2.6 Our Experience in Some Chemoenzymatic Projects
We have implemented enzymatic syntheses in some of our work. Our approach to developing and implementing large scale production of chemicals, intermediates and active pharmaceutical ingredients is achieved by principally following different approaches with various degrees of complexity:
- Follow a purely chemical strategy.
- Use a chemoenzymatic route combining chemical and biocatalytic steps. In this case, the biocatalyst is given preference to be used for performing the key reaction(s) requiring high selectivity or specificity or to replace environmentally intolerable reaction steps.
- Use a total biological synthesis by fermentation or multistep biotransformation.
To develop a particular chemoenzymatic process, the following general steps are followed:
- Test and evaluate inexpensive commercially available (bulk) biocatalysts or suitable microorganisms. Enzymes that do not require coenzymes, such as hydrolases (lipases, esterases, proteases), are still preferred biocatalysts for the preparation of optically active compounds.
- Search and screen for suitable novel microbial biocatalysts from natural sources, e.g. by selective enrichment techniques from environmental samples, starting from soil or sewage samples, or alternatively, and in rare cases, by testing extracts of plant or animal tissues. This laborious and more demanding approach is essential when no suitable enzymes or microbes are commercially accessible for the conversion of a defined substrate into a defined product using a particular reaction type. Both approaches, 1 and 2, are complementary.
- Search for completely novel enzymes catalyzing difficult reaction types, where no indications or prior knowledge on their existence is available.
Furthermore, to set up a practical bioconversion process, screening and optimization work is required at the following different levels:
- Screening for suitable microorganisms or plant cells, and so on, and/or enzymes with the required catalytic properties and selectivities.
- Screening for the optimum conditions for culture growth and production of the desired enzyme(s). This can be very demanding work in cases where enzyme expression is not constitutive. In this case, the best and most economical process parameters, such as the conditions for enzyme induction and the right harvesting time, must be identified.
- Screening for the optimum reaction conditions with whole cells, crude, or purified enzymes including engineering and technological aspects such as cofactor regeneration, immobilization, type of reactor, and so on.
- Optimization of the biocatalyst by directed evolution or any other enzyme engineering technologies, to design the ideal biocatalyst under the given process conditions.
To perform bio-reactions, the biocatalyst can be applied under various process conditions:
- batch or fed-batch application of whole cells in free or immobilized form in aqueous environment;
- application of whole cells in two-liquid phase, multiphase systems, or in micelles;
- use of acetone-dried or permeabilized cells;
- batch or fed-batch application of free crude or purified enzymes in aqueous or organic environment;
- use of modified enzymes in organic solvents;
- continuous application of immobilized enzymes;
- use of enzyme membrane reactors – the method of choice for systems with cofactor recycling or for reactions with expensive enzymes.
Finally, with respect to the choice of a reactor for biocatalytic reactions, no special equipment is needed for biocatalysis in many cases, and ‘ordinary’ stirred tanks, used in large-scale chemical synthesis with temperature and pH control, are sufficient, thus fitting a biocatalytic process in existing facilities & equipment.
2.6.1 Protease-mediated Synthesis of Valganciclovir Intermediate135
Valganciclovir, an antiviral drug, is used to treat cytomegalovirus infections.135–141 It is a valyl ester of ganciclovir (a prodrug) which provides better bioavailability of ganciclovir when administered orally to cytomegalovirus infected patients. This prodrug for ganciclovir (valganciclovir) is found in the form of mixture of two diastereomers. According to the FDA specifications, the diastereomeric ratio of valganciclovir should be maintained in the range 55 : 45 to 45 : 55. After its administration, these diastereomers are rapidly converted into ganciclovir by hepatic and intestinal esterase. The biocatalytic step involved the prochiral hydrolysis catalyzed by hydrolases (Figure 2.1).
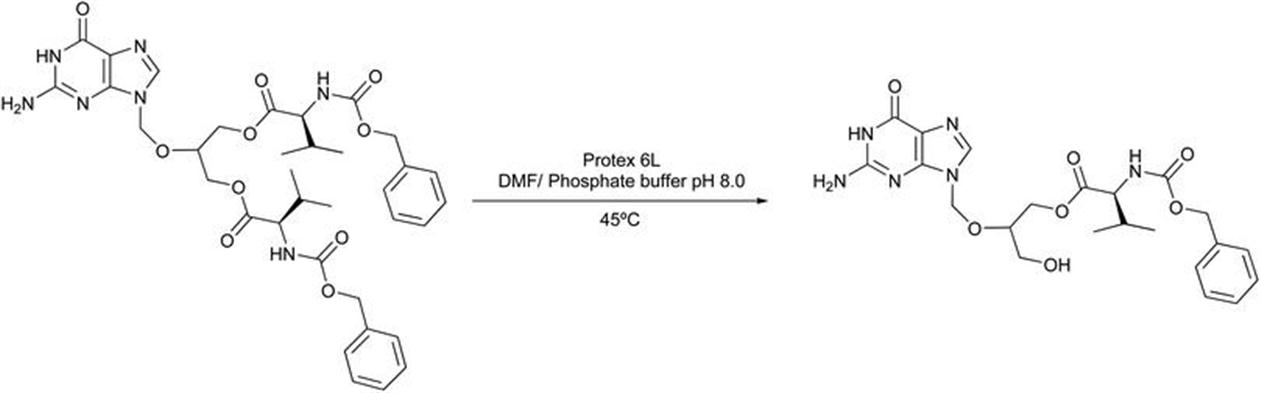
Figure 2.1 Enzymatic scheme representing the enzymatic synthesis of valganciclovir intermediate.
To this end, various hydrolases were screened towards the mono-hydrolysis of the di-valine ester of ganciclovir (e.g. lipases, CAL-A, CAL-B, proteases, esterases, etc.). The starting material was sparingly soluble in water, and hence solvents were also screened (dioxane, dimethylformamide, methyl alcohol, methylene dichloride, acetone, tetrahydrofuran, acetonitrile, ethyl alcohol, toluene, isopropyl alcohol, 2-methoxyethanol, diglyme, tert-amyl alcohol, 2-Me-THF, 2-BuOH, 1-BuOH, 1-propanol). The reaction was found to occur in solvent–water mixtures, and the enzyme Protex 6L (provided by Genencor) displayed positive results in a water–dimethylformamide mixture at pH 6.0–8.0 and temperature of 45–50 °C. Once these conditions were set, a scale-up reaction was carried out in a 500 mL round-bottom flask with an overhead stirrer and pH stat connection. The reaction mixture consisted of 20 g of the starting material in 180 mL of DMF and 120 mL of 0.1 M phosphate buffer with pH 8.0. The pH was maintained using 0.5 M NaOH. After completion of reaction, the product was isolated, giving a yield of 25% and a purity >95%. The above process suggested that Protex 6L was a versatile biocatalyst that was able to hydrolyze the starting material with a good yield and purity of >95%.
2.6.2 Chemoenzymatic Synthesis of Optically Pure Rivastigmine Intermediate Using ADH from Baker’s Yeast and KRED142,143
The chemoenzymatic synthesis of chiral intermediate [(S)-N-ethyl-N-methylcarbamic acid 3-(1-hydroxyethyl)phenyl ester] – a precursor of rivastigmine – was developed using alcohol dehydrogenase from Baker's yeast (Figure 2.2).
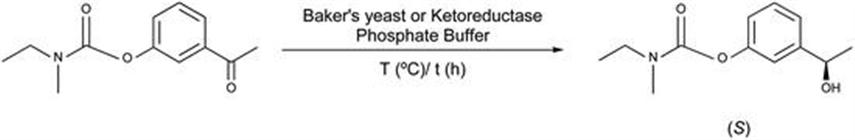
Figure 2.2 Enzymatic synthesis of rivastigmine intermediate.
To that end, N-ethyl-N-methylcarbamoyl acetophenone – synthesized by condensation of N-ethyl-N-methylcarbamoyl chloride with 3-hydroxyacetophenone – was screened for the stereoselective reduction by means of alcohol dehydrogenases. An in-process purification of crude alcohol dehydrogenase from Baker's yeast144 has been adopted and used in an enzymatic step. Various references are available on the purification of alcohol dehydrogenase and stereoselective reduction catalyzed by alcohol dehydrogenases.145–159 Apart from Baker's yeast, a number of alcohol dehydrogenases from various sources like Thermoanaerobium brockii (in recombinant form in E. coli), Parvibaculum lavamentivorans, or Deinococcus radiodurans (in recombinant form in E. coli) were screened towards enantioselective reduction of ketone to alcohol.
2.6.3 Preparation of Deoxynojirimycin, Key Intermediate of an Anti-diabetic Drug
Type 2 diabetes has posed a severe threat to humankind worldwide. Insulin resistance is one of the primary cause as seen in type II diabetes. Even though the exact cause of the insulin resistance remains unknown, it can be partially attributed to obesity and associated lipotoxicity. N-Nonyl and N-hydroxyethyl (miglitol) of nojirimycin are drugs used in the treatment of diabetes type II symptoms and certain other metabolic disorders like Gaucher’s disease. Treatment of type-2 diabetes with miglitol (along with other drugs) is mainly prescribed for people whose diabetes cannot be controlled by dietary controls. Its mechanism of action involves retarding the breakdown and adsorption of table sugar/oligosaccharides in the small intestine, thus blood sugar levels following meals are lowered.160
Azasugars – also known as iminosugars – are mimetics of native sugars whereby the ring oxygen is replaced by a nitrogen atom. Due to their structural similarity to native carbohydrates, azasugars are able to act as glycosidase inhibitors and therefore have enormous therapeutic potential in the treatment of a variety of diseases including viral infection, bacterial infection, lysosomal storage disorders, cancer and diabetes. In particular 1-deoxyazasugars – such as 1-deoxynojirimycin and 1-deoxygalactonojirimycin – have been reported to inhibit various glycosidases in a reversible or competitive manner due to their structural resemblances to the sugar moieties of natural substrates.161,162 For the synthesis of deoxynojirimycin, aminosorbitol (1-amino-1-deoxyglucitol) is a key intermediate.
As depicted in Figure 2.3, anhydrous dextrose, benzylamine and Raney nickel in methanol were heated to 50 °C to form MGL-IA, simultaneously hydrogenated under 10–12 kg of hydrogen gas pressure to give MGL-IB. After completion of the reaction, water was added to the reaction mixture which was then filtered, palladium carbon was added to the filtrate, and hydrogenated at 50 °C, under 10–12 kg of hydrogen gas pressure at 50 °C to give aminosorbitol. The first step for either the reductive amination or the reductive alkylation is the reversible formation of the imine intermediate and water. The imine is then hydrogenated to the amine. Herein, a possible by-product formation results from the addition of the product amine to the imine, resulting in a dimer-like product with loss of ammonia (Figure 2.4).
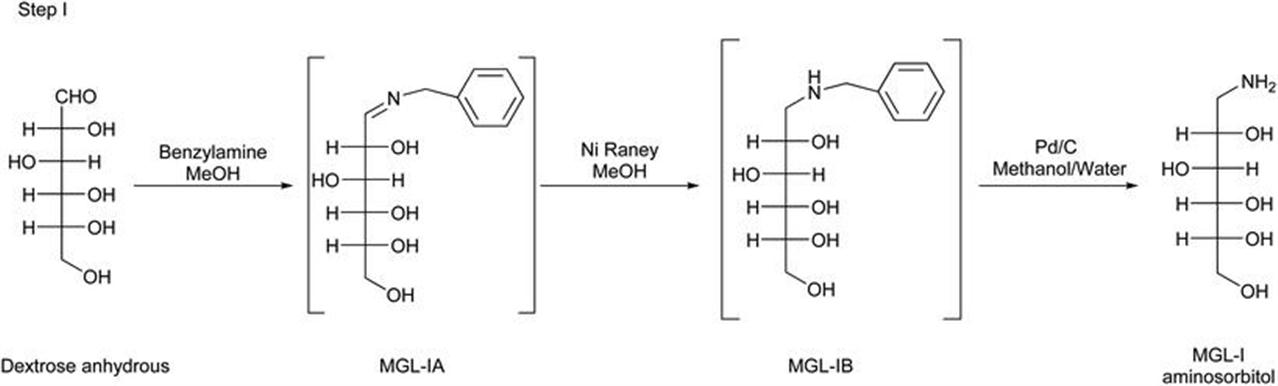
Figure 2.3 Step I reactions to obtain aminosorbitol (MGL-I), intermediate in the synthesis of deoxynojirimycin.
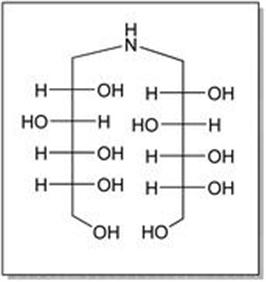
Figure 2.4 Dimer structure formed as by-product in the formation of MGL-I.
Filtration is a key parameter in this process, as heavy metals like nickel and palladium are used together. To remove these metals they have to be filtered through a Celite® bed. Extreme precaution has to be taken while filtering both nickel and palladium, as both could catch fire. If the filtration is not properly conducted, then a heavy metal residue may be found in the filtrate. Subsequently, the reaction carried out between aminosorbitol and methyl formate in methanol (at reflux) gives MGL-II (Figure 2.5). The solid is filtered off and washed with methanol. The amount of dimer by-product – formed in the previous stage – is reduced during this filtration, presumably due to higher dimer solubility in methanol.
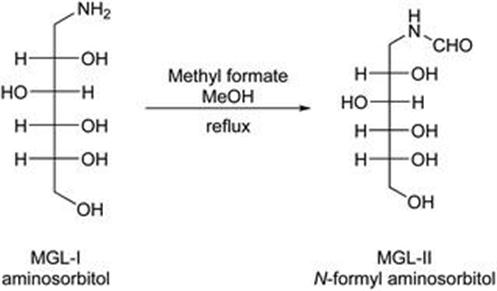
Figure 2.5 Step II, showing the formation of MGL-II by formylation of aminosorbitol.
Later on, the pH of the solution of N-formyl aminosorbitol in water was adjusted to 4–6 using orthophosphoric acid and then oxidation was performed with Gluconobacter oxydans DSM2003 whole cells with oxygen gas purging, resulting in N-formyl aminosorbose. Subsequently, reaction with sodium hydroxide results in 1,2-dehydro-2-(methylhydroxy)piperidine-3,4,5-triol, which further reacts with sodium borohydride followed by neutralization with hydrochloric acid, rendering crude deoxynojirimycin (Figure 2.6). Final purification proceeds by means of Indion 225 H resin, ammonia solution, and crystallization in 2-methoxyethanol/water to yield pure deoxynojirimycin.
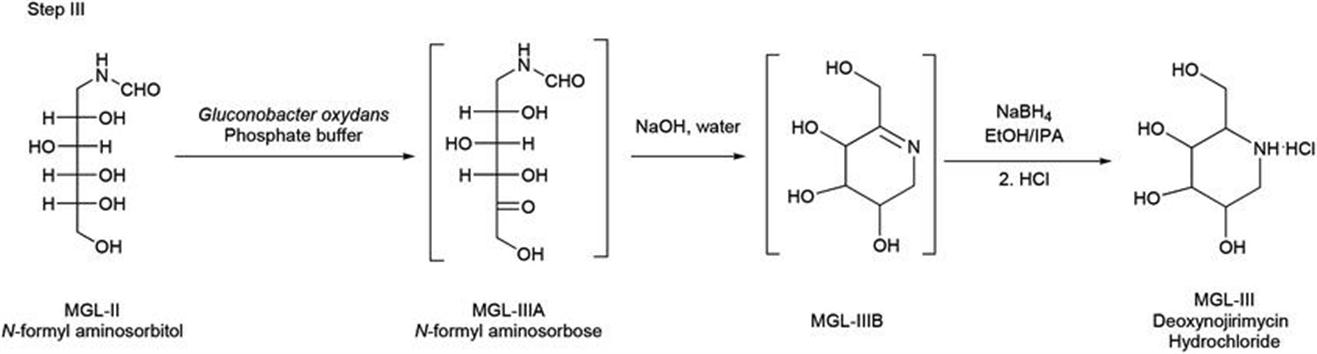
Figure 2.6 Step III, showing the formation of deoxynojirimycin hydrochloride (MGL-III) from formyl aminosorbitol.
The reaction mixture of deoxynojirimycin contains several organic and inorganic impurities. Among the organic ones, the dimer impurity of amino-sorbitol (Figure 2.4) and the formylated dimer impurity of amino-sorbitol may be pinpointed. With regard to inorganic impurities, sodium chloride, sodium borohydride, boric acid, residual palladium, residual nickel and iron may be found. As the solubility of deoxynojirimycin is very high in water, it is very tedious to remove these water soluble inorganic salts. For their removal from deoxynojirimycin, resin purification was done, using an Indion 225 H resin packed in a proper column. Organic impurities such as dimer impurity of aminosorbitol and formylated dimer impurity of aminosorbitol do not bind to resin and hence are removed from deoxynojirimycin. The complete binding of deoxynojirimycin from the reaction mixture is assured by assay of elute. The resin is then washed with water until elute is free of inorganic impurities. Deoxynojirimycin is then eluted form the resin using ammonia solution. Elute is subsequently distilled and crystallized to get pure deoxynojirimycin base with a purity greater than 96%. Inorganic impurities and heavy metals were also removed during this process of resin purification. The isolated free base is then converted into hydrochloride salt in quantitative yield and purity greater than 99.9%.
As described above, separation of the inorganic impurities is tedious work while preparing these compounds of high purity, especially in highly regulated markets like Japan, where the total impurity level requirements may not exceed more than 0.1%, and other inorganic impurities cannot exceed 20 ppm. Preparing compounds for such a market represents a challenge for the synthetic community, especially at the purification steps. Apart from the above-described resin method, several other methods have been tried for purification of deoxynojirimycin, such as membrane dialysis, carboxymethyl-Sepharose chromatography, preparative thin-layer chromatography,163 as well as using hydrochloric acid.164 Of all of them, purification using resin seems to be the most viable. The better resin for the preparation of deoxynojirimycin was Lewatit l SP 112 H, which is a strong acid exchange resin. However, the availability of this resin prompted a search for another one that could be reusable, easily available and cheap. Of all the methods used for the preparation of deoxynojirimycin and their salts,164–176 the biocatalytic one involving the preparation of N-formyl-aminosorbitol, conversion into N-formyl aminosorbose through biotransformation, and finally cyclization and reduction to yield deoxynojirimycin seems to be the best from a commercial point of view.
For this project, apart from the successful biocatalyst employed (see above), various routes and enzymes were assessed previously. In the following, some selected examples are discussed. In these cases, a number of tests were run with locally available Baker's yeast, as well as with yeasts from Saccharomyces cerevisiae Type I obtained from Sigma Aldrich. Thus, locally available Baker's yeast was grown on D-sorbitol media containing 6% D-sorbitol, 2.4% yeast extract, and 4.8% KH2PO4 in Milli-Q water for 48 h. The cells obtained were then centrifuged, washed with 0.02 M MgSO4, and stored below 4 °C for future use. Wet cells were then inoculated in autoclaved reaction media containing 2% 1-amino-1-deoxy-D-glucitol solution containing 4% of yeast extract, 20% of D-sorbitol and 2% potassium dihydrogen phosphate in Milli-Q water, pH 5.0 (adjusted using 2.0 M HCl). The reaction media was stirred for 72 h, centrifuged and separated by column chromatography using Amberlyst-15 H+ resin. The acid wash was distilled off and the residue obtained was precipitated with ethanol to obtain white crystals of 1-deoxynorjorimycin. In another line, Saccharomyces cerevisiae Type I from Sigma Aldrich was inoculated in a reaction buffer pH 7.0 containing 1% 1-amino-1-deoxy-D-glucitol, 1% NAD+, 5% D-glucose in Milli-Q water, 100 mM potassium phosphate. The reaction media was stirred for 72 h, centrifuged and separated by column chromatography using Amberlyst-15 H+ resin. The acid wash was distilled off and the residue obtained was precipitated with ethanol to obtain white crystals of 1-deoxynorjorimycin.
Various lessons arose during the process development:
Absorbance study of bacterial growth is mandatory. It should be carried out both for the shake flask method and at a mini-fermenter scale.
Significant variability was observed in parameters in large-scale versus lab-scale batches. Example 1: In lab scale batches, OD absorbance (600 nm) after inoculation was found to be 0.03–0.05 while in large-scale fermenter batches the OD absorbance (610 nm) after inoculation was found to be 0.16.
Example 2: The shake flask method gave rise to maximum OD of 1.2–1.8 in 36–40 h with a viability of 89% whereas in large-scale fermenter batches enormous growth was observed within 6 h with an absorbance of 2.4 and viability of 75%.
Before inoculating into a fermenter, a performance test should be carried out at lab scale level and the cells harvested should be reactive with the substrate. Optimization of various reaction parameters was carried and statistically their significance was also determined in the finalized process.
During the biotransformation process, i.e. during oxidation of N-formyl using Gluconobacter oxydans DSM2003 whole cells, three main unknown impurities peaks (with defined retention times) were observed in a HPLC chromatogram while reaction monitoring. Since higher levels of impurities affect the yield of the process, efforts were carried out to study the factors that can reduce the formation of process impurities. Thus, a study of the effect of pH during the stage III reaction of preparation of deoxynojirimycin base was conducted. Experiments involved reaction of N-formyl aminosorbitol in the presence of water, oxygen and Gluconobacter oxydans DSM2003 whole cells, followed by addition of sodium hydroxide and sodium borohydride to give deoxynojirimycin. Post work-up and subsequent crystallization with 2-methoxyethanol yielded deoxynojirimycin base. In this experiment the pH of the reaction was varied to study its effect during the reaction. A regression analysis was also performed using Minitab to obtain clarity about the role of the pH during the reaction. It was observed that at the extreme pH limits, e.g. at pH 2.0, reaction did not occur, whereas at pH 8.0 reaction did not reach completion. The pH range 4–6 showed a certain effect on yield and purity. Interestingly, it was observed that the pH had a negative correlation with one of the impurities (number 3). Thus, when a null hypothesis p-test was carried out, no significant effect of pH was to be found on product purity, impurity 1 and impurity 2, but a significant influence was seen in minimizing impurity 3. Furthermore, large-scale batches were statistically analyzed to achieve better understanding of the influence of the list of parameters on the output obtained. The parameters pH, RPM, and oxygen cylinders consumed along the course of the reaction were studied during stage III of the reaction described above. Their effect on the output and reaction completion time was studied. It was seen that only RPM showed a statistically significant effect on the reaction completion time while the rest of the factors did not contribute to any significant effect on the output or reaction completion time.