Bristol-Myers Squibb: Preparation of Chiral Intermediates for the Development of Drugs and APIs
RAMESH N. PATEL
SLRP Associates,LLC, Consultation in Biotechnology, 572 Cabot Hill Road, Bridgewater, NJ 08807 USA
References
1. H. Zhao, K. Chockalingom and Z. Chen, Curr. Opin. Biotechnol., 2002, 13, 104.
2. R. N. Patel, Stereoselective Biocatalysis, Marcel Dekker, Inc., New York, 2000.
3. G. W. Huisman and J. J. Lalonde, in Biocatalysis in the Pharmaceutical and Biotechnology Industries, ed. R. N. Patel, CRC Press, Boca Raton, Florida, 2007, p. 717.
4. U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185.
5. R. N. Patel, ACS Catal., 2011, 1, 1056.
6. M. T. Reetz, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH Verlag GmbH, Weinheim, Germany, 3rd edn, 2012, p. 214.
7. R. N. Patel, Biomolecules, 2013, 3, 741.
8. N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285.
9. H.-P. Meyer, E. Eichhorn, S. Hanlon, S. Lutz, M. Schürmann, R. Wohlgemuth and R. Coppolecchia, Catal. Sci. Technol., 2013, 3, 29.
10. R. N. Patel, in Biocatalysis in the Pharmaceutical and Biotechnology Industries, ed. R. N. Patel, CRC Press, Boca Raton, Florida, 2007, p. 207.
11. C. Simons, U. Hanefeld, I. W. C. E. Arends, T. Maschmeyer and R. A Sheldon, Top. Catal., 2006, 40, 35.
12. M. Hall, C. Stueckler, H. Ehammer, E. Pointner, G. Oberdorfer, K. Gruber, B. Hauer, R. Stuermer, W. Kroutil, P. Macheroux and K. Faber, Adv. Synth. Catal., 2008, 350, 411.
13. A. S. Wells, G. L. Finch, P. C. Michaels and J. W. Wong, Org. Process Res. Dev., 2012, 16, 1986.
14. G. W. Huisman and S. J. Collier, Curr. Opin. Chem. Biol., 2013, 17, 284.
15. R. N. Patel, in Science of Synthesis: Biocatalysis in Organic Synthesis 3, ed. K. Faber, W. D. Fessner and N. J. Turner, Georg Thieme Verlag, Stuttgart, 2015, p. 403.
16. R. N. Patel, Green Biocatalysis, John Wiley & Sons, Inc., Hoboken, NJ, 2016.
17. S. B. Roberts, Adv. Drug Delivery Rev., 2002, 54, 1579.
18. K. W. Gillman, J. E. Jr Starrett, M. F. Parker, K. Xie, J. J. Bronson, L. R. Marcin, K. E. McElhone, C. P. Bergstrom, R. A. Mate, R. Williams, J. E. Meredith, C. R. Burton, D. M. Barten, J. H. Toyn, S. B. Roberts, K. A. Lentz, J. G. Houston, R. Zaczek, C. F. Albright, C. P. Decicco, J. E. Macor and R. E. Olson, ACS Med. Chem. Lett., 2010, 1, 120.
19. I. Ojima, K. Koji and K. Nakahashi, J. Org. Chem., 1989, 54, 4511.
20. A. Goswami, S. L. Goldberg, R. M. Johnston, R. L. Hanson and W. L. Parker, WO/2012/106579, 2012.
21. R. L. Hanson, R. M. Johnston, S. L. Goldberg, W. L. Parker and A. Goswami, Org. Process Res. Dev., 2013, 17, 693.
22. B. D. Roth, Prog. Med. Chem., 2002, 40, 1.
23. S. Y. Sit, R. A. Parker, I. Motoc, W. Han, N. Balasubramanian, J. D. Catt, P. J. Brown, W. E. Harte, M. D. Thompson and J. J. Wright, J. Med. Chem., 1990, 33(11), 2982.
24. R. N. Patel, C. M. McNamee and L. J. Szarka, Appl. Microbiol. Biotechnol., 1992, 38, 56.
25. N. Balasubramanian, P. J. Brown, J. D. Catt, W. T. Han, R. A. Parker, S. Y. Sit and J. J. Wright, J. Med. Chem., 1989, 32(9), 2038.
26. R. N. Patel, A. Banerjee, C. G. McNamee, D. B. Brzozowski, R. L. Hanson and L. J. Szarka, Enzyme Microb. Technol., 1993, 15, 1014.
27. Z. Guo, Y. Chen, A. Goswami, R. L. Hanson and R. N. Patel, Tetrahedron: Asymmetry, 2006, 17, 1589.
28. S. Goldberg, Z. Guo, S. Chen, A. Goswami and R. N. Patel, Enzyme Microb. Technol., 2008, 43, 544.
29. P. V. Chaturvedula, L. Chen, R. Civiello, A. P. Degnan, G. M. Dubowchik, X. Han, J. J. Jiang, J. E. Macor, G. S. Poindexter, G. O. Tora and G. Luo, US Pat. 2007/0149503 A1, 2007.
30. X. Han, R. L. Civiello, C. M. Conway, D. A. Cook, C. D. Davis, R. Macci, S. S. Pin, S. X. Ren, R. Schartman, L. J. Signor, G. Thalody, K. A. Widmann, C. Xu, P. V. Chaturvedula, J. E. Macor and G. M. Dubowchik, Bioorg. Med. Chem. Lett., 2012, 22, 4723.
31. P. V. Chaturvedula, S. E. Mercer, S. S. Pin, G. Thalody, C. Xu, C. M. Conway, D. Keavy, L. Signor, G. H. Cantor, N. Mathias, P. Moench, R. Denton, R. Macci, R. Schartman, V. Whiterock, C. Davis, J. E. Macor and G. M. Dubowchik, Bioorg. Med. Chem. Lett., 2013, 23, 3157.
32. R. L Hanson, B. L. Davis, S. L. Goldberg, R. M. Johnston, W. L. Parker, T. P. Tully, M. A. Montana and R. N. Patel, Org. Process Res. Dev., 2008, 12, 1119.
33. E. M. Sinclair and D. J. Drucker, Curr. Opin. Endocrinol., Diabetes Obes., 2005, 12, 146.
34. D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, A. Khanna, J. G. Robertson, A. Wang, L. M. Simpkins, P. Taunk, Q. Huang, S.-P. Han, B. Abboa-Offei, M. Cap, L. Xin, L. Tao, E. Tozzo, G. E. Welzel, D. M. Egan, J. Marcinkeviciene, S. Y. Chang, S. A. Biller, M. S. Kirby, R. A. Parker and L. G. Hamann, J. Med. Chem., 2005, 48, 5025.
35. T. C. Vu, D. B. Brzozowski, R. Fox, J. D. Jr Godfrey, R. L. Hanson, S. V. Kolotuchin, J. A. Jr Mazzullo, R. N. Patel, J. Wang, K. Wong, J. Yu, J. Zhu, R. D. Magnin, D. J. Augeri and L.
G. Hamann, WO 2004052850, 2004.
36. H. G. Cheon, S.-S. Kim, K.-R. Kim, S.-D. Rhee, S. D. Yang, J. H. Ahn, J. H. Park, S.-D. Lee, J. M. Jung, W. H. Lee and H. Y. Kim, Biochem. Pharmacol., 2005, 70, 22.
37. I. Brandt, J. Joossens, X. Chen, M.-B. Maes, S. Scharpé, I. De Meester and A. M. Lambeir, Biochem. Pharmacol., 2005, 70, 134.
38. R. L. Hanson, S. L. Goldberg, D. B. Brzozowski, T. P. Tully, D. Cazzulino, W. L. Parker, O. K. Lyngberg, T. C. Vu, M. K. Wong and R. N. Patel, Adv. Synth. Catal., 2007, 349, 1369.
39. R. L. Hanson, R. M. Johnston, S. L Goldberg, W. L. Parker and R. N. Patel, Enzyme Microb. Technol., 2011, 48, 445.
40. M. Kunishima, C. Kawachi, K. Hioki, S. Terao and S. Tani, Tetrahedron, 2001, 57, 1551.
41. I. Gill and R. N. Patel, Bioorg. Med. Chem. Lett., 2006, 16, 705.
42. J. J. Holst, C. Orskov, O. V. Nielsen and T. W. Schwartz, FEBS Lett., 1987, 211, 169.
43. T. S. Haque, W. R. Ewing, C. Mapelli, V. G. Lee, R. B. Sulsky. D. J. Riexinger and R. L. Martinez, Y. Z. Zhu, WO2007082264, 2007.
44. C. Mapelli, S. I. Natarajan, J.-P. Meyer, M. M. Bastod, M. S. Bernatowicz, V. G. Lee, J. Pluscec, D. J. Riexinger, E. S. Sieber-McMaster, K. L. Constantine, C. A. Smith-Monroy, R. Golla, Z. Ma, D. A. Longhi, D. Shi, L. Xin, J. R. Taylor, B. Koplowitz, C. L. Chi, A. Khanna, G. W. Robinson, S. Seethala, H. A. Antal-Zimanyi, R. H. Stoffel, S. Han, J. M. Whaley, C. S. Huang, J. Krupinski and W. R. Ewing, J. Med. Chem., 2009, 52, 7788.
45. Y. Chen, S. L. Goldberg, R. L. Hanson, W. L. Parker, I. Gill, T. P. Tully, M. Montana, A. Goswami and R. N. Patel, Org. Process Res. Dev., 2011, 15, 241.
46. J. Robl, C. Sun, J. Stevenson, D. Ryono, L. Simpkins, M. Cimarusti, T. Dejneka, W. Slusarchyk, S. Chao, L. Stratton, R. Misra, M. Bednarz, M. Asaad, H. Cheung, B. Aboa-Offei, P. Smith, P. Mathers, M. Fox, T. Schaeffer, A. Seymour and N. Trippodo, J. Med. Chem., 1997, 40, 1570.
47. R. L. Hanson, M. D. Schwinden, A. Banerjee, D. B. Brzozowski, B.-C. Chen, B. P. Patel, C. G. McNamee, G. A. Kodersha, D. R. Kronenthal, R. N. Patel and L. J. Szarka, Bioorg. Med. Chem., 1999, 7, 2247.
48. R. L. Hanson, J. Howell, T. LaPorte, M. Donovan, D. Cazzulino, V. Zannella, M. Montana, V. Nanduri, S. Schwarz, R. Eiring, S. Durand, J. Wasylyk, L. Parker, M. Liu, F. Okuniewicz, B.-C. Chen, J. Harris, K. Natalie, K. Ramig, S. Swaminathan, V. Rosso, S. Pack, B. Lotz, P. Bernot, A. Rusowicz, D. Lust, K. Tse, J. Venit, L. Szarka and R. N. Patel, Enzyme Microb. Technol., 2000, 26, 348.
49. R. N. Patel, Biomol. Eng., 2001, 17, 167.
50. R. N. Patel, A. Banerjee, V. Nanduri, S. Goldberg, R. Johnston, R. Hanson, C. McNamee, D. Brzozowski, T. P. Tully, R. Ko, T. LaPorte, D. Cazzulino, S. Swaminathan, L. Parker and J. Venit, Enzyme Microb. Technol., 2000, 27, 376.
51. M. Ondetti and D. Cushman, J. Med. Chem., 1981, 24, 355.
52. M. Ondetti, B. Rubin and D. Cushman, Science, 1977, 196, 441.
53. D. Cushman and M. Ondetti, in Progress in Medicinal Chemistry, ed. G. P. Ellis and G. B. West, Elsevier/North Holland, Amsterdam, The Netherlands, 1980, vol. 17, p. 42.
54. C. Goodhue and J. Schaeffer, Biotechnol. Bioeng., 1971, 13, 203.
55. R. N. Patel, J. Howell, A. Banerjee, K. Fortney and L. Szarka, Appl. Microbiol. Biotechnol., 1991, 36, 29.
56. J. Moniot, U.S. Patent Application: CN 88–100862, 1988.
57. M. Ondetti, A. Miguel and J. Krapcho, US Pat. 4316906, 1982.
58. G. Bold, A. Faessler, H.-G. Capraro, R. Cozens, T. Klimkait, J. Lazdins, J. Mestan, B. Poncioni, J. Roesel, D. Stover, M. Tintelnot-Blomley, F. Acemoglu, W. Beck, E. Boss, M. Eschbach, T. Huerlimann, E. Masso, S. Roussel, K. Ucci-Stoll, D. Wyss and M. Lang, J. Med. Chem., 1998, 41, 3387.
59. B. S. Robinson, K. A. Riccardi, Y. F. Gong, Q. Guo, D. A. Stock, W. S. Blair, B. J. Terry, C. A. Deminie, F. Djang, R. J. Colonno and P.-F. Lin, Antimicrob. Agents Chemother., 2000, 44, 2093.
60. B. Degertekin and A. S. Lok, Curr. Opin. Gastroenterol., 2008, 24, 306.
61. F. G. Njoroge, K. X. Chen, N. Y. Shih and J. J. Piwinski, Acc. Chem. Res., 2008, 41, 50.
62. U. Kragl, D. Nasic-Racki and C. Wandrey, Bioprocess Eng., 1996, 14, 291.
63. A. S. G. Krix, A. S. Bommarius, K. Kottenhahn, M. Schwarm and M. R. Kula, J. Biotechnol., 1997, 53, 29.
64. A. Galkin, L. Kulakova, T. Yoshimura, K. Soda and N. Esaki, Appl. Environ. Microbiol., 1997, 63, 4651.
65. A. Menzel, H. Werner, J. Altenbuchner and H. Gröger, Eng. Life Sci., 2004, 4, 573.
66. R. N. Patel, L. Chu and R. H. Mueller, Tetrahedron: Asymmetry, 2003, 14, 3105.
67. N. I. Bowers, P. M. Skonezny, G. L. Stein, T. Franceschini, S.-J. Chiang, W. L. Anderson, L. You and Z. Xing, US Pat. 2009/0286303, 2009.
68. Z. Xu, J. Singh, M. D. Schwinden, B. Zheng, T. P. Kissick, B. Patel, M. J. Humora, F. Quiroz, L. Dong, D.-M. Hsieh, J. E. Heikes, M. Pudipeddi, M. D. Lindrud, S. K. Srivastava, D. R. Kronenthal and R. H Mueller, Org. Process Res. Dev., 2002, 6, 323.
69. Y. K. Bong, M. Vogel, S. J. Collier, V. Mitchell and J. Mavinahalli, Patent Application WO/2011/005527, 2011.
70. H. Jajoo, R. Mayol, J. LaBudde and I. Blair, Drug Metab. Dispos., 1989, 17, 634.
71. R. Mayol, US Pat., 6150365 2000, 2000.
72. J. Yevich, J. New, W. Lobeck, P. Dextraze, E. Bernstein, D. Taylor, F. Yocca, M. Eison and D. Jr Temple, J. Med. Chem., 1992, 35, 4516.
73. J. Yevich, R. Mayol, J. Li and F. Yocca, US Pat., 2003022899, 2003.
74. R. L. Hanson, W. L. Parker, D. B. Brzozowski, T. P. Tully, A. Kotnis and R. N. Patel, Tetrahedron: Asymmetry, 2005, 16, 2711.
75. R. N. Patel, L. Chu, V. Nanduri, L. Jianqing, A. Kotnis, W. Parker, M. Liu and R. H. Mueller, Tetrahedron: Asymmetry, 2005, 16, 2778.
76. S. Goldberg, V. Nanduri, L. Chu, R. Johnston and R. N. Patel, Enzyme Microb. Technol., 2006, 39, 1441.
77. S. F. Innaimo, M. Seifer, G. S. Bisacchi, D. N. Standring, R. Zahler and R. J. Colonno, Antimicrob. Agents Chemother., 1997, 41, 1444.
78. E. V. Genovesi, L. Lamb, I. Medina, D. Taylor, M. Seifer, S. Innaimo, R. J. Colonno, D. N. Standring and J. M. Clark, Antimicrob. Agents Chemother., 1998, 42(12), 3209.
79. Y. R. Pendri, C.-P. Chen, S. Patel, J. M. Evans, J. Liang, D. R. Kronenthal, D. Krishnamurty, M. X. Zhou and P. Vemishetti, PCT Int. Appl. WO 2004052310 A2 20040624 CAN 141:38813, 2004.
80. D. A. Griffith and S. J. Danishefsky, J. Am. Chem. Soc., 1996, 118, 9526.
81. S. J. Danishefsky, M. P. Cabal and K. Chow, J. Am. Chem. Soc., 1989, 111, 3456.
82. R. N. Patel, A. Banerjee, Y. R. Pendri, J. Liang, C.-P. Chen and R. Mueller, Tetrahedron: Asymmetry, 2006, 17, 175.
83. R. J. Cherney, P. Carter, J. V. Duncia, D. S. Gardner and J. B. Santella, PCT Int. Appl. WO/2004/071460 A2, 2004.
84. R. J. Cherney, J. B. Brogan, R. Mo, Y. C. Lo, C. Yang, P. B. Miller, P. A. Scherle, B. F. Molino, P. H. Carter and C. P. Decicco, Bioorg. Med. Chem. Lett., 2009, 19, 597.
85. P. H. Carter, Expert Opin. Ther. Pat., 2013, 23, 549–568.
86. Y. Chen, S. Tian and L. Deng, J. Am. Chem. Soc., 2000, 122, 9542.
87. C. Bolm, I. Schiffers, C. L. Dinter and A. Gerlach, J. Org. Chem., 2000, 65, 6984.
88. W.-M. Dai, K. K. Y. Yeung, C. W. Chow and I. D. Williams, Tetrahedron: Asymmetry, 2001, 12, 1603.
89. S. Kobayashi, K. Kamiyama, T. Iimori and M. Ohno, Tetrahedron Lett., 1984, 25, 2557.
90. A. Goswami and T. P. Kissick, Org. Process Res. Dev., 2009, 13, 483.
91. R. Holton, P. Biediger and P. Joatman, Taxol: Science and Application, ed. M. Suffness, CRC Press, New York, 1995, p. 97.
92. D. Kingston, Taxol: Science and Application, ed. M. Suffness, CRC Press, New York, 1995, p. 287.
93. R. Hanson, J. Wasylyk, V. Nanduri, D. Cazzulino, R. N. Patel and L. Szarka, J. Biol. Chem., 1994, 269, 22145.
94. V. Nanduri, R. Hanson, T. LaPorte, R. Ko, R. N. Patel and L. Szarka, Biotechnol. Bioeng., 1995, 48, 547.
95. R. N. Patel, Annu. Rev. Microbiol., 1995, 98, 361.
96. R. Hanson, J. Howell, D. Brzozowski, S. Sullivan, R. N. Patel and L. Szarka, Biotechnol. Appl. Biochem., 1997, 26, 153.
97. R. N. Patel, A. Banerjee, J. Howell, C. McNamee, D. Brozozowski, D. Mirfakhrae, V. Nanduri, J. Thottathil and L. Szarka, Tetrahedron: Asymmetry, 1993, 4, 2069.
98. R. N. Patel, A. Banerjee, R. Ko, J. Howell, W.-S. Li, F. Comezoglu, R. Partyka and L. Szarka, Biotechnol. Appl. Biochem., 1994, 20, 23.
99. E. Baloglu and D. Kingston, J. Nat. Prod., 1999, 62, 1068.
100. F. S. Gibson, US Pat. 6307071B1, 2001.
101. W. Rose, B. Long, C. Fairchild, F. Lee and J. Kadow, Clin. Cancer Res., 2001, 7, 2016.
102. H. Mastalerz, D. Cook, C. R. Fairchild, S. Hansel, W. Johnson, J. F. Kadow, B. H. Long, W. C. Rose, J. Tarrant, M.-J. Wu, Q. Xue, G. Zhang, M. Zoeckler and D. M. Vyas, Bioorg. Med. Chem., 2003, 11, 4315.
103. D. G. I. Kingston and Z.-Y. Zhao, US Pat. 5059699, 1991.
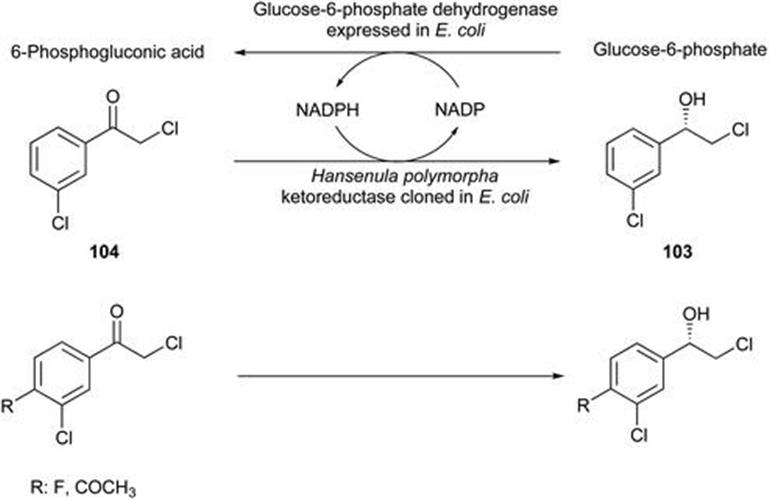
104. R. N. Patel, J. Howell, R. Chidambaram, S. Benoit and J. Kant, Tetrahedron: Asymmetry, 2003, 14, 3673.
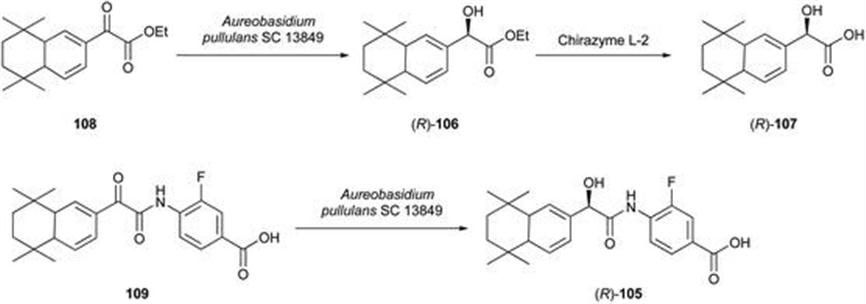
105. R. L. Hanson, W. L. Parker and R. N. Patel, Biotechnol. Appl. Biochem., 2006, 45, 81.
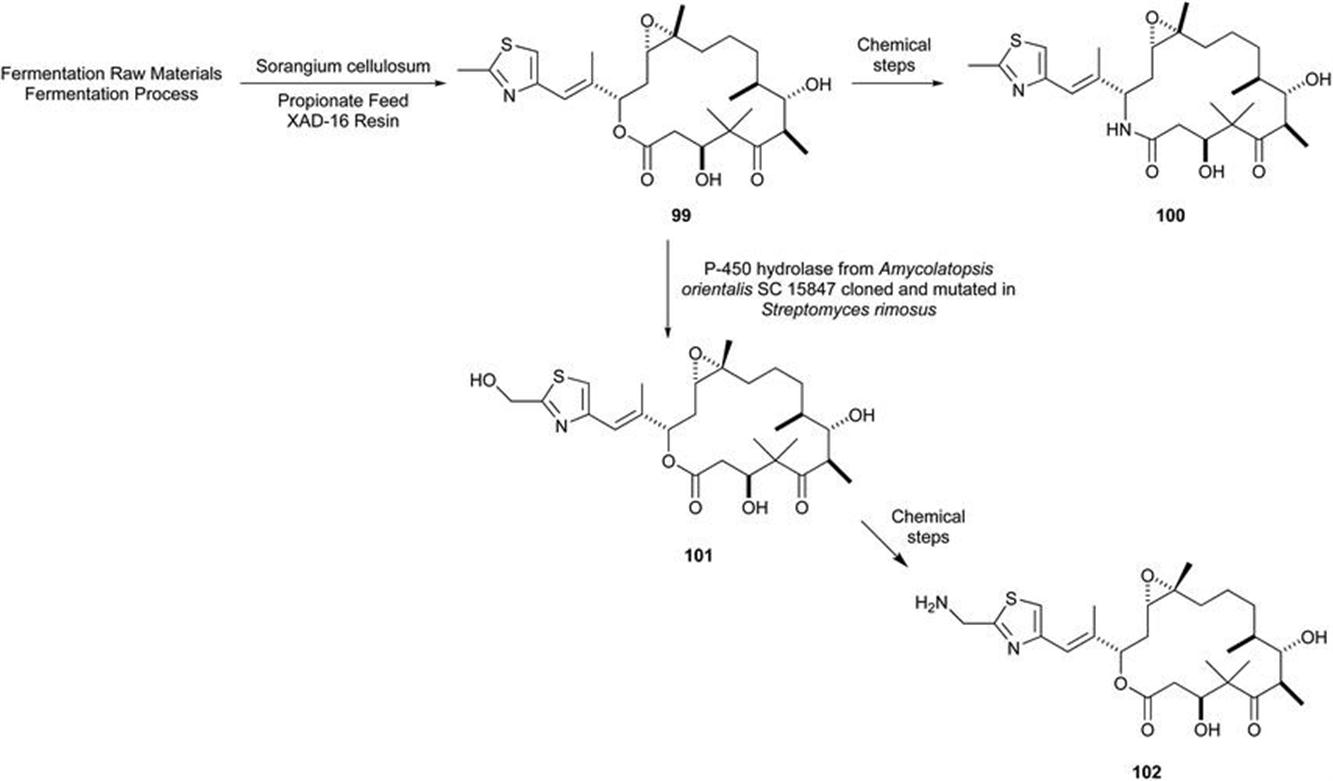
106. K. Gerth, S. Pradella, O. Perlova, S. Beyer and R. Muller, J. Biotechnol., 2003, 106, 233.
107. D. Benigni, R. Stankavage, S.-J. Chiang, H. Hou, B. Eagan, D. Gu, D. Hou, L. Mintzmyer, T. P. Tully, D. L. Davis, I. Hargro, M. Mascari, G. Galvin, G. Stein, C. W. McConlogue and F. T. Comezoglu, PCT Int. Appl. 85 pp WO 2004026254 A2 20040401 CAN 140:286248 A 2004:270015, 2004.
108. S. Goodin, M. P. Kane and E. H. Rubin, J. Clin. Oncol., 2004, 2, 2015.
109. K. C. Nicolaou, F. Roschangar and D. Vourloumis, Angew. Chem., Int. Ed., 1998, 37, 2014.
110. K.-H. Altmann, Org. Biomol. Chem., 2004, 2, 2137.
111. E. N. Boddy, K. Hotta, M. L. Tse, R. E. Watts and C. Khosla, J. Am. Chem. Soc., 2004, 126, 7436.
112. N. Lin, K. Brakora and M. Seiden, Curr. Opin. Invest. Drugs, 2003, 4, 746.
113. J. D. Basch, S.-J. Chiang, S.-W. Liu, A. Nayeem and Y.-L. Sun, PCT Int. Appl. 192 pp. WO 2004078978, A1 20040916, CAN 141:255532, AN 2004:756881, 2004.
114. J. Basch and S.-J. Chiand, J. Ind. Microbiol. Biotechnol., 2007, 34, 171.
115. J. M. Carboni, W. W. Hurlburt and M. M. Gottardis, PCT Int. Appl. 39 pp. WO 2004030625, A2 20040415, CAN 140:315050, AN 2004:308360, 2004.
116. F. Beaulieu, C. Ouellet, K. Zimmermann, U. Velaparthi and M. D. Wittman, PCT Int. Appl., 65 pp. WO 2004063151, A2 20040729, CAN 141:140461, AN 2004:606437, 2004.
117. R. L. Hanson, S. Goldberg, A. Goswami, T. P. Tully and R. N. Patel, Adv. Synth. Catal., 2005, 347, 1073.
118. H. Kagechika, E. Kawachi, Y. Hashimoto, K. Shudo and T. Himi, J. Med. Chem., 1988, 31, 182.
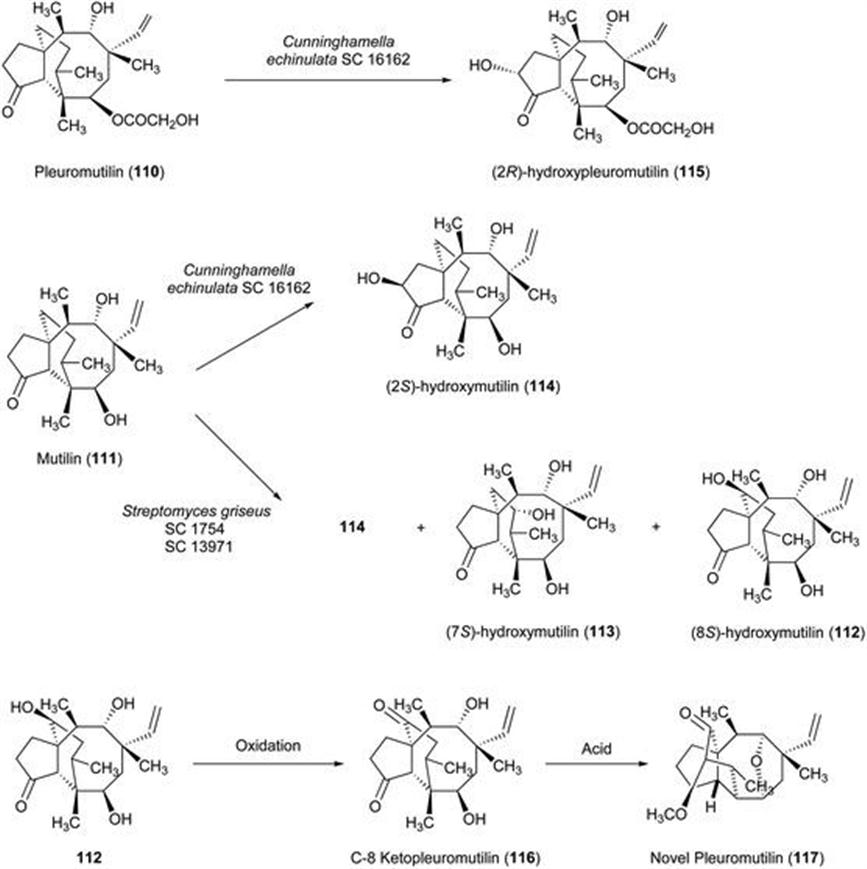
119. H. Kagechika and K. Shudo, J. Med. Chem., 2005, 48, 5875.
120. G. M. Morriss-Kay, Adv. Organ Biol., 1979, 3, 79.
121. R. C. Moon and L. M. Itri, Retinoids, 1984, 2, 327.
122. R. N. Patel, L. Chu, R. Chidambaram, J. Zhu and J. Kant, Tetrahedron: Asymmetry, 2002, 13, 349.
123. G. Hoegenauer, Antibiotics, 1979, 5, 344.
124. H. Berner, H. Vyplel, G. Schulz and P. Stuchlik, Tetrahedron, 1983, 39, 1317.
125. R. Hanson, J. Matson, D. Brzozowski, T. LaPorte, D. Springer and R. N. Patel, Org. Process Res. Dev., 2002, 6, 482.
126. D. Springer, M. Sorenson, S. Huang, T. Connolly, J. Bronson, J. Matson, R. Hanson, D. Brzozowski, T. LaPorte and R. N. Patel, Bioorg. Med. Chem. Lett., 2003, 13, 1751.