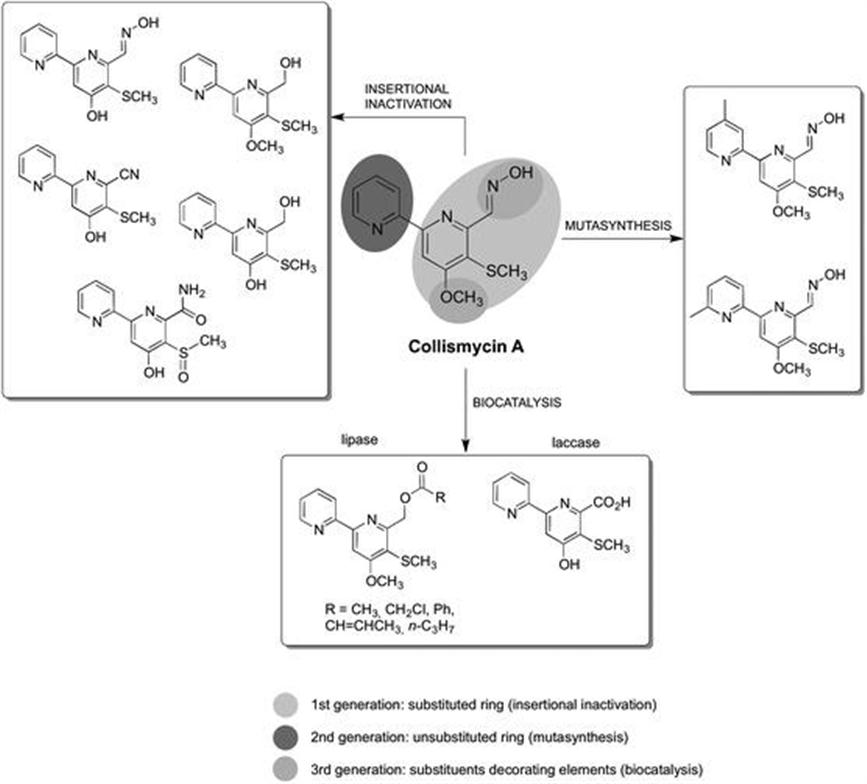
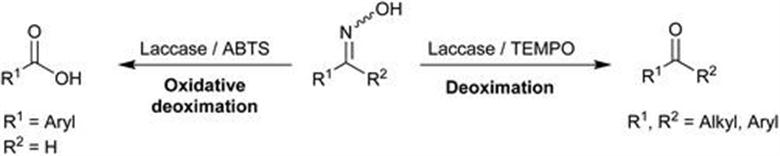
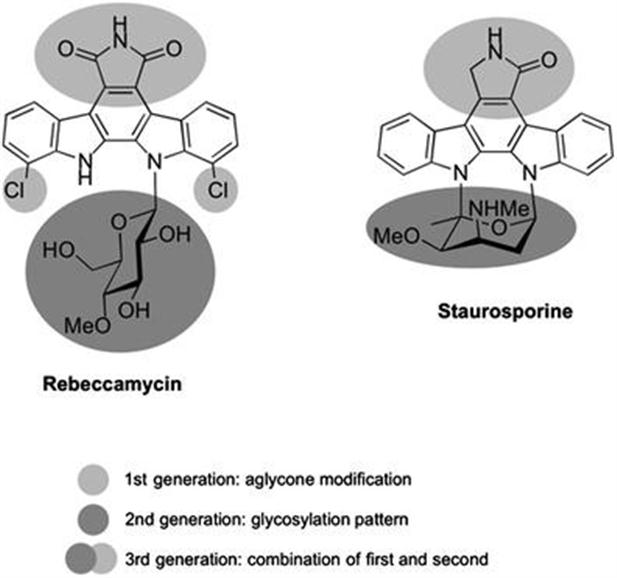
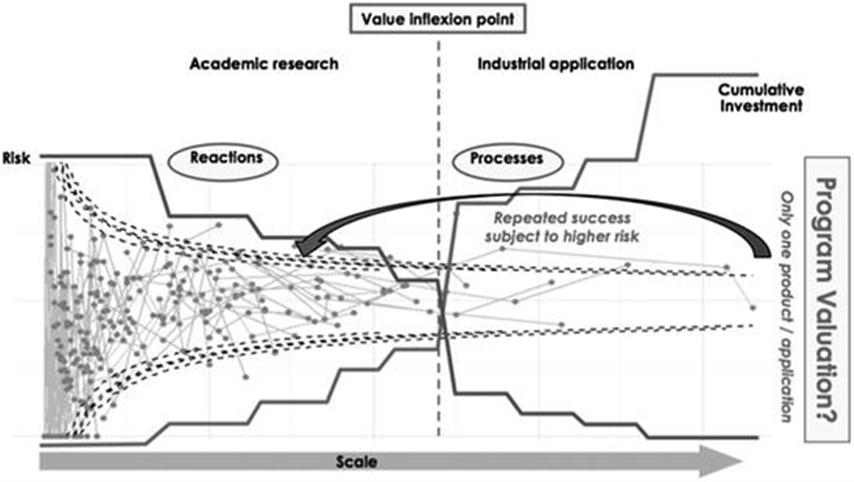
4.2 Biocatalysis
4.2.1 Enantiopure Chiral Building Blocks
Chiral building blocks are essential to create sophisticated libraries in medicinal chemistry or as raw materials for process development of chiral APIs. EntreChem's technology, based on green biocatalytic technology offered by lipases, ketoreductases and transaminases, enables a growing offer of chiral products, especially chiral amino alcohols and diamines of high enantiomeric and chemical purity. The processes used for the laboratory synthesis are fully scalable, to ensure satisfactory re-supply of any of the isomers listed in Figure 4.1. As can be seen, we have developed chemoenzymatic routes for every enantiomer of both cis- and trans-1,2-cyclopentyl- and 1,2-cyclohexyl amino-alcohols and diamines, including monoprotected derivatives to facilitate the use of these building blocks in medicinal chemistry.
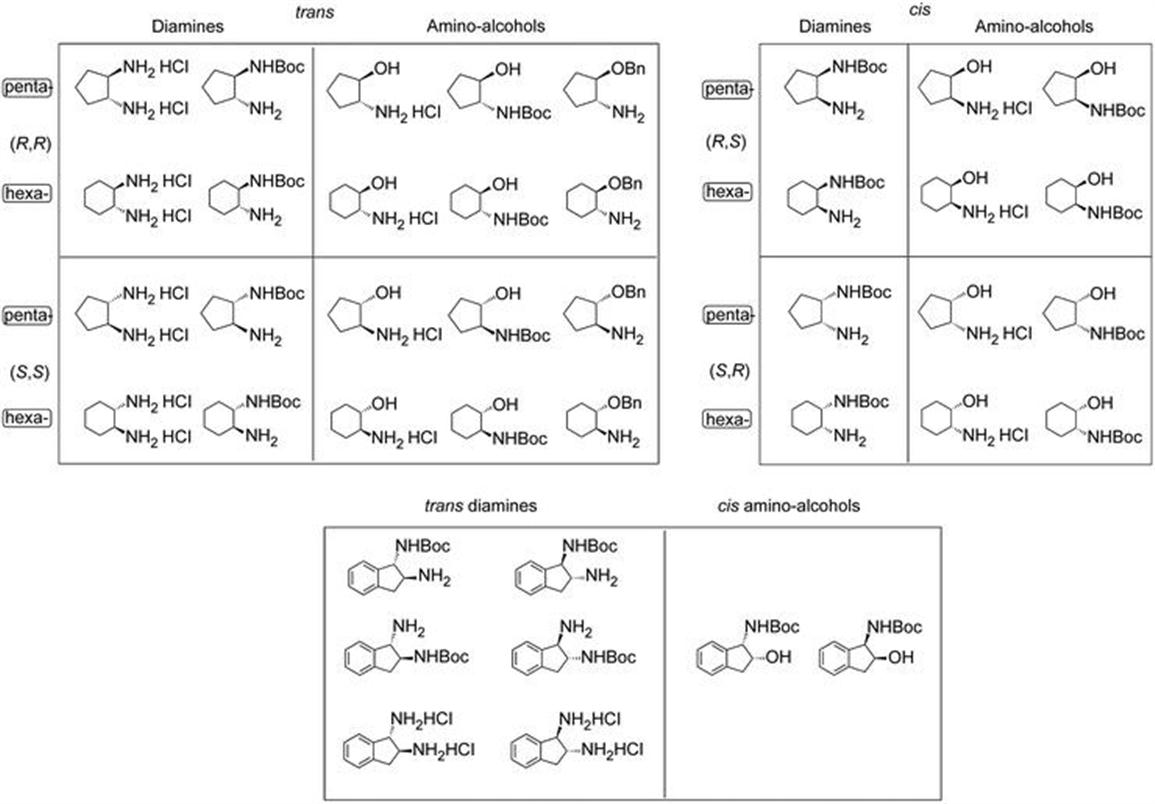
Figure 4.1 Portfolio of building blocks offered by EntreChem.
In this sense, enzymatic syntheses are an exciting and emerging opportunity in sustainable chemistry due to the intrinsic green features of enzymes which have water (a safe, non-toxic, biorenewable and cheap solvent) as their natural environment, enhancing the expectations of biocatalysis-based chemical manufacturing.
EntreChem also provides custom services to clients, in order to prepare other building blocks similar to those of Figure 4.1 or to develop novel chemoenzymatic routes to key pharmaceutical intermediates. A recently disclosed example is the synthesis of the heart-rate reducing drug ivabradine (Scheme 4.1).1–3 Introducing chirality in the intermediate is challenging because the amino hot spot is primary and therefore remote to the chiral center. However, this was solved by a lipase-catalyzed alkoxycarbonylation with diethyl carbonate, and to kill two birds with one stone the carbonate product itself can be reduced to install the methylamino group present in the historical chemical processes of the sponsor.4 This is particularly convenient since it does not alter the end-game of the synthesis, the use of the same penultimate molecule as in the original process helps keep regulatory changes to a minimum. However, despite being operationally simple, this approach suffered from important drawbacks such as the recalcitrant 50% maximum yield inherent to KRs, the low enantioselectivity and the need for flash chromatography (enantiopure ivabradine in 30% overall yield for a three-step sequence). Alternatively, a route based on transaminases (TAs) or ketoreductases (KREDs) starting from an aldehyde precursor provided a chiral amine or alcohol, respectively. The amine would require two steps to the final product (including the same penultimate as above), but the alcohol could be converted in one step into the final ivabradine. Meanwhile the bioreduction displayed null enantioselectivity, the biocatalytic amination, performed at 25 mM and laboratory scale and involving a DKR, led to the target ivabradine in 50% yield in a four-step sequence without the need of chromatographic purification.
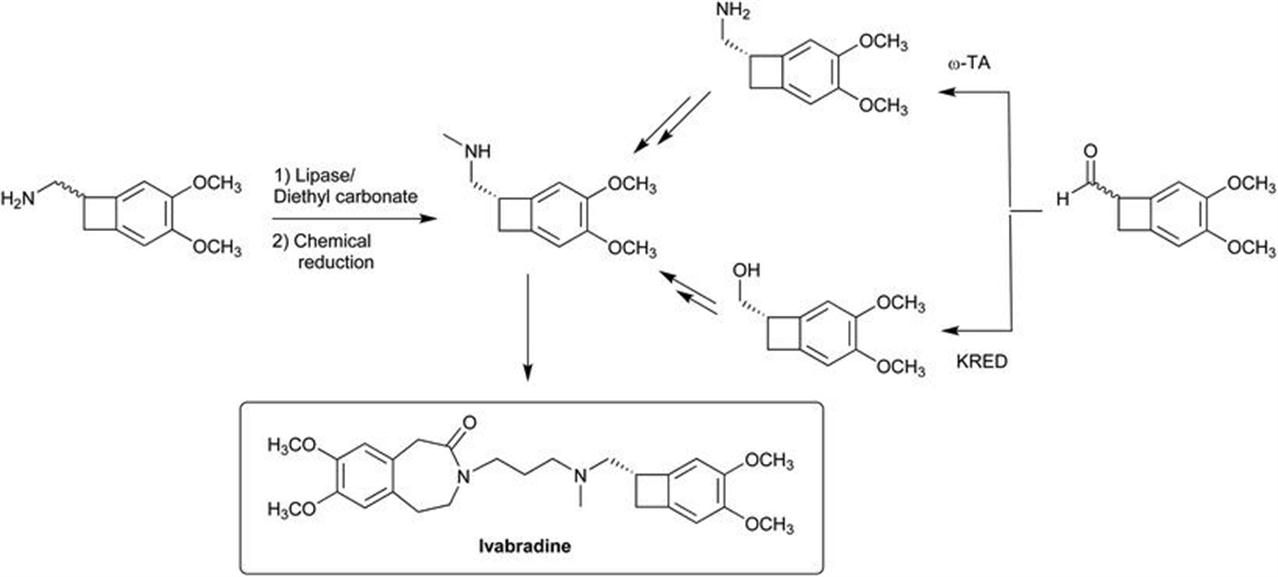
Scheme 4.1 Chemoenzymatic routes to ivabradine.
4.2.2 Cascade Processes Taking Advantage of Biocatalysis
Cascade and one-pot reactions represent an exciting recent development in white biotechnology.5–9 Although the concept of performing enzymatic multistep syntheses in a concurrent fashion is not very recent (multistep reactions in whole cells were used as early as the 1980s for the production of amino acids),10 it has received increased attention in the past few years. Especially from a green chemistry point of view, cascades represent a very promising approach, particularly due to the avoidance of intermediate downstream and purification steps. These often contribute significantly to the overall environmental impact of a reaction. Furthermore, workup and isolation of intermediate products binds production capacities and resources thereby also significantly contributing to production costs on industrial scale. On a more fundamental level, multistep cascades can be considered as biomimetic approaches. Although metabolic engineering was developed as a successful technology for the targeted manipulation of naturally evolved metabolic pathways (see next section), the synthesis of complex, high-value pharmaceutical compounds often lies beyond the alteration of existing pathways.
Figure 4.2 summarizes the concept behind the design of successful cascade processes. The idea is to combine cascade units (individual reactions) in a one-pot, if not fully concurrent, procedure. Typically, each unit will rely on a catalytic system, which could be based on biocatalysis (isolated or whole cells), metal-catalysis or organocatalysis. Each cascade unit must be carefully studied in terms of key parameter robustness (e.g., temperature, solvent, concentrations of enzyme and substrate, cofactors or other reagents), not only to find the optimal ones, but to identify the upper and lower limits of each parameter, since the assembly of the cascade may require operating a particular cascade unit outside optimal parameters when practiced as a standalone reaction. Importantly, not only “within-unit” parameters are to be considered, but also parameters from other cascade units that could cause incompatibility of operation between those units. This is the case for cofactors or auxiliary reagents needed in one but perhaps very detrimental for another unit, not to mention the presence of the other catalysts themselves. The compatibility of such cross-parameters determines more often than not the final operation of the cascade, which might need to sacrifice the all-concurrent operational feasibility and settle for the one-pot operation. In any case, the advantages of such a setting as mentioned above with respect to step-wise, or even telescoped, processes are still significant.
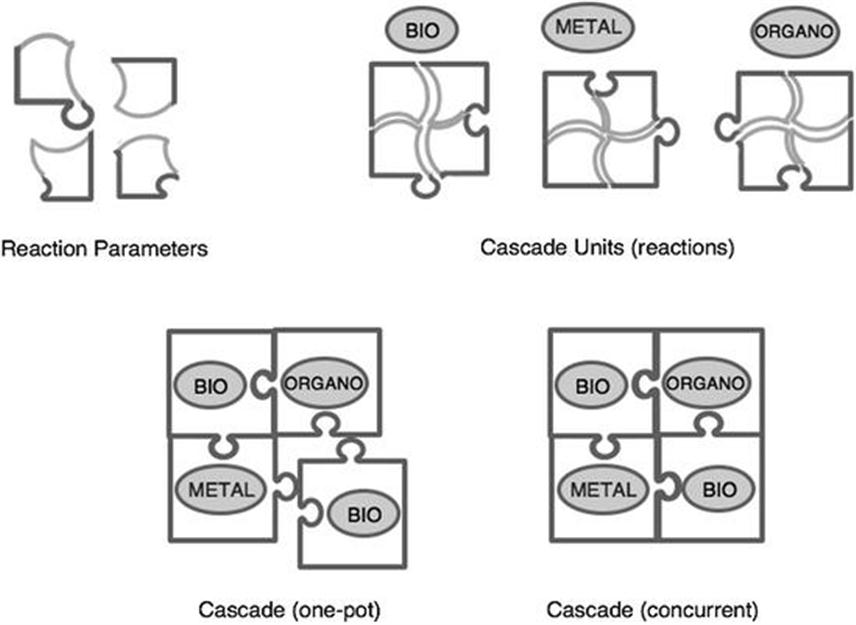
Figure 4.2 Concept of cascade processes involving bio-, metal- and organo-catalysis.
As for the possibilities to combine several isolated enzyme classes to facilitate a one-pot cascade, the use of redox enzymes has been successfully demonstrated in recent years.5,6 In this context, EntreChem has developed very recently an enzymatic cascade in water consisting of a stereoselective bioreduction of cyclic β-ketonitriles concurrently coupled to a whole cell-catalyzed nitrile hydrolysis in one-pot (Scheme 4.2).11 The first step, mediated by NADPH-dependent ketoreductases (KREDs), involved a dynamic reductive kinetic resolution (DYRKR),12 which led to β-hydroxynitriles in very high enantio- and diastereomeric ratios. Then, the simultaneous exposure to nitrile hydratase and amidase from whole cells of Rhodococcus rhodochrous provided the corresponding cyclic β-hydroxyacids with excellent overall yield and optical purity for the all-enzymatic cascade.13
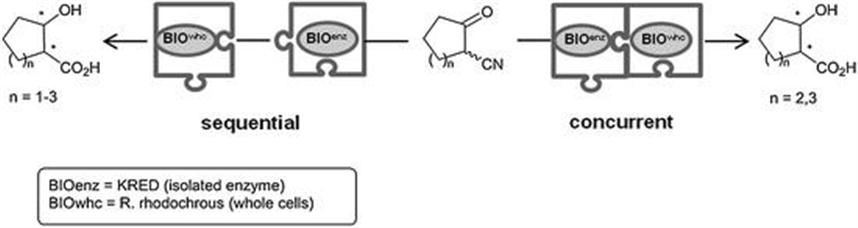
Scheme 4.2 Enzymatic cascade syntheses of cyclic β-hydroxyacids.
The success of the strategy lay both in the synchronization between a fast racemization of the epimerizable stereocenter before ketoreduction in the first step and a microorganism that converts the cyano group of the β-hydroxynitrile rather than that contained in the starting β-ketonitrile. Interestingly, the KREDs showed marked cis-diastereoselectivity, complementing the existing methodologies, mostly aimed at trans-diastereoisomers, readily available from epoxides and aziridines. The strategy was particularly effective with the cyclohexyl and cycloheptyl substrates, and the adequate selection of stereocomplementary KREDs delivered both antipodes of the cis-2-hydroxycycloxane- and cis-2-hydroxycycloheptane-carboxylic acids in >95% yield and total selectivity of >99 : 1 dr and >99% ee at 20 mM substrate concentration and lab-scale (Scheme 4.3). Although operationally viable, the concurrent process for the five-membered ring homologue was challenging since the optimal selectivity in the ketoreduction was achieved at pH 5.0, which inhibits the activity of R. rhodochrous. This issue was circumvented by means of a sequential setup and, once the bioreduction was completed, the only experimental setting consisted of a slight pH increase to 7.0 and further addition of the bacterial suspension.
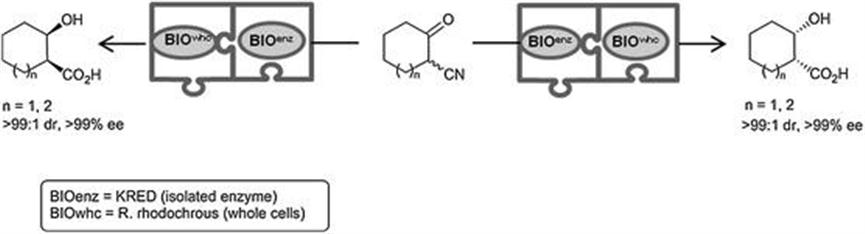
Scheme 4.3 Biocascades towards 2-hydroxycycloahexane- and cyclopentanecarboxylic acids.
Besides enzymatic cascades, the integration of enzymatic and chemocatalysis in one-pot sequences represents a more attractive and unexplored challenge since chemists have historically focused on combining reactions belonging to one of these technologies exclusively.14–16 Actually, examples of metal catalysts (or organocatalysts) working hand in hand with enzymes are still very scarce, especially in aqueous media,17 and imposes several challenges, namely (i) catalyst compatibility and stability, (ii) cross-reactivity, (iii) shifting the reaction equilibrium to the product side, and (iv) scalability. Despite these facts, the pool of metal catalysts able to operate in water has expanded enormously in recent years, providing novel opportunities to combine with enzymes which have water as their natural environment.
In this context, EntreChem developed the first combination of ω-transaminases (ω-TAs), which catalyze the transfer of an amino group from an amino donor onto a carbonyl moiety, with a metal-catalyzed reaction, namely the ruthenium-promoted isomerization of allylic alcohols which delivers carbonyl compounds (Scheme 4.4).18 Actually, this widely-studied isomerization had been recently performed in non-conventional solvents such as water or deep eutectic solvents, enabling such a combined process in aqueous media. Once both steps had been independently optimized, studies of compatibility revealed that both ω-TA and cofactor (pyridoxal-5′-phosphate, PLP) impacted negatively the metal catalyst. Consequently, the process was accomplished in a sequential fashion at lab-scale. Indeed, the aqueous medium from the metal-catalyzed reaction (200 mM substrate concentration) was used directly to feed the bioamination with the only telescopic adjustment of a ten-fold dilution of the medium before adding the ω-TA and PLP. As a result, and taking advantage of the available ω-TAs, both antipodes of chiral amines were isolated in very high yield and enantiomeric excess without the need for further purification.
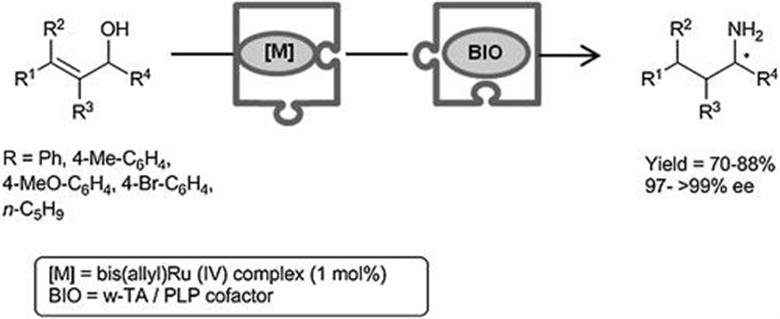
Scheme 4.4 Ruthenium-catalyzed allylic alcohol isomerization combined with an enantioselective enzymatic amination in a one-pot sequential process.
In a further study, the metal-catalyzed allylic alcohol isomerization was also combined with a bioreduction mediated by KREDs, enabling in this case the isolation of the analogue chiral alcohols (Scheme 4.5).19 On this occasion, both reaction conditions and reagents of each step were fully compatible and a concurrent process was efficiently implemented at 200 mM substrate concentration at lab-scale. Thus, the chemical and the enzymatic step proceeded simultaneously from the beginning in the same pot to furnish the products in high yield and enantioselectivity. Furthermore, a detailed study of the reaction kinetics revealed a rapid deactivation of the enzyme under the reaction conditions and, consequently, the faster the isomerization takes place, the higher the overall yield of the cascade. Thus, and despite a slight drop in the yield compared to a parallel sequential process, this first example of a genuine concurrent metal and biocatalyzed reaction in water is an important contribution to the field of cascade processes, as it does not require site-isolating techniques such as compartmentalization or encapsulation.
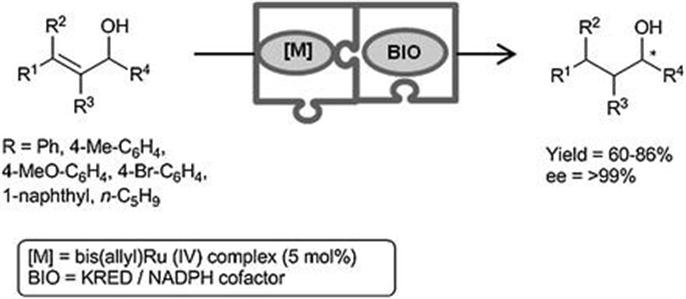
Scheme 4.5 Ruthenium-catalyzed allylic alcohol isomerization combined with an enantioselective bioreduction in a one-pot concurrent process.