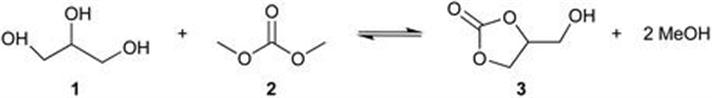
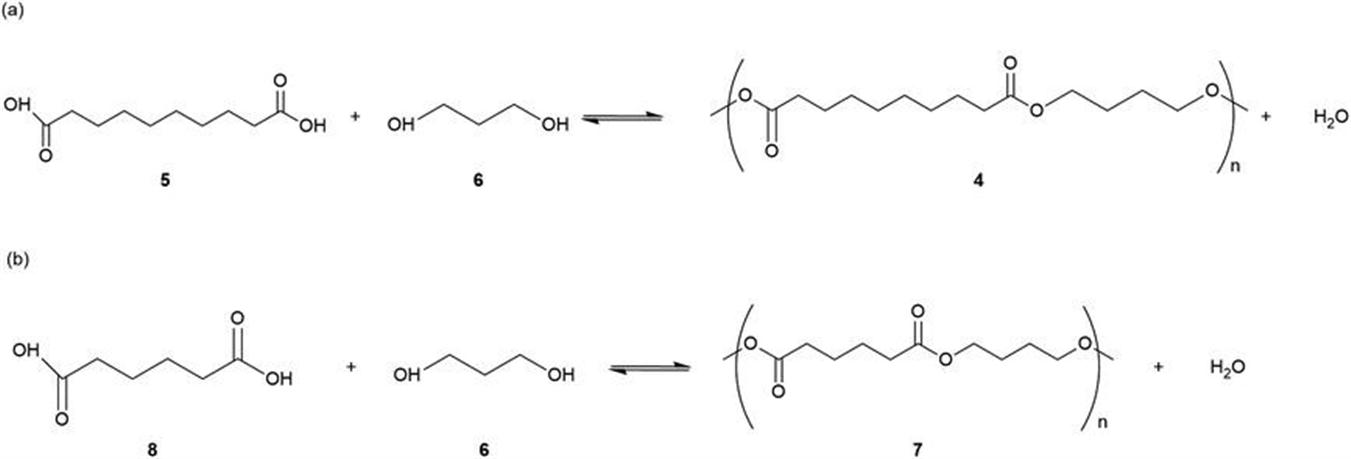
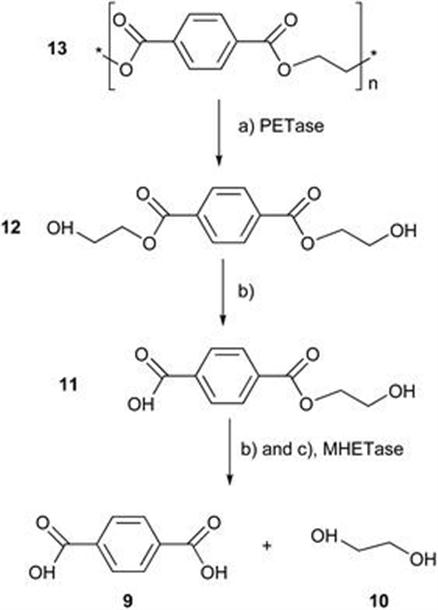
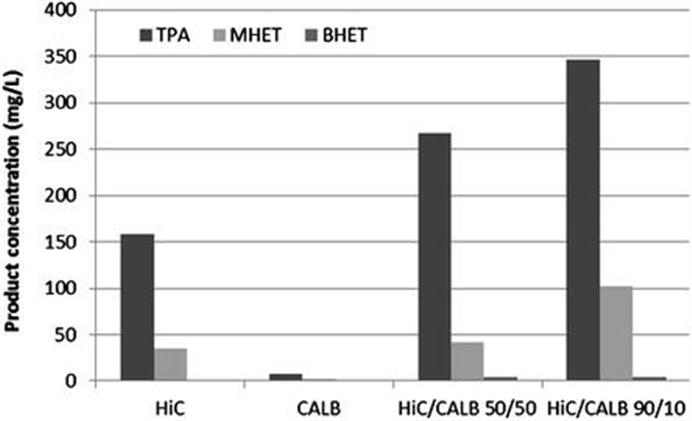
10.2 Hydrolysis of Lignocellulosic and Starchy Biomass
Sugarcane bagasse is one of the main agroindustry lignocellulosic biomasses in the world. In Brazil alone, its generation at mills has been over 100 million tons since 2000.3 This biomass can be used for either the production of tailor-made enzyme cocktails or as substrate for enzymatic hydrolysis to generate fermentable sugars. Agroindustrial lignocellulosic biomass is mainly constituted of three fractions: cellulose (15–55 wt%), hemicellulose (25–50 wt%) and lignin (10–40 wt%).4 In most cases, hemicellulose is removed from the solid fraction and often depolymerized during physico-chemical pretreatments,5 resulting in a solid that consists mostly of cellulose and lignin, which is named cellulignin by some authors.6–8 The deconstruction of cellulose present in cellulignin involves primarily the action of three groups of enzymes: cellobiohydrolases (EC 3.2.1.91), endoglucanases (EC 3.2.1.4) and β-glucosidases (EC 3.2.1.21) (Figure 10.1(a)), with specificities for different parts of the polysaccharide.3 These enzymes act synergistically for the release of the final fermentable sugar, glucose. Figure 10.1(b) represents a type of synergy between cellobiohydrolases and β-glucosidases with sequential release of cellobiose (glucose β-1,4-linked disaccharide) and then glucose, whereas Figure 10.1(c) shows synergy between endoglucanases and β-glucosidases, indicating that the last enzymes can also catalyze hydrolysis of oligossacharides.3,9
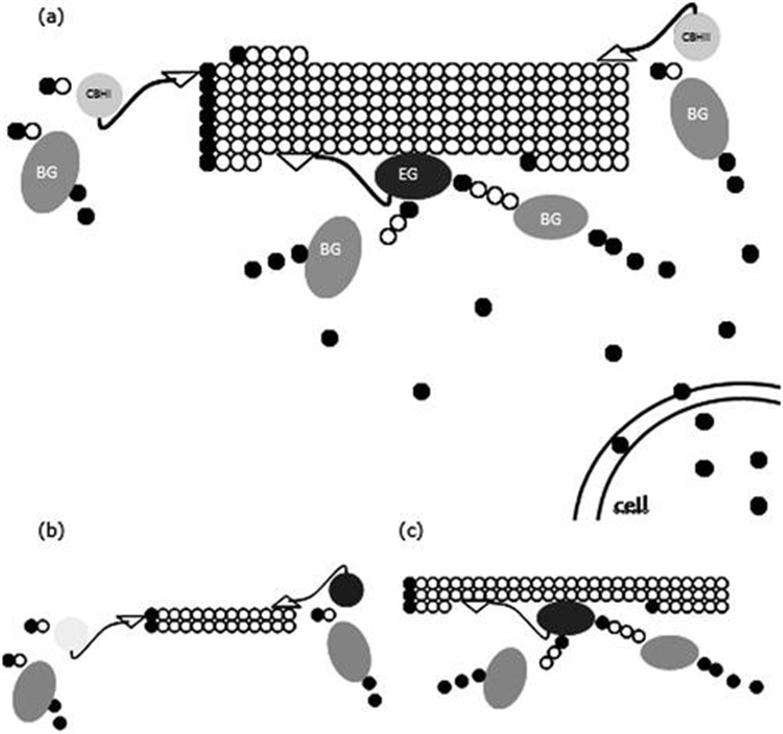
Figure 10.1 Representation of cellulases synergistic action. (a) Global synergy; (b) CBH/BG synergy, and (c) EG/BG synergy. EG: endoglucanase; BG: β-glucosidase; CBHI: cellobiohydrolase I; CBHII: cellobiohydrolase II. Black and white circles correspond to reducing and non-reducing glucose units, respectively. Figure adapted with permission from ref. 3.
Cenpes’ research for deconstruction of sugarcane bagasse started in 2004, in partnership with the Federal University of Rio de Janeiro. This biomass was used for the production of cellulases by several fungi,10 with promising results with Penicillium funiculosum,11,12Trichoderma harzianum13,14 and Aspergillus niger,13 each one with a distinct profile of cellulolytic activities (Figure 10.2).
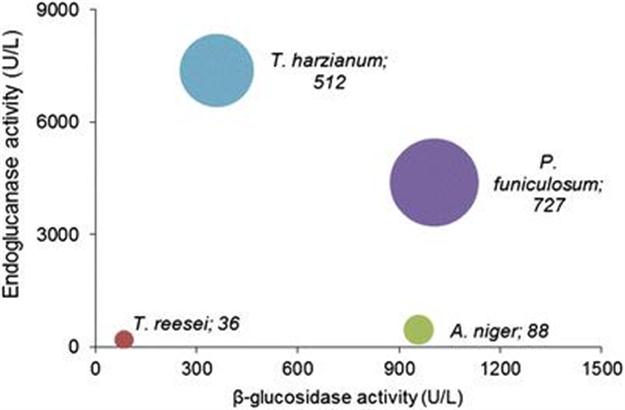
Figure 10.2 Activity of cellulolytic enzymes produced by filamentous fungi. Bubble size corresponds to cellobiohydrolase activity (numerical activity is given after the fungus name).
As a part of cooperation with PETROBRAS, a preliminary study investigated cellulase production by P. funiculosum using either untreated or pretreated (only acid, only alkali and acid plus alkali pretreatments) sugarcane bagasse as carbon source and inducer.11 Acid pretreatment increased substantially carboxymethyl cellulose (CMCase, endoglucanase) activity, whereas filter paper (FPase) and β-glucosidase activities were mainly improved by alkali pretreatment. The highest FPase, β-glucosidase and CMCase activities were 354, 1835 and 3588 U L−1, respectively. Later, Maeda et al.15 optimized the culture conditions of this same strain and the CMCase activity increased to 6917 U L−1, with significant improvements in productivity, achieved within 72 h of fermetnation.
More recently, studies on lignocellulose deconstruction carried out by PETROBRAS included accessory proteins, such as swollenins and lytic polysaccharide monooxygenases (LPMOs),9,16 as well as cellulosomes9,17 and addition of other non-hydrolytic proteins, such as bovine serum albumin combined with surfactants.18 Rocha16 expressed a T. harzianum swollenin in A. niger, achieving a protein concentration of 197 mg L−1, which is around 93 times higher than that found in the parent strain. The use of this T. harzianum protein increased by two-fold the hydrolysis efficiency of a commercial cellulase cocktail, on bench-scale preliminary tests.19
The enzymes produced by selected filamentous fungi using sugarcane bagasse as inducer were applied in cellulose hydrolysis. The cocktail from P. funiculosum boosted the activity of the commercial cellulase preparation Multifect©, and the combined enzymatic hydrolysis was increased by 50%.20 In another set of experiments, an experimental mixture design was applied for optimization of acid and alkali pretreated sugarcane bagasse hydrolysis. By using a proportion of 15% of T. harzianum IOC-3844, 50% of P. funiculosum ATCC11797 and 35% of A. niger ATCC 1004 cocktails, a global hydrolysis yield of 91% was achieved after 48 h.21
The one-pot conversion of sugarcane bagasse into ethanol in a consolidated bioprocessing strategy was preliminarily studied by Groposo et al.17 The strain Clostridium thermocellum ATCC27405 was able to produce a slightly higher concentration of the alcohol when cultivated in the presence of raw sugarcane bagasse (21.9 mM) than that observed when the corresponding acid pretreated biomass was used (19.6 mM), after 72 h of fermentation. This proof of concept opens new perspectives, since it indicates that biomass pretreatment is not always necessary for second-generation ethanol production.
Finally, PETROBRAS is also involved in the conversion of the sugars from enzymatic hydrolysis for the production of molecules of interest by industry, via fermentation. Initially, the interest was focused on ethanol. Many publications were reported as a result of this interest, including pretreatment strategies22–24 and fermentation.25 More recently, other bioproducts were also targeted, such as succinic acid,26 butanol,27 and propionic acid.28 At Cenpes facilities, biomass deconstruction studies have been carried out since 2004 at bench scale and since 2007 at a small pilot scale. PETROBRAS has a patent portfolio related to lignocellulose deconstruction. The granted patents are summarized in Table 10.1. In addition, there are other three patent applications under consideration (PI 0803782-5, PCT/BR2010/000216 and PCT/BR2010/000334).
Table 10.1 Granted patents related to lignocellulose deconstruction
| Company | Company |
|---|---|
| Process for producing ethanol from a hydrolysate of the hemicellulose fraction of sugarcane bagasse in a press reactor | PI0505299-8 (Brazil), US8642289 B2 (USA), EP2167672 B1 (EPO), ES2382001 (Spain), DK2167672 (Denmark) |
| Process for the fermentative production of ethanol from solid lignocellulosic material comprising a step of treating a solid lignocellulosic material with alkaline solution in order to remove the lignin | DK178525 (Denmark), JP5325793 (Japan), SE535902 C2 (Sweden), CA2660673 (Canada), GB2454119 (Great Britain), US8232082 B2 (USA) |
| Process for production of an enzymatic preparation for hydrolysis of cellulose from lignocellulosic residues and application thereof in the production of ethanol | EP2373787 B1 (EPO) |
For the production of biomolecules from lignocellulosic biomass, there are still some challenges that need to be addressed. From the technical point of view, improvements in pretreatment procedures for a reliable industrial operation must be considered. An efficient pretreatment also tends to influence both hydrolysis (by means of cellulose accessibility to enzymes) and fermentation efficiencies (regarding an inhibitor’s – acetic acid, furfural, hydroxymethylfurfural – concentration). The exploration of the synergy between cellulolytic enzymes, oxidative enzymes and accessory proteins, and the production of a cocktail at low cost, is equally important and may drastically impact process economics. Finally, the use of microbial strains (either recombinant or not) with the ability to produce efficiently the bioproducts from both C5 (xylose, arabinose) and C6 (glucose) sugars, under a stable metabolic activity, is of paramount importance to achieve feasible technical and economic targets. PETROBRAS has been also working on this topic, in collaboration with the University of Brasilia.29
It is worth mentioning that PETROBRAS also had a strong development regarding ethanol production from starchy biomass. This interest actually started during the 1970s and then returned early this century with two approaches. One was the application of commercial enzymes for sequential liquefaction and saccharification of castor bean cake (Ricinus communis L.) followed by alcoholic fermentation, in which a hydrolysis efficiency of 91.4% and ethanol titer of 34.5 g L−1 were obtained.30–32 The other was the production of combined tailor-made amylolytic/proteolytic cocktails from agricultural cakes and brans33–39 and their use for cold starch hydrolysis (hydrolysis at sub-gelatinization temperature) and ethanol production, in which an ethanol titer of 78 g L−1 was obtained.40–42