Chapter III.1.2
Sterilization of Implants and Devices
Introduction
Successful sterilization of biomaterials used in implants and devices is a critical prerequisite for their successful clinical application. It requires knowledge of sterility concepts and sterilization technologies that render products sterile. Understanding the effect of sterilization processes on the biomaterials themselves is also increasingly important. These topics are the focus of this chapter.
Delivering a sterile product is the responsibility of a hospital, a manufacturer of an aseptically processed device or a manufacturer of a terminally sterilized device. This chapter is largely focused on the latter: industrial terminal sterilization. Equipping the biomaterials scientist to find a terminal sterilization solution for a product, if possible, and thereby avoiding aseptic processing of the product, is a desired outcome of the chapter.
The current regulatory expectation of the term “sterile” for blood-contacting medical devices and implants is to produce only one non-sterile device out of one million. If you think about it, this is an extreme target. Fortunately, it is normally attainable, and clearly a patient heading to the hospital appreciates this high safety target. The patient, however, would cease to be pleased if that sterile device failed during treatment because the sterilization cycle was so rigorous in killing microbes that it also damaged the product. The challenge for the biomaterials scientist responsible for defining a terminal sterilization process is an optimization problem – to determine and define a cost effective sterilization process window where sterility is achieved, and yet deleterious material effects are minimized. Many traditional medical devices are made with materials that are compatible with multiple sterilization modalities. In this scenario, finding a terminal sterilization solution is straightforward. However, the optimization problem is much more complex for biologics and combination devices, both of which are important and rapidly growing markets.
To navigate these challenges, it is useful for the biomaterials scientist to have a concrete understanding of common terminal sterilization technologies (see below, Sterilization Technologies), which are the basis of both achieving sterility and any product material effects. Material compatibility constraints often drive the choice of sterilization modality. The next section of the chapter, therefore, provides an overview of sterilization material compatibility challenges and guidance (see below, Material Compatibility). Robust sterilization validation methods embodied in national and international standards are the foundation for the strong patient safety record of terminal sterilization. However, some materials, especially biologics, may have undesirable responses to the techniques used, and then may require special sterilization methods. Sterility assurance concepts are, therefore, reviewed in sufficient detail to give the biomaterials scientist confidence in evaluating all options for finding a sterilization solution (see below, Sterility and Patient Safety). The chapter closes with a view toward exciting challenges on the terminal sterilization horizon. The combination device market offers compelling patient benefits, and equally compelling challenges for terminal sterilization. Awareness of these challenges and knowledge to meet them will serve the biomaterials scientist well (see below, Summary and Future Challenges).
Sterilization Technologies
How is microbial contamination on fully packaged devices reduced by 9, 12 or even greater orders of magnitude? This section provides a concrete understanding of industrial sterilization technologies that deliver these enormous microbial reduction levels. Two terminal sterilization modalities, radiation sterilization and ethylene oxide sterilization, dominate the industrial (non-hospital) terminal sterilization market. These robust workhorse terminal sterilization technologies utilize well-established validation processes (see below, Sterility and Patient Safety) that, in most instances, far exceed regulatory requirements for a one in a million sterility assurance level. Under well-controlled and validated conditions, neither of these modalities inflicts significant material degradation damage on many traditional devices. In addition, both are capable of processing high volumes of product at low cost. These important technologies are reviewed in some detail, along with a brief overview of a number of other methodologies that are less frequently utilized.
Radiation Sterilization
Safety First
Radiation sterilization doses are lethal. Terminal sterilization doses typically range from 8 to 35 kGy. An acute lethal dose to man is approximately 0.01 kGy, requiring an exposure time of only a fraction of a second in some processes. To ensure worker safety in these high radiation environments, significant shielding, robust interlocks, and the utmost care are required in radiation processing facilities. Another safety concern is exposure to ozone, a toxic gas, produced by radiation as it passes through air. Appropriate ventilation and ozone monitors are required to provide fresh air, and ensure the safety of personnel prior to entering a processing cell.
Technology Overview
Terminal sterilization by radiation sterilization is elegantly simple. Fully packaged medical devices are exposed to a validated dose from a radiation source that emits electrons or photons that penetrate through the final packaging and inactivate the device’s microbial load. One parameter, radiation dose, correlates directly with microbial kill, and this is easily measured to provide process control. The microbial kill mechanism involves radiation-induced scission of DNA chains to stop microbial reproduction. While radiation destroys the ability of most microorganisms to reproduce, the resistance of viruses such as HIV-1 remains a concern (Smith et al., 2001).
International radiation sterilization standards (ISO 11137-1; see Table III.1.2.1) call out three radiation sterilization modalities: gamma, electron beam, and X-ray. Gamma and electron beam dominate the radiation sterilization market, and are described in enough detail below to provide a working knowledge of the technologies. Sources of the radiation, processing facility layouts, mechanism of radiation energy deposition into materials, and key processing parameters are described. Cycle times range from minutes to half a day. Technologies available for R&D, pilot, and low volume processing are also described. These small-scale systems will play an increasingly important role in qualifying materials and providing special processes to meet demanding material compatibility needs of combination devices.
TABLE III.1.2.1 Sterilization Standards

AAMI: Association for the Advancement of Medical Instrumentation
ANSI:American National Standards Institute
EU: European Standard
EU GMP: European Union Good Manufacturing Practice
ISO: International Organization for Standardization
Gamma Sterilization
Cobalt-60 gamma sterilization accounts for approximately 80% of the radiation sterilization market, 40% of the overall terminal sterilization industrial market. 60Co is a radioactive element that undergoes nuclear decay producing useful gamma radiation. Note: while the gamma source itself is radioactive, the product sterilized by the exposure to the radiation does not become radioactive in the slightest measure.
High product volume 60Co processing plants are relatively simple. Small 60Co pellets are doubly encapsulated in rods, which are arranged into racks and stored in a pool of water, with some 20 feet of water acting as shielding to keep the constantly emitted gamma radiation away from the processing room when product is not being processed. When the room is cleared, interlocks satisfied, and the facility is ready to process product, a mechanical elevator system raises the radiation source from the pool into the room. A conveyor system moves many totes of fully packaged product into the room and around the racks of 60Co, often passing by multiple times, as seen in Figure III.1.2.1.
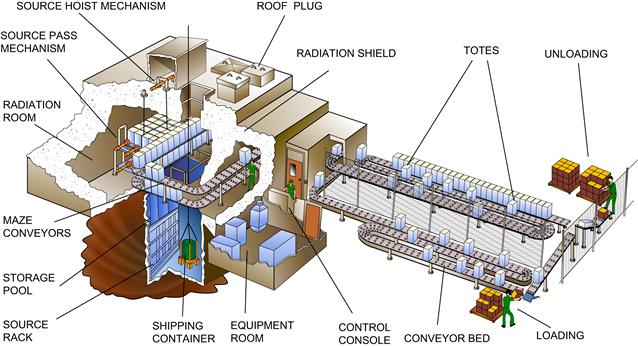
FIGURE III.1.2.1 High volume production gamma irradiation facility
(courtesy of MDS Nordion, Canada).
60Co gamma particles have no charge and penetrate uniformly through a very long distance of material. This is a significant processing benefit. Configurations of product within shipper boxes and processing load configurations are rarely a limitation due to the high gamma penetration. Product is then brought back out of the processing room. Product is released for distribution after dosimeters are read and documented to confirm that the product received the proper dose.
The beneficial sterilization effects of 60Co gamma rays (along with any deleterious material effects) come as a result of the energy of the photons being deposited into the microbes and materials. By definition, the energy deposited per unit weight is the radiation dose (typical units are kJ/kg, i.e., kilogray or kGy). Energy deposition from a gamma ray into a material occurs through a Compton Scattering interaction that generates high energy electrons (0.5 MeV). These primary electrons travel through the material and deposit their energy in 60–100 electron volt bundles as they generate secondary electrons. It is these secondary electrons that cause all microbial kill. They do so by forming oxidizing free radicals. In addition to causing scission of DNA, the radicals can also cause scission or crosslinking of polymer chains (see Material Compatibility).
Electron Beam Sterilization
Electron beam (e-beam) sterilization accounts for approximately 20% of the radiation sterilization market, 10% of the overall terminal sterilization industrial market. High energy electrons for e-beam sterilization are generated by a variety of technologies that take 200 volt electrons from the power grid and accelerate them to 0.2 million to 10 million electron volts (0.2–10 MeV, typically 5–10 MeV). At these energy levels, no part of the e-beam processing plant (or the product irradiated) is or becomes radioactive; e-beam accelerators turn off like a light bulb. Before turning on the accelerator, however, the processing room must be cleared and safety interlocks satisfied to avoid human exposure to the high doses of radiation and ozone that are generated. Accelerated electrons for device sterilization are typically magnetically focused into a 1–5 cm diameter beam, and magnetically scanned at high frequency across the conveyor width, typically 12–48 inches.
The conveyor system transports product through the shielding and in front of the beam. Typically, one product, one shipper box or one thin tote of product at a time passes by the beam’s scan horn. Electrons from accelerators do not penetrate nearly as far as photons from gamma sources. A rule of thumb is that the maximum penetration (in centimeters) from a two-sided e-beam process is 0.8 times the beam energy (in MeV) divided by the density of product (in g/cm3). Resulting maximum penetration distances are only a fraction of a meter, with higher beam energies resulting in higher penetration. Limited penetration can lead to inefficient processing and/or large distributions of delivered dose. In practice, this requires significant dosimetry work to develop load configurations that ensure an appropriate range of e-beam doses. Due to the limited penetration of accelerated electrons, products need to be carefully “dose-mapped,” as their size and shape provide shielding, scattering interfaces, and incident angles which impact dose distribution. These differences provide a less homogeneous dose profile than that of 60Co.
Product is then brought back out of the processing room. Product is released for distribution after dosimeters are read and documented to confirm that the product received the proper dose. E-beam processing facilities can either be in a processing room as described above for a gamma facility or be of a self-shielded design, as shown in Figure III.1.2.2.

FIGURE III.1.2.2 Medium volume self-shielded production electron beam irradiation facility.
The mechanism of energy deposition from e-beams is that the high-energy electrons journey through the material and deposit their energy in 60–100 electron volt bundles as they generate secondary electrons. Like gamma processing, the resulting secondary electrons cause microbial kill, as well as all product material effects. The main advantage of e-beam relative to gamma in terms of material compatibility is that the dose is delivered very quickly. Products being treated in gamma sterilization processes often have dwell times of several hours, whereas products typically dwell in an e-beam process for only minutes. The short processing time limits oxidative degradation, since oxygen does not have time to diffuse into the product. The main disadvantage of e-beams versus 60Co is that the negatively charged electrons do not penetrate deeply into a material compared with neutral gamma rays, which penetrate through the product being sterilized.
X-ray Sterilization
X-ray sterilization accounts for a small fraction of the radiation sterilization market. It is a hybrid between gamma sterilization and e-beam sterilization. Radiation is generated from high-energy electrons from accelerators, typically 5 MeV electrons, impinging on a high atomic number (“z”) target, often tungsten, to generate X-rays. The X-ray photons behave nearly identically to photons from gamma sources in terms of energy deposition and high penetration capabilities. Like e-beam plants, no part of the X-ray processing plant is or becomes radioactive if X-rays are generated by electrons with energies no greater than 5 MeV.
Important Processing Parameters
Radiation sterilization processing parameters that are of practical importance to the biomaterials scientist are those that affect the maximum dose to the product, and those that affect the throughput of product through the process. The maximum dose to the product is dependent on:
• sterilization dose (minimum dose to the product);
• dose uniformity during the radiation sterilization process; and
• number of sterile cycles for which the device will be qualified.
Dose uniformity ratio (DUR) is a measure of the range of doses delivered to product by a given radiation sterilization process. DUR is defined, for a given load configuration and density, as the ratio of the maximum to the minimum dose received by any one product. The DUR of a radiation sterilization process is always greater than 1.0; radiation sterilization does not provide a single dose, but a distribution of doses delivered throughout any one product. Dose uniformity is dependent on the radiation source (gamma is typically significantly more uniform than e-beam), product density, product shipper box or pallet size, sterilizer configuration, and for e-beam, energy of the electrons. High density products with large boxes, resulting in large distances through which the radiation must travel, result in a high distribution of doses, e.g., a DUR greater than 2.
Throughput for a radiation sterilization process is primarily dependent on the radiation source intensity, i.e., for gamma sterilizers the number of curies of 60Co, and for e-beam and X-ray sterilizers the number of electrons, the current. Secondarily, throughput is a function of product density and quantity of product processed at a given time. For example, while it may be beneficial to reduce the quantity of product processed at a given time to reduce the dose distribution, and hence the overall dose to product, this change also reduces throughput efficiency.
R&D, Pilot, and Low Volume Technologies
Small radiation sources are very powerful tools for the biomaterial scientist and his/her team. An R&D gamma cell like that shown in Figure III.1.2.3 provides a narrow dose range, very close to a DUR of 1.0. Along with delivering precise doses for the sterilization validation process (see below, Sterility and Patient Safety), the biomaterials scientist will be well-served by these narrow dose ranges during material qualification exercises, especially if the source is easily accessible to provide fast iterations during the product development process.

FIGURE III.1.2.3 Low volume self-contained R&D gamma cell manufactured by MDS Nordion
(courtesy of IAEA, STI/PUB/1313).
In addition, small self-shielded low energy e-beam accelerators are available to process single units. The low energy of the beam, typically 0.5–2.0 MeV, may only penetrate a single product or a sample portion of a product at a time. This low energy keeps the accelerator to a manageable size and cost, and allows for rapid product development iterations.
Ethylene Oxide (EO) Technologies
Safety First
Ethylene oxide gas is toxic, carcinogenic, and very explosive. EO sterilization cycles must be designed to avoid conditions or parameters that may be explosive. Precautions must be taken to avoid any equipment, tools, etc., that may spark in the vicinity of EO chambers. Explosions can be horrific in magnitude, and extreme care must be taken. After an EO process is complete, employees must take appropriate precautions to avoid exposure to EO gas, e.g., use of respirators when working in the vicinity of product that has not been fully aerated to dissipate the gas. Finally, the manufacturer of EO sterilized product is responsible for following international standards to ensure that product is allowed to aerate in order to get EO residual levels below permissible limits for distribution (ISO 10993-7; see Table III.1.2.1).
Technology Overview
Ethylene oxide sterilization accounts for approximately 50% of the industrial terminal sterilization market, and is another conceptually simple terminal sterilization process. Fully packaged devices are placed into an ethylene oxide chamber and exposed to a validated combination of humidity, ethylene oxide gas, temperature, and time. Microbial-resistant sterile barrier packaging must be used, to allow ingress and egress of EO and water vapor within a defined load configuration. Following the sterilization process, EO levels are brought below acceptable levels through completion of a validated in-chamber vacuum purge process or a post-sterilization aeration process. Product is released for distribution following review and documentation of routine monitoring parameters and, in many instances, biologic indicator test results. Total cycle times range from six hours to several days.
The international ethylene oxide sterilization standard, ISO 11135-1 (see Table III.1.2.1) refers to the use of both ethylene oxide, and mixtures of ethylene oxide and a diluent. Diluents were commonly used as a deterrent to explosions, but are less common today since many common diluents are HCFCs and their use has been curtailed. Instead, nitrogen blanketing and other cycle design features are used to reduce the presence of air/oxygen and keep EO non-flammable. Diluents do not affect the principles of operation and validation of an EO cycle.
EO sterilization principles and practical aspects are described below in enough detail to provide a working knowledge of the technology. Chamber dynamics, processing plant layout, mechanism of microbial kill, and key processing parameters are described. Cycle times range from several hours to several days. Technologies available for R&D, pilot, and low volume processing are also described.
Ethylene oxide is a highly reactive cyclic ether with two carbons and one oxygen, CH2CH2O (Figure III.1.2.4). It is a gas at room temperature, with a boiling point of 11°C. It is pressurized and stored as a liquid for use in EO processing plants. The mechanism of microbial kill is alkylation of the amine groups of DNA. Moisture facilitates microbial kill; product has to be exposed to a humid environment before EO exposure. EO kill rate is a function of temperature and concentration of EO gas.

FIGURE III.1.2.4 Ethylene oxide molecule.
In practice, approximately three cubic meters of product is placed on pallets in well-defined configurations. Product is humidified, either in a dedicated preconditioning area (typically 6–24 hours at 50–80% relative humidity (RH) and mild temperatures) or within the EO processing chamber with dynamic vacuum/steam pulsing. One to 40 pallets of product are placed into industrial EO chambers. A three-pallet chamber is shown in Figure III.1.2.5.

FIGURE III.1.2.5 Medium volume ethylene oxide production sterilization chamber manufactured by Getinge
(Courtesy of Getinge Inc.).
Along with humidification, early phases often include evacuation to remove oxygen, and injection of nitrogen to keep the cycle non-explosive. Evacuation levels can be down to 2 torr in some cycles, with very fast evacuation rates. EO gas is injected into the chamber with the humidified product, and allowed to dwell for a validated period of time, EO concentration, temperature, and humidity. Typical in-chamber exposure times are 6–24 hours at processing temperatures of 40–65°C and RH of 30–90%. The EO is then purged, for example, with dynamic vacuum/steam pulsing followed by vacuum/nitrogen pulsing. If EO residual levels do not meet regulatory requirements for product release at the end of the cycle, product is placed into a dedicated forced air heated aeration room.
Important Processing Parameters
Ethylene oxide sterilization processing parameters that are of practical importance to the biomaterials scientist are those that can affect the product and package, and those that affect throughput. Deep vacuum levels, rate of vacuum cycles, chamber temperature and concentration, and time of exposure to humidity, EO gas, and any diluents utilized can all potentially affect packaging and/or device material components, especially pharmaceuticals and biologics. The size of the chamber, the length of the cycle, and any preconditioning and/or aeration steps affect cycle times and therefore product throughput.
R&D, Pilot, and Low Volume Technologies
Small ethylene oxide sterilizers are very powerful tools for the biomaterials scientist and his team. As well as providing a vehicle for well-controlled microbial kill challenge testing, they can be used to facilitate finding material compatibility solutions and qualifying product at most challenging parameters. Special low temperature and/or low humidity EO cycles can be developed and validated to avoid material compatibility concerns. A well-controlled eight cubic foot R&D EO chamber is shown in Figure III.1.2.6. Small EO chambers are also used extensively within the hospital setting.

FIGURE III.1.2.6 Low volume ethylene oxide R&D sterilization chamber
(Courtesy of Getinge Inc.).
Other Terminal Sterilization Technologies
Moist heat, i.e., steam, sterilization has long been a workhorse technology for reusable devices in hospitals and certain industrial applications. The high temperatures limit its use for most devices with plastic materials, including most single-use devices. Moist heat sterilization, like other major industrial and hospital processes, is well-characterized in national and international sterilization standards (see Table III.1.2.1). Gas phase hydrogen peroxide sterilization has, in the last decade, come into broad application in hospitals, but has limited industrial applications. This was the first technology to drive the generic ISO 14937 “General Criteria” sterilization standard (see Table III.1.2.1). A number of additional gas chemical sterilization technologies have been developed, but have not found significant industrial terminal sterilization application, including chlorine dioxide, ozone, nitrogen dioxide, supercritical carbon dioxide, and propylene oxide. In addition, dry heat sterilization has limited applications.
Liquid chemical sterilization is sometimes used for sterilization, in applications either with animal or human tissues that are not compatible with terminal sterilization options or as a high level disinfection for reuse of certain devices, such as blood dializers. The process involves the immersion of the device into formulations of either an aldehyde or an oxidizing agent (e.g., gluteraldehyde, hydrogen peroxide or peracetic acid), sometimes with buffers, anti-corrosive agents, and detergents. The method does not provide the process control or sterility assurance levels of terminal sterilization processes (Chamberlain et al., 1999). Automation of the process, however, has led to significant success in providing safe tissue product.
Material Compatibility
The attention of this chapter will now shift to the response of the materials being terminally sterilized, especially the molecular effects which in turn can affect physical, chemical, and mechanical properties of the material. Specific guidance is provided for the two major industrial sterilization technologies: radiation and EO. Many of the principles and processes outlined, however, are applicable to all sterilization technologies. In addition, there are material compatibility resources available in the literature for some of the less utilized technologies, for example, for hydrogen peroxide and ozone (AAMI, 2008), nitrogen dioxide (Kulla et al., 2009), and supercritical carbon dioxide (White et al., 2006).
To reap the high patient safety benefits of robust terminal sterilization technologies, the biomaterials scientist needs to manage potential material compatibility concerns. This is necessary in order to ensure a high quality product, and to claim compliance to international sterilization standards that require assessment of material compatibility with terminal sterilization.
Requirements in Sterilization Standards to Assess Material Compatibility
International sterilization standards require the assessment of material compatibility under worst case sterilization conditions. For radiation sterilization, ISO 11137-1 (see Table III.1.2.1) requires the following:
8.1.1 When treated with the maximum acceptable dose, product shall meet its specified functional requirements throughout its defined lifetime.
For ethylene oxide sterilization, ISO 11135-1: 2006 (see Table III.1.2.1) requires the following:
7.2.1 It shall be confirmed that the product and its packaging meet specified requirements for safety, quality, and performance following the application of the defined sterilization process at the most challenging process parameters for the product/package. The influence of the tolerances for the process parameters shall be taken into consideration.
Effect of Terminal Sterilization Modalities on Materials
Knowledge of a sterilization modality’s processing parameters and how they interact with materials (see above, Sterilization Technologies) is a key element in assessing the compatibility of materials with a given terminal sterilization process. A summary of material effects with six sterilization modalities is given in AAMI TIR17 (see Table III.1.2.1); radiation and ethylene oxide sterilization are reviewed below.
Radiation Sterilization
Radiation chemistry initiated by secondary electrons (see above, Sterilization Technologies) produces material effects during radiation sterilization. These effects include generation of free radicals that can cause polymer chain scission or cross-linking, as well as interaction with oxygen that can do further damage to the synthetic polymer or biologic product being sterilized. The radiation chemistry of polymeric materials and industrial applications have been studied and extensively reviewed (e.g., Forsythe and Hill, 2000; Clough, 2001; AAMI, 2008). Many device materials are essentially inert to the relatively low doses applied during radiation sterilization. Some of these materials are actually cross-linked by the radiation, e.g., polyethylene (PE), with a neutral or positive effect on functional properties. Some polymers of medical device interest, however, tend toward scission and are mechanically degraded to a significant extent following radiation sterilization, in particular polytetrafluoroethylene (PTFE), fluorinated ethylene propylene (FEP), polyacetals, and natural polypropylene (PP). In some cases, the presence of air can accelerate chain scission via a radiation-induced oxidation process. Degrading materials may be acceptable in certain applications, but must be evaluated carefully after exposure to a maximum dose to ensure clinically acceptable performance over the shelf-life of the device. In glassy polymers like polymethylmethacrylate (PMMA), low energy electrons may be trapped within the polymer, causing yellow or brownish discoloration, a challenge for radiation sterilization of contact lenses. Also, in cross-linking materials like PE (UHMWPE [Ultra High Molecular Weight Polyethylene] in orthopedic implants) it may be important to anneal out the trapped free radicals, to avoid undesirable, long-term free radical-induced reactions. Bioabsorbable polymers, e.g., polylactide (PLA) and PLGA or poly(lactic-co-glycolic acid), show significant molecular weight (MW) reduction as a function of sterilization dose. This change in initial MW of the polymer may affect in vivo polymer degradation dynamics, leading to a change in device performance, such as drug release kinetics from degradable PLGA microparticles. In special cases, it may be possible to account for this MW decrease by increasing the initial pre-sterile molecular weight. In such cases, radiation would be the sterilization modality of choice, since irradiations can be done in a refrigerated state to maintain structural integrity by keeping well below the glass transition temperature of the polymer.
Secondary effects in material may arise from or be avoided by managing the radiation sterilization process. The temperature of a device will be elevated during irradiation, for a few seconds for e-beam sterilization (typically to 50°C with polymeric materials) or for a few hours for gamma sterilization (typically 30–40°C). For low glass temperature bioabsorbable materials, this may be significant. If so, it can be managed by processing the device at a reduced temperature. If oxygen and humidity contribute to deleterious material interactions with radiation, the device can be packaged in an inert packaging environment to manage the problem.
Biologics and Human-Based Tissue: Compatibility with Radiation Sterilization.
Progress has been made achieving compatibility of tissue products with radiation sterilization. Sterilization process concerns are addressed in AAMI TIR37: Sterilization of health care products – Radiation – Guidance on sterilization of human tissue-based products (see Table III.1.2.1). Low dose radiation sterilization validation methods (see Sterility and Patient Safety) offer promise of expanded use of radiation sterilization with tissues and other biologic products. There are also other interesting cases where biologics are terminally sterilized with radiation after special processing to produce sterile biologics, e.g., sterile liposomal vaccines and human insulin. Special processing may include lyophilization, cryoprotection, and addition of free radical scavengers or radio-protectors (e.g., the anti-oxidant, ascorbic acid) (Mohammed et al., 2006; Terryn et al., 2007). Processes have also been developed using radical scavengers and other tools to improve radiation compatibility of tissue products. These developments offer promise for the expanding market of tissue engineering scaffolds.
Ethylene Oxide Sterilization
Alkylation chemistry, the microbial kill mechanism for DNA, may produce other material effects, but in general the effects are small. Other EO process parameters, i.e., temperature, humidity, and evacuation cycles, can also affect device materials. Materials must be able to withstand the most challenging range of conditions used in a given EO cycle. As described previously, this may include humidity preconditioning before, and aeration cycles afterwards, for several days at 40°C; EO cycles for 6–24 hours at processing temperatures of 40–65°C; RH in the range of 30% to 90%; and evacuation levels down to 2 torr in some cycles with very fast evacuation rates.
Bioabsorbable polymers may be difficult to process under these conditions, in particular if structure integrity is required. Both the temperature and the humidity can degrade bioabsorable material properties. Packaging materials need to be able to withstand the evacuation rates and pressures. Materials with known deleterious responses to typical single EO sterilization cycles include polyacrylates, e.g., polymethylmethacrylate (PMMA), and some styrene resins, e.g., polystyrene (PS) and styrene acrylonitrile (SAN). These materials may be acceptable in certain applications, but must be evaluated carefully after exposure to worse case cycle (“most challenging parameters”) to ensure clinically acceptable performance over the shelf-life of the device. It is not expected that EO sterilization will affect device performance over time.
Pharmaceuticals and Biologics: Compatibility with EO Sterilization.
In the pharmaceutical/biologics and human-based tissue industries, the issue of sterilization can take on a variety of special conditions and concerns. In the pharmaceutical industry, the predominant use of EO (and radiation) sterilization is used for packaging components that enter the aseptic processes (bottles, plugs, caps). EO is also used to sterilize some dry pharmaceutical active components. Liquids and temperature-sensitive drugs will likely not be compatible. Few biologics are compatible with EO sterilization due to concerns regarding temperature and EO residues, as well as the ability for EO to alkylate chemically reactive species such as amines or proteins. This is analogous to the reaction of EO with DNA to sterilize product.
Other Sterilization Modalities
For heat sterilization modalities, material compatibility challenges arise from the temperatures to which the devices are exposed and, for moist heat, from hydrolysis reactions. For oxidative sterilization technologies, e.g., hydrogen peroxide, ozone, and chlorine dioxide, the oxidative mechanism of microbial reduction is also the mechanism by which devices are damaged.
Optimizing Chances for Finding Material Compatibility Solutions
Starting with the best candidate materials is the surest way to optimize material compatibility with terminal sterilization. Guidance on selecting material compatible with six terminal sterilization modalities is available (AAMI, 2008). The most challenging material selection scenario is the combination of a difficult-to-sterilize primary material, which is central to the clinical use of the device, with secondary materials that are also sensitive to sterilization, e.g., an active agent (pharmaceutical or biologic), a bioabsorbable material, and/or active electronics. The best approach is to select a sterilization modality that is compatible with the primary material, and then try to select secondary materials that are compatible with the selected sterilization modality.
It is also important to avoid material processing errors that lead to performance failures caused by sterilization. Both extrusion and molding processes can build in significant polymer stresses, leading to product testing failures post-sterilization. This can be identified by testing product pre-sterilization as a control to compare with post-sterilization testing.
Choosing or developing clinically relevant test methods is also important. It can be tempting to use easily available test methods to demonstrate material performance before and after sterilization. If the test method does not correlate well with clinical performance, the test may not be an appropriate indicator of material compatibility. Clinically-relevant device attributes and test methods vary enormously depending on the device. Physical, analytical, and microbiological tests need to be considered. To characterize drug release kinetics and other combination device properties, micro- and nano-characterization of drug–polymer matrices may be required (Ding et al., 2009). Material biocompatibility can change as a function of sterilization modality and process. Device endotoxin levels are typically not sensitive to the sterilization modality, and testing may often be performed before or after sterilization. The effect of sterilization on the surface of biomaterials should also be considered. Radiation typically has a minimal effect on biomaterial surfaces, while EO, oxidative gases, and liquid chemical sterilants can have either a positive or negative surface effect depending on the application of the biomaterial (Chamberlain et al., 1999).
Ensuring adequate device performance throughout its intended shelf-life is the final material compatibility challenge for the biomedical scientist. The most conservative route is to age the materials in the final product format at room temperature, and confirm the desired functionality throughout the desired shelf-life. If the product is designed to be stored in refrigerator or freezer conditions, this approach can require exceptionally long development timeframes, and can limit the availability of important technologies to the market. As a result, accelerated aging models have been developed to safely and conservatively estimate device performance over time. The cornerstone of all models, however, is confirmation of the model with aging at storage conditions.
Arrhenius (semi-logarithmic) temperature-based accelerated aging models have been used extensively in the medical device industry for product (AAMI, 2008) and packaging (ASTM, 2007). Practical methods have been developed that can accelerate aging by two-fold to greater than ten-fold. Basic conservative aging models that are simple and cost-effective have been shown to be appropriate for most materials (Lambert and Tang, 2000). With additional resources, more sophisticated correlations between accelerated and real-time conditions can be developed to provide more aggressive aging factors.
For devices combined with pharmaceutical materials, regulatory agencies expect to have accelerated stability protocols based on ICH (International Conference on Harmonisation) guidelines. Distinguishing between accelerated device-aging models and pharmaceutical stability requirements is both challenging and important.
Sterility and Patient Safety
For a device with significant sterilization material compatibility challenges, a robust understanding of sterility concepts and associated patient safety issues may be the key to discovering a cost-effective sterilization solution. At one end of the spectrum of sterilization options is overkill terminal sterilization. This approach has for decades served the industry well for devices with limited material compatibility concerns. The other end of the spectrum includes liquid chemical sterilization of biologics, and aseptic processing of combination devices, relatively costly options from many perspectives. Between these extremes are creative terminal sterilization validation methodologies, and terminal sterilization to non-traditional specifications. Without the ability to skillfully navigate this continuum of options, the biomaterials scientist may not be able to cost-effectively bring a product to market.
This section begins with a review of sterilization-related product and patient safety issues. This is followed by an overview of basic terminal sterilization principles related to logarithmic microbial reduction, validation of sterility, and maintaining sterility over time through sterile barrier packaging. The end of the section reviews efforts to provide creative sterilization validation methods to reduce material compatibility concerns, and differentiates terminal sterilization from sterilization achieved by aseptic processing.
Product and Patient Safety Issues
Review of the US Food and Drug Administration (FDA) product recalls and the Center for Disease Control (CDC) publications indicates very few instances related to inadequate industry terminal sterilization practices (Favero, 2001). Therefore, it appears the existing standards and medical device manufacturers, and associated contractors, are providing safe and effective devices in terms of product sterility. In the following case studies we will take a look at where the industry safety concerns may lie by examining a few sterilization-related incidents with subsequent patient infections or injury.
Three case studies described here in the text illustrate how small changes in product design and/or the materials used, in combination with the sterilization protocols used, caused significant medical problems. The cases are examples which had significant patient safety issues. Case 1 involved a highly respected major device and pharmaceutical manufacturer utilizing a moist heat terminal sterilization process with cooling water. Case 2 is from a medical supply company totally unaware of the requirements related to the GMP (Good Manufacturing Practice) and aseptically processed products or drugs. Case 3 highlights an instance where a sterilizer and sterilization technology was sold to hospitals without regulatory approval. Sterilization technologies need to be evaluated by the manufacturers as being suitable for their devices and the materials used. Hospitals should not use sterilization technologies that are not indicated in the device manufacturer’s directions for use. These instances, although significant, are relatively rare.
Case 1
From October 1970 until March 1, 1971, eight US hospitals encountered 150 cases of bacteremias associated with Enterobacter cloacae or Erwinia, a genus of Enterobacteriaceae which is gram-negative. All hospitals had used the same intraveneous (IV) fluids provided by a major manufacturer. The CDC studies for IV systems indicate that at least 6% of all IV tubing or bottles are contaminated after the system has been in use. A higher risk of contamination is attributable to systems that are not changed after 48 hours. However, in these cases the incidence was much higher. CDC sampling and culturing of fluids directly indicated no contamination. Bottles that were opened by unscrewing the bottle cap and closing again revealed contamination. After further investigation, it was determined that the manufacturer was in the process of implementing a new closure system and that the contamination was associated with the new cap design. The older cap design contained a red rubber disc and a Glisonite wafer which was pressed against the bottle opening. It was discovered that the red rubber had antibacterial properties against the contaminating organisms. The new cap design allowed these organisms to be drawn up into the lined cap while cooling after being autoclaved, but the red rubber had been eliminated and no longer provided an antibacterial effect. The organisms were prevalent in the manufacturing environment due to spillage of solutions during manufacturing. Recalls were initiated for all affected products. Ulitimately, the epidemiological studies estimated the total outbreak of bloodstream infections to be between 2000 and 8000 episodes caused by these contaminated IV fluids. Approximately 10% of the case patients in the hospitals studies died while bacteremic or shortly thereafter (CDC, 1997).
Case 2
From December 12, 2004 until February 15, 2005, local health departments and the CDC identified 36 cases of Pseudomonas fluorescens infections in patients that were previously administered a heparin-saline flush from prefilled syringes. The majority of the patients had central venous catheters and lines which required removal and treatment with antibiotics. Prefilled syringes cultured by a few of the hospitals involved all grew the contaminating organism. In almost all cases blood cultures from the patients grew Pseudomonas fluorescens. The investigation performed by the CDC indicated that seven of nine product lots tested were contaminated. The company filling the syringes had contracted a pharmacy to prepare a heparin concentrate which was returned to the manufacturer and then added to saline bags, and was subsequently used to fill the syringes. Although preparation of drug products to fill prescriptions is considered pharmaceutical compounding and not manufacturing, in this instance the supplier was acting like a manufacturer and subject to the FDA Good Manufacturing Practices. This manufacturer did not perform final product sterility testing of the finished products, which could have detected the contamination and prevented the product distribution (CDC, 2005).
Case 3
April 13, 1998, the FDA issued a safety alert regarding the use of the AbTox Plazlyte™ sterilization system. The Plazlyte™ sterilizers utilized a proprietary, low temperature gas plasma, along with vaporized peracetic acid. Although the system provided sufficient lethality, the FDA had not cleared the safety, performance or instructions for use of this sterilizer. The sterilizers were sold directly to hospitals seeking alternatives for ethylene oxide sterilizers. The warning was initiated due to serious eye injuries to corneal endothelial cells which resulted in corneal transplantation in some patients. The problem occurred when surgical instruments were sterilized, and copper and zinc salts formed on the surfaces of the sterile instruments. The copper compound residues were toxic to corneal endothelia and resulted in blindness (FDA, 1998). During the time period of January 8–14, 1998, six of eight patients undergoing intraocular surgery incurred corneal edema and opacification of the cornea (CDC, 1998).
The majority of patient safety issues do not come from non-sterile, aseptically-processed product, or from improperly sterilized products, regardless of the delivered Sterility Assurance Level (SAL). Patient safety issues develop from the introduction of foreign materials into the body during patient treatment at hospitals. There is a relatively high incidence of surgical site infections (SSI), estimated to be at 2% for all procedures. In 2002 there were approximately 1.7 million hospital-acquired infections (HAIs). Of these, 22% were urinary tract infections, 14% were bloodstream infections, 15% were pneumonia, 22% were surgical site infections, and 17% were related to other issues (Klevens et al., 2007).
As can be seen from these examples, patient safety goes well beyond the ability to provide a sterile product. Nonetheless, the validation of product sterility is an absolute must, and part of the regulatory submission and approval process. Principles of terminal sterilization validation are the next topics covered.
Terminal Sterilization Validation Principles
Logarithmic Microbial Reduction
An amazing feature of terminal sterilization processes is the ability to enormously reduce microbial contamination levels on fully packaged product, with exceptionally high process control to meet regulatory requirements for sterility. The word sterile is defined as “free from viable microorganisms.” Sterilization is defined as a “validated process used to render product free from viable microorganisms” (ISO/TS 11139, 2006). These definitions are problematic, as they imply an absolute condition. Sterilization processes are based on microbial inactivation which is exponential in nature, in most cases, and follows first order kinetics. Therefore, the sterility of a product is expressed in terms of probability. While the probability can be reduced to a very low number, it can never be reduced to zero. The probability of a non-sterile unit associated with inactivation of a microbial population is quantified by the term Sterility Assurance Level (SAL).
Logarithmic microbial reduction to achieve various sterility assurance levels is illustrated in Figure III.1.2.7. The SAL required for regulatory purposes is 10−3 or 10−6, the probability of one in one thousand units as non-sterile or one in one million units as non-sterile, respectively. The SAL of 10−6 is a lesser number, but provides a greater assurance of sterility.

FIGURE III.1.2.7 Sterility Assurance Levels: Illustration of logarithmic microbial reduction.
Sterilization Validations and Consensus Standards
If at all possible, regulators require a terminal sterilization process over the risk associated with aseptic processing. The strong track record of terminally sterilized product in regards to patient safety owes much to robust sterilization validation methods. It is, therefore, important to understand the basic principles surrounding terminal sterilization validations. The terminology of international sterilization validation standards is provided in this section to equip the biomaterials scientist to interface effectively with regulatory and sterilization vendor personnel.
Sterilization is a special process where the results of the processing cannot be demonstrated by routine testing. For instance, in order to demonstrate an SAL of 10−6 it would require the sterility testing of approximately one million products with only one positive. This is neither possible nor practical. Therefore, sterilization processes are validated. EN 556-1 2001 indicates the following:
Evidence that a medical device is sterile comes from: (1) the initial validation of the sterilization process and subsequent revalidations that demonstrate the acceptability of the process; and (2) information gathered during routine control and monitoring which demonstrates that the validated process has been delivered in practice.
The achievement of sterility is predicted from the bioburden level on products, the resistance of the micro-organisms comprising that bioburden, and the extent of treatment imposed during sterilization.
How are these sterilization validations attained? The medical device and pharmaceutical industries, in conjunction with regulatory authorities, have cooperatively developed industry standards. Device manufacturers, contract sterilization providers, regulatory authorities, and academic resources are all part of the process of developing a consensus standard. These standards provide requirements and guidance for the performance of the sterilization validation. Table III.1.2.1 provides a listing of standards documents utilized for validating and controlling a particular sterilization process.
Sterilization equipment and the process must be qualified and validated. The following paragraphs provide the general steps outlined in the standards for the validation of the sterilization process. Terminology used in the standards is introduced as it is recognized by regulatory bodies around the world. The first considerations are termed “Product Definition” and “Process Definition.” For a radiation process this includes defining product families, establishing the maximum acceptable dose (the highest dose the device can withstand and still function properly), and establishing the sterilization dose (the validated minimum dose the device can receive to ensure sterility). The considerations for an ethylene oxide process include defining product families, confirming that the product design allows EO and humidity to penetrate the product and package, defining the hardest to kill location within the product, and determining the microbial rate of inactivation.
Sterilization equipment and systems go through an Installation Qualification (IQ). The IQ entails the calibration of measurement equipment. Utilities and services are verified that they are provided and capable of supporting the equipment requirements. The system software and control hardware are tested and qualified as appropriate for the process. In the case of contract sterilization services, the IQ and Operational Qualification are generally performed by the contract facility, and must be reassessed on a regular basis for inadvertent changes and degradation of equipment performance.
The IQ is followed by an Operational Qualification (OQ). The OQ tests the installed equipment to determine that it operates and performs within predetermined specifications. Procedures for the operation of equipment are established. For example, the OQ for a gamma irradiator would entail dose distribution studies in varying density loads, for all possible modes of operation, and determining the impact of system restarts. The OQ for gaseous sterilization systems (e.g., EO, steam, dry heat) include empty chamber temperature distribution studies, rates and depth of evacuation, injection rates and temperatures of gases, etc.
The next element of the sterilization validation is the Performance Qualification (PQ). ISO 14937 (see Table III.1.2.1) defines the PQ as the: “process of obtaining and documenting evidence that the equipment, as installed and operated in accordance with operational procedures, consistently performs in accordance with predetermined criteria, and thereby yields product meeting its specification.” For radiation processes this entails the demonstration of the dose distribution for the product-specific loading configuration, establishing the minimum and maximum dose location(s), and demonstrating that the product can be routinely processed within the range of the minimum and maximum allowable doses. In the case of ethylene oxide sterilization, the PQ would include a microbial qualification under subnominal conditions, and measurement of product temperature and humidity distributions. The reduction or dissipation of sterilant residuals in full cycle worse case conditions (highest gas concentration and longest exposure time) is also required.
Maintaining Sterility Over Time: Primary Package Validation
If the sterilization process is controlled to the extent that the standards require, e.g., such that the process delivers to the product a 9 to 12 or greater log reduction in microbial load, then what do we have to worry about? The package. The product packaging and sterile barrier system is critical to maintaining the safety and integrity of the sterilized product. Device packaging must maintain the product sterility throughout the expected shelf-life. As indicated previously in the section Material Compatibility, this would require accelerated and real-time aging studies. Packaging failures (i.e., open seals, pinholes in materials, material degradation, etc.) represent one of the most common reasons for product recalls. Compromised packaging jeopardizes the safety and integrity of the sterile product, and becomes a patient safety issue. The standards for packaging validation provide the industry with the requirements for validation of device packaging (ISO 11607-1; see Table III.1.2.1).
Packaging failures represent one of the most common reasons for product recalls. Examples of possible causes of such failures include pinholes worn into the sterile barrier from device protrusions, cracks in rigid packaging components, dislodged devices from the packaging fixtures, product damage or wear, and dust generated by device components rubbing against other materials.
Packaging materials used for the sterilization of gaseous sterilants must allow for the ingress of the sterilant to the device surfaces, while packaging for irradiated products can be a vapor barrier. A breathable package may be desirable for irradiated products, as some materials (polyethylene, PVC, and polyurethane used in tubing, drapes, and gowns) tend to generate gases during radiation sterilization. If the gases are trapped within the package, they may present potentially offensive odors when opened. Odor generation may be reduced incorporating antioxidants or by using higher molecular weight materials and breathable packaging materials (Hemmerich, 2000). For product material compatibility purposes, and to protect the product from the environment (i.e., water vapor, oxygen, etc.), one may have to provide packaging with a very low rate of gas transmission.
Product and packaging are subject to an accelerated aging program, and a simulation of the transportation environment. The shipping simulation can be performed according to one of several approaches, either ASTM D4169-05 Standard practice for shipping containers and systems, or by an appropriate International Safe Transit Association (ISTA) procedure such as ISTA 2A – Packaged-Products 150 lb (68 kg) or less. Following performance of the shipping simulation and aging, the packaging and products are inspected for packaging failures. Examples of possible failures include pinholes worn into the sterile barrier from device protrusions, cracks in rigid packaging components, dislodged devices from the packaging fixtures, product damage or wear, and dust generated by device components rubbing against other materials.
Alternative Sterilization Validation Methodologies and Specifications
Where does the biomaterials scientist go if standard terminal sterilization processes and validation methodologies do not meet the product need? Straight to liquid chemical sterilization or aseptic processing? These are relatively expensive options that may not provide as high a level of process control as terminal sterilization. It is, therefore, hoped that the biomaterials scientist will explore creative sterilization validation methods, and ensure that the sterility assurance specification being used is appropriate.
Validation Considerations for Minimizing Process Impact
In the majority of sterilization validations, the sterilization process delivers a significantly greater amount of lethality than is actually required. Typically, for devices composed of materials and packaging that are compatible with the sterilization process, the sterilization scientist uses a sterilization process that is easy to validate, such as substantiation of a standard sterilization dose of 25 kGy or an “overkill” approach. EO processes often use half-cycle overkill approaches that deliver SAL values of at least 10−12, and often values of 10−20 to 10−40, an overkill of enormous proportions. If product and or package functionality are a potential issue, one should explore options of minimizing the conditions of the terminal sterilization process in order to overcome material compatibility challenges.
EO sterilization process optimization can be achieved by exploring the possibilities of using a bioburden-based validation process and minimizing the conditions of exposure to the process (Annex A of ISO 11135-1). Figure III.1.2.8 illustrates the benefit of simply using actual product bioburden (orange line; assuming a realistic but high bioburden of 1000) as opposed to using the overkill method where an initial bioburden of 1,000,000 is assumed (purple line). The times required to achieve the desired SAL are considerably less. Using the overkill assumption, a 12 log reduction down to an SAL of 10−6 is achieved at about 120 minutes, whereas with an initial population of 1000 a 9 log reduction to attain the 10−6 SAL is achieved in approximately 95 minutes. If the microbial resistance of the actual product bioburden is not as robust as the severe biological indicator challenge assumed in the overkill method, and the slope of the actual bioburden line (orange line) decreases, the time to achieve an SAL of 10−6 is reduced even further.

FIGURE III.1.2.8 Microbial contamination level versus EO processing time: High and very high initial contamination levels.
Even though radiation sterilization validation processes are based on natural product bioburden, as opposed to an overkill method, the sterilization validation may not be optimized to deliver the lowest dose, but an acceptable or convenient dose. New radiation sterilization validation methods have recently been developed (See Table III.1.2.1: Method VDMAX, AAMI TIR33; Modified Method 2, AAMI TIR40) that allow for the lowest possible doses to be achieved for a given product. Along with optimizing the minimum sterilization dose, the biomaterials scientist may want to reduce the range of doses provided in the radiation process. An unnecessarily high distribution of doses throughout one product leads to high product doses and, therefore, significant overkill and potential damage to product. Achieving a low sterilization dose and a narrow distribution of doses may lower the overall product dose to allow for successful radiation sterilization of a sensitive product. In addition, as mentioned in the section Material Compatibility, cold processing and processing with an inert environment within the product packaging may also help reduce material degradation even further.
Application of Sterility Assurance Level (SAL)
The typical SAL for most devices distributed in Europe and the US is 10−6 (EN 556-1; see Table III.1.2.1). Historically the US permitted a greater SAL value of 10−3 for items such as surgical drapes and gowns (ST67; see Table III.1.2.1). However, with the requirement for CE marking at the start of 1998, and in recognition of certain Pharmacopeia requirements, the SAL value decreased to a greater assurance of sterility, 10−6. This is not to say that a device cannot be submitted and approved by the regulatory authorities with an SAL value greater than 10−6. Regulatory approval for these lower sterility assurance levels, such as 10−3, may be granted if it is shown that the device functionality cannot be achieved with a SAL of 10−6. Alternatively, if the biomaterials scientists, the microbiologists, and sterilization personnel cannot demonstrate that the device is compatible from a product performance perspective with any sterilization process, they may need to move the product to an aseptic process where contamination rates may not be greater than 0.1%. With either of these scenarios, it must also be shown that the benefits of the device for the target population outweigh the risks (potential harms and severity of these harms to the patient) associated with the use of the device. Another major perceived concern for a greater sterility assurance level such as 10−3 is the potential use of the device on elderly patients, immuno-compromised patients, those with HIV, transplant patients, burn patients, newborns, and children. This perception is countered by the fact that these high risk patients routinely are treated with aseptically processed products.
Terminal Sterilization Versus Aseptic Processing
In order to put terminal sterilization and sterility assurance levels in perspective, it is useful to compare terminal sterilization with aseptic processing. In aseptic processing there is no inactivation of the product bioburden. These products are not exposed to a sterilization process in their final packaging. Aseptic processing is defined as “handling of sterile product, containers, and/or devices in a controlled environment, in which the air supply, materials, equipment, and personnel are regulated to maintain sterility” (ISO 13408-1, 2008). Aseptic processing includes compounding, filtration, and filling. Through simulations or media fills of the aseptic process, the manufacturer must demonstrate the effectiveness of the controls over the risks of contamination associated with each step of the process. The control over potential contamination in an aseptic process results in a frequency of a non-sterile unit occurring of less than one in one thousand (0.1%). The sterility of the final product is based on the filter efficiency and integrity, the lack of available contamination in the surrounding environment, and controlling a series of operations in order to “maintain” sterility.
Since better process control and patient safety is achieved through terminal sterilization processes, the current regulatory environment requires medical device manufacturers to demonstrate that a product cannot be terminally sterilized before they can convert to an aseptic process. The extent of this demonstration is not well defined. To serve the needs of the industry when a terminal sterilization option is not achievable, there is current activity within the ISO TC198 Working Group 9 on development of a standard for solid medical devices (ISO/Committee Draft 13408-7, Aseptic processing of health care products – Part 7: Aseptic qualification of solid medical devices and combination medical devices). It is hoped that the creative terminal sterilization options provided in this chapter will permit some biomaterials scientists to avoid this option.
Summary and Future Challenges
The medical device industry has been well served by robust workhorse terminal sterilization technologies, radiation and ethylene oxide, in addition to many other technologies that meet lesser needs. The important processing parameters and mechanisms of microbial kill are well established. This information provides foundational insights into the interactions of the sterilization modality with a device’s materials. This information, along with available guidance on the compatibility of materials subject to sterilization, allows for the biomaterials scientist to overcome material compatibility challenges in order to reap the benefits of terminal sterilization relative to aseptic processing. Patient safety is ensured by terminal sterilization, as a result of the very high assurance of sterility specifications utilized, strong process control, and robust sterilization validation methodologies that have been incorporated into national and international standards.
The challenge for the biomaterials scientist is to skillfully apply terminal sterilization technologies to as many products as possible, especially the more recent combination devices. There are many material compatibility challenges that will make this a formidable challenge. Examples include drug delivery systems with active biological agents, bioresorbable temperature-sensitive polymers, and radiation-sensitive electronics. The ability to find terminal sterilization solutions to avoid aseptic processing will allow the device industry to provide these innovative therapies at the lowest price, and with the highest associated process control and patient safety.
The solution to these challenges will be an effort from a combination of disciplines. Material scientists and engineers need to develop increasingly robust materials that can withstand terminal sterilization processes. Even so, biomaterials scientists will need to carefully choose a sterilization modality that is compatible with key primary materials; this may require the utilization or development of novel sterilization technologies. Once a technology is defined that is compatible with the most sensitive materials of the device, supporting materials must be selected, processed, and tested to optimize chances for success with the sterilization modality. Optimal accelerated aging techniques to ensure product functionality over time are also required. In addition, to optimize chances of success with sensitive materials, the sterilization community needs to continue to develop and publish creative and gentle sterilization validation methodologies (e.g., minimizing radiation dose; ethylene oxide concentration, humidity, and temperature). Finally, regulatory authorities, industry forums, and international bodies involved in writing standards need to continue to challenge the bases for sterility assurance levels to assure that the terminal sterilization requirement for combination devices is not being set to an arbitrarily high standard.
Bibliography
1. See Table III.1.2.1 for additional Sterilization Standard references.
2. AAMI. TIR17 Compatibility of materials subject to sterilization AAMI. 2008.
3. ASTM. F1980–07 Standard guide for accelerated aging of sterile barrier systems for medical devices ASTM. 2007.
4. CDC, Center for Disease Control. Epidemiologic notes and reports, nosocomial bacteremias associated with intraveneous fluid therapy – USA. MMWR 1997; December 26, 46(51) 1227–1233.
5. CDC, Center for Disease Control. 1998; Corneal Decompensation after Intraocular Ophthalmic Surgery FDA Safety Alert: Warning Regarding the Use of the AbTox Plazlyte™ Sterilization System. MMWR, April 24,47(15),306–308 http://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/PublicHealthNotifications/ucm062297.htm?utm_source-fdaSearch_utm_medium_website_utm_term-AbTox_utm-content-1; 1998.
6. CDC, Center for Disease Control. Pseudomonas bloodstream infections associated with a heparin/saline flush – Missouri, New York, Texas, and Michigan, 2004–2005 MMWR 2005; March 25, 54(11), 269–272.
7. Chamberlain VC, Lambert BJ, Tang FW, et al. Sterilization Effects. In: von Recum AF, ed. Handbook of Biomaterials Evaluation. Taylor and Francis, Columbus, OH 1999:253–261.
8. Clough RL. High-energy radiation and polymers: A review of commercial processes and emerging applications. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 2001;185:8–33.
9. Ding N, Pacitti SD, Tang FW, et al. XIENCE V™ stent design and rationale. Journal of Interventional Cardiology. 2009;22(Suppl. 1):S18–S27.
10. EN556-1. Sterilization of medical devices – Requirements for medical devices to be designated “STERILE” – Part 1: Requirements for terminally sterilized medical devices BSI: British Standards Institute. 2001.
11. Favero MS. Sterility Assurance: Concepts for Patient Safety. In: Rutala WA, ed. Disinfection, Sterilization and Antisepsis: Principles and Practices in Healthcare Facilities. Association for Professionals in Infection Control and Epidemiology 2001;110–119.
12. FDA, US Food and Drug Administration. Safety alert: Warning regarding the use of the AnTox PlazlyteTM sterilization system. 1998; In: http://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/PublicHealthNotifications/UCM062297; 1998.
13. Forsythe JS, Hill DJ T. The radiation chemistry of fluoropolymers. Progress in Polymer Science. 2000;25(1):101–136.
14. Klevens M, Edwards JR, Richards Jr CL, Horan TC, Gaynes RP, et al. Estimating health care-associated infections and deaths in U.S hospitals, 2002. Public Health Reports. 2007;122 March–April.
15. Hemmerich KJ. Polymer materials selection for radiation-sterilized products. Medical Device and Diagnostic Industry 2000; February.
16. ISO 13408-1. Aseptic processing of health care products – Part 1: General requirements ISO. 2008.
17. ISO/TS 11139. Sterilization of healthcare products – Vocabulary ISO. 2006; .
18. Kulla J, Reich R, Bioedel SJr, et al. Sterilising combination products using oxides of nitrogen. Medical Device and Diagnostic Industry. 2009;31 http://www.mddionline.com/article/sterilizing-combination-products-using-oxides-nitrogen; 2009; March.Accessed 24.05.10.
19. Lambert BJ, Tang FW. Rationale for practical medical device accelerated aging programs in AAMI TIR17. Radiation Physics and Chemistry. 2000;57:349–353.
20. Mohammed AR, Bramwell VW, Coombes AG, Perrie Y. Lyophilisation and sterilisation of liposomal vaccines to produce stable and sterile products. Methods. 2006;40:30–38.
21. Smith RA, Ingels J, Lochemes JJ, Dutkowsky JP, Pifer LL. Gamma irradiation of HIV-1. Journal of Orthopaedic Research. 2001;19(5):815–819.
22. Terryn H, Maquille A, Houée-Levin C, Tilquin B. Irradiation of human insulin in aqueous solution, first step towards radiosterilization. International Journal of Pharmaceutics. 2007;343:4–11.
23. White A, Burns D, Christensen TW, et al. Effective terminal sterilisation using supercritical carbon dioxide. Journal of Biotechnology. 2006;123(4):504–515.