B.4
Polymer–Drug Conjugates
Introduction
The concept of synthetic polymer–drug conjugates was developed in the 1970s. In the late 1960s and early 1970s, Frank Davis at Rutgers University NJ, USA conceived of the idea of reducing the potential immunogenicity of the new genetically-engineered recombinant proteins by PEGylating them (Davis, 2002). Helmut Ringsdorf published a prescient article on polymer–drug conjugates in 1975 that included concepts of conjugation of cell ligands and “trafficking” molecules (e.g., lipids and polyethylene glycol (PEG)) to the polymer backbone (Ringsdorf, 1975). In the mid-1970s Jindrich Kopecek and colleagues in Prague prepared poly(HPMA)–drug conjugates with a pendant, degradable tetrapeptide spacer to the drug (Kopecek et al., 1977). Later, in a collaboration with Ruth Duncan, they designed and synthesized a hydroxypropyl methacrylamide (HPMA) copolymer with doxorubicin conjugated to the HPMA backbone by a tetrapeptide spacer that was a substrate for the lysosomal enzyme, cathepsin B (Duncan, 2009). They also later conjugated a targeting ligand (galactose) to the same spacer, producing one of the first examples of a targeted, polymer–drug conjugate. This section reviews many of the different polymer–drug conjugates other than PEG, which was discussed earlier in this section.
Poly(HPMA) as a Drug Carrier
The first synthetic polymer drug conjugate to be tested clinically was an HPMA copolymer (N-2-hydroxypropyl methacrylamide) conjugated to doxorubicin (Figure B.4.1A). HPMA polymers were developed by Kopecek and Bazilova (Kopecek and Bazilova, 1973), and used by Duncan and co-workers for drug delivery applications due to its biocompatibility and low blood protein-binding properties (Duncan, 2009). Functionalized copolymers were synthesized by copolymerization of HPMA with a methacryloylated-peptidyl-nitrophenylester; this co-monomer was used for drug conjugation through an aminolysis reaction. Many clinically-tested conjugates use the Gly-Phe-Leu-Gly peptidyl linker introduced through the second co-monomer. This peptide sequence is a substrate for lysosomal thiol-dependent proteases (e.g., cathepsin B), so that drug release is designed to occur after endocytosis of the drug conjugate and trafficking to the lysosomal compartment. Because the HPMA backbone is not biodegradable, the molecular weight of the polymers is typically limited to <40 k, so that materials can be cleared renally.
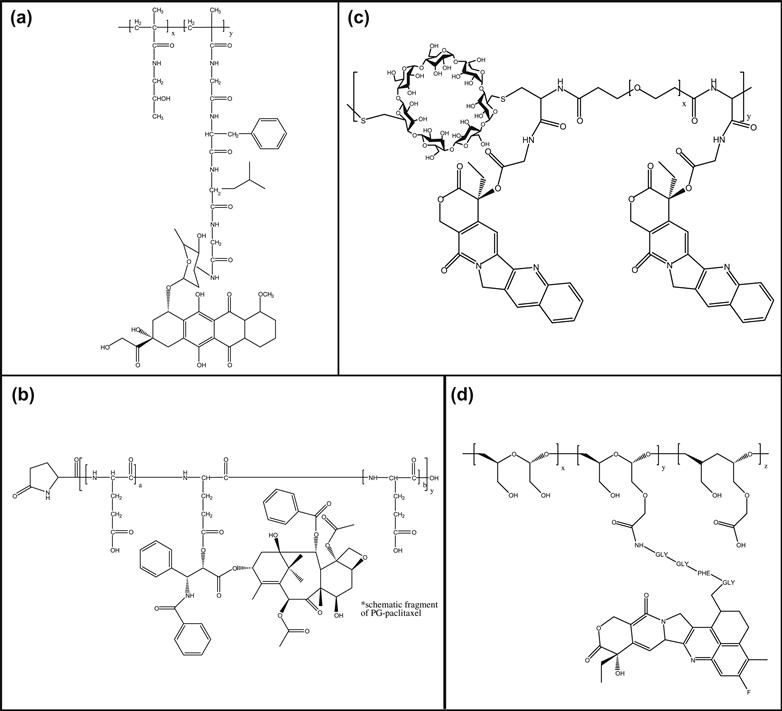
FIGURE B.4.1 Four polymer-drug conjugates: (A) Poly(hydroxypropyl methacrylamide) (PHPMA) conjugated to doxorubicin; (B) Poly(glutamic acid) (PG) conjugated to paclitaxel; (C) Cyclodextrin backbone polymer conjugated to camptothecin; (D) Polyacetal conjugated to camptothecin.
Several other chemotherapeutic drugs, such as doxorubicin, paclitaxel, camptothecin, and platinates, have been conjugated to HPMA and evaluated in clinical trials with varying success. The paclitaxel and camptothecin conjugates developed by Pharmacia were halted after Phase I trials due to toxicity issues. Doxorubicin conjugates licensed to Pharmacia entered Phase II trials, and a galactose-modified copolymer was also evaluated for targeted delivery to hepatocarcinoma. Prolonged plasma circulation was achieved with this conjugate compared with free doxorubicin, and tumor accumulation was observed in metastatic breast cancer patients (Duncan, 2009). An HPMA–platinate conjugate has been evaluated in Phase II clinical trials by Access Pharmaceuticals. They reported drug efficacy with favorable safety profiles in patients (Nowotnik and Cvitkovic, 2009).
Poly(glutamic acid) (PG) as a Drug Carrier
Poly(L-glutamic acid) (PG) is a biodegradable polymer that is degraded by the lysosomal enzyme cathepsin B (Figure B.4.1B). PG has been conjugated to various anticancer agents, including doxorubicin, paclitaxel, and camptothecin (Li, 2002). Several drug–polymer linkages were also tested, including amides, hydrolyzable ester, hydrazone bonds, and enzymatically degradable peptide spacers. Drug activity was optimized by varying the release kinetics from the degrading polymer and evaluating drug efficacy.
Two PG–drug conjugates, PG–paclitaxel and PG–camptothecin, were evaluated clinically by Cell Therapeutics, Inc. The first to enter the clinic was a PG conjugated to paclitaxel via a glycine linker. The polymers had an average molecular weight of 38 kD with 37% by weight paclitaxel conjugated in the 2′ position of the glutamic acid by an ester bond. The drug conjugate was shown to be relatively stable in the blood circulation, and to release active drug primarily through lysosomal cathepsin B degradation of the polymer backbone after cellular uptake (Paz-Ares et al., 2008). The PG–paclitaxel conjugate delivered similar amounts of active paclitaxel compared with equivalent doses of standard paclitaxel (the “area under the curve” or AUC values were similar), but the prolonged distribution of the polymer–drug conjugate resulted in decreased maximum plasma concentration (Cmax) compared with the standard paclitaxel dose, and also reduced myelosuppression and alopecia (Bonomi, 2007). Interestingly, Phase III studies showed that the overall survival of patients treated with PG–paclitaxel did not differ significantly from patients treated with free paclitaxel. However, a subset of patients, specifically premenopausal women, responded more favorably to the drug conjugates, perhaps due to higher estrogen levels that correlate with cathepsin B activity. An application was submitted for marketing to the European Medicines Agency by Cell Therapeutics in 2008 for this formulation; PG–paclitaxel may therefore be the first polymer–anticancer drug conjugate to be marketed. Cell Therapeutics also investigated clinically a PG-camptothecin conjugate. However, development of this material was halted after Phase I/II studies.
Cyclodextrin Polymers as Drug Carriers
Cyclodextrins (Figure B.4.1C) are cyclic oligomers of glucose, and have been used as drug solubilizers in US Food and Drug Administration (FDA)-approved formulations. Linear polymers of cyclodextrin synthesized by condensation polymerization of bifunctionalized cyclodextrin monomers with a second comonomer have been investigated as drug carriers in clinical trials. The first material evaluated clinically by Insert Therapeutics is a cyclodextrin polymer–camptothecin conjugate (IT-101; Figure B.4.1C) (Davis, 2009a,b). Camptothecin was conjugated to the carboxylate groups of the cyclodextrin polymer through a glycine linker, resulting in a hydrolyzable ester bond (Cheng, 2003). IT-101 has molecular weight ~70 k, and is renally excreted. In aqueous solutions, IT-101 self-assembles into particles with sizes ~30–40 nm in diameter. Unlike the micellar structure depicted in Figure B.4.1, the self-assembly of IT-101 particles is likely driven by inclusion complex formation between the cyclodextrin and camptothecin molecules. Phase I clinical trials of IT-101 for treatment of advanced solid tumors showed long half-life of IT-101 in humans (~40 hours) and disease stabilization in several patients.
Polyacetals as Drug Carriers
A biodegradable polyacetal poly(1-hydroxymethylethylene hydroxymethylformal) (Figure B.4.1D) conjugated to camptothecin (XMT-1001) has also been investigated in clinical trials. This molecular carrier, under development by Mersana Therapeutics, delivers camptothecin via a two-phase drug release mechanism (Yurkovetskiy et al., 2004). A camptothecin derivative is first released from the polymeric backbone; this lipophilic derivative is active but not as potent as camptothecin. This derivative is then hydrolyzed into camptothecin. It is hypothesized that the dual-release approach may result in improved tumor delivery. XMT-1001 entered Phase I clinical trials in 2007.
Bibliography
1. Bonomi P. Paclitaxel poliglumex (PPx, CT-2103): Macromolecular medicine for advanced non-small-cell lung cancer. Exp Rev Anticancer Ther. 2007;7:415–422.
2. Cheng J, Khin KT, Jensen GS, Liu A, Davis ME. Synthesis of linear, β-cyclodextrin-based polymers and their camptothecin conjugates. Bioconj Chem 14 2003:1007–1017.
3. Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol Pharm. 2009a;6(3):659–668.
4. Davis ME. Design and development of IT-101, a cyclodextrin-containing polymer conjugate of camptothecin. Adv Drug Del Rev. 2009;61:1189–1192.
5. Davis FF. The origin of pegnology. Adv Drug Del Rev. 2002;54:457–458.
6. Duncan R. Development of HPMA copolymer-anticancer conjugates: Clinical experience and lessons learnt. Adv Drug Del Rev. 2009;61:1131–1148.
7. Kopecek J, Bazilova H. Poly[HPMA] 1 Radical polymerization and copolymerization. Eur Polym J. 1973;9:7–14.
8. Kopecek J, Ulbrich K, Vacik J, et al. Copolymers based on n-substituted acrylamides and methacrylamides, and n,n disubstituted acrylamides, and the method of their manufacturing. US Patent 4,062,831 1977; Dec 13, 1977.
9. Li C. Poly(L-glutamic acid)-anticancer drug conjugates. Adv Drug Del Rev. 2002;54:695–713.
10. Nowotnik DP, Cvitkovic E. ProLindac (AP5346): A review of the development of an HPMA DACH platinum polymer therapeutic. Adv Drug Del Rev. 2009;61:1214–1219.
11. Paz-Ares L, Ross H, O’Brien H, et al. Phase III trial comparing paclitaxel poliglumex vs docetaxel in the second-line treatment of non-small-cell lung cancer. Br J Cancer. 2008;98:1608–1613.
12. Ringsdorf H. Structure and properties of pharmacologically active polymers. J Polym Sci Polym Symp. 1975;51:135–153.
13. Yurkovetskiy AV, Hiller A, Syed S, et al. Synthesis of a macromolecular camptothecin conjugate with dual phase drug release. Mol Pharm. 2004;5:375–382.