34.1 Introduction
The ductus arteriosus (DA) is an essential fetal vessel connecting the main pulmonary artery and aorta. In utero, high pulmonary vascular resistance coupled with relatively low systemic resistance promotes right-to-left blood flow through the DA, allowing blood oxygenated by the placenta to bypass the developing lungs [1, 2]. Soon after birth, the DA must close in order to facilitate proper perfusion of the newly inflated lungs, a process normally complete by 12–48 h in full-term neonates.
In some cases, the DA fails to close after birth, a condition termed patent ductus arteriosus (PDA). PDA is a common congenital heart defect, occurring in up to 80% of premature infants weighing <1000 g [3]. Several comorbidities including neurodevelopmental impairment, intraventricular hemorrhage, pulmonary hemorrhage, respiratory distress syndrome, bronchopulmonary dysplasia, necrotizing enterocolitis, and spontaneous intestinal perforation have been attributed to or associated with prolonged patency of the DA [4]. In contrast, keeping the DA open to preserve pulmonary or systemic circulation is essential in certain cardiovascular conditions where blood flow to the lungs or body is disrupted [5]. Unfortunately, there are surprisingly few therapies currently available to promote DA closure or maintain vessel patency. Pharmacology-based therapeutics are non-specific and relatively inefficient. While surgical ligation, catheter-based closure, and stent implantation are effective alternatives, these mechanical approaches come with their own risks and are sometimes limited by the anatomical size constraints of extremely premature newborns [6].
34.2 Molecular Regulation of the DA
Molecular regulators of DA tone
Promote DA patency | Promote DA constriction |
|---|---|
Blood flow Nitric oxide (cGMP/PKG) Prostaglandin E2 (cAMP/PKA) Adenosine (cAMP) Atrial naturetic peptide (cGMP) Voltage-gated K+ channels: Kv1.2, Kv1.5, Kv2.1 Large-conductance voltage-dependent and calcium-activated (BKCa) channels ATP-gated K+ (KATP) channels | Oxygen Endothelin 1 8-iso-PGF2α Reactive oxygen species (H2O2) TRPM3 Angiotensin II RhoA/B, Rock1/2 Bradykinin Acetylcholine, norepinephrine |
While nitric oxide (NO) and prostaglandin E2 (PGE2) are typically considered the primary mediators of DA dilation, other factors clearly play a role. Potassium (K+) channels are well-characterized DA dilators. Voltage-gated K+ channels, large-conductance voltage-dependent and calcium-activated K+ (BKCa) channels, and ATP-gated K+ (KATP) channels are all specifically enriched in the DA compared to other vascular beds [9]. KATP channels are hetero-octameric complexes of pore-forming inward rectifier K+ channel subunits (Kir6.1 or Kir6.2) and regulatory sulfonylurea receptor subunits (SUR1, SUR2a, or SUR2b) [10]. Different combinations of these subunits are expressed in a tissue specific manner and exhibit different pharmacological properties, thereby making them attractive targets for DA-specific therapies. This notion is supported by animal studies demonstrating that activating or inhibiting KATP channels directly modulates DA tone [11, 12].
At birth, the DA constricts in response to a sharp increase in oxygen (O2) tension coupled with a loss of vasodilators. Several mechanisms have been proposed to explain the DA’s unique ability to sense and respond to O2. These include cytochrome P450-mediated induction of endothelin 1 and production of “constrictor” isoprostanes (8-iso-PGF2α), and mitochondrial-mediated reactive oxygen species inhibition of Kv1.5 and Kv2.1 [8]. O2 can also inhibit KATP channels resulting in membrane depolarization, activation of voltage-dependent calcium (Ca2+) channels, and increased intracellular Ca2+ accumulation [12]. Other factors known to regulate DA contraction are listed in Table 34.1 [2, 6–8]. While it is clear that multiple mechanisms are involved, all pathways eventually converge on Ca2+-mediated phosphorylation of myosin light chain, leading to actin/myosin interaction and ultimately DA smooth muscle cell contraction [2, 8].
34.3 Genetic Regulation of the DA
Genetic regulators of DA tone
Mouse models of PDA | Human syndromes | Non-syndromic SNPs |
|---|---|---|
Smooth muscle related: Tfap2b−/− Myocd−/− Myh11−/− Prostaglandin pathway: Ptgs2−/− Ptgs1/Ptgs2 double−/− EP4−/− Sloco2a1−/− Hpdg−/− | Char (TFAP2B) Mowat-Wilson (SMADIP1) Loeys-Dietz (TGFBR1/2) Noonan (PTPN11) Holt-Oram (TBX5) Rubinstein-Taybi (CREBP) DiGeorge (TBX1) Periventricular heterotopia (FLNA) Cantu (ABCC9, KCNJ8) | Increased risk of PDA: TFAP2B (rs987237) TRAF1 (rs1056567) AGTR1 (rs5186) Decreased risk of PDA: PTGIS (rs493694, rs693649) ESR1 (rs2234693) IFN-γ (rs2430561) |
Others: Notch2−/− Notch2/Notch3 double−/− Jag1−/− Brg1−/− Bmp9−/− with loss of Bmp10 Nfe2−/− Itga2b−/− |
Syndromes featuring PDA (Table 34.2) can also be informative regarding genetic regulation of DA development and function [6, 13]. For instance, clinically significant PDA occurs in 50% of patients with Cantu syndrome, a condition caused by gain-of-function mutations in KATP channel genes, KCNJ8 and ABCC9, which encode the vascular-specific KATP channel subtype Kir6.1/SUR2B. In these cases, PDA is resistant to indomethacin therapy and often requires surgical ligation to achieve closure [14].
While only 10% of PDA cases are associated with chromosomal abnormalities [7], identifying single-nucleotide polymorphisms (SNPs) associated with non-syndromic PDA may be more informative regarding the more common sporadic cases of PDA (Table 34.2). Variants in TFAP2β, TRAF1 (TNF receptor-associated factor 1), and AGTR1 (Angiotensin II type 1 receptor) were associated with an increased risk of PDA while SNPs in PTGIS (prostaglandin I2 synthase), ESR1 (estrogen receptor-α), and IFN-γ (interferon-γ) were found to be protective [6]. Unfortunately, replicating many of these findings in other cohorts has proved difficult to this point [15].
34.4 Pharmacological Regulation of the DA
Only three drugs are currently available to treat PDA (indomethacin, ibuprofen, and acetaminophen). All are non-selective cyclooxygenase inhibitors that suppress prostaglandin signaling. Alternately, infusion of prostaglandin E1 (PGE1) is the only pharmacologic option used to maintain DA patency in cases of ductus-dependent congenital heart defects. None of these therapies specifically target the DA, leading to off-target effects on other vascular beds. Furthermore, indomethacin and ibuprofen have been associated with spontaneous intestinal perforation and necrotizing enterocolitis while PGE1 has been associated with apnea, fever, and other physiologic disturbances [16, 17].
Pharmacological regulators of DA tone
Maternal drugs | Neonatal drugs |
|---|---|
Increased risk of PDA: Tocolytics (indomethacin, nifedipine, magnesium sulfate) ACE inhibitors Antihistamines Anticonvulsants (valproic acid, phenytoin) Amphetamines Alcohol | Dilates DA: PGE1 Gentamicin PDE inhibitors (milrinone, amrinone, sildenafil) Cimetidine Heparin Diazoxide |
Cause fetal DA constriction: NSAIDs SSRIs antidepressants (sertraline, fluoxetine) Polyphenols Betamethasone | Constricts DA: NSAIDs Caffeine KATP inhibitors (glibenclamide) |
Furthermore, drugs commonly administered to neonates often have vasoactive properties and may inadvertently affect postnatal DA closure [18]. Several drugs have been shown to specifically dilate the DA or cause resistance to indomethacin therapy (Table 34.3). Of note, DA reopening has been reported after exposure to diazoxide, a KATP channel activator used to treat neonatal hyperinsulinism [20].
34.5 Clinical Implications and Future Directions
PDA remains a significant problem that is inefficiently managed with currently available therapies. Therefore, a greater emphasis must be placed on identifying other factors that can be targeted to regulate DA tone.
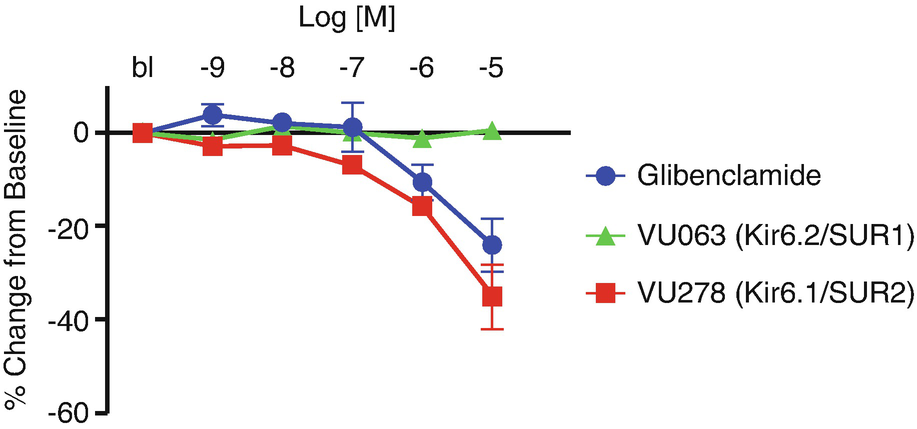
Term-gestation mouse DAs were treated with increasing concentrations of glibenclamide, VU063, or VU278. Changes in lumen diameter were measured and plotted as a percent change from the initial lumen diameter reading under baseline (bl) conditions. Glibenclamide and VU279 caused DA constriction while VU063 had no effect
In conclusion, an emerging body of genetic, physiological, and pharmacological evidence paints a bright future for the treatment of PDA. Developing a comprehensive understanding of the molecular mechanisms and signaling pathways that regulate DA patency, coupled with focused efforts to develop specific pharmacological modulators of these new targets, is creating unprecedented opportunities for improving PDA outcomes. Among several potential drug targets emerging from these studies, vascular-specific Kir6.1/SUR2 channels hold significant promise due to their enriched expression in the DA and unique pharmacological properties. Ongoing efforts by our laboratories will explore the therapeutic potential of Kir6.1/SUR2B channels in regulating DA patency.
This work was supported by R21HL132805 and AHA15SDG25280015 awarded to E.L.S. and by NIHR01DK082884 awarded to J.S.D.

Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.