Chapter 17
Eliminations

A so-called β-elimination reaction occurs when two groups are lost from adjacent atoms so that a new double1 (or triple) bond is formed. In general, the atom bearing a leaving group is the α, and the other the β atom. In an α-elimination, both groups are lost from the same atom to give a carbene (or a nitrene):

In a γ-elimination, a three-membered ring is formed:

Some of these processes were discussed in Chapter 10. Another type of elimination involves the expulsion of a fragment from within a chain or ring (X–Y–Z → X–Z + Y). Such reactions are called extrusion reactions. This chapter discusses β-elimination and extrusion reactions (see Sec. 2.F.vi); however, β-elimination in which both X and W are hydrogen atoms are oxidation reactions. They are treated in Chapter 19.
17.A. Mechanisms And Orientation
β-Elimination reactions may be divided into two types: one type taking place largely in solution, the other (pyrolytic eliminations) mostly in the gas phase. In the reactions, one group leaves with its electrons and the other without (i.e., it is pulled off), the latter most often being hydrogen. In these cases, the former leaves as the leaving group or nucleofuge. For pyrolytic eliminations, there are two principal mechanisms, one pericyclic and the other a free radical pathway. A few photochemical eliminations are also known (the most important is Norrish-type II cleavage of ketones, Sec. 7.A.vii), but these are not generally of synthetic importance2 and will not be discussed further. In most β-eliminations, the new bonds are C=C or C C. The discussion of mechanisms is largely confined to these cases.3 Mechanisms in solution (E2, E1)4 and E1cB are discussed first. While standard methods are used to examine elimination reactions, new techniques (e.g., the velocity map ion imaging technique) have been used to study ultrafast elimination reactions.5
C. The discussion of mechanisms is largely confined to these cases.3 Mechanisms in solution (E2, E1)4 and E1cB are discussed first. While standard methods are used to examine elimination reactions, new techniques (e.g., the velocity map ion imaging technique) have been used to study ultrafast elimination reactions.5
17.A.i. The E2 Mechanism
In the E2 mechanism (elimination, bimolecular), the proton on the β carbon is pulled off by a base, leading to near-simultaneous expulsion of the leaving group (nucleofuge):

The mechanism takes place in one step and is kinetically second order: first order in substrate and first order in base. An ab initio study has produced a model for the E2 transition state geometry.6 The IUPAC designation is AxHDHDN, or more generally (to include cases where the electrofuge is not hydrogen), AnDEDN. It often competes with the SN2 mechanism (Sec. 10.A.i). With respect to the substrate, the difference between the two pathways is whether the species with the unshared pair attacks the carbon (and thus acts as a nucleophile) or the hydrogen (and thus acts as a base). As in the case of the SN2 mechanism, the leaving group may be positive or neutral and the base may be negatively charged or neutral.
Evidence for the existence of the E2 mechanism includes (1) the reaction displays the proper second-order kinetics; (2) when the hydrogen is replaced by deuterium in second-order eliminations, there is an isotope effect of from 3 to 8, consistent with breaking of this bond in the rate-determining step.7 However, neither of these results alone could prove an E2 mechanism, since both are compatible with other mechanisms also (e.g., see E1cB, Sec. 17.A.iii). The most compelling evidence for the E2 mechanism is found in stereochemical studies.8
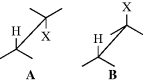
As will be illustrated in the examples below, the E2 mechanism is stereospecific: The five atoms involved (including the base) in the transition state must be in one plane. There are two ways for this to happen. The H and X may be trans to one another (A) with a dihedral angle of 180°, or they may be cis (B) with a dihedral angle of 0°.9 Conformation A is called antiperiplanar, and this type of elimination, in which H and X depart in opposite directions, is called anti elimination. Conformation B is syn periplanar, and this type of elimination, with H and X leaving in the same direction, is called syn elimination. Many examples of both kinds have been discovered. In the absence of special effects (discussed below), anti elimination is usually greatly favored over syn elimination, probably because A is a staggered conformation (Sec. 4.O.i) and the molecule requires less energy to reach this transition state than it does to reach the eclipsed transition state B. Solvent effects play an important role in the conformational preference. A few of the many known examples of predominant or exclusive anti elimination follow:
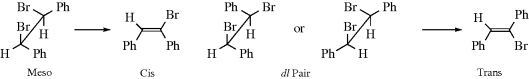
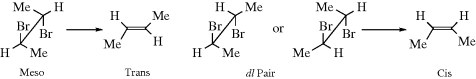
CCO2H, but the trans isomer reacts ~ 50 times faster than the cis compound.14Some examples of syn elimination have been found in molecules where H and X could not achieve an antiperiplanar conformation.
The examples given so far illustrate two points. (1) Anti elimination requires a dihedral angle of 180°. When this angle cannot be achieved, anti elimination is greatly slowed or prevented entirely. (2) For the simple systems so far discussed, syn elimination is not found to any significant extent unless anti elimination is greatly diminished by failure to achieve the 180° angle.
The concept of vinylogy was introduced in Section 6.B and in Reaction 10-68, category 4. Using this concept, a 1,2-elimination can be extended to give a 1,x-elimination when π bonds are incorporated between the carbon bearing the acidic proton and the leaving group (e.g., X-C-C=C-C, X-C-C=C-C=C-C or X-C-CC-C).18
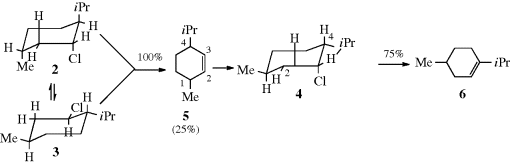
As noted in Section 4.Q.ii, six-membered rings are the only ones among rings of 4–13 members in which strain-free antiperiplanar conformations can be achieved. It is not surprising, therefore, that syn elimination is least common in six-membered rings. Cycloalkyltrimethylammonium hydroxides were subjected to elimination (Reaction 17-7) and the following percentages of syn elimination were found with each ring size: four-membered, 90%; five-membered, 46%; six-membered, 4% seven-membered, 31–37%.19 Note that the NMe3+ group has a greater tendency to syn elimination than do other common leaving groups (e.g., OTs, Cl, and Br).
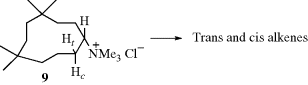
Other examples of syn elimination have been found in medium-ring compounds, where both cis and trans alkenes are possible (Sec. 4.K.i). As an illustration, elimination of 1,1,4,4-tetramethyl-7-cyclodecyltrimethylammonium chloride (9)20 gave mostly trans- but also some cis-tetramethylcyclodecenes as products. Note that trans-cyclodecenes, although stable, are less stable than the cis isomers. In order to determine the stereochemistry of the reaction, the elimination was repeated, this time using deuterated substrates. When 9 was deuterated in the trans position (Ht = D), there was a substantial isotope effect in the formation of both cis and trans alkenes, but when 9 was deuterated in the cis position (Hc = D), there was no isotope effect in the formation of either alkene. Since an isotope effect is expected for an E2 mechanism,21 these results indicated that only the trans hydrogen (Ht) was lost, whether the product was the cis or the trans isomer.22 This in turn means that the cis isomer must have been formed by anti elimination and the trans isomer by syn elimination. Anti elimination could take place from approximately the conformation shown, but for syn elimination the molecule must twist into a conformation in which the C–Ht and C–NMe3+ bonds are syn periplanar. Other types of evidence have also demonstrated this remarkable result, called the syn–anti dichotomy.23 The fact that syn elimination in this case predominates over anti (as indicated by the formation of trans-isomer in greater amounts than cis) has been explained by conformational factors.24 The syn–anti dichotomy has also been found in other medium-ring systems (8–12 membered),25 although the effect is greatest for 10-membered rings. With leaving groups,26 the extent of this behavior decreases in the order +NMe3 > OTs > Br > Cl, which parallels steric requirements. When the leaving group is uncharged, syn elimination is favored by strong bases and by weakly ionizing solvents.27
Syn elimination and the syn–anti dichotomy have also been found in open-chain systems, although to a lesser extent than in medium-ring compounds. For example, in the conversion of 3-hexyl-4-d-trimethylammonium ion to 3-hexene with potassium sec-butoxide, ~ 67% of the reaction followed the syn-anti dichotomy.28 In general, syn elimination in open-chain systems is only important in cases where certain types of steric effect are present. One such type is compounds in which substituents are found on both the β′ and the γ carbons (the unprimed letter refers to the branch in which the elimination takes place). The factors that cause these results are not completely understood, but the following conformational effects have been proposed as a partial explanation.29 The two anti- and two syn-periplanar conformations are, for a quaternary ammonium salt:
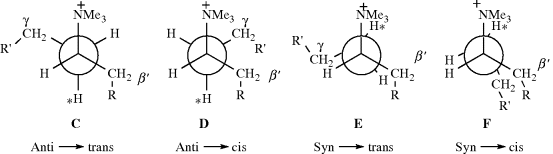
In order for an E2 mechanism to take place, a base must approach the proton marked ∗. In C, this proton is shielded on both sides by R and R′. In D, the shielding is on only one side. Therefore, when anti elimination does take place in such systems, it should give more cis product than trans. Also, when the normal anti elimination pathway is hindered sufficiently to allow the syn pathway to compete, the anti → trans route should be diminished more than the anti → cis route. When syn elimination begins to appear, it seems clear that E, which is less eclipsed than F, should be the favored pathway and syn elimination should generally give the trans-isomer. In general, deviations from the synanti dichotomy are greater on the trans side than on the cis. Thus, trans-alkenes are formed partly or mainly by syn elimination, but cis-alkenes are formed entirely by anti elimination. Predominant syn elimination has also been found in compounds of the form R1R2CHCHDNMe3+, where R1 and R2 are both bulky.30 In this case, the conformation leading to syn elimination (H) is also less strained than G, which gives anti elimination. The G compound has three bulky groups (including NMe3+) in the gauche position to each other.
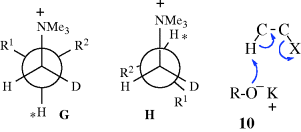
It was mentioned above that weakly ionizing solvents promote syn elimination when the leaving group is uncharged. This is probably caused by ion pairing, which is greatest in nonpolar solvents.31 Ion pairing can cause syn elimination with an uncharged leaving group by means of the transition state shown in 10. This effect was graphically illustrated by elimination from 1,1,4,4-tetramethyl-7-cyclodecyl bromide.32 The ratio of syn to anti elimination when this compound was treated with t-BuOK in the nonpolar benzene was 55.0. When the crown ether dicyclohexano-18-crown-6 was added (this compound selectively removes K+ from the t-BuO− K++ ion pair and thus leaves t-BuO− as a free ion), the syn/anti ratio decreased to 0.12. Large decreases in the syn/anti ratio on addition of the crown ether were also found with the corresponding tosylate and with other nonpolar solvents.33 However, with positively charged leaving groups the effect is reversed. Here, ion pairing increases the amount of anti elimination.34 In this case, a relatively free base (e.g., PhO−) can be attracted to the leaving group (see 11), putting it in a favorable position for attack on the syn β hydrogen, while ion pairing would reduce this attraction.
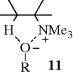
It can be concluded that anti elimination is generally favored in the E2 mechanism, but that steric (inability to form the antiperiplanar transition state), conformational, ion pairing, and other factors cause syn elimination to intervene (and even predominate) in some cases.
17.A.ii. The E1 Mechanism
The E1 mechanism is a two-step process in which the rate-determining step is ionization of the substrate to give a carbocation that rapidly loses a β proton to a base, usually the solvent:
The IUPAC designation is DN + DE (or DN + DH). This mechanism normally operates without an added base. Just as the E2 mechanism competes with the SN2,35 so the E1 mechanism competes with the SN1. In fact, the first step of the E1 is exactly the same as that of the SN1 mechanism. The second step differs in that the solvent pulls a proton from the β carbon of the carbocation rather than attacking it at the positively charged carbon, as in the SN1 process. In a pure E1 reaction (without ion pairs, etc.), the product should be completely nonstereospecific, since bond rotation is possible in the carbocation before deprotonation.
Some of the evidence for the E1 mechanism is as follows:
17.A.iii. The E1cB Mechanism40
In the E1 mechanism, X leaves first and then H is removed. In the E2 mechanism, H is removed, which triggers the expulsion of X. There is a third possibility: The H is removed first to form 12, and then X leaves. This reaction is a two-step process, called the E1cB mechanism,41 or the carbanion mechanism, since the intermediate is a carbanion, (12). The
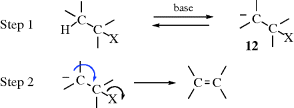
name E1cB comes from the fact that it is the conjugate base of the substrate that is giving up the leaving group (see the SN1cB mechanism, Sec. 10.G.iii, category 1). The IUPAC designation is AnDE + DN or AxhDH + DN (see Sec. 9.F). Three limiting cases can be distinguished: (1) The carbanion returns to starting material faster than it forms product: step 1 is reversible; step 2 is slow. (2) Step 1 is the slow step, and formation of product is faster than return of the carbanion to starting material. In this case, step 1 is essentially irreversible. (3) Step 1 is rapid, and the carbanion goes slowly to product. This case occurs only with the most stable carbanions. Here, too, step 1 is essentially irreversible. These cases have been given the designations: (1) (E1cB)R, (2) (E1cB)I (or E1cBirr), and (3) (E1)anion. Their characteristics are listed in Table 17.1.42 Investigations of the reaction order are generally not very useful (except for case 3, which is first order), because cases 1 and 2 are second order and thus difficult or impossible to distinguish from the E2 mechanism by this procedure.43 The greatest likelihood of finding the E1cB mechanism is expected in substrates that have (a) a poor nucleofuge and (b) an acidic hydrogen. In addition, most investigations have concerned such substrates. The following is some of the evidence in support of the E1cB mechanism:
Table 17.1 Kinetic Predictions for Base-Induced β-Eliminationsa

CCl. When the reaction was stopped before completion, there was deuterium in the recovered alkene.44 A similar result was found for pentahaloethanes.45 These substrates are relatively acidic. In both cases, the electron-withdrawing halogens increase the acidity of the hydrogen, and in the case of trichloroethylene there is the additional factor that a hydrogen on an sp2 carbon is more acidic than one on an sp3 carbon (Sec. 8.F, category 7). Thus, the E1cB mechanism is more likely to be found in eliminations yielding triple bonds than in those giving double bonds. Another likely place for the E1cB mechanism should be in reaction of a substrate like PhCH2CH2Br, since the carbanion is stabilized by resonance with the phenyl group. Nevertheless, no deuterium exchange was found here.46 If this type of evidence is a guide, then it may be inferred that the (E1cB)R mechanism is quite rare, at least for eliminations with common leaving groups (e.g., Br, Cl, or OTs), which yield C=C double bonds.
N bonds, the initial step is loss of a positive group (normally a proton) from the oxygen or nitrogen. These may also be regarded as E1cB processes.
17.A.iv. The E1–E2–E1cB Spectrum
In the three mechanisms so far considered, the similarities are greater than the differences. In each case, there is a leaving group that comes off with its pair of electrons and another group (usually hydrogen) that comes off without them. The only difference is in the order of the steps. It is now generally accepted that there is a spectrum of mechanisms ranging from one extreme, in which the leaving group departs well before the proton (pure E1), to the other extreme, in which the proton is removed first and then, after some time, the leaving group follows (pure E1cB). The pure E2 case would be somewhere in the middle, with both groups leaving simultaneously. However, most E2 reactions are not exactly in the middle, but somewhere to one side or the other. For example, the nucleofuge might depart just before the proton. This case may be described as an E2 reaction with a small amount of E1 character. The concept can be expressed by the question: In the transition state, which bond (C–H or C–X) has undergone more cleavage?61
Note that in both E1 and E2 reactions, removal of the hydrogen atom is an acid–base reaction, requiring a base. A stronger base is required for the E2, and a weaker base for E1. Further, the E1 reaction requires a solvent that facilitates ionization to a carbocation (e.g., aqueous media), whereas the E2 reaction is usually done in a protic solvent (e.g., an alcohol).
One way to determine just where a given reaction stands on the E1–E2–E1cB spectrum is to study isotope effects, which ought to tell something about the behavior of bonds in the transition state.62 For example, CH3CH2NMe3+ showed a nitrogen isotope effect (k14/k15) of 1.017, while PhCH2CH2NMe3+ gave a corresponding value of 1.009.63 It would be expected that the phenyl group would move the reaction toward the E1cB side of the line, which means that for this compound the C–N bond is not as greatly broken in the transition state as it is for the unsubstituted one. The isotope effect bears this out, for it shows that in the phenyl compound, the mass of the nitrogen has less effect on the reaction rate than it does in the unsubstituted compound. Similar results have been obtained with SR2+ leaving groups by the use of 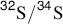 isotope effects64 and with Cl (
isotope effects64 and with Cl (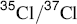 ).65 The position of reactions along the spectrum has also been studied from the other side of the newly forming double bond by the use of H/D and H/T isotope effects,66 although interpretation of these results is clouded by the fact that β hydrogen isotope effects are expected to change smoothly from small to large to small again as the degree of transfer of the β hydrogen from the β carbon to the base increases67 (in Sec. 6.B, it was noted that isotope effects are greatest when the proton is half-transferred in the transition state), by the possibility of secondary isotope effects (e.g., the presence of a β deuterium or tritium may cause the leaving group to depart more slowly), and by the possibility of tunneling.68 Other isotope-effect studies have involved labeled α or β carbon, labeled α hydrogen, or labeled base.58
).65 The position of reactions along the spectrum has also been studied from the other side of the newly forming double bond by the use of H/D and H/T isotope effects,66 although interpretation of these results is clouded by the fact that β hydrogen isotope effects are expected to change smoothly from small to large to small again as the degree of transfer of the β hydrogen from the β carbon to the base increases67 (in Sec. 6.B, it was noted that isotope effects are greatest when the proton is half-transferred in the transition state), by the possibility of secondary isotope effects (e.g., the presence of a β deuterium or tritium may cause the leaving group to depart more slowly), and by the possibility of tunneling.68 Other isotope-effect studies have involved labeled α or β carbon, labeled α hydrogen, or labeled base.58
Another way to study the position of a given reaction on the spectrum involves the use of β aryl substitution. Since a positive Hammet ρ value is an indication of a negatively charged transition state, the ρ value for substituted β aryl groups should increase as a reaction moves from E1-to-E1cB-like along the spectrum. This has been shown to be the case in a number of studies;69 for example, ρ values of ArCH2CH2X increase as the leaving-group ability of X decreases. A typical set of ρ values was X = I, 2.07; Br, 2.14; Cl, 2.61; SMe2+, 2.75; F, 3.12.70 As seen previously, decreasing leaving-group ability correlates with increasing E1cB character.
Still another method measures volumes of activation.71 These are negative for E2 and positive for E1cB mechanisms. Measurement of the activation volume therefore provides a continuous scale for deciding just where a reaction lies on the spectrum.
17.A.v. The E2C Mechanism72
Certain alkyl halides and tosylates undergo E2 eliminations faster when treated with such weak bases as Cl− in polar aprotic solvents or PhS− than with the usual E2 strong bases (e.g., RO− in ROH).73 In order to explain these results, it was proposed74 that there is a spectrum75 of E2 transition states in which the base can interact in the transition state with the α carbon, as well as with the β hydrogen. At one end of this spectrum is a mechanism (called E2C) in which, in the transition state, the base interacts mainly with the carbon. The E2C mechanism is characterized by strong nucleophiles that are weak bases. At the other extreme is the normal E2 mechanism, here called E2H to distinguish it from E2C, characterized by strong bases. Transition state 18 represents a transition state between these extremes. Additional evidence76 for the E2C mechanism is derived from Br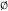 nsted equation considerations (Sec. 8.D), from substrate effects, from isotope effects, and from the effects of solvents on rates.
nsted equation considerations (Sec. 8.D), from substrate effects, from isotope effects, and from the effects of solvents on rates.
However, the E2C mechanism has been criticized, and it has been contended that all the experimental results can be explained by the normal E2 mechanism.77 McLennan and Lim78 suggested that the transition state is that shown as 19. An ion-pair mechanism has also been proposed.79 Although the actual mechanisms involved may be a matter of controversy, there is no doubt that a class of elimination reactions exists that is characterized by second-order attack by weak bases.80 These reactions also have the following general characteristics:81 (1) they are favored by good leaving groups; (2) they are favored by polar aprotic solvents; (3) the reactivity order is tertiary > secondary > primary, the opposite of the normal E2 order (Sec. 17.D.i); (4) the elimination is always anti (syn elimination is not found), but in cyclohexyl systems, a diequatorial anti elimination is about as favorable as a diaxial anti elimination (unlike the normal E2 reaction, Sec. 17.A.i, categories 2,3); (5) they follow Zaitsev's rule (see below), where this does not conflict with the requirement for anti elimination.
17.B. Regiochemistry of the Double Bond
With some substrates, a β hydrogen is present on only one carbon and (barring rearrangements) there is no doubt as to the identity of the product. For example, PhCH2CH2Br can give only PhCH=CH2. However, in many other cases two or three alkenyl products are possible. In the simplest such case, a sec-butyl compound can give either 1- or 2-butene. There are a number of rules that enable a prediction, in many instances, of which product will predominantly form.82
17.C. Stereochemistry of the Double Bond
When elimination takes place on a compound of the form CH3–CABX or CHAB–CGGX, the new alkene does not have cis–trans isomerism, but for compounds of the form CHEG–CABX (E and G not H) (24) and CH2E–CABX (25), cis and trans isomers are possible. When the anti E2 mechanism is in operation, 24 gives the isomer
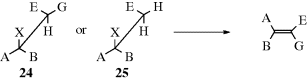
arising from trans orientation of X and H. As seen previously (Sec. 17.A.i), an erythro compound gives the cis alkene and a threo compound gives the trans. For 25, two conformations are possible for the transition state; these lead to different isomers and often both are obtained. However, the one that predominates is often determined by an eclipsing effect.103 For example, Zaitsev elimination from 2-bromopentane can occur as follows: In conformation I, the ethyl group is between Br and Me, while in J it is between Br and H. This means that J is more stable, and most of the elimination should occur from this conformation. This is indeed what happens, and 51% of the trans isomer is formed (with KOEt) compared to 18% of the cis (the rest is the Hofmann product).104 These effects become larger with increasing size of groups A, B, and E.
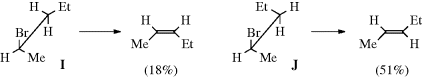
However, eclipsing effects are not the only factors that affect the cis/trans ratio in anti E2 eliminations. Other factors are the nature of the leaving group, the base, the solvent, and the substrate. Not all of these effects are completely understood.105
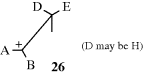
For E1 eliminations, if there is a free carbocation (26), it is free to rotate, and no matter the geometry of the original compound, the more stable situation is the one where the larger of the D–E pair is opposite the smaller of the A–B pair and the corresponding alkene should form. If the carbocation is not completely free, then to that extent, E2-type products are formed. Similar considerations apply in E1cB eliminations.106
17.D. Reactivity
In this section, we examine the effects of changes in the substrate, base, leaving group, and medium on (1) overall reactivity, (2) E1 versus E2 versus E1cB,107 and (3) elimination versus substitution.
17.D.i. Effect of Substrate Structure
Table 17.2 The Effect of α and β Branching on the Rate of E2 Elimination and the Amount of Alkene Formeda
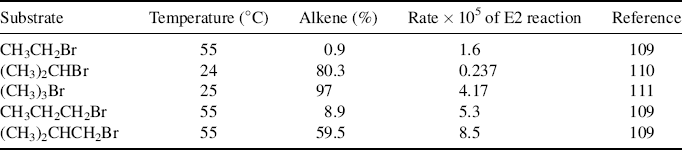
aThe reactions were between the alkyl bromide and 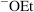 . The rate for isopropyl bromide was actually greater than that for ethyl bromide, if the temperature difference is considered. Neopentyl bromide, the next compound in the β-branching series, cannot be compared because it has no β hydrogen and cannot give an elimination product without rearrangement.
. The rate for isopropyl bromide was actually greater than that for ethyl bromide, if the temperature difference is considered. Neopentyl bromide, the next compound in the β-branching series, cannot be compared because it has no β hydrogen and cannot give an elimination product without rearrangement.
17.D.ii. Effect of the Attacking Base
17.D.iii. Effect of the Leaving Group
17.D.iv. Effect of the Medium
17.E. Mechanisms and Orientation in Pyrolytic Eliminations
17.E.i. Mechanisms128
Several types of compound undergo elimination on heating, with no other reagent present. Reactions of this type are often run in the gas phase. The mechanisms are obviously different from those already discussed, since all those require an external base, which may be the solvent, in one of the steps, and there is no external base or solvent present in pyrolytic elimination. Two mechanisms have been found to operate. One involves a cyclic transition state, which may be four, five, or six membered. Examples of each size are
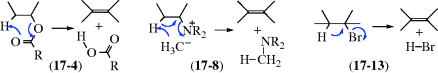
In this mechanism, the two groups leave at about the same time and bond to each other as they are doing so. The designation is Ei in the Ingold terminology and cyclo-DEDNAn in the IUPAC system. The elimination must be syn and, for the four- and five-membered transition states, the four or five atoms making up the ring must be coplanar. Coplanarity is not required for the six-membered transition state, since there is room for the outside atoms when the leaving atoms are staggered.
As in the E2 mechanism, it is not necessary that the C–H and C–X bond be broken simultaneously in the transition state. In fact, there is also a spectrum of mechanisms here, ranging from a mechanism in which C–X bond breaking is a good deal more advanced than C–H bond breaking to one in which the extent of bond breaking is virtually identical for the two bonds. Evidence for the existence of the Ei mechanism includes:
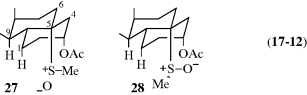
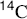 isotope effects for the Cope elimination (17-9) show that both the C–H and C–N bonds have been extensively broken in the transition state.134
isotope effects for the Cope elimination (17-9) show that both the C–H and C–N bonds have been extensively broken in the transition state.134Where a pyrolytic elimination lies on the mechanistic spectrum seems to depend mostly on the leaving group. When this is halogen, all available evidence suggests that in the transition state the C–X bond is cleaved to a much greater extent than the C–H bond; that is, there is a considerable amount of carbocation character in the transition state. This observation is in accord with the fact that a completely nonpolar four-membered cyclic transition state violates the Woodward–Hoffmann rules (see the similar case of Reaction 15-63). Evidence for the carbocation-like character of the transition state when halide is the leaving group is that relative rates are in the order I > Br > Cl135 (see Sec. 10.G.iii), and that the effects of substituents on reaction rates are in accord with such a transition state.136 Rate ratios for pyrolysis of some alkyl bromides at 320 °C were ethyl bromide, 1; isopropyl bromide, 280; tert-butyl bromide, 78,000. Also, α-phenylethyl bromide had about the same rate as tert-butyl bromide. On the other hand, β-phenylethyl bromide was only slightly faster than ethyl bromide.137 This result indicates that C–Br cleavage was much more important in the transition state than C–H cleavage, since the incipient carbocation was stabilized by a alkyl and α-aryl substitution, while there was no incipient carbanion to be stabilized by β aryl substitution. These substituent effects, as well as those for other groups, are very similar to the effects found for the SN1 mechanism and thus in very good accord with a carbocation-like transition state.
For carboxylic esters, the rate ratios were much smaller,138 although still in the same order, so that this reaction is closer to a pure Ei mechanism, although the transition state still has some carbocationic character. Other evidence for a greater initial C–O cleavage with carboxylic esters is that a series of 1-arylethyl acetates followed σ+ rather than σ, showing carbocationic character at the 1 position.139 The extent of E1 character in the transition state increases in the following order of ester types: acetate < phenylacetate < benzoate < carbamate < carbonate.140 Cleavage of xanthates (Reaction 17-5), cleavage of sulfoxides (Reaction 17-12), the Cope Reaction (17-9), and Reaction 17-8 are probably very close to straight Ei mechanisms.141
The second type of pyrolysis mechanism is completely different and involves free radicals. Initiation occurs by pyrolytic homolytic cleavage. The remaining steps may vary, and a few are shown below. Free radical mechanisms are mostly found in pyrolyses of polyhalides and of primary monohalides,142 although they also have been postulated in pyrolysis of certain carboxylic esters.143 β-Elimination of tosyl radicals is known.144 Much less is known about these mechanisms and we will not consider them further. Free radical eliminations in solution are also known, but are rare.145
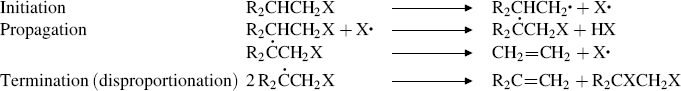
17.E.ii. Orientation in Pyrolytic Eliminations
As in the E1–E2–E1cB mechanistic spectrum, Bredt's rule applies; and if a double bond is present, a conjugated system will be preferred, if sterically possible. Apart from these considerations, the following statements can be made for Ei eliminations:
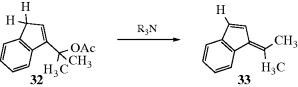
17.E.iii. 1,4-Conjugate Eliminations151
1,4-eliminations of the type H-C-C=CC-X → C=C-C=C are much rarer than conjugate additions (Chapter 15), but some examples are known.152 One such is the conversion of 32 to 33.153
17.F. Reactions
Reactions in which a C=C or a CC bond is formed will be considered first. From a synthetic point of view, the most important reactions for the formation of double bonds are 17-1 (usually by an E1 mechanism), 17-7, 17-13, and 17-22 (usually by an E2 mechanism), and 17-4, 17-5, and 17-9 (usually by an Ei mechanism). The only synthetically important method for the formation of triple bonds is 17-13.154 In the second section, reactions in which CN bonds and C=N bonds are formed will be considered, and then eliminations that give C=O bonds and diazoalkanes. Finally, extrusion reactions will be discussed.
17.F.i. Reactions in which C=C and CC Bonds are Formed
A. Reactions in which Hydrogen is Removed from One Side
In Reactions 17-1–17-6, the other leaving atom is oxygen. In Reactions 17-7–17-11, it is nitrogen. For reactions in which hydrogen is removed from both sides, see 19-1–19-6.
17-1 Dehydration of Alcohols
Hydro-hydroxy-elimination
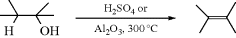
Dehydration of alcohols can be accomplished in several ways. Both H2SO4 and H3PO4 are common reagents, but formation of an intermediate carbocation can lead to rearrangement products and to ether formation (Reaction 10-12). If the alcohol is volatile, vapor-phase elimination over Al2O3 is an excellent method since side reactions are greatly reduced. This method has even been applied to such high-molecular-weight alcohols as 1-dodecanol.155 Other metallic oxides (e.g., Cr2O3, TiO2, WO3) have been used, as have been sulfides, other metallic salts, and zeolites. The presence of an electron-withdrawing group usually facilitates elimination of water, as in the aldol condensation (Reaction 16-34). Similarly, 2-nitroalcohols (products of the Henry reaction, 16-37) give conjugated nitro compounds when heated with zeolite Y–Y.156 Treating a 4-hydroxy lactam with DMAP and Boc anhydride leads to the conjugated lactam.157 Elimination of serine derivatives to α-alkylidene amino acid derivatives was accomplished with (EtO)2POCl.158 Another method of avoiding side reactions is the conversion of alcohols to esters, followed by pyrolysis (17-4–17-6). The ease of dehydration increases with α branching, and tertiary alcohols are dehydrated so easily with only a trace of acid that it sometimes happens even when the investigator desires otherwise. Indeed, the initial alcohol products of many base-catalyzed condensations dehydrate spontaneously after an acid workup (Chapter 16) because the new double bond can be in conjugation with one already there.
Many other dehydrating agents159 have been used on occasion: P2O5, I2, and PPh3–I2,160 BF3–etherate, DMSO, SiO2–Cl/Me3SiCl,161 KHSO4 anhydrous CuSO4, and phthalic anhydride, among others. Secondary and tertiary alcohols can also be dehydrated, without rearrangements, simply on refluxing in HMPA.162 With nearly all reagents, dehydration follows Zaitsev's rule. An exception involves the passage of hot alcohol vapors over thorium oxide at 350–450 °C, under which conditions Hofmann's rule is followed.163
Transition metals can induce the dehydration of certain alcohols. β-Hydroxy ketones are converted to conjugated ketones by treatment with CeCl3 and NaI.164 In the presence of a Pd complex, alkyl cyclopropanols undergo a dehydration reaction to give a conjugated ketone.165 A δ-hydroxy-α,β-unsaturated aldehyde was converted to a dienyl aldehyde with a Hf catalyst.166 β-Hydroxy esters are converted to conjugated esters when treated with 2 molar equivalents of SmI2.167 The reaction of a β-hydroxy nitrile with MeMgCl168 or with MgO169 leads to a conjugated nitrile. In another variation of the dehydration reaction, vicinal bromohydrins are converted to alkenes upon treatment with In, InCl3, and a Pd catalyst.170 Chlorohydrins react similarly when treated with Sm, and then diiodomethane.171
Carboxylic acids can be dehydrated by pyrolysis to give a ketene: RCH2CO2H → RCH=C=O. Ketene itself is commercially prepared in this manner. Carboxylic acids have been converted to ketenes by treatment with certain reagents, including TsCl,172 dicyclohexylcarbodiimide,173 and 1-methyl-2-chloropyridinium iodide (Mukaiyama's reagent).174 Analogously, amides can be dehydrated with P2O5, pyridine, and Al2O3 to give ketenimines:175
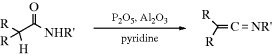
There is no way in which dehydration of alcohols can be used to prepare triple bonds: gem-diols and vinylic alcohols are not normally stable compounds and vic-diols176 give either conjugated dienes or lose only 1 equiv of water to give an aldehyde or ketone. Dienes can be prepared, however, by heating alkynyl alcohols with triphenylphosphine.177
When proton acids catalyze alcohol dehydration, the mechanism is E1.178 The principal process involves conversion of ROH to ROH2+ and cleavage of the latter to R+ and H2O, although with some acids a secondary process probably involves conversion of the alcohol to an inorganic ester and ionization of that ester (illustrated for H2SO4):
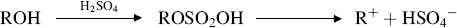
Note that these mechanisms are the reverse of those involved in the acid-catalyzed hydration of double bonds (Reaction 15-3), in accord with the principle of microscopic reversibility. With anhydrides (e.g., P2O5, phthalic anhydride), as well as with some other reagents (e.g., HMPA),179 it is likely that an ester is formed, and the leaving group is the conjugate base of the corresponding acid. In these cases, the mechanism can be E1 or E2. The mechanism with Al2O3 and other solid catalysts has been studied extensively, but is poorly understood.180
Magnesium alkoxides (formed by ROH + Me2Mg → ROMgMe) have been decomposed thermally, by heating at 195–340 °C to give the alkene, CH4, and MgO.181 Syn elimination is found and an Ei mechanism is likely. Similar decomposition of aluminum and zinc alkoxides has also been accomplished.182
OS I, 15, 183, 226, 280, 345, 430, 473, 475; II, 12, 368, 408, 606; III, 22, 204, 237, 312, 313, 353, 560, 729, 786; IV, 130, 444, 771; V, 294; VI, 307, 901; VII, 210, 241, 363, 368, 396; VIII, 210, 444. See also, OS VII, 63; VIII, 306, 474. No attempt has been made to list alkene-forming dehydration reactions accompanying condensations or rearrangements.
17-2 Cleavage of Ethers to Alkenes
Hydro-alkoxy-elimination
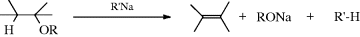
Alkenes can be formed by the treatment of ethers with very strong bases (e.g., alkylsodium or alkyllithium183 compounds, sodium amide,184 or LDA),185 although there are side reactions with many of these reagents. The reaction is aided by electron-withdrawing groups in the β position, and, for example, EtOCH2CH(COOEt)2 can be converted to CH2=C(COOEt)2 without any base at all, but simply by heating.186 tert-Butyl ethers are cleaved more easily than others. Several mechanisms are possible. In many cases, the mechanism is probably E1cB or on the E1cB side of the mechanistic spectrum,187 since the base required is so strong, but it has been shown (by the use of PhCD2OEt) that PhCH2OEt reacts by the five-membered Ei mechanism:188 Propargylic benzyl ethers are converted to conjugated dienes by heating with a Ru catalyst.189 Ethers also have been converted to alkenes and alcohols by passing vapors over hot P2O5 or Al2O3 (this method is similar to Reaction 17-1), but this is not a general reaction.
Cyclic ethers (e.g., THF) react slowly with organolithium reagents with cleavage that produces a C=C unit.190 Fragmentation of 2,5-dihydrofuran with ethylmagnesium chloride and a chiral Zr catalyst leads to a chiral, homoallylic alcohol.191 Acetals can be converted to enol ethers (34) in this manner. When ketals react with 2 molar equivalents of tri-isobutylaluminum, the product is a vinyl ether.192 This can also be done at room temperature by treatment with trimethylsilyl triflate and a tertiary amine193 or with Me3SiI in the presence of hexamethyldisilazane.194
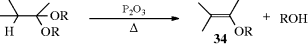
Conversion of a carbonyl compound to an enol phosphate195 or triflate196 allows a subsequent elimination reaction to give an alkyne. Conversion of an aldehyde to the vinyl nonaflate (nonafluorobutane-1-sulfonyl) was followed by reaction with a phosphazene base to give the alkyne.197
Enol ethers can be pyrolyzed to alkenes and aldehydes in a manner similar to that of Reaction 17-4
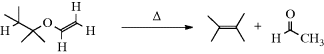
The rate of this reaction for R–O–CH=CH2 increased in the order Et < iPr < t-Bu.198 The mechanism is similar to that of Reaction 17-4.
OS IV, 298, 404; V, 25, 642, 859, 1145; VI, 491, 564, 584, 606, 683, 948; VIII, 444.
17-3 The Conversion of Epoxides and Episulfides to Alkenes
epi-Oxy-elimination
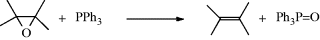
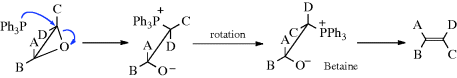
Epoxides can be converted to alkenes199 by treatment with triphenylphosphine200 or triethylphosphite P(OEt)3.201 The first step of the mechanism is nucleophilic substitution (Reaction 10-35), followed by a four-center elimination. Since inversion accompanies the substitution, the overall elimination is anti; that is, if two groups A and C are cis in the epoxide, they will be trans in the alkene:
Alternatively, the epoxide can be treated with lithium diphenylphosphide (Ph2PLi), and the product quaternized with methyl iodide.202 Alkenes have also been obtained from epoxides by reaction with a large number of reagents,203 among them Li in THF,204 trimethylsilyl iodide,205 F3COOH–NaI,206 and compounds of Sm,207 Mo, In,208 and the W reagents mentioned in Reaction 17-18. Some of these methods give syn elimination. Treatment of cyclooctane oxide with Ph3P–OPPh3 and NEt3 gave cyclooctadiene.209 Sodium amalgam with a Co–salen complex converted epoxides to alkenes.210
Epoxides can be converted to allylic alcohols211 by treatment with several reagents, including sec-butyllithium,212 and iPr2NLi–t-BuOK (the LIDAKOR reagent).213 These bases remove the proton from the adjacent carbon, leading to formation of a C=C unit and opening of the epoxide to give an alkoxide. Phenyllithium reacts with epoxides in the presence of LTMP to give a trans-alkene.214 Sulfur ylids (e.g., Me2S=CH2) also convert epoxides to allylic alcohols.215 Bromomethyl epoxides react with InCl3/NaBH4 to give an allylic alcohol216 or with Me3S+Br− and butyllithium to give a dienyl alcohol.217 α,β-Epoxy ketones are converted to conjugated ketones by treatment with NaI in acetone in the presence of Amberlyst 15,218 or with 2.5 molar equivalents of SmI2.219
When an optically active reagent is used, optically active allylic alcohols can be produced from achiral epoxides.220 Sparteine and sec-butyllithium generate a chiral base that leads to formation of chiral allylic alcohols.221 Chiral diamines react with organolithium reagents to produce chiral bases that convert epoxides to allylic alcohols with good enantioselectivity.222 Chiral diamines with a mixture of LDA and DBU (Reactions 15-32 and 17-13) give similar results.223
Episulfides224 can be converted to alkenes.225 However, in this case the elimination is syn, so the mechanism cannot be the same as that for conversion of epoxides. The phosphite attacks sulfur rather than carbon. Among other reagents that convert episulfides to alkenes are certain Rh complexes,226 LiAlH4227 (this compound behaves quite differently with epoxides, see Reaction 19-35), and MeI.228 Episulfoxides can be converted to alkenes and sulfur monoxide simply by heating.229
17-4 Pyrolysis of Carboxylic Acids and Esters of Carboxylic Acids
Hydro-acyloxy-elimination

Direct elimination of a carboxylic acid (decarboxylation) to an alkene has been accomplished by heating in the presence of Pd catalysts.230 Carboxylic esters that bear an alkyl group with a β hydrogen can be pyrolyzed, most often in the gas phase, to give the corresponding acid and an alkene.231 No solvent is required. Since rearrangement and other side reactions are few, the reaction is synthetically very useful and is often carried out as an indirect method of accomplishing 17-1. The yields are excellent and the workup is easy. Many alkenes have been prepared in this manner. For higher alkenes (above ~ C10) a better method is to pyrolyze the alcohol in the presence of acetic anhydride.232
The mechanism is Ei (see Sec. 17.E.i). Lactones can be pyrolyzed to give unsaturated acids, provided that the six-membered transition state required for Ei reactions is available (it is not for five- and six-membered lactones, but it is for larger rings233). Amides give a similar reaction, but require higher temperatures.
Allylic acetates give dienes when heated with certain Pd234 or Mo235 compounds.
OS III, 30; IV, 746; V, 235; IX, 293.
17-5 The Chugaev Reaction
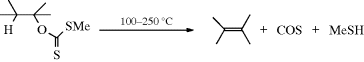
Methyl xanthates are prepared by treatment of alcohols with NaOH and CS2 to give RO–C(=S)–SNa, followed by treatment of this with iodomethane.236 Pyrolysis of the xanthate to give the alkene, COS, and the thiol is called the Chugaev reaction.237 The reaction is like 17-4; an indirect method of accomplishing 17-2. The temperatures required with xanthates are lower than with ordinary esters, which is advantageous because possible isomerization of the resulting alkene is minimized. The mechanism is Ei, similar to that of Reaction 17-4. For a time, there was doubt as to which sulfur atom closed the ring, but now there is much evidence, including the study of 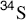 and
and 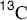 isotope effects, to show that it is the C=S sulfur (see 35).238 In a structural variation of this reaction, heating a propargylic xanthate with 2,4,6-trimethylpyridinium trifluoromethyl sulfonate leads to formation of an alkene.239
isotope effects, to show that it is the C=S sulfur (see 35).238 In a structural variation of this reaction, heating a propargylic xanthate with 2,4,6-trimethylpyridinium trifluoromethyl sulfonate leads to formation of an alkene.239
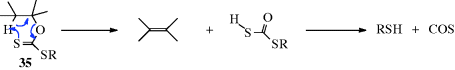
The mechanism is thus exactly analogous to that of Reaction 17-5.
OS VII, 139.
17-6 Decomposition of Other Esters
Hydro-tosyloxy-elimination
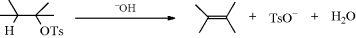
Several types of inorganic ester can be cleaved to alkenes by treatment with bases. Esters of sulfuric, sulfurous, and other acids undergo elimination in solution by E1 or E2 mechanisms, as do tosylates and other esters of sulfonic acids.240 It has been shown that bis(tetra-n-butylammonium) oxalate, (Bu4N+)2 (COO−)2, is an excellent reagent for inducing tosylates to undergo elimination rather than substitution.241 Aryl sulfonates have also been cleaved without a base. Esters of 2-pyridinesulfonic acid and 8-quinolinesulfonic acid gave alkenes in high yields simply on heating, without a solvent.242 Phosphonate esters have been cleaved to alkenes by treatment with Lawesson's reagent243 (see Reaction 16-11).
OS, VI, 837; VII, 117.
17-7 Cleavage of Quaternary Ammonium Hydroxides
Hydro-trialkylammonio-elimination
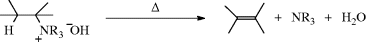
Cleavage of quaternary ammonium hydroxides is the final step of the process known as Hofmann exhaustive methylation or Hofmann degradation or just Hofmann elimination.244 In the first step, a primary, secondary, or tertiary amine is treated with enough iodomethane to convert it to the quaternary ammonium iodide (Reaction 10-31). In the second step, the iodide counterion is converted to the hydroxide counterion by treatment with silver oxide. In the cleavage step, an aqueous or alcoholic solution of the ammonium hydroxide is distilled, often under reduced pressure. The decomposition generally takes place between 100 and 200 °C. Alternatively, the solution can be concentrated to a syrup by distillation or freeze-drying.245 When the syrup is heated at low pressures, the cleavage reaction takes place at lower temperatures than are required for the reaction in the ordinary solution, probably because the base (HO− or RO−) is less solvated.246 The reaction has never been an important synthetic tool, but has been used in the determination of the structure of unknown amines, especially alkaloids. In many of these compounds, the nitrogen is in a ring, or even at a ring junction, and in such cases formation of the alkene is incomplete. Repetitions of the process are required to remove the nitrogen completely, as in the conversion of 2-methylpiperidine to 1,5-hexadiene by two rounds of exhaustive methylation followed by pyrolysis.
A side reaction involving nucleophilic substitution to give an alcohol (R4N+ −OH → ROH + R3N) generally accompanies the normal elimination reaction,247 but seldom causes trouble. However, when none of the four groups on the nitrogen has a β hydrogen, substitution is the only reaction possible. On heating Me4N+ −OH in water, methanol is obtained, although without a solvent the product is not methanol, but dimethyl ether.248
The mechanism of elimination is usually E2 in protic solvents. Hofmann's rule is generally obeyed by acyclic and Zaitsev's rule by cyclohexyl substrates (Sec. 17.B, category 4). In certain cases, where the molecule is highly hindered or if the ammonium hydroxide is heated without solvent (neat), a five-membered Ei mechanism, similar to that in Reaction 17-8, has been shown to operate. That is, the hydroxide in these cases does not attract the β hydrogen, but instead removes one of the methyl hydrogen atoms (see 36), which removes a proton from the less substituted β-carbon atom to give the less substituted alkene with loss of the amine. It is also possible that the hydroxide (rather than the N-ylid) removes the β-hydrogen atom via an eclipsed rotamer in which the hydroxide is tethered to the ammonium unit and a syn-transition state is lower in energy.
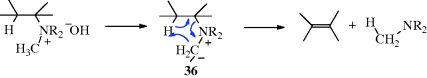
The obvious way to distinguish between this mechanism and the ordinary E2 mechanism is by the use of deuterium labeling. For example, if the reaction is carried out on a quaternary hydroxide deuterated on the β carbon (R2CDCH2NMe3+ −OH), the fate of the deuterium indicates the mechanism. If the E2 mechanism were in operation, the trimethylamine produced would contain no deuterium, which would be found only in the water. But if the mechanism is Ei, the amine would contain deuterium. In the case of the highly hindered compound (Me3C)2CDCH2NMe3+ −OH, the deuterium did appear in the amine, demonstrating an Ei mechanism for this case.249 With simpler compounds, the mechanism is E2, since here the amine was deuterium-free.250
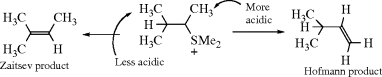
When the nitrogen bears more than one group possessing a β hydrogen, which group cleaves? The Hofmann rule says that within a group the hydrogen on the least alkylated carbon cleaves. This tendency is also carried over to the choice of which group cleaves: thus ethyl with three β-hydrogen atoms cleaves more readily than any longer n-alkyl group, all of which have two β-hydrogen atoms. “The β hydrogen is removed most readily if it is located on a methyl group, next from RCH2, and least readily from R2CH.”251 In fact, the Hofmann rule as first stated252 in 1851 applied only to which group cleaved, not to the orientation within a group. The latter could not have been specified in 1851, since the structural theory of organic compounds was not formulated until 1857–1860. Of course, the Hofmann rule (applied to which group cleaves or to orientation within a group) is superseded by conjugation possibilities. Thus PhCH2CH2N+Me2Et −OH gives mostly styrene instead of ethylene.
Triple bonds have been prepared by pyrolysis of 1,2-bis(ammonium) salts.253
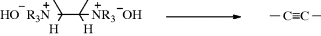
OS IV, 980; V, 315, 608; VI, 552. Also see, OS V, 621, 883; VI, 75.
17-8 Cleavage of Quaternary Ammonium Salts with Strong Bases
Hydro-trialkylammonio-elimination
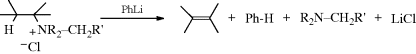
When quaternary ammonium halides are treated with strong bases (e.g., PhLi, KNH2 in liquid NH3254), an elimination can occur that is similar in products, although not in mechanism, to Reaction 17-7. This is an alternative to Reaction 17-7 and is done on the quaternary ammonium halide, so that it is not necessary to convert this to the hydroxide. The mechanism is Ei:
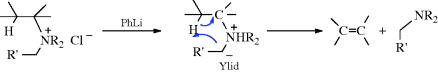
An α′ hydrogen is obviously necessary in order for the ylid to be formed. This type of mechanism is called α′,β elimination, since a β hydrogen is removed by the α′ carbon. The mechanism has been confirmed by labeling experiments similar to those described at Reaction 17-7,255 and by isolation of the intermediate ylids.256 An important synthetic difference between this and most instances of Reaction 17-7 is that syn elimination is observed here and anti elimination in Reaction 17-7, so products of opposite configuration are formed when the alkene exhibits cis–trans isomerism.
An alternative procedure that avoids the use of a very strong base is heating the salt with KOH in polyethylene glycol monomethyl ether.257
Benzotriazole has been shown to be a good leaving group for elimination reactions. The reaction of an allylic benzotriazole (3-benzotriazoyl-4-trimethylsilyl-1-butene) with n-butyllithium, and then an alkyl halide leads to an alkylated 1,3-diene upon heating.258
17-9 Cleavage of Amine Oxides
Hydro-(Dialkyloxidoammonio)-elimination
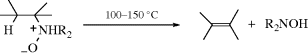
Cleavage of amine oxides to produce an alkene and a hydroxylamine is called the Cope reaction or Cope elimination (not to be confused with the Cope rearrangement, 18-32). It is an alternative to Reactions 17-7 and 17-8.259 The reaction is usually performed with a mixture of amine and oxidizing agent (see 19-29) without isolation of the amine oxide. Because of the mild conditions, side reactions are few, and the alkenes do not usually rearrange. The reaction is thus very useful for the preparation of many alkenes. A limitation is that it does not open six-membered rings containing nitrogen, although it does open rings of 5 and 7–10 members.260 Rates of the reaction increase with increasing size of α- and β-substituents.261 The reaction can be carried out at room temperature in dry Me2SO or THF.262 The influence of solvent effects has been examined.263 The elimination is a stereoselective syn process,264 and the five-membered Ei mechanism operates:
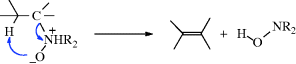
Almost all evidence indicates that the transition state must be planar. Deviations from planarity as in Reaction 17-4 (see Sec. 17.E.i) are not found here, and indeed this is why six-membered heterocyclic nitrogen compounds do not react. Because of the stereoselectivity of this reaction and the lack of rearrangement of the products, it is useful for the formation of trans-cycloalkenes (eight-membered and higher). A polymer-bound Cope elimination reaction has been reported.265
OS IV, 612.
17-10 Pyrolysis of Keto-ylids
Hydro-(oxophosphoryl)-elimination
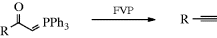
Phosphorus ylids are quite common (see Reaction 16-44) and keto-phosphorus ylids (RCOCH=PPh3) are also known. When these compounds are heating (flash vacuum pyrolysis, FVP) to > 500 °C, alkynes are formed. Simple alkynes266 can be formed as well as keto-alkynes267 and en-ynes.268 Rearrangement from ylids derived from tertiary amines and α-diazo ketones is also known.269
17-11 Decomposition of Toluene-p-sulfonylhydrazones
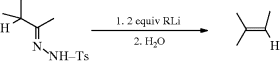
Treatment of the tosylhydrazone of an aldehyde or a ketone with a strong base leads to the formation of an alkene, the reaction being formally an elimination accompanied by a hydrogen shift.270 The reaction (called the Shapiro reaction) has been applied to tosylhydrazones271 of many aldehydes and ketones. The most useful synthetic method involves treatment of the substrate with at least 2 molar equivalents of an organolithium compound272 (usually MeLi) in ether, hexane, or tetramethylenediamine.273 This procedure gives good yields of alkenes without side reactions and, where a choice is possible, predominantly gives the less highly substituted alkene. Tosylhydrazones of α,β-unsaturated ketones give conjugated dienes.274 The mechanism275 has been formulated as that shown.
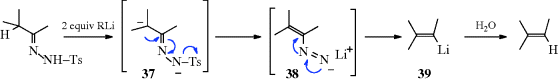
Evidence for this mechanism is (1) two molar equivalents of RLi are required; (2) the hydrogen in the product comes from the water and not from the adjacent carbon, as shown by deuterium labeling;276 and (3) the intermediates 37–39 have been trapped.277 This reaction, when performed in tetramethylenediamine, can be a synthetically useful method278 of generating vinylic lithium compounds (39), which can be trapped by various electrophiles279 (e.g., D2O, to give deuterated alkenes), CO2 (to give α,β-unsaturated carboxylic acids, Reaction 16-30), or DMF (to give α,β-unsaturated aldehydes, Reaction 16-82). Treatment of N-aziridino hydrazones with LDA leads to alkenes with high cis selectivity.280
The reaction also takes place with other bases (e.g., NaH, LiH,281 Na in ethylene glycol, NaNH2) or with smaller amounts of RLi, but in these cases side reactions are common and the orientation of the double bond is in the other direction (to give the more highly substituted alkene). The reaction with Na in ethylene glycol is called the Bamford–Stevens reaction.282 For these reactions, two mechanisms are possible: a carbenoid and a carbocation mechanism.283 The side reactions found are those expected of carbenes and carbocations. In general, the carbocation mechanism is chiefly found in protic solvents and the carbenoid mechanism in aprotic solvents. Both routes involve formation of a diazo compound (40), which in some cases can be isolated. In fact, this reaction has been used as a synthetic method for the preparation of diazo compounds.284 In the absence of protic solvents, 36 loses N2, and hydrogen migrates, to give the alkene product. The migration of hydrogen may immediately follow, or be simultaneous with, the loss of N2. In a protic solvent, 40 becomes protonated to give the diazonium ion 41, which loses N2 to give the corresponding carbocation, that may then undergo elimination or give other reactions characteristic of carbocations. A diazo compound is an intermediate in the formation of alkenes by treatment of N-nitrosoamides with a Rh(II) catalyst.285
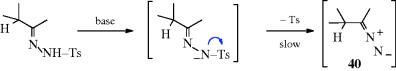
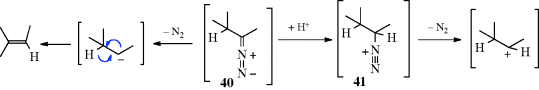
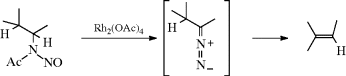
OS VI, 172; VII, 77; IX, 147. For the preparation of a diazo compound, see OS VII, 438.
17-12 Cleavage of Sulfoxides, Selenoxides, and Sulfones
Hydro-alkylsulfinyl-elimination
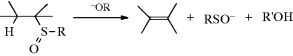
Hydro-alkylsulfinyl-elimination
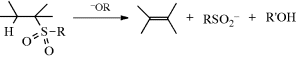
Sulfonium compounds (–C–+SR2) undergo elimination similar to that of their ammonium counterparts (Reactions 17-7 and 17-8) in scope and mechanism, but this reaction is not of great synthetic importance. These syn-elimination reactions are related to the Cope eln (Reaction 17-9) and Hofmann elimination (Reaction 17-7).286
Sulfones and sulfoxides287 with a β hydrogen, on the other hand, undergo elimination on treatment with an alkoxide or, for sulfones,288 even with hydroxide.289 Sulfones also eliminate in the presence of an organolithium reagent and a Pd catalyst.290 Mechanistically, these reactions belong on the E1–E2–E1cB spectrum.291 Although the leaving groups are uncharged, the orientation follows Hofmann's rule, not Zaitsev's. Sulfoxides (but not sulfones) also undergo elimination upon pyrolysis at ~ 80 °C in a manner analogous to Reaction 17-9. The mechanism is also analogous, being the five-membered Ei mechanism with syn elimination.292
Selenoxides293 and sulfinate esters (R2CH–CHR–SO–OMe)294 also undergo elimination by the Ei mechanism, and the selenoxide reaction takes place at room temperature. The reaction with selenoxides has been extended to the formation of triple bonds.295
Both α-keto selenoxides296 and sulfoxides297 have been used in a method for the conversion of ketones, aldehydes, and carboxylic esters to their α,β-unsaturated derivatives. Allylic sulfoxides undergo 1,4-elimination to give dienes.298
A radical elimination reaction generates alkenes from sulfoxides. The reaction of a 2-bromophenyl alkylsulfoxide with Bu3SnH and AIBN (see Sec. 14.A.i for a discussion of these standard radical conditions) leads to an alkene.299
OS VI, 23, 737; VIII, 543; IX, 63.
17-13 Dehydrohalogenation of Alkyl Halides
Hydro-halo-elimination
The elimination of HX from an alkyl halide is a very general reaction and can be accomplished with chlorides, fluorides, bromides, and iodides.300 Hot alcoholic KOH is the most frequently used base, although stronger bases301 (−OR, −NH2, etc.) or weaker ones (e.g., amines) are used where warranted.302 The bicyclic amidines 1,5-diazabicyclo[3.4.0]non-5-ene (DBN)303 and DBU304 are good reagents for difficult cases.305 Solvation by HMPA promotes LDA mediated dehydrobromination.306
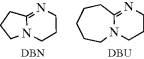
Dehydrohalogenation with the non-ionic base (Me2N)3P=N-P(NMe3)2=NMe is even faster.307 A Co catalyst with dimethylphenylsilylmethylmagnesium chloride leads to formation of terminal alkenes from 2° alkyl bromides.308 Phase-transfer catalysis has been used with hydroxide as base.309 As previously mentioned (Sec. 17.A.v), certain weak bases in dipolar aprotic solvents are effective reagents for dehydrohalogenation. Among those most often used for synthetic purposes are LiCl or LiBr-LiCO3 in DMF.310 Dehydrohalogenation occurs by simply heating the alkyl halide in HMPA with no other reagent present.311 As in nucleophilic substitution (Sec. 10.G.iii), the order of leaving group reactivity is I > Br > Cl > F.312 Tertiary halides undergo elimination most easily. Eliminations of chlorides, bromides, and iodides follow Zaitsev's rule, except for a few cases where steric effects are important (e.g., see Sec. 17.B, category 4). Eliminations of fluorides follow Hofmann's rule (Sec. 17.B, category 4).
This reaction is by far the most important way of introducing a triple bond into a molecule.313 Alkyne formation can be accomplished with substrates of the types:314
When the base is NaNH2, 1-alkynes predominate (where possible), because this base is strong enough to form the salt of the alkyne, shifting any equilibrium between 1- and 2-alkynes. When the base is −OH or −OR, the equilibrium tends to be shifted to the internal alkyne, which is thermodynamically more stable. If another hydrogen is suitably located (e.g., –CRH–CX2–CH2–), allene formation can compete, although alkynes are usually more stable. Tetrabutylammonium fluoride mediates the dehydrobromination of vinyl bromide to terminal alkynes.315 Treatment of 1,1-dibromo-1-alkenes with a Pd-catalyst, followed by reaction with tetrabutylammonium hydroxide gives an internal alkyne.316

1,1-Dibromoalkenes are converted to alkynes when treated with n-butyllithium.317 This transformation is a modification of the Fritsch–Buttenberg–Wiechell rearrangement in which a 1,1-diaryl vinyl bromide (42) gives a diaryl alkyne upon treatment with base.318 Vinyl sulfoxides that contain a leaving group (e.g., chloride) on the double bond react with tert-butyllithium to give a lithio alkyne, and hydrolysis leads to the final product, an alkyne.
Dehydrohalogenation is generally carried out in solution, with a base, and the mechanism is usually E2, although the E1 mechanism has been demonstrated in some cases. However, elimination of HX can be accomplished by pyrolysis of the halide, in which case the mechanism is Ei (Sec. 17.E.i) or, in some instances, the free radical mechanism (Sec. 17.E.i). Pyrolysis is normally performed without a catalyst at ~ 400 °C. The pyrolysis reaction is not generally useful synthetically, because of its reversibility. Less work has been done on pyrolysis with a catalyst319 (usually a metallic oxide or salt), but the mechanisms here are probably E1 or E2.

In the special case of the prochiral carboxylic acids (43), dehydrohalogenation with an optically active lithium amide gave an optically active product with %ee as high as 82%.320
OS I, 191, 205, 209, 438; II, 10, 17, 515; III, 125, 209, 270, 350, 506, 623, 731, 785; IV, 128, 162, 398, 404, 555, 608, 616, 683, 711, 727, 748, 755, 763, 851, 969; V, 285, 467, 514; VI, 87, 210, 327, 361, 368, 427, 462, 505, 564, 862, 883, 893, 954, 991, 1037; VII, 126, 319, 453, 491; VIII, 161, 173, 212, 254; IX, 191, 656, 662. Also see, OS VI, 968.
17-14 Dehydrohalogenation of Acyl Halides and Sulfonyl Halides
Hydro-halo-elimination

Ketenes can be prepared by treatment of acyl halides with tertiary amines321 or with NaH and a crown ether.322 The scope is broad, and most acyl halides possessing an α hydrogen give the reaction, but if at least one R is hydrogen, only the ketene dimer, not the ketene, is isolated. However, if a reactive ketene must be used in a reaction with a given compound, the ketene can be generated in situ in the presence of the given compound.323
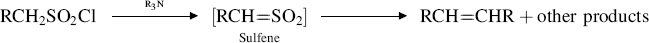
Closely related is the reaction of tertiary amines with sulfonyl halides that contain an α hydrogen. In this case, the initial product is the highly reactive sulfene, which cannot be isolated but reacts further to give products, one of which may be the alkene that is the dimer of RCH.324 Reactions of sulfenes in situ are also common (e.g., see 16-48).
OS IV, 560; V, 294, 877; VI, 549, 1037; VII, 232; VIII, 82.
17-15 Elimination of Boranes
Hydro-boranetriyl-elimination

Trialkylboranes are formed from an alkene and BH3 (Reaction 15-16). When the resulting borane is treated with another alkene, an exchange reaction occurs.325 This is an equilibrium process that can be shifted by using a large excess of alkene, by using an unusually reactive alkene, or by using an alkene with a higher boiling point than the displaced alkene and removing the latter by distillation. The reaction is useful for shifting a double bond in the direction opposite to that resulting from normal isomerization methods (12-2). This cannot be accomplished simply by treatment of a borane with an alkene, because elimination in this reaction follows Zaitsev's rule: It is in the direction of the most stable alkene. However, heating borane (44) leads to 45 (Reaction 18-11) and when 45 is heated with a higher-boiling alkene (e.g., 1-decene), the exchange reaction gives 46. These isomerizations proceed essentially without rearrangement. The mechanism is probably the reverse of borane addition (Reaction 15-16).
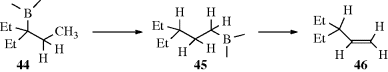
A similar reaction, but irreversible, has been demonstrated for alkynes.326
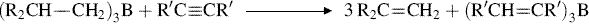
17-16 Conversion of Alkenes to Alkynes
Hydro-methyl-elimination
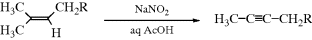
Alkenes of the form shown lose the elements of methane when treated with sodium nitrite in acetic acid and water, to form alkynes in moderate-to-high yields.327 The R may contain additional unsaturation, as well as OH, OR, OAc, C=O, and other groups, but the Me2C=CHCH2– portion of the substrate is necessary for the reaction to take place. The mechanism is complex, beginning with a nitration that takes place with allylic rearrangement [Me2C=CHCH2R → H2C=CMeCH(NO2)CH2R], and involving several additional intermediates.328 The CH3 lost from the substrate appears as CO2, as demonstrated by the trapping of this gas.328
17-17 Decarbonylation of Acyl Halides or Aldehydes
Hydro-chloroformyl-elimination
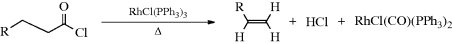
Acyl chlorides containing an α hydrogen are smoothly converted to alkenes, with loss of HCl and CO, upon heating with chlorotris(triphenylphosphine)rhodium, with metallic Pt, or with certain other catalysts.329 The mechanism probably involves conversion of RCH2CH2COCl to RCH2CH2–RhCO(Ph3P)2Cl2 followed by a concerted syn elimination of Rh and H330 (see also, Reactions 14-32 and 19-12).
Aldehydes are decarbonylated to give the corresponding hydrocarbon in the presence of an Ir catalyst and triphenylphosphine.

B. Reactions in which Neither Leaving Atom is Hydrogen
17-18 Deoxygenation of Vicinal Diols
Dihydroxy-elimination
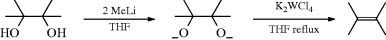
vic-Diols can be deoxygenated by treatment of the dilithium dialkoxide with the tungsten halide (K2WCl6), or with certain other tungsten reagents, in refluxing THF.331 Tetrasubstituted diols react most rapidly. The elimination is largely, but not entirely, syn. Several other methods have been reported,332 in which the diol is deoxygenated directly, without conversion to the dialkoxide. These include treatment with Ti metal,333 with TsOH–NaI,334 and by heating with CpReO3,335 where Cp is cyclopentadienyl.
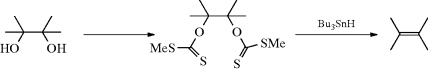
vic-Diols can also be deoxygenated indirectly, through sulfonate ester derivatives. For example, vic-dimesylates and vic-ditosylates have been converted to alkenes by treatment, respectively, with naphthalene–sodium336 and with NaI in DMF.337 In another procedure, the diols are converted to bis(dithiocarbonates) [bis(xanthates)], which undergo elimination (probably by a free radical mechanism) when treated with tri-n-butylstannane in toluene or benzene.338vic-Diols can also be deoxygenated through cyclic derivatives (Reaction 17-19).
17-19 Cleavage of Cyclic Thionocarbonates
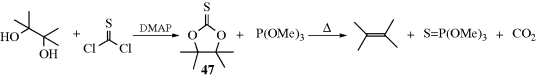
Cyclic thionocarbonates (47) can be cleaved to alkenes (the Corey–Winter reaction)339 by heating with trimethyl phosphite340 or other trivalent phosphorus compounds341 or by treatment with bis(1,5-cyclooctadiene)nickel.342 The thionocarbonates (e.g., 47) can be prepared by treatment of 1,2-diols with thiophosgene and DMAP.343 The elimination is of course syn, so the product is sterically controlled. Alkenes that are not sterically favored can be made this way in high yield (e.g., cis-PhCH2CH=CHCH2Ph).344 Certain other five-membered cyclic derivatives of 1,2-diols can also be converted to alkenes.345
17-20 The Ramberg–Bäcklund Reaction
Ramberg–Bäcklund halosulfone transformation
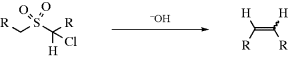
The reaction of an α-halo sulfone with a base to give an alkene is called the Ramberg–Bäcklund reaction.346 The reaction is quite general for α-halo sulfones with an α′ hydrogen, despite the unreactive nature of α-halo sulfones in normal SN2 reactions (Sec. 10.G.i, category 6). Halogen reactivity is in the order I > Br 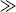 Cl. Phase-transfer catalysis has been used.347 In general, mixtures of cis and trans isomers are obtained, but usually the less stable cis isomer predominates. The mechanism involves formation of an episulfone, and then elimination of SO2. There is much evidence for this mechanism,348
Cl. Phase-transfer catalysis has been used.347 In general, mixtures of cis and trans isomers are obtained, but usually the less stable cis isomer predominates. The mechanism involves formation of an episulfone, and then elimination of SO2. There is much evidence for this mechanism,348
including the isolation of the episulfone intermediate,349 and also the preparation of episulfones in other ways and the demonstration that they give alkenes under the reaction conditions faster than the corresponding α-halo sulfones.350 Episulfones synthesized in other ways (e.g., Reaction 16-48) are reasonably stable compounds, but eliminate SO2 to give alkenes when heated or treated with base.
If the reaction is run on the unsaturated bromosulfones (RCH2CH=CHSO2CH2Br, prepared by reaction of BrCH2SO2Br with RCH2CH=CH2 followed by treatment with Et3N), RCH=CHCH=CH2 is produced in moderate-to-good yields.351 The compound mesyltriflone (CF3SO2CH2SO2CH3) can be used as a synthon for the tetraion 2−C=C2−. Successive alkylation (Reaction 10-67) converts it to CF3SO2CR1R2SO2CHR3R4 (anywhere from one to four alkyl groups can be put in), which, when treated with base, gives R1R2C=CR3R4.352 The nucleofuge here is the CF3SO2− ion. There is an example of a Ramberg–Bäcklund reaction that is induced by a Michael addition.353
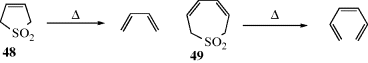
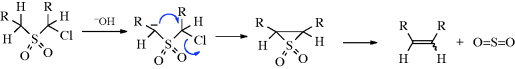
2,5-Dihydrothiophene-1,1-dioxides (48) and 2,17-dihydrothiepin-1,1-dioxides (49) undergo analogous 1,4- and 1,6-eliminations, respectively (see also, Reaction 17-36). These are concerted reactions and, as predicted by the orbital-symmetry rules (Reaction 15-50. A, and immediately preceding pages), the former354 is a suprafacial process and the latter355 an antarafacial process. The rules also predict that elimination of SO2 from episulfones cannot take place by a concerted mechanism (except antarafacially, which is unlikely for such a small ring), and evidence shows that this reaction occurs by a nonconcerted pathway.356 The eliminations of SO2 from 48 and 49 are examples of cheletropic reactions,357 which are defined as reactions in which two σ bonds that terminate at a single atom (in this case the sulfur atom) are made or broken in concert.358
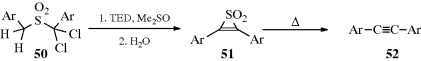
α,α-Dichlorobenzyl sulfones (50) react with an excess of the base triethylenediamine (TED) in DMSO at room temperature to give 2,3-diarylthiiren-1,1-dioxides (51), which can be isolated.359 Thermal decomposition of 51 gives the alkynes 52.360
A Ramberg–Bäcklund-type reaction has been carried out on the α-halo sulfides (ArCHClSCH2Ar), which react with t-BuOK and PPh3 in refluxing THF to give the alkenes (ArCH=CHAr).361 Cyclic sulfides lead to ring-contracted cyclic alkenes upon treatment with NCS in CCl4 followed by oxidation with m-chloroperoxybenzoic acid.362
The Ramberg–Bäcklund reaction can be regarded as a type of extrusion reaction (see Sec. 17.F.vi).
OS V, 877; VI, 454, 555; VIII, 212.
17-21 The Conversion of Aziridines to Alkenes
epi-Imino-elimination

Aziridines not substituted on the nitrogen atom react with nitrous acid to produce alkenes.363 An N-nitroso compound is an intermediate (Reaction 12-50); other reagents that produce such intermediates also give alkenes. The reaction is stereospecific: cis-aziridines give cis-alkenes and trans-aziridines give trans-alkenes.364 Aziridines carrying N-alkyl substituents can be converted to alkenes by treatment with ferrous iodide365 or with m-chloroperoxybenzoic acid.366 An N-oxide intermediate (Reaction 19-29) is presumably involved in the latter case. N-Tosylaziridnes give allylic sulfonamides when treated with butyllithium.367N-Tosyl aziridines are converted to N-tosyl imines when treated with boron trifluoride.368 2-Tosylmethyl N-tosylaziridines react with Te2− in the presence of Adogen 464 to give allylic N-tosyl amines.369 2-Halomethyl N-tosyl aziridines also react with In metal in methanol to give N-tosyl allylic amines.370
17-22 Elimination of Vicinal Dihalides
Dihalo-elimination
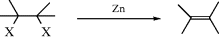
Dehalogenation has been accomplished with many reagents, the most common being Zn, Mg, and iodide ion.371 Heating in HMPA is often enough to convert a vic-dibromide to an alkene.372 Among reagents used less frequently have been phenyllithium, phenylhydrazine, CrCl2, Na2S in DMF,373 and LiAlH4.374 Electrochemical reduction has also been used.375 Treatment with In376 or Sm377 metal in methanol, InCl3/NaBH4,378 heating with Zn in acetic acid,379 or reaction of a Grignard reagent and Ni(dppe)Cl2 (dppe = 1,2-diphenylphosphinoethane).380 When the reagent is Zn, antistereospecificity has been observed in some cases,381 but not in others.382 Microwave irradiation of a vic-dibromide in an ionic liquid leads to the alkene.383 The reaction of a vicinal dibromide with triethylamine and DMF with microwave irradiation leads to vinyl bromide.384 α,β-Dibromo amides are converted to conjugated amides upon photolysis in methanol.385
One useful feature of this reaction is that there is no doubt about the position of the new double bond, so that it can be used to give double bonds exactly where they are wanted. For example, allenes, which are not easily prepared by other methods, can be prepared from X–C–CX2–C–X or X–C–CX=C– systems.386 Cumulenes have been obtained from 1,4-elimination:
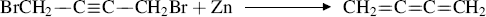
Cumulenes have also been prepared by treating alkynyl epoxides with boron trifluoride.387 1,4-Elimination of BrC–C=C–CBr has been used to prepare conjugated dienes C=C–C=C.388 Allenes are formed by heating propargylic alcohols with arylboronic acids (Reaction 12-28) and a Pd catalyst.389 Allenes are also formed from propargylic amines using a CuI and a Pd catalyst.390 In addition, allenes are formed from lithium bromocyclopropylidenoids.391
The reaction can be carried out for any combination of halogens, except where one is fluorine. Mechanisms are often complex and depend on the reagent and reaction conditions.392 For different reagents, mechanisms involving carbocations, carbanions, and free radical intermediates, as well as concerted mechanisms, have been proposed.
OS III, 526, 531; IV, 195, 268; V, 22, 255, 393, 901; VI, 310, VII, 241. Also see, OS IV, 877, 914, 964.
17-23 Dehalogenation of α-Halo Acyl Halides
Dihalo-elimination
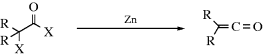
Ketenes can be prepared by dehalogenation of α-halo acyl halides with zinc or with triphenylphosphine.393 The reaction generally gives good results when the two R groups are aryl or alkyl, but not when either one is hydrogen.394
OS IV, 348; VIII, 377.
17-24 Elimination of a Halogen and a Hetero Group
Alkoxy-halo-elimination
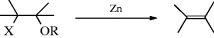
The elimination of OR and halogen from β-halo ethers is called the Boord reaction. It can be carried out with Zn, Mg, Na, or certain other reagents.395 The yields are high and the reaction is of broad scope. β-Halo acetals readily yield vinylic ethers, X–C–C(OR)2 → C=C–OR and 2 molar equivalents of SmI2 in HMPA is effective.396 Besides β-halo ethers, the reaction can also be carried out on compounds of the formula Z–C–C–Z where X is halogen and Z is OCOR, OTs,397 NR2,398 or SR.399 When X = Cl and Z = OAc, heating in THF with an excess of SmI2 followed by treatment with dilute aq HCl gives an alkene.400 When Z = I and the other Z is an oxygen of an oxazolone (a carbamate unit), heating with In metal in methanol leads to an allylic amine.401 The Z group may also be OH, but then X is limited to Br and I.402 Like Reaction 17-22, this method ensures that the new double bond will be in a specific position. The fact that Mg causes elimination in these cases limits the preparation of Grignard reagents from these compounds. It has been shown that treatment of β-halo ethers and esters with Zn gives nonstereospecific elimination,403 so the mechanism was not E2. An E1cB mechanism was postulated because of the poor leaving-group ability of OR and OCOR. Bromohydrins can be converted to alkenes (elimination of Br, OH) in high yields by treatment with LiAlH4-TiCl3.404
OS III, 698, IV, 748; VI, 675.
17.F.ii. Fragmentations
When carbon is the positive leaving group (the electrofuge) in an elimination, the reaction is called fragmentation.405 These processes occur on substrates of the form W–C–C–X, where X is a normal nucleofuge (e.g., halogen, OH2+, OTs, NR3+) and W is a positive-carbon electrofuge. In most of the cases, W is HO–C– or R2N–C–, so that the positive charge on the carbon atom is stabilized by the unshared pair of the oxygen or nitrogen, for example,
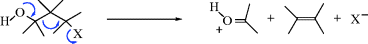
The mechanisms are mostly E1 or E2. We will discuss only a few fragmentations, since many are possible and not much work has been done on most of them. Reactions 17-25–17-28 and 17-30 may be considered fragmentations (see also, Reactions 19-12 and 19-13).
17-25 1,3-Fragmentation of γ-Amino, γ-Hydroxy Halides and 1,3-Diols
Dialkylaminoalkyl-halo-elimination, and so on
Hydroxyalkyl-hydroxy-elimination
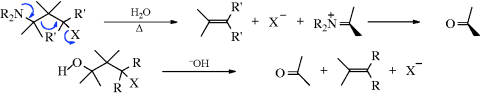
γ-Dialkylamino halides undergo fragmentation when heated with water to give an alkene and an iminium salt, which under the reaction conditions is hydrolyzed to an aldehyde or ketone (16-2).406 γ-Hydroxy halides and tosylates are fragmented with base. In this instance, the base does not play its usual role in elimination reactions, but instead serves to remove a proton from the OH group, which enables the carbon leaving group to come off more easily:

Prelog and Zalán407 first observed this type of fragmentation in work that solved the structure of quinine and other Cinchona alkaloids in a 1,3-elimination ring-opening reaction. Subsequent work by Grob408 elucidated the mechanism of the reaction, and this 1,3-elimination is often referred to as Grob fragmentation.409 The mechanism of these reactions is often E1, but an E2 mechanism also operates.410 It has been shown that stereoisomers of cyclic γ-amino halides and tosylates in which the two leaving groups can assume an antiperiplanar conformation react by the E2 mechanism, while those isomers in which the groups cannot assume such a conformation either fragment by the E1 mechanism or do not undergo fragmentation at all, but in either case give rise to side products characteristic of carbocations.411 An example of a Grob fragmentation is the conversion of 53 to 54.412
γ-Dialkylamino alcohols do not give fragmentation, since for ionization the OH group must be converted to +OH2 and this would convert NR2 to +NR2H, which does not have the unshared pair necessary to form the double bond with the carbon.413
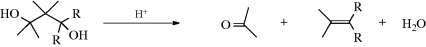
1,3-Diols in which at least one OH group is tertiary or is located on a carbon with aryl substituents can be cleaved by acid treatment.414 The reaction is most useful synthetically when at least one of the OH groups is on a ring.415
17-26 Decarboxylation of β-Hydroxy Carboxylic Acids and of β-Lactones
Carboxy-hydroxy-elimination

An OH and a CO2H group can be eliminated from β-hydroxy carboxylic acids by refluxing with excess DMF dimethyl acetal.416 Mono-, di-, tri-, and tetrasubstituted alkenes have been prepared by this method in good yields.417 There is evidence that the mechanism involves E1 or E2 elimination from the zwitterionic intermediate −O2C–C–C–O–C=N+Me2.418 The reaction has also been accomplished419 under extremely mild conditions (a few seconds at 0 °C) with PPh3 and EtO2C–N=N–CO2Et, diethyl azodicarboxylate.420 In a related procedure, β-lactones undergo thermal decarboxylation to give alkenes in high yields. The reaction has been shown to be stereospecific syn elimination.421 There is evidence that this reaction also involves a zwitterionic intermediate.422
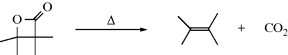
There are no OS references, but see OS VII, 172, for a related reaction.
17-27 Fragmentation of α,β-Epoxy Hydrazones
Eschenmoser–Tanabe ring cleavage
Cyclic α,β-unsaturated ketones423 can be cleaved by treatment with base of their epoxy tosylhydrazone derivatives to give acetylenic ketones,424 in what is known as the Eschenmoser–Tanabe ring cleavage. The reaction can be applied to the formation of acetylenic aldehydes (R = H) by using the corresponding, 2,4-dinitro-tosylhydrazone derivatives.425 Hydrazones (e.g., 55) prepared from epoxy ketones and ring-substituted N-aminoaziridines undergo similar fragmentation when heated.426

OS VI, 679.
17-28 Elimination of CO and CO2 from Bridged Bicyclic Compounds
seco-Carbonyl-1, 4-elimination
On heating, bicyclo[2.2.1]hept-2,3-en-17-ones (56) usually lose CO to give cyclohexadienes,427 in a type of reverse Diels–Alder reaction. Bicyclo[2.2.1]heptadienones (57) undergo the reaction so readily (because of the stability of the benzene ring produced) that
they cannot generally be isolated. The parent (57) has been obtained at 10–15 K in an Ar matrix, where its spectrum could be studied.428 Both 56 and 57 can be prepared by a Diels-Alder reactions between a cyclopentadienone and an alkyne or alkene, so that this reaction is a useful method for the preparation of specifically substituted benzene rings and cyclohexadienes.429 Unsaturated bicyclic lactones of the type 58 can also undergo the reaction, losing CO2 (see also, 17-35).
OS III, 807; V, 604, 1037.
Reversal of the Diels–Alder reaction may be considered a fragmentation (see 15-50).
17.F.iii. Reactions in which CN or C=N Bonds are Formed
17-29 Dehydration of Oximes and Similar Compounds
C- Hydro- N- hydroxy-elimination; C- Acyl- N- hydroxy-elimination
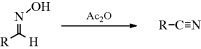
Aldoximes can be dehydrated to nitriles430 by many dehydrating agents, of which acetic anhydride is the most common. Among reagents that are effective under mild conditions431 (room temperature) are Ph3P–CCl4,432 PPh3–I2,433 Pd(OAc)2/PPh3 in refluxing CH3CN,434 ferric sulfate,435 Cu(II) with ultrasound,436 and ZnO/CH3COCl under solvent- free conditions.437N,N-Dimethylformamide catalyzes the thermal dehydration of aldoximes.438 Heating an oxime with a Ru catalyst gives the nitrile.439 Heating with the Burgess reagent [Et3N+−SO2N-CO2Me] in PEG is effective for this transformation.440 Sulfuric acid impregnated silica gel441 gives the nitrile, as does microwave irradiation of an oxime with tetrachloropyridine on alumina.442 Aldehydes can be converted to oximes in situ and microwave irradiation on alumina443 or with ammonium acetate444 gives the nitrile. Solvent-free reactions are known.445 The reaction is most successful when the H and OH are anti. Various alkyl and acyl derivatives of aldoximes (RCH=NOR,446 RCH=NOCOR, RCH=NOSO2Ar, etc.), also give nitriles, as do chlorimines (RCH=NCl; the latter with base treatment).447N,N-Dichloro derivatives of primary amines give nitriles upon pyrolysis: RCH2NCl2 → RCN.448
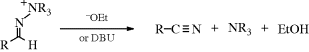
Quaternary hydrazonium salts (derived from aldehydes) give nitriles when treated with −OEt449 or DBU (see Reaction 17-13):450 as do dimethylhydrazones (RCH=NNMe2), when treated with Et2NLi and HMPA.451 All these are methods of converting aldehyde derivatives to nitriles. For the conversion of aldehydes directly to nitriles, without isolation of intermediates (see Reaction 16-16).
Hydroxylamines that have an α-proton are converted to nitrones when treated with a Mn–salen complex.452
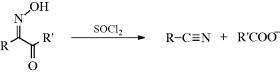
Certain ketoximes can be converted to nitriles by the action of proton or Lewis acids.453 Among these are oximes of α-diketones (illustrated above), α-keto acids, α-dialkylamino ketones, α-hydroxy ketones, β-keto ethers, and similar compounds.454 These are fragmentation reactions, analogous to 17-25. For example, α-dialkylamino ketoximes also give amines and aldehydes or ketones besides nitriles:455
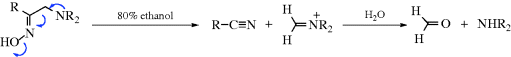
The reaction that normally occurs on treatment of a ketoxime with a Lewis or proton acid is the Beckmann rearrangement (18-17); fragmentations are considered side reactions, often called “abnormal” or “second-order” Beckmann rearrangements.456 Obviously, the substrates mentioned are much more susceptible to fragmentation than are ordinary ketoximes, since in each case an unshared pair is available to assist in removal of the group cleaving from the carbon. However, fragmentation is a side reaction even with ordinary ketoximes457 and, in cases where a particularly stable carbocation can be cleaved, may be the main reaction:458
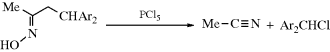
There are indications that the mechanism at least in some cases first involves a rearrangement and then cleavage. The ratio of fragmentation to Beckmann rearrangement of a series of oxime tosylates [RC(=NOTs)Me] was not related to the solvolysis rate but was related to the stability of R+ (as determined by the solvolysis rate of the corresponding RCl), which showed that fragmentation did not take place in the rate-determining step.459 It may be postulated then that the first step in the fragmentation and in the rearrangement is the same and that this is the rate-determining step. The product is determined in the second step:
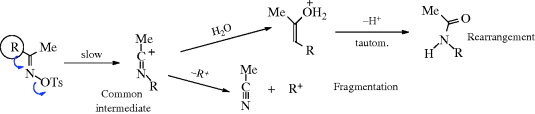
However, in other cases the simple E1 or E2 mechanisms operate.460
OS V, 266; IX, 281; OS II, 622; III, 690.
17-30 Dehydration of Unsubstituted Amides
N,N- Dihydro- C- oxo-bielimination
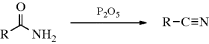
Unsubstituted amides can be dehydrated to nitriles.461 Phosphorous pentoxide is the most common dehydrating agent for this reaction, but many others, including POCl3, PCl5, CCl4–Ph3P,462 HMPA,463 LiCl with a Zr catalyst,464 MeOOCNSO2NEt3 (the Burgess reagent),465 Me2N=CHCl+ Cl−,466 AlCl3/KI/H2O,467 PPh3/NCS,468 oxalyl chloride/DMSO/ −78 °C469 (Swern conditions, see Reaction 19-3), o-iodoxybenzoic acid/Et4NBr,470 PdCl2 in aq media,471 TBAF and hydrosilanes,472 Fe complexes,473 and SOCl2 have also been used.474 Heating an amide with paraformaldehyde and formic acid gives the nitrile.475 It is possible to convert an acid to the nitrile, without isolation of the amide, by heating its ammonium salt with the dehydrating agent,476 or by other methods.477N,N-Disubstituted ureas give cyanamides (R2N–CO–NH2 → R2N–CN) when dehydrated with CHCl3–NaOH under phase-transfer conditions.478 Treatment of an amide with aq NaOH and ultrasound leads to the nitrile.479
N-Alkyl-substituted amides can be converted to nitriles and alkyl chlorides by treatment with PCl5. This is called the von Braun reaction (not to be confused with the other von Braun reaction, 10-54).

OS I, 428; II, 379; III, 493, 535, 584, 646, 768; IV, 62, 144, 166, 172, 436, 486, 706; VI, 304, 465.
17-31 Conversion of N-Alkylformamides to Isonitriles (Isocyanides)
CN-Dihydro-C-oxo-bielimination
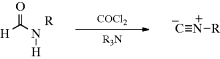
Isocyanides (isonitriles) can be prepared by elimination of water from N-alkylformamides480 with phosgene and a tertiary amine.481 Other reagents, among them TsCl in quinoline, POCl3 and a tertiary amine,482 triflic anhydride/(iPr)2NEt,483 2,4,6-Trichloro[1,3,5]triazine (cyanuric chloride, TCT), with microwave irradiation),484 and Ph3P–CCl4–Et3N485 have also been employed. Formamides react with thionyl chloride (two sequential treatments) to give an intermediate that gives an isonitrile upon electrolysis in DMF with LiClO4.486
A variation of this process uses carbodiimides,487 which can be prepared by the dehydration of N,N′-disubstituted ureas with various dehydrating agents,488 among which are TsCl in pyridine, POCl3, PCl5, P2O5–pyridine, and TsCl (with phase-transfer catalysis).489 Hydrogen sulfide can be removed from the corresponding thioureas by treatment with HgO, NaOCl, or diethyl azodicarboxylate–triphenylphosphine.490
OS V, 300, 772; VI, 620, 751, 987. See also, OS VII, 27. For the carbodiimide/thiourea dehydration, see OS V, 555; VI, 951.
17.F.iv. Reactions in which C=O Bonds are Formed
Many elimination-type reactions in which C=O bonds are formed were considered in Chapter 16, along with their more important reverse reactions (also see, Reactions 12-40 and 12-41).
17-32 Pyrolysis of β-Hydroxy Alkenes
O- Hydro-C-allyl-elimination
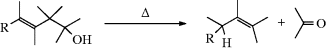
When pyrolyzed, β-hydroxy alkenes cleave to give alkenes and aldehydes or ketones.491 Alkenes produced this way are quite pure, since there are no side reactions. The mechanism has been shown to be pericyclic, primarily by observations that the kinetics are first order492 and that, for ROD, the deuterium appeared in the allylic position of the new alkene.493 This mechanism is the reverse of that for the oxygen analogue of the ene synthesis (Reaction 16-54). β-Hydroxyacetylenes react similarly to give the corresponding allenes and carbonyl compounds.494 The mechanism is the same despite the linear geometry of the triple bonds.

In a related reaction, pyrolysis of allylic ethers that contain at least one α hydrogen gives alkenes and aldehydes or ketones. The mechanism is also pericyclic495
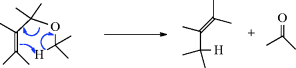
17.F.v. Reactions in which N=N Bonds are Formed
17-33 Eliminations to Give Diazoalkanes
N-Nitrosoamine-diazoalkane transformation
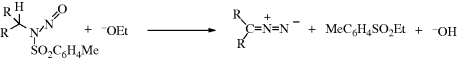
Various N-nitroso-N-alkyl compounds undergo elimination to give diazoalkanes.496 One of the most convenient methods for the preparation of diazomethane involves base treatment of N-nitroso-N-methyl-p-toluenesulfonamide (illustrated above, with R = H).497 However, other compounds commonly used are (base treatment is required in all cases):
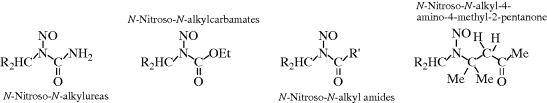
All of these compounds can be used to prepare diazomethane, although the sulfonamide, which is commercially available, is most satisfactory. N-Nitroso-N-methylcarbamate and N-nitroso-N-methylurea give good yields, but are highly irritating and carcinogenic.498 For higher diazoalkanes, the preferred substrates are nitrosoalkylcarbamates.
Most of these reactions probably begin with a 1,3 nitrogen-to-oxygen rearrangement, followed by the actual elimination (illustrated for the carbamate):
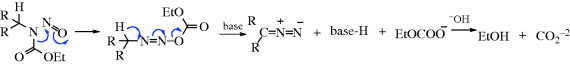
OS II, 165; III, 119, 244; IV, 225, 250; V, 351; VI, 981.
17.F.vi. Extrusion Reactions
We consider an extrusion reaction499 to be one in which an atom or group Y connected to two other atoms X and Z is lost from a molecule, leading to a product in which X is bonded directly to Z.

Reactions 14-32 and 17-20 also fit this definition. Reaction 17-28 does not fit the definition, but is often also classified as an extrusion reaction. A scale of extrusion facility has been developed, showing that the ease of extrusion of the common Y groups is in the order: –N=N– > –COO– > –SO2– > –CO–.500
17-34 Extrusion of N2 from Pyrazolines, Pyrazoles, and Triazolines
Azo-extrusion
1-Pyrazolines (59) can be converted to cyclopropane and N2 on photolysis501 or pyrolysis.502 The tautomeric 2-pyrazolines (60), which are more stable than 59, also give the reaction, but in this case an acidic or basic catalyst is required, the function of which is to convert 60 to 59.503 In the absence of such catalysts, 60 does not react.504 In a similar manner, triazolines (61) are converted to aziridines.505 Side reactions are frequent with both 59 and 61, and some substrates do not give the reaction at all. However, the reaction has proved synthetically useful in many cases. In general, photolysis gives better yields and fewer side reactions than pyrolysis with both 59 and 61. 3H-Pyrazoles506 (62) are stable to heat, but in some cases can be converted to cyclopropenes on photolysis,507 although in other cases other types of products are obtained.
There is much evidence that the mechanism508 of the 1-pyrazoline reactions generally involves diradicals, although the mode of formation and detailed structure (e.g., singlet vs triplet) of these radicals may vary with the substrate and reaction conditions. The reactions of the 3H-pyrazoles (62) have been postulated to proceed through a diazo compound that loses N2 to give a vinylic carbene.509
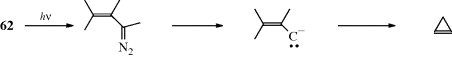
OS V, 96, 929. See also, OS VIII, 597.
17-35 Extrusion of CO or CO2
Carbonyl-extrusion
Although the reaction is not general, certain cyclic ketones can be photolyzed to give ring-contracted products.510 In the example above, the cyclobutanone (63) was photolyzed to give 64.511 This reaction was used to synthesize tetra-tert-butyltetrahedrane, (65).512
The mechanism probably involves a Norrish-type I cleavage (Sec. 7.A.vii), loss of CO from the resulting radical, and recombination of the radical fragments.
Certain lactones extrude CO2 on heating or on irradiation (e.g., the pyrolysis of 66).513
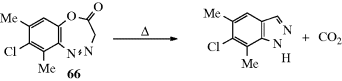
Decarboxylation of β-lactones (see 17-26) may be regarded as a degenerate example of this reaction. Unsymmetrical diacyl peroxides (RCO–OO–COR′) lose two molecules of CO2 when photolyzed in the solid state to give the product RR′.514 Electrolysis was also used, but yields were lower. This is an alternative to the Kolbe reaction (11-34) (see also, 17-28 and 17-38).
There are no OS references, but see OS VI, 418, for a related reaction.
17-36 Extrusion of SO2
Sulfonyl-extrusion
In a reaction similar to 17-35, certain sulfones, both cyclic and acyclic,515 extrude SO2 on heating or photolysis to give ring-contracted products.516 An example is the preparation of naphtho(b)cyclobutene shown above.517 In a different kind of reaction, five-membered cyclic sulfones can be converted to cyclobutenes by treatment with butyllithium followed by LiAlH4,518 for example,
This method is most successful when both the α and α′ position of the sulfone bear alkyl substituents (see also, Reaction 17-20). Treating four-membered ring sultams with SnCl2 led to aziridine products via loss of SO2.519
OS VI, 482.
17-37 The Story Synthesis
When cycloalkylidine peroxides (e.g., 60) are heated in an inert solvent (e.g., decane), extrusion of CO2 takes place; the products are the cycloalkane containing three carbon atoms less than the starting peroxide and the lactone containing two carbon atoms less520 (the Story synthesis).521 The two products are formed in comparable yields, usually ~ 15–25% each. Although the yields are low, the reaction is useful because there are not many other ways to prepare large rings. The reaction is versatile, having been used to prepare rings of every size from 8 to 33 members.
Both dimeric and trimeric cycloalkylidine peroxides can be synthesized522 by treatment of the corresponding cyclic ketones with H2O2 in acid solution.523 The trimeric peroxide is formed first and is subsequently converted to the dimeric compound.524
17-38 Alkene synthesis by Twofold Extrusion
Carbon dioxide,thio-extrusion
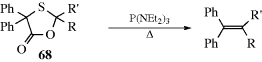
4,4-Diphenyloxathiolan-5-ones (68) give good yields of the corresponding alkenes when heated with tris(diethylamino)phosphine.525 This reaction is an example of a general type: alkene synthesis by twofold extrusion of X and Y from a molecule of the type 69.526 Other examples are photolysis of 1,4-diones527 (e.g., 70) and treatment of acetoxy sulfones [RCH(OAc)CH2SO2Ph] with Mg/EtOH and a catalytic amount of HgCl2.528 68 can be prepared by the condensation of thiobenzilic acid [Ph2C(SH)COOH] with aldehydes or ketones.
OS V, 297.
Notes
1. See Williams, J.M.J. Preparation of Alkenes, A Practical Approach, Oxford Univ. Press, Oxford, 1996.
2. See Neckers, D.C.; Kellogg, R.M.; Prins, W.L.; Schoustra, B. J. Org. Chem. 1971, 36, 1838.
3. Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973. For reviews, see Gandler, J.R. in Patai, S. Supplement A: The Chemistry of Double-bonded Functional Groups, Vol. 2, pt. 1, Wiley, NY, 1989, pp. 733–797; Cockerill, A.F.; Harrison, R.G. in Patai, S. The Chemistry of Functional Groups, Supplement A pt. 1, Wiley, NY, 1977, pp. 153–221; More O'Ferrall, R.A. in Patai, S. The Chemistry of the Carbon–Halogen Bond, pt. 2, Wiley, NY, 1973, pp. 609–675; Cockerill, A.F. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9; Elsevier, NY, 1973, pp. 163–372; Saunders, Jr., W.H. Acc. Chem. Res. 1976, 9, 19; Bordwell, F.G. Acc. Chem. Res. 1972, 5, 374; Fry, A. Chem. Soc. Rev. 1972, 1, 163; LeBel, N.A. Adv. Alicyclic Chem. 1971, 3, 195; Bunnett, J.F. Surv. Prog. Chem. 1969, 5, 53; in Patai, S. The Chemistry of Alkenes, Vol. 1, Wiley, NY, 1964, the articles by Saunders, Jr., W.H. pp. 149–201 (eliminations in solution); and by Maccoll, A. pp. 203–240 (pyrolytic eliminations); Köbrich, G. Angew. Chem. Int. Ed. 1965, 4, 49, pp. 59–63 (for the formation of triple bonds).
4. Thibblin, A. Chem. Soc. Rev. 1993, 22, 427.
5. Roeterdink, W.G.; Rijs, A.M.; Janssen, M.H.M. J. Am. Chem. Soc. 2006, 128, 576.
6. Schrder, S.; Jensen, F. J. Org. Chem. 1997, 62, 253. See Wu, W.; Shaik, S.; Saunders, Jr., W.H. J. Org. Chem. 2010, 75, 3722.
7. See Shiner, Jr., V.J.; Smith, M.L. J. Am. Chem. Soc. 1961, 83, 593. For a review of isotope effects, see Fry, A. Chem. Soc. Rev. 1972, 1, 163.
8. Bartsch, R.A.; Závada, J. Chem. Rev. 1980, 80, 453; Sicher, J. Angew. Chem. Int. Ed. 1972, 11, 200; Pure Appl. Chem. 1971, 25, 655; Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 105–163; Cockerill, A.F. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, pp. 217–235; More O'Ferrall, R.A. in Patai, S. The Chemistry of the Carbon–Halogen Bond, pt. 2, Wiley, NY, 1973, pp. 630–640.
9. DePuy, C.H.; Morris, G.F.; Smith, J.S.; Smat, R.J. J. Am. Chem. Soc. 1965, 87, 2421.
10. Pfeiffer, P. Z. Phys. Chem. 1904, 48, 40.
11. Winstein, S.; Pressman, D.; Young, W.G. J. Am. Chem. Soc. 1939, 61, 1645.
12. Cristol, S.J.; Hause, N.L.; Meek, J.S. J. Am. Chem. Soc. 1951, 73, 674.
13. Hughes, E.D.; Ingold, C.K.; Rose, J.B. J. Chem. Soc. 1953, 3839.
14. Michael, A. J. Prakt. Chem. 1895, 52, 308. See also, Marchese, G.; Naso, F.; Modena, G. J. Chem. Soc. B 1968, 958.
15. Kwart, H.; Takeshita, T.; Nyce, J.L. J. Am. Chem. Soc. 1964, 86, 2606.
16. See Sicher, J.; Pánkova, M.; Závada, J.; Kniezo, L.; Orahovats, A. Collect. Czech. Chem. Commun. 1971, 36, 3128; Bartsch, R.A.; Lee, J.G. J. Org. Chem. 1991, 56, 212, 2579.
17. Cristol, S.J.; Hause, N.L. J. Am. Chem. Soc. 1952, 74, 2193.
18. See Werner, C.; Hopf, H.; Dix, I.; Bubenitschek, P.; Jone, P.G. Chemistry: European J. 2007, 13, 9462.
19. Cooke, Jr., M.P.; Coke, J.L. J. Am. Chem. Soc. 1968, 90, 5556. See also, Coke, J.L.; Smith, G.D.; Britton, Jr., G.H. J. Am. Chem. Soc. 1975, 97, 4323.
20. Závada, J.; Svoboda, M.; Sicher, J. Collect. Czech. Chem. Commun. 1968, 33, 4027.
21. Other possible mechanisms [e.g., E1cB, Sec. 17.A.iii or α′, β-elimination (Reaction 17-8)], were ruled out in all these cases by other evidence.
22. This conclusion has been challenged by Coke, J.L. Sel. Org. Transform 1972, 2, 269.
23. Sicher, J.; Závada, J. Collect. Czech. Chem. Commun. 1967, 32, 2122; Závada, J.; Sicher, J. Collect. Czech. Chem. Commun. 1967, 32, 3701. For a review, see Bartsch, R.A.; Závada, J. Chem. Rev. 1980, 80, 453.
24. Bartsch, R.A.; Závada, J. Chem. Rev. 1980, 80, 453; Coke, J.L. Sel. Org. Transform. 1972, 2, 269; Sicher, J. Angew. Chem. Int. Ed. 1972, 11, 200; Pure Appl. Chem. 1971, 25, 655.
25. See Coke, J.L.; Mourning, M.C. J. Am. Chem. Soc. 1968, 90, 5561.
26. See Sicher, J.; Jan, G.; Schlosser, M. Angew. Chem. Int. Ed. 1971, 10, 926; Závada, J.; Pánková, M. Collect. Czech. Chem. Commun. 1980, 45, 2171 and references cited therein.
27. See Sicher, J.; Závada, J. Collect. Czech. Chem. Commun. 1968, 33, 1278.
28. Bailey, D.S.; Saunders, Jr., W.H. J. Am. Chem. Soc. 1970, 92, 6904. See Schlosser, M.; An, T.D. Helv. Chim. Acta 1979, 62, 1194; Pánková, M.; Kocián, O.; Krupicka, J.; Závada, J. Collect. Czech. Chem. Commun. 1983, 48, 2944.
29. Chiao, W.; Saunders, Jr., W.H. J. Am. Chem. Soc. 1977, 99, 6699.
30. Dohner, B.R.; Saunders, Jr., W.H. J. Am. Chem. Soc. 1986, 108, 245.
31. Bartsch, R.A.; Závada, J. Chem. Rev. 1980, 80, 453; Bartsch, R.A. Acc. Chem. Res. 1975, 8, 239.
32. Svoboda, M.; Hapala, J.; Závada, J. Tetrahedron Lett. 1972, 265.
33. See Baciocchi, E.; Ruzziconi, R.; Sebastiani, G.V. J. Org. Chem. 1979, 44, 3718; Croft, A.P.; Bartsch, R.A. Tetrahedron Lett. 1983, 24, 2737; Kwart, H.; Gaffney, A.H.; Wilk, K.A. J. Chem. Soc. Perkin Trans. 2 1984, 565.
34. Borchardt, J.K.; Saunders, Jr., W.H. J. Am. Chem. Soc. 1974, 96, 3912.
35. See Villano, S.M.; Eyet, N.; Lineberger, W.C.; Bierbaum, V.M. J. Am. Chem. Soc. 2009, 131, 8227.
36. Baciocchi, E.; Clementi, S.; Sebastiani, G.V.; Ruzziconi, R. J. Org. Chem. 1979, 44, 32.
37. Cooper, K.A.; Hughes, E.D.; Ingold, C.K.; MacNulty, B.J. J. Chem. Soc. 1948, 2038.
38. See Thibblin, A. J. Am. Chem. Soc. 1987, 109, 2071; J. Phys. Org. Chem. 1989, 2, 15.
39. Sneen, R.A. Acc. Chem. Res. 1973, 6, 46; Thibblin, A.; Sidhu, H. J. Chem. Soc. Perkin Trans. 2 1994, 1423. See, however, McLennan, D.J. J. Chem. Soc. Perkin Trans. 2 1972, 1577.
40. Cockerill, A.F.; Harrison, R.G. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, pp. 158–178; Hunter, D.H. Intra-Sci. Chem. Rep. 1973, 7(3), 19; McLennan, D.J. Q. Rev. Chem. Soc. 1967, 21, 490. For a general discussion, see Koch, H.F. Acc. Chem. Res. 1984, 17, 137.
41. See Ryberg, P.; Matsson, O. J. Org. Chem. 2002, 67, 811.
42. This table, which appears in Cockerill, A.F.; Harrison, R.G. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, p. 161, was adapted from a longer one in Bordwell, F.G. Acc. Chem. Res. 1972, 5, 374, see p. 375.
43. The (E1cB)I mechanism cannot be distinguished from E2 by this means, because it has the identical rate law: Rate = k[substrate][B−]. The rate law for (E1cB)R is different: Rate = k[substrate][B−]/[BH], but this is often not useful because the only difference is that the rate is also dependent (inversely) on the concentration of the conjugate acid of the base, and this is usually the solvent, so that changes in its concentration cannot be measured.
44. Houser, J.J.; Bernstein, R.B.; Miekka, R.G.; Angus, J.C. J. Am. Chem. Soc. 1955, 77, 6201.
45. Hine, J.; Wiesboeck, R.; Ghirardelli, R.G. J. Am. Chem. Soc. 1961, 83, 1219; Hine, J.; Wiesboeck, R.; Ramsay, O.B. J. Am. Chem. Soc. 1961, 83, 1222.
46. Skell, P.S.; Hauser, C.R. J. Am. Chem. Soc. 1945, 67, 1661.
47. Keeffe, J.R.; Jencks, W.P. J. Am. Chem. Soc. 1983, 105, 265.
48. For a borderline E1cB–E2 mechanism, see Jia, Z.S.; Rudzinsci, J.; Paneth, P.; Thibblin, A. J. Org. Chem. 2002, 67, 177.
49. Cann, P.F.; Stirling, C.J.M. J. Chem. Soc. Perkin Trans. 2 1974, 820. For other examples; see Kurzawa, J.; Leffek, K.T. Can. J. Chem. 1977, 55, 1696.
50. Patai, S.; Weinstein, S.; Rappoport, Z. J. Chem. Soc. 1962, 1741. See also, Hilbert, J.M.; Fedor, L.R. J. Org. Chem. 1978, 43, 452.
51. Bordwell, F.G. Acc. Chem. Res. 1972, 5, 374.
52. Banait, N.S.; Jencks, W.P. J. Am. Chem. Soc. 1990, 112, 6950.
53. Ölwegård, M.; McEwen, I.; Thibblin, A.; Ahlberg, P. J. Am. Chem. Soc. 1985, 107, 7494.
54. Baciocchi, E.; Ruzziconi, R.; Sebastiani, G.V. J. Org. Chem. 1982, 47, 3237.
55. See Gula, M.J.; Vitale, D.E.; Dostal, J.M.; Trometer, J.D.; Spencer, T.A. J. Am. Chem. Soc. 1988, 110, 4400; Garay, R.O.; Cabaleiro, M.C. J. Chem. Res. (S), 1988, 388; Gandler, J.R.; Storer, J.W.; Ohlberg, D.A.A. J. Am. Chem. Soc. 1990, 112, 7756.
56. Bunting, J.W.; Toth, A.; Heo, C.K.M.; Moors, R.G. J. Am. Chem. Soc. 1990, 112, 8878. See also, Bunting, J.W.; Kanter, J.P. J. Am. Chem. Soc. 1991, 113, 6950.
57. Alunni, S.; De Angelis, F.; Ottavi, L.; Papavasileiou, M.; Tarantelli, F. J. Am. Chem. Soc. 2005, 127, 15151. See also, Mosconi, E.; De Angelis, F.; Belpassi, L.; Tarantelli, F.; Alunni, S. Eur. J. Org. Chem. 2009, 5501.
58. Bordwell, F.G.; Yee, K.C.; Knipe, A.C. J. Am. Chem. Soc. 1970, 92, 5945.
59. See Berndt, A. Angew. Chem. Int. Ed. 1969, 8, 613; Albeck, M.; Hoz, S.; Rappoport, Z. J. Chem. Soc. Perkin Trans. 2 1972, 1248; 1975, 628.
60. Kwok, W.K.; Lee, W.G.; Miller, S.I. J. Am. Chem. Soc. 1969, 91, 468. See also, Petrillo, G.; Novi, M.; Garbarino, G.; Dell'Erba, C.; Mugnoli, A. J. Chem. Soc. Perkin Trans. 2 1985, 1291.
61. See Cockerill, A.F.; Harrison, R.G. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, pp. 178–189; Saunders, Jr., W.H. Acc. Chem. Res. 1976, 9, 19; Bunnett, J.F. Surv. Prog. Chem. 1969, 5, 53; Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 47–104; Bordwell, F.G. Acc. Chem. Res. 1972, 5, 374.
62. Fry, A. Chem. Soc. Rev. 1972, 1, 163. See also, Hasan, T.; Sims, L.B.; Fry, A. J. Am. Chem. Soc. 1983, 105, 3967; Pulay, A.; Fry, A. Tetrahedron Lett. 1986, 27, 5055.
63. Ayrey, G.; Bourns, A.N.; Vyas, V.A. Can. J. Chem. 1963, 41, 1759. Also see, Simon, H.; Müllhofer, G. Pure Appl. Chem. 1964, 8, 379, 536; Smith, P.J.; Bourns, A.N. Can. J. Chem. 1970, 48, 125.
64. Wu, S.; Hargreaves, R.T.; Saunders, Jr., W.H. J. Org. Chem. 1985, 50, 2392 and references cited therein.
65. Grout, A.; McLennan, D.J.; Spackman, I.H. J. Chem. Soc. Perkin Trans. 2 1977, 1758.
66. See Thibblin, A. J. Am. Chem. Soc. 1988, 110, 4582; Smith, P.J.; Amin, M. Can. J. Chem. 1989, 67, 1457.
67. However, see Blackwell, L.F. J. Chem. Soc. Perkin Trans. 2 1976, 488.
68. See Miller, D.J.; Saunders, Jr., W.H. J. Org. Chem. 1981, 46, 4247 and previous papers in this series. See also, Amin, M.; Price, R.C.; Saunders, Jr., W.H. J. Am. Chem. Soc. 1990, 112, 4467.
69. Blackwell, L.F.; Buckley, P.D.; Jolley, K.W.; MacGibbon, A.K.H. J. Chem. Soc. Perkin Trans. 2 1973, 169; Smith, P.J.; Tsui, S.K. J. Am. Chem. Soc. 1973, 95, 4760; Can. J. Chem. 1974, 52, 749.
70. DePuy, C.H.; Bishop, C.A. J. Am. Chem. Soc. 1960, 82, 2532, 2535.
71. Brower, K.R.; Muhsin, M.; Brower, H.E. J. Am. Chem. Soc. 1976, 98, 779. For a review, see van Eldik, R.; Asano, T.; le Noble, W.J. Chem. Rev. 1989, 89, 549.
72. McLennan, D.J. Tetrahedron 1975, 31, 2999; Ford, W.T. Acc. Chem. Res. 1973, 6, 410.
73. See Hayami, J.; Ono, N.; Kaji, A. Bull. Chem. Soc. Jpn. 1971, 44, 1628.
74. Parker, A.J.; Ruane, M.; Biale, G.; Winstein, S. Tetrahedron Lett. 1968, 2113.
75. This is apart from the E1-E2-E1cB spectrum.
76. See Kwart, H.; Wilk, K.A. J. Org. Chem. 1985, 50, 3038.
77. See Bunnett, J.F.; Migdal, C.A. J. Org. Chem. 1989, 54, 3037, 3041 and references cited therein.
78. McLennan, D.J.; Lim, G. Aust. J. Chem. 1983, 36, 1821. For an opposing view, see Kwart, H.; Gaffney, A. J. Org. Chem. 1983, 48, 4502.
79. Ford, W.T. Acc. Chem. Res. 1973, 6, 410.
80. For convenience, these are called E2C reactions, although the actual mechanism is in dispute.
81. Beltrame, P.; Biale, G.; Lloyd, D.J.; Parker, A.J.; Ruane, M.; Winstein, S. J. Am. Chem. Soc. 1972, 94, 2240; Beltrame, P.; Ceccon, A.; Winstein, S. J. Am. Chem. Soc. 1972, 94, 2315.
82. See Hückel, W.; Hanack, M. Angew. Chem. Int. Ed. 1967, 6, 534.
83. Often given the German spelling: Saytzeff, or Saytseff, or Saytzev.
84. de la Mare, P.B.D. Prog. StereoChem. 1954, 1, 112.
85. See Cram, D.J.; Sahyun, M.R.V. J. Am. Chem. Soc. 1963, 85, 1257.
86. Ho, I.; Smith, J.G. Tetrahedron 1970, 26, 4277.
87. See Feit, I.N.; Saunders, Jr., W.H. J. Am. Chem. Soc. 1970, 92, 5615.
88. See Booth, H.; Franklin, N.C.; Gidley, G.C. J. Chem. Soc. C 1968, 1891; Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 192–193.
89. See Ingold, C.K. Proc. Chem. Soc. 1962, 265.
90. Bunnett, J.F. Surv. Prog. Chem. 1969, 5, 53.
91. Brown, H.C.; Wheeler, O.H. J. Am. Chem. Soc. 1956, 78, 2199.
92. See Bartsch, R.A. J. Org. Chem. 1970, 35, 1334; Charton, M. J. Am. Chem. Soc. 1975, 97, 6159.
93. See Banthorpe, D.V.; Hughes, E.D.; Ingold, C.K. J. Chem. Soc. 1960, 4054.
94. Saunders, Jr., W.H.; Fahrenholtz, S.R.; Caress, E.A.; Lowe, J.P.; Schreiber, M.R. J. Am. Chem. Soc. 1965, 87, 3401; Brown, H.C.; Klimisch, R.L. J. Am. Chem. Soc. 1966, 88, 1425.
95. Bartsch, R.A.; Bunnett, J.F. J. Am. Chem. Soc. 1968, 90, 408.
96. Froemsdorf, D.H.; Robbins, M.D. J. Am. Chem. Soc. 1967, 89, 1737. See also, Feit, I.N.; Breger, I.K.; Capobianco, A.M.; Cooke, T.W.; Gitlin, L.F. J. Am. Chem. Soc. 1975, 97, 2477.
97. Bartsch, R.A.; Roberts, D.K.; Cho, B.R. J. Org. Chem. 1979, 44, 4105.
98. Bartsch, R.A.; Read, R.A.; Larsen, D.T.; Roberts, D.K.; Scott, K.J.; Cho, B.R. J. Am. Chem. Soc. 1979, 101, 1176.
99. Angelini, G.; Lilla, G.; Speranza, M. J. Am. Chem. Soc. 1989, 111, 7393.
100. Sicher, J.; Svoboda, M.; Pánková, M.; Závada, J. Collect. Czech. Chem. Commun. 1971, 36, 3633; Bailey, D.S.; Saunders, Jr., W.H. J. Am. Chem. Soc. 1970, 92, 6904.
101. Muir, D.M.; Parker, A.J. J. Org. Chem. 1976, 41, 3201.
102. Lloyd, D.J.; Muir, D.M.; Parker, A.J. Tetrahedron Lett. 1971, 3015.
103. See Cram, D.J.; Greene, F.D.; DePuy, C.H. J. Am. Chem. Soc. 1956, 78, 790; Cram, D.G. in Newman, M.S. Steric Effects in Organic Chemistry, Wiley, NY, 1956, pp. 338–345.
104. Brown, H.C.; Wheeler, O.H. J. Am. Chem. Soc. 1956, 78, 2199.
105. Alunni, S.; Baciocchi, E. J. Chem. Soc. Perkin Trans. 2 1976, 877; Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 165–193.
106. See Redman, R.P.; Thomas, P.J.; Stirling, C.J.M. J. Chem. Soc., Chem. Commun. 1978, 43.
107. See Cockerill, A.F.; Harrison, R.G. in Patai, S. The Chemistry of Functional Groups, Supplement A, pt. 1, Wiley, NY, 1977, pp. 178–189.
108. See Butskus, P.F.; Denis, G.I. Russ. Chem. Rev. 1966, 35, 839.
109. Hughes, E.D.; Ingold, C.K.; Maw, G.A. J. Chem. Soc. 1948, 2072; Hughes, E.D.; Ingold, C.K.; Woolf, L.I. J. Chem. Soc. 1948, 2084.
110. Brown, H.C.; Berneis, H.L. J. Am. Chem. Soc. 1953, 75, 10.
111. Dhar, M.L.; Hughes, E.D.; Ingold, C.K.; Masterman, S. J. Chem. Soc. 1948, 2055.
112. Dhar, M.L.; Hughes, E.D.; Ingold, C.K. J. Chem. Soc. 1948, 2058.
113. Hughes, M.L.; Ingold, C.K.; Maw, G.A. J. Chem. Soc. 1948, 2065.
114. See van Zeist, W.-J.; Bickelhaupt, F.M. Org. Biomol. Chem. 2010, 8, 3118; de Jong, G.Th.; Bickelhaupt, F.M. ChemPhysChem 2007, 8, 1170.
115. See Bickelhaupt, F.M. J. Comput. Chem. 1999, 20, 114. Prof. F.M. Bickelhaupt, Vrije Universiteit Amsterdam, personal communication.
116. Baciocchi, E. Acc. Chem. Res. 1979, 12, 430. See also, Baciocchi, E.; Ruzziconi, R.; Sebastiani, G.V. J. Org. Chem. 1980, 45, 827.
117. This list is from Banthorpe, D.V. Elimination Reactions, Elsevier, NY, 1963, p. 4.
118. Loupy, A.; Seyden-Penne, J. Bull. Soc. Chim. Fr. 1971, 2306.
119. See Ono, N. in Feuer, H.; Nielsen, A.T. Nitro Compounds; Recent Advances in Synthesis and Chemistry, VCH, NY, 1990, pp. 1–135, pp. 86–126.
120. These lists are from Banthorpe, D.V. Elimination Reactions, Elsevier, NY, 1963, pp. 4, 7.
121. See Stirling, C.J.M. Acc. Chem. Res. 1979, 12, 198. See also, Varma, M.; Stirling, C.J.M. J. Chem. Soc., Chem. Commun. 1981, 553.
122. See Wright, D.G. J. Chem. Soc., Chem. Commun. 1975, 776. See, however, Cavazza, M. Tetrahedron Lett. 1975, 1031.
123. Veeravagu, P.; Arnold, R.T.; Eigenmann, E.W. J. Am. Chem. Soc. 1964, 86, 3072.
124. Cooper, K.A.; Dhar, M.L.; Hughes, E.D.; Ingold, C.K.; MacNulty, B.J.; Woolf, L.I. J. Chem. Soc. 1948, 2043.
125. Aksnes, G.; Stensland, P. Acta Chem. Scand., 1989, 43, 893, and references cited therein.
126. Jones, M.E.; Ellison, G.B. J. Am. Chem. Soc. 1989, 111, 1645. For a different result with other reactants, see Lum, R.C.; Grabowski, J.J. J. Am. Chem. Soc. 1988, 110, 8568.
127. Cooper, K.A.; Hughes, E.D.; Ingold, C.K.; Maw, G.A.; MacNulty, B.J. J. Chem. Soc. 1948, 2049.
128. Taylor, R. in Patai, S. The Chemistry of Functional Groups, Supplement B pt. 2, Wiley, NY, 1979, pp. 860–914; Smith, G.G.; Kelly, F.W. Prog. Phys. Org. Chem. 1971, 8, 75, pp. 76–143, 207–234; in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 5, Elsevier, NY, 1972, the articles by Swinbourne, E.S. pp. 149–233 (pp. 158–188), and by Richardson, W.H.; O'Neal, H.E. pp. 381–565 (pp. 381–446); Maccoll, A. Adv. Phys. Org. Chem. 1965, 3, 91. See Egger, K.W.; Cocks, A.T. in Patai, S. The Chemistry of the Carbon–Halogen Bond, pt. 2, Wiley, NY, 1973, pp. 677–745; Maccoll, A. Chem. Rev. 1969, 69, 33.
129. O'Connor, G.L.; Nace, H.R. J. Am. Chem. Soc. 1953, 75, 2118.
130. Barton, D.H.R.; Head, A.J.; Williams, R.J. J. Chem. Soc. 1953, 1715.
131. See, however, Briggs, W.S.; Djerassi, C. J. Org. Chem. 1968, 33, 1625; Smissman, E.E.; Li, J.P.; Creese, M.W. J. Org. Chem. 1970, 35, 1352.
132. Jones, D.N.; Saeed, M.A. Proc. Chem. Soc. 1964, 81. See also, Goldberg, S.I.; Sahli, M.S. J. Org. Chem. 1967, 32, 2059.
133. See Bailey, W.J.; Bird, C.N. J. Org. Chem. 1977, 42, 3895.
134. Wright, D.R.; Sims, L.B.; Fry, A. J. Am. Chem. Soc. 1983, 105, 3714.
135. Maccoll, A., in Patai, S. The Chemistry of Alkenes, Vol. 1, Wiley, NY, 1964, pp. 215–216.
136. For reviews of such studies, see Maccoll, A. Chem. Rev. 1969, 69, 33.
137. See Chuchani, G.; Rotinov, A.; Dominguez, R.M.; Martin, I. Int. J. Chem. Kinet. 1987, 19, 781.
138. Scheer, J.C.; Kooyman, E.C.; Sixma, F.L.J. Recl. Trav. Chim. Pays-Bas 1963, 82, 1123. See also, Louw, R.; Vermeeren, H.P.W.; Vogelzang, M.W. J. Chem. Soc. Perkin Trans. 2 1983, 1875.
139. Taylor, R. J. Chem. Soc. Perkin Trans. 2 1978, 1255. See also, August, R.; McEwen, I.; Taylor, R. J. Chem. Soc. Perkin Trans. 2 1987, 1683, and other papers in this series; Al-Awadi, N.A. J. Chem. Soc. Perkin Trans. 2 1990, 2187.
140. Taylor, R. J. Chem. Soc. Perkin Trans. 2 1975, 1025.
141. For a review of the mechanisms of Reaction 17-12 and 17-9, and the pyrolysis of sulfilimines, see Oae, S.; Furukawa, N. Tetrahedron 1977, 33, 2359.
142. See Barton, D.H.R.; Howlett, K.E. J. Chem. Soc. 1949, 155, 165.
143. See Louw, R.; Kooyman, E.C. Recl. Trav. Chim. Pays-Bas 1965, 84, 1511.
144. Timokhin, V.I.; Gastaldi, S.; Bertrand, M.P.; Chatgilialoglu, C. J. Org. Chem. 2003, 68, 3532.
145. Boothe, T.E.; Greene, Jr., J.L.; Shevlin, P.B. J. Org. Chem. 1980, 45, 794; Stark, T.J.; Nelson, N.T.; Jensen, F.R. J. Org. Chem. 1980, 45, 420; Kochi, J.K. Organic Mechanisms and Catalysis, Academic Press, NY, 1978, pp. 346–349; Kamimura, A.; Ono, N. J. Chem. Soc., Chem. Commun. 1988, 1278.
146. Froemsdorf, D.H.; Collins, C.H.; Hammond, G.S.; DePuy, C.H. J. Am. Chem. Soc. 1959, 81, 643; Haag, W.O.; Pines, H. J. Org. Chem. 1959, 24, 877.
147. DePuy, C.H.; King, R.W. Chem. Rev. 1960, 60, 431, with tables showing product distributions.
148. Bailey, W.J.; Baylouny, R.A. J. Am. Chem. Soc. 1959, 81, 2126.
149. Botteron D.G.; Shulman, G.P. J. Org. Chem. 1962, 27, 2007.
150. See Bamkole, T.; Maccoll, A. J. Chem. Soc. B 1970, 1159.
151. Taylor, R. in Patai, S. The Chemistry of Functional Groups, Supplement B, pt. 2, Wiley, NY, 1979, pp. 885–890; Smith, G.G.; Mutter, L.; Todd, G.P. J. Org. Chem. 1977, 42, 44; Chuchani, G.; Dominguez, R.M. Int. J. Chem. Kinet. 1981, 13, 577; Hernández, A.; Chuchani, G. Int. J. Chem. Kinet. 1983, 15, 205.
152. See Wakselman, M. Nouv. J. Chem. 1983, 7, 439.
153. Ölwegård, M.; Ahlberg, P. Acta Chem. Scand., 1990, 44, 642. See also, Ölwegård, M.; Ahlberg, P. J. Chem. Soc., Chem. Commun. 1989, 1279.
154. Friedrich, K. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C pt. 2, Wiley, NY, 1983; pp. 1376–1384; Ben-Efraim, D.A. in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, pt. 2, Wiley, NY, 1978, pp. 755–790. For a comparative study of various methods, see Mesnard, D.; Bernadou, F.; Miginiac, L. J. Chem. Res. (S) 1981, 270, and referrences cited therein.
155. Spitzin, V.I.; Michailenko, I.E.; Pirogowa, G.N. J. Prakt. Chem. 1964, [4] 25, 160; Bertsch, H.; Greiner, A.; Kretzschmar, G.; Falk, F. J. Prakt. Chem. 1964, [4] 25, 184.
156. Anbazhagan, M.; Kumaran, G.; Sasidharan, M. J. Chem. Res. (S) 1997, 336.
157. Mattern, R.-H. Tetrahedron Lett. 1996, 37, 291.
158. Berti, F.; Ebert, C.; Gardossi, L. Tetrahedron Lett. 1992, 33, 8145.
159. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 291–294.
160. Alvarez-Manzaneda, E.J.; Chahboun, R.; Torres, E.C.; Alvarez, E.; Alvarez-Manzaneda, R.; Haidour, A.; Ramos, J. Tetrahedron Lett. 2004, 45, 4453.
161. Firouzabadi, H.; Iranpoor, N.; Hazarkhani, H.; Karimi, B. Synth. Commun. 2003, 33, 3653.
162. Monson, R.S.; Priest, D.N. J. Org. Chem. 1971, 36, 3826; Lomas, J.S.; Sagatys, D.S.; Dubois, J.E. Tetrahedron Lett. 1972, 165.
163. Lundeen, A.J.; Van Hoozer, R. J. Org. Chem. 1967, 32, 3386. See also, Davis, B.H. J. Org. Chem. 1982, 47, 900; Iimori, T.; Ohtsuka, Y.; Oishi, T. Tetrahedron Lett. 1991, 32, 1209.
164. Bartoli, G.; Bellucci, M.C.; Petrini, M.; Marcantoni, E.; Sambri, L.; Torregiani, E. Org. Lett. 2000, 2, 1791.
165. Okumoto, H.; Jinnai, T.; Shimizu, H.; Harada, Y.; Mishima, H.; Suzuki, A. Synlett 2000, 629.
166. Saito, S.; Nagahara, T.; Yamamoto, H. Synlett 2001, 1690.
167. Concellón, J.M.; Pérez-Andrés, J.A.; Rodríguez-Solla, H. Angew. Chem. Int. Ed. 2000, 39, 2773.
168. Fleming, F.F.; Shook, B.C. Tetrahedron Lett. 2000, 41, 8847.
169. Fleming, F.F.; Shook, B.C. J. Org. Chem. 2002, 67, 3668.
170. Cho, S.; Kang, S.; Keum, G.; Kang, S.B.; Han, S.-Y.; Kim, Y. J. Org. Chem. 2003, 68, 180.
171. Concellón, J.M.; Rodríguez-Solla, H.; Huerta, M..; Pérez-Andrés, J.A. Eur. J. Org. Chem. 2002, 1839.
172. Brady, W.T.; Marchand, A.P.; Giang, Y.F.; Wu, A. Synthesis 1987, 395; J. Org. Chem. 1987, 52, 3457.
173. Olah, G.A.; Wu, A.; Farooq, O. Synthesis 1989, 568.
174. Brady, W.T.; Marchand, A.P.; Giang, Y.F.; Wu, A. J. Org. Chem. 1987, 52, 3457; Funk, R.L.; Abelman, M.M.; Jellison, K.M. Synlett 1989, 36.
175. Stevens, C.L.; Singhal, G.H. J. Org. Chem. 1964, 29, 34.
176. See Bartók, M.; Molnár, A. in Patai, S. The Chemistry of Functional Groups, Supplement E, pt. 2, Wiley, NY, 1980, pp. 721–760.
177. Guo, C.; Lu, X. J. Chem. Soc., Chem. Commun. 1993, 394.
178. Vinnik, M.I.; Obraztsov, P.A. Russ. Chem. Rev. 1990, 59, 63; Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 221–274, 317–331; Knözinger, H. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 2, Wiley, NY, 1971, pp. 641–718.
179. See Kawanisi, M.; Arimatsu, S.; Yamaguchi, R.; Kimoto, K. Chem. Lett. 1972, 881.
180. Beránek, L.; Kraus, M. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 20, Elsevier, NY, 1978, pp. 274–295; Noller, H.; Andréu, P.; Hunger, M. Angew. Chem. Int. Ed. 1971, 10, 172; Berteau, P.; Ruwet, M.; Delmon, B. Bull. Soc. Chim. Belg. 1985, 94, 859.
181. Ashby, E.C.; Willard, G.F.; Goel, A.B. J. Org. Chem. 1979, 44, 1221.
182. Brieger, G.; Watson, S.W.; Barar, D.G.; Shene, A.L. J. Org. Chem. 1979, 44, 1340.
183. Tayama, E.; Sugai, S. Synlett 2006, 849.
184. For a review, see Maercker, A. Angew. Chem. Int. Ed. 1987, 26, 972.
185. Fleming, F.F.; Wang, Q.; Steward, O.W. J. Org. Chem. 2001, 66, 2171.
186. Feely, W.; Boekelheide, V. Org Synth. IV, 298.
187. For a gas phase investigation, see DePuy, C.H.; Bierbaum, V.M. J. Am. Chem. Soc. 1981, 103, 5034.
188. Letsinger, R.L.; Pollart, D.F. J. Am. Chem. Soc. 1956, 78, 6079.
189. Yeh, K.-L.; Liu, B.; Lo, C.-Y.; Huang, H.-L.; Liu, R.-S. J. Am. Chem. Soc. 2002, 124, 6510.
190. See Cohen, T.; Stokes, S. Tetrahedron Lett. 1993, 34, 8023.
191. Morken, J.P.; Didiuk, M.T.; Hoveyda, A.H. J. Am. Chem. Soc. 1993, 115, 6997.
192. Cabrera, G.; Fiaschi, R.; Napolitano, E. Tetrahedron Lett. 2001, 42, 5867.
193. Gassman, P.G.; Burns, S.J. J. Org. Chem. 1988, 53, 5574.
194. Miller, R.D.; McKean, D.R. Tetrahedron Lett. 1982, 23, 323. For another method, see Marsi, M.; Gladysz, J.A. Organometallics 1982, 1, 1467.
195. Negishi, E.; King, A. O.; Klima, W. L.; Patterson, W.; Silveira, A. J. Org. Chem. 1980, 45, 2526.
196. Clasby, M. C.; Craig, D. Synlett 1992, 825.
197. Lyapkalo, I.M.; Vogel, M.A.K.; Boltukhina, E.V.; Vav ík, J. Synlett 2009, 55.
ík, J. Synlett 2009, 55.
198. McEwen, I.; Taylor, R. J. Chem. Soc. Perkin Trans. 2 1982, 1179. See also, Taylor, R. J. Chem. Soc. Perkin Trans. 2 1988, 737.
199. For reviews, see Wong, H.N.C.; Fok, C.C.M.; Wong, T. Heterocycles 1987, 26, 1345; Sonnet, P.E. Tetrahedron 1980, 36, 557, p. 576.
200. Wittig, G.; Haag, W. Chem. Ber. 1955, 88, 1654.
201. Scott, C.B. J. Org. Chem. 1957, 22, 1118.
202. Vedejs, E.; Fuchs, P.L. J. Am. Chem. Soc. 1971, 93, 4070; 1973, 95, 822.
203. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 272–277.
204. Gurudutt, K.N.; Ravindranath, B. Tetrahedron Lett. 1980, 21, 1173.
205. Denis, J.N.; Magnane, R.; Van Eenoo, M.; Krief, A. Nouv. J. Chim. 1979, 3, 705. See Caputo, R.; Mangoni, L.; Neri, O.; Palumbo, G. Tetrahedron Lett. 1981, 22, 3551.
206. Sarma, D.N.; Sharma, R.P. Chem. Ind. (London) 1984, 712.
207. Matsukawa, M.; Tabuchi, T.; Inanaga, J.; Yamaguchi, M. Chem. Lett. 1987, 2101.
208. Mahesh, M.; Murphy, J.A.; Wessel, H.P. J. Org. Chem. 2005, 70, 4118.
209. Hendrickson, J.B.; Walker, M.A.; Varvak, A.; Hussoin, Md.S. Synlett 1996, 661.
210. Isobe, H.; Branchaud, B.P. Tetrahedron Lett. 1999, 40, 8747.
211. Smith, J.G. Synthesis 1984, 629, pp. 637–642; Crandall, J.K.; Apparu, M. Org. React. 1983, 29, 345. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 231–233. See also, Okovytyy, S.; Gorb, L.; Leszczynski, J. Tetrahedron 2001, 57, 1509.
212. Doris, E.; Dechoux, L.; Mioskowski, C. Tetrahedron Lett. 1994, 35, 7943.
213. Thurner, A.; Faigl, F.; Töke, L.; Mordini, A.; Valacchi, M.; Reginato, G.; Czira, G. Tetrahedron 2001, 57, 8173.
214. Hodgson, D.M.; Fleming, M.J.; Stanway, S.J. J. Org. Chem. 2007, 72, 4763.
215. Alcaraz, L.; Cridland, A.; Kinchin, E. Org. Lett. 2001, 3, 4051.
216. Ranu, B.C.; Banerjee, S.; Das, A. Tetrahedron Lett. 2004, 45, 8579.
217. Alcaraz, L.; Cox, K.; Cridland, A.P.; Kinchin, E.; Morris, J.; Thompson, S.P. Org. Lett. 2005, 7, 1399.
218. Righi, G.; Bovicelli, P.; Sperandio, A. Tetrahedron 2000, 56, 1733.
219. Concellón, J.M.; Bardales, E. J. Org. Chem. 2003, 68, 9492. In a similar manner, epoxy amides are converted to conjugated amides, see Concellón, J.M.; Bardales, E. Eur. J. Org. Chem. 2004, 1523.
220. Su, H.; Walder, L.; Zhang, Z.; Scheffold, R. Helv. Chim. Acta 1988, 71, 1073, and references cited therein. Also see, Brookes, P.C.; Milne, D.J.; Murphy, P.J.; Spolaore, B. Tetrahedron 2002, 58, 4675.
221. Alexakis, A.; Vrancken, E.; Mangeney, P. J. Chem. Soc. Perkin Trans. 1 2000, 3354.
222. Equey, O.; ALexakis, A. Tetrahedron Asymmetry 2004, 15, 1069.
223. Bertilsson, S.K.; Södergren, M.J.; Andersson, P.G. J. Org. Chem. 2002, 67, 1567; Bertilsson, S.K.; Andersson, P.G. Tetrahedron 2002, 58, 4665.
224. See Sonnet, P.E. Tetrahedron 1980, 36, 557, see p. 587; Goodman, L.; Reist, E.J. in Kharasch, N.; Meyers, C.Y. The Chemistry of Organic Sulfur Compounds, Vol. 2, Pergamon, Elmsford, NY, 1966, pp. 93–113.
225. Neureiter, N.P.; Bordwell, F.G. J. Am. Chem. Soc. 1959, 81, 578.
226. Calet, S.; Alper, H. Tetrahedron Lett. 1986, 27, 3573.
227. See Latif, N.; Mishriky, N.; Zeid, I. J. Prakt. Chem. 1970, 312, 421.
228. See Helmkamp, G.K.; Pettitt, D.J. J. Org. Chem. 1964, 29, 3258.
229. Aalbersberg, W.G.L.; Vollhardt, K.P.C. J. Am. Chem. Soc. 1977, 99, 2792.
230. Gooßen, L.J.; Rodríguez, N. Chem. Commun. 2004, 724.
231. See DePuy, C.H.; King, R.W. Chem. Rev. 1960, 60, 431, p. 432; Jenneskens, L.W.; Hoefs, C.A.M.; Wiersum, U.E. J. Org. Chem. 1989, 54, 5811, and references cited therein.
232. Aubrey, D.W.; Barnatt, A.; Gerrard, W. Chem. Ind. (London) 1965, 681.
233. See Bailey, W.J.; Bird, C.N. J. Org. Chem. 1977, 42, 3895.
234. Heck, R.F. Palladium Reagents in Organic Synthesis, Academic Press, NY, 1985, pp. 172–178. See Cheng, H.-Y.; Sun, C.-S.; Hou, D.-R. J. Org. Chem. 2007, 72, 2674.
235. Trost, B.M.; Lautens, M.; Peterson, B. Tetrahedron Lett. 1983, 24, 4525.
236. See Nagle, A.S.; Salvataore, R.N.; Cross, R.M.; Kapxhiu, E.A.; Sahab, S.; Yoon, C.H.; Jung, K.W. Tetrahedron Lett. 2003, 44, 5695.
237. DePuy, C.H.; King, R.W. Chem. Rev. 1960, 60, 431, see p. 444; Nace, H.R. Org. React. 1962, 12, 57.
238. Bader, R.F.W.; Bourns, A.N. Can. J. Chem. 1961, 39, 348.
239. Fauré-Tromeur, M.; Zard, S.Z. Tetrahedron Lett. 1999, 40, 1305.
240. For a list of reagents used for sulfonate cleavages, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 294–295.
241. Corey, E.J.; Terashima, S. Tetrahedron Lett. 1972, 111.
242. Corey, E.J.; Posner, G.H.; Atkinson, R.F.; Wingard, A.K.; Halloran, D.J.; Radzik, D.M.; Nash, J.J. J. Org. Chem. 1989, 54, 389.
243. Shimagaki, M.; Fujieda, Y.; Kimura, T.; Nakata, T. Tetrahedron Lett. 1995, 36, 719.
244. Bentley, K.W. in Bentley, K.W.; Kirby, G.W. Elucidation of Organic Structures by Physical and Chemical Methods, 2nd ed. (Vol. 4 of Weissberger, A. Techniques of Chemistry), pt. 2, Wiley, NY, 1973, pp. 255–289; White, E.H.; Woodcock, D.J. in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, pp. 409–416; Cope, A.C.; Trumbull, E.R. Org. React. 1960, 11, 317.
245. Archer, D.A. J. Chem. Soc. C 1971, 1327.
246. Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 4–5.
247. Baumgarten, R.J. J. Chem. Educ. 1968, 45, 122.
248. See Musker, W.K.; Stevens, R.R. J. Am. Chem. Soc. 1968, 90, 3515.
249. Cope, A.C.; Mehta, A.S. J. Am. Chem. Soc. 1963, 85, 1949. See also, Baldwin, M.A.; Banthorpe, D.V.; Loudon, A.G.; Waller, F.D. J. Chem. Soc. B 1967, 509.
250. Cope, A.C.; LeBel, N.A.; Moore, P.T.; Moore, W.R. J. Am. Chem. Soc. 1961, 83, 3861.
251. Cope, A.C.; Trumbull, E.R. Org. React. 1960, 11, 317, see p. 348.
252. Hofmann, A.W. Liebigs Ann. Chem. 1851, 78, 253.
253. See Franke, W.; Ziegenbein, W.; Meister, H. Angew. Chem. 1960, 72, 391, see pp. 397–398.
254. Bach, R.D.; Bair, K.W.; Andrzejewski, D. J. Am. Chem. Soc. 1972, 94, 8608; J. Chem. Soc., Chem. Commun. 1974, 819.
255. Bach, R.D.; Knight, J.W. Tetrahedron Lett. 1979, 3815.
256. Wittig, G.; Burger, T.F. Liebigs Ann. Chem. 1960, 632, 85.
257. Hünig, S.; Öller, M.; Wehner, G. Liebigs Ann. Chem. 1979, 1925.
258. Katritzky, A.R.; Serdyuk, L.; Toader, D.; Wang, X. J. Org. Chem. 1999, 64, 1888.
259. See Cope, A.C.; Trumbull, E.R. Org. React. 1960, 11, 317, see p. 361; DePuy, C.H.; King, R.W. Chem. Rev. 1960, 60, 431, see pp. 448–451.
260. Cope, A.C.; LeBel, N.A. J. Am. Chem. Soc. 1960, 82, 4656; Cope, A.C.; Ciganek, E.; Howell, C.F.; Schweizer, E.E. J. Am. Chem. Soc. 1960, 82, 4663.
261. Závada, J.; Pánková, M.; Svoboda, M. Collect. Czech. Chem. Commun. 1973, 38, 2102.
262. Cram, D.J.; Sahyun, M.R.V.; Knox, G.R. J. Am. Chem. Soc. 1962, 84, 1734.
263. Acevedo, O.; Jorgensen, W.L. J. Am. Chem. Soc. 2006, 128, 6141.
264. See, for example, Bach, R.D.; Andrzejewski, D.; Dusold, L.R. J. Org. Chem. 1973, 38, 1742.
265. Sammelson, R.E.; Kurth, M.J. Tetrahedron Lett. 2001, 42, 3419.
266. Aitken, R.A.; Atherton, J.I. J. Chem. Soc. Perkin Trans. 1 1994, 1281.
267. Aitken, R.A.; Hérion, H.; Janosi, A.; Karodia, N.; Raut, S.V.; Seth, S.; Shannon, I.J.; Smith, F.C. J. Chem. Soc. Perkin Trans. 1 1994, 2467.
268. Aitken, R.A.; Boeters, C.; Morrison, J.J. J. Chem. Soc. Perkin Trans. 1 1994, 2473.
269. DelZotto, A.; Baratta, W.; Miani, F.; Verardo, G.; Rigo, P. Eur. J. Org. Chem. 2000, 3731.
270. See Adlington, R.M.; Barrett, A.G.M. Acc. Chem. Res. 1983, 16, 55; Shapiro, R.H. Org. React. 1976, 23, 405.
271. See Barluenga, J.; Moriel, P.; Valdés, C.; Aznar, F. Angew. Chem. Int. Ed. 2007, 46, 5587.
272. Shapiro, R.H. Tetrahedron Lett. 1968, 345; Meinwald, J.; Uno, F. J. Am. Chem. Soc. 1968, 90, 800.
273. Stemke, J.E.; Bond, F.T. Tetrahedron Lett. 1975, 1815.
274. See Dauben, W.G.; Rivers, G.T.; Zimmerman, W.T. J. Am. Chem. Soc. 1977, 99, 3414.
275. For a review of the mechanism, see Casanova, J.; Waegell, B. Bull. Soc. Chim. Fr. 1975, 922.
276. See Ref. 272; Shapiro, R.H.; Hornaman, E.C. J. Org. Chem. 1974, 39, 2302.
277. Lipton, M.F.; Shapiro, R.H. J. Org. Chem. 1978, 43, 1409.
278. See Traas, P.C.; Boelens, H.; Takken, H.J. Tetrahedron Lett. 1976, 2287; Stemke, J.E.; Chamberlin, A.R.; Bond, F.T. Tetrahedron Lett. 1976, 2947.
279. For a review, see Chamberlin, A.R.; Bloom, S.H. Org. React. 1990, 39, 1.
280. Maruoka, K.; Oishi, M.; Yamamoto, H. J. Am. Chem. Soc. 1996, 118, 2289.
281. Biellmann, J.F.; Pète, J. Bull. Soc. Chim. Fr. 1967, 675.
282. Bamford, W.R.; Stevens, R.R. J. Chem. Soc. 1952, 4735. For a tandem Bamford–Stevens–Claisen rearrangement, see May, J.A.; Stoltz, B.M. J. Am. Chem. Soc. 2002, 124, 12426.
283. See Nickon, A.; Werstiuk, N.H. J. Am. Chem. Soc. 1972, 94, 7081.
284. See Regitz, M.; Maas, G. Diazo Compounds, Academic Press, NY, 1986, pp. 257–295. For an improved procedure, see Wulfman, D.S.; Yousefian, S.; White, J.M. Synth. Commun. 1988, 18, 2349.
285. Godfrey, A.G.; Ganem, B. J. Am. Chem. Soc. 1990, 112, 3717.
286. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 161–171.
287. See Cubbage, J.W.; Guo, Y.; McCulla, R.D.; Jenks, W.S. J. Org. Chem. 2001, 66, 8722.
288. See Yoshida, T.; Saito, S. Chem. Lett. 1982, 165.
289. Hofmann, J.E.; Wallace, T.J.; Argabright, P.A.; Schriesheim, A. Chem. Ind. (London) 1963, 1234.
290. Gai, Y.; Jin, L.; Julia, M.; Verpeaux, J.-N. J. Chem. Soc., Chem. Commun. 1993, 1625.
291. Hofmann, J.E.; Wallace, T.L.; Schriesheim, A. J. Am. Chem. Soc. 1964, 86, 1561.
292. Schmitz, C.; Harvey, J.N.; Viehe, H.G. Bull. Soc. Chim. Belg. 1994, 103, 105.
293. Back, T.G. in Patai, S. The Chemistry of Organic Selenium and Telurium Compounds, Vol. 2, Wiley, NY, 1987, pp. 91–213, pp. 95–109; Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis, Pergamon, Elmsford, NY, 1986, pp. 132–143; Reich, H.J. Acc. Chem. Res. 1979, 12, 22, in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. C, Academic Press, NY, 1978, pp. 15–101; Sharpless, K.B.; Gordon, K.M.; Lauer, R.F.; Patrick, D.W.; Singer, S.P.; Young, M.W. Chem. Scr. 1975, 8A; 9. See Liotta, D. Organoselenium Chemistry, Wiley, NY, 1987.
294. Jones, D.N.; Higgins, W. J. Chem. Soc. C 1970, 81.
295. Reich, H.J.; Willis Jr., W.W. J. Am. Chem. Soc. 1980, 102, 5967.
296. Reich, H.J.; Renga, J.M.; Reich, I.L. J. Am. Chem. Soc. 1975, 97, 5434 and references cited therein; Crich, D.; Barba, G.R. Org. Lett. 2000, 2, 989. For lists of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 287–290.
297. Trost, B.M.; Salzmann, T.N.; Hiroi, K. J. Am. Chem. Soc. 1976, 98, 4887. For a review of this and related methods, see Trost, B.M. Acc. Chem. Res. 1978, 11, 453.
298. de Groot, A.; Jansen, B.J.M.; Reuvers, J.T.A.; Tedjo, E.M. Tetrahedron Lett. 1981, 22, 4137.
299. Imboden, C.; Villar, F.; Renaud, P. Org. Lett. 1999, 1, 873.
300. See Baciocchi, E. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement D, pt. 2, Wiley, NY, 1983, pp. 1173–1227.
301. See Anton, D.R.; Crabtree, R.H. Tetrahedron Lett. 1983, 24, 2449.
302. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 256–258.
303. Vogel, E.; Klärner, F. Angew. Chem. Int. Ed. 1968, 7, 374.
304. Oediger, H.; Möller, F. Angew. Chem. Int. Ed. 1967, 6, 76; Wolkoff, P. J. Org. Chem. 1982, 47, 1944.
305. See Oediger, H.; Möller, F.; Eiter, K. Synthesis 1972, 591.
306. Clayden, J. Organolithiums: Selectivity for Synthesis, Pergamon, New York, 2002.; For a mechanistic evaluation and an analysis of the influence of HMPA, see Ma, Y.; Ramirez, A.; Singh, K.J.; Keresztes, I.; Collum, D.B. J. Am. Chem. Soc. 2006, 128, 15399.
307. Schwesinger, R.; Schlemper, H. Angew. Chem. Int. Ed. 1987, 26, 1167.
308. Kobayashi, T.; Ohmiya, H.; Yorimitsu, H.; Oshima, K. J. Am. Chem. Soc. 2008, 130, 11276.
309. Halpern, M.; Zahalka, H.A.; Sasson, Y.; Rabinovitz, M. J. Org. Chem. 1985, 50, 5088. See also, Barry, J.; Bram, G.; Decodts, G.; Loupy, A.; Pigeon, P.; Sansoulet, J. J. Org. Chem. 1984, 49, 1138.
310. See Fieser, L.F.; Fieser, M. Reagents for Organic Syntheses, Vol. 1, Wiley, NY, 1967, pp. 606–609; Yakobson, G.G.; Akhmetova, N.E. Synthesis 1983, 169, see pp. 170–173.
311. See Hoye, T.R.; van Deidhuizen, J.J.; Vos, T.J.; Zhao, P. Synth. Commun. 2001, 31, 1367.
312. Matsubara, S.; Matsuda, H.; Hamatani, T.; Schlosser, M. Tetrahedron 1988, 44, 2855.
313. Ben-Efraim, D.A. in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, pt. 2, Wiley, NY, 1978, p. 755; Köbrich, G.; Buck, P. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 100–134; Köbrich, G. Angew. Chem. Int. Ed. 1965, 4, 49, see pp. 50–53.
314. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 569–571.
315. Okutani, M.; Mori, Y. Tetrahedron Lett. 2007, 48, 6856; Okutani, M.; Mori, Y. J. Org. Chem. 2009, 74, 442.
316. Chelucci, G.; Capitta, F.; Baldino, S. Tetrahedron 2008, 64, 10250.
317. Chernick, E.T.; Eisler, S.; Tykwinski, R.R. Tetrahedron Lett. 2001, 42, 8575.
318. Fritsch, P. Ann. 1894, 279, 319; Buttenberg, W.P. Ann., 1894, 279, 324; Wiechell, H. Ann. 1894, 279, 337. For a review, see Stang, P.J. Chem. Rev. 1978, 78, 383. See Jahnke, E.; Tykwinski, R.R. Chem. Commun. 2010, 3235; Pratt, L.M.; Nguyen, N.V.; Kwon, O. Chem. Lett. 2009, 38, 574.
319. For a review, see Noller, H.; Andréu, P.; Hunger, M. Angew. Chem. Int. Ed. 1971, 10, 172.
320. Duhamel, L.; Ravard, A.; Plaquevent, J.C.; Plé, G.; Davoust, D. Bull. Soc. Chim. Fr. 1990, 787.
321. See Tidwell, T.T. Ketenes, Wiley, NY, 1995.
322. Taggi, A.E.; Wack, H.; Hafez, A.M.; France, S.; Lectka, T. Org. Lett. 2002, 4, 627.
323. See Luknitskii, F.I.; Vovsi, B.A. Russ. Chem. Rev. 1969, 38, 487.
324. See King, J.F. Acc. Chem. Res. 1975, 8, 10; Nagai, T.; Tokura, N. Int. J. Sulfur Chem. Part B 1972, 207; Truce, W.E.; Liu, L.K. Mech. React. Sulfur Compd. 1969, 4, 145; Opitz, G. Angew. Chem. Int. Ed. 1967, 6, 107; Wallace, T.J. Q. Rev. Chem. Soc. 1966, 20, 67.
325. Brown, H.C.; Bhatt, M.V.; Munekata, T.; Zweifel, G. J. Am. Chem. Soc. 1967, 89, 567; Taniguchi, H. Bull. Chem. Soc. Jpn. 1979, 52, 2942.
326. Hubert, A.J. J. Chem. Soc. 1965, 6669.
327. Abidi, S.L. Tetrahedron Lett. 1986, 27, 267; J. Org. Chem. 1986, 51, 2687.
328. Corey, E.J.; Seibel, W.L.; Kappos, J.C. Tetrahedron Lett. 1987, 28, 4921.
329. For a review, see Tsuji, J.; Ohno, K. Synthesis 1969, 157. For extensions to certain other acid derivatives, see Minami, I.; Nisar, M.; Yuhara, M.; Shimizu, I.; Tsuji, J. Synthesis 1987, 992.
330. Lau, K.S.Y.; Becker, Y.; Huang, F.; Baenziger, N.; Stille, J.K. J. Am. Chem. Soc. 1977, 99, 5664.
331. Sharpless, K.B.; Umbreit, M.A.; Nieh, T.; Flood, T.C. J. Am. Chem. Soc. 1972, 94, 6538.
332. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 297–299.
333. McMurry, J.E. Acc. Chem. Res. 1983, 16, 405 and references cited therein.
334. Sarma, J.C.; Sharma, R.P. Chem. Ind. (London) 1987, 96.
335. Cook, G.K.; Andrews, M.A. J. Am. Chem. Soc. 1996, 118, 9448.
336. Carnahan, Jr., J.C.; Closson, W.D. Tetrahedron Lett. 1972, 3447.
337. Dafaye, J. Bull. Soc. Chim. Fr. 1968, 2099.
338. Barrett, A.G.M.; Barton, D.H.R.; Bielski, R. J. Chem. Soc. Perkin Trans. 1 1979, 2378.
339. See Block, E. Org. React. 1984, 30, 457; Sonnet, P.E. Tetrahedron 1980, 36, 557, pp. 593–598; Mackie, R.K. in Cadogan, J.I.G. Organophosphorus Reagents in Organic Synthesis, Academic Press, NY, 1979, pp. 354–359.
340. Corey, E.J.; Winter, R.A.E. J. Am. Chem. Soc. 1963, 85, 2677.
341. Corey, E.J. Pure Appl. Chem. 1967, 14, 19, see pp. 32–33.
342. Semmelhack, M.F.; Stauffer, R.D. Tetrahedron Lett. 1973, 2667. For another method, see Vedejs, E.; Wu, E.S.C. J. Org. Chem. 1974, 39, 3641.
343. Corey, E.J.; Hopkins, P.B. Tetrahedron Lett. 1982, 23, 1979.
344. Corey, E.J.; Carey, F.A.; Winter, R.A.E. J. Am. Chem. Soc. 1965, 87, 934.
345. See Beels, C.M.D.; Coleman, M.J.; Taylor, R.J.K. Synlett 1990, 479.
346. Paquette, L.A. Org. React. 1977, 25, 1; Mech. Mol. Migr. 1968, 1, 121; Acc. Chem. Res. 1968, 1, 209; Meyers, C.Y.; Matthews, W.S.; Ho, L.L.; Kolb, V.M.; Parady, T.E. in Smith, G.V. Catalysis in Organic Synthesis, Academic Press, NY, 1977, pp. 197–278; Rappe, C. in Patai, S. The Chemistry of the Carbon-Halogen Bond, pt. 2, Wiley, NY, 1973, pp. 1105–1110; Bordwell, F.G. Acc. Chem. Res. 1970, 3, 281.
347. Hartman, G.D.; Hartman, R.D. Synthesis 1982, 504.
348. See Bordwell, F.G.; Wolfinger, M.D. J. Org. Chem. 1974, 39, 2521; Bordwell, F.G.; Doomes, E. J. Org. Chem. 1974, 39, 2526, 2531.
349. Sutherland, A.G.; Taylor, R.J.K. Tetrahedron Lett. 1989, 30, 3267.
350. Bordwell, F.G.; Williams, Jr., J.M.; Hoyt, Jr., E.B.; Jarvis, B.B. J. Am. Chem. Soc. 1968, 90, 429; Bordwell, F.G.; Williams, Jr., J.M. J. Am. Chem. Soc. 1968, 90, 435.
351. Block, E.; Aslam, M.; Eswarakrishnan, V.; Gebreyes, K.; Hutchinson, J.; Iyer, R.; Laffitte, J.; Wall, A. J. Am. Chem. Soc. 1986, 108, 4568.
352. Hendrickson, J.B.; Boudreaux, G.J.; Palumbo, P.S. J. Am. Chem. Soc. 1986, 108, 2358.
353. Vasin, V.A.; Bolusheva, I.Yu.; Razin, V.V. Russ. J. Org. Chem. 2010, 46, 758.
354. Mock, W.L. J. Am. Chem. Soc. 1966, 88, 2857; McGregor, S.D.; Lemal, D.M. J. Am. Chem. Soc. 1966, 88, 2858.
355. Mock, W.L. J. Am. Chem. Soc. 1969, 91, 5682.
356. Bordwell, F.G.; Williams, Jr., J.M.; Hoyt, Jr., E.B.; Jarvis, B.B. J. Am. Chem. Soc. 1968, 90, 429; Bordwell, F.G.; Williams, Jr., J.M. J. Am. Chem. Soc. 1968, 90, 435. See also, Vilsmaier, E.; Tropitzsch, R.; Vostrowsky, O. Tetrahedron Lett. 1974, 3987.
357. See Mock, W.L. in Marchand, A.P.; Lehr, R.E. Pericyclic Reactions, Vol. 2, Academic Press, NY, 1977, pp. 141–179.
358. Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry, Academic Press, NY, 1970, pp. 152–163.
359. Philips, J.C.; Swisher, J.V.; Haidukewych, D.; Morales, O. Chem. Commun. 1971, 22.
360. Philips, J.C.; Morales, O. J. Chem. Soc., Chem. Commun. 1977, 713.
361. Mitchell, R.H. Tetrahedron Lett. 1973, 4395. For a similar reaction without base treatment, see Pommelet, J.; Nyns, C.; Lahousse, F.; Merényi, R.; Viehe, H.G. Angew. Chem. Int. Ed. 1981, 20, 585.
362. MacGee, D.I.; Beck, E.J. J. Org. Chem. 2000, 65, 8367.
363. See Sonnet, P.E. Tetrahedron 1980, 36, 557, see p. 591; Dermer, O.C.; Ham, G.E. Ethylenimine and other Aziridines, Academic Press, NY, 1969, pp. 293–295.
364. See Carlson, R.M.; Lee, S.Y. Tetrahedron Lett. 1969, 4001.
365. Imamoto, T.; Yukawa, Y. Chem. Lett. 1974, 165.
366. Heine, H.W.; Myers, J.D.; Peltzer, III, E.T. Angew. Chem. Int. Ed. 1970, 9, 374.
367. Hodgson, D.M.; Štefane, B.; Miles, T.J.; Witherington, J. J. Org. Chem. 2006, 71, 8510.
368. Sugihara, Y.; Iimura, S.; Nakayama, J. Chem. Commun. 2002, 134.
369. Chao, B.; Dittmer, D.C. Tetrahedron Lett. 2001, 42, 5789.
370. Yadav, J.S.; Bandyapadhyay, A.; Reddy, B.V.S. Synlett 2001, 1608.
371. Baciocchi, E. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement D, pt. 1, Wiley, NY, 1983, pp. 161–201. Also see, Bosser, G.; Paris, J. J. Chem. Soc. Perkin Trans. 2 1992, 2057.
372. Khurana, J.M.; Bansal, G.; Chauhan, S. Bull. Chem. Soc. Jpn. 2001, 74, 1089.
373. Fukunaga, K.; Yamaguchi, H. Synthesis 1981, 879. See also, Nakayama, J.; Machida, H.; Hoshino, M. Tetrahedron Lett. 1983, 24 3001; Landini, D.; Milesi, L.; Quadri, M.L.; Rolla, F. J. Org. Chem. 1984, 49, 152.
374. For a lists of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 259–263.
375. See Shono, T. Electroorganic Chemistry as a New Tool in Organic Synthesis, Springer, NY, 1984, pp. 145–147; Fry, A.J. Synthetic Organic Electrochemistry, 2nd ed., Wiley, NY, 1989, pp. 151–154.
376. Ranu, B.C.; Guchhait, S.K.; Sarkar, A. Chem. Commun. 1998, 2113.
377. Yanada, R.; Negoro, N.; Yanada, K.; Fujita, T. Tetrahedron Lett. 1996, 37, 9313.
378. Ranu, B.C.; Das, A.; Hajra, A. Synthesis 2003, 1012.
379. Gaenzler, F.C.; Smith, M.B. Synlett 2007, 1299.
380. Malanga, C.; Aronica, L.A.; Lardicci, L. Tetrahedron Lett. 1995, 36, 9189.
381. See Gordon, M.; Hay, J.V. J. Org. Chem. 1968, 33, 427.
382. See Sicher, J.; Havel, M.; Svoboda, M. Tetrahedron Lett. 1968, 4269.
383. Ranu, B.C.; Jana, R. J. Org. Chem. 2005, 70, 8621.
384. Kuang, C.; Senboku, H.; Tokuda, M. Tetrahedron Lett. 2001, 42, 3893.
385. Aruna, S.; Kalyanakumar, R.; Ramakrishnan, V.T. Synth. Commun. 2001, 31, 3125.
386. See Schuster, H.F.; Coppola, G.M. Allenes in Organic Synthesis, Wiley, NY, 1984, pp. 9–56; Landor, P.D. in Landor, S.R. The Chemistry of the Allenes, Vol. 1, Academic Press, NY, 1982; pp. 19–233; Taylor, D.R. Chem. Rev. 1967, 67, 317.
387. Wang, X.; Ramos, B.; Rodriguez, A. Tetrahedron Lett. 1994, 35, 6977.
388. Engman, L.; Byström, S.E. J. Org. Chem. 1985, 50, 3170.
389. Yoshida, M.; Gotou, T.; Ihara, M. Tetrahedron Lett. 2004, 45, 5573.
390. Nakmura, H.; Kamakura, T.; Ishikura, M.; Biellmann, J.-F. J. Am. Chem. Soc. 2004, 126, 5958.
391. Azizoglu, A.; Balci, M.; Mieusset, J.-L.; Brinker, U.H. J. Org. Chem. 2008, 73, 8182.
392. See Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 332–368; Baciocchi, W. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement D, pt. 2, Wiley, NY, 1983, p. 161.
393. Darling, S.D.; Kidwell, R.L. J. Org. Chem. 1968, 33, 3974.
394. See McCarney, C.C.; Ward, R.S. J. Chem. Soc. Perkin Trans. 1 1975, 1600. See also, Masters, A.P.; Sorensen, T.S.; Ziegler, T. J. Org. Chem. 1986, 51, 3558.
395. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 263–267, for reagents that produce olefins from β-halo ethers and esters, and from halohydrins.
396. Park, H.S.; Kim, S.H.; Park, M.Y.; Kim, Y.H. Tetrahedron Lett. 2001, 42, 3729.
397. Reeve, W.; Brown, R.; Steckel, T.F. J. Am. Chem. Soc. 1971, 93, 4607.
398. Gurien, H. J. Org. Chem. 1963, 28, 878.
399. Amstutz, E.D. J. Org. Chem. 1944, 9, 310.
400. Concellón, J.M.; Bernad, P.L.; Bardales, E. Org. Lett. 2001, 3, 937.
401. Yadav, J.S.; Bandyopadhyay, A.; Reddy, B.V.S. Tetrahedron Lett. 2001, 42, 6385.
402. Concellón, J.M.; Pérez-Andrés, J.A.; Rodríguez-Solla, H. Chem. Eur. J. 2001, 7, 3062.
403. House, H.O.; Ro, R.S. J. Am. Chem. Soc. 1965, 87, 838.
404. McMurry, J.E.; Hoz, T. J. Org. Chem. 1975, 40, 3797.
405. Becker, K.B.; Grob, C.A. in Patai, S. The Chemistry of Functional Groups, Supplement A, pt. 2, Wiley, NY, 1977, pp. 653–723; Grob, C.A. Angew. Chem. Int. Ed. 1969, 8, 535; Grob, C.A.; Schiess, P.W. Angew. Chem. Int. Ed. 1967, 6, 1.
406. Grob, C.A.; Ostermayer, F.; Raudenbusch, W. Helv. Chim. Acta 1962, 45, 1672.
407. Prelog, V.; Zalán, E. Helv. Chim. Acta 1944, 27, 535; Prelog, V.; Häfliger, O. Helv. Chim. Acta 1950, 33, 2021.
408. Grob, C.A. Angew. Chem. Int. Ed. 1969, 8, 535 and references cited therein; Grob, C.A.; Kiefer, H.R.; Lutz, H.J.; Wilkens, H.J. Helv. Chim. Acta 1967, 50, 416.
409. See Prantz, K.; Mulzer, J. Chem. Rev. 2010, 110, 3741.
410. Fischer, W.; Grob, C.A. Helv. Chim. Acta 1978, 61, 2336 and references cited therein.
411. Geisel, M.; Grob, C.A.; Wohl, R.A. Helv. Chim. Acta 1969, 52, 2206 and references cited therein.
412. Chass, D.A.; Buddhsukh, D.; Magnus, P.D. J. Org. Chem. 1978, 43, 1750.
413. Grob, C.A.; Hoegerle, R.M.; Ohta, M. Helv. Chim. Acta 1962, 45, 1823.
414. Zimmerman, H.E.; English Jr., J. J. Am. Chem. Soc. 1954, 76, 2285, 2291, 2294.
415. For a review, see Caine, D. Org. Prep. Proced. Int. 1988, 20, 1.
416. Hara, S.; Taguchi, H.; Yamamoto, H.; Nozaki, H. Tetrahedron Lett. 1975, 1545.
417. See Rüttimann, A.; Wick, A.; Eschenmoser, A. Helv. Chim. Acta 1975, 58, 1450.
418. Mulzer, J.; Brüntrup, G. Tetrahedron Lett. 1979, 1909.
419. For another method, see Tanzawa, T.; Schwartz, J. Organometallics 1990, 9, 3026.
420. Mulzer, J.; Lammer, O. Angew. Chem. Int. Ed. 1983, 22, 628.
421. See Adam, W.; Martinez, G.; Thompson, J.; Yany, F. J. Org. Chem. 1981, 46, 3359.
422. Mulzer, J.; Zippel, M.; Brüntrup, G. Angew. Chem. Int. Ed. 1980, 19, 465; Mulzer, J.; Zippel, M. Tetrahedron Lett. 1980, 21, 751. See also, Moyano, A.; Pericàs, M.A.; Valentí, E. J. Org. Chem. 1989, 573.
423. See MacAlpine, G.A.; Warkentin, J. Can. J. Chem. 1978, 56, 308, and references cited therein.
424. Eschenmoser, A.; Felix, D.; Ohloff, G. Helv. Chim. Acta 1967, 50, 708; Tanabe, M.; Crowe, D.F.; Dehn, R.L.; Detre, G. Tetrahedron Lett. 1967, 3739; Tanabe, M.; Crowe, D.F.; Dehn, R.L. Tetrahedron Lett. 1967, 3943.
425. Corey, E.J.; Sachdev, H.S. J. Org. Chem. 1975, 40, 579.
426. Felix, D.; Müller, R.K.; Horn, U.; Joos, R.; Schreiber, J.; Eschenmoser, A. Helv. Chim. Acta 1972, 55, 1276.
427. See Stark, B.P.; Duke, A.J. Extrusion Reactions, Pergamon, Elmsford, NY, 1967, pp. 16–46.
428. Birney, D.M.; Wiberg, K.B.; Berson, J.A. J. Am. Chem. Soc. 1988, 110, 6631.
429. See Ogliaruso, M.A.; Romanelli, M.G.; Becker, E.I. Chem. Rev. 1965, 65, 261, pp. 300–348. For references to this and related reactions, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 207–213.
430. Friedrich, K. in Patai, S.; Rappoport, Z. The Chemistry of the Carbon-Carbon Triple Bond, pt. 2, Wiley, NY, 1978, pp. 1345–1390; Friedrich, K.; Wallenfels, K. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 92–96; Fatiadi, K. in Friedrich, K. in Patai, S.; Rappoport, Z. The Chemistry of the Carbon–Carbon Triple Bond, pt. 2, Wiley, NY, 1978, pp. 1057–1303.
431. Attanasi, O.; Palma, P.; Serra-Zanetti, F. Synthesis 1983, 741; Jursic, B. Synth. Commun. 1989, 19, 689.
432. Kim, J.N.; Chung, K.H.; Ryu, E.K. Synth. Commun. 1990, 20, 2785.
433. Narsaiah, A.V.; Sreenu, D.; Nagaiah, K. Synth. Commun. 2006, 36, 137.
434. Kim, H.S.; Kim, S.H.; Kim, J.N. Tetrahedron Lett. 2009, 50, 1717.
435. Desai, D.G.; Swami, S.S.; Mahale, G.D. Synth. Commun. 2000, 30, 1623.
436. Jiang, N.; Ragauskas, A.J. Tetrahedron Lett. 2010, 51, 4479.
437. Hosseini Sarvari, M. Synthesis 2005, 787.
438. Supsana, P.; Liaskopoulos, T.; Tsoungas, P.G.; Varvounis, G. Synlett 2007, 267.
439. Yang, S.H.; Chang, S. Org. Lett. 2001, 3, 4209.
440. Miller, C.P.; Kaufman, D.H. Synlett 2000, 1169.
441. Sarvari, M.H. Synthesis 2005, 787.
442. Lingaiah, N.; Narender, R. Synth. Commun. 2002, 32, 2391.
443. Bose, D.S.; Narsaiah, A.V. Tetrahedron Lett. 1998, 39, 6533.
444. Das, B.; Ramesh, C.; Madhusudhan, P. Synlett 2000, 1599.
445. See Sharghi, H.; Sarvari, M.H. Synthesis 2003, 243.
446. Anand, N.; Owston, N.A.; Parker, A.J.; Slatford, P.A.; Willia, J.M.J. Tetrahedron Lett. 2007, 48, 7761.
447. Hauser, C.R.; Le Maistre, J.W.; Rainsford, A.E. J. Am. Chem. Soc. 1935, 57, 1056; Pyun, S.Y.; Lee, D.C.; Seung, Y.J.; Cho, B.R. J. Org. Chem. 2005, 70, 5327.
448. Roberts, J.T.; Rittberg, B.R.; Kovacic, P. J. Org. Chem. 1981, 46, 4111.
449. See Ioffe, B.V.; Zelenina, N.L. J. Org. Chem. USSR, 1968, 4, 1496.
450. Moore, J.S.; Stupp, S.I. J. Org. Chem. 1990, 55, 3374.
451. Cuvigny, T.; Le Borgne, J.F.; Larchevêque, M.; Normant, H. Synthesis 1976, 237.
452. Cicchi, S.; Cardona, F.; Brandi, A.; Corsi, M.; Goti, A. Tetrahedron Lett. 1999, 40, 1989.
453. Gawley, R.E. Org. React. 1988, 35, 1; McCarty, C.G. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 416–439; Casanova, J. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 915–932.
454. See Olah, G.A.; Vankar, Y.D.; Berrier, A.L. Synthesis 1980, 45; Conley, R.T.; Ghosh, S. Mech. Mol. Migr. 1971, 4, 197.
455. Fischer, H.P.; Grob, C.A. Helv. Chim. Acta 1963, 46, 936.
456. See Ferris, A.F. J. Org. Chem. 1960, 25, 12.
457. See Hill, R.K.; Conley, R.T. J. Am. Chem. Soc. 1960, 82, 645.
458. Hassner, A.; Nash, E.G. Tetrahedron Lett. 1965, 525.
459. Grob, C.A.; Fischer, H.P.; Raudenbusch, W.; Zergenyi, J. Helv. Chim. Acta 1964, 47, 1003.
460. Grob, C.A.; Sieber, A. Helv. Chim. Acta 1967, 50, 2520; Green, M.; Pearson, S.C. J. Chem. Soc. B 1969, 593.
461. Bieron J.F.; Dinan, F.J. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 274–283; Friedrich, K.; Wallenfels, K. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 96–103; Friedrich, K. in Patai, S.; Rapoport, Z. The Chemistry of Functional Groups, Supplement C, pt. 2, Wiley, NY, 1978, p. 1345.
462. Harrison, C.R.; Hodge, P.; Rogers, W.J. Synthesis 1977, 41.
463. Monson, R.S.; Priest, D.N. Can. J. Chem. 1971, 49, 2897.
464. Ruck, R.T.; Bergman, R.G. Angew. Chem. Int. Ed. 2004, 43, 5375.
465. Claremon, D.A.; Phillips, B.T. Tetrahedron Lett. 1988, 29, 2155.
466. Barger, T.M.; Riley, C.M. Synth. Commun. 1980, 10, 479.
467. Boruah, M.; Konwar, D. J. Org. Chem. 2002, 67, 7138.
468. Iranpoor, N.; Firouzabadi, H.; Aghapoor, G. Synth. Commun. 2002, 32, 2535.
469. Nakajima, N.; Ubukata, M. Tetrahedron Lett. 1997, 38, 2099.
470. Bhalerao, D.S.; Mahajan, U.S.; Chaudhari, K.H.; Akamanchi, K.G. J. Org. Chem. 2007, 72, 662.
471. Maffioli, S.I.; Marzorati, E.; Marazzi, A. Org. Lett. 2005, 7, 5237.
472. Zhou, S.; Junge, K.; Addis, D.; Das, S.; Beller, M. Org. Lett. 2009, 11, 2461.
473. Zhou, S.; Addis, D.; Das, S.; Junge, K.; Beller, M. Chem. Commun. 2009, 4883.
474. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1983–1985.
475. Heck, M.-P.; Wagner, A.; Mioskowski, C. J. Org. Chem. 1996, 61, 6486.
476. See Imamoto, T.; Takaoka, T.; Yokoyama, M. Synthesis 1983, 142.
477. For a list of methods, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1949–1950.
478. Schroth, W.; Kluge, H.; Frach, R.; Hodek, W.; Schädler, H.D. J. Prakt. Chem. 1983, 325, 787.
479. Sivakumar, M.; Senthilkumar, P.; Pandit, A.B. Synth. Commun. 2001, 31, 2583.
480. See Creedon, S.M.; Crowley, H.K.; McCarthy, D.G. J. Chem. Soc. Perkin Trans. 1 1998, 1015.
481. Hoffmann, P.; Gokel, G.W.; Marquarding, D.; Ugi, I. in Ugi, I. Isonitrile Chemistry, Academic Press, NY, 1971, pp. 10–17; Ugi, I.; Fetzer, U.; Eholzer, U.; Knupfer, H.; Offermann, K. Angew. Chem. Int. Ed. 1965, 4, 472; Newer Methods Prep. Org. Chem. 1968, 4, 37.
482. See Obrecht, R.; Herrmann, R.; Ugi, I. Synthesis 1985, 400.
483. Baldwin, J.E.; O'Neil, I.A. Synlett 1991, 603.
484. Porcheddu, A.; Giacomelli, G.; Salaris, M. J. Org. Chem. 2005, 70, 2361.
485. Appel, R.; Kleinstück, R.; Ziehn, K. Angew. Chem. Int. Ed. 1971, 10, 132.
486. Guirado, A.; Zapata, A.; Gómez, J.L.; Trebalón, L.; Gálvez, J. Tetrahedron 1999, 55, 9631.
487. Bocharov, B.V. Russ. Chem. Rev. 1965, 34, 212;Williams, A.; Ibrahim, I.T. Chem. Rev. 1981, 81, 589.
488. Also see Kim, S.; Yi, K.Y. J. Org. Chem. 1986, 51, 2613, Tetrahedron Lett. 1986, 27, 1925.
489. Jászay, Z.M.; Petneházy, I.; Töke, L.; Szajáni, B. Synthesis 1987, 520.
490. Mitsunobu, O.; Kato, K.; Tomari, M. Tetrahedron 1970, 26, 5731.
491. Arnold, R.T.; Smolinsky, G. J. Am. Chem. Soc. 1959, 81, 6643. For a review, see Marvell, E.N.; Whalley, W. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 2, Wiley, NY, 1971, pp. 729–734.
492. Voorhees, K.J.; Smith, G.G. J. Org. Chem. 1971, 36, 1755.
493. Arnold, R.T.; Smolinsky, G. J. Org. Chem. 1960, 25, 128; Smith, G.G.; Taylor, R. Chem. Ind. (London) 1961, 949.
494. Viola, A.; Proverb, R.J.; Yates, B.L.; Larrahondo, J. J. Am. Chem. Soc. 1973, 95, 3609.
495. Kwart, H.; Slutsky, J.; Sarner, S.F. J. Am. Chem. Soc. 1973, 95, 5242; Egger, K.W.; Vitins, P. Int. J. Chem. Kinet. 1974, 6, 429.
496. Regitz, M.; Maas, G. Diazo Compounds; Academic Press, NY, 1986, pp. 296–325; Black, T.H. Aldrichimica Acta 1983, 16, 3. See Cowell, G.W.; Ledwith, A. Q. Rev. Chem. Soc. 1970, 24, 119, pp. 126–131; Smith, P.A.S. Open-chain Nitrogen Compounds, W. A. Benjamin, NY, 1966, pp. 257–258, 474–475, in Vol. 2.
497. de Boer, T.J.; Backer, H.J. Org. Synth. IV, 225, 250; Hudlicky, M. J. Org. Chem. 1980, 45, 5377.
498. Searle, C.E. Chem. Br. 1970, 6, 5.
499. Stark, B.P.; Duke, A.J. Extrusion Reactions, Pergamon, Elmsford, NY, 1967. For a review of extrusions that are photochemically induced, see Givens, R.S. Org. Photochem. 1981, 5, 227.
500. Paine, A.J.; Warkentin, J. Can. J. Chem. 1981, 59, 491.
501. Van Auken, T.V.; Rinehart, Jr., K.L. J. Am. Chem. Soc. 1962, 84, 3736.
502. Adam, W.; De Lucchi, O. Angew. Chem. Int. Ed. 1980, 19, 762; Stark, B.P.; Duke, A.J. Extrusion Reactions, Pergamon, Elmsford, NY, 1967, pp. 116–151. See Mackenzie, K. in Patai, S. The Chemistry of the Hydrazo, Azo, and Azoxy Groups, pt. 1, Wiley, NY, 1975, pp. 329–442.
503. See Jones, W.M.; Sanderfer, P.O.; Baarda, D.G. J. Org. Chem. 1967, 32, 1367.
504. McGreer, D.E.; Wai, W.; Carmichael, G. Can. J. Chem. 1960, 38, 2410; Kocsis K.; Ferrini, P.G.; Arigoni, D.; Jeger, O. Helv. Chim. Acta 1960, 43, 2178.
505. For a review, see Scheiner, P. Sel. Org. Transform. 1970, 1, 327.
506. See Sammes, M.P.; Katritzky, A.R. Adv. Heterocycl. Chem. 1983, 34, 2.
507. See Pincock, J.A.; Morchat, R.; Arnold, D.R. J. Am. Chem. Soc. 1973, 95, 7536.
508. Engel, P.S. Chem. Rev. 1980, 80, 99; Engel, P.S.; Nalepa, C.J. Pure Appl. Chem. 1980, 52, 2621; Reedich, D.E.; Sheridan, R.S. J. Am. Chem. Soc. 1988, 110, 3697.
509. Pincock, J.A.; Morchat, R.; Arnold, D.R. J. Am. Chem. Soc. 1973, 95, 7536.
510. See Redmore, D.; Gutsche, C.D. Adv. Alicyclic Chem. 1971, 3, 1, see pp. 91–107; Stark, B.P.; Duke, A.J. Extrusion Reactions, Pergamon, Elmsford, NY, 1967, pp. 47–71.
511. Ramnauth, J.; Lee-Ruff, E. Can. J. Chem. 1997, 75, 518. See also, Ramnauth, J.; Lee-Ruff, E. Can. J. Chem. 2001, 79, 114.
512. Maier, G.; Pfriem, S.; Schäfer, U.; Matusch, R. Angew. Chem. Int. Ed. 1978, 17, 520.
513. Ried, W.; Wagner, K. Liebigs Ann. Chem. 1965, 681, 45.
514. Lomölder, R.; Schäfer, H.J. Angew. Chem. Int. Ed. 1987, 26, 1253.
515. Gould, I.R.; Tung, C.; Turro, N.J.; Givens, R.S.; Matuszewski, B. J. Am. Chem. Soc. 1984, 106, 1789.
516. Vögtle, F.; Rossa, L. Angew. Chem. Int. Ed. 1979, 18, 515; Stark, B.P.; Duke, A.J. Extrusion Reactions, Pergamon, Elmsford, NY, 1967, pp. 72–90; Kice, J.L. in Kharasch, N.; Meyers, C.Y. The Chemisry of Organic Sulfur Compounds, Vol. 2, Pergamon, Elmsford, NY, 1966, pp. 115–136. For a review of extrusion reactions of S, Se, and Te compounds, see Guziec, Jr., F.S.; SanFilippo, L.J. Tetrahedron 1988, 44, 6241.
517. Cava, M.P.; Shirley, R.L. J. Am. Chem. Soc. 1960, 82, 654.
518. Photis, J.M.; Paquette, L.A. J. Am. Chem. Soc. 1974, 96, 4715.
519. Kataoka, T.; Iwama, T. Tetrahedron Lett. 1995, 36, 5559.
520. Sanderson, J.R.; Story, P.R.; Paul, K. J. Org. Chem. 1975, 40, 691; Sanderson, J.R.; Paul, K.; Story, P.R. Synthesis 1975, 275.
521. See Story, P.R.; Busch, P. Adv. Org. Chem. 1972, 8, 67, see pp. 79–94.
522. See Paul, K.; Story, P.R.; Busch, P.; Sanderson, J.R. J. Org. Chem. 1976, 41, 1283.
523. Kharasch, M.S.; Sosnovsky, G. J. Org. Chem. 1958, 23, 1322; Ledaal, T. Acta Chem. Scand., 1967, 21, 1656. For another method, see Sanderson, J.R.; Zeiler, A.G. Synthesis 1975, 125.
524. Story, P.R.; Lee, B.; Bishop, C.E.; Denson, D.D.; Busch, P. J. Org. Chem. 1970, 35, 3059. See also, Sanderson, J.R.; Wilterdink, R.J.; Zeiler, A.G. Synthesis 1976, 479.
525. Barton, D.H.R.; Willis, B.J. J. Chem. Soc. Perkin Trans. 1 1972, 305.
526. See Guziec, Jr., F.S.; SanFilippo, L.J. Tetrahedron 1988, 44, 6241.
527. Turro, N.J.; Leermakers, P.A.; Wilson, H.R.; Neckers, D.C.; Byers, G.W.; Vesley, G.F. J. Am. Chem. Soc. 1965, 87, 2613.
528. Lee, G.H.; Lee, H.K.; Choi, E.B.; Kim, B.T.; Pak, C.S. Tetrahedron Lett. 1995, 36, 5607.