Chapter I.2.11
Applications of “Smart Polymers” as Biomaterials
Introduction
Stimulus-responsive, “intelligent” polymers are polymers that respond to small changes in physical or chemical conditions near a critical condition with sharp and relatively large phase or property changes. They are also known as “smart,” stimuli-responsive or “environmentally-sensitive” polymers. These polymers can take many forms; for example, they may be dissolved in aqueous solutions, adsorbed or grafted on aqueous–solid interfaces, or cross-linked in the form of hydrogels. They may also be combined chemically or physically with other molecules, especially a variety of bioactive molecules. A number of reviews over the past 20–25 years have highlighted applications of smart polymers in the biomedical field (Hoffman, 1987, 1995, 1997; Hoffman et al., 2000; Okano et al., 2000; Roy and Gupta, 2003; Roy et al., 2010).
Many different stimuli-responsive polymers have been investigated, and the various stimuli that researchers have used are listed in Table I.2.11.1. Typically, when the polymer’s smart response is stimulated, the resultant behavior will depend on the initial state of the polymer (Figure I.2.11.1). Three examples of this are described below:
• When a smart polymer is dissolved in an aqueous solution, it will exhibit a sudden onset of turbidity as it phase separates, and if its concentration is high enough it may convert from a viscous solution to a gel.
• When a smart polymer is chemically grafted to an aqueous–solid interface and is stimulated to phase separate, it will collapse, converting that interface from a hydrophilic to a hydrophobic interface. If the smart polymer is dissolved in solution and is stimulated to phase separate in the presence of a solid–aqueous interface, it may physically adsorb at the interface, especially when the surface composition has a balance of hydrophobic and polar groups similar to the phase-separating smart polymer.
• A smart polymer may be chemically cross-linked into a network. When it is swollen in an aqueous solution below its critical condition, it may be called a “smart hydrogel.” When the polymer network chains are stimulated to phase separate at their critical condition, the hydrogel will collapse and release much of its swelling solution as a burst.
TABLE I.2.11.1 Environmental Stimuli
Physical
Temperature
Ionic strength
Solvents
Radiation (UV, visible)
Electric fields
Mechanical stress
High pressure
Sonic radiation
Magnetic fields
Chemical
pH
Specific ions
Chemical agents
Biochemical
Enzyme substrates
Affinity ligands
Other biological agents
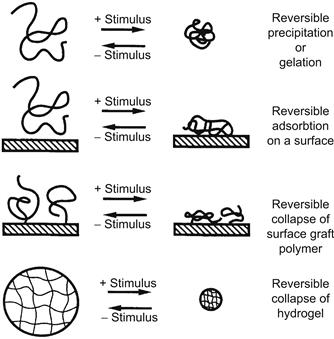
FIGURE I.2.11.1 Schematic illustration showing the different types of responses of “intelligent” polymer systems to environmental stimuli. Note that all systems are reversible when the stimulus is reversed.
(Hoffman et al., (2000) Journal of Biomedical Materials Research (JBMR): Reproduced with permission of JBMR, J. Wiley, Publisher.)
These phenomena all reverse when the stimulus is reversed. The phase separation of a water-solvated smart polymer chain is driven by the release of the hydrophobically bound water molecules on the polymer backbone, which creates a large gain in entropy in the system. Often the rate of reversion back to the hydrated state is slower than the collapse, because in the reverse process the hydrophobic groups of the polymer have to be rehydrated, and that process is thermodynamically opposed by the resultant decline in entropy of the system (the reswelling process is favored by the exothermic hydration and expansion of the polymer chains). The rate of collapse of smart polymer systems and its reversal can also depend on the dimensions of the smart polymer system. Rates will be more rapid for systems with smaller dimensions, e.g., nanoscale versus microscale dimensions.
The rate of collapse of smart polymer systems and its reversal can depend on the dimensions of the smart polymer system. Rates will be more rapid for systems with smaller dimensions, e.g., nanoscale versus microscale dimensions.
Smart polymers may be physically mixed with, complexed with, or chemically conjugated to biomolecules to yield a large and diverse family of polymer–biomolecule “biohybrid” systems that can respond to biological, as well as to physical and chemical, stimuli. Sometimes such biohybrids are called “doubly smart.” Biomolecules that may be combined with smart polymer systems include proteins and peptides, sugars and polysaccharides, single and double-stranded oligonucleotides, RNA and DNA, simple lipids and phospholipids, and a wide spectrum of recognition ligands and synthetic drug molecules. In addition, polyethylene glycol (PEG) may be conjugated to the smart polymer backbone to provide it with “stealth” properties (Figure I.2.11.2 schematically shows many different molecules, one or several together, which may be conjugated to a single smart polymer chain). The reader is referred to recent reviews of the synthesis and applications of smart polymer-protein conjugates (e.g., Hoffman, et al., 2000; Heredia and Maynard, 2007; Hoffman and Stayton, 2010; Grover and Maynard, 2010; Broyer et al., 2011).

FIGURE I.2.11.2 Schematic illustration showing the variety of natural or synthetic biomolecules that may be conjugated to a smart polymer. In some cases, only one molecule may be conjugated, such as a recognition protein, which may be linked to the protein at a reactive terminal group of the polymer, or it may be linked at a reactive pendant group along the polymer backbone. In other cases more than one molecule may be conjugated along the polymer backbone, such as a targeting ligand along with many drug molecules.
(Hoffman et al., (2000) Journal of Biomedical Materials Research (JBMR): Reproduced with permission of JBMR, J. Wiley, Publisher.)
Among the most studied of these smart polymer–biomolecule systems are conjugates of the smart polymer with drugs, enzymes, and antibodies (see below). Such smart bioconjugates may be utilized as free conjugates in solution, or they may be physically adsorbed or chemically immobilized at solid interfaces. A free enzyme, antibody or drug may also be physically or chemically entrapped within smart hydrogels or smart polymer phase-separated masses (phase-separated polymer–drug masses in vivo are known as “depot” drug delivery systems). There have been a number of successful applications in both medicine and biotechnology for such smart polymer–biomolecule systems and, as such, they represent an important and exciting extension of polymeric biomaterials beyond their well-known uses in implants, medical devices, and drug delivery (see Chapter II.5.16 on Drug Delivery Systems for more details).
Smart polymer–biomolecule conjugates in solution, on surfaces, and within hydrogels represent an important and exciting extension of polymeric biomaterials beyond their well-known uses in implants, medical devices, and drug delivery.
Smart Polymers in Solution
There are many polymers that exhibit thermally-induced precipitation out of aqueous solutions (Table I.2.11.2), and the polymer that has been the most extensively studied is poly(N-isopropyl acrylamide), or PNIPAAm. This polymer is soluble in water below 32°C, and it precipitates sharply as temperature is raised above 32°C (Heskins and Guillet, 1968; Schild, 1992). The precipitation temperature is called the lower critical solution temperature, or LCST. If the solution contains buffer and salts the LCST may be reduced several degrees. If NIPAAm monomer is copolymerized with more hydrophilic monomers, such as acrylamide, the LCST will increase and may even disappear. If NIPAAm monomer is copolymerized with more hydrophobic monomers, such as n-butylacrylamide, the LCST decreases (Figure I.2.11.3) (Priest et al., 1987). NIPAAm may also be copolymerized with pH-sensitive monomers, leading to random copolymers with both temperature- and pH-responsive behavior (Murthy, et al., 1999; Zareie et al., 2000; Murthy et al., 2003a,b; Yin, et al., 2006). Tirrell, and more recently, Stayton, Hoffman, and co-workers have studied the behavior of pH-sensitive alpha-alkylacrylic acid polymers in solution (Tirrell, 1987; Lackey et al., 1999; Murthy et al., 1999; Stayton et al., 2000; Murthy et al., 2003a,b; Yin et al., 2006; Stayton and Hoffman, 2008). As pH is lowered, these polymers become increasingly protonated and hydrophobic, and eventually phase separate; this transition can be sharp, resembling the phase transition at the LCST. If a polymer such as poly(ethylacrylic acid) (PEAA) or poly(propylacrylic acid) (PPAA) is in the vicinity of a lipid bilayer membrane as pH is lowered, the polymer will interact with the membrane and disrupt it. These polymers have been applied by Stayton and Hoffman and co-workers to stimulate endosomal escape of the drug (with or without its nanocarrier) to the cytosol. This action is a consequence of the drop in pH in the endosome, which causes PPAA to become hydrophobic and disrupt the endosomal membrane (e.g., Stayton and Hoffman, 2008; also see detailed discussion and referencing of this work in Chapter II.5.16 on Drug Delivery Systems). Bae and co-workers have also developed interesting temperature- and pH-sensitive polymers for endosomal release of drug formulations (e.g., Na et al., 2006; Kang and Bae, 2007).
TABLE I.2.11.2 Some Polymers and Surfactants that Exhibit Thermally-Induced Phase Separation in Aqueous Solutions
Polymers/Surfactants with Ether Groups
Poly(ethylene oxide) (PEO)
Poly(ethylene oxide/propylene oxide) random copolymers [poly(EO/PO)]
PEO–PPO–PEO triblock surfactants (Polyoxamers or Pluronics)
PLGA–PEO–PLGA triblock polymers
Alkyl–PEO block surfactants (Brij)
Poly(vinyl methyl ether)
Polymers with Alcohol Groups
Poly(hydropropyl acrylate)
Hydroxypropyl cellulose
Methylcellulose
Hydroxypropyl methylcellulose
Poly(vinyl alcohol) derivatives
Polymers with Substituted Amide Groups
Poly(N-substituted acrylamides)
Poly(N-acryloyl pyrrolidine)
Poly(N-acryloyl piperidine)
Poly(acryl-l-amino acid amides)
Others
Poly(methacrylic acid)
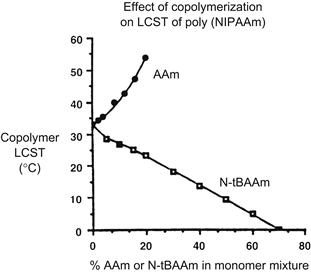
FIGURE I.2.11.3 Copolymerization of a thermally sensitive polymer, PNIPAAm, with a more hydrophilic comonomer, AAm, raises the LCST of the copolymer, whereas copolymerization with a more hydrophobic comonomer, N-tBAAm, lowers the LCST.
(Hoffman et al., (2000) Journal of Biomedical Materials Research (JBMR): Reproduced with permission of JBMR, J. Wiley, Publisher.)
NIPAAm has been copolymerized with pH-responsive macromonomers of acrylic acid (AAc), which yielded graft copolymers that independently exhibit two distinct stimuli responses, one for the backbone PNIPAAm and one for the side chain PAAc (Chen and Hoffman, 1995). Block copolymers of two such smart polymers also exhibit two distinct stimuli responses if the two individual blocks are each long enough.
A family of thermally gelling and biodegradable triblock copolymers has been developed by Sung Wan Kim and co-workers for injectable formulations that form degradable, drug depot masses at body conditions (Vernon et al., 2000; Lee et al., 2001; Jeong et al., 2002). The formulation is a medium viscosity, injectable solution at room temperature, and a phase-separated hydrogel mass at 37°C. These polymers are based on blocks of hydrophobic, degradable polyesters combined with blocks of PEO. The copolymers are triblocks with varying MW segments of PLGA and PEO. Typical compositions are PEO–PLGA–PEO and PLGA–PEO–PLGA. They are discussed further in Chapter II.5.16 on Drug Delivery Systems.
Doo Sung Lee, You Han Bae, and co-workers have developed similar biocompatible and degradable A–B–A triblock copolymers having both pH and temperature sensitivity; they are based on two blocks of poly(caprolactone-co-lactic acid) copolymers (PCLA), combined with a central PEG block. The pH sensitivity is derived from short blocks containing the sulfonamide group (OSM) that are attached at each end of the triblock. Typical formulae of the copolymers are PCLA–PEG–PCLA and, when modified with oligomers containing the sulfonamide group at each end, OSM–PCLA–PEG–PCLA–OSM (Shim et al., 2005, 2006).
Smart Polymer–Protein Bioconjugates in Solution
Smart polymers may be conjugated randomly to proteins by binding the reactive end of the polymer or reactive pendant groups along the polymer backbone to reactive sites on the protein. One may utilize functionalized free radical initiator or chain transfer agents to synthesize oligomers with one functional end group, which can then be derivatized to form a reactive group that can be conjugated to the protein. For example, NIPAAm has been copolymerized with reactive monomers such as N-hydroxysuccinimide acrylate (NHS acrylate) to yield a random copolymer with pendant NHS groups, which have then been conjugated to the protein. (These synthesis methods are described in Cole et al., 1987; Monji and Hoffman, 1987; Chen et al., 1990; Chen and Hoffman, 1990; Yang et al., 1990; Takei et al., 1993a; Chen and Hoffman, 1994; Ding et al., 1996, 1998.) Vinyl monomer groups may also be conjugated to proteins to provide “comonomer” vinyl groups for copolymerization with free monomers such as NIPAAm (Shoemaker et al., 1987).
Normally the lysine amino groups are the most reactive protein sites for random polymer conjugation to proteins, and N-hydroxysuccinimide (NHS) attachment chemistry is often utilized. Other possible sites include –COOH groups of aspartic or glutamic acid residues, –OH groups of serine or tyrosine residues, –SH groups of cysteine residues, and terminal amino groups. The most likely attachment site will be determined by the reactive group on the polymer and the reaction conditions, especially the pH. Because these conjugations are generally carried out in a nonspecific way, the conjugated polymer can interfere sterically with the protein’s active site or modify its microenvironment, typically reducing the bioactivity of the protein. Although PEG is not a smart polymer like PNIPAAm, in the special case of PEGylation of the new recombinant protein drugs, it is critical to attach the PEG molecule in a way so as to minimize such steric hindrance. In this situation, the pH may be lowered to favor conjugation of PEG–NHS to the terminal amino group, rather than to randomly located lysine amino groups. On rare occasions the conjugation of a polymer may increase the activity of the protein (e.g., Ding et al., 1998). Figure I.2.11.4 shows various ways that polymers may be directly conjugated to proteins or formed by growth from the protein (Hoffman and Stayton, 2010).

FIGURE I.2.11.4 Various methods for synthesizing polymer–protein bioconjugates.
Random conjugation of temperature-sensitive (mainly) and pH-sensitive (occasionally) polymers to proteins has been extensively investigated, and applications of these conjugates have been focused on immunoassays, affinity separations, enzyme recovery, and drug delivery. One of the earliest applications of a smart polymer–biomolecule conjugate was an immunoassay developed by Hoffman, along with co-workers at a Seattle company called Genetic Systems Corp. They conjugated PNIPAAm to an antibody and used it to capture an antigen, then added a second, labeled antibody to the antigen, and warmed to phase separate the labeled immune complex sandwich. The assay was similar to an ELISA assay, but was much faster and was as accurate as ELISA. This concept was first presented by Hoffman at a PIMS meeting in 1986 (Hoffman et al., 1986), published in 1987 (Monji and Hoffman, 1987), and patented in 1988 (Monji et al., 1988). Other interesting work on soluble–insoluble polymer–biomolecule conjugates can be found in the following references: Schneider et al., 1981; Okamura et al., 1984; Nguyen and Luong, 1989; Taniguchi et al., 1989; Chen and Hoffman, 1990; Monji et al., 1990; Pecs et al., 1991; Taniguchi et al., 1992; Takei et al., 1993b; Galaev and Mattiasson, 1993; Takei et al., 1994a; Fong et al., 1999; Anastase-Ravion et al., 2001.
In some cases the “smart” polymer may be a polyligand, such as polybiotin or poly(glycosyl methacrylate), which is used to phase separate target molecules by complexation to multiple binding sites on target proteins, such as streptavidin and Concanavalin A, respectively (Larsson and Mosbach, 1979; Morris et al., 1993; Nakamae et al., 1994).
Wu et al. have synthesized PNIPAAm–phospholipid conjugates for use in drug delivery formulations as components of thermally sensitive composites and liposomes (Wu et al., 1992, 1993).
There has been some concern about the toxicity of PNIPAAm, related to the toxic nature of the acrylamide monomer which is similar to NIPAAm. If any NIPAAm monomer residues are present in the polymer product, it is most likely they can be washed out. Although there are no significant reports of PNIPAAm toxicity, this issue has not been adequately investigated to date.
Although there are no significant reports of PNIPAAm toxicity, that issue has not been adequately investigated to date.
Smart Polymers and their Biomolecule Conjugates on Surfaces
Smart polymers, especially PNIPAAm, have been coated on various surfaces by physical adsorption, chemical conjugation, affinity complexation, radiation-induced polymerization, direct or chain-transfer initiated polymerization from the surface, and plasma discharge deposition. These methods and various applications of them are discussed in this section.
Smart polymers, especially PNIPAAm, have been coated on various surfaces by physical adsorption, chemical conjugation, affinity complexation, radiation-induced polymerization, direct or chain-transfer initiated polymerization from the surface, and plasma discharge deposition.
One may covalently graft PNIPAAm to a surface by exposing the surface to ionizing radiation in the presence of the monomer (and in the absence of air) or by pre-irradiating the polymer surface in air, and later contacting the surface with the monomer solution and heating in the absence of air to expose trapped radicals. These surfaces exhibit stimulus-responsive changes in wettability (e.g., Uenoyama and Hoffman, 1988; Takei et al., 1994b; Kidoaki et al., 2001). Okano and Yamato and co-workers have been pioneers in the area of smart polymer cell culture surfaces. They applied the radiation grafting technique to form surfaces having a surface layer of grafted PNIPAAm (Yamato and Okano, 2001; Shimizu et al., 2003). They cultured cells to confluent sheets on these surfaces at 37°C, which is above the LCST of the polymer. When the PNIPAAm collapses, the interface becomes hydrophobic and leads to physical adsorption of cell adhesion proteins from the culture medium, enhancing the cell culture process. Then, when the temperature is lowered, the interface becomes hydrophilic as the PNIPAAm chains rehydrate, and the cell sheets are released from the surface along with the cell adhesion proteins, which remain bound to the cell surfaces. The cell sheet can be recovered and used in tissue engineering, e.g., for artificial corneas and heart patches, and in periodontal applications. These exciting new applications of smart surface cell sheets are being pursued in the clinic. Patterned PNIPAAm surfaces have also been prepared for cell culture (Yamato et al., 2001).
Ratner and co-workers have used a gas plasma discharge technique to deposit temperature-responsive coatings from a NIPAAm monomer vapor phase plasma discharge. When cells are cultured on these surfaces, they also form reversible cell sheets in a manner similar to Okano and Yamato’s PNIPAAm-grafted surfaces (Pan et al., 2001).
Smart polymers may also be grafted to surfaces by polymerizing the monomer directly from the surface. This may be done by first attaching to the surface either a radical initiator, such as an azo or peroxide initiator, or a chain transfer agent (CTA) for controlled radical polymerizations such as reversible addition-fragmentation chain transfer polymerization (RAFT) (e.g., Chiefari et al., 1998; McCormick and Lowe, 2004; Moad et al., 2008) or atom transfer radical polymerization (ATRP) (e.g., Wang and Matyjaszewski, 1995; Matyjaszewski and Xia, 2001; Matyjaszewski and Tsarevsky, 2009). (The ATRP of the smart polymer from a biomolecule such as a protein to form the bioconjugate directly was discussed above in this chapter.) Then the polymerization is initiated at the surface or in the surrounding solution, and the result is a smart polymer grafted to the surface. In the case of RAFT or ATRP controlled radical polymerizations, the molecular weights of the grafted polymer can be controlled to a narrow range. In a recent example, Golden and co-workers conjugated a RAFT CTA to the surface of a microporous, hydroxylated nylon membrane, and graft polymerized NIPAAm directly from the surface, forming a graft polymer of around 9000 MW (Golden et al., 2010). Then this surface was heated, and the collapsed PNIPAAm-coated porous membrane was used in a microfluidic channel to sequester dilute PNIPAAm-antibody conjugates that had earlier captured a biomarker antigen in a blood test sample. In this way, the captured biomarker was concentrated on the membrane surface and later released by cooling to provide a concentrated pulse flowing downstream in the channel for this smart polymer microfluidic immunoassay.
The research team of Stayton and Hoffman, and co-workers have also developed a variety of nanoparticles (nps) coated with PNIPAAm for various applications in diagnostic immunoassays. They include polystyrene latex nanoparticles, magnetic nanoparticles, and gold nanoparticles. In the case of the latex nps, Malmstadt and co-workers functionalized aminated polystyrene latex nps with PNIPAAm by conjugating an NHS-terminated PNIPAAm chain to the surface amino groups. The aminated beads were also functionalized with an NHS–PEG–biotin polymer, and streptavidin was bound to the biotin groups. The beads were localized in a microfluidic channel by raising the temperature in one section of the channel. The PNIPAAm- and antibody-functionalized beads were then used to capture labeled, biotinylated antigens in a test sample, to produce a smart polymer competition immunoassay carried out in a microfluidic device (Malmstadt et al., 2003a,b, 2004).
The research team of Lai and Nash and co-workers developed PNIPAAm and PNIPAAm-antibody coatings on magnetic and gold nps, which were used to capture dilute antigen biomarkers in a test sample, then later were captured on the PNIPAAm-grafted porous membrane surface described above (Golden et al., 2010). After that they were released by cooling, forming a concentrated pulse of labeled nps for assay downstream in a microfluidic device. PNIPAAm-coated magnetic and gold nps were also applied in lateral flow, “dipstick” assay by the Stayton–Hoffman group.
In a process for coating magnetic nps with PNIPAAm, Lai and co-workers formed magnetic nps in the presence of a dodecyl-terminated PNIPAAm. The dodecyl-terminated PNIPAAm polymer formed micelles that concentrated the pentacarbonyl iron precursor of magnetic iron oxides in the dodecyl core of the micelle before it was converted at high temperature to magnetic iron oxide (Lai et al., 2007, 2009). To coat the gold nps, Nash bound RAFT-polymerized PNIPAAm chains to the gold surface via sulfur’s affinity to gold, utilizing the sulfur-containing groups in the dithioester or trithio carbonate terminal groups from the original CTA of the RAFT polymer chain (Nash et al., 2010a,b,c). The coated magnetic particles were mainly used for separating and concentrating the captured antigen, and the coated gold particles were mainly used as color (red) labels to indicate the approximate concentration of the captured antigen.
PNIPAAm may also be physically adsorbed to surfaces. Work by Miura and Hoffman and co-workers established the importance of matching the smart polymer composition with the surface composition in order to enhance the stimulus-driven adsorption of the smart polymer on the surface (Miura et al., 1994).
Smart polymers may also be grafted in gradients to surfaces to provide surfaces of gradually varying hydrophilicity and hydrophobicity as a function of the polymer composition and conditions. This phenomenon has been applied by Okano, Kikuchi, and co-workers to prepare chromatographic column packing for use in eluate-free (“green”) chromatographic separations (Kobayashi et al., 2001; Kikuchi and Okano, 2002). Ishihara et al. (1982, 1984) developed photo-responsive coatings and membranes that reversibly change surface wettability or swelling due to the photo-induced isomerization of an azobenzene-containing polymer.
Site-Specific Smart Polymer Bioconjugates
Conjugation of a responsive polymer to a specific site near the “activity” pocket of a genetically engineered protein is a powerful new concept initially demonstrated by Chilkoti et al. (1994) (see also later work by Stayton and Hoffman et al., 2000). Such site-specific protein–smart polymer conjugates can permit sensitive environmental control of the protein’s recognition process, which controls all living systems. They conjugated the smart polymer to a reactive –SH thiol group from a cysteine that had been inserted by site-specific mutagenesis at a site nearby the active site of the protein (Figure I.2.11.5). To do this, they first used cassette mutagenesis to insert a site-specific mutation into the DNA sequence of the protein, and then cloned the mutant in cell culture. This method is applicable only to proteins whose complete peptide sequence is known. The preparation of the reactive smart polymer is similar to methods described above, but in this case, the reactive end or pendant groups and the reaction conditions were specifically designed to favor conjugation to –SH groups rather than to –NH2 groups. Typical SH-reactive polymer end groups include maleimide and vinyl sulfone groups.
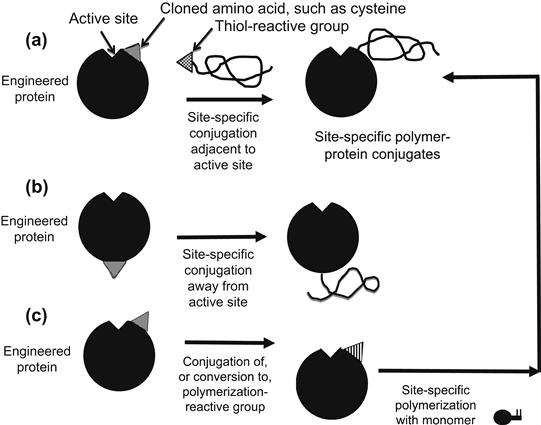
FIGURE I.2.11.5 Schematic illustration of various processes for preparing a site-specific conjugate of a polymer with a genetically-engineered, mutant protein.
The specific site for polymer conjugation can also be located far away from the active site (Chilkoti et al., 1994), in order to avoid interference with the biological functioning of the protein, or nearby or even within the active site, in order to control the ligand–protein recognition process, and the resulting biological activity of the protein (Figure I.2.11.5), (see also Hoffman and Stayton, 2010). Temperature-, pH-, and light-sensitive smart polymers have been used to form such novel, “really smart” bioconjugates (Ding et al., 1999; Bulmus et al., 1999; Stayton et al., 2000; Ding et al., 2001; Shimoboji et al., 2001, 2002a,b, 2003). Since the objective is to control the activity of the protein, and not to phase separate it, in practice these smart polymer-engineered protein bioconjugates would be immobilized on the surfaces of microarray chips, microbeads or nanobeads.
Site-specific conjugates of PNIPAAm to protein mutants of streptavidin and the enzyme endocellulase have been extensively studied by the Stayton–Hoffman group. Site-specific bioconjugates of PNIPAAm with streptavidin have been used to control access of biotin to its binding site on streptavidin (Stayton et al., 1995), and have enabled separation of biotinylated proteins according to the size of the protein (Ding et al., 2001). Ding, Stayton, and Hoffman et al. (1999) also found that raising the temperature to induce the collapse of the PNIPAAm chain “triggered” the release of the bound biotin molecules (perhaps due to a reduced biotin “off” rate compared to the “on” rate) (Ding et al., 1999). For the site-specific enzyme conjugates, a combined temperature- and light-sensitive polymer was conjugated to specific sites on endocellulase, which provided “on-off” control of the enzyme activity with either light or temperature (Shimoboji et al., 2001, 2002a,b, 2003). Bulmus et al. (1999) conjugated a pH- and temperature-responsive PNIPAAm copolymer to a specific site on streptavidin.
Triggered release of bound ligands by the smart polymer-engineered protein bioconjugates could be used to release therapeutics, such as for topical drug delivery to the skin or mucosal surfaces of the body, and also for localized delivery of drugs within the body by stimulated release at pretargeted sites using noninvasive, focused stimuli such as ultrasound or delivery of light stimuli from catheters. Triggered release could also be used to release and recover affinity-bound ligands from chromatographic and other supports in eluate-free conditions, including capture and release of specific cell populations to be used in stem cell and bone marrow transplantation. These processes could involve two different stimuli-responsive polymers with sensitivities to the same or different stimuli. For delicate target ligands such as peptides and proteins, recovery could be affected without the need for time-consuming and harsh elution conditions. Triggered release could also be used to remove inhibitors, toxins or fouling agents from the recognition sites of enzymes and affinity molecules, such as those used in biosensors, diagnostic assays or affinity separations. This could be used to “regenerate” such recognition proteins for extended process use. Light-controlled binding and release of site-specific protein conjugates may be utilized as a molecular switch for various applications in biotechnology, medicine, and bioelectronics, including hand-held diagnostic devices, biochips, and lab-on-a-chip devices.
Fong, Stayton, and Hoffman (Fong et al., 1999) have developed an interesting construct to control the distance of the PNIPAAm from the active site. For this purpose, they conjugated one sequence of complementary nucleotides to a specific site near the binding pocket of streptavidin, and a second sequence to the end of a PNIPAAm chain. Then, by controlling the location and length of the complementary sequence, the self-assembly via hybridization of the two single-chain DNA sequences could be used to control the distance of the polymer from the streptavidin binding site.
Smart Polymer Hydrogels
When a smart polymer is cross-linked to form a gel, it will collapse and reswell in water as a stimulus raises or lowers it through its critical condition. PNIPAAm hydrogels have been extensively studied, starting with the pioneering work of Toyoichi Tanaka in 1981 (Tanaka, 1981). In the mid- to late-1980s, Hoffman and co-workers were among the first to recognize the potential of PNIPAAm hydrogels as biomaterials; they showed that smart gels could be used to entrap enzymes and cells, and then by inducing cyclic collapse and swelling of the gel, the enzymes (or enzymes within the cells) could be turned “on” and “off.” Park and Hoffman also showed enhanced enzyme efficiency for an entrapped enzyme in a packed bed, hydrogel bead reactor, since the collapse delivered product faster than it could diffuse out, and the reswelling imbibed substrate faster than it could diffuse in. They also could deliver drugs or remove toxic biomolecules, by stimulus-induced collapse or swelling. Dong and Hoffman developed an interesting pH- and temperature-responsive gel that swelled linearly at 37°C and pH 7.4, at rates depending on the AAc monomer content in the gel. This work with PNIPAAm gels in drug delivery systems (DDS) is discussed in detail in Chapter II.5.16 on Drug Delivery Systems (Dong and Hoffman, 1986, 1987, 1990, 1991; Park and Hoffman, 1988, 1990a,b,c, 1994) (Figure I.2.11.6).
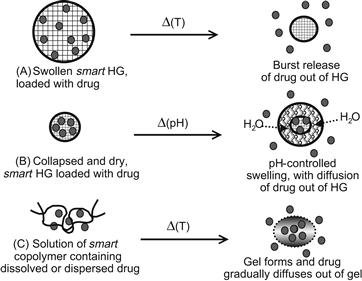
FIGURE I.2.11.6 Schematic illustration showing three ways that smart gel formulations may be stimulated to release bioactive agents: (A) thermally-induced collapse, which is relevant to skin or mucosal drug delivery; (B) pH-induced swelling, which is relevant to oral drug delivery, where the swelling is induced by the increase in pH in going from the gastric to enteric regions; and (C) sol-to-gel formation, which is relevant to injectable or topical formulations of a triblock copolymer solution that are thermally gelled at body temperature. For in vivo uses, the block copolymer is designed to be degradable. The first two apply to cross-linked gels applied topically or orally, and the third is relevant to thermally-induced formation of gels from polymer solutions that are delivered topically or by injection.
(See also Chapter II.5.16 Drug Delivery Systems.)
The research team of Kim, Okano, Bae, and their co-workers were also actively studying smart hydrogels in the late 1980s and 1990s. For example, they investigated smart gels containing entrapped cells that could be used as artificial organs (Vernon et al., 2000). Their drug delivery work with smart hydrogels is discussed in detail in Chapter II.5.16 on Drug Delivery Systems.
Since then, the properties of PNIPAAm hydrogels have been widely investigated in the form of beads, slabs or discs, and multilamellar laminates (Park and Hoffman, 1992a,b; Hu et al., 1995, 1998; Mitsumata et al., 2001; Gao and Hu, 2002; Kaneko et al., 2002).
Okano and co-workers designed and synthesized PNIPAAm gels that collapse much more rapidly than PNIPAAm gels that are simply cross-linked with a divinyl comonomer. Yoshida, Okano, and co-workers copolymerized NIPAAm monomer with a PNIPAAm macromer and a cross-linker, which yielded a gel with PNIPAAm chains grafted to the PNIPAAm network. The grafted chains collapsed first at the LCST, enhancing the rate of collapse of the network, along with the related rate of expulsion of water from the gel (Yoshida et al., 1995; Masahiko et al., 2003).
Peppas and co-workers (e.g., Peppas et al., 2000; Robinson and Peppas, 2002) have extensively studied pH-sensitive acrylic acid-acrylate copolymer smart gels. This work is discussed in more detail in Chapter I.2.5 on Hydrogels (see also Peppas, 1997, 2001; Peppas et al., 1999, 2000).
Smart hydrogel compositions have been developed that are both thermally gelling and biodegradable (Zhong et al., 2002; Yoshida et al., 2003). These sol–gel systems have been used to deliver drugs by in vivo injections, and are discussed in the section on Smart Polymers in Solution (see also Chapter I.2.5 on Hydrogels, and Chapter II.5.16 on Drug Delivery Systems).
Matsuda and co-workers incorporated PNIPAAm into physical mixtures with natural polymers such as hyaluronic acid and gelatin, for use as tissue engineering scaffolds (Ohya et al., 2001a,b). Park and Hoffman developed a unique process for synthesizing mm-size spherical beads of PNIPAAm (Park and Hoffman, 1992c).
Nakamae, Hoffman, and co-workers developed novel compositions of smart gels containing phosphate groups that were used to bind cationic proteins as model drugs, and then to release them by a combination of thermal stimuli and ion exchange (Nakamae et al., 1992, 1997; Miyata et al., 1994).
A number of smart hydrogel drug delivery systems have been designed to respond to biologic signals in a feedback manner. Many of these gels contain an immobilized enzyme. They are discussed in detail in the Chapter II.5.16 on Drug Delivery Systems.
Conclusions
Smart polymers in solution, on surfaces, and as hydrogels have been utilized in many interesting ways, especially in combination with biomolecules such as proteins and drugs. Important applications include affinity separations, enzyme processes, immunoassays, drug delivery, and toxin removal. These smart polymer–biomolecule systems represent an important extension of polymeric biomaterials beyond their well-known uses in implants and medical devices.
Bibliography
1. Anastase-Ravion S, Ding Z, Pelle A, Hoffman AS, Letourneur D. New antibody purification procedure using a thermally-responsive polyNIPAAm-dextran derivative conjugate. J Chromatogr B. 2001;761:247–254.
2. Broyer RM, Grover GN, Maynard HD. Emerging synthetic techniques for protein–polymer conjugations. Chem Commun. 2011;47:2212–2226.
3. Bulmus V, Ding Z, Long CJ, Stayton PS, Hoffman AS. Design, synthesis and site-specific conjugation of a pH- and temperature-sensitive polymer to streptavidin for pH-controlled binding and triggered release of biotin. Bioconj Chem. 1999;11:78–83.
4. Chen G, Hoffman AS. Synthesis of carboxylated poly(NIPAAm) oligomers and their application to form thermo-reversible polymer-enzyme conjugates. J Biomater Sci Polymer Edn. 1994;5:371–382.
5. Chen G, Hoffman AS. Graft copolymer compositions that exhibit temperature-induced transitions over a wide range of pH. Nature. 1995;373:49–52.
6. Chen JP, Hoffman AS. Polymer-protein conjugates II Affinity precipitation of human IgG by poly(N-isopropyl acrylamide)-protein A conjugates. Biomaterials. 1990;11:631–634.
7. Chen JP, Yang HJ, Hoffman AS. Polymer–protein conjugates I Effect of protein conjugation on the cloud point of poly(N-isopropyl acrylamide). Biomaterials. 1990;11:625–630.
8. Chiefari J, Chong YK, Ercole F, Krstina J, Jeffery J, et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer: The RAFT process. Macromolecules. 1998;31(16):5559–5562. doi 10.1021/ma9804951.
9. Chilkoti A, Chen G, Stayton PS, Hoffman AS. Site-specific conjugation of a temperature-sensitive polymer to a genetically-engineered protein. Bioconj Chem. 1994;5:504–507.
10. Cole CA, Schreiner SM, Priest JH, Monji N, Hoffman AS. N-isopropyl acrylamide and N-acryl succinimide copolymers: A thermally reversible water soluble activated polymer for protein conjugation. In: Russo P, ed. Washington, DC: ACS; 1987;245–254. Reversible Polymeric Gels and Related Systems, ACS Symposium Series. Vol. 350.
11. Ding ZL, Chen G, Hoffman AS. Synthesis and purification of thermally-sensitive oligomer–enzyme conjugates of poly(NIPAAm)–trypsin. Bioconj Chem. 1996;7:121–125.
12. Ding ZL, Chen G, Hoffman AS. Properties of polyNIPAAm–trypsin conjugates. J Biomed Mater Res. 1998;39:498–505.
13. Ding ZL, Long CJ, Hayashi Y, Bulmus EV, Hoffman AS, et al. Temperature control of biotin binding and release with a streptavidin–polyNIPAAm site-specific conjugate. J Bioconj Chem. 1999;10:395–400.
14. Ding ZL, Shimoboji T, Stayton PS, Hoffman AS. A smart polymer shield that controls the binding of different size biotinylated proteins to streptavidin. Nature. 2001;411:59–62.
15. Dong LC, Hoffman AS. Thermally reversible hydrogels: III Immobilization of enzymes for feedback reaction control. J Contr Rel. 1986;4:223–227.
16. Dong LC, Hoffman AS. Thermally reversible hydrogels: Swelling characteristics and activities of copoly(NIPAAm-AAm) gels containing immobilized asparaginase. In: Russo P, ed. Washington, DC: ACS; 1987;236–244. Reversible Polymeric Gels and Related Systems, ACS Symposium Series. Vol 350.
17. Dong LC, Hoffman AS. Synthesis and application of thermally-reversible heterogels for drug delivery. J Contr Release. 1990;13:21–32.
18. Dong LC, Hoffman AS. A novel approach for preparation of pH- and temperature-sensitive hydrogels for enteric drug delivery. J Contr Release. 1991;15:141–152.
19. Fong RB, Ding ZL, Long CJ, Hoffman AS, Stayton PS. Thermoprecipitation of streptavidin via oligonucleotide-mediated self-assembly with poly(NIPAAm). Bioconj Chem. 1999;10:720–725.
20. Galaev IY, Mattiasson B. Affinity thermoprecipitation: Contribution of the efficiency and access of the ligand. Biotechnol Bioeng. 1993;41:1101–1106.
21. Gao J, Hu ZB. Optical properties of N-isopropylacrylamide microgel spheres in water. Langmuir. 2002;18:1360–1367.
22. Golden AL, Battrell CF, Pennell S, Hoffman AS, Lai JJ, et al. Simple fluidic system for purifying and concentrating diagnostic biomarkers using stimuli-responsive antibody conjugates and membranes. Bioconj Chem. 2010;21:1820–1826.
23. Grover GN, Maynard HD. Protein–polymer conjugates: Synthetic approaches by controlled radical polymerizations and interesting applications. Current Opin Chem Biol. 2010;14:818–827.
24. Heredia KL, Maynard HD. Synthesis of protein–polymer conjugates. Organic & Biomol Chem. 2007;5:45–53.
25. Heskins H, Guillet JE. Solution properties or poly(N-isopropyl acrylamide. J Macromol Sci Chem. 1968;A2(6):1209.
26. Hoffman AS. Applications of thermally reversible polymers and hydrogels in therapeutics and diagnostics. J Contr Rel. 1987;6:297–305.
27. Hoffman AS. Intelligent polymers in medicine and biotechnology. Macromol Symp. 1995;98:645–664.
28. Hoffman AS. Intelligent polymers in medicine and biotechnology. In: Park K, ed. Controlled Drug Delivery. Washington, DC: ACS Publications; 1997.
29. Hoffman AS, Stayton PS. Conjugates of stimuli-responsive polymers and proteins. Prog Polym Sci. 2010;32:922–932.
30. Hoffman AS, et al. Novel Application of Polymers in Bioseparations and Diagnostics. Proceedings of Polymers in Medicine and Surgery (PIMS) V: Holland 1986; 37(1), 2.
31. Hoffman AS, Stayton PS, Bulmus V, Chen G, Chen J, et al. Really smart bioconjugates of smart polymers and receptor proteins. J Biomed Mater Res. 2000;52:577–586.
32. Hu ZB, Zhang XM, Li Y. Synthesis and application of modulated polymer gels. Science. 1995;269:525.
33. Hu ZB, Chen YY, Wang CJ, Zheng YY, Li Y. Polymer gels with engineered environmentally responsive surface patterns. Nature. 1998;393:149.
34. Ishihara K, Okazaki A, Negishi N, Shinohara I, Okano T, et al. Photo-induced change in wettability and binding ability of azoaromatic polymer. J Appl Polymer Sci. 1982;27:239–245.
35. Ishihara K, Hamada N, Kato S, Shinohara I. Photo-induced swelling control of amphiphilic azoaromatic polymer membrane. Polymer Sci (Polymer Chem. Ed.). 1984b;22:21–128.
36. Jeong B, Kim SW, Bae YH. Thermosensitive sol–gel reversible hydrogels. Adv Drug Delivery Rev. 2002;54:37–51.
37. Kaneko D, Gong JP, Osada Y. Polymer gels as soft and wet chemomechanical systems: An approach to artifical muscles. J Mater Chem. 2002;12:2169–2177.
38. Kang HC, Bae YH. pH-Tunable endosomolytic oligomers for enhanced nucleic acid delivery. Advanced Functional Materials. 2007;17:1263–1272.
39. Kidoaki S, Ohya S, Nakayama Y, Matsuda T. Thermoresponsive structural change of a PNIPAAm graft layer measured with AFM. Langmuir. 2001;17:2402–2407.
40. Kikuchi A, Okano T. Intelligent thermoresponsive polymeric stationary phases for aqueous chromatography of biological compounds. Progr Polymer Sci. 2002;27:1165–1193.
41. Kobayashi J, Kikuchi A, Sakai K, Okano T. Aqueous chromatography utilizing pH-/temperature-responsive polymers as column matrix surfaces for separation of ionic bioactive compounds. Anal Chem. 2001;73:2027–2033.
42. Lackey CA, Murthy N, Press OW, Tirrell DA, Hoffman AS, et al. Hemolytic activity of pH-responsive polymer–streptavidin bioconjugates. Bioconj Chem. 1999;10:401–405.
43. Lai J, Hoffman AS, Stayton PS. Dual magnetic-temperature responsive nanoparticles for microfluidic separations and assays. Langmuir. 2007;23:7385–7391.
44. Lai JJ, Nelson KE, Nash MA, Hoffman AS, Yager P, et al. Dynamic bioprocessing and microfluidic transport control with smart magnetic nanoparticles in laminar-flow devices. Lab Chip. 2009;9:1997–2002.
45. Larsson PO, Mosbach K. Affinity precipitation of enzymes. FEBS Lett. 1979;98:333–338.
46. Lee DS, Shim MS, Kim SW, Lee H, Park I, et al. Novel thermoreversible gelation of biodegradable PLGA-block-PEO-block-PLGA triblock copolymers in aqueous solution. Macromol Rapid Commun. 2001;22:587–592.
47. Masahiko A, Matsuura T, Kasai M, Nakahira T, Hara Y, et al. Preparation of comb-type N-isopropylacrylamide hydrogel beads and their application for size-selective separation media. Biomacromolecules. 2003;4:395–403.
48. Malmstadt N, Yager P, Hoffman AS, Stayton PS. A Smart Microfluidic Affinity Chromatography Matrix Composed of Poly(N-isopropylacrylamide)-Coated Beads. Anal Chem. 2003a;75:2943–2949.
49. Malmstadt N, Hyre D, Ding Z, Hoffman AS, Stayton PS. Affinity Thermoprecipitation and Recovery of Biotinylated Biomolecules via a Mutant Streptavidin-Smart Polymer Conjugate. Bioconj Chem. 2003b;14:575–580.
50. Malmstadt N, Hoffman AS, Stayton PS. Smart Mobile Affinity Matrix for Microfluidic Immunoassays. Lab Chip. 2004;4:412–415.
51. Matyjaszewski K, Tsarevsky NV. Nanostructured functional materials prepared by atom transfer radical polymerization. Nature Chemistry. 2009;1:276–288. doi 10.1038/NCHEM.257.
52. Matyjaszewski K, Xia J. Atom transfer radical polymerization. Chem Rev. 2001;101:2921–2990. doi 10.1021/cr940534g ISSN 0009-2665. PMID 11749397.
53. McCormick C, Lowe AB. Aqueous RAFT polymerization: Recent developments in synthesis of functional water-soluble (co)polymers with controlled structures. Accounts of Chemical Research. 2004;37:312–325. doi 10.1021/ar0302484.
54. Mitsumata T, Gong JP, Osada Y. Shape memory functions and motility of amphiphilic polymer gels. Polymer Adv Technol. 2001;12:136–150.
55. Miura M, Cole CA, Monji N, Hoffman AS. Temperature-dependent adsorption/desorption behavior of lcst polymers on various substrates. J Biomtls Sci (Polymer Ed). 1994;5:555–568.
56. Miyata T, Nakamae K, Hoffman AS, Kanzaki Y. Stimuli-sensitivities of hydrogels containing phosphate groups. Macromol Chem Phys. 1994;195:1111–1120.
57. Moad G, Rizzardo E, Thang SH. Radical addition-fragmentation chemistry in polymer synthesis. Polymer. 2008;49:1079–1131. doi 10.1016/j.polymer.2007.11.020.
58. Monji N, Hoffman AS. A novel immunoassay system and bioseparation process based on thermal phase separating polymers. Appl Biochem Biotechnol. 1987;14:107–120.
59. Monji, N., Hoffman, A. S., Priest, J. H. & Houghton, R. L. (1988). Thermally-Induced Phase Separation Immunoassay. U.S. Patent 4,780,409 (10/25/88).
60. Monji N, Cole CA, Tam M, Goldstein L, Nowinski RC, et al. Application of a thermally-reversible polymer–antibody conjugate in a novel membrane-based immunoassay. Biochem Biophys Res Commun. 1990;172:652–660.
61. Morris JE, Hoffman AS, Fisher RR. Affinity precipitation of proteins by polyligands. Biotechnol Bioeng. 1993;41:991–997.
62. Murthy N, Stayton PS, Hoffman AS. The design and synthesis of polymers for eukaryotic membrane disruption. J Controlled Release. 1999;61:137–143.
63. Murthy N, Campbell J, Fausto N, Hoffman AS, Stayton PS. Bioinspired polymeric carriers that enhance intracellular delivery of biomolecular therapeutics. Bioconj Chem. 2003a;14:412–419.
64. Murthy N, Campbell J, Fausto N, Hoffman AS, Stayton PS. Design and synthesis of pH-responsive polymeric carriers that target uptake and enhance the intracellular delivery of oligonucleotides to hepatocytes. J Contr Rel. 2003b;89(3):365–374.
65. Na K, Lee DH, Hwang DJ, Lee KH, Bae YH. pH-Sensitivity and pH-dependent structural change in polymeric nanoparticles of poly(vinyl sulfadimethoxine)-deoxycholic acid conjugate. Eur Polym J. 2006;42:2581–2588.
66. Nakamae K, Miyata T, Hoffman AS. Swelling behavior of hydrogels containing phosphate groups. Macromol Chem. 1992;193:983–990.
67. Nakamae K, Miyata T, Jikihara A, Hoffman AS. Formation of poly(glucosyloxyethyl methacrylate)-concanavalin a complex and its glucose sensitivity. J Biomtls Sci (Polymer Ed). 1994;6:79–90.
68. Nakamae K, Nizuka T, Miyata T, Furukawa M, Nishino T, et al. Lysozyme loading and release from hydrogels carrying pendant phosphate groups. J Biomater Sci (Polymer Ed). 1997;9:43–53.
69. Nash MA, Yager P, Hoffman AS, Stayton PS. Mixed stimuli-responsive magnetic and gold nanoparticle system for rapid purification, enrichment and detection of biomarkers. Bioconj Chem. 2010a;21:2197–2204.
70. Nash MA, Lai JJ, Hoffman AS, Yager P, Stayton PS. “Smart” diblock copolymers as templates for magnetic-core gold-shell nanoparticle synthesis. Nano Lett. 2010b;10:85–91.
71. Nash MA, Hoffman JM, Stevens DY, Hoffman AS, Stayton PS, et al. Laboratory-scale protein striping system for patterning biomolecules onto paper-based immunochromatographic test strips. Lab Chip. 2010c;10:2279–2282.
72. Nguyen AL, Luong JHT. Syntheses and application of water soluble reactive polymers for purification and immobilization of biomolecules. Biotechnol Bioeng. 1989;34:1186–1190.
73. Ohya S, Nakayama Y, Matsuda T. Thermoresponsive artificial extracellular matrix for tissue engineering: Hyaluronic acid bioconjugated with poly(N-isopropylacrylamide) grafts. Biomacromolecules. 2001a;2:856–863.
74. Ohya S, Nakayama Y, Matsuda T. Material design for an artificial extracellular matrix: Cell entrapment in poly(N-isopropylacrylamide) (PNIPAM)-grafted gelatin hydrogel. J Artif Organs. 2001b;4:308–314.
75. Okamura K, Ikura K, Yoshikawa M, Sakaki R, Chiba H. Soluble–insoluble interconvertible enzymes. Agric Biol Chem. 1984;48:2435–2440.
76. Okano T, Kikuchi A, Yamato M. Intelligent hydrogels and new biomedical applications. In: Biomaterials and Drug Delivery toward the New Millennium. Seoul, Korea: Han Rim Won Publishing Co; 2000;77–86.
77. Pan YV, Wesley RA, Luginbuhl R, Denton DD, Ratner BD. Plasma-polymerized N-isopropylacylamide: Synthesis and characterization of a smart thermally responsive coating. Biomacromolecules. 2001;2:32–36.
78. Park TG, Hoffman AS. Effect of temperature cycling on the activity and productivity of immobilized β-galactosidase in a thermally reversible hydrogel bead reactor. Appl Biochem Biotechnol. 1988;19:1–9.
79. Park TG, Hoffman AS. Immobilization and characterization of β-galactosidase in thermally reversible hydrogel beads. J Biomed Mater Res. 1990a;24:21–38.
80. Park TG, Hoffman AS. Immobilization of A. simplex cells in a thermally-reversible hydrogel: Effect of temperature cycling on steroid conversion. Biotech Bioeng. 1990b;35:52–159.
81. Park TG, Hoffman AS. Immobilized biocatalysts in reversible hydrogels. In: Tanaka A, ed. 1990c;588–593. Enzyme Engineering X Ann NY Acad Sci. 613.
82. Park TG, Hoffman AS. Preparation of large, uniform size temperature-sensitive hydrogel beads. J Polymer Sci A Polymer Chem. 1992a;30:505–507.
83. Park TG, Hoffman AS. Synthesis and characterization of pH- and/or temperature-sensitive hydrogels. J Appl Polymer Sci. 1992b;46:659–671.
84. Park TG, Hoffman AS. Preparation of large, uniform size temperature-sensitive hydrogel beads. J Poly Sci A., Poly Chem. 1992c;30:505–507.
85. Park TG, Hoffman AS. Estimation of temperature-dependent pore sizes in poly(NIPAAm) hydrogel beads. Biotechnol Progr. 1994;10:82–86.
86. Pecs M, Eggert M, Schügerl K. Affinity precipitation of extracellular microbial enzymes. J Biotechnol. 1991;21:137–142.
87. Peppas NA. Hydrogels and drug delivery. Critical Opinion in Colloid and Interface Science. 1997;2:531–537.
88. Peppas NA. Gels for drug delivery. In: Encyclopedia of Materials: Science and Technology. Amsterdam: Elsevier; 2001;3492–3495.
89. Peppas NA, Keys KB, Torres-Lugo M, Lowman AM. Poly(ethylene glycol)-containing hydrogels in drug delivery. J Controlled Release. 1999;62:81–87.
90. Peppas NA, Huang Y, Torres-Lugo M, Ward JH, Zhang J. Physicochemical foundations and structural design of hydrogels in medicine and biology. Ann Revs Biomed Eng. 2000;2:9–29.
91. Priest JH, Murray S, Nelson RG, Hoffman AS. LCSTs of aqueous copolymers of N-isopropyl acrylamide and other N-substituted acrylamides. In: Russo P, ed. Washington, DC: ACS; 1987;255–264. Reversible Polymeric Gels and Related Systems, ACS Symposium Series. Vol. 350.
92. Robinson DN, Peppas NA. Preparation and characterization of pH-responsive poly(methacrylic acid-g-ethylene glycol) nanospheres. Macromolecules. 2002;35:3668–3674.
93. Roy D, Cambre JN, Sumerlin BS. Future perspectives and recent advances in stimuli-responsive materials. Prog in Polymer Sci. 2010;35:278–301.
94. Roy I, Gupta MN. Smart polymeric materials: Emerging biochemical applications. Chemistry & Biology. 2003;10:1161–1171.
95. Schild HG. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog Polym Sci. 1992;17:163–249.
96. Schneider M, Guillot C, Lamy B. The affinity precipitation technique: Application to the isolation and purification of trypsin from bovine pancreas. Ann NY Acad Sci. 1981;369:257–263.
97. Shim WS, Yoo JS, Bae YH, Lee DS. Novel injectable pH and temperature sensitive block copolymer hydrogel. Biomacromolecules. 2005;6:2930–2934.
98. Shim WS, Kim JH, Park H, Kim K, Kwon IC, et al. Biodegradability and biocompatibility of a pH- and thermo-sensitive hydrogel formed from a sulfonamide-modified poly(e-caprolactone-co-lactide)–poly(ethylene glycol)–poly (e-caprolactone-co-lactide) block copolymer. Biomaterials. 2006;27:5178–5185.
99. Shimizu T, Yamato M, Kikuchi A, Okano T. Cell sheet engineering for myocardial tissue reconstruction. Biomaterials. 2003;24:2309–2316.
100. Shimoboji T, Ding Z, Stayton PS, Hoffman AS. Mechanistic investigation of smart polymer–protein conjugates. Bioconj Chem. 2001;12:314–319.
101. Shimoboji T, Ding ZL, Stayton PS, Hoffman AS. Photoswitching of ligand association with a photoresponsive polymer–protein conjugate. Bioconj Chem. 2002a;13:915–919.
102. Shimoboji T, Larenas E, Fowler T, Kulkarni S, Hoffman AS, et al. Photoresponsive polymer–enzyme switches. Proc Natl Acad Sci USA. 2002b;99:16592–16596.
103. Shimoboji T, Larenas E, Fowler T, Hoffman AS, Stayton PS. Temperature-induced switching of enzyme activity with smart polymer–enzyme conjugates. Bioconj Chem. 2003;14:517–525.
104. Shoemaker S, Hoffman AS, Priest JH. Synthesis of vinyl monomer-enzyme conjugates. Appl Biochem and Biotechnology. 1987;15:11.
105. Stayton PS, Hoffman AS. Smart pH-responsive carriers for intracellular delivery of Biomolecular Drugs. In: Torchilin V, ed. Multifunctional Pharmaceutical Nanocarriers. New York, NY: Springer Publishers; 2008.
106. Stayton PS, Shimoboji T, Long C, Chilkoti A, Chen G, et al. Control of protein–ligand recognition using a stimuli-responsive polymer. Nature. 1995;378:472–474.
107. Stayton PS, Hoffman AS, Murthy N, Lackey C, Cheung C, et al. Molecular engineering of proteins and polymers for targeting and intracellular delivery of therapeutics. J Contr Rel. 2000;65:203–220.
108. Takei YG, Aoki T, Sanui K, Ogata N, Okano T, et al. Temperature-responsive bioconjugates 1 Synthesis of temperature-responsive oligomers with reactive end groups and their coupling to biomolecules. Bioconj Chem. 1993a;4:42–46.
109. Takei YG, Aoki T, Sanui K, Ogata N, Okano T, et al. Temperature-responsive bioconjugates 2 Molecular design for temperature-modulated bioseparations. Bioconj Chem. 1993b;4:341–346.
110. Takei YG, Matsukata M, Aoki T, Sanui K, Ogata N, et al. Temperature-responsive bioconjugates 3 Antibody-poly(N-isopropylacrylamide) conjugates for temperature-modulated precipitations and affinity bioseparations. Bioconj Chem. 1994a;5:577–582.
111. Takei YG, Aoki T, Sanui K, Ogata N, Sakurai Y, et al. Dynamic contact angle measurements of temperature-responsive properties for PNIPAAm grafted surfaces. Macromolecules. 1994b;27:6163–6166.
112. Tanaka T. Gels. Scientific American. 1981;244:124.
113. Taniguchi M, Kobayashi M, Fujii M. Properties of a reversible soluble–insoluble cellulase and its application to repeated hydrolysis of crystalline cellulose. Biotechnol Bioeng. 1989;34:1092–1097.
114. Taniguchi M, Hoshino K, Watanabe K, Sugai K, Fujii M. Production of soluble sugar from cellulosic materials by repeated use of a reversibly soluble-autoprecipitating cellulase. Biotechnol Bioeng. 1992;39:287–292.
115. Tirrell D. Macromolecular switches for bilayer membranes. J Contr Rel. 1987;6:15–21.
116. Uenoyama S, Hoffman AS. Synthesis and characterization of AAm/NIPAAm grafts on silicone rubber substrates. Radiat Phys Chem. 1988;32:605–608.
117. Vernon B, Kim SW, Bae YH. Thermoreversible copolymer gels for extracellular matrix. J Biomed Mater Res. 2000;51:69–79.
118. Wang J, Matyjaszewski K. Controlled/“living” radical polymerization Atom transfer radical polymerization in the presence of transition-metal complexes. J Am Chem Soc. 1995;117:5614–5615. doi 10.1021/ja00125a035.
119. Wu XS, Hoffman AS, Yager P. Conjugation of phosphatidylethanolamine to poly(NIPAAm) for potential use in liposomal drug delivery systems. Polymer. 1992;33:4659–4662.
120. Wu XS, Hoffman AS, Yager P. Synthesis of and insulin release from erodible polyNIPAAm-phospholipid composites. J Intell Mater Syst Struct. 1993;4:202–209.
121. Yamato M, Okano T. Cell sheet engineering for regenerative medicine. Macromol Chem Symp. 2001;14(2):21–29.
122. Yamato M, Kwon OH, Hirose M, Kikuchi A, Okano T. Novel patterned cell co-culture utilizing thermally responsive grafted polymer surfaces. J Biomed Mater Res. 2001;55:137–140.
123. Yang HJ, Cole CA, Monji N, Hoffman AS. Preparation of a thermally phase-separating copolymer with a controlled number of active ester groups per polymer chain. J Polymer Sci A Polymer Chem. 1990;28:219–226.
124. Yin X, Stayton PS, Hoffman AS. Temperature- and pH-responsiveness of poly(n-isopropylacrylamide-co-propylacrylic acid) copolymers prepared by RAFT polymerization. Biomacromol. 2006;7:1381–1385.
125. Yoshida R, Uchida K, Kaneko Y, Sakai K, Kikuchi A, et al. Comb-type grafted hydrogels with rapid de-swelling response to temperature changes. Nature. 1995;374:240–242.
126. Yoshida T, Aoyagi T, Kokufuta E, Okano T. Newly designed hydrogel with both sensitive thermoresponse and biodegradability. J Polymer Sci A Polymer Chem. 2003;41:779–787.
127. Zareie HM, Bulmus V, Gunning PA, Hoffman AS, Piskin E, et al. Investigation of a pH- and temperature-sensitive polymer by AFM. Polymer. 2000;41:6723–6727.
128. Zhong Z, Dijkstra PJ, Feijen J, Kwon Y-Mi, Bae YH, et al. Synthesis and aqueous phase behavior of thermoresponsive biodegradable poly(d, l-3-methyl glycolide)-b-poly(ethylene glycol)-b-poly(d, l-3-methyl glycolide) triblock copolymers. Macromol Chem Phys. 2002;203:1797–1803.