr. Chem. 1988, 149, 211; 1987, 140, 43; Davies, J.E.; Finocchiaro, P.; Herbstein, F.H. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 2, Academic Press, NY, 1984, pp. 407–453.
176. For a review, see Giglio, E. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 2, Academic Press, NY, 1984, pp. 207–229.
177. See Miki, K.; Masui, A.; Kasei, N.; Miyata, M.; Shibakami, M.; Takemoto, K. J. Am. Chem. Soc. 1988, 110, 6594.
178. Barbour, L.J.; Caira, M.R.; Nassimbeni, L.R. J. Chem. Soc., Perkin Trans. 2 1993, 2321. Also see, Barbour, L.J.; Caira, M.R.; Nassimbeni, L.R. J. Chem. Soc., Perkin Trans. 2 1993, 1413.
179. Lämsä, M.; Suorsa, T.; Pursiainen, J.; Huuskonen, J.; Rissanen, K. Chem. Commun. 1996, 1443.
180. Sherman, J.C.; Knobler, C.B.; Cram, D.J. J. Am. Chem. Soc. 1991, 113, 2194.
181. van Wageningen, A.M.A.; Timmerman, P.; van Duynhoven, J.P.M.; Verboom, W.; van Veggel, F.C.J.M.; Reinhoudt, D.N. Chem. Eur.J. 1997, 3, 639; Fraser, J.R.; Borecka, B.; Trotter, J.; Sherman, J.C. J. Org. Chem. 1995, 60, 1207; Place, D.; Brown, J.; Deshayes, K. Tetrahedron Lett. 1998, 39, 5915. See also: Jasat, A.; Sherman, J.C. Chem. Rev. 1999, 99, 931.
182. Chapman, R.G.; Sherman, J.C. J. Org. Chem. 2000, 65, 513.
183. See Bender, M.L.; Komiyama, M. Cyclodextrin Chemistry, Springer, NY, 1978. For reviews, see in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Academic Press, NY, 1984, the reviews, by Saenger, W. Vol. 2, pp. 231–259, Bergeron, R.J. Vol. 3, pp. 391–443, Tabushi, I. Vol. 3, pp. 445–471, Breslow, R. Vol. 3, pp. 473–508; Croft, A.P.; Bartsch, R.A. Tetrahedron 1983, 39, 1417; Tabushi, I.; Kuroda, Y. Adv. Catal., 1983, 32, 417; Tabushi, I. Acc. Chem. Res. 1982, 15, 66; Saenger, W. Angew. Chem. Int. Ed. 1980, 19, 344; Bergeron, R. J. Chem. Ed. 1977, 54, 204; Griffiths, D.W.; Bender, M.L. Adv. Catal. 1973, 23, 209.
184. García-Río, L.; Hall, R.W.; Mejuto, J.C.; Rodriguez-Dafonte, P. Tetrahedron 2007, 63, 2208.
185. Szejtli, J. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 3, Academic Press, NY, 1984, p. 332; Nickon, A.; Silversmith, E.F. The Name Game, Pergamon, Elmsford, NY, p. 235.
186. Modified from Saenger, W.; Beyer, K.; Manor, P.C. Acta Crystallogr. Sect. B, 1976, 32, 120.
187. For reviews, see Pagington, J.S. Chem. Br., 1987, 23, 455; Szejtli, J. in Atwood, J.L.; Davies, J.E.; MacNicol, D.D. Inclusion Compounds, Vol. 3, Academic Press, NY, 1984, pp. 331–390.
188. See Saenger, W. Angew. Chem. Int. Ed. 1980, 19, 344.
189. Engeldinger, E.; Armspach, D.; Matt, D. Chem. Rev. 2003, 103, 4147.
190. For a monograph, see Schill, G. Catenanes, Rotaxanes, and Knots, Academic Press, NY, 1971. For a review, see Schill, G. in Chiurdoglu, G. Conformational Analysis, Academic Press, NY, 1971, pp. 229–239.
191. Solladié, N.; Chambron, J.-C.; Sauvage, J.-P. J. Am. Chem. Soc. 1999, 121, 3684.
192. Sasabe, H.; Kihara, N.; Furusho, Y.; Mizuno, K.; Ogawa, A.; Takata, T. Org. Lett. 2004, 6, 3957.
193. Safarowsky, O.; Vogel, E.; Vögtle, F. Eur. J. Org. Chem. 2000, 499.
194. Amabilino, D.B.; Ashton, P.R.; Balzani, V.; Boyd, S.E.; Credi, A.; Lee, J.Y.; Menzer, S.; Stoddart, J.F.; Venturi, M.; Williams, D.J. J. Am. Chem. Soc. 1998, 120, 4295.
195. Chiu, S.-H.; Rowan, S.J.; Cantrill, S.J.; Ridvan, L.; Ashton, R.P.; Garrell, R.L.; Stoddart, J.-F. Tetrahedron 2002, 58, 807; Roh, S.-G.; Park, K.-M.; Park, G.-J.; Sakamoto, S.; Yamaguchi, K.; Kim, K. Angew. Chem. Int. Ed. 1999, 38, 638.
196. See Onagi, H.; Easton, C.J.; Lincoln, S.F. Org. Lett. 2001, 3, 1041; Cantrill, S.J.; Youn, G.J.; Stoddart, J.F.; Williams, D.J. J. Org. Chem. 2001, 66, 6857.
197. Chiu, S.-H.; Pease, A.R.; Stoddart, J.F.; White, A.J.P.; Williams, D.J. Angew. Chem. Int. Ed. 2002, 41, 270.
198. Schwierz, H.; Vögtle, F. Synthesis 1999, 295.
199. Elizarov, A.M.; Chiu, S.-H.; Stoddart, J.-F. J. Org. Chem. 2002, 67, 9175.
200. MacLachlan, M. J.; Rose, A.; Swager, T. M. J. Am. Chem. Soc. 2001, 123, 9180.
201. Amabilino, D.B.; Ashton, P.R.; Boyd, S.E.; Gómez-López, M.; Hayes, W.; Stoddart, J.F. J. Org. Chem. 1997, 62, 3062.
202. For discussions, see Schill, G. Catenanes, Rotaxanes, and Knots, Academic Press, NY, 1971. For a review, see Schill, G. in Chiurdoglu, G. Conformational Analysis, Academic Press, NY, 1971, pp. 229–239; Walba, D.M. Tetrahedron 1985, 41, 3161.
203. Fujita, M.; Ibukuro, F.; Seki, H.; Kamo, O.; Imanari, M.; Ogura, K. J. Am. Chem. Soc. 1996, 118, 899.
204. Harrison, I.T.; Harrison, S. J. Am. Chem. Soc. 1967, 89, 5723; Ogino, H. J. Am. Chem. Soc. 1981, 103, 1303; Harrison, I.T. J. Chem. Soc., Perkin Trans. 1 1974, 301; Schill, G.; Beckmann, W.; Schweikert, N.; Fritz, H. Chem. Ber. 1986, 119, 2647. See also, Agam, G.; Graiver, D.; Zilkha, A. J. Am. Chem. Soc. 1976, 98, 5206.
205. For a directed synthesis of a rotaxane, see Schill, G.; Zürcher, C.; Vetter, W. Chem. Ber. 1973, 106, 228.
206. Deleuze, M. S.; Leigh, D. A; Zerbetto, F. J. Am. Chem. Soc. 1999, 121, 2364.
207. For the synthesis of a doubly interlocking [2]catenane, see Ibukuro, F.; Fujita, M.; Yamaguchi, K.; Sauvage, J.-P. J. Am. Chem. Soc. 1999, 121, 11014.
208. See Lukin, O.; Godt, A.; Vögtle, F. Chem. Eur. J., 2004, 10, 1879.
209. Frisch, H.L.; Wasserman, E. J. Am. Chem. Soc. 1961, 83, 3789.
210. Molecular Catenanes, Rotaxanes and Knots (Eds., Sauvage, J.-P.; Dietrich-Buchecker, C.O.) Wiley-VCH, Weinheim, 1999; Ashton, P.R.; Bravo, J.A.; Raymo, F.M.; Stoddart, J.F.; White, A.J.P.; Williams, D.J. Eur. J. Org. Chem. 1999, 899; Mitchell, D.K.; Sauvage, J.-P. Angew. Chem. Int. Ed. 1988, 27, 930; Nierengarten, J.-F.; Dietrich-Buchecker, C.O.; Sauvage, J.-P. J. Am. Chem. Soc. 1994, 116, 375; Chen, C.-T.; Gantzel, P.; Siegel, J.S.; Baldridge, K.K.; English, R.B.; Ho, D.M. Angew. Chem. Int. Ed. 1995, 34, 2657.
211. Kaida, T.; Okamoto, Y.; Chambron, J.-C.; Mitchell, D.K.; Sauvage, J.-P. Tetrahedron Lett. 1993, 34, 1019.
212. Schmieder, R.; Hübner, G.; Seel, C.; Vögtle, F. Angew. Chem. Int. Ed. 1999, 38, 3528.
213. Prelog, V.; Gerlach, H. Helv. Chim. Acta 1964, 47, 2288; Gerlach, H.; Owtischinnkow, J.A.; Prelog, V. Helv. Chim. Acta 1964, 47, 2294; Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds, Wiley, NY, 1994, pp. 1176–1181; Chorev, M.; Goodman, M. Acc. Chem. Res. 1993, 26, 266; Mislow, K. Chimia, 1986, 40, 395.
214. For an example, see Anelli, P.L.; Spencer, N.; Stoddart, J.F. J. Am. Chem. Soc. 1991, 113, 5131.
215. Oshikiri, T.; Takashima, Y.; Yamaguchi, H.; Harada, A. J. Am. Chem. Soc. 2005, 127, 12186.
216. Anelli, P.L.; Spencer, N.; Stoddart, J.F. J. Am. Chem. Soc. 1991, 113, 5131. For a review of the synthesis and properties of molecules of this type, see Philp, D.; Stoddart, J.F. Synlett 1991, 445.
217. Diederich, F.; Dietrich-Buchecker, C.O.; Nierengarten, S.-F.; Sauvage, J.-P. J. Chem. Soc., Chem. Commun. 1995, 781.
218. Dietrich-Buchecker, C.O.; Nierengarten, J.-F.; Sauvage, J.-P. Tetrahedron Lett. 1992, 33, 3625. See Dietrich-Buchecker, C.O.; Guilhem, J.; Pascard, C.; Sauvage, J.-P. Angew. Chem. Int. Ed. 1990, 29, 1154.
219. Liu, L.F.; Depew, R.E.; Wang, J.C. J. Mol. Biol. 1976, 106, 439.
220. Williams, A.R.; Northrop, B.N.; Chang, T.; Stoddart, J.F.; White, A.J.P.; Williams, D.J. Angew. Chem. Int. Ed. 2006, 45, 6665.
221. Day, A.I.; Blanch, R.J.; Arnold, A.P.; Lorenzo, S.; Lewis, G.R.; Dance, I. Angew. Chem. Int. Ed,. 2002, 41, 275.
222. Mock, W.L. Top. Curr. Chem. 1995, 175, 1; Mock, W.L. in Comprehensive Supramolecular Chemistry, Vol. 2 (Eds.: Atwood, J.L.; Davies, J.E.D.; MacNicol, D.D.; Vogtle, F.), Pergamon, Oxford, 1996, pp. 477–493; Day, A.; Arnold, A.P.; Blanch, R.J.; Snushall, B. J. Org. Chem. 2001, 66, 8094. For cucurbit[10]uril, see Liu, S.; Zavalij, P.Y.; Isaacs, L. J. Am. Chem. Soc. 2005, 127, 16798.