10
Putting the Zombies to Rest
![]()
As we age, we build up an increasing collection of “death-resistant” cells in our tissues. This is one part of our biochemical program to avoid cancer: shutting down the activity of potentially cancerous cells before they can cause trouble. Unfortunately, rather than simply remaining silent and harmless, such cells still manage to damage surrounding tissue through chemical signals gone awry. But by taking a leaf from the world of new, targeted cancer therapies, we can foresee the development of safe, effective methods to remove these senescent cells from the picture.
 So far, I’ve mostly been talking about specific forms of damage that happen at the molecular level to our cells and their components, and how we can restore functionality to our cells and tissues by undoing, or rendering harmless, that damage. But there are a few cases where the aging body accumulates cells that are damaged in such a way that they don’t just stop contributing to the economy of the body, but actually become toxic to the system that supports them.
So far, I’ve mostly been talking about specific forms of damage that happen at the molecular level to our cells and their components, and how we can restore functionality to our cells and tissues by undoing, or rendering harmless, that damage. But there are a few cases where the aging body accumulates cells that are damaged in such a way that they don’t just stop contributing to the economy of the body, but actually become toxic to the system that supports them.
I’ve already discussed one such case, back in Chapter 5: cells that have been taken over by mutant mitochondria. When mitochondria lose the ability to process fuels, as a result of mutations to their internal DNA, what ultimately causes us harm is (in my view) not the resulting failure of these organelles to carry out their job. Rather, it’s the maladaptive way that their host cell alters its metabolism in order to survive that failure. This metabolic alteration keeps such cells limping along by dumping oxidative stress outside their membranes and on to far-flung areas of the body.
At first glance, one might think that the best thing for the body to do with such cells would be to kill them off, thereby saving the rest of the body from their toxic influence. But the nature of the specific cells that develop this problem makes any attempt at simple removal fraught. The most notable case, arguably, is skeletal muscle. The design of muscle means that destroying a single muscle cell-like structure will snap the entire fiber in which it’s embedded. Loss of muscle cells to aging (rather than to disuse) is already a major source of age-related frailty; we can’t afford to add to that problem by killing off more of them in self-defense.
So in this particular case, as I described in Chapter 6, what seems to make the most sense is to find a way to preserve and restore normal metabolic activity in affected cells in the face of their colonization by mutant mitochondria, instead of killing them.
However, there are plenty of other cases in which the costs of destroying a toxic cell are negligible, and the benefits clear and direct. Everyone is familiar with one such case—cancer—and no one disputes that destroying cancer cells is unambiguously positive. I won’t be discussing cancer in this chapter, however (except for the applicability beyond cancer of some existing anti-cancer treatments), because that disease poses such unique challenges that I’ve devoted a whole separate chapter (Chapter 12) to it. Instead, I’ll focus on three cell types that pose a much less catastrophic threat than cancer, but that still make a substantial collective contribution to age-related descent into illness, frailty, and death. From what I can see, there is no reason to attempt to rehabilitate these cells: it seems best that they, like cancer cells, be destroyed. I’m choosing to discuss them together because of the similarity of the threats that they pose and of the strategies that I advocate for dealing with them.
![]() Attack of the Clones
Attack of the Clones
The decline of the immune system is one of the most deadly effects of aging. Infections that young people shake off as mere inconveniences are commonly fatal in the biologically old. Influenza, for instance, puts 114,000 Americans into the hospital each year, and flu and flu-related illnesses claim the lives of about 51,000. But the disease burden is dramatically skewed toward people who have previously suffered the ravages of aging (see Figure 1). Deaths from influenza and influenza-associated pneumonia are almost unheard-of in adults until the seventh decade, after which rates climb exponentially. In the United States, over 90 percent of all deaths from the two diseases are in people over sixty-five years of age.

Figure 1. Aging vulnerability to pneumonia and flu. (a) Hospitalization rates by age, Connecticut, 1993–1997 (females). Redrawn.1 (b) Death rates by age. Redrawn from CDC data.2
We could, of course, do something about the death toll through vaccination. But not much: between 30 percent and 75 percent of older people fail to respond to flu shots, compared with just 10 percent of young adults. Add to this the facts that we sometimes vaccinate against the wrong strain of the flu, limiting the effects of even successful vaccination. There are various reasons for the weakening of the immune system that happens with aging, some of which are ultimately downstream effects of aging elsewhere in the body (like the systemic increase in free radical stress spread by mutant mitochondria). But one of the most profound—and unexpected—factors underlying our inability to mount a defense against infections that young people shrug off without ever suffering a sniffle is, believe it or not, a form of immunological overcrowding.
![]() State of the Forces Report
State of the Forces Report
There are two main branches to the immune system. One is the so-called innate immune system, whose “innateness” comes from the fact that its job is so general that it doesn’t have to “learn” to identify a specific enemy. Its job is similar to that of regular soldiers on patrol in a demilitarized zone, trying to maintain order but unsure of who might be the enemy, ready to confront anything suspicious-looking that they happen upon. There seem to be very few changes in the innate immune system with aging, and those that have been reported appear to be secondary to other aspects of aging and age-related disease (and we’ll be discussing a few of those later on), or to factors that are common in the elderly but not a result of biological aging at all, such as vitamin and mineral deficiencies.3
The other main branch is the adaptive immune system. The adaptive immune system is more like a division of highly trained special forces units, each of them expert in waging targeted, tactically sophisticated warfare against specific enemies. This branch is responsible for the ability of the immune system to learn about invaders—and, thus, for the effectiveness of vaccines.
Within the adaptive immune system are the B cells and T cells. B cells are mostly responsible for defending us against pathogens like bacteria and parasites that are purely foreign to the body, and that can therefore be targeted directly for destruction. B cells recognize specific markers (antigens) on the surface of such an invader that reveal it as foreign, and churn out antibodies to them. I talked about antibodies back in Chapter 8, in discussing vaccination as a way of clearing out amyloids: they destroy alien cells by binding to the antigens of the organisms they’re intended to fight, acting like homing beacons that attract “missiles” fired by other components of the immune system, or blocking receptors and other proteins that are needed for the pathogen’s survival.
By contrast, cytotoxic T cells (also called CD8 cells because of the characteristic receptor they bear) are responsible for rooting out the enemy within: cells that are native to the body but that have now been turned against it, such as cancer cells or cells hijacked by viruses. (There are other types of T cell in addition to CD8 cells—more on them later.) CD8 cells also use antigens to target their foes, but because the targets are hiding in—or in the case of cancer, as—the body’s own cells, CD8s don’t get the chance to catch the pathogens’ calling cards on their own surfaces, and can’t target the invaders directly. Instead, CD8 cells pick up antigens on the surfaces of host cells that have been infected by a pathogen, or more often on other cells in the immune system that act like reconnaissance agents, scooping up copies of the antigen left in the wreckage of cells destroyed by the enemy and reporting their findings back to CD8 cells to alert them to the threat. Having spotted enemy colors, CD8 cells seek out and destroy the infected host cells, eliminating their threat to the rest of the body.
The problem, once again, begins with the body’s need to balance competing priorities in its metabolic processes—and to do so in the face of limited resources. On the one hand, it’s critical that the immune system be able to identify and fight off infectious agents that it’s never seen before, so it needs to have a reserve of CD8 cells that are ready to respond to new threats, “learn” about their key antigens, and then mount an attack; these are called naïve CD8 cells. On the other hand, the process of ferreting out an enemy that you don’t recognize takes time, during which an invader could gain a life-threatening foothold in the body, so we also have a complement of memory CD8 cells—veterans of old immunological battles, which remember the enemy that they defeated and stand ready to identify and fight them off again.
![]() Balanced Budgets
Balanced Budgets
This would be fine if we could keep on hand as many T cells as we might like, including plenty of naïve cells and large contingents of memory cells specific to each of the many pathogens that our body has racked up in its rogues’ gallery over the course of the years. But producing and maintaining these armies is a resource-intensive investment, and as with everything else, the body’s “budget” for the immune system is limited. To avoid going into deficit on its “military” spending, the body maintains a strict policy of balanced budgets—a limited amount of “immunological space” (as it’s been called) for the entire T cell population in aggregate. The immune system ruthlessly maintains a cap on the total number of naïve and memory cells combined in the body at any given time, although the specific makeup of that population is in constant flux, shifting dynamically as the body responds to the threat of the moment.
When this system is working well—as it does in most young people—it is the very model of the kind of flexible, low-cost, highly mobile, well-trained army that many of today’s generals and world leaders dream of constructing. During an infection with a particular pathogen, there is a rapid redeployment of forces to meet the threat on the ground. Whether it’s memory cells mobilizing against an enemy that they’ve seen before, or naïve cells uncovering and mounting an attack on a brand-new threat, CD8 cells appropriate to the enemy at hand expand their numbers, dividing rapidly in a process called clonal expansion, and then fan out, identifying and destroying cells bearing the foreign protein markers against which they specialize. (This use of the term “clone” is one of several in biology; it must not be confused, let me stress, with the popular nonscientific use of the word. I’ll have more to say about the various meanings of “cloning” in the next chapter.)
But once an enemy has been defeated, maintaining huge numbers of CD8 cells whose only mission is to wage war on a foe that has just been driven away would be a waste of limited resources. With the body’s iron discipline on its immunological budget, it can’t afford to have so much of its army be specialized for combating just one opponent if that enemy isn’t actually in the process of waging a campaign. So the body initiates a rapid and massive scaling back of these cells, ordering the bulk of the veterans to engage in a carefully orchestrated self-destruct program (apoptosis), after which it can rebalance its deployment of forces to a more generic defensive posture. But a few veterans of the recent conflict are kept on after the cessation of hostilities as memory cells, on the lookout for signs of a renewed attack from the invaders that they know so well. The small numbers required to maintain the body’s vigilance against a known enemy make this expense quite tolerable, so that the cost of keeping these cells on the payroll never puts significant strain on the “budget” of the immune system. That’s the plan, anyway.
![]() Old Soldiers Never Die…
Old Soldiers Never Die…
Unfortunately, this model of fiscal and military discipline only works well for infections that can be totally eliminated from the body. It begins to break down when the body faces enemies that it can fight to a standstill but not quite wipe out entirely. One class of such enemies is viruses of the herpes family: not just the infections commonly called “herpes” (herpes simplex of the mouth or genitals), but also Epstein-Barr virus (the one that usually causes glandular fever), varicella zoster (which causes chicken pox), and most especially a little-known infection called cytomegalovirus (CMV). All these viruses can be beaten back enough to put an end to active, symptomatic disease, but they are never completely defeated. A few copies of the virus continue to lurk hidden in some hard-to-reach corner of the body, dormant and out of sight of the immune system, waiting for the day when the tissue or the body as a whole is in such a weakened state that they can flare up again. In fact, the very name “herpes” is taken from the Greek herpein, “to creep,” in reference to their ability to sneak about the body while they await conditions favorable to their reactivation.
You may never have heard of CMV, even though the odds are good that you are carrying it (up to 85 percent of adults over the age of forty do). That’s because CMV rarely causes a recognizable illness, even briefly: about half of those undergoing CMV infection or reactivation suffer no symptoms at all, while the other half are afflicted only with hard-to-diagnose, nonspecific complaints such as general malaise, fever, and sweats.
But new research is showing us how CMV (and probably some other viruses) can also cause serious long-term harm to those of us who only suffer mild and transient activation and reactivation of the virus. Because the body can never quite consolidate its victory against these viruses, anti-CMV memory cells get called up to active duty again and again, and over successive iterations they gradually begin to ignore the apoptotic signal that is supposed to scale back their forces at the cessation of hostilities. There are various theories as to why this might happen, but I think it’s most likely to be part of a complex adaptation to protect us against uncontrolled cell division (i.e., cancer) in these cells.4 Whatever its origin, the inability to recall these veteran troops progressively weakens the immune system’s ability to fight other infections, new or old. The iron limitations on the “immunological space” or “military budget” ensure that, when the body can’t cull unneeded T cells specific to CMV or other infections, it has to make up the numbers with other immunological soldiers. As a result, the numbers of naïve cells available to keep the body ready to face new threats, and of memory cells for other pathogens, dwindle to dangerously low levels.5
![]() …They Just Fade Away
…They Just Fade Away
It’s bad enough that death-resistant anti-CMV veterans refuse to take their scheduled retirement, preventing needed redeployments and the hiring of new recruits. But the situation is actually worse than this. These problematic clonal expansions don’t just refuse to make room for other soldiers to do their job: like crippled or aged fighters, these weakened (the immunologist’s term is anergic) T cells can’t even carry out their own duties.6
One of the most important factors crippling these anergic T cells appears to be the loss of a key cell-surface receptor called CD28—an effect that has been observed in humans7 and animals.8 T cells are alerted to the presence of enemy forces by antigen-presenting cells (APCs), the immune system’s reconnaissance teams, which identify enemy combatants’ antigens through direct encounters with them or by digging through the rubble of old battlegrounds (the remains of cells ravaged by them). When T cells lose CD28, APCs can’t recognize them to alert them to the danger, and their intelligence report gets filed away unread. CMV-specific CD8 cells are unusually susceptible to this loss.
Another problem with anergic CD8 cells is that, having staked out a huge territory for themselves and thereby squeezed out other T-cell populations, they simultaneously lose the ability to reproduce themselves. Memory T cells normally express a receptor called KLRG1, which is there to keep them from proliferating when no infection is present. But healthy cells bearing KLRG1 are able to reproduce when a threat is actually present. Usually. Anergic CD8 cells have KLRG1 present on their surfaces,9 but they also have another marker on their surfaces called CD57, which is lacking in normal memory cells.10 When CD57 is present at the same time as KLRG1, the cells’ ability to reproduce themselves is locked down tight, so that they still can’t transform their reserve division into a full-fledged army when their sworn enemy is swarming over the ramparts.
Also contributing to this cellular “infertility” is the fact that the same cells have short telomeres—the long stretches of nonsense DNA that cap our chromosomes’ ends. Anergic T cells hit this problem because, unlike most immune cells, they have lost the effective activity of the enzyme telomerase, which is required to renew telomeres. Most cells don’t express telomerase, but it’s essential to the healthy functioning of CD8 cells because they are called upon to expand their numbers quickly and frequently over the life span in response to new infections. So the lack of strong telomerase action is a further mechanism of the enfeeblement of these cells. See the sidebar, “Backgrounder on Telomeres and Telomerase,” for more information, which will be amplified in Chapter 12.
BACKGROUNDER ON TELOMERES AND TELOMERASE
Every time a cell divides, it must make a new copy of its DNA. The enzyme responsible for doing this—called the DNA polymerase—is a bit like a molecular monorail train, zipping for ward along the “guide rail” provided by the DNA strand that it is to replicate. As it travels along the “guide rail,” the polymerase en zyme makes a letter-by-letter copy of the strand beneath it, spooling the new, replicated strand out to the side as it goes.
DNA polymerase has a fundamental shortcoming, however. For reasons whose details needn’t concern us here, the machinery never quite manages to replicate the entire strand of DNA. A small amount of a chromosome’s DNA material is therefore lost with every round of cell division, leaving the copied strand shorter than the original. Eventually, the end of the chromosome is eaten away.
A second problem that our cells have to solve regarding our chromosomes is that they often break, due to radiation and other stressors. The cell needs to repair such breaks. But what it must scrupulously avoid doing is stitching two intact chromosomes together end-to-end, mistaking the unjoined ends of the chromosome for the unjoined ends of a broken chromosome. Thus, it needs a way to recognize that the bona fide end of a chromosome is not just one side of a chromosome break.
Telomeres are half of Nature’s solution to both these problems. Telomeres contain no genetic information—they are extremely boring DNA consisting of many copies of a short sequence—and they are present at the ends of all our chromosomes. If this repeated sequence gets a bit shorter during successive rounds of cell division and DNA copying, no harm is done until it is mostly eroded. The other half of the solution is the enzyme telomerase, which is able to add copies of that sequence to the end of a DNA strand. This solves both problems: cells expressing telomerase can compensate for the telomere shortening that occurs during cell division, and cells with or without active telomerase can avoid stitching chromosomes end-to-end because the break-repairing machinery recognizes the distinctive telomeric sequence and leaves it alone.
Humans and some other species have made ingenious use of the telomere/telomerase system to protect themselves against cancer. Cancer can only kill us by its cells dividing a lot; this is impossible without telomerase, because without a way to renew the telomere, it will slowly erode away, the chromosome ends will be come indistinguishable from chromosome breaks, and the cancer cell will be stopped in its tracks by a joining-together of some of its chromosomes. Humans therefore turn off their telomerase genes as thoroughly as they dare, so that a lot of mutation is needed to turn telomerase on again and thereby allow a cancer to divide often enough to kill us.
Although there’s less research on it, old CMV carriers also suffer from the expansion of defective CD4 cells—the “T-helper” cells that help other immune cells to ramp up their counteroffensive when pathogens first invade. Outwardly healthy older carriers of CMV infection have the same large clonal expansions of CMV-targeting but CD28-lacking CD4 cells as are seen in their CD8 populations, leading to the same crowding-out of other T-cell specialists and lack of responsiveness to activation by antigen-presenting cells.11
As with their CD8 cousins, CD4 cells stripped of CD28 can’t respond to antigen-presenting cells by deploying CD8 and other immune cells to face the threat. Put this together with the inability of those very CD8 cells to attack their targets effectively, and CMV would be left to run rampant, generating yet more clonal expansions and wider immune dysfunction.
Clonally expanded anti-CMV CD8 cells are anergic (ineffectual) in other ways, too. When they are first infected with their species’ version of CMV, young mice produce very effective CD8 cells targeting the virus, which recognize at least twenty-four proteins specific to it; but after the infection becomes chronic, their anti-CMV forces become restricted to clones that recognize an average of only five such proteins.12 And older CMV-infected humans’ anergic CD8 cells mount a weaker response to the threat than do younger infectees’ cells, producing significantly lower amounts of interferon gamma, a key chemical messenger responsible for ramping up the T-cell response to the virus.13,14
![]() When Bad Generals Lead Good Armies
When Bad Generals Lead Good Armies
The failure of anergic T cells to clear out CMV infections then probably leads to many of the other failures of immune function commonly seen in the frail elderly that can’t be chalked up to any direct effect of aging of the cells in question. Some of these effects might be expected to flow from changes in the production of cytokines by such cells, which influence the activity of many other soldiers in the adaptive and innate immune systems, but others exert much more lasting changes than just problems in signaling.
Notably, it’s now widely accepted that the aging of T cells is responsible for the age-related losses in effectiveness seen in our B cells—the immune cells that produce antibodies to foreign antigens, which flag pathogens for destruction by other cells. B cells rely on signals from CD4 (T-helper) cells to mature and to develop antibodies, so it was only a matter of time before someone confirmed that old T cells cause declines in the development and effectiveness of B cells, independent of the aging of the B cells themselves. 15,16,17 Unfortunately, no one has (to my knowledge) yet directly looked to see if these effects are due to the effects of CMV-induced clonal expansion that are so central to other aspects of T-cell aging, so we don’t know how much the specific phenomenon of anergic T cells contributes to these declines. I’d be very interested in the results of such experiments.
Moving beyond the mechanistic studies and molecular biology, the real impact of the creeping takeover of the immune system by anergic CD8 clones on the health of people bearing them is also becoming clear as scientists begin to study its influence. Animal studies show that age-related clonal expansion of specific CD8 populations reduces the variety of T cells present in their bodies and compromises their ability to mount an effective immune defense.18 The parallel in humans can be seen in findings such as a poorer CD8 response to flu shots19 and a blunting of the reinforcement of T cell immunity to Epstein-Barr virus that can otherwise occur later in life,20 in people with clonal expansions of anti-CMV memory cells.
![]() Taking the Full Toll
Taking the Full Toll
If the cost of anergic T-cell clones to the body were limited to the increase in deaths and disabilities that can be directly chalked up to infectious disease, that would be plenty enough reason to want to do something about them. But there’s considerable evidence that anergic CD8 cells contribute to age-related morbidity and mortality from causes with no obvious immunological link.
For starters, when you throw an influenza or influenza-induced pneumonia attack onto an aged body, you wind up with shocking long-term consequences that can greatly accelerate other disease process and hasten a person’s slide into helplessness and the grave.21 A significant body of evidence shows that influenza in the elderly increases deaths from unexpected sources like heart attacks, strokes, and seemingly unrelated respiratory disorders; it also worsens the course of congestive heart failure.
Also, the fact that it takes biologically old people so long to recover from the flu, when overlaid on the general frailty induced by other aspects of aging, probably contributes to serious, often permanent functional decay and disability. A bout of influenza often lays an older person in a hospital bed for as much as three weeks, and studies show that for each day that they spend “resting” this way, elderly people lose up to 5 percent of their muscle power and 1 percent of their aerobic capacity. But no one thinks of influenza or immunological aging when they see an elderly woman struggle to open the doors at the mall, or slip on the ice and break her hip.
There are other age-related diseases in which anergic T-cell clones appear to play an important role, but where the evidence is not nearly so clear-cut. One is osteoporosis. Older women who have suffered an osteoporotic fracture have been found to carry higher levels of anergic CD8 cells than matched women with no bone disease, and there is a molecular basis for thinking that defective CD8 cells are actually a cause, rather than an effect, of the underlying thinning of the women’s bones.22
Additionally, albeit more speculatively, even the course of atherosclerosis could be affected by the creeping “clonalisation” of the T-cell population, by leading to a state of chronic inflammation that could hasten a heart attack. In support of this hypothesis, patients with coronary artery disease have higher levels of anergic CD8 cells than otherwise matched healthy people—a fact that is independently related both to CMV infection and also to the presence of the disease itself.23 Thus, the weakening of the immune system appears to be both facilitating and also the result of arterial infections, which may in turn be the itchy trigger finger toying with the loaded gun of atherosclerotic arteries.
As I said, the evidence for many of these downstream effects of anergic T-cell clones is still not conclusive. But a couple of remarkable studies now coordinated through the European Union T-CIA (T Cell Immunity and Ageing) project have gone some way towards giving us a clearer picture of the total cost, in deaths, of this driver of immunological aging, whatever may ultimately wind up written on the death certificate.
These researchers hunted through two cohorts of Sweden’s “oldest old” (people in their eighties24 and nineties,25,26), selecting only people who were particularly healthy compared to most people of their chronological age: free of preexisting serious diseases of the heart, brain, liver, or kidney; without diabetes or cancer or signs of existing, active infection or chemical markers of inflammation; and not taking any drugs that have significant effects on the immune system, including recent vaccination. The European team found that even amongst these relatively healthy but old people, a few are silently suffering a complex of immunological defects (the “immune risk phenotype”) including several forms of aging damage that can be caused by CMV infection—not least, the clonal expansions of anergic anti-CMV CD8 cells.
The facts that the resulting study population was healthy but very calendar-old (by today’s standards), and that some did and some did not harbor anergic T-cell clones, allowed the T-CIA team to study their effects “cleanly,” in a population where its presence could really be said to predict, rather than follow, preexisting disease, over the course of the next two years.
It was hardly a surprise that having the immune risk phenotype increased these people’s chances of death—but the size of the effect was a shock. The effect was especially powerful in the population in their nineties, amongst whom its presence could predict 57 percent of the deaths. This, remember, from the immunological aging damage induced by a virus whose active infection state is passed through without any notice in many people, and even in the rest of us usually causes only low-level malaise and fevers.
It’s important to see the full implications of this finding. The impact of having the immune risk phenotype was seen at the level of all-cause mortality, not just in risk of death from infectious disease. While pathogens do claim many very biologically old people’s lives, such deaths can’t entirely account for the result.
![]() Slash-and-Burn for New Growth
Slash-and-Burn for New Growth
As more and more evidence has accumulated fingering anti-CMV CD8 cell clones in the age-related enfeebling of the immune system, immunologists have begun to see the hopeful side of the phenomenon. If so much of the aging of the immune system is indeed the result of this overreaching expansionism, then preventing or reversing it should (respectively) protect or restore a youthful immune system in chronologically aged people. Vaccines would again be as effective in people at presently advanced ages as they were in their youth, and the enormous burden of suffering caused in the old by infections that young people escape after an unpleasant day or two home from work or school would be lifted.
One option for prevention, advocated by many immunologists, is vaccination against CMV. Even before we understood that CMV infection is a central driver of the age-related weakening of the immune system, a 1999 report on the sluggish pace of development of new vaccines put out by the Institute of Medicine (IOM) of the National Academy of Sciences ranked the pursuit of an effective anti-CMV vaccine as the highest priority item on the list, based only on the then-known lifetime human and financial costs of the virus. The U.S. National Vaccine Program Office would later agree, calling for more government dollars to go into CMV vaccine research. Today, confronted with the strong evidence condemning CMV infection as a major reason for the aging of the immune system, many immunologists are sounding the call for such investments even more loudly.
Though the merits of this case seem strong, it’s worth noting that this strategy is fundamentally preventative. While it can reduce the risk of CMV infection, and possibly improve the immune response of existing infectees to the virus, vaccination can’t eliminate it—and it certainly can’t reverse the accumulated effects that a lifetime of CMV infection has had on the immune system. Thus, a CMV vaccine might save a relatively small number of babies from suffering tragic birth defects, and prevent the deaths of many AIDS and transplant patients, but it would do little for the many millions of people already suffering with the chronic infections and continuous vulnerability of having immune systems worn down by clonally expanded anergic CD8 cells.
Other proposals, which would at least have the potential to undo some aspects of immunological aging, involve trying to remedy the defects of the existing anergic T cells using gene therapy. The idea is that by delivering copies of genes for proteins that are either missing or underactive in these cells (such as those for the CD28 receptor or telomerase), we could restore their effectiveness at doing their particular job, and prevent their suppressive effects on other T-cell populations. While there is some merit to these proposals, there are also limits to their likely effectiveness and a lot of uncertainties to their path to clinical development. And in the case of telomerase, we’d still face the big worry that has to be taken seriously when contemplating the introduction of telomerase into any cell, let alone one that we know is wracked with aging damage: cancer. I’ll talk about this problem much more in Chapter 12, but here’s a brief taster: because cells require a minimum telomere length in order to keep reproducing themselves, and because each cell division shaves off a little nub from the cell’s telomeres, cells with potentially carcinogenic mutations require a way to renew their telomeres if they are going to go on to become full-blown malignancies. Nearly all cancer cells accomplish this by wrenching out the self-imposed parking brake from their telomerase genes. Do we really want to introduce this gene into defective cells—or worse, into “bystander” cells in which telomerase should never be turned on, should some of the gene therapy vector “infect” them too?
No: the solution here is not to try to rehabilitate these cells, but to get rid of them. Older CMV infectees appear to have no shortage of functional T cells targeting cells infected by the virus: it’s just that these cells are suppressed by the crowding influence of huge populations of anergic ones. And remember that even if we could restore all of these defective T cells to their full immunological power, they would still cause problems so long as they continue to sprawl out over precious, limited immunological real estate, preventing the retention of both naïve and memory cells needed to protect us from other pathogens.
The solution to this is conceptually simple. Remove the anergic T-cell clones, and immunological space will be opened up for healthy cells of other types and specificities to move in—and the repressive effects of the anergic clones on their healthier anti-CMV cousins will be lifted.
The problem, of course, is how to purge anergic T cells from our systems while leaving behind all (or, at least, nearly all) the healthy memory and naïve cells that we’re trying to liberate from the former’s repressive domination. While oncologists can to some extent increase the effectiveness—and decrease the toxicity—of drugs or radiation by applying them as narrowly as possible to a relatively large lump at a fairly well-defined spot in the body, we can’t do the same against anergic T cells, which are spread all over the body rather than being concentrated in one place. The same feature rules out surgery: tumors can often be removed (or at least beaten back) with the knife, with varying degrees of safety and clinical benefit, but we will not be in a position to pluck individual anergic T cells from the body one-by-one for the foreseeable future.
But even though the cancer therapies of the recent past don’t offer a good model for the development of the required biotechnology, the most exciting of the currently available and imminent treatments for cancer suggest a path to therapies that would indeed selectively eliminate the burden of cells that will not die.
![]() Smells like Gleevec
Smells like Gleevec
Even if no one you know has cancer, there’s a good chance you’ve heard about Gleevec (a.k.a. STI–571 or imatinib), Iressa (ZD1839 or gefitinib), Herceptin (trastuzumab), and others less famous or still working their way through the approval process. These so-called “targeted cancer therapies” have been rightly hailed as breakthroughs; even the language of “miracles,” although absurdly overused in popular books on health, seems justified to many people who have seen tumors disappear from their own bodies or from those of their loved ones, without the horrific side effects associated with radiation and chemotherapy. Even so, these drugs are not completely without side effects—no drug that “messes with metabolism” can be. Herceptin, for example, targets a growth receptor called HER-2: by tying up HER-2, it prevents the excessive growth of cancer cells that get their growth-stimulus fix by producing too much HER-2 on their surfaces. But other, healthy cells rely on a low level of HER-2 stimulation to proliferate normally. Because of this, Herceptin users can suffer deadly congestive heart failure—a side effect that recent research has also uncovered in a small number of users of Gleevec, which was thought to be an extremely clean drug precisely because it only targets an abnormal form of a growth-signal transducer.27
In the same way, interfering with anergic T cells’ resistance to apoptosis might lead to their death, but this still leaves open the question of how to undo this resistance without killing needed cells elsewhere in the body.
I am confident that we can perform reverse-engineering, to adapt the new targeted cancer therapies—and even newer ones that are now in various stages of clinical development—to develop the ability to create “smart bombs” that will destroy anergic T cells (and also the other kinds of toxic cells that we’ll be discussing later on) with minimal harm to healthy ones.28 We can foresee the ability to couple carefully chosen toxins to molecules that home in selectively on the tell-tale signatures of anergic clones and thereby to directly, decisively kill them flat out instead of just interfering with their metabolism.
![]() Light Kills Vampires
Light Kills Vampires
One cancer treatment that suggests ways to take out anergic T cells is photodynamic therapy (PDT). PDT starts with a drug that, when illuminated with laser light, either heats up greatly or produces a massive burst of free radicals. Drugs exist that have this feature and are also taken up selectively by cancer cells, allowing oncologists to cause a lot of photosensitizing drug to accumulate in the target cells while avoiding much uptake by normal cells. By themselves, PDT drugs are harmless, having no effects as long as the patient is kept away from the light. Similarly, low-energy red laser lights is harmless to people who have not received such drugs: the rays pass harmlessly through the body. But when such a laser beam penetrates cells that contain a photodynamic drug, the agent’s photosensitizing properties are revealed in a searing targeted blaze of heat or a maelstrom of free radicals that destroys the tumor cells while leaving all but their very close neighbors unharmed.
The first PDT drug, Photofrin, was approved in industrialized countries as a treatment for advanced lung, digestive tract, and urinary tract cancers in the early 1990s, and more advanced versions are now in use clinically or are in late stages of development. The most interesting of these, Pc-4, accumulates more in some kinds of cancer cells than in healthy ones because it dissolves well in fats, and these particular cancers have an unusually high fat content. Once it enters the cell, certain features of Pc-4’s structure allow it to insert itself into the cancer cell’s energy factories, the mitochondria of which I’ve said so much in Chapters 5 and 6. Turn on the laser, and the free radical bombardment begins, either taking the cell out cleanly via radical-induced apoptosis, or at worst leaving some debris as the cell dies the nasty way instead, when the free radicals tear through the cell, cross-linking its proteins, turning its lipid membranes rancid, and wracking its DNA with mutations.
![]() The Molecular Swiss Army Knife
The Molecular Swiss Army Knife
On the frontiers of medicine, we are now seeing the emergent use of nanotechnology—engineering performed at the molecular level—to destroy cancer cells selectively, again providing us with a road map toward the development of a targeted therapy for toxic cells such as anergic T-cell clones. One such technology is dendrimers: tiny particles with exquisitely complex branching structures that extend outward like bushes, forming a spherical shape (see Figure 2). Dendrimers’ branches are engineered in a way that allows us to bind a wide range of molecules to them. This makes them like nanotechnological Swiss Army knives: several useful tools can be united into one compact little package. Dendrimers can carry one molecule to target a given cell type, one or more deadly drugs or other poisons to kill target cells once they’re located, and (if desired) a molecule that will allow researchers or doctors to track the progress of the whole package as it moves through the body.
One dendrimer under experimental development combines folic acid (yes, the vitamin) with the established anti-cancer drug methotrexate and a fluorescent compound called fluorescein. The folic acid is there to target the dendrimer to cancer cells. Many cancers suck up massive amounts of this vitamin because it is required for the production of new DNA—and because the cell needs to create a whole new copy of its DNA blueprints every time it divides, cancers have high metabolic requirements for folic acid to support the feverish pace of their proliferation. To keep their reserves of the vitamin topped up, many cancer cells “learn” to sprout veritable forests of folic acid receptors on their surface.
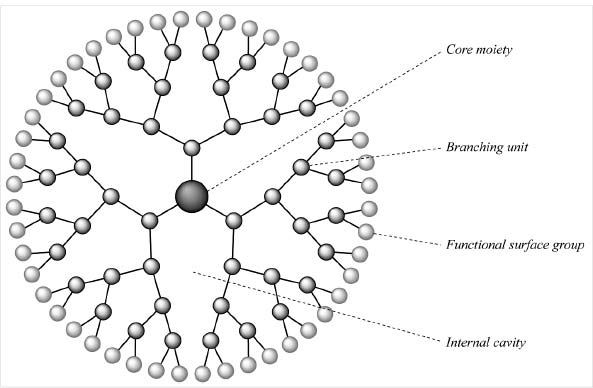
Figure 2. An “unloaded” dendrimer.
This dendrimer was tested in mice that had been injected with a human nasopharyngeal cancer line. Their tumors had grown quickly, reaching a plateau at about fifty days. Giving one group of animals a low dose of plain methotrexate had hardly any effect at all on the growth of the tumors (see Figure 3). A dose more than four times as high (the “medium dose” in the figure) reduced the growth rate quite significantly, but didn’t actually benefit the animals much: half of them were dead of either the tumors or the side effects of the drug within thirty-nine days. Increasing the dose by a further 50 percent (the “high” dose) brought cancer growth down to almost nothing—but without doing the animals any good, because the drug’s toxicity caused the rapid loss of a third of the animals’ body weight, and again either allowed or caused the death of half of them just over a month into the experiment.
But look at what was achieved with the same drug targeted using a dendrimer! When targeted using this new technology, a dose of methotrexate equivalent to the lowest untargeted methotrexate dose was as effective at slowing tumor growth as a dose of plain methotrexate over four times higher. Moreover, the dendrimer-targeted methotrexate appeared to have very low toxicity.30
In a follow-up study, the same group compared the effects of the low-dose methotrexate to those of the same dose delivered using the targeted dendrimer, this time for an extended period of ninety-nine days. Left untreated, the cancer-bearing mice began dying quickly—about fifty days into the experiment—and low-dose methotrexate improved survival only modestly. But by the end of the ninety-nine-day study, three out of eight of the animals getting the dendrimer-targeted drug were still alive—and impressively, one of these animals was completely cured of its cancer by day thirty-nine. Again, the dendrimer-targeted drug was nontoxic.
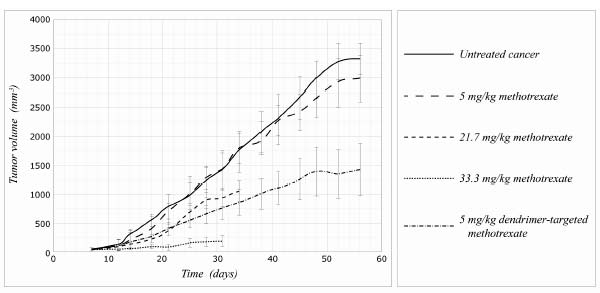
Figure 3. Targeted dendrimer technology against cancer growth in mice. Redrawn.29
![]() Switch the Hacksaw with the Toothpick
Switch the Hacksaw with the Toothpick
The beauty of dendrimers, like Swiss Army knives, is that they are so readily customized. Researchers are experimenting with dendrimers bearing many different targeting molecules and cancer-killing agents. One very clever application under development is a “dendrimerized” version of a one-two anticancer punch known as boron neutron capture therapy (BNCT)—a neat idea that was first proposed over fifty years ago, but that until now no one has ever quite managed to make work.
The idea behind BNCT is similar to photodynamic therapy. First, you inject the patient with a form of the mineral boron. Once enough of the mineral has accumulated in cancer cells, you flood them with low-energy beams of neutrons. The boron itself is harmless, and so (more or less) is the neutron beam. But when incoming neutrons hit the form of boron used in BNCT, it absorbs them into its atomic nucleus and suddenly becomes extremely unstable, releasing radioactive alpha particles. Alpha particles have enough energy to nuke the cell in which the original boron is located and a few of its neighbors, but they run out of energy quickly and the low-energy leftovers are harmless. Thus, the boron-loaded cells are killed but no widespread damage ensues.
The trick, of course, is to find a way to target the boron selectively to cancer cells, so that when you flip on the neutron beams you don’t destroy healthy brain and other tissue. Scientists have been trying to turn BNCT into a viable clinical therapy for a rare, extremely aggressive, and hard-to-treat brain cancer called glioblastoma multiforme since 1951, with some limited success, but they’ve never achieved good enough responses to justify using it as a standard treatment for the disease.
But scientists have recently reported very promising results in an animal model of glioblastoma multiforme that was treated with a BNCT using dendrimer-targeted boron. Human glioblastoma cells bearing a mutated version of the epidermal growth factor receptor (EGFR) called EGFRvIII that is implicated in the majority of these cancers were implanted into the brains of rats. The researchers then heavily loaded their dendrimers with boron, and then sent them hunting for the gliomas by attaching a monoclonal antibody to the mutated EGFR. To get a handle on just how promising the dendrimer really was, they compared its effects not only to what happens in animals given no therapy at all, but also to animals given p-boronophenylalanine (BPA—the most promising preparation of boron being used in BNCT clinical trials), or else the boron-loaded dendrimer in combination with (but not bound to) BPA. Within a day, about 60 percent of the injected BPA-bearing dendrimer had homed in on tumors carrying the mutant receptor, achieving concentrations that were about triple those of BPA alone; the uptake by normal tissues was negligible. Impressed by its homing ability, the researchers waited to see whether it could actually cure the animals of their gliomas.
The results were decisive (see Figure 4). Untreated animals lived an average of just twenty-six days. Animals who received BPA could expect to survive for forty days: a significant improvement, but still a grim prognosis. But animals given the dendrimer-targeted boron lived an average of seventy days, with 10 percent of them surviving for six months—considered a “cure” in the same way that five-year survival is considered a “cure” in humans, since healthy rats have a life expectancy of about thirty months. And animals that were lucky enough to get the dendrimer along with BPA survived, on average, for a remarkable 85.5 days, more than three times the life expectancy of the untreated animals, and more than double the survivorship of animals getting the best experimental therapy available. Plus, an impressive one in five of the BPA-plus-dendrimer-treated animals achieved a “cure” as just defined.
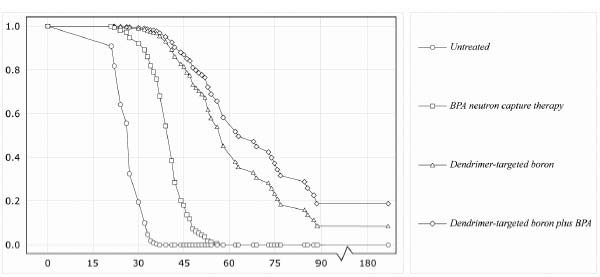
Figure 4. Targeted dendrimer technology dramatically improves the effectiveness of BNCT. Redrawn.31
Other targeting molecules, tumor types, and cancer-killing agents have been successfully treated by dendrimers in experimental models. These first-generation devices are turning out to be a very effective, versatile way of creating specific, lethal missiles for seeking out tumors that express known cell-surface receptors as hallmarks—and they offer promise for anergic T cells, too.
![]() PRO-Suicide Counseling
PRO-Suicide Counseling
Today, gene therapy is a routine practice in mice, used to do everything from testing experimental gene-based therapies, to investigating the effects of turning genes on and off in an organism, to creating new models of human disease by modifying animal cells to be more like human ones. Gene therapy for humans is still highly experimental, but it’s clearly only a matter of time before we master it: the need for cures for congenital diseases, and the potential utility of gene therapy in medical challenges as wide-ranging as rheumatoid arthritis, trauma, dental tissue engineering and AIDS (to name just a few), is providing the impetus for the basic and clinical science needed to bring it into our therapeutic armory.32
One option that gene therapy will furnish us with is the ability to build a new suicide mechanism into our T cells that would cause them to self-destruct should they ever turn anergic. Scientists have for some time been able to introduce into mice (and other laboratory animals) genes that will only turn on in the presence of a particular factor, such as an antibiotic, UV light, a sugar, or even a signalling factor like calcium. This allows us to turn such genes on and off at will, simply by administering the relevant factor.
The ability to introduce a gene that is only expressed when researchers want it to be has been a powerful new tool for studying those genes’ effects. But these techniques are also now being turned to medical purposes. If, instead of designing these genes to be turned on in response to externally supplied factors, we instead make their activation dependent on the presence of a particular protein whose internal synthesis is diagnostic of a cell that we want to be rid of, then we have yet another way to selectively target cells for destruction.
Just as with the other targeting technologies I’ve discussed, the first work in this direction has been in the cancer field. As I’ve noted, the one absolute requirement for a cancer cell to threaten us is that it have a way to keep renewing its telomeres: otherwise, its furious growth will grind to a halt when it reaches the end of the telomeric line, which is long before it can meaningfully threaten our health. Usually, this is accomplished by activating the repressed gene for the telomerase enzyme—a gene that all of our cells contain, but which is turned off in healthy cells most or all of the time. So by “infecting” the cells of a patient with a “suicide gene” that would be activated in the presence of high levels of telomerase, cancer cells could be killed from within. This would eliminate the need to target a drug or the immune system to the offending cell: every cell would hold within it the seeds of its own destruction should it ever turn to the dark side.
In principle, we could generate a literal “suicide gene” that would destroy the cell in the presence of the tell-tale protein. Indeed, this has already been done in animal models of cancer, using genes that regulate apoptosis;33 anergic T cells are resistant to apoptotic signaling, but this resistance might be overcome by bombarding them with insistent messages to shrivel and die. But there’s an even better alternative that’s under more advanced development. This uses the somewhat more readily controlled—and therefore safer—technique of installing the gene for a protein that is largely harmless in itself, but which activates an inactive form of a deadly drug—a so-called “prodrug.”
Prodrugs are substances that are inactive and harmless until they are metabolized in some way, whereupon they are chemically transformed into a pharmacologically active product. Most prodrugs are activated by enzymes in our livers and are then released in active form to the rest of the body, but others act more like molecular “sleeper agents,” going about the body unobtrusively, minding their own business and blending in with their environment, until a prearranged signal is given—and their hidden purpose suddenly becomes revealed in the form of a precision strike on their target.
Several antiviral drugs, such as the herpes drug ganciclovir (Cytovene/Cymevene), work somewhat along these lines. Ganciclovir stops viruses from using the DNA-replication machinery of their host cells to reproduce themselves. It does this by interfering with the action of the virus’s unusual version of thymidine kinase (TK), an enzyme that is required for the synthesis of DNA.
Thymidine kinase’s job is to make thymine (a “letter” in the DNA code’s “alphabet”) available to be added onto the new DNA chain, by joining thymidine to phosphate molecules taken from the “energy currency molecule” ATP. Ganciclovir acts like a molecular impersonator on a mission to sabotage an enemy factory. It first uses its strong structural resemblance to thymidine to fool the viral TK into thinking that it is that molecule. Duped, TK hands over thymidine’s rightful phosphate group to the drug.
Ganciclovir then uses its shiny new phosphate group to perpetuate its identity theft, presenting its phony credentials to the cell’s DNA synthesis machinery, which unknowingly inserts it into the emerging DNA strand in thymine’s place. At this point, the sabotage of the hijacked equipment is accomplished, because while the machinery can slide ganciclovir onto the DNA strand, it can’t add any further genetic letters onto ganciclovir once it’s in place. Without the ability to copy its DNA, the virus can’t replicate itself, and its expansion campaign is brought to an abrupt end; all that’s left is for the immune system to besiege and ultimately destroy the cells in which the remaining virus is holed up.
If ganciclovir were as good at tricking the version of the TK enzyme used by our own cells as it is against the viral version, it would potentially be a very effective cancer-killer: again, cancer can only survive by keeping up the insane pace of its growth, and turning this off by shutting down its DNA synthesis capacity quickly tames tumors. But, of course, such a drug would come with some pretty serious side effects, because it would shut down the growth of normal cells at the same time. This might make it an acceptable therapeutic bargain for cancer—the effect would wear off after the drug was withdrawn, allowing patients to recover—but it would make it totally unacceptable for its present use as a herpes treatment.
In fact, however, ganciclovir is rather poor at mimicking the human TK enzyme, and its effects are thus mostly restricted to turning off virus replication—though it does have some negative impact on the body’s ability to regenerate its blood cells and on the production of sperm. But a team of Japanese and American scientists recently realized that they could in principle use the viral TK/ganciclovir combination to shut down cancers if they could introduce the enzyme into cancer victims’ cells using gene therapy, but turn it on exclusively in cancer cells.
As I’ve already indicated, there is one obvious way to distinguish cancer cells from normal ones which could provide a mechanism for controlling the activation of viral TK: active telomerase. By designing a version of the viral TK gene that would be attached to a “trigger” (promoter) that would turn the gene on only in the presence of telomerase, the researchers realized that they could set the enzyme to work in a cancer patient’s malignant cells, while leaving it dormant almost everywhere else in the body.
At this point the flow chart of what they were designing was beginning to look like the biotech equivalent of one of those exceedingly convoluted, multistep devices that players build up in the board game “Mousetrap.” The scientists would set the “trap” by first seeding a copy of the gene for the viral TK enzyme, complete with its special telomerase “trigger,” into every cell in the patient’s body. The patient would then swallow some ganciclovir tablets, which would penetrate all of his or her cells indiscriminately.
In most cells, the drug would have no effect, because nearly all cells have their telomerase enzyme firmly turned off. But when ganciclovir entered a cancer cell, the trap would be sprung. The cancer’s abundant telomerase enzyme would flip on the viral TK enzyme; the TK would scoop up the ganciclovir, adding on the phosphate group needed by DNA “letters” for insertion into the emerging DNA copy strand; the next time it reached for the relevant “letter,” the DNA-copying machinery would grab the phosphorylated ganciclovir by mistake, jamming it into the “letter’s” place on the strand. At that point, you would almost hear the cry of “Mousetrap!” as the DNA synthesis machines seized up, cell division came to a screeching halt, and the cancer shut down. See Figure 5.

Figure 5. How ganciclovir enables viral thymidine kinase to kill mammalian cancer cells.
It was a crazy, convoluted solution—but it worked in the test-tube against liver, kidney, pancreas, and thyroid cancer cells. Moreover, the setup proved largely harmless to normal rat thyroid and human skin cells.34 So the team took the next step in bringing something off a lab bench and into a clinic: a careful study in laboratory animals.
The researchers first cooked up two batches of customized viral TK genes: one with the telomerase-activated “on” switch, and another with a switch that could be expected to be flipped in healthy and cancerous cells alike. They then slid these genes into viruses from the same family as the common cold, allowing them to literally infect animals with the gene constructs. They first tried these constructs out on healthy animals, to see what the potential was for side effects. As expected, animals that got viral TK under the control of the nonselective promoter suffered nasty liver damage upon injection with ganciclovir, while putting in the same gene under a telomerase promoter appeared to be basically harmless, since there were no telomerase-expressing cancer cells present to activate it.
At this point, the researchers decided that their experimental therapy was ready to hit the next stage: a test in animals that had been injected with implanted human thyroid carcinoma cells. Giving such animals a copy of the viral TK driven by a promoter that did not rely on the presence of telomerase to activate it brought tumor growth to a complete standstill—but, as expected, it also prevented normal cells from reproducing themselves, leading to nasty liver damage.
But when scientists tried the selective targeting of viral TK to cancer cells by using the telomerase promoter, ganciclovir shut down tumor growth just as completely as it had when the TK was controlled by the nonselective promoter—and without the latter’s toxic effects. The apparent safety of this highly selective intervention is all the more convincing when you remember that the effect can be turned on and off at will, by administering or withdrawing the ganciclovir.
![]() Know Your Enemy
Know Your Enemy
We’ve seen that biologists can be just as creative as weapons engineers holed up in the Skunk Works at finding new ways to target and kill cancer cells selectively, i.e., leaving healthy cells unharmed. It is foreseeable that the same methods in use or under development against cancer could be used to target anergic T cells. In this case, we’re blessed with an enemy that is walking around with a bull’s-eye painted right on its chest. The same dysfunctional receptor profile that strips anergic T cells of their ability to recognize their target antigens (absence of CD28) and to proliferate in response to infection (presence of KLRG1 and CD57), possibly along with some other markers (such as reduced levels of CD154, implicated in the failure of old T cells to support B cell development) already allows scientists to identify these cells, and could also be used to target such cells for destruction.
Unleashing the wrath of the immune system against its oppressors would be poetic justice, but vaccination (either passive or active) against these cells might be tricky. For one thing, their most prominent antigenic feature is the lack of a cell-surface protein (CD28), and while we could target the combination of KLRG1 and CD57, it’s not yet clear whether all unwanted cells express these two proteins, nor whether other, desirable cells do. As it happens, immunologists already do identify anergic cells using a combination of immune proteins—but these could not easily be used for vaccination purposes. Also, after all, the problem that we’re looking to resolve is characterized by a poor response to vaccination, so an “anergic T-cell shot” might only be effective in relatively immunologically “young” people. So while this approach is promising for many kinds of toxic cells, it may be less so for CD8s.
But that still leaves us with a lot of options. Dendrimer-based targeting approaches seem the most straightforward, because they allow for the targeting of cells via multiple identification criteria, and also because they can introduce any number of poisons into the cells that they select, from outright toxins to boron for BNCT.
And while using vaccines to target anergic T cells might be problematic, there could be another way to use the immune system as a proxy army, cooperating with it to restore the sovereignty of the immune system’s government-in-exile. Remember that anergic CD8 cells first become a problem because they stop listening to the apoptotic order to scale back their forces that’s sent out after their target pathogen has been routed from the body’s frontiers. They are able to ignore these orders because they produce high levels of bcl-2, a protein that blocks apoptotic signalling.
This suggests the possibility of restoring normal apoptotic signaling in such cells by delivering “antisense RNA” for bcl-2’s blueprints to them—strips of genetic material matched to the transcribed DNA instructions for the protein, preventing the encoded bcl-2 from actually being produced in the cell. With bcl-2 production brought down to normal or nearly nonexistent levels, anergic CD8 cells would finally hear their curtain call and bow out.
Would purging the immunological “space” of anergic T cells be enough to rejuvenate the immune system completely? I can’t say for sure, because it hasn’t been done—and, as I am well aware, the body is an incredibly complex machine whose parts have not yet all been identified, let alone their purposes and interactions. Existing research tells us pretty clearly that the direct and indirect immunosuppressive effects of these cells are powerful enough that a thorough spring cleaning of them will profoundly improve T cell-mediated immunity, and also very probably the functioning of other aspects of the immune system that T cells support and govern. But we’ll only know just how profoundly once we’ve done it.
I can tell you right now that there is at least one aspect of immune aging that the removal of anergic T cell clones will not address: thymic involution. The thymus is a gland located just behind your breastbone. It’s where immune cells first produced in your bone marrow go to learn to become T cells. As we age, the thymus loses cells and shrinks away, and in the process its output of naïve T cells plummets. This, of course, imposes further limits on the body’s ability to respond to new threats.
In principle, there is a fairly straightforward way of dealing with this, however: stem cell therapy. This is foreseeable biotechnology, as we will see in the next chapter (see the sidebar there, titled “Rebuilding the Thymus”).35 The accomplishment of such a goal would entail significant advances in the stem cell field, including mastering the art of turning embryonic stem cells into the progenitors of the different cells of the body, and then engineering new tissue to rejuvenate the old one, renewing old tissues with pristine new cells—but these are just the same problems that are being solved quite rapidly for tissues all over the body, so there is ample reason for optimism.
That’s all I have to say about the immune zombies; now it’s time to look at some other types of supernumerary cells.
![]() Deadly Combat in the Battle of the Bulge
Deadly Combat in the Battle of the Bulge
The second kind of toxic cell that we’ll want to rid ourselves of is excess fat tissue—most important, the so-called visceral fat that surrounds your internal organs, as opposed to the subcutaneous fat that lies under your skin all over the body. It’s widely believed that, as people get older, they just “naturally” become more resistant to the effects of the hormone insulin, whose job it is to move carbohydrates and amino acids into fat and muscle cells. This change causes a range of threatening metabolic changes, the most extreme of which manifest in people with full-blown type II (“adult onset”) diabetes. It’s also common wisdom that older people “naturally” enter into a more inflammatory state, with the body slowly burning up from within because of an excessive production of inflammatory signal molecules.
When you take in more calories than you expend, your body hangs on to them rather than allowing them to go to waste. This is not the result of perversity on the part of evolution, but a survival strategy: until very recently (by evolutionary standards) there was a good chance that quite soon you’d be in a period of famine, when those stored calories would be your lifeline. If your body isn’t under the kinds of challenges (like weight-bearing exercise) that signal the body to build up metabolically expensive muscle or bone tissue, it will take the easy way out by storing the calories as fat. But in an environment where feast is never followed by famine, and where exercise is almost entirely a voluntary affair, we fail to shed that extra fat tissue, and it slowly accumulates as we age. Because this accumulation is a difference of aging versus healthy young bodies, it qualifies as “aging damage” under my engineering definition, even though it might not be considered as such from a purely theoretical point of view.
It’s long been known that this damage—in the form of being overweight or obese—puts you at greater risk of diabetes, heart disease, and various other ailments, but it’s only recently become clear why and how. You may have heard that fat stored in different parts of the body has different health implications: having an “apple shape” (fat centralized in your midsection, as in a “beer belly”) puts you at great risk for diabetes and heart disease, while having a “pear shape” (fat clumped on your bottom or thighs) is unsightly but much less hazardous to your health. To the extent that there is some biomedical basis for this distinction, it lies in the difference in the locations of visceral and subcutaneous fat. Visceral fat is most visible around your middle because it clumps around major internal organs like the liver and kidneys. By contrast, subcutaneous fat just lies under your skin—and of course, there’s skin all over your body, though there are more prominent depots for this fat type in some places than others.
Recent studies have found that nearly all observed age-related insulin resistance, and much of the age-related pro-inflammatory signaling shift, can be attributed to the accumulation of excess visceral fat, which precedes and predicts the development of all the elements of the metabolic storm known as syndrome X: insulin resistance, low HDL (“good”) cholesterol, and high blood pressure, triglycerides (blood fats), and blood sugar.
Even more tellingly, when you compare people of different ages, you find that the difference in insulin effectiveness between young and old disappears when you account for the difference in fat, and visceral fat in particular.36, 37
In striking studies at the Albert Einstein College of Medicine, aging animals have had most of their resistance to insulin and other hormones reversed by highly invasive surgery that scrapes out most of their visceral fat, making their body compositions similar to much younger animals, or to animals of the same age that had been subjected to calorie restriction (among whose benefits are dramatic reductions in age-related insulin resistance and inflammatory signaling).38 This latter result was especially striking because, when the scientists looked at the distribution of fat in the calorie-restricted animals, they found that they actually had more subcutaneous fat than their young counterparts, but less visceral fat—and their insulin effectiveness was almost the same.
Further pinning the blame on visceral fat, a recent study confirmed that undergoing liposuction—whose invasiveness and risks of trauma are kept low by removing only the relatively easy-to-access but cosmetically significant subcutaneous fat, while leaving the much harder-to-remove visceral fat behind—does not improve the insulin resistance associated with the original obesity.39 On the other side of the same coin, several studies have now shown that putting overweight people on low-calorie diets or exercise programs significantly improves their insulin resistance quite early on—well before it has had a chance to impact their overall weight by much, but after it has had time to reduce their level of visceral fat, which (fortunately) is the first thing to go when energy needs aren’t being met.
The reasons for all of this have become clear as scientists have increasingly come to understand the nature of fat itself. Fat tissue was once considered to be just inert storage space, like carrying around a spare tank of gas on your derriere. Instead, we now know that it is a metabolically active, dynamic tissue that secretes and responds to a range of hormonal and other signaling molecules. We also now appreciate that fatty tissue is not composed only of “fat cells” (adipocytes), but is a mixture of different cell types, including supporting connective tissue, nerves, and blood vessels, as well as immune cells—notably, macrophages. In fact, adipocytes are actually derived from the same precursors as macrophages, and secrete many of the same immune-system regulating molecules, such as the coagulation-enhancing enzyme plasminogen activator inhibitor-1, and pro-inflammatory signaling molecules (cytokines) such as tumor necrosis factor alpha, monocyte chemoattractant protein-1, and interleukin-6.
As the size of the fat depot increases, adipocytes begin pumping out more and more of these inflammatory molecules, some of which promote infiltration of the tissue by macrophages and some of which signal the precursor cells that give rise to both adipocytes and macrophages to take the path toward the latter instead of the former. Macrophages, in turn, produce even more inflammatory messenger-molecules, creating a self-reinforcing inflammatory feedback loop.
The most exciting finding in the emerging science of fat in the last ten years has been the discovery that these signaling molecules not only cause a potentially pathological increase in systemic inflammation, but also increase the body’s resistance to insulin. This conclusion is supported by studies showing that isolated muscle and fat cells become insulin-resistant when bombarded with the very inflammatory mediators that adipocytes and macrophages produce, and that chubby lab rodents’ insulin resistance is relieved by aspirin treatment, in part via blocking the effect of cytokines. And the relationship doesn’t just hold up under the artificial conditions of the laboratory: during sepsis (the inflammatory storm generated in response to severe infection), human patients often exhibit very severe insulin resistance as part of the immune response.
Evidently, these and related questions will keep an army of basic and clinical researchers in the diabetes field busy for decades, resolving paradoxes, isolating deeply intertwined metabolic pathways, and double-checking their results in different models. But for engineering purposes, fortunately, we don’t have to wait to have the results of these investigations: we just need to observe the presence of damage, and fix it.
![]() Doing It the Old-fashioned Way
Doing It the Old-fashioned Way
In this case, of course, there are two very simple, inexpensive solutions that don’t involve advanced biotechnology: diet and exercise. But unfortunately, as decades of research and centuries of anecdotal experience have shown, most of us find it very difficult to lose weight once we put it on. As little as a 100-calorie daily energy imbalance—about what you get in a single medium-sized biscuit—accounts for the standard weight gain that creeps up on the average person in the decades between high school and middle age, and while it’s easy to load it on, it’s hard for most people to shed those extra pounds and keep them off. The situation is not nearly as dire as is often made out—studies show that about one overweight person in five successfully achieves long-term weight loss, and research is clarifying what it takes to get there—but the metabolic consequences of excess visceral fat are far too deadly, and the magnitude of the current obesity epidemic far too staggering, to leave the cure of this form of aging damage to self-help programs or public health measures designed to remediate our present “toxic food environment.”40 Realistically, unless we are ready to leave the fate of millions to a sudden wave of greater personal or political responsibility, we must look for biomedical solutions to visceral fat.
![]() Fat for the Fire
Fat for the Fire
One option that we might pursue is to shrink away visceral fat by causing it to burn off its excess stored energy. Scientists have of course been trying to develop drugs to do this for decades, but so far the only moderately effective such drugs are amphetamines, and their side effects and addictiveness clearly don’t fit our remit.
For several years, the appetite-regulating hormone leptin seemed to offer the chance of shrinking fat and maintaining insulin sensitivity. An extremely rare genetic mutation that leads to a congenital lack of leptin makes both rodent and human victims monstrously obese, and injecting these mice with leptin leads to dramatic weight loss. Causing these rodents to produce more leptin inside their fat cells (using genetic engineering) makes them eat 30 percent to 50 percent less food, leading to more insulin sensitivity and an almost complete disappearance of their body fat. Moreover, the effects are stronger than can be accounted for by their newfound light dining habits alone.41 Fat cells of animals with the extra leptin gene were expressing other genes that activate mitochondria, turning them into little “fat burning machines.”42 Paradoxically, however, while injections of leptin lead to rapid fat loss in both normal and obese rodents, the same level of leptin that would peel away the pounds in lighter mice circulates naturally in the bodies of their fat cousins, necessitating a much higher level of leptin to achieve similar weight loss. This is in part because leptin is, ironically, produced by the very fat cells whose swelling with stored energy it was supposed to inhibit, so that chubbier rodents naturally produce more leptin, not less. And indeed, after drugs giant Hoffmann-La Roche invested a fortune to develop a way to mass-produce human leptin in genetically modified bacteria, they found the hormone to be a miserable failure as a weight-loss treatment.43
This led scientists to speculate that overweight members of both species come to suffer “leptin resistance” in the same way that they suffer insulin resistance: levels of the hormone are high, but cells stop responding properly to its signals to turn off the appetite and turn on fat-burning. Recent studies by Roger Unger, the researcher who had originally raised hopes for leptin by showing its powerful effects in mice, show how this can happen at the molecular level. When you overfeed mice a high-fat, high-calorie chow, their fat cells scale back the expression of the genes that tell the cell how to build leptin receptor “doors” on their surfaces, so that the cell stops hearing the signal.44 Similarly, unpublished studies of the fat tissue of grossly overweight people show that their expression of the leptin receptor gene is consistently so low as to be undetectable, while lean, young people may have anywhere from quite low to extremely high levels of gene expression at any given time.45
Unfortunately, as with leptin itself, the path to using leptin receptor gene therapy as a way to reverse the negative effects of visceral fat is not a clear one. Mice with the extra leptin gene may have stayed slim in the face of an overly rich diet, but they did not fully escape its consequences: they still suffered the same “ectopic” (mislocalized) fat infiltration of their livers, muscle, and heart as did mice lacking the extra leptin receptors eating the same chow, and their insulin resistance—the key negative effect of excess visceral fat that we need to address—was just as bad.
While we might find some way of avoiding some of this by turning the gene on and off again, thereby restoring our insulin sensitivity, it seems unclear how we would deal with the ectopic fat. We might expect that as soon as we turned the gene off the calorie imbalance that had led to the initial overgrowth of fat cells would begin again; while we would shrink fat cells back down with each round of therapy, we would have no way to eliminate the cells themselves, and over the course of a greatly extended life a visceral cavity full of large numbers of even relatively small fat cells might still lead to metabolic mayhem.
![]() Really Trimming the Fat
Really Trimming the Fat
No—what seems most likely to solve the problem of visceral fat is not to try to tame it, but to cull it: actually to remove a substantial number of the bloated, excess cells. The same sorts of cell-specific targeting of cancer or anergic T cells we’ve been discussing seem likely to be applicable to visceral fat too; and while, unlike in those other cases, no specific markers of visceral fat cells have been identified, we don’t need to be nearly so specific. Unlike cancer or anergic T cells, some fat—including some visceral fat—is not only metabolically harmless, it’s necessary for carrying on the business of life. Aside from being a spare tank of metabolic fuel that we draw upon and refill every day, those metabolic factors we’ve been talking about—energy-regulating hormones, inflammatory peptides, and others—also have healthful uses. As with all of metabolism, this was built into us by evolution for our benefit. As anti-aging engineers, it’s not our job to interfere with it—just to prevent the damage that it causes in aging bodies.
![]() Night of the Living Dead
Night of the Living Dead
The last class of toxic cells that I want to address in this chapter are so-called “senescent” cells.46 They got this name because of the (rather dubious) analogy drawn between these cells and aging humans by their codiscoverer, Dr. Leonard Hayflick, then of the Wistar Institute in Philadelphia. These cells, like the others that we’ve been discussing, begin their lives as normal constituents of the skin, joints, and other tissues. They are normally quiescent, not dividing regularly, but they remain capable of reproducing themselves on demand, as part of their normal function (unlike “postmitotic” cells, which lose the ability to divide ever again once they reach their mature form and are only replaced by new cells coming from the body’s stem cell pools—if they’re replaced at all).
The defining feature of senescent cells is that they, like postmitotic cells, have lost the ability to divide. Hayflick observed, contrary to the dogma of the day, that cells from these tissues would not keep reproducing in the petri dish indefinitely: they would seem normal for several successive periods of replication, but would then suddenly enter into a twilight state in which they didn’t die, but became in various ways abnormal. Their appearance became blotchy, their shapes irregular. They failed to form the neatly whorled colonies of mutually adhering cells that were the norm in younger cultures. And above all, they stopped reproducing themselves.
The use of the word “senescent” to describe these cells is, however, a bit misleading. When people hear about these cells, they often assume that cellular “senescence” is the ultimate fate of all of the cells in the body with age, and that the entry of “young” cells into this senescent state is the underlying cause of aging. Moreover, the term evokes an image of these cells as somnambulant old has-beens, sleepwalking through the remaining days of the rest of the body’s life, not contributing anything to the organs in which they reside but also not doing us any positive harm. Their only downside, we might think, is a crime of omission: inability to replenish aging organs.
In fact, senescent cells are generally held to be extremely rare even in very aged people.47 However, their possible role in aging has turned out to be far more complex—and far more active—than we at first imagined.
The most obvious characteristic of senescent cells is, as mentioned, their loss of the ability to reproduce. But, like worn-out lechers, senescent cells desperately try to stimulate themselves into activity—pumping out substances that, though essential to their healthy function back when they were contributing members of a healthy tissue, may promote the development of cancer when present in excess. Various mechanisms are involved.
For a start, some of the most common signal molecules overproduced by senescent cells are chemical messengers like epidermal growth factor that directly spur cell division in their neighbors.
As a second example, many senescent cells also overproduce protein-digesting enzymes such as matrix metalloproteinases (MMPs), which are the “demolition teams” of tissue remodelling. These enzymes perform the essential function of clearing away the old, damaged “scaffolding” in which cells are embedded in a tissue, making space for new growth. But, just as having the outside wall of your house knocked down during a renovation would leave you vulnerable to burglary, so an excessive or uncontrolled MMP activity can enable cancerous cells to escape from the restraints of the tissue in which they were originally embedded, and/or to work their way into new tissues far removed from the original cancer site—the metastasis process.
And most recently, scientists have found yet another way that senescent cells potentially roll out the welcome mat for nascent cancers: by churning out dangerous overdoses of vascular endothelial growth factor (VEGF)48 and stromal cell-derived factor 1 (SDF1),49 which promote the growth of new blood vessels.
As you can see, the picture turns out to be a lot more complicated than we had once thought. As I’ll discuss later on, the senescence phenomenon is probably an evolved response to DNA damage, helping to prevent that damage from becoming cancerous. From the point of view of that one cell, this is an extremely effective short-term protection against cancer, because it shuts down the cell proliferation that is the very heart of the disease. But it seems likely that the “unlifestyle” that a senescent cell leads ultimately facilitates cancer progression in the long term by destabilising its neighbors. So—you guessed it—they’ve got to go.
![]() Putting the Zombies to Rest
Putting the Zombies to Rest
One way to eliminate senescent cells is to make them no longer senescent. In cell culture experiments, this has been achieved in a variety of ways, such as by relengthening exhausted telomeres with telomerase or by depleting proteins associated with senescence. But reversing senescence would run the risk of cancer, because senescent cells typically get that way as a response to potentially carcinogenic changes to the cell, such as damaged DNA, hyperactive cancer-promoting genes, or (again) very short telomeres, which promote a mutagenic state. Restoring proliferative capacity in such cells could potentially take us out of the frying pan of senescence and into the fire of cancer.
Similarly, approaches based on turning off the dangerous metabolic abnormalities of senescent cells carry risks, because other, healthy cells depend upon these same pathways for normal function. Chronically jamming growth signals, enzymes, and inflammatory messengers might well prevent senescent cells from watering the seeds of cancer, but it would also lead to “crop failures” of cells all across the body.
As usual, the engineering approach to this dilemma is to rewrite the rulebook. We will maintain the body’s evolved capacity to shut down cells in danger of going cancerous, by leaving the metabolic regulation of senescence as it is. Indeed, as we’ll see in Chapter 12, we will ultimately need to transform the body in ways that will make it almost immune from cancer by ensuring that all cells run out of steam long before they would threaten us with uncontrolled cell growth. But we will eliminate the threat posed by those cells that do senesce by eliminating the cells themselves.
![]() Silver Bullets
Silver Bullets
The first significant development in this area came in 1995, in a lab at the Lawrence Berkeley National Laboratory headed by Dr. Judith Campisi, one of my coauthors on the original SENS scientific manifesto. Campisi and coworkers found that a relatively easy, reliable test for the activity of an enzyme called senescence-associated beta-galactosidase (SA-beta-gal) could identify senescent cells not only in petri dishes, but in skin samples taken from older humans.
Unfortunately, SA-beta-gal is not a perfectly selective marker for senescence. As subsequent studies showed, the enzyme does occur in nonsenescent cells—usually at very low levels, but sometimes in high concentration. It turns out that, contrary to the simple interpretation of the Campisi lab’s findings, this enzyme is actually identical to one that is normally found in all of our lysosomes—the cellular waste incinerators, whose clogging (you’ll recall from Chapter 7) underlies many of the worst pathologies of aging. The change into the senescent state does not suddenly trigger the secretion of SA-beta-gal into the main body of the cell out of nowhere: instead, it appears that there is always some low level of SA-beta-gal floating around in even normal cells, as can be detected with techniques that assess the level of the enzyme itself in the cell—but the level is so low that its activity is barely (if at all) detectable by the methods that Campisi’s lab initially used, which are unfavorable to the enzyme’s functioning.50,51,52
But as a cell goes through round after round of replication—thereby drawing ever closer to senescence—its SA-beta-gal levels rise.53 Probably this is because the cell begins to overproduce the enzyme in response to the stresses of aging—notably, the need for more lysosomes as they become less and less effective at doing their job (and also as their cell division rate, hence garbage dilution rate, slows and the job thereby becomes intrinsically more challenging). Eventually the level gets so high that its activity is noticeable even under suboptimal conditions.
SA-beta-gal activity is, notably, found at abnormally high levels in cells taken from tissues where cells are under stress, due to inflammatory diseases that fuel cell proliferation (such as chronic hepatitis C, atherosclerotic plaques, and venous ulcers). Most interesting is the finding that levels of the enzyme shoot up in cells undergoing “crisis,”54,55 a period in which cells that have somehow escaped senescence are still undergoing cell division and the erosion of their telomeres. Such cells usually just run out of steam, but occasionally they undergo a mutation that removes the clamp from their telomerase genes, making full transformation into malignancy almost inevitable.
What’s emerging, then, is a picture of SA-beta-gal as an enzyme that appears at high level in the main bodies of cells undergoing some kind of stress that may ultimately threaten their neighbors. This might mean that by using high levels of SA-beta-gal as an identifier for the destruction of senescent cells, we would simultaneously take out some useful “targets of opportunity.”
However, we may be able to establish a system of double-checks, to help us weed out more genuinely senescent cells while leaving more innocent (but suspicious-looking) cells unmolested. This is because, in addition to SA-beta-gal, senescent cells also produce abnormally high levels of other molecules involved in the programmed senescence response. Senescent baboon skin cells, for example, contain an activated form of the protein ATM kinase, which responds to DNA damage by activating several tumor suppressor genes, including the famous p53. Senescent cells also exhibit high levels of p53, as well as the binding protein (53BP1) by which its gene interacts with ATM kinase, and p21, a senescence regulator that works under p53’s command.56 Some senescent cells also contain high levels of p16, the other main regulator of the process. Levels of this protein, for reasons as yet unknown, also climb slowly with age in nonsenescent cells, making it an unreliable marker for senescence when taken in isolation; but it—like these other features—could still potentially be used as part of a double-checking mechanism, with multiple proteins being used to distinguish genuinely senescent cells from those expressing only one of them for unrelated reasons.57
This chapter has focused on the accumulation of toxic cells with age, and the foreseeable biotechnology with which we should be able to purge ourselves of those cells as part of our platform for the rejuvenation of our bodies—restoring the immune system, alleviating metabolic mayhem, and protecting our cells from being goaded on to cancer. In the next chapter, I’ll look at the opposite problem: cell loss with age, and the scientific—and, just as importantly, political—hurdles that we face in bringing forward the ability to renew our tissues with fresh, new replacements.