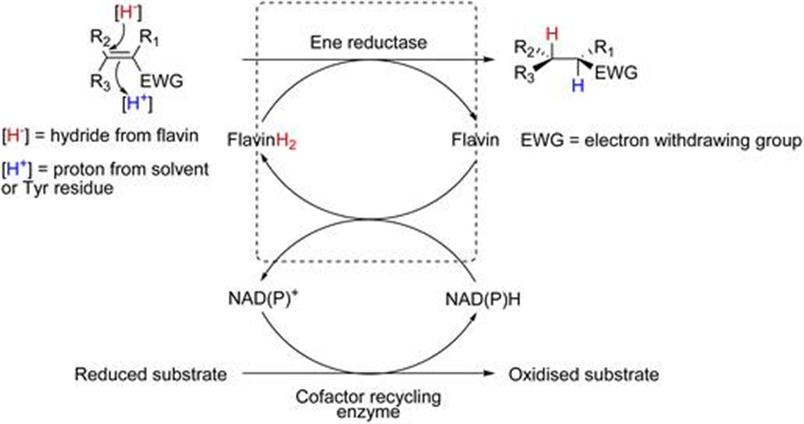
Almac Sciences, Department of Biocatalysis and Isotope Chemistry, 20 Seagoe Industrial Estate, Craigavon BT63 5QD, Northern Ireland, UK
8.4 Examples of Ene Reductase Reactions Reported in the Literature
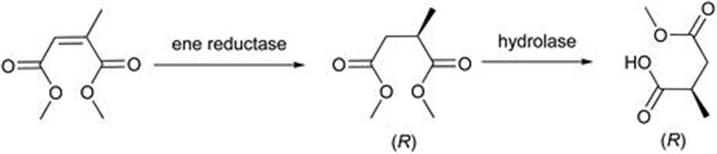
To demonstrate that ene reductases are receiving serious attention from industrial pharmaceutical process chemistry groups one can cite the work that Pfizer Inc. have published, pertinent to the synthesis of pregabalin.23,24 This is a γ-aminobutyric acid mimic used in the treatment of CNS disorders such as epilepsy. The key bioreduction utilises an ene reductase cloned into E. coli, from Lycopersicon esculentum, that can reduce both geometric isomers in high enantiomeric excess as shown in Figure 8.2. The enzyme was considerably faster for the (E)-isomer. Both acid and ester were substrates. Preparative scale reduction of the (E)-isomer was achieved (>99% ee in 69% yield) using NADPH as the cofactor, with recycling of the cofactor enabled by Lactobacillus brevis alcohol dehydrogenase and isopropanol as the hydrogen source. Whilst the authors noted that issues such as enzyme activity and stability still need to be addressed before an effective industrial reduction of the geometric isomer mixture can be realised, reaction and enzyme engineering offers the potential to achieve this.
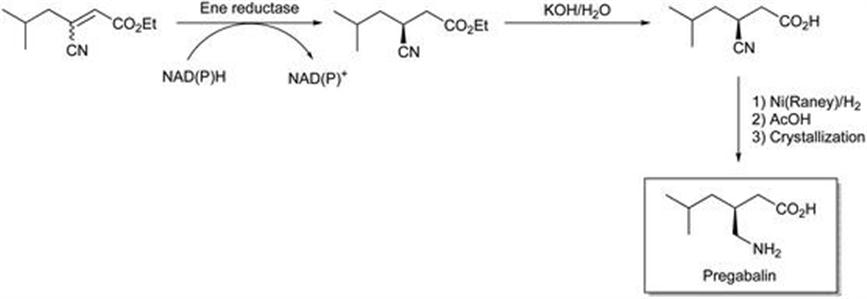
Figure 8.2 Synthesis of pregabalin using an ene reductase.
Porto et al.25 reported for the first time the ene-reduction of aromatic malononitriles with electron withdrawing substituents (Figure 8.3a) by biotransformation using whole cells marine-derived Penicillium citrinum CBMAI 1186, with the products obtained in good yields (>93%). With electron donating substituents (Figure 8.3b) the yield of desired products was much lower. It is likely that the OH substituent, which results in a 12% yield, disfavours biohydrogenation. The methoxy substituent resulted in an 84% yield and the OH and OMe functionalised substrate returned a 66% yield. The reduction of the C=C bond by CBMAI is shown to depend on the electronic effects promoted by the attached substituents.
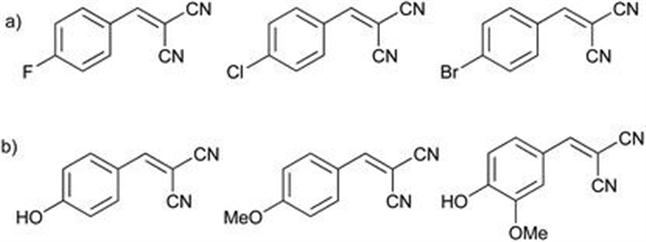
Figure 8.3 Substrates with (a) electron withdrawing and (b) electron donating substituents for bioreduction with CBMAI 1186.
Utilising biocatalysts in the synthesis of menthol is highly desirable, as the monoterpenoids are of high commercial value, and mint crop prices can be volatile due to reliance on harvest yields with the process including expensive steam distillation and filtration processes.26–28 The optimisation strategies for biocatalytic terpenoid synthesis focus on an expression host and the metabolic engineering of a biosynthesis pathway. Scrutton et al.29 have demonstrated a one-pot biotransformation of (1R,2S,5R)-(−)-menthol and (1S,2S,5R)-(+)-neomenthol from pulegone, a commonly found flavour in perfume products. They developed a strategy using recombinant E. coli extracts containing the biosynthetic genes for an ene reductase (NtDBR from Nicotiana piperita) and two menthone dehydrogenases (MMR and MNMR from Mentha piperita) with moderately pure menthol (79.1%) and neomenthol (89.9%) obtained. The advantages of the one-pot strategy are the ability to optimise each enzymatic step and the ability to generate libraries of pure compounds for use in high-throughput screening. Further to this work the authors also pinpointed a mechanistic switch between ketoreduction and ene-reductase activity in the short-chain dehydrogenases/reductases family which could potentially afford ene-reductases from the transformation of SDR ketoreductases.30
Characterisation of two OYEs from the genome of Chryseobacterium sp. CA49 for their ene reductase activities showed that Chr-OYE1 had broad substrate scope (reduced 18 out of 19 substrates studied) and excellent stereoselectivity, while Chr-OYE2 had limited activity towards activated alkenes. The mutant Chr-OYE2-M183Y displayed improved activity and stereoselectivity (>99%) when compared with the wildtype catalyst.31
8.4.1 Ene Reductases as Part of a Reaction Sequence
Whilst still at an early stage, a greater level of sophistication is emerging in the operation of biocatalysis, where more than one biotransformation is performed in sequence. This in effect creates an artificial metabolic pathway, and can take a net conversion of substrate to product well beyond what might be achieved simply through chemical methods alone. The phrase ‘reaction cascade’ has been coined to describe such sequences. Ene reductases have been demonstrated as useful players in this emerging technology. As an example, the biotransformation of allylic alcohols into lactones was reported.32,33Figure 8.4 shows the scheme for the biotransformation of carveol into a related lactone. The sequence consisted of an oxidation by alcohol dehydrogenase to form an enone, the stereoselective hydrogenation of the alkene by an ene reductase, and finally the lactone formation by a Baeyer–Villiger monooxygenase. Overall, a 60% yield was obtained with an excellent >99% diastereomeric excess. This approach was shown to work for a range of unsaturated alcohols. Furthermore, the three biocatalysts were expressed simultaneously in E. coli, so that a single whole biocatalyst was able to perform the illustrated sequence, representing an operationally simple procedure, with the full redox balance of cofactors effected by internal metabolism of the E. coli cells.
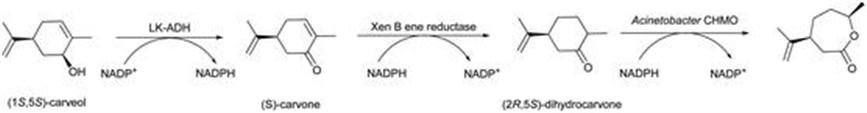
Figure 8.4 Transformation of allylic alcohol into lactone.
A second example reported the one-pot conversion of (R)- and (S)-carvone into carveol.34 This synthesis utilised the two-enzyme sequence shown in Figure 8.5. In the first step carbon–carbon double bond reduction, ene reductase LacER from Lactobacillus casei was used. For the second step, a carbonyl reductase from Sporobolomyces salmonicolor (SSCR) or Candida magnolia (CMCR) was employed to perform the asymmetric ketone reduction. The aim was to develop a more effective process for the conversion of carvones into enantiomerically pure dihydrocarveols, compounds of high interest due to their application as fragrance ingredients. A one-pot process was demonstrated at 0.1 M substrate concentration, yielding in the case of (R)-carvone a product with 99% diastereomeric excess, and in the case of (S)-carvone 86% de.
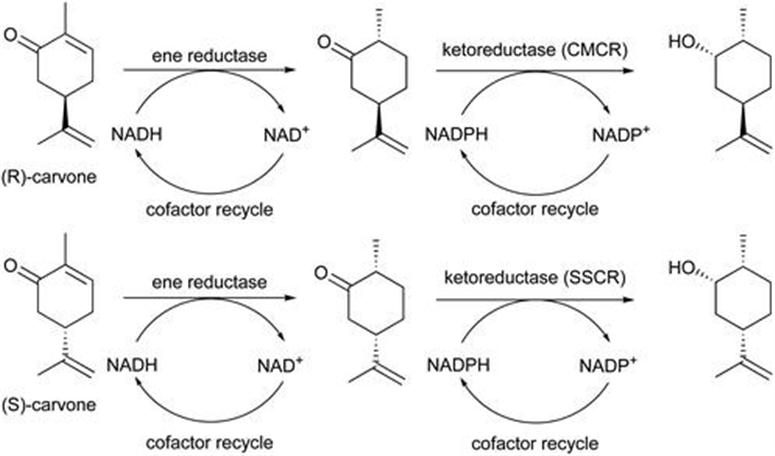
Figure 8.5 Biotransformation of carvone enantiomers.
A similar approach using an ene reductase followed by alcohol dehydrogenase was described for the conversion of a range of enals and enones into primary and secondary alcohols, respectively.35 The target alcohols are intermediates for subsequent amine formation in CNS drugs. In the system developed here, for which one example is shown in Figure 8.6, the ene reductase (Old Yellow Enzyme 2) from Saccharomyces cerevisiae was cloned into E. coli and used in tandem with horse liver alcohol dehydrogenase, giving the product in 89% isolated yield with 99% ee.
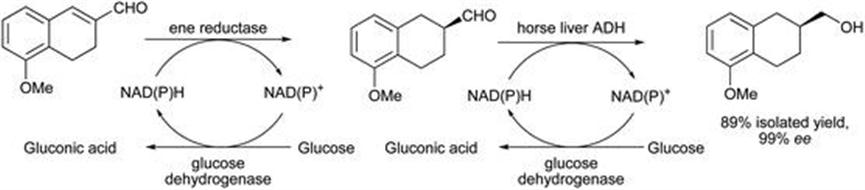
Figure 8.6 Conversion from enal into primary alcohol dual enzyme sequence.
The system encompasses an ene-reductase (from the Old Yellow Enzyme family) with an alcohol dehydrogenase (ADH), for the reduction of either α,β-unsaturated aldehyde or ketone to give the alcohol in both high yields and optical purity. Although ene-reductases significantly improve chemoselectivity and conversions, optically pure α-substituted aldehydes can spontaneously racemise under typical biotransformation conditions even at neutral pH (Figure 8.7).36 Two factors have been shown to minimise the loss of optical purity – using a biphasic system37,38 or applying the in situ substrate feeding product removal (SFPR) technology,2,39–41 ensuring that the unstable saturated aldehyde is immediately reduced to the more stable alcohol.
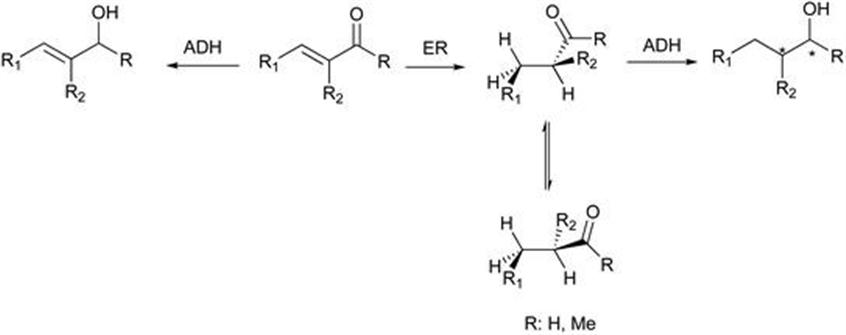
Figure 8.7 Typical chemical path of α,β-unsaturated aldehyde/ketone reductions with isolated enzymes or microorganisms.
Coupled use of ene reductases and ω-transaminases have been shown to result in diastereomerically enriched (R)- and (S)-amine derivatives in one-pot sequential and cascade processes (Figure 8.8) as demonstrated on a β-substituted cyclic enone.42 The one-pot synthesis required no modifications to reaction conditions, and a high chemoselectivity of the ω-TAs was shown in the cascade reaction. Ene reductases from the Old Yellow Enzyme family coupled with commercially available ω-TAs gave >99% conversion and de for a variety of α- or β-substituted unsaturated ketones as substrates.
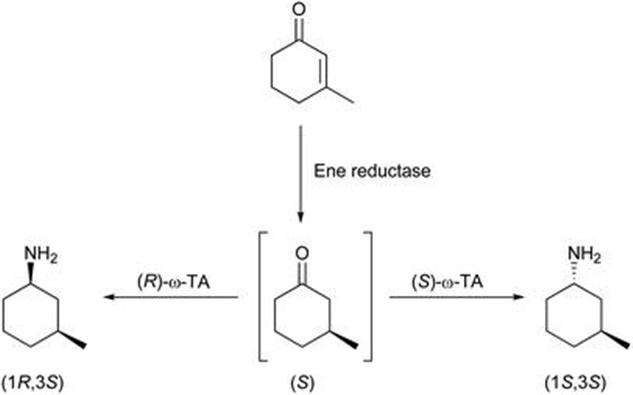
Figure 8.8 Examples of ER/ω-TA cascade reactions.
A further example from Hauer et al.43 reports the enzymatic reduction of allylic alcohols, which are normally not directly reduced by enzymes such as Old Yellow Enzymes as they do not contain an electron withdrawing group to activate the C=C bond. It was found that the ene reductase nicotinamide-dependent cyclohex-2-en-1-one reductase (NCR) did not catalyse the reaction but the morphinone reductase (MR) from Pseudomonas putida M10 and OYE1 from Saccharomyces pastorianus gave good activity towards allylic substrates. The grafting of loop A and loop B regions from OYE1 and MR into the inactive NCR scaffold resulted in active variants for the cascade reduction (Figure 8.9).
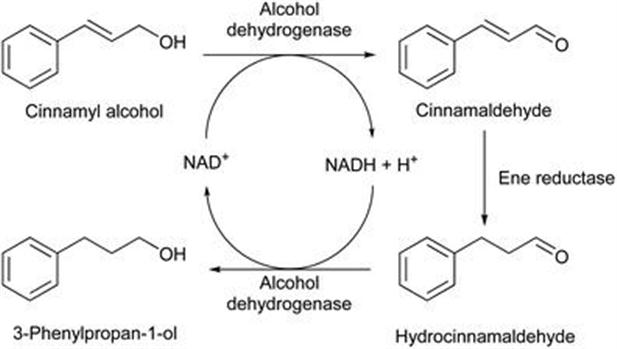
Figure 8.9 Bienzymatic three-step cascade reaction for the reduction of cinnamyl alcohol through the coupling of an alcohol dehydrogenase (ADH) with ene reductase wild-type enzymes and loop-grafted variants.
Moran et al.44 reported that several α-acetoxymethyl enones participated in bioreduction cascades. The substrate is reduced by an OYE with the loss of the acetoxy group, forming a new enone that is subsequently reduced by an OYE to form an enantiomerically enriched ketone (Figure 8.10). The acetoxy group is an adequate leaving group for the enones investigated containing electron-rich aromatic rings, thus avoiding the hydrolysis that occurs with the α-halomethyl enones. The main biocatalytic cascade pathway for α-acetoxymethyl enones with EREDs is most likely an allylic substitution followed by a hydrogenation.
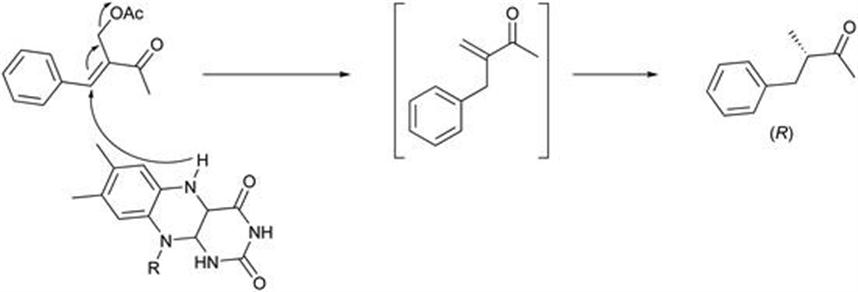
Figure 8.10 SN2′ type mechanism proposed for ERED catalysed reaction of α-acetoxymethyl enones.
More recently, Zhao et al.45 report a one-pot sequential chemoenzymatic system for the formation of 2-aryl-succinate derivatives. The sequence consisted of Rh-catalysed diazo-coupling, giving more than 9 : 1 selectivity for heterocoupling of the two diazo-esters, and a reduction mediated by an ene reductase with up to 99% ee (Figure 8.11). There was a preferential generation of the (E)-alkene from the diazo-coupling reaction giving high yield and enantioselectivity. The ene-reductase selectively reduced the (E)-alkene in a mixture of (E) and (Z) isomers. The combination of organometallic and enzymatic catalysis allows unusual transformations without the need to purify and isolate intermediates.
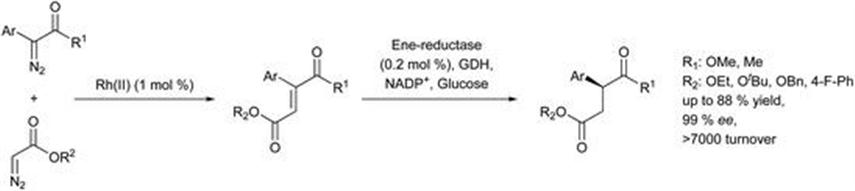
Figure 8.11 One-pot sequential chemoenzymatic system.
To encourage the uptake of this type of approach by industry more literature examples are urgently needed, and it is important to emphasise the need for multidisciplinary collaboration between practitioners of synthetic biology to produce and characterise new enzymes and chemists to apply these in enzyme cascades, with shared understanding of specific challenges such as cofactor and equilibrium demands. The academic sector has a key role to play in this process of enzyme discovery, development and demonstration, not just for ene reductases but many other enzyme types also.
8.4.2 Ene Reductases and Solvents
The use of organic solvents is an integral feature of biocatalysis, where they serve a number of purposes. These include solubilisation of substrates, in situ removal of products and modulation of rate or selectivity. Process development of biotransformations will frequently involve screening for optimisation of an appropriate co-solvent. In some cases the solvent may even serve as the substrate. Ene reductases have been shown to demonstrate good solvent tolerance.38 The ene reductases KYE1 from Kluyveromyces lactis and YersER from Yersinia bercovieri (both derived from the Old Yellow Enzyme family) were shown to be active against a broad range of substrates including cyclic enones, cyclic enol-ethers, alkene esters/diesters and nitrostyrenes. In this work, glucose dehydrogenase/glucose was used as the reducing source and recycling system for nicotinamide cofactor. When the enzymes were tested with a range of co-solvents the rate and selectivity was maintained surprisingly well, with little drop in performance in 20% v/v ethylene glycol, DMSO, hexane and toluene, representing both water miscible and immiscible solvents. In biphasic systems with toluene or hexane, the reaction was not impaired even at 70% v/v solvent.
(2R,5R)-Dihydrocarvone is a key intermediate in the production of natural products, antimalarial drugs and valuable chiral building blocks.46 However, its synthesis by whole-cell biotransformation often results in by-products (Figure 8.12). It has been reported that the ene-reductase NostocER1 from the cyanobacterium Nostoc sp. PCC 7120 and a NADP+ accepting mutant of the formate dehydrogenase (FDH) from Mycobacterium vaccae can be applied to the whole-cell batch bioreduction of (R)-carvone.47 In aqueous medium, the biotransformation resulted in a low conversion of 27.2% and 81.7% de of (2R,5R)-dihydrocarvone. The introduction of a second phase was found to have an impact on the biotransformation. Three key observations were made:
- The second phase served as a substrate reservoir for the poorly water soluble (R)-carvone.48
- Ionic liquids or adsorbent resins were found to improve substrate conversion at concentrations ≥50 mM. Carvones are known for their antimicrobial activity and toxic effects on the biocatalyst are reduced by lowering the substrate concentration present in aqueous phase.
- The stereoselectivity improves due to efficient product extraction from the aqueous phase and the concomitant protection from isomerisation by the E. coli cells.
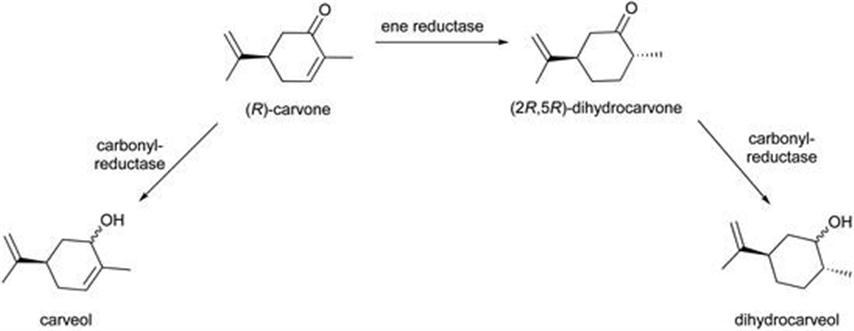
Figure 8.12 Asymmetric reduction of (R)-carvone to (2R,5R)-dihydrocarvone.
With the use of a biphasic system the conversion increased to >85% and high stereoselectivities were observed (96.0–99.2% de). Notably the undesired formation of carveols or dihydrocarveols as side products was not observed. The batch biotransformation was demonstrated at litre scale and shows potential for production of (2R,5R)-dihydrocarvone at larger scales.
Thus, the use of co-solvents during ene reductase process development should routinely be considered where appropriate, if they can bring a benefit to the reaction.
8.4.3 Challenges of Co-factor Recycle
The catalysis of ene reductases utilises nicotinamide cofactors (NADH, NADPH) in a manner similar to carbonyl reductases. Stoichiometric use of these natural coenzymes is not viable economically, and their instability can hinder catalytic processes that employ coenzyme recycling. Whilst this presents additional challenges for operation and cost, these challenges should in no way be viewed as insurmountable by pharmaceutical chemists. This reasoning is derived from the extensive success of carbonyl reductases now being realised for ketone into alcohol biotransformations in pharmaceutical intermediate manufacture, where cofactor requirements are routinely addressed. Whilst ene reductases as an enzyme class can utilise both NADH and NADPH as reducing hydride equivalents, they tend to have a preference for NADPH over NADH.
Various cofactor recycling systems may be used, utilising methods and enzyme sources developed for carbonyl reductases. The most commonly used approaches are in Figure 8.13.
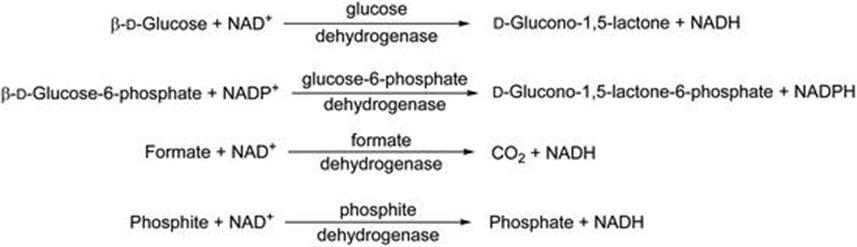
Figure 8.13 Commonly used cofactor recycling systems.
Of these approaches, the first using glucose/glucose dehydrogenase is the most frequently used. In carbonyl reductase biocatalysis cofactor recycle is often achieved using isopropanol as the hydrogen donor for cofactor recycle. Furthermore, the same enzyme may be used both for ketone reduction and isopropanol oxidation, with equilibrium driven by using an excess of the alcohol solvent. The same methodology, where a solvent stable alcohol dehydrogenase is used to oxidise isopropanol has been established for ene reductase systems.49 In this system, the oxidation of the sacrificial 2-propanol to acetone by the solvent-stable alcohol dehydrogenase (ADHA) from Rhodococcus ruber has been coupled to the bioreduction of activated alkenes using ene reductases as shown in Figure 8.14.
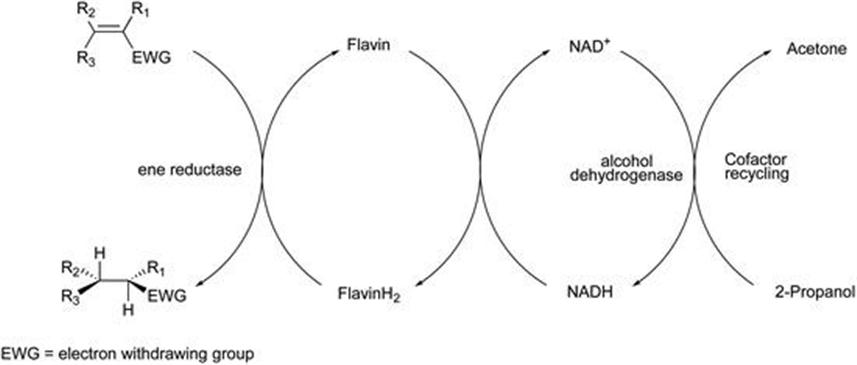
Figure 8.14 Co-factor recycling with isopropanol.
A various typical ene reductase substrates were reduced by this method, and performance was generally similar to other recycling methods based on glucose/glucose dehydrogenase or formate/formate dehydrogenase.
The key message is that for a given ene reductase biotransformation cofactor regeneration may be achieved by different enzymes and substrates. Crucially though, the literature reports that different systems may give different results in terms of yield and selectivity for a specific reaction, and choice of the cofactor recycling system should be an integral part of biotransformation development for large scale application.
8.4.4 Avoiding the Use of Nicotinamide Co-factors
In some cases it may be desirable to avoid the requirement for regeneration of the nicotinamide cofactor by a second enzyme-catalysed redox cycle. To address this desire a coupled substrate approach was developed.50,51 This demonstrates a simpler concept whereby a low cost substrate such as a 2-enone or 1,4-dione serves as the hydrogen donor for the direct recycling of the flavin cofactor. The system uses only a single enzyme and is independent of nicotinamide cofactor. During this recycle process the co-substrate is desaturated, and the reaction is driven thermodynamically by the rapid aromatisation of the desaturated product. This is illustrated in Figure 8.15, where cyclohexenone is used as the co-substrate for reduction of 4-ketoisophorone. A wide range of enones were demonstrated as suitable co-substrates for the reduction of 4-ketoisophorone to form (R)-levodione. This appears to be a promising approach, and from an industrial perspective has an appealing degree of simplicity over dual enzyme systems requiring nicotinamide cofactors. It is anticipated that more examples based on this approach will emerge in due course.
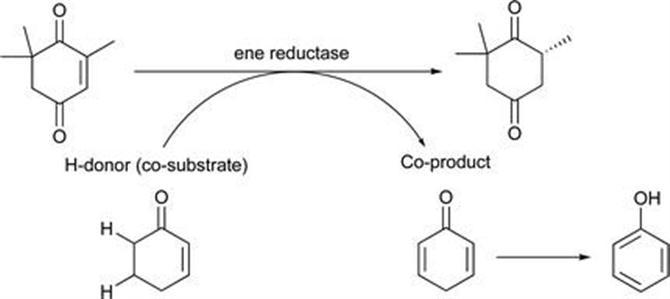
Figure 8.15 Bypassing cofactor requirement by use of a co-substrate.
Industrial interest in this approach is evident through a patent issued to BASF.52 This patent is based on the bioreduction of various enones by the YqjM gene product of Bacillus subtilis and the FCC248 gene product (an estrogen binding protein).
An alternative approach to address the cofactor issue is by the use of synthetic mimics of NAD(P)H.53 Several synthesised analogues are shown in Figure 8.16.
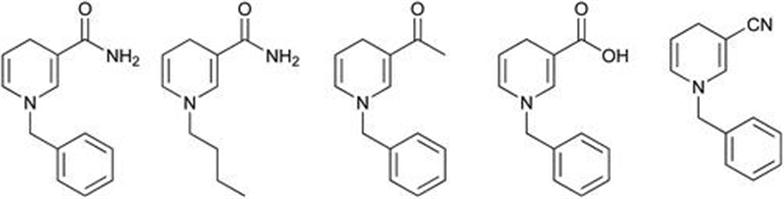
Figure 8.16 Synthetic mimics of nicotinamide cofactor.
These were tested against a range of ene reductases for the reduction of ketoisophorone to levodione. Good activity was found, and in some cases performance exceeded that of the natural cofactor. Recycling of the cofactor mimics was also demonstrated using formate as the reductant and a rhodium-based catalyst. It is claimed that these mimics are inexpensive to synthesise and have greater stability than their natural counterparts, making them an intriguing potential alternative to use of the natural cofactors.
Until more recently synthetic mimics were not reported to significantly enhance enzyme activity; however, kinetic investigations with the ene-reductase Old Yellow Enzyme found the flavin-reducing step to be slower than the substrate reduction. Hauer et al.54 reasoned that varying the electrochemical potential of the utilized cofactor could improve the rate of flavin-reduction. The introduction of aromatic residues could serve as electron donating residues when attached to the nitrogen atom thus altering electrochemical properties (Figure 8.17). When compared with NADH, cyclic voltammetry identified that the lowered oxidation potential of HPNAH represented a higher ability for hydride donation. The enzyme 2-cyclohexen-1-one reductase from Zymomonas mobilis (NCR)55 was a highly active cofactor enzyme pair with HPNAH, with a sixfold higher vmax relative to NADH most likely due to the ability of HPNAH to reduce the flavin faster.
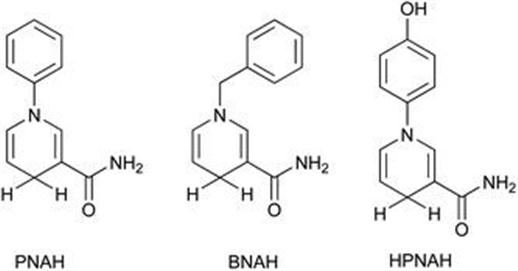
Figure 8.17 Synthetic cofactors, PNAH (1-phenyl-1,4-dihydronicotinamide), BNAH (1-benzyl-1,4-dihydronicotinamide) and HPNAH (1-(4-hydroxyphenyl)1,4-dihydronicotinamide).
There are further reports of biomimetics (1–5) that can outperform natural coenzymes in biotransformations (Figure 8.18).53 The synthetic coenzymes are stable relative to biological equivalents, inexpensive to manufacture and have been proven to work with a wide range of ene-reductases. The biomimetics can be used in only catalytic amounts at the expense of formate.
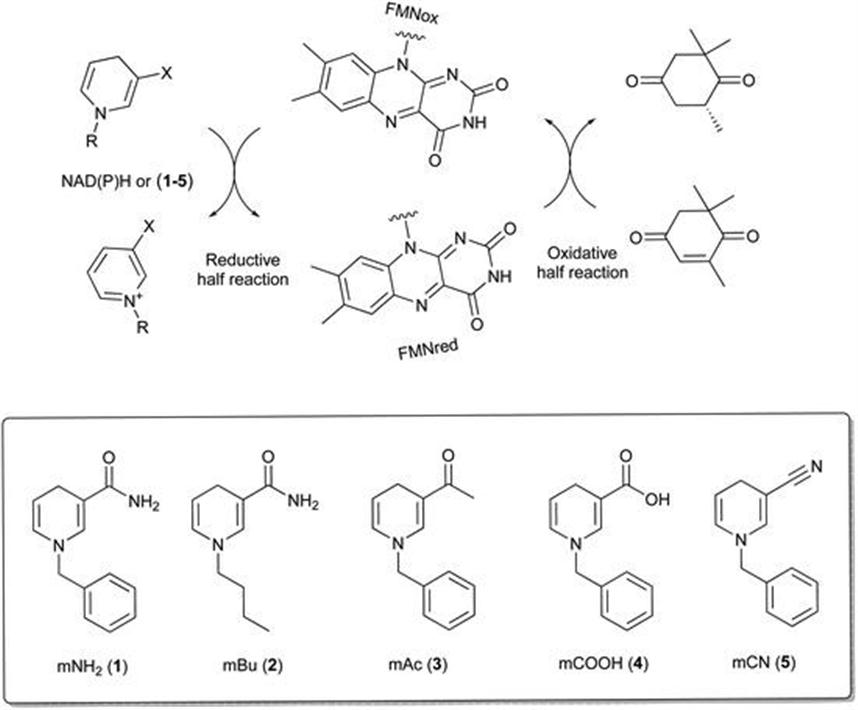
Figure 8.18 Synthetic nicotinamide biomimetic mNADHs (1–5) and the catalytic cycle of ER-catalysed reactions.
Scrutton et al.56 further demonstrated an approach using photosensitive transition metal complexes of Ru(II) or Ir(II) as electron donors for OYE-catalysed α,β-unsaturated alkene reduction. The light driven biocatalytic systems are a cheaper alternative to costly redox coenzymes and potentially avoid the need for enzyme-based cofactor regeneration systems. As an alternative to NADPH as the hydride donor, the yields and enantioselectivity for C-terminally histidine-tagged enzymes PETNR-His8 and TOYE-His6 were comparable for a broad range of substrates. With cyclohexen-2-one there were high levels of product accumulation with the main electron transfer pathway via photoexcitation of the photosensitiser. This is followed by successive reductive and oxidative steps between TEA, methyl viologen and enzyme bound flavin (FMN) to the alkene substrate (Figure 8.19).
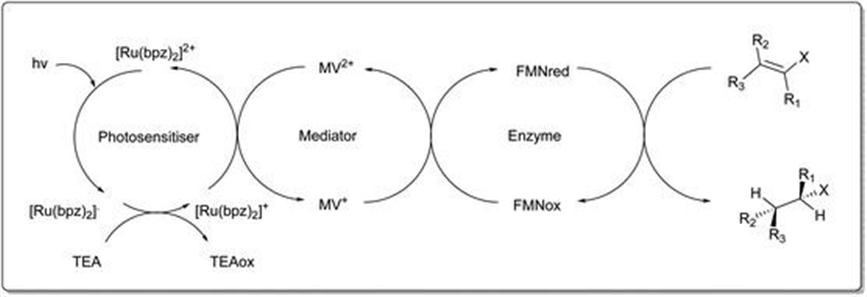
Figure 8.19 Electron-transfer processes that contribute to enzyme reduction in light-driven biocatalytic cycles of PETNR and TOYE.
Finally, artificial electron donors, such as reduced methyl viologen, can be used as an alternative to NAD(P)H in ene reductase biotransformations. However, this donor, perhaps better known as the herbicide paraquat, is highly toxic and therefore not a desirable reagent for utilisation in biocatalytic products such as foods and pharmaceuticals.57
Since the cost contribution of enzymes for NAD(P)+ reduction and recycle can be demanding from an industrial perspective it is essential that cofactor recycle or methods that avoid cofactor altogether are evaluated carefully during the process of scale up and development. It is apparent that there are numerous approaches that can be evaluated, and for industrial ene reductase biocatalysis this is a crucial factor for consideration in the development of a viable process.
8.4.5 Impact of Synthetic Biology
The recent impact of synthetic biology on multiple aspects of biocatalysis is striking, and is manifested by greatly decreasing the cost, time and risk associated with cloning enzymes. Furthermore, the ability to utilise published data relating to enzyme amino acid sequences and three-dimensional structure to semi-rationally alter enzyme performance is unprecedented. These techniques have filtered through to ene reductases, with good examples in the literature where enzyme performance has been improved and new enzymes have been discovered.
Gene shuffling, where the gene sequences of a small number of similar enzymes are cut into fragments then recombined together to give new chimeric ‘shuffled’ proteins is a methodology practised by companies such as Codexis Inc. The technique is one method of several for altering the properties of enzymes. Codexis demonstrated this approach by creating new ene reductases from three Old Yellow Enzymes derived from Saccharomyces cerevisiae and Saccharomyces pastorianus.58 Some of the mutants showed dramatic improvements in the level of activity against tested substrates. Two examples are shown in Figure 8.20. In the reduction of (Z)-ethyl 2-cyano-3-phenylbut-2-enoate several mutants were found where the activity was improved more than 20-fold relative to the parent enzyme. With 8a-methyl-3,4,8,8a-tetrahydronaphthalene-1,6(2H,7H)-dione several enzymes demonstrated over ten-fold increase in activity relative to the parent enzyme.
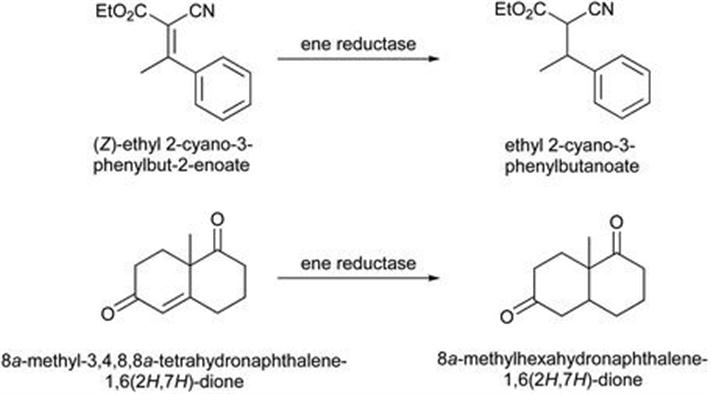
Figure 8.20 Substrates for engineered ene reductases.
These results show that the activity levels of ene reductases can be altered substantially by synthetic biology and the application of routine random mutagenesis methods. Recently, Hauer and coworkers59 achieved a deeper understanding of the >99% (S)-selective reduction of both isomers of citral, where the reaction is catalysed by NADH-dependent cyclohexenone ene reductase (NCR) from Zymomonas mobilis, using active-site mutational studies and docking simulation. The selectivity demonstrated, by the structurally similar (E/Z)-isomers, was shown to be dependent on the introduced mutations. It was possible to invert (E)-citral reduction enantioselectivity from >99% (S) to an ee of 46% (R) with the introduction of mutation W66A. For (Z)-citral the ee remained as >88% for all single residue variants. The studies concluded that W66 offers a leverage position which can induce (R) selectivity in NCR-catalysed citral reduction with reversed citral binding modes (Figure 8.21).
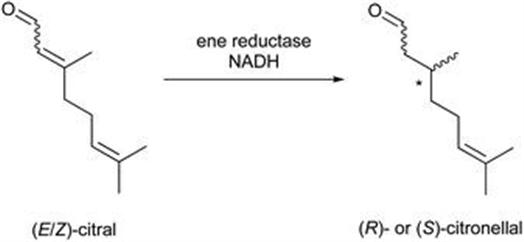
Figure 8.21 Hydrogenation of (E/Z)-citral to (R/S)-citronellal catalysed by ene reductase.
Iterative saturation mutagenesis is a more rational approach where, based on known protein structure, key amino acid residues are systematically changed to other amino acids in order to improve key enzyme properties such as selectivity or stability. This approach was exploited for an ene reductase YqjM, cloned from Bacillus subtilis.60 Using the known structure of this enzyme, 20 amino acid sites were subjected to alteration, and the resulting altered enzyme tested for reduction of various cyclopentenone and cyclohexenone substrates. For the biotransformation shown in Figure 8.22, the wild-type enzyme catalyses reduction to the (R)-isomer with 99% ee.
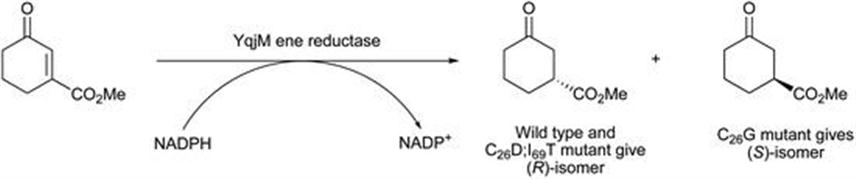
Figure 8.22 Reversal of stereoselectivity in ene reductases by mutation.
Two mutations of this enzyme (cysteine to aspartate at position 26 and isoleucine to threonine at position 69) improved the reaction rate by about one-third, whilst still catalysing a selective reduction of 99% ee. However, a different mutant, where just the cysteine at position 26 was changed to glycine, demonstrated completely reversed enantioselectivity, with the product now 98% ee (S)-isomer. This work demonstrated that both reaction rate and selectivity can be altered for ene reductases.
New methods for enzyme discovery continue to emerge, such as the recently reported catalophore method.61 In this approach the protein structure of known ene reductases was used as a starting point to define the three-dimensional spatial positioning of a few key active site enzyme residues. This ‘catalophore’ map was then used to interrogate a database of many different enzymes with known structure, and was able to identify new enzymes with ene reductase activity despite having very different amino acid sequences and enzyme topology to established enzymes, and with different stereopreference to known enzymes.
It is important that the academic sector continues to develop enzyme discovery methods, exploiting the full potential of synthetic biology and use these methods to characterise new ene reductase enzymes. Further data on structure, properties and biotransformation performance will allow industrial scientists to increase their ene reductase competencies.
8.4.6 Ene Reductases in Reverse: Oxidation
An interesting and potentially important advance in ene reductase technology was reported where the enzyme functions in the reverse direction.62 Instead of alkene reduction the authors demonstrated that a range of cyclic ketones could be desaturated by oxidation to the corresponding enones, with an example shown in Figure 8.23.
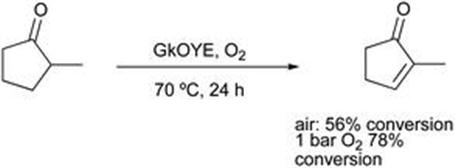
Figure 8.23 Oxidation by ene reductases.
Unlike the reductive process the reaction did not require a nicotinamide cofactor, and it was demonstrated that oxygen functioned as the oxidant. The enzyme cycle was completed by the aerobic reoxidation of the flavin FMNH2. The reactions were found to work best at higher temperature, around 70 °C. In the case of racemic ketones, the potential for chiral products was evident. The use of pure oxygen was shown to increase the extent of reaction.
The synthesis of substituted naphthols by an efficient enzymatic method from the corresponding tetralones has been reported.63 This utilises the ability of the ene-reductases of the Old Yellow Enzyme family to work in reverse. Screening of Almac’s selectAZyme panel of EREDs resulted in over 60% of the enzymes yielding 2-naphthol when 2-tetralone was used as the substrate. Moderate to excellent conversions (up to >99%) were reported for the selected EREDs for the production of a set of substituted naphthols (Figure 8.24). The robustness of the process was demonstrated with a 2.0 g scale reaction which gave a 91% isolated yield.
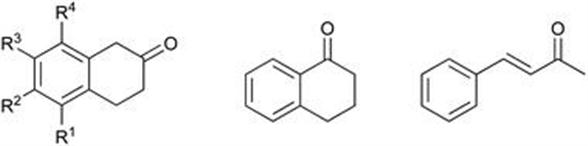
Figure 8.24 Range of tetralones tested for the production of naphthols using EREDs.
Whilst the application of ene reductases as oxidative catalysts is very much embryonic, there is good potential for reaction and enzyme engineering to advance this mode of application towards routine viability. The main challenges to be addressed include broadening the range of substrates amenable to oxidation, dealing with limited oxygen solubility at higher temperature and increasing enzyme stability, turnover and selectivity.
8.4.7 Thermophilic Ene-reductases
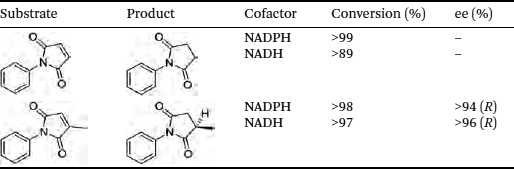
Ene-reductases that come from extremophiles are gaining importance due to their higher stability. The genome of the acidophilic iron-oxidising bacterium Ferrovum sp. JA12 was reported as the first thermophilic-like ene-reductase (FOYE-1).64 It is closely related to three mesophilic ene-reductases, namely DrOYE, RmOYE and OYERo2. FOYE-1 was found to be highly efficient in the transformation of different maleimides creating stereoselective succinimides. High conversions were achieved using FOYE-1 for the reduction of N-phenylmaleimide (>99% with NADPH) and also on N-phenyl-2-methylmaleimide (>98%, using NADPH, Table 8.1). The stability of the enzyme was studied between 20 and 70 °C with the optimum activity of FOYE-1 observed at 50 °C (160 U mg−1). At the upper end of the temperature range studied the activities were reduced (39–51 U mg−1).
Table 8.1 Asymmetric bioreduction of activated maleimides using FOYE-1
8.4.8 Alternative Screening Methods
Typically, for screening ene reductase enzymes, either GC or HPLC is used for analysis, which is advantageous as both stereoselectivity and conversion can be assessed. For primary screening, an alternative and potentially quicker colorimetric method reported by Monti et al.65 allows for the determination of conversion in a substrate independent way (FRED, fast and reliable ene reductase detection). To validate the FRED assay, the bioreduction of substrates by OYE1 were analysed by both FRED and by GC (Figure 8.25), with a comparable determination of conversion. There is a wide range of possible applications for this technology, not least for quick, reliable primary screening of libraries.
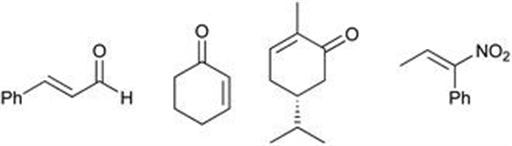
Figure 8.25 Substrate scope for validation of FRED assay.
Quertinmont and Lutz have explored the potential alternative methods to evaluate libraries of OYE variants.66 They developed a protocol – RAPPER (Rapid Parallel Protein EvaluatoR)67 – that allows for systematic protein engineering studies of OYE1. RAPPER enables fast and more efficient, semi-quantitative evaluation of enzyme variants as an initial screening protocol to discover tailored biocatalysts, but does have limitations such as small sample sizes.
References
1. D. J. Pollard and J. M. Woodley, Trends Biotechnol., 2007, 25, 66.
2. R. N. Patel, Biomolecules, 2013, 3, 741.
3. A. Wells and H. P. Meyer, ChemCatChem, 2014, 6, 918.
4. G. W. Huisman and S. J. Collier, Curr. Opin. Chem. Biol., 2013, 17, 284.
5. A. M. Bezborodov and N. A. Zagustina, Appl. Biochem. Microbiol., 2016, 52, 237.
6. M. S. Malik, E.-S. Park and J.-S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163.
7. T. S. Moody, S. Mix, G. Brown and D. Beecher, in Science of Synthesis, Biocatalysis in Organic Synthesis Vol. 2, ed. K. Faber, N. J. Turner and W. D. Fessner, Georg Thieme Verlag, Stuttgart, 2015, p. 421.
8. A. McMordie and T. S. Moody, Natural enzymes – key to 21st century process design, Manufacturing Chemist, 11th October 2013, available at https://www.manufacturingchemist.com/technical/article_page/Natural_enzymes_key_to_21st_century_process_design/92185.
9. A. S. Wells, J. W. Wong, P. C. Michels, D. A. Entwistle, K. Fandrick, G. L. Finch, A. Goswami, H. Lee, S. Mix and T. S. Moody, Org. Process Res. Dev., 2016, 20(3), 594.
10. H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892.
11. H. S. Toogood and N. S. Scrutton, Curr. Opin. Chem. Biol., 2014, 19, 107.
12. Y. Fu, K. Castiglione and D. Weuster-Botz, in Industrial Biocatalysis, Pan Stanford Series on Biocatalysis, Vol. 1, ed. P. Grunwald, Pan Stanford Publishing Pte. Ltd, Singapore, 2015, p. 631.
13. K. Durchschein, M. Hall and K. Faber, Green Chem., 2013, 15(7), 1764.
14. G. Oberdorfer, K. Gruber, K. Faber and M. Hall, Synlett, 2012, 23(13), 1857.
15. B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070.
16. B. Dominguez, U. Schnell, S. Bisagni and T. Kalthoff, Johnson Matthey Technol. Rev., 2016, 60(4), 243.
17. C. K. Winkler, G. Tasnadi, D. Clay, M. Hall and K. Faber, J. Biotechnol., 2012, 162, 381.
18. T. Ohshima, H. Tadaoka, K. Hori, N. Sayo and K. Mashima, Chem.–Eur. J., 2008, 14, 2060.
19. J. B. Tuttle, S. G. Ouellet and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 12662.
20. S. Ehira, H. Teramoto, M. Inui and H. Yukawa, Microbiology, 2010, 156, 1335.
21. J. Z. Cheng, C. M. Coyle, D. G. Panaccione and S. E. O’Connor, J. Am. Chem. Soc., 2010, 132, 1776.
22. H. Sobajima, M. Takeda, M. Sugimori, N. Kobashi, K. Kiribuchi, E. M. Cho, C. Akimoto, T. Yamaguchi, E. Minami, N. Shibuya, F. Schaller, E. W. Weiler, T. Yoshihara, H. Nishida, H. Nojiri, T. Omori, M. Nishiyama and H. Yamane, Planta, 2003, 216(4), 692.
23. C. K. Winkler, D. Clay, S. Davies, P. O’Neill, P. McDaid, S. Debarge, J. Steflik, M. Karmilowicz, J. W. Wong and K. Faber, J. Org. Chem., 2013, 78, 1525.
24. S. Debarge, P. McDaid, P. O’Neill, J. Frahill, J. W. Wong, D. Carr, A. Burrell, S. Davies, M. Karmilowicz and J. Steflik, Org. Process Res. Dev., 2014, 18, 109.
25. D. E. Q. Jimenez, I. M. Ferreira, W. G. Birolli, L. P. Fonseca and A. L. M. Porto, Tetrahedron, 2016, 72, 7317.
26. J. Alonso-Gutierrez, R. Chan, T. S. Batth, P. D. Adams, J. D. Keasling, C. J. Petzold and T. S. Lee, Metab. Eng., 2013, 19, 33.
27. J. L. Bicas, A. P. Dionisio and G. M. Pastore, Chem. Rev., 2009, 109, 4518.
28. J. Bohlmann, C. L. Steele and R. Croteau, J. Biol. Chem., 1997, 272, 21784.
29. H. S. Toogood, A. N. Cheallaigh, S. Tait, D. J. Mansell, A. Jervis, A. Lygidakis, L. Humphreys, E. Takano, J. M. Gardiner and N. S. Scrutton, ACS Synth. Biol., 2015, 4, 1112.
30. A. Lygidakis, V. Karuppiah, R. Hoeven, A. Ni Cheallaigh, D. Leys, J. M. Gardiner, H. S. Toogood and N. S. Scrutton, Angew. Chem., Int. Ed., 2016, 55, 9596.
31. X.-Q. Pei, M.-Y. Xu and Z.-L. Wu, J. Mol. Catal. B: Enzym., 2016, 123, 91.
32. N. Oberleitner, C. Peters, J. Muschiol, M. Kadow, S. Saß, T. Bayer, P. Schaaf, N. Iqbal, F. Rudroff, M. D. Mihovilovic and U. T. Bornscheuer, ChemCatChem, 2013, 3524.
33. N. Oberleitner, C. Peters, F. Rudroff, U. T. Bornscheuer and M. D. Mihovilovic, J. Biotechnol., 2014, 192, 393.
34. X. Chen, X. Gao, Q. Wu and D. Zhu, Tetrahedron: Asymmetry, 2012, 23, 734.
35. E. Brenna, F. G. Gatti, L. Malpezzi, D. Monti, F. Parmeggiani and A. Sacchetti, J. Org. Chem., 2013, 78, 4811.
36. M. Bechtold, E. Brenna, C. Femmer, F. G. Gatti, S. Panke, F. Parmeggiani and A. Sacchetti, Org. Process Res. Dev., 2012, 16, 269.
37. C. Stueckler, N. J. Mueller, C. K. Winkler, S. M. Glueck, K. Gruber, G. Steinkellner and K. Faber, Dalton Trans., 2010, 39, 8472.
38. Y. Yanto, C. K. Winkler, S. Lohr, M. Hall, K. Faber and A. S. Bommarius, Org. Lett., 2011, 13, 2540.
39. J. T. Vicenzi, M. J. Zmijewski, M. R. Reinhard, B. E. Landen, W. L. Muth and P. G. Marler, Enzyme Microb. Technol., 1997, 20, 494.
40. B. A. Anderson, M. M. Hansen, A. R. Harkness, C. L. Henry, J. T. Vicenzi and M. J. Zmijewski, J. Am. Chem. Soc., 1995, 117, 12358.
41. P. D’Arrigo, C. Fuganti, G. Pedrocchi Fantoni and S. Servi, Tetrahedron, 1998, 54, 15017.
42. D. Monti, M. C. Forchin, M. Crotti, F. Parmeggiani, F. G. Gatti, E. Brenna and S. Riva, ChemCatChem, 2015, 7, 3106.
43. S. Reich, B. M. Nestl and B. Hauer, ChemBioChem, 2016, 17, 561.
44. B. R. S. Paula, D. Zampieri, J. A. R. Rodrigues and P. J. S. Moran, Adv. Synth. Catal., 2016, 358, 3555.
45. Y. Wang, M. J. Bartlett, C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catal., 2017, 7, 2548.
46. C. K. Winkler, G. Tasnádi, D. Clay, M. Hall and K. Faber, J. Biotechnol., 2012, 162, 381.
47. K. Castiglione, Y. Fu, I. Polte, S. Leupold, A. Meo and D. Weuster-Boltz, Biochem. Eng. J., 2017, 117, 102.
48. I. Fichan, C. Larroche and J. B. Gros, J. Chem. Eng. Data, 1999, 44, 56.
49. K. Tauber, M. Hall, W. Kroutil, W. M. F. Fabian, K. Faber and S. M. Glueck, Biotechnol. Bioeng., 2011, 108, 1462.
50. C. Stueckler, T. C. Reiter, N. Baudendistel and K. Faber, Tetrahedron, 2010, 66, 663.
51. C. K. Winkler, D. Clay, M. Entner, M. Plank and K. Faber, Chem.–Eur. J., 2014, 20, 1403.
52. S. Maurer, B. Hauer, M. Bonnekessel, K. Faber and C. Stückler, US8709767B2, 2014.
53. T. Knaus, C. E. Paul, C. W. Levy, S. de Vries, F. G. Mutti, F. Hollmann and N. S. Scrutton, J. Am. Chem. Soc., 2016, 138, 1033.
54. S. A. Löw, I. M. Löw, M. J. Weissenborn and B. Hauer, ChemCatChem, 2016, 8, 911.
55. A. Müller, B. Hauer and B. Rosche, Biotechnol. Bioeng., 2007, 98, 22.
56. M. K. Peers, H. S. Toogood, D. J. Heyes, D. Mansell, B. J. Coe and N. S. Scrutton, Catal. Sci. Technol., 2016, 6, 169.
57. H. Simon, J. Bader, H. Gunther, S. Neumann and J. Thanos, Angew. Chem., Int. Ed. Engl., 1985, 24, 539.
58. C. Savile, V. Mitchell, X. Zhang and G. Huisman, US8329438B2, 2012.
59. N. Kress, J. Rapp and B. Hauer, ChemBioChem, 2017, 18, 717.
60. D. J. Bougioukou, S. Kille, A. Taglieber and M. T. Reetz, Adv. Synth. Catal., 2009, 351, 3287.
61. G. Steinkellner, C. C. Gruber, T. Pavkov-Kellerm, A. Binter, K. Steiner, C. Winkler, A. Lyskowski, O. Schwamberger, M. Oberer, H. Schwab, K. Faber, P. Macheroux and K. Gruber, Nat. Commun., 2014, 5(4150), 1.
62. M. Schittmayer, A. Glieder, M. K. Uhl, A. Winkler, S. Zach, J. H. Schrittwieser, W. Kroutil, P. Macheroux, K. Gruber, S. Kambourakis, J. D. Rozzell and M. Winkler, Adv. Synth. Catal., 2011, 353, 268.
63. P. P. Kelly, D. Lipscomb, D. J. Quinn, K. Lemon, J. Caswell, J. Spratt, B. Kosjek, M. Truppo and T. S. Moody, Adv. Synth. Catal., 2016, 358, 731.
64. A. Scholtissek, S. R. Ullrich, M. Mϋhling, M. Schlömann, C. E. Paul and D. Tischler, Appl. Microbiol. Biotechnol., 2017, 101, 609.
65. M. C. Forchin, M. Crotti, F. G. Gatti, F. Parmeggiani, E. Brenna and D. Monti, ChemBioChem, 2015, 16, 1571.
66. L. T. Quertinmont and S. Lutz, Tetrahedron, 2016, 72, 7282.
67. L. T. Quertinmont, R. Orru and S. Lutz, Chem. Commun., 2015, 51, 122.
68. D. Mangan, I. Miskelly and T. S. Moody, Adv. Synth. Catal., 2012, 354, 2185.
69. K. Tauber, M. Hall, W. Kroutil, W. M. F. Fabian, K. Faber and S. M. Glueck, Biotechnol. Bioeng., 2011, 108, 1462.
70. T. Stilger, M. Bönits, M. V. Filho and A. Liese, Chem. Ing. Tech., 2002, 74, 1035.
71. K. Goldberg, K. Edegger, W. Kroutil and A. Liese, Biotechnol. Bioeng., 2006, 95, 192.
72. T. Daussmann, T. C. Rosen and P. Dϋnkelmann, Eng. Life Sci., 2006, 6, 125.
73. C. K. Winkler, G. Tasnádia, D. Clay, M. Hall and K. Faber, J. Biotechnol., 2012, 162, 381.