38
The Lazarus Mouse
A substantial part of the immune system revolves around the way information is communicated. Molecules send and receive signals, urging immune cells to attack, do further surveillance, withdraw, implode, lurk, help new tissue grow. In the broadest sense, this information is transmitted in two different formats, or media.
Some of the communications are known as soluble, or fluid-like, and involve interleukins. These molecules are released and can travel around and infuse other cells with instructions.
The second type, which I’ve already also described and will elaborate on here, involves molecules or proteins that appear on the surface of cells and that connect, or bind, to a molecule or protein on another cell. These are like antibodies. They travel the body not in fluid form but attached to a cell, and then they connect to another cell at an extremely specific spot. These are mates in a puzzle that require physical proximity.
The concept is important because it helped save Jason’s life. To show how, I need to delve a little more deeply into the science.
Typically, one piece of the puzzle is called a ligand, and the other is a receptor. (Ligand comes from the Latin ligare, meaning to bind.) A ligand binds to a receptor.
Through the 1980s and the 1990s, immunologists did a lot of looking for molecules on the surface of immune cells—basic archaeology—and then tried to find their mates. One reason they would hunt for these pairs was in hopes of finding a match that would help explain what each molecule was doing on the cell surface in the first place. What did the individual pieces of the puzzle do, and what happened when you put them together?
“Every piece builds a story. It’s like getting to know a friend. Same thing through a series of encounters through molecules,” said Matthew “Max” Krummel, an immunologist who was there at the moment of one of the twentieth century’s major scientific moments—when CD80 and CD86 met their mates.
This is the story.
In the late 1980s, work had been done that identified two ligands that are expressed on the surface of two major immune system cells, B cells and dendritic cells. Scientists had discovered that these ligands bind to specific molecules on the surface of T cells.
As these various immune cells circulated in the festival of our lives, they would bump into one another. If the surface of a B cell had the right ligand and the surface of the T cell had the right receptor, the two molecules would bind to each other. This would set off a reaction.
Okay, fine, but so what? What was the reaction?
So we’re trying to cure cancer here, give it a sec, will you? Stick with me.
In plainer English still:
T cells can attack invaders and organize attacks. Researchers discovered molecules on the surface of T cells that connect to molecules from other parts of the immune system, namely B cells and dendritic cells. In other words, scientists had found pieces of a puzzle that fit together but without knowing what the puzzle looked like—or exactly what that puzzle meant. They discovered that one of the key molecules on the surface of the T cell was called CTLA-4. Another was CD28.
One more bit of deep trivia that is not at all trivial: CTLA-4 and CD28 both bind to ligands called B7-1 and B7-2—also known as CD80 and CD86.
Okay, so then what?
Around 1989, CTLA-4 was being explored jointly by two eventual all-star academics in the immune sciences, James Allison, who was at Berkeley at the time, and Jeffrey Bluestone, who was at the University of Chicago and eventually the University of California at San Francisco. There was a third researcher named Peter Linsley, from Bristol-Myers Squibb, a pharmaceutical company, doing related work.
Bluestone and Allison weren’t particularly interested in cancer, or rather, that wasn’t their central focus. They were concerned with the immune system overall.
At Berkeley, a PhD student in Allison’s lab performed an experiment that entailed taking a tumor from a mouse, putting it into a test tube, and then injecting it with foreign genes. The impact of injecting these genes was to cause the tumor cells to present the molecule called B7-1, the ligand that binds to receptors on T cells called CTLA-4 and CD28. The researchers then injected T cells into the test tube, and lo and behold, the T cells attacked the tumor in force, attracted by B7-1, and they wiped out the malignancy.
One more time, gently, for good measure. Researchers figured out how to display a puzzle piece that attracted another puzzle piece and this stimulated an immune system response to wipe out a tumor.
Good news, right?
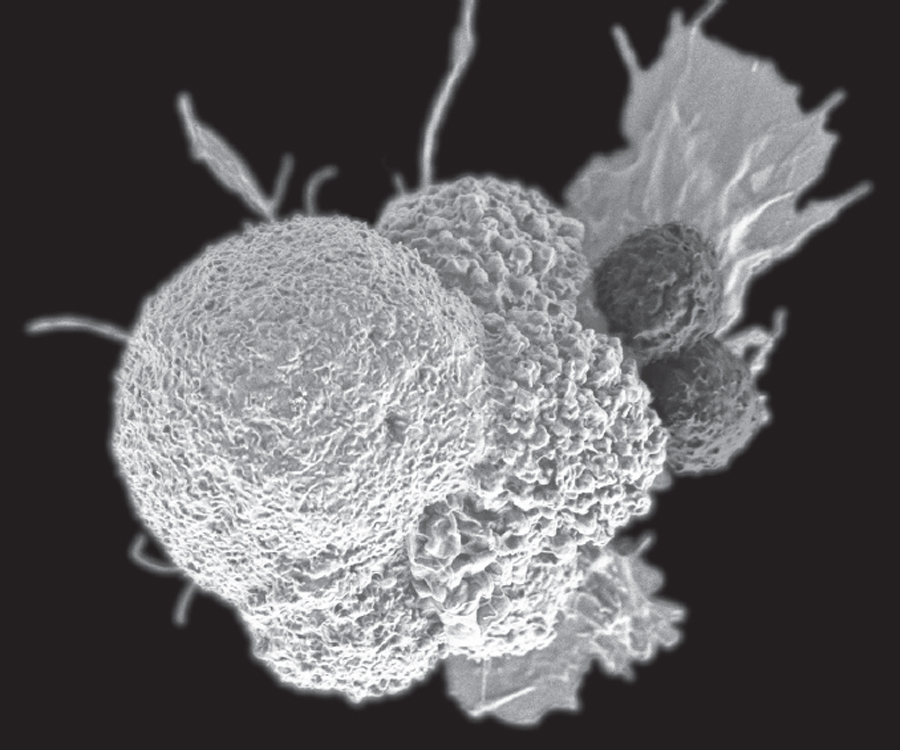
Two T cells (right) attack a cancer cell. (NCI/NIH)
Yes, a huge step in the right direction. But this wasn’t yet the Holy Grail. The steps were too artificial, like tailor-making a tumor by putting foreign genes into it so that it could be targeted. Plus, this whole thing had happened in a test tube. This wasn’t yet a solution to allow manipulation of the human immune system. But it was a powerful indication that such a solution was possible.
This is where Krummel began his collaboration with Allison, who won the 2018 Nobel Prize for what happened next.
Allison and Krummel decided to experiment further with CTLA-4, the other molecule that had bound with B7-1 and B7-2. They soon noticed a curious thing. When CTLA-4 was attracted to and bound to a ligand, the immune system didn’t ramp up as it had in the mouse experiment. Instead, the immune system seemed to be dampened or to have no effect at all.
“I thought, we gotta figure out what CTLA-4 does,” Allison reflected. Something about it nagged at him.
Krummel and Allison asked a question: If CD28 causes T cells to multiply, but CTLA-4 seems to have no effect, what might happen if you combined these agents?
What they discovered was a turning point. Stimulating CD28 led to an increase in T cells, and a heightened immune response. But when CTLA-4 was mixed in, it brought down the level of T cell response. Not only that: The more CTLA-4 was added, the fewer T cells proliferated. That suggested that CTLA-4, rather than causing the immune system response to increase, was causing it to turn down or even off.
They sensed they were on to something big.
Krummel devised a chemical process that would allow him to create varying levels of CD28 and CTLA-4 such that he could begin fine-tuning the amount of T cells created. The year was 1994.
“We could turn T cells up and down like turning a stereo up and down,” Krummel said. Or, if you prefer a different metaphor: “We found a hot tap and a cold tap. We immediately had this whiteboard discussion,” he said. What did this mean and what could they do with it?
They started experimenting, trying all kinds of combinations, one after the next. “In the course of nine months, we went from volume control—hot and cold—to every single animal model I could touch, pushing T cells to grow faster, watch them grow slower. That’s when Jim brought in the tumor model.”
Allison, by now steeped in this as virtually no other scientist, turned ideas over and over in his mind, trying to make sense of it all. What did these molecular interactions add up to? He joked with me about the pieces finally coming together one night in 1994, while his mind was wandering after “too much wine.” He thought he might understand how cancer was playing a trick on the immune system, allowing the disease to evade our defenses. And he had an idea how to reverse the trick.
Allison had invited a postdoc named Dana Leach into the lab. Leach brought the rodents with the tumors, which now were out of the test tube and into actual critters. The vet injected rats with several fast-growing cancers. The researchers let the cancers blossom. Then they injected the mice with a molecule—an antibody—that was aimed at disrupting any connection that the cancer cells might be trying to make to the CTLA-4.
The idea was to see if they could keep the cancer from turning on the brakes of the immune system by disrupting the communication between the cancer and the immune system.
“We were just trying things out,” Krummel said.
A few days later, Allison came in to check out the progress. “I went, ‘Holy shit! It cured all the mice.’”
The previous experiment had entailed isolating the tumor tissue in a test tube and then modifying its genetics so that it would stimulate a T cell response. This was ultimately impractical.
But in the new experiment, the researchers did nothing at all to the tumor. It was just a tumor, like one that might be growing inside any of us, eventually inside Jason. It was in its natural state.
Instead of changing the tumor, the researchers added antibodies to interrupt cancer’s trick and stimulate a response from the immune system. Specifically, they inserted an antibody that bound to the immune system so that it would take off the brakes in our elegant defense.
“What was surprising is that we hadn’t given the immune system any new information about the tumor,” Krummel said. “There was a set of preexisting cells”—the T cells—“and they were raring to go.”
When Allison looks back, he thinks of the immune system in a vastly different way than we conceived of it for the longest time. He doesn’t think of it merely as a powerful killing machine, not at all. Instead, he thinks of it as sharing killing powers with extraordinary powers of restraint. One of the chief jobs of the immune system is to shut down its attacks, hit the off button. Screeching brakes get applied to the T cells.
“They get a signal. They kill themselves. If it didn’t work, people would get diabetes, multiple sclerosis, lupus,” he said. “By far this negative selection is the central tolerance, to get rid of T cells; 90 percent of every T cell that’s developed gets killed.”
He’d figured out what CTLA-4 did. “CTLA-4 is there to protect you from being killed by your immune system.”
Whoa.
But isn’t cancer killing people? Why would our bodies allow the brakes to come on in the face of a deadly tumor?
The answer is related to a trade-off with wound healing, which is one of the most important functions of both the body and the immune system.