Chapter 7
Irradiation Processes in Organic Chemistry
Most reactions carried out in organic chemistry laboratories take place between molecules, all of which are in their ground electronic states. In a photochemical reaction,1 however, a reacting molecule has been previously promoted to an electronically excited state by absorption of light. A molecule in an excited state must lose its extra energy in some manner; it cannot remain in the excited state for long. The subject of electronic spectra is closely related to photochemistry. A chemical reaction is not the only possible means of relinquishing the extra energy in a photochemical process. In this chapter, electronically excited states and the processes of promotion to these states will be discussed. Reactions of such molecules are called photoreactions. There are enantioselective organocatalytic photoreactions, but they will not be discussed here.2 Two other methods are available to facilitate chemical reactions: sonochemistry and microwave chemistry. Although the physical processes involved are not the same excitation processes observed in photochemistry, irradiation with ultrasound or with microwaves have a significant influence on chemical reactivity, and both are widely used. For that reason, they are included in this chapter.
7.A. Photochemistry3
7.A.i. Excited States and the Ground State
Electrons can move from the ground-state energy level of a molecule to a higher level (i.e., an unoccupied orbital of higher energy) if outside energy is supplied. In a photochemical process, this energy is in the form of light. Light of any wavelength has an energy value associated with it given by E = hν, where n is the frequency of the light (n = velocity of light c divided by the wavelength λ), and h is Planck's constant. Since the energy levels of a molecule are quantized, the amount of energy required to raise an electron in a given molecule from one level to a higher one is a fixed quantity. Only light with exactly the frequency corresponding to this amount of energy will cause the electron to move to the higher level. If light of another frequency (too high or too low) is sent through a sample, it will pass out without a loss in intensity, since the molecules will not absorb it. However, if light of the correct frequency is passed into a sample, molecules will use that energy for electron promotion, and the light that leaves the sample will be diminished in intensity or altogether gone. A spectrophotometer is an instrument that allows light of a given frequency to pass through a sample and that detects (by means of a phototube) the amount of light that has been transmitted, that is, not absorbed. A spectrophotometer compares the intensity of the transmitted light with that of the incident light. Automatic instruments gradually and continuously change the frequency, and an automatic recorder plots a graph of absorption versus frequency or wavelength.
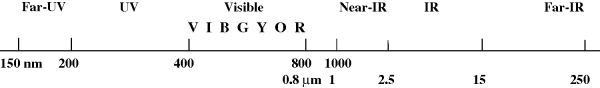
The energy of electronic transitions corresponds to light in the vis, UV, and far-UV regions of the spectrum (Fig. 7.1). Absorption positions are normally expressed in wavelength units, usually nanometers (nm).4 If a compound absorbs in the visible, it is colored, possessing a color complementary to that absorbed.5 Thus a compound absorbing in the violet has a yellow color. Organic chemists study the far-UV region less often than the vis or ordinary UV regions because special vacuum instruments are required, owing to the fact that oxygen and nitrogen absorb in these regions.
Fig. 7.1 The UV, vis, and IR portions of the electromagnetic spectrum.
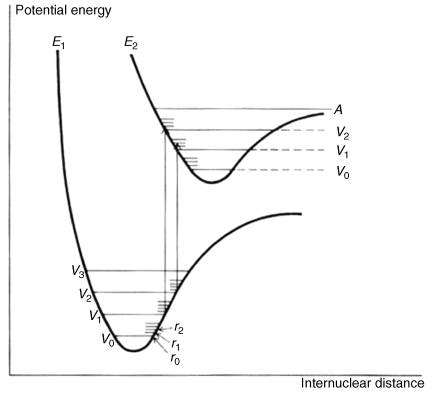
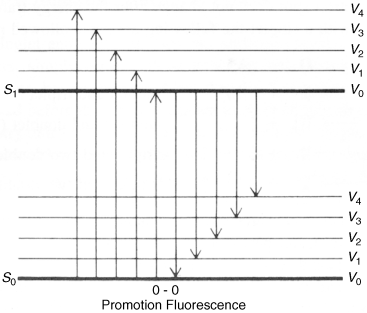
From these considerations it would seem that an electronic spectrum should consist of one or more sharp peaks, each corresponding to the transfer of an electron from one electronic level to another. Under ordinary conditions the peaks are seldom sharp. In order to understand why, it is necessary to realize that molecules are constantly vibrating and rotating and that these motions are also quantized. A molecule at any time is not only in a given electronic state, but also in a given vibrational and rotational state. The difference between two adjacent vibrational levels is much smaller than the difference between adjacent electronic levels, and the difference between adjacent rotational levels is smaller still. A typical situation is shown in Fig. 7.2. When an electron moves from one electronic level to another, it moves from a given vibrational and rotational level within that electronic level to some vibrational and rotational level at the next electronic level. A given sample contains a large number of molecules, and even if all of them are in the ground electronic state, they are still distributed among the vibrational and rotational states (though the ground vibrational state, Vo, is most heavily populated). This means that not just one wavelength of light will be absorbed, but a number of them close together, with the most probable transition causing the most intense peak. But in molecules containing more than a few atoms, there are so many possible transitions, and these are so close together that what is observed is a relatively broad band. The height of the peak depends on the number of molecules making the transition and is proportional to log ε, where ε is the extinction coefficient. The extinction coefficient can be expressed by ε = E/cl, where c is the concentration in moles per liter, l is the cell length in centimeters, and E = log Io/I, where Io is the intensity of the incident light and I of the transmitted light. The wavelength is usually reported as λmax, meaning that this is the top of the peak. Purely vibrational transitions (e.g., between Vo and V1 of E1), which require much less energy, are found in the IR region and are the basis of IR spectra. Purely rotational transitions are found in the far-IR and microwave (beyond the far-IR) regions.
Fig. 7.2 Energy curves for a diatomic molecule. Two possible transitions are shown. When an electron has been excited to the point marked A, the molecule may cleave (Sec. 7.A.v).
A UV or vis absorption peak is caused by the promotion of an electron in one orbital (usually ground-state) to a higher orbital. Normally, the amount of energy necessary to make this transition depends mostly on the nature of the two orbitals involved and much less on the rest of the molecule. Therefore, a simple functional group (e.g., the C=C double bond) always causes absorption in the same general area. A group that causes absorption is called a chromophore.
7.A.ii. Singlet and Triplet States: “Forbidden” Transitions
In most organic molecules, all electrons in the ground state are paired, with each member of a pair possessing opposite spin, as demanded by the Pauli principle. When one of a pair of electrons is promoted to an orbital of higher energy, the two electrons no longer share an orbital, and the promoted electron may, in principle, have the same spin as its former partner or the opposite spin. As seen in Chapter 5, a molecule in which two unpaired electrons have the same spin is called a triplet,6 while one in which all spins are paired is a singlet. Thus, at least in principle, for every excited singlet state there is a corresponding triplet state. In most cases, the triplet state has a lower energy than the corresponding singlet, which is in accord with Hund's rule. Therefore, a different amount of energy, and hence a different wavelength, is required to promote an electron from the ground state (which is almost always a singlet) to an excited singlet than to the corresponding triplet state.
It would thus seem that promotion of a given electron in a molecule could result either in a singlet or a triplet excited state depending on the amount of energy added. However, this is often not the case because transitions between energy levels are governed by selection rules, which state that certain transitions are “forbidden”. There are several types of “forbidden” transitions, two of which are more important than the others.
The word “forbidden” is in quotation marks because these transitions are not actually forbidden, but only highly improbable. In most cases, promotions from a singlet ground state to a triplet excited state are so improbable that they cannot be observed, and it is safe to state that in most molecules only singlet–singlet promotions take place. However, this rule does break down in certain cases, most often when a heavy atom (e.g., iodine) is present in the molecule, in which cases it can be shown from spectra that singlet–triplet promotions are occurring.7 Symmetry-forbidden transitions can frequently be observed, though usually with low intensity.
7.A.iii. Types of Excitation
When an electron in a molecule is promoted (normally only one electron in any molecule), it usually goes into the lowest available vacant orbital, though promotion to higher orbitals is also possible. For most organic molecules, there are consequently four types of electronic excitation:
The four excitation types above are listed in what is normally the order of decreasing energy. Thus light of the highest energy (in the far-UV) is necessary for σ → σ∗ excitation, while n → π∗ promotions are caused by ordinary UV light. However, the order may sometimes be altered in some solvents.
In 1,3-butadiene (and other compounds with two conjugated double bonds), there are two π and two π∗ orbitals (Sec. 2.C). The energy difference between the higher π (χ2) and the lower π∗ (χ3) orbital is less than the difference between the π and π∗ orbitals of ethylene. Therefore 1,3-butadiene requires less energy than ethylene, and thus light of a higher wavelength, to promote an electron. This is a general phenomenon, and it may be stated that, in general, the more conjugation in a molecule, the more the absorption is displaced toward higher wavelengths (see Table 7.1).10 When a chromophore absorbs at a certain wavelength and the substitution of one group for another causes absorption at a longer wavelength, a bathochromic shift is said to have occurred. The opposite kind of shift is called hypsochromic.
Table 7.1 Ultraviolet Absorption10 of CH3–(CH=CH)n–CH3 for Some Values of n
| n | nm |
| 2 | 227 |
| 3 | 263 |
| 6 | 352 |
| 9 | 413 |
Of the four excitation types listed above, the π → π∗ and n → π∗ are far more important in organic photochemistry than the other two. Compounds containing C=O groups can be excited in both ways, giving rise to at least two peaks in the UV.
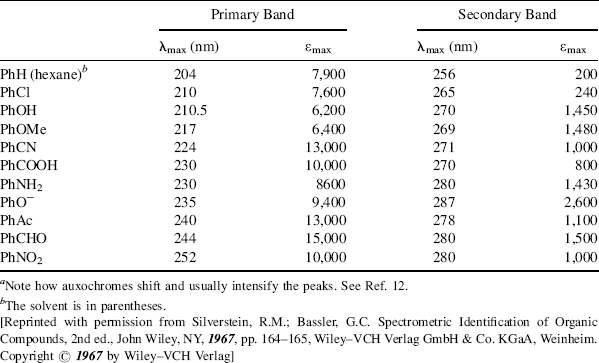
As seen above, a chromophore is a group that causes a molecule to absorb light. Examples of chromophores in the vis or UV are C=O, N=N,11 Ph, and NO2. Some chromophores in the far-UV (beyond 200 nm) are C=C, C≡C, Cl, and OH. An auxochrome is a group that displaces (through resonance) and usually intensifies the absorption of a chromophore present in the same molecule. Groups (e.g., Cl, OH, and NH2) are generally regarded as auxochromes since they shift (usually bathochromically) the UV and vis bands of chromophores (e.g., Ph or C=O; see Table 7.2).12 Since auxochromes are themselves chromophores (to be sure, generally in the far-UV), it is sometimes difficult to decide which group in a molecule is an auxochrome and which is a chromophore. For example, in acetophenone (PhCOMe) is the chromophore Ph or C=O? In such cases, the distinction becomes practically meaningless.
Table 7.2 Some UV Peaks of Substituted Benzenesa
7.A.iv. Nomenclature and Properties of Excited States
An excited state of a molecule can be regarded as a distinct chemical species, different from the ground state of the same molecule and from other excited states. It is obvious that some method of naming excited states is required. Unfortunately, there are several methods in use, depending on whether one is primarily interested in photochemistry, spectroscopy, or MO theory.13 One of the most common methods simply designates the original and newly occupied orbitals, with or without a superscript to indicate singlet or triplet. Thus the singlet state arising from promotion of a π to a π∗ orbital in ethylene would be the 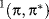 state or the π,π∗ singlet state. Another very common method can be used even in cases where one is not certain which orbitals are involved. The lowest-energy excited state is called S1, the next is S2, and so on. Triplet states are similarly labeled T1, T2, T3, and so on. In this notation, the ground state is So. Other notational systems exist, but this text shall discuss only the two types just mentioned.
state or the π,π∗ singlet state. Another very common method can be used even in cases where one is not certain which orbitals are involved. The lowest-energy excited state is called S1, the next is S2, and so on. Triplet states are similarly labeled T1, T2, T3, and so on. In this notation, the ground state is So. Other notational systems exist, but this text shall discuss only the two types just mentioned.
The properties of excited states are not easy to measure because of their generally short lifetimes and low concentrations, but enough work has been done for us to know that they often differ from the ground state in geometry, dipole moment, and acid or base strength.14 For example, acetylene, which is linear in the ground state, has a trans geometry in the excited state with ~sp2 carbons in the 1(π,π∗) state.15 Similarly, the 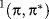 and the
and the 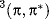 states of ethylene have a perpendicular and not a planar geometry,16 and the
states of ethylene have a perpendicular and not a planar geometry,16 and the 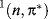 and
and 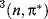 states of formaldehyde are both pyramidal.17 Triplet species tend to stabilize themselves by distortion, which relieves interaction between the unpaired electrons. Obviously, if the geometry is different, the dipole moment will probably differ also and the change in geometry and electron distribution often results in a change in acid or base strength.18 For example, the S1 state of 2-naphthol is a much stronger acid (pK = 3.1) than the ground state (S0) of the same molecule (pK = 9.5).19
states of formaldehyde are both pyramidal.17 Triplet species tend to stabilize themselves by distortion, which relieves interaction between the unpaired electrons. Obviously, if the geometry is different, the dipole moment will probably differ also and the change in geometry and electron distribution often results in a change in acid or base strength.18 For example, the S1 state of 2-naphthol is a much stronger acid (pK = 3.1) than the ground state (S0) of the same molecule (pK = 9.5).19
7.A.v. Photolytic Cleavage
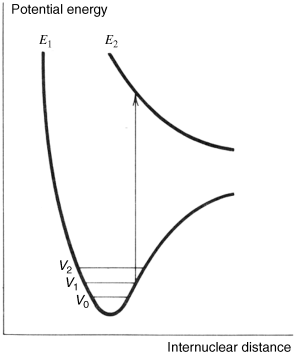
As stated above, when a molecule absorbs a quantum of light it is promoted to an excited state. Actually, that is not the only possible outcome. Because the energy of vis and UV light is of the same order of magnitude as that of covalent bonds (Table 7.3), another possibility is that the molecule may cleave into two parts, a process known as photolysis. There are three situations that can lead to cleavage:
Table 7.3 Typical Energies for Some Covalent Single Bondsa and the Corresponding Approximate Wavelengths.
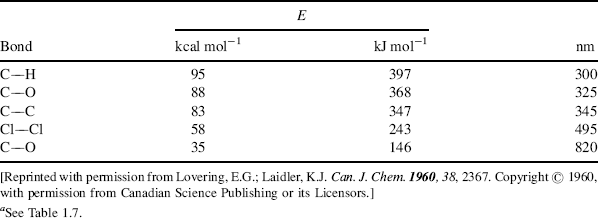
Fig. 7.3 Promotion to a dissociative state results in bond cleavage.
A photolytic cleavage can break the molecule into two smaller molecules or into two free radicals (see Sec. 7.A.vii). Cleavage into two ions, though known, is rare. Once free radicals are produced by a photolysis, they behave like free radicals produced in any other way (Chap 5) except that they may be in excited states, and this can cause differences in behavior.20
7.A.vi. The Fate of the Excited Molecule: Physical Processes
When a molecule has been photochemically promoted to an excited state, it does not remain in the excited state for long. Most promotions are from the S0 to the S1 state. As seen previously, promotions from So to triplet states are “forbidden”. Promotions to S2 and higher singlet states take place, but in liquids and solids these higher states usually drop very rapidly to the S1 state (~10−13 to ~10−11 s). The energy lost when an S2 or S3 molecule drops to S1 is given up in small increments to the environment by collisions with neighboring molecules. Such a process is called an energy cascade. In a similar manner, the initial excitation and the decay from higher singlet states initially populate many of the vibrational levels of S1, but these also cascade, down to the lowest vibrational level of S1. Therefore, in most cases, the lowest vibrational level of the S1 state is the only important excited singlet state.21 This state can undergo various physical and chemical processes. In the following list, we describe the physical pathways open to molecules in the S1 and excited triplet states. These pathways are also shown in a modified Jablonski diagram (Fig. 7.4) and in Table 7.4.
Fig. 7.4 Modified Jablonski diagram showing transitions between the excited and the ground state. Radiative processes are shown by straight lines, radiationless processes by wavy lines. vc = vibrational cascade; hνf = fluorescence; hνp = phosphorescence.
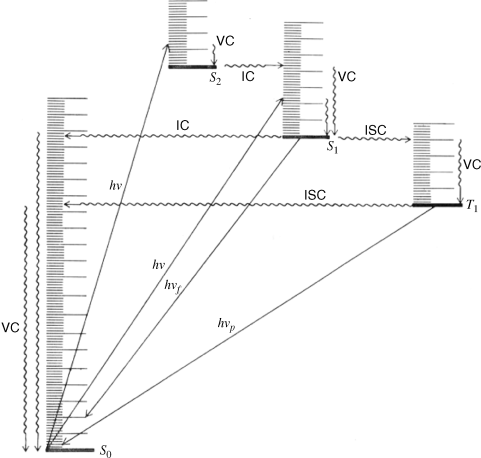
Table 7.4 Physical Processes Undergone by Excited Moleculesa
So + hν →  |
Excitation |
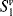 |
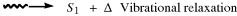 |
| S1 → S1 + hν | Fluorescence |
| S1 | 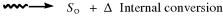 |
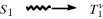 |
Intersystem crossing |
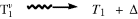 |
Vibrational relaxation |
| T1 → So + hν | Phosphorescence |
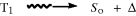 |
Intersystem crossing |
 |
Singlet–singlet transfer (photosensitization) |
 |
Triplet–triplet transfer (photosensitization) |
| a. The superscript v indicates vibrationally excited state: excited states higher than S1 or T1 are omitted. | |
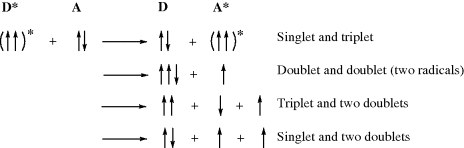
Fig. 7.5 Promotion and fluorescence between S1 and S0 states.
7.A.vii. The Fate of the Excited Molecule: Chemical Processes
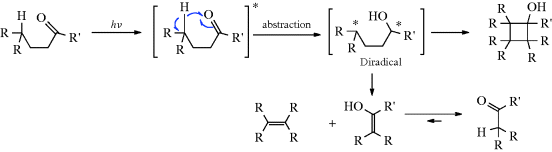
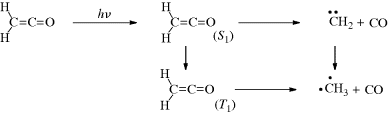
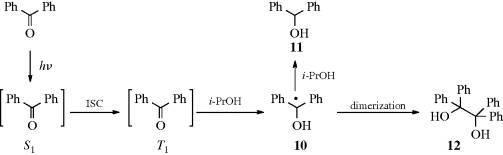
Although both excited singlet and triplet species can undergo chemical reactions, they are much more common for triplets, simply because these generally have much longer lifetimes. Excited singlet species, in most cases, have a lifetime of <10−10 s and undergo one of the physical processes already discussed before they have a chance to react chemically. Therefore, photochemistry is largely the chemistry of triplet states.41 Table 7.542 lists many of the possible chemical pathways that can be taken by an excited molecule.43 The first four of these are unimolecular reactions; the others are bimolecular. In the case of bimolecular reactions, it is rare for two excited molecules to react with each other (because the concentration of excited molecules at any one time is generally low); reactions are between an excited molecule and an unexcited molecule of either the same or another species. The reactions listed in Table 7.5 are primary processes. Secondary reactions often follow, since the primary products are frequently radicals or carbenes; even if they are ordinary molecules, they are often in upper vibrational levels and so have excess energy. In almost all cases, the primary products of photochemical reactions are in their ground states, though exceptions are known.44 Of the reactions listed in Table 7.5, the most common are cleavage into radicals (1), decomposition into molecules (2), and (in the presence of a suitable acceptor molecule) photosensitization (7), which we have already discussed. The following are some specific examples of reaction categories (1)–(6). Other examples are discussed in Part II.45,46
Table 7.5 Primary Photochemical Reactionsa of an Excited Molecule A–B–Cb
| Reactions | Reaction Type | Example Number |
| (A–B–C) → A–B• + C• | Simple cleavage into radicals46 | (1) |
| (A–B–C) → E + F | Decomposition into molecules | (2) |
| (A–B–C) → A–C–B | Intramolecular rearrangement | (3) |
| (A–B–C) → A–B–C′ | Photoisomerization | (4) |
| (A–B–C) + RH → A–B–C–H + R• | Hydrogen-atom abstraction | (5) |
| (A–B–C) → (ABD)2 | Photodimerization | (6) |
| (A–B–C) + A → ABX + A∗ | Photosensitization | (7) |
| a. Examples are given in the text; the most common are (1), (2), and, in the presence of a suitable acceptor molecule (7). | ||
| b. See Ref. 42. | ||
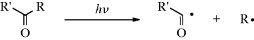
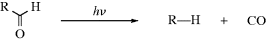
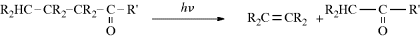
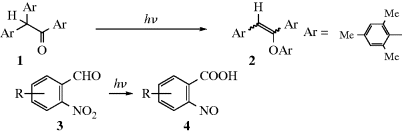
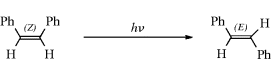
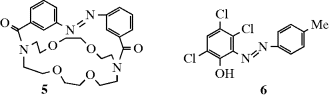
 ion in the dark compartment, the process reverses the normal reaction whereby these ions neutralize each other.64 Thus the energy of light is used to do chemical work.65 Another example of a category 4 reaction is the conversion of bicyclo[2.2.1]hept-2,5-diene to 7.58 The thermal isomerization of dibenzosemibullvalene (9) to the corresponding dibenzodihydropentalenofuran (8) in quantitative yield was known,66 but in another example of a category 4 reaction the photochemical isomerization of 8 to 9 has now been reported.67
ion in the dark compartment, the process reverses the normal reaction whereby these ions neutralize each other.64 Thus the energy of light is used to do chemical work.65 Another example of a category 4 reaction is the conversion of bicyclo[2.2.1]hept-2,5-diene to 7.58 The thermal isomerization of dibenzosemibullvalene (9) to the corresponding dibenzodihydropentalenofuran (8) in quantitative yield was known,66 but in another example of a category 4 reaction the photochemical isomerization of 8 to 9 has now been reported.67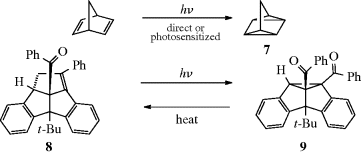
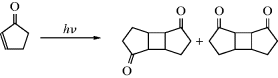
7.A.viii. The Determination of Photochemical Mechanisms69
The methods used for the determination of photochemical mechanisms are largely the same as those used for organic mechanisms in general (Chapter 6): product identification, isotopic tracing, the detection and trapping of intermediates, and kinetics. There are, however, a few new factors: (1) there are generally many products in a photochemical reaction, as many as 10 or 15; (2) in measuring kinetics, there are more variables, since it is possible to study the effect on the rate of the intensity or the wavelength of light; (3) in the detection of intermediates by spectra the technique of flash photolysis can be used, which can detect extremely short-lived intermediates.
In addition to these methods, there are two additional techniques.
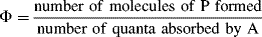
7.B. Sonochemistry
Sonochemistry (chemical events induced by exposure to ultrasound) occupies an important place in organic chemistry.70 The chemical effects of high-intensity ultrasound were extensively studied in aqueous solutions for many years,71 but are now applied to a variety of organic solvents. The origin of sonochemistry is acoustic cavitation: the creation, growth, and implosive collapse of gas vacuoles in solution by the sound field. Acoustic cavitation is the phenomenon by which intense ultrasonic waves induce the formation, oscillation, and implosion of gas bubbles in liquids.72 Liquids irradiated with high-power ultrasound undergo chemical decomposition and emit light.73 These phenomena occur near the end of the collapse of bubbles expanded many times their equilibrium sizes. Chemistry (sonochemistry), light emission (sonoluminescence), and cavitation noise often accompany the process of acoustic cavitation.74
Sonochemistry generates gas vacuoles in situ. The collapse of gas vacuoles generates transient hot spots with local temperatures of several thousand Kelvin, and pressures of hundreds of atmospheres. A sonochemical hot spot forms where the gas- and liquid-phase reaction zones have effective temperatures of 5200 and 1900 K, respectively.75 The high temperatures and pressures that are achieved in the bubbles during the quasiadiabatic collapse76 lead to the generation of chemistry and to the emission of light, most probably coming from molecular excited states and molecular recombination. Note that work has been done that shows the commonly held view that bubbles are filled with saturated gas is inconsistent with a realistic estimate of condensation rates.77 The alternative view of extensive solvent vapor supersaturation in bubbles uniformly heated to a few thousand kelvin, depending on the conditions, is in accord with sonochemical rates and products.78
There is a correlation between sonochemical and sonoluminescence measurements, which is usually not observed. Sonoluminescence is the consequence that both the sonochemical production (under air) of oxidizing species, and the emission of light reflect the variations of the primary sonochemical acts, which are themselves due to variations of the number of “active” bubbles.79 Pulsed ultrasound in the high-frequency range (>1 MHz) is extensively used in medical diagnosis, and the effects of pulsed ultrasound in the 20-kHz range using an immersed titanium horn has been reported.80
The chemical effects of ultrasound have been studied for >50 years,81 and applied to colloid chemistry in the 1940s.82 Modern interest in the chemical uses of ultrasound involves chemistry in both homogeneous83 and heterogeneous84 systems. Organic solvents (e.g., alkanes) support acoustic cavitation and the associated sonochemistry. This leads to carbon–carbon bond cleavage and radical rearrangements, with the peak temperatures reached in such cavities controlled by the vapor pressure of the solvent.85
It is often difficult to compare the sonochemical results reported from different laboratories (the reproducibility problem in sonochemistry).86 The sonochemical power irradiated into the reaction system can be different for different instruments. Several methods are available to estimate the amount of ultrasonic power entered into a sonochemical reaction,86 the most common being calorimetry. This experiment involves measurement of the initial rate of a temperature rise produced when a system is irradiated by power ultrasound. It has been shown that calorimetric methods combined with the Weissler reaction can be used to standardize the ultrasonic power of individual ultrasonic devices.87
Sonochemistry has been used to facilitate or assist many organic reactions,88 and there are other applications.89 The scope of reactions studied is beyond this work, but some representative examples will be listed. Ultrasound has been used to promote lithiation of organic compounds,90 for the generation of carbenes,91 and reactions of metal carbonyls where sonochemical ligand dissociation has been observed, which often produces multiple CO substitution.92 The influence of ultrasound on phase-transfer catalyzed thioether synthesis has been studied.93
Sonochemistry has been applied to acceleration of the Reformatsky reaction,94 Diels–Alder reactions,95 the arylation of active methylene compounds96 nucleophilic aromatic substitution of haloarenes,97 and to hydrostannation and tin hydride reduction.98 Other sonochemical applications involve the reaction of benzyl chloride and nitrobenzene,99 an SRN1 reaction in liquid ammonia at room temperature,100 and Knoevenagel condensation of aromatic aldehydes.101 Iodination of aliphatic hydrocarbons can be accelerated,102 and oxyallyl cations have been prepared from α,α′-diiodoketones using sonochemistry.103 Sonochemistry has been applied to the preparation of carbohydrate compounds.104 When sonochemistry is an important feature of a chemical reaction, this fact will be noted in the reactions presented in Chapters 10–19.
7.C. Microwave Chemistry
In 1986, independent work by Gedye et al.,105 as well as Majetich and co-workers106 reported the use of microwave irradiation for organic reactions. Gedye described four different types of reactions, including the hydrolysis of benzamide to benzoic acid under acidic conditions, and all reactions showed significant rate enhancements when compared to the same reactions done at reflux conditions.107 Majetich, Giguere and co-workers106 reported rate enhancements for microwave-promoted Diels–Alder, Claisen, and ene reactions. Many publications108 have appeared that describe chemical synthesis promoted by microwave irradiation, including many review articles109 and books.110
Microwaves are electromagnetic waves (see Sec. 7.A.i) and there are electric and magnetic field components. Charged particles start to migrate or rotate as the electric field is applied,111 which leads to further polarization of polar particles. Because the concerted forces applied by the electric and magnetic components of microwaves are rapidly changing in direction (2.4 × 109 s−1), warming occurs.111 In general, the most common frequencies used for microwave dielectric heating112 are 918 MHz and 2.45 GHz113 (wavelengths of 33.3 and 12.2 cm, respectively), which are in the region between the IR and radiowave wavelengths in the electromagnetic spectrum. For chemical reactions done with microwave irradiation, rapid heating is usually observed,114 and if a solvent is used superheating of that solvent was always observed.112 Agitation is usually important.115 In the early days of microwave chemistry, reactions were often done in open vessels, but also in sealed Teflon or glass vessels using unmodified domestic household ovens.116 Dielectric heating is direct so if the reaction matrix has a sufficiently large dielectric loss tangent, and contains molecules possessing a dipole moment, a solvent is not required. The use of dry-reaction microwave chemistry is increasingly popular.117
Microwave dielectric heating was initially categorized by thermal effects and nonthermal effects.118 “Thermal effects are those which are caused by the different temperature regime which can be created due to microwave dielectric heating. Nonthermal effects are effects,119 which are caused by effects specifically inherent to the microwaves and are not caused by different temperature regimes.”111 Some claimed special effects120 in microwave chemistry, such as lowering of Gibbs energy of activation, but later study under careful temperature control indicated no special rate effects.121 When conventional microwave ovens were used, temperature control was difficult, particularly when reactions are carried out in closed reaction vessels. The main contributing factor to any rate acceleration caused by microwave dielectric heating seems to be due to a thermal effect. The thermal effect may be due to a faster initial heating rate or to the occurrence of local regions with higher temperatures.111
Conventional microwave ovens are used less often for microwave chemistry today. Microwave reactors for chemical synthesis are commercially available and widely used in academia and in industry. These instruments have built-in magnetic stirring, direct temperature control of the reaction mixture, shielded thermocouples or IR sensors, and the ability to control temperature and pressure by regulating microwave output power.
The applications of microwave chemistry to organic chemistry are literally too numerous to mention. A few representative examples will be given to illustrate the scope and utility. The combined use of microwaves and ultrasound is important in process chemistry and organic synthesis.122 Microwave chemistry is widely used in synthesis,123 including organocatalyzed asymmetric reactions.124 Examples include the Heck reaction (Reaction 13-10),125 the Suzuki reaction (Reaction 13-12),126 the Sonogashira reaction (Reaction 13-13),127Ullman-type couplings (Reaction 13-3),128 cycloaddition reactions (Reactions 15-58–15-66),129 dihydroxylation (Reaction 15-48),130 and the Mitsunobu reaction (Reaction 10-23).131 There are a multitude of other reactions types from earlier literature that can be found in the cited review articles. When microwave chemistry is an important feature of a chemical reaction, this fact will be noted in the reactions presented in Chapters 10–19.
Notes
1. See Michl, J.; Bona i
i -Koutecky, V. Electronic Aspects of Organic Photochemistry, Wiley, NY, 1990; Scaiano, J.C. Handbook of Organic Photochemistry, 2 vols., CRC Press, Boca Raton, FL, 1989; Coxon, J.M.; Halton, B. Organic Photochemistry, 2nd ed., Cambridge University Press, Cambridge, 1987; Coyle, J.D. Photochemistry in Organic Synthesis, Royal Society of Chemistry, London, 1986, Introduction to Organic Photochemistry, Wiley, NY, 1986; Horspool, W.M. Synthetic Organic Photochemistry, Plenum, NY, 1984; Margaretha, P. Preparative Organic Photochemistry, Top. Curr. Chem. 1982, 103; Turro, N.J. Modern Molecular Photochemistry, W.A. Benjamin, NY, 1978; Rohatgi-Mukherjee. K.K. Fundamentals of Photochemistry, Wiley, NY, 1978; Barltrop, J.A.; Coyle, J.D. Principles of Photochemistry, Wiley, NY, 1978; Scaiano, J.; Johnston, L.J. Org. Photochem. 1989, 10, 309. For a history of photochemistry, see Roth, H.D. Angew. Chem. Int. Ed. 1989, 28, 1193; Braslavsky, S.E.; Houk, K.N. Pure Appl. Chem. 1988, 60, 1055. See also, the series, Advances in Photochemistry, Organic Photochemistry, and Excited States.
-Koutecky, V. Electronic Aspects of Organic Photochemistry, Wiley, NY, 1990; Scaiano, J.C. Handbook of Organic Photochemistry, 2 vols., CRC Press, Boca Raton, FL, 1989; Coxon, J.M.; Halton, B. Organic Photochemistry, 2nd ed., Cambridge University Press, Cambridge, 1987; Coyle, J.D. Photochemistry in Organic Synthesis, Royal Society of Chemistry, London, 1986, Introduction to Organic Photochemistry, Wiley, NY, 1986; Horspool, W.M. Synthetic Organic Photochemistry, Plenum, NY, 1984; Margaretha, P. Preparative Organic Photochemistry, Top. Curr. Chem. 1982, 103; Turro, N.J. Modern Molecular Photochemistry, W.A. Benjamin, NY, 1978; Rohatgi-Mukherjee. K.K. Fundamentals of Photochemistry, Wiley, NY, 1978; Barltrop, J.A.; Coyle, J.D. Principles of Photochemistry, Wiley, NY, 1978; Scaiano, J.; Johnston, L.J. Org. Photochem. 1989, 10, 309. For a history of photochemistry, see Roth, H.D. Angew. Chem. Int. Ed. 1989, 28, 1193; Braslavsky, S.E.; Houk, K.N. Pure Appl. Chem. 1988, 60, 1055. See also, the series, Advances in Photochemistry, Organic Photochemistry, and Excited States.
2. Wessig, P. Angew. Chem. Int. Ed. 2006, 45, 2168.
3. Zimmerman, H.E. Pure Appl. Chem. 2006, 78, 2193.
4. Formerly, millimicrons (mμ) were frequently used; numerically they are the same as nanometers.
5. For monographs, see Zollinger, H. Color Chemistry, VCH, NY, 1987; Gordon, P.F.; Gregory, P. Organic Chemistry in Colour, Springer, NY, 1983; Griffiths, J. Colour and Constitution of Organic Molecules, Academic Press, NY, 1976. See also, Fabian, J.; Zahradník, R. Angew. Chem. Int. Ed. 1989, 28, 677.
6. See Kurreck, H. Angew. Chem. Int. Ed. 1993, 32, 1409.
7. See Koziar, J.C.; Cowan, D.O. Acc. Chem. Res. 1978, 11, 334.
8. An n electron is one in an unshared pair.
9. See Malkin, Yu.N.; Kuz'min, V.A. Russ. Chem. Rev. 1985, 54, 1041.
10. Bohlmann, F.; Mannhardt, H. Chem. Ber. 1956, 89, 1307.
11. For a review of the azo group as a chromophore, see Rau, H. Angew. Chem. Int. Ed.1973, 12, 224.
12. These values are from Silverstein, R.M.; Bassler, G.C. Spectrometric Identification of Organic Compounds, 2nd ed., John Wiley, NY, 1967, pp. 164–165. Also see Jaffé, H.H.; Orchin, M. Theory and Applications of Ultraviolet Spectroscopy, Wiley, NY, 1962, p. 257.
13. See Pitts, Jr., J.N.; Wilkinson, F.; Hammond, G.S. Adv. Photochem. 1963, 1, 1; Porter, G.B.; Balzani, V.; Moggi, L. Adv. Photochem. 1974, 9, 147; Braslavsky, S.E.; Houk, K.N. Pure Appl. Chem. 1988, 60, 1055.
14. For reviews of the structures of excited states, see Zink, J.I.; Shin, K.K. Adv. Photochem. 1991, 16, 119; Innes, K.K. Excited States 1975, 2, 1; Hirakawa, A.Y.; Masamichi, T. Vib. Spectra Struct. 1983, 12, 145.
15. Ingold, C.K.; King, G.W. J. Chem. Soc. 1953, 2702, 2704, 2708, 2725, 2745. For a review of acetylene photochemistry, see Coyle, J.D. Org. Photochem. 1985, 7, 1.
16. Merer, A.J.; Mulliken, R.S. Chem. Rev. 1969, 69, 639.
17. Garrison, B.J.; Schaefer III, H.F.; Lester Jr., W.A. J. Chem. Phys. 1974, 61, 3039; Streitwieser Jr., A.; Kohler, B. J. Am. Chem. Soc. 1988, 110, 3769. For reviews of excited states of formaldehyde, see Buck, H.M. Recl. Trav. Chim. Pays-Bas 1982, 101, 193, 225; Moule, D.C.; Walsh, A.D. Chem. Rev. 1975, 75, 67.
18. See Ireland, J.F.; Wyatt, P.A.H. Adv. Phys. Org. Chem. 1976, 12, 131.
19. Weller, A. Z. Phys. Chem. (Frankfurt am Main) 1955, 3, 238, Discuss. Faraday Soc. 1959, 27, 28.
20. Lubitz, W.; Lendzian, F.; Bittl, R. Acc. Chem. Res. 2002, 35, 313.
21. See Turro, N.J.; Ramamurthy, V.; Cherry, W.; Farneth, W. Chem. Rev. 1978, 78, 125.
22. See Lin, S.H. Radiationless Transitions, Academic Press, NY, 1980. For reviews, see Kommandeur, J. Recl. Trav. Chim. Pays-Bas 1983, 102, 421; Freed, K.F. Acc. Chem. Res. 1978, 11, 74.
23. For other exceptions, see Sugihara, Y.; Wakabayashi, S.; Murata, I.; Jinguji, M.; Nakazawa, T.; Persy, G.; Wirz, J. J. Am. Chem. Soc. 1985, 107, 5894, and references cited therein. See also, Turro, N.J.; Ramamurthy, V.; Cherry, W.; Farneth, W. Chem. Rev. 1978, 78, 125, see pp. 126–129.
24. Also see Li, R.; Lim, E.C. Chem. Phys. 1972, 57, 605; Sharf, B.; Silbey, R. Chem. Phys. Lett. 1970, 5, 314; Schlag, E.W.; Schneider, S.; Fischer, S.F. Annu. Rev. Phys. Chem. 1971, 22, 465, pp. 490. There is evidence that ISC can also occur from the S2 state of some molecules: Samanta, A. J. Am. Chem. Soc. 1991, 113, 7427; Ohsaku, M.; Koga, N.; Morokuma, K. J. Chem. Soc. Perkin Trans. 2 1993, 71.
25. Moore, W.M.; Hammond, G.S.; Foss, R.P. J. Am. Chem. Soc. 1961, 83, 2789.
26. See Lower, S.K.; El-Sayed, M.A. Chem. Rev. 1966, 66, 199. For a review of physical and chemical processes of triplet states see Wagner, P.J.; Hammond, G.S. Adv. Photochem. 1968, 5, 21.
27. See Albini, A. Synthesis, 1981, 249; Turro, N.J.; Dalton, J.C.; Weiss, D.S. Org. Photochem. 1969, 2, 1. Ionic liquids may be soluble photosensitizers. See Hubbard, S.C.; Jones, P.B. Tetrahedron 2005, 61, 7425.
28. In certain cases excited states can be produced directly in ordinary reactions. See White, E.H.; Miano, J.D.; Watkins, C.J.; Breaux, E.J. Angew. Chem. Int. Ed. 1974, 13, 229.
29. For another table of this kind, see Calvert, J.G.; Pitts, Jr., J.N. Photochemistry, Wiley, NY, 1966, p. 89.
30. See Bennett, R.G.; Schwenker, R.P.; Kellogg, R.E. J. Chem. Phys. 1964, 41, 3040; Ermolaev, V.L.; Sveshnikova, E.B. Opt. Spectrosc. (USSR) 1964, 16, 320.
31. SeeKavarno, G.J.; Turro, N.J. Chem. Rev. 1986, 86, 401; Mariano, P.S. Org. Photochem. 1987, 9, 1.
32. For a discussion of pyrylogens as an electron-transfer sensitizer, see Clennan, E.L.; Liao, C.; Ayokosok, E. J. Am. Chem. Soc. 2008, 130, 7552.
33. See Engel, P.S.; Monroe, B.M. Adv. Photochem. 1971, 8, 245.
34. Verhoeven, J.W. Pure Appl. Chem. 1996, 68, 2223 (see p. 2268).
35. See Samanta, S.; Mishra, B.K.; Pace, T.C.S.; Sathyamurthy, N.; Bohne, C.; Moorthy, J.N. J. Org. Chem. 2006, 71, 4453.
36. See Aspari, P.; Ghoneim, N.; Haselbach, E.; von Raumer, M.; Suppan, P.; Vauthey, E. J. Chem. Soc., Faraday Trans. 1996, 92, 1689; Cohen, S.G.; Parola, A.; Parsons, Jr., G.H. Chem. Rev. 1973, 73, 141; von Raumer, M.; Suppan, P.; Haselbach, E. Helv. Chim. Acta 1997, 80, 719.
37. Inbar, S.; Linschitz, H.; Cohen, S.G. J. Am. Chem. Soc. 1982, 104, 1679; Bobrowski, K.; Marciniak, B.; Hug, G.L. J. Photochem. Photobiol. A: Chem. 1994, 81, 159; Wakasa, M.; Hayashi, H. J. Phys. Chem. 1996, 100, 15640.
38. Marciniak, B.; Bobrowski, K.; Hug, G.L. J. Phys. Chem. 1993, 97, 11937.
39. Becker, H.-D. J. Org. Chem. 1967, 32, 2115; 2124; 2140.
40. Lathioor, E.C.; Leigh, W.J.; St. Pierre, M.J. J. Am. Chem. Soc. 1999, 121, 11984.
41. See Wagner, P.J.; Hammond, G.S.; Wagner, P.J.; Hammond, G.S. Adv. Photochem. 1968, 5, 21. For other reviews of triplet states, see Top. Curr. Chem. 1975, Vols. 54 and 55.
42. Adapted from Calvert, J.G.; Pitts, Jr., J.N. Photochemistry, Wiley, NY, 1966, p. 367.
43. For a different kind of classification of photochemical reactions, see Dauben, W.G.; Salem, L.; Turro, N.J. Acc. Chem. Res. 1975, 8, 41. For reviews of photochemical reactions where the molecules are geometrically constrained, see Ramamurthy, V. Tetrahedron 1986, 42, 5753; Ramamurthy, V.; Eaton, D.F. Acc. Chem. Res. 1988, 21, 300; Turro, N.J.; Cox, G.S.; Paczkowski, M.A. Top. Curr. Chem. 1985, 129, 57.
44. Turro, N.J.; Lechtken, P.; Lyons, A.; Hautala, R.T.; Carnahan, E.; Katz, T.J. J. Am. Chem. Soc. 1973, 95, 2035.
45. See Ninomiya, I.; Naito, T. Photochemical Synthesis, Academic Press, NY, 1989; Coyle, J.D. Photochemistry in Organic Synthesis, Royal Society of Chemistry, London, 1986; Schönberg, A. Preparative Organic Photochemistry, Springer, Berlin, 1968.
46. See DeLuca, L.; Giacomelli, G.; Porcu, G.; Taddei, M. Org. Lett. 2001, 3, 855.
47. For reviews, see Jackson, W.M.; Okabe, H. Adv. Photochem. 1986, 13, 1; Kresin, V.Z.; Lester, Jr., W.A. Adv. Photochem. 1986, 13, 95.
48. See Formosinho, S.J.; Arnaut, L.G. Adv. Photochem. 1991, 16, 67; Newton, R.F. in Coyle, J.D. Photochemistry in Organic Synthesis, Royal Society of Chemistry, London, 1986, pp. 39–60; Lee, E.K.C.; Lewis, R.S. Adv. Photochem. 1980, 12, 1; Coyle, J.D.; Carless, H.A.J. Chem. Soc. Rev. 1972, 1, 465; Bérces, T. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 5; Elsevier, NY, 1972, pp. 277–380; Turro, N.J.; Dalton, J.C.; Dawes, K.; Farrington, G.; Hautala, R.; Morton, D.; Niemczyk, M.; Shore, N. Acc. Chem. Res. 1972, 5, 92; Wagner, P.J. Top. Curr. Chem. 1976, 66, 1. Also see Weiss, D.S. Org. Photochem. 1981, 5, 347; Rubin, M.B. Top. Curr. Chem. 1985, 129, 1; 1969, 13, 251; Childs, R.F. Rev. Chem. Intermed. 1980, 3, 285. C=S compounds, see Coyle, J.D. Tetrahedron 1985, 41, 5393; Ramamurthy, V. Org. Photochem. 1985, 7, 231. C=N compounds, see Mariano, P.S. Org. Photochem. 1987, 9, 1.
49. See Adam, W.; Oppenländer, T. Angew. Chem. Int. Ed. 1986, 25, 661; Dürr, H.; Ruge, B. Top. Curr. Chem. 1976, 66, 53; Drewer, R.J. in Patai, S. The Chemistry of the Hydrazo, Azo, and Azoxy Groups, pt. 2, Wiley, NY, 1975, pp. 935–1015.
50. See Wagner, P.J. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 3, Academic Press, NY, 1980, pp. 381–444; Acc. Chem. Res. 1971, 4, 168. See Niu, Y.; Christophy, E.; Hossenlopp, J.M. J. Am. Chem. Soc. 1996, 118, 4188 for a new view of Norrish Type II elimination.
51. See Wilson, R.M. Org. Photochem. 1985, 7, 339, pp. 349–373; Scaiano, J.C.; Lissi, E.A.; Encina, M.V. Rev. Chem. Intermed. 1978, 2, 139. Also see Wagner, P.J. Acc. Chem. Res. 1989, 22, 83.
52. This mechanism was proposed by Yang, N.C.; Yang, D.H. J. Am. Chem. Soc. 1958, 80, 2913. The diradical intermediate has been trapped: Wagner, P.J.; Zepp, R.G. J. Am. Chem. Soc. 1972, 94, 287; Wagner, P.J.; Kelso, P.A.; Zepp, R.G. J. Am. Chem. Soc. 1972, 94, 7480; Adam, W.; Grabowski, S.; Wilson, R.M. Chem. Ber. 1989, 122, 561. See also, Caldwell, R.A.; Dhawan, S.N.; Moore, D.E. J. Am. Chem. Soc. 1985, 107, 5163.
53. See Casey, C.P.; Boggs, R.A. J. Am. Chem. Soc. 1972, 94, 6457.
54. For a review of the photochemistry of carboxylic acids and acid derivatives, see Givens, R.S.; Levi, N. in Patai, S. The Chemistry of Acid Derivatives, pt. 1; Wiley, NY, 1979, pp. 641–753.
55. See Hwu, J.R.; Chen, B.-L.; Huang, L.W.; Yang, T.-H. J. Chem. Soc. Chem. Commun. 1995, 299.
56. Wagner, P.J.; Zhou, B. J. Am. Chem. Soc. 1988, 110, 611.
57. See Morrison, H.A. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, pt. 1, Wiley, NY, 1969, pp. 165–213, 185–191; Kaupp, G. Angew. Chem. Int. Ed. 1980, 19, 243. See also, Yip, R.W.; Sharma, D.K. Res. Chem. Intermed. 1989, 11, 109.
58. See Sonnet, P.E. Tetrahedron 1980, 36, 557; Schulte-Frohlinde, D.; Görner, H. Pure Appl. Chem. 1979, 51, 279; Saltiel, J.; Charlton, J.L. in de Mayo, P. Rearrangements in Grund and Excited States, Vol. 3, Academic Press, NY, 1980, pp. 25–89; Saltiel, J.; Chang, D.W.L.; Megarity, E.D.; Rousseau, A.D.; Shannon, P.T.; Thomas, B.; Uriarte, A.K. Pure Appl. Chem. 1975, 41, 559; Saltiel, J.; D'Agostino, J.; Megarity, E.D.; Metts, L.; Neuberger, K.R.; Wrighton, M.; Zafiriou, O.C. Org. Photochem. 1979, 3, 1. Also see Leigh, W.J.; Srinivasan, R. Acc. Chem. Res. 1987, 20, 107; Steinmetz, M.G. Org. Photochem. 1987, 8, 67; Adam, W.; Oppenländer, T. Angew. Chem. Int. Ed. 1986, 25, 661; Johnson, R.P. Org. Photochem. 1985, 7, 75.
59. For a review of the photoisomerization of stilbenes, see Waldeck, D.H. Chem. Rev. 1991, 91, 415.
60. Kawamura, Y.; Takayama, R.; Nishiuchi, M.; Tsukayama, M. Tetrahedron Lett. 2000, 41, 8101.
61. Deyrup, J.A.; Betkouski, M. J. Org. Chem. 1972, 37, 3561.
62. Akabori, S.; Kumagai, T.; Habata, Y.; Sato, S. J. Chem. Soc. Perkin Trans. 1 1989, 1497; Shinkai, S.; Yoshioka, A.; Nakayama, H.; Manabe, O. J. Chem. Soc. Perkin Trans. 2 1990, 1905. For a review, see Shinkai, S.; Manabe, O. Top. Curr. Chem. 1984, 121, 67.
63. Haberfield, P. J. Am. Chem. Soc. 1987, 109, 6177.
64. Haberfield, P. J. Am. Chem. Soc. 1987, 109, 6178.
65. See Beer, P.D. Chem. Soc. Rev. 1989, 18, 409. For an example not involving a macrocycle, see Feringa, B.L.; Jager, W.F.; de Lange, B.; Meijer, E.W. J. Am. Chem. Soc. 1991, 113, 5468.
66. Sajimon, M.C.; Ramaiah, D.; Muneer, M.; Rath, N.P.; George, M.V. J. Photochem. Photobiol. A Chem. 2000, 136, 209.
67. Sajimon, M.C.; Ramaiah, D.; Thomas, K.G.; George, M.V. J. Org. Chem. 2001, 66, 3182.
68. Eaton, P.E. Acc. Chem. Res. 1968, 1, 50. For a review of the photochemistry of α,β-unsaturated ketones, see Schuster, D.I. in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 2, Wiley, NY, 1989, pp. 623–756.
69. For a review, see Calvert, J.G.; Pitts, Jr., J.N. Photochemistry, Wiley, NY, 1966, pp. 580–670.
70. Mason, T.J., Ed. Advances in Sonochemistry, JAI Press, NY, 1990–1994; Vols. 1–3, Price, G.J., Ed. Current Trends in Sonochemistry, Royal Society of Chemistry, Cambridge, UK, 1992; Suslick, K.S. Science 1990, 247, 1439; Suslick, K.S. Ultrasound: Its Chemical, Physical, and Biological Effects, VCH, NY, 1988; Young, F.R. Cavitation, McGraw-Hill, NY, 1989; Brennen, C.E. Cavitation and Bubble Dynamics, Oxford University Press, Oxford, UK, 1995; Anbar, M. Science 1968, 161, 1343. For a discussion of ultrasound in the chemistry of heterocycles, see Cella, R.; Stefani, H.A. Tetrahedron 2009, 65, 2619.
71. Apfel, R.E., in Edmonds, P. Methods in Experimental Physics, Academic Press, New York, 1981; Vol. 19; Makino, K.; Mossoba, M.M.; Riesz, P. J. Am. Chem. Soc. 1982, 104, 3537.
72. Stottlemeyer, T.R.; Apfel, R.E. J. Acoust. Soc. Am. 1997, 102, 1413.
73. Suslick, K.S.; Crum, L.A. In Sonochemistry and Sonoluminescence, Handbook of Acoustics, Crocker, M.J., Ed., Wiley, NY, 1998; Chapter 23; Leighton, T.G. The Acoustic Bubble, Academic Press, London, 1994; Chapter 4; Brennen, C.E. Cavitation and Bubble Dynamics, Oxford University Press, 1995, Chapters 1–4; Hua, I.; Hoffmann, M.R. Environ. Sci. Technol. 1997, 31, 2237.
74. Suslick, K.S.; Didenko, Y.T.; Fang, M.M.; Hyeon, T.; Kolbeck, K.J.; McNamara, III, W.B.; Mdleleni, M.M.; Wong, M. Philos. Trans. R. Soc. London A 1999, 357, 335. For problems of sonochemistry and cavitation, see Margulis, M.A. Ultrasonics Sonochemistry, 1994, 1, S87.
75. Suslick, K.S.; Hammerton, D.A.; Cline, Jr., R.E. J. Am. Chem. Soc. 1986, 108, 5641.
76. Didenko, Y.T.; McNamara, III, W.B.; Suslick, K.S. J. Am. Chem. Soc. 1999, 121, 5817.
77. Colussi, A. J.; Hoffmann, M.R. J. Phys. Chem. A. 1999, 103, 11336.
78. Colussi, A.J.; Weavers, L.K.; Hoffmann, M.R. J. Phys. Chem. A 1998, 102, 6927.
79. Segebarth, N.; Eulaerts, O.; Reisse, J.; Crum, L.A.; Matula, T.J. J. Phys. Chem. B. 2002, 106, 9181.
80. Dekerckheer, C.; Bartik, K.; Lecomte, J.-P.; Reisse, J. J. Phys. Chem. A. 1998, 102, 9177.
81. Elpiner, I. E. Ultrasound: Physical, Chemical, and Biological Effects, Consultants Bureau, NY, 1964.
82. Sollner, K. Chem. Rev. 1944, 34, 371.
83. Suslick, K.S.; Schubert, P.F.; Goodale, J.W. J. Am. Chem. Soc. 1981,103, 7342; Sehgal, C.; Yu, T.J.; Sutherland, R.G.; Verrall, R.E. J. Phys. Chem. 1982, 86, 2982; Sehgal, C.M.; Wang, S.Y. J. Am. Chem. Soc. 1981, 103, 6606.
84. Han, B.-H.; Boudjouk, P. J. Org. Chem. 1982, 47, 5030; Boudjouk, P.; Han, B.-H. Tetrahedron Lett. 1981, 22, 3813; Han, B.-H.; Boudjouk, P. J. Org. Chem. 1982, 47, 751; Boudjouk, P.; Han, B.-H.; Anderson, K.R. J. Am. Chem. Soc. 1982, 104, 4992; Boudjouk, P.; Han, B.-H. J. Catal. 1983, 79, 489; Racher, S.; Klein, P. J. Org. Chem. 1981, 46, 3558; Regen, S.L.; Singh, A. J. Org. Chem. 1982, 47, 1587; Kegelaers, Y.; Eulaerts, O.; Reisse, J.; Segebarth, N. Eur. J. Org. Chem. 2001, 3683.
85. Suslick, K.S.; Gawienowski, J.J.; Schubert, P.F.; Wang H.H. J. Phys. Chem. 1983, 87, 2299.
86. Mason, T.J. Practical Sonochemistry: User's Guide to Applications in Chemistry and Chemical Engineering, Ellis Horwood, West Sussex, 1991, pp. 43–46; Broeckaert, L.; Caulier, T.; Fabre, O.; Maerschalk, C.; Reisse, J.; Vandercammen, J.; Yang, D.H.; Lepoint, T.; Mullie, F. Current Trends in Sonochemistry, Price, G.J., Ed., Royal Society of Chemistry, Cambridge, 1992, p. 8; Mason, T.J.; Lorimer, J.P.; Bates, D.M.; Zhao, Y. Ultrasonics Sonochemistry 1994, 1, S91; Mason, T.J.; Lorimer, J.P.; Bates, D.M. Ultrasonics 1992, 30, 40.
87. Kimura, T.; Sakamoto, T.; Leveque, J.-M.; Sohmiya, H.; Fujita, M.; Ikeda, S.; Ando, T. Ultrasonics Sonochemistry 1996, 3, S157.
88. Synthetic Organic Sonochemistry Luche, J.-L. (Universite de Savoie, France), Plenum Press, NY. 1998; Luche, J.-L. Ultrasonics Sonochemistry, 1996, 3, S215.
89. Adewuyi, Y.G. Ind. Eng. Chem. Res. 2001, 40, 4681.
90. Boudjouk, P.; Sooriyakumaran, R.; Han, B.H. J. Org. Chem. 1986, 51, 2818, and Ref. 1 therein.
91. Regen, S.L.; Singh, A. J. Org. Chem. 1982, 47, 1587.
92. Suslick, K.S.; Goodale, J.W.; Schubert, P.F.; Wang, H.H. J. Am. Chem. Soc. 1983, 105, 5781.
93. Wang, M.-L.; Rajendran, V. J. Mol. Catalysis A: Chemical 2005, 244, 237.
94. Han, B.H.; Boudjouk, P. J. Org. Chem. 1982, 47, 5030.
95. Nebois, P.; Bouaziz, Z.; Fillion, H.; Moeini, L.; Piquer, Ma.J.A.; Luche, J.-L.; Riera, A.; Moyano, A.; Pericàs, M.A. Ultrasonics Sonochemistry 1996, 3, 7.
96. Me¡ iarová, M.; Kiripolsky, M.; Toma, Š. Ultrasonics Sonochemistry 2005, 12, 401.
97. Me¡ iarová, M.; Toma, S.; Magdolen, P. Ultrasonics Sonochemistry 2003, 10, 265.
98. Nakamura, E.; Machii, D.; Inubushi, T. J. Am. Chem. Soc. 1989, 111, 6849.
99. Vinatoru, M.; Stavrescua, R.; Milcoveanu, A.B.; Toma, M.; Mason, T.J. Ultrasonics Sonochemistry 2002, 9, 245.
100. Manzo, P.G.; Palacios, S.M.; Alonso, R.A. Tetrahedron Lett. 1994, 35, 677.
101. McNulty, J.; Steere, J.A.; Wolf, S. Tetrahedron Lett. 1998, 39, 8013.
102. Kimura, T.; Fujita, M.; Sohmiya, H.; Ando, T. Ultrasonics Sonochemistry 2002, 9, 205.
103. Montaña, A.M.; Grima, P.M. Tetrahedron Lett. 2001, 42, 7809.
104. Kardos, N.; Luche, J.-L. Carbohydrate Res. 2001, 332, 115.
105. Gedye, R. N.; Smith, F. E.; Westaway, K. C. Can. J. Chem. 1987, 66, 17.
106. Giguere, R.J.; Bray, T.; Duncan, S.M.; Majetich, G. Tetrahedron Lett. 1986, 27, 4945.
107. Taken from Horeis, G.; Pichler, S.; Stadler, A.; Gössler, W.; Kappe, C.O. Microwave-Assisted Organic Synthesis - Back to the Roots, Fifth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-5), 2001. (available at http://www.mdpi.org/ecsoc-5.htm).
108. Kappe, C. O. Angew. Chem. Int. Ed. 2004, 43, 6250.
109. Majetich, G.; Karen, W. in Kingston, H.M.; Haswell, S.J. Microwave-Enhanced Chemistry. Fundamentals, Sample Preparation, and Applications, American Chemical Society, Washington, DC, 1997, p. 772; Bose, A.K.; Manhas, M.S.; Banik, B.K.; Robb, E.W. Res. Chem. Intermed. 1994, 20, 1; Majetich, G.; Hicks, R. Res. Chem. Intermed. 1994, 20, 61; Strauss, C.R.; Trainor, R.W. Aust. J. Chem. 1995, 48, 1665; Caddick, S. Tetrahedron 1995, 51, 10403; Mingos, D M.P. Res. Chem. Intermed. 1994, 20, 85; Berlan, J. Rad. Phys. Chem. 1995, 45, 581; Fini, A.; Breccia, A. Pure Appl. Chem. 1999, 71, 573.
110. Kingston, H.M.; Haswell, S.J. Microwave-Enhanced Chemistry. Fundamentals, Sample Preparation, and Applications, American Chemical Society, 1997; Loupy, A. Microwaves in Organic Synthesis, Wiley-VCH, Weinheim, 2002; Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light, CEM Publishing, Matthews, NC, 2002; Lidström, P., Tierney, J.P. Microwave-Assisted Organic Synthesis, Blackwell Scientific, 2005; Kappe, C.O.; Stadler, A. Microwaves in Organic and Medicinal Chemistry, Wiley–VCH, Weinheim, 2005.
111. Galema, S.A. Chem. Soc. Rev. 1997, 26, 233.
112. Gabriel, C.; Gabriel, S.; Grant, E.H.; Halstead, B.S.J.; Mingos, D.M.P. Chem. Soc. Rev. 1998, 27, 213.
113. This frequencey is usually applied in domestic microwave ovens.
114. See Hoogenboom, R.; Wilms, T.F.A.; Erdmenger, T.; Schubert, U.S. Austr. J. Chem. 2009, 62, 236.
115. Moseley, J.D.; Lenden, P.; Thomson, A.D.; Gilday, J.P. Tetrahedron Lett. 2007, 48, 6084.
116. Caddick, S. Tetrahedron 1995, 51, 10403.
117. Varma, R. S. Green Chem. 1999, 43; Kidawi, M. Pure Appl. Chem. 2001, 73, 147; Varma, R. S. Pure Appl. Chem. 2001, 73, 193.
118. Langa, F.; de la Cruz, P.; de la Hoz, A.; Díaz-Ortiz, A.; Díez-Barra, E. Contemp. Org. Synth. 1997, 4, 373. Also see Schmink, J.R.; Leadbeater, N.E. Org. Biomol. Chem., 2009, 7, 3842.
119. See Kuhnert, N. Angew. Chem. Int. Ed. 2002, 41, 1863.
120. Laurent, R.; Laporterie, A.; Dubac, J.; Berlan, J.; Lefeuvre, S.; Audhuy, M. J. Org. Chem. 1992, 57, 7099 and references therein.
121. Raner, K.D.; Strauss, C.R.; Vyskoc, F.; Mokbel, L. J. Org. Chem. 1993, 58, 950, and references cited therein.
122. Cravotto, G.; Cintas, P. Chemistry: European J. 2007, 13, 1902.
123. See Larhed, M.; Moberg, C.; Hallberg, A. Acc. Chem. Res. 2002, 35, 717; Nüchter, M.; Ondruschka, B.; Bonrath, W.; Gum, A. Green Chem. 2004, 6, 128; Roberts, B.A.; Strauss, C.R. Acc. Chem. Res. 2005, 38, 653; Kuznetsov, D.V.; Raev, V.A.; Kuranov, G.L.; Arapov, O.V.; Kostikov, R.R. Russ. J. Org. Chem. 2005, 41, 1719. For a discussion of microwave-assisted organic synthesis in near critical water, see Kremsner, J.M.; Kappe, C.O. Eur. J. Org. Chem. 2005, 3672.
124. Mossé, S.; Alexakis, A. Org. Lett. 2006, 8, 3577.
125. Larhed, M.; Moberg, C.; Hallberg, A. Acc. Chem. Res. 2002, 35, 717; Olofsson, K.; Larhed, M. in Lidström, P.; Tierney, J.P. Microwave-Assisted Organic Synthesis, Blackwell, Oxford, 2004, Chap. 2., Andappan, M.M.S.; Nilsson, P.; Larhed, M. Mol. Diversity 2003, 7, 97.
126. Nuteberg, D.; Schaal, W.; Hamelink, E.; Vrang, L.; Larhed, M. J. Comb. Chem. 2003, 5, 456; Miller, S.P.; Morgan, J.B.; Nepveux, F.J.; Morken, J.P. Org. Lett. 2004, 6, 131; Kaval, N.; Bisztray, K.; Dehaen, W.; Kappe, C.O.; Van der Eycken, E. Mol. Diversity 2003, 7, 125; Gong, Y.; He, W. Heterocycles 2004, 62, 851; Leadbeater, N.E.; Marco, M. J. Org. Chem. 2003, 68, 888; Bai, L.; Wang, J.-X.; Zhang, Y. Green Chem. 2003, 5, 615; Leadbeater, N.E.; Marco, M. J. Org. Chem. 2003, 68, 5660.
127. Kaval, N.; Bisztray, K.; Dehaen, W.; Kappe, C.O.; Van der Eycken, E. Mol. Diversity 2003, 7, 125; Gong, Y.; He, W. Heterocycles 2004, 62, 851; Leadbeater, N.E.; Marco, M.; Tominack, B.J. Org. Lett. 2003, 5, 3919; Appukkuttan, P.; Dehaen, W.; Van der Eycken, E. Eur. J. Org. Chem. 2003, 4713.
128. Wu, Y.-J.; He, H.; L'Heureux, A. Tetrahedron Lett. 2003, 44, 4217; Lange, J.H.M.; Hofmeyer, L.J.F.; Hout, F.A.S.; Osnabrug, S.J.M.; Verveer, P.C.; Kruse, C.G.; Feenstra, R.W. Tetrahedron Lett. 2002, 43, 1101.
129. See Van der Eycken, E.; Appukkuttan, P.; De Borggraeve, W.; Dehaen, W.; Dallinger, D.; Kappe, C.O. J. Org. Chem. 2002, 67, 7904; Pinto, D.C.G.A.; Silva, A.M.S.; Almeida, L.M.P.M.; Carrillo, J.R.; D'az-Ortiz, A.; de la Hoz, A.; Cavaleiro, J.A.S. Synlett 2003, 1415.
130. Dupau, P.; Epple, R.; Thomas, A.A.; Fokin, V.V.; Sharpless, K.B. Adv. Synth. Catal. 2002, 344, 421.
131. Raheem, I.T.; Goodman, S.N.; Jacobsen, E.N. J. Am. Chem. Soc. 2004, 126, 706.