12
Nuclear Mutations and the Total Defeat of Cancer
![]()
Apart from the small amount in our mitochondria, all our DNA is housed in the nucleus of our cells. Like mitochondrial DNA, it accumulates damage throughout our life, and this can theoretically lead to innumerable health problems. However, I believe that in practice only one of those problems—cancer—arises within what we currently consider a normal lifetime. Thus, if we could really thoroughly defeat cancer, nuclear mutations would be harmless. The most audacious component of SENS is just that—a way to defeat cancer altogether.
 In a few places in earlier chapters, particularly Chapter 10, I’ve whetted your appetite with regard to telomeres and telomerase. I know you have that appetite, because when someone asks me what I do and I say I work on combating aging, the commonest response (apart from the just slightly predictable “Hurry up!”) is “Ah, telomeres.” And indeed, telomeres and telomerase play a very prominent role in SENS. But not the role that most of you are probably expecting.
In a few places in earlier chapters, particularly Chapter 10, I’ve whetted your appetite with regard to telomeres and telomerase. I know you have that appetite, because when someone asks me what I do and I say I work on combating aging, the commonest response (apart from the just slightly predictable “Hurry up!”) is “Ah, telomeres.” And indeed, telomeres and telomerase play a very prominent role in SENS. But not the role that most of you are probably expecting.
As I’ve stressed throughout these chapters, the “engineering” approach to combating aging is fundamentally different from conventional thinking about aging and what to do about it, in that it focuses on the actual damage that the aging organism accrues, rather than the metabolic processes that cause that damage to accumulate.
This operational definition of aging makes the problem tractable. In the old-school, “gerontological” approach, the number of potential contributors to the aging process is legion, and getting control of all of them is a paralyzingly daunting task. It requires us to have a detailed understanding of an enormous number of complex pathways, interference with any of which is both difficult and bound to cause unwanted side effects as the normal function of those pathways is perturbed.
Anti-aging engineering sets us largely free of these problems. We leave metabolism to carry out its necessary but messy work, and find ways to undo or render harmless the relatively small number of fixed changes—molecular damage, in other words—that occur in the actual structure of the aging organism as a result of those processes. We are left with a field of just seven classes of damage to deal with—classes for which solutions are foreseeable, and whose repair is unlikely in itself to cause any negative side effects. All that we intend to remove is initially inert but eventually pathogenic (pathology-causing) damage—aspects of the aged body that the young organism does just fine without.
There is, however, one apparently gargantuan hole in this logic, and that is the question of damage to the DNA code in the cell’s nucleus (as opposed to the DNA held in the mitochondria, which I discussed back in Chapters 5 and 6). Whereas mitochondrial DNA is responsible only for the production of the energy factories in which it is housed, nuclear DNA is the master blueprint from which our entire biological structure is built up and maintained over time. The proteins1 that it encodes not only make up essential structural features of the body, from the lens of the eye to the pumping muscles of the heart and the miles and miles of arteries that carry blood to our cells, but also include tiny enzymatic machines that do everything from detoxifying poisons to building up fatty membranes and carrying chemical signals from one cell to the next. Damage the DNA, and you corrupt the code of our genetic program, or render perfectly good genetic instructions unreadable by the machinery that transcribes them into orders to be sent out to the body’s protein-making “factories.”
And your genes do suffer accumulating damage over time. The DNA in the nucleus is subject to a continuous assault on its structure. Each cell’s nuclear DNA takes about a million damaging “hits” every day, caused by everything from ultraviolet radiation and environmental toxins to the free radical by-products of its metabolic processes. And even brand-new DNA isn’t necessarily pristine: when the cell replicates itself, errors perpetrated by the machinery that copies the cell’s genetic information often create production flaws of varying degrees of seriousness.
Much of this mischief is quickly fixed by the cell’s elaborate quality control system for DNA, but some of it is irreparable by its nature. Some other damage is potentially reparable, but becomes indelible if the cell divides before repairs are made. Such permanent changes are mutations, and while mutations that occur elsewhere than in sperm and egg cells (and their progenitors) will not be passed on to the organism’s progeny, they will be perpetuated in the cell in which they occur and in any of its “descendents.”
On top of damage to nuclear DNA itself, there is damage to the so-called epigenetic structures of our chromosomes—the “scaffolding” that is anchored to our DNA. Epigenetic structures contribute important information by determining which genes are turned on in a cell and which are turned off, allowing the same overall DNA to be used to create cells as diverse as liver, heart, and kidney cells. Because of this, changes in the epigenetic scaffolding of a cell’s DNA ultimately have the same range of functional effects on the cell as changes to the genes themselves: By turning on genes that should be turned off (or vice-versa), or increasing or decreasing their activity, these “epimutations” change the complement of proteins produced by the cell. Because they are operationally equivalent in terms of their impact on cell function, I’ll allow myself a little terminological sloppiness to avoid belaboring the point with extra verbiage. From here on, I’ll mostly be using “mutations” to refer to both these kinds of genetic damage—true mutations and epimutations.
Because they occur on an occasional, random basis and are permanent, mutations accumulate with age—and therefore, they qualify as “aging damage” by the definition embraced by the anti-aging engineer. The implication, then, is that we will have to either fix them or render them harmless if we’re going to keep the body from progressively declining into pathology over time.
![]() It Is Broke. We Can’t Fix It
It Is Broke. We Can’t Fix It
Having read this far, you may be expecting that I’ll propose a fix for mutations similar to those I’ve suggested for AGE cross-links, unwanted cells, or lysosomes: just get rid of the junk. But a moment’s reflection should show that this can’t be done. Damaged genes may be dysfunctional, but we can’t afford to just do without them: a cell with anything other than a functional gene is still damaged. And that presents a daunting challenge—so daunting, in fact, that for a time I despaired of its solution, and feared that mutations would act as ship-smashing cliffs to any ark that we might build to survive the deluge of metabolism and emerge into an ageless future.
So, you may think, If we can’t afford to destroy defective genes, can’t we repair them instead? Unfortunately, this is a technical near-impossibility in the foreseeable future, for the simple reason that there are so many different genes in the nuclear DNA. The human nucleus has two copies of nearly all our genes; the aggregate size of one copy (the “haploid genome”) is about three billion “letters” of DNA. Exactly how much of this is really actual instructions for building and regulating the body is a matter of some debate, but certainly there are plenty of different places where damage to a “letter” could cause the misspelling of a “word,” leading to a loss of information that could harm the function of the cell. That’s a lot of potential damage that we would expect to need to fix.
The problem is how to do that for so many distinct genes. Any mechanism we might use to fix a damaged gene would have to somehow “know” how to distinguish an intact gene from a damaged one. True, the body already does this, by comparing the damaged DNA to its complementary strand in the double helix (or sometimes to the homologous chromosome, i.e. the other copy of the relevant stretch of DNA—remember I just told you that most genes are present in two copies in each cell), but it’s not clear that this helps us much. It’s hard to see how we could improve on our existing, inbuilt ability to repair DNA, which in humans is already amazingly effective.
We could, in principle, solve this problem if our DNA-repair system relied on a blueprint independent of the genetic information within the cell. But this would require a molecular-level tool that would carry in itself a master copy for each of the many tens of thousands of genes (each of which is typically a thousand or more DNA letters in length). It’s conceivable that advanced nanotechnology might somehow be able to provide this level of detail to machines that could then carry out the repairs,2 but only in the very distant future; we’re nowhere near that level of proficiency today.
In fact, this summary actually understates the difficulty of the job we would face if our solution to mutations were to fix them one at a time. You’re probably visualizing that these double-copy gene mutations are nice, clean breaks cutting across the two strands of the DNA helix—and sometimes they are. But sometimes, the DNA is either initially hit, or is mistakenly “fixed” by the body, in a way that leaves two distinct errors on either strand, separated by a dozen or so DNA letters. This leaves gaps whose repair, even given a proper blueprint, would entail first fixing the damage to both sides independently and then realigning the two strands and zipping them back together.
This one really had me stumped back in July 2000, at the conference where the engineering approach to developing anti-aging biomedicine first crystallised in my mind. If we can’t just throw out damaged DNA, and there’s no clear way to fix all of the possible forms of permanent DNA damage that accumulate with age, aren’t we stuck with a mechanism of aging that we can’t do anything about—and that will therefore kill us even if we repair or obviate every other molecular and cellular change that contributes to aging? At first I essentially ignored the problem, relying that other approaches to cancer would suffice, but I became increasingly doubtful that they would.
![]() Who’s Afraid of the Big Bad Mutations?
Who’s Afraid of the Big Bad Mutations?
Throughout these chapters, I’ve always explained how a given kind of molecular or cellular change (“damage”) probably contributes to aging pathology. But in most cases, it’s not totally clear to what extent a given kind of alteration actually adds to the loss of function, increased disease risk, and exponential rise in death rates that characterise biological aging. In all the cases we’ve discussed so far, however, the answer to this question doesn’t really matter. Aging damage is, by definition, not a part of a healthy, youthful body, so removing or obviating that damage certainly won’t do us any harm—and, from what we can tell, will almost certainly do us significant good.
In this case, however, I couldn’t see a clear way either to repair or to render harmless mutations in the nuclear DNA. So I had to take a step back and ask a more fundamental question: Do we, as a practical matter, actually have to worry about nuclear DNA damage?
Even to ask such a question sounds a bit crazy to some of my colleagues. Because the nuclear DNA is so clearly critical to cell structure and function, it seems beyond debate that mutations are a contributor to aging. This concept was first formally proposed in the late 1950s, before we really even understood what genes were, and it has become almost universally accepted by both scientists and the public at large.
But, however initially intuitive it may sound, this notion has never been justified with direct (or even good indirect, i.e., correlative) evidence. The only way to rule out something’s involvement with aging is to speed it up and observe no effect on life span or age-related pathology.3 This has been accomplished for nuclear mutations, but not yet well enough to be decisive. In mice, deleting a gene that normally fixes up free radical hits to genes before they have a chance to go on to become actual, fixed mutations greatly increases the steady-state level of such damage in the nuclear DNA. Yet these animals appear to suffer no pathology as a result, and have normal life spans.4 However, we can’t read too much into this result because the mutation rate is only elevated rather modestly. Similarly, a study was recently performed using four strains of mutant mice, each with a knockout of a different DNA-repair gene. In one such strain, mutations accumulated more than in normal mice—and yet, the effects on lifespan were unclear.5
A very good, and equally direct, test for showing that something is a key contributor to aging is to slow it down or arrest it and observe a direct anti-aging effect: an increase in the “natural limits” to the organism’s life span and the preservation over time of youthful functionality. Of course, no one has ever done this with nuclear mutations, or indeed with anything else. Unfortunately for scientific clarity, all the successful anti-aging interventions that we currently know of in mammals change many things about the organism, from antioxidant enzymes, to maintenance of proteins, to the activity of the cell’s garbage-disposal system. This prevents us from isolating any one of those changes as the dominant cause of the anti-aging effect, and thus isolating any particular type of damage as being the dominant contributor to aging. Calorically restricted animals, for instance, lose a significant amount of bone mass, but no one thinks that this loss is responsible for CR’s anti-aging effect.
A close approximation of such a test, however, was published a couple of years ago, in the form of mice that had been given genes allowing them to produce extra amounts of an antioxidant enzyme (catalase), specifically targeted to different parts of their bodies.6,7 I mentioned these mice in earlier chapters, but the study is worth a recap. Catalase detoxifies an abundant oxidizing molecule (hydrogen peroxide), so it can potentially protect things it’s close to from at least one form of damage. Putting catalase into these animals’ mitochondria, which significantly reduced the development of mitochondrial DNA deletion mutations, reduced their vulnerability to several age-related diseases, and extended their maximum life span by about 20 percent—the first unambiguous case of an antioxidant genetic intervention with an effect on this key measure of aging in mammals. Yet giving these organisms catalase targeted to the nucleus, which reduced nuclear mutations, provided no benefit in terms of lifespan.
Another type of evidence that is often raised in support of the idea of nuclear genetic damage as a contributor to aging is the existence of so-called “accelerated aging” models whose symptoms arise from accelerated rates of nuclear mutation accumulation. These are animals either with various inborn mutations, or subjected to outside assaults (like bombardment with toxic chemicals or X-ray radiation), that increase the accumulation of nuclear DNA damage as they age, either by increasing the rate at which it is formed or by knocking out the machinery that repairs it. This includes such human genetic diseases as Hutchinson-Gilford syndrome (“progeria”) and Werner’s syndrome.
Victims of these diseases, whether they walk on two feet or four, do often look in many ways like the elderly, suffering pathology that can be eerily parallel to that seen as animals get older, from bone diseases and failing hearts to scruffy fur and cataracts. But the fact that the symptoms of an abnormal pathology look a lot like the symptoms of “normal” aging doesn’t prove that the mechanisms of one underlie the mechanisms of the other, any more than a wet lawn proves that the same mechanisms govern rainfall patterns and sprinkler systems. Almost anything that messes up normal equilibrium in the body but takes a while to kill you will look like “premature aging;” the question is what if any relationship a given change bears to aging in the rest of us. Evolutionary biologist Michael Rose, a geneticist at the University of California who has himself bred remarkably slow-aging, long-lived fruit flies, puts the point succinctly: “A lot of people can kill things off sooner, by screwing around with various mechanisms, but to me that’s like killing mice with hammers: it doesn’t show that hammers are related to aging.”
Because we don’t have this kind of direct evidence, we normally rely on more indirect, correlative types of evidence of a phenomenon’s involvement with aging. One is to compare the rate of accumulation of some kind of damage in animals that age at different rates (that is, operationally: whose “oldest old” finally succumb to “natural” death, after first progressively losing youthful functionality, at different chronological ages). While all aging animals do accumulate nuclear mutations, the rate at which longer-lived animals suffer free radical damage to the nuclear DNA doesn’t correlate well with their maximum life spans (unlike the corresponding rate in mitochondrial DNA, which does).
Frustrating, isn’t it? But the good news is that, in my view anyway, the available evidence allows us to conclude with some confidence that nonspecific nuclear DNA damage is not meaningful contributors to aging. I briefly summarized the argument for this conclusion back in Chapter 4; here’s the full story.
What gives force to the idea that nuclear mutations are a cause of aging is the fact that the mutations we inherit from our parents are major risk factors for a wide range of diseases, including age-related diseases from cancer to heart attacks to Alzheimer’s. The same is sometimes true of mutations that occur very early in development, when there are so few cells in the little ball that will eventually transform itself into a human body that damage to the nuclear DNA of just one of them can infect almost our entire being with the same flaw.
But the situation is quite different after we’re born, which is when new mutations occur and accumulate with the passage of time—and thus, where we have to think about an effect of nuclear mutations as a form of aging damage (as opposed to as a source of inborn vulnerability to aging diseases). This is because mutations only spread from one cell to another when the second cell’s DNA is actually derived from the DNA of the first, as happens when (and only when) a cell divides and passes a copy of its DNA on to its progeny. Thus, whereas inherited mutations infect all of the cells in our mature bodies (because all of our mature cells derive from a single, mutated fertilized egg), age-related mutations happen in our mature bodies one cell at a time, as a result of random events like the radiation that comes into a plane at high altitude, or toxins produced by invisible mold spores in your food. Any such mutations can only affect the one cell in which they occur, and its descendents.
This fact greatly limits the potential of age-related mutations to become prevalent enough in our tissues to impair function. A lot of our cells—including the ones in tissues where the impact of aging is most clear, like the brain and heart—don’t divide at all once we mature, so any mutations in such cells go no further. And even in cells that do divide—skin cells, say, or the cells lining your gut—the rate of cell division after we mature is balanced by the short life of the cells’ descendents, so that an individual cell’s mutations are present in only a few cells in the body at any instant and thus get little chance to “take over” the tissue in which the mutation occurs.
Also, even before we had hard numbers on the subject, we knew that complete “knockout” mutations would be relatively rare occurrences. For one thing, most mutations in DNA create errors in the encoded proteins that, if they affect proteins at all, only make them less effective, rather than completely dysfunctional. But even in cases where a gene is damaged so badly that either it can’t be used as the basis for protein manufacture at all, or else the product that it produces will be useless—or even toxic—still generally no harm will ensue, because the damage will be to a gene that isn’t even being used in that cell! Remember that the DNA of each cell includes not just the genes needed for that cell to do its specialised job, but the entire instruction manual for producing every cell type in your body. Individual cells acquire their specialized function—heart cell, liver cell, kidney cell, skin—by turning off most of their DNA, leaving active only those genes that they require to perform their particular function. In a typical cell, only about one tenth of a person’s full gene complement is active. So about 90 percent of the genes in a cell could be irreparably damaged without affecting the cell’s function one bit.
It gets better yet. Even though evolution is far too thrifty to allow cells to go around wasting their resources in producing proteins that aren’t important to cell function, the cell can nonetheless suffer the loss of many proteins and still limp along without harming the body, even contributing in some degree to its internal economy. Remember, many people are born with such mutations affecting every single one of their cells, including all the cells where that protein is normally needed to do their jobs, and these people still live for decades. The same mutation occurring in only a small fraction of one’s cells might not even be noticed in a normal lifetime, being compensated for by the fact that the rest of one’s cells are fully functional.
![]() Mutations: Few, and Not Far Between
Mutations: Few, and Not Far Between
And sure enough, the actual number of mutations that accumulate with aging does indeed appear to be rather low—too low to have any serious effect on the aging of the tissue in which they appear. We know this thanks in large part to the work of a team headed by physiology professor Jan Vijg, now at the Buck Institute for Age Research. Vijg’s group came up with a very ingenious way to obtain a representative sample of both the amount of mutation that accumulates in different tissues with age, and the general kind of damage that occurs.
What Vijg’s group found was surprising to them and to a lot of other researchers. They discovered, first, that there were not nearly enough relatively minor point mutations (“one-letter” mutations in DNA “words” or “sentences”) to realistically have a meaningful impact on overall tissue function or on the aging of the organism as a whole.
More important, the number of mutations in a typical cell doesn’t even increase between early and late adulthood in our most critical tissue (the brain), and the total burden of mutations only goes up by a factor of two to three in any tissue, even in old age.8,9 This may sound like a lot, until you remember how many genes there are in such a cell and how few mutations are present in young people. Doubling or tripling a small number of initial mutations still leaves the cell with only a small number of them; and when you consider how few of those are occurring in genes actually used by the cell in which they occur, how few of that subset actually disable (as opposed to merely inhibit) a critical function of the cell in question, and how little the loss of a particular activity in a particular cell can really affect the overall function of a tissue, an organ, or an entire animal, it becomes clear that an increase like this is actually more of an inconvenience than a crippling blow to the aging organism.
But Vijg immediately appreciated that this was not a knockout blow to the involvement of nuclear mutations in aging. This is because some of the mutations identified by his group may be much more severe than the disabling of a single gene. Some of them, for instance, could take the form of deletions—the total removal of large stretches of DNA, annihilating many genes at once even though the event is, strictly speaking, only a single mutational event. Deletions can occur when two widely separated breaks occur at the same time in the same chromosome, and Vijg’s data showed that a significant proportion of the mutations that accumulate with age do involve such breaks, which at first glance suggests there are a significant number of deletions amongst the modest increase in the total number of mutations that happens with aging. This would have a much greater impact than would be suggested by a simple tallying-up of mutation frequencies.
Luckily, however, it turns out that most of the cases in which a cell suffers two distinct breakages of DNA don’t actually lead to deletions.10 Fully half of them are actually cases of fractures happening on two separate chromosomes, which are not physically linked to one another and thus do not lead to a deletion—much as if you cut two separate pieces of string across their respective middles and switch the pieces’ partners, rather than making two snips in the same length of material and discarding the portion between the two cuts. And even in the remaining cases, where both breaks occur on the same chromosome, many of the incidents are equally harmless, leading to mutations called inversions that shuffle chromosomes around while typically leaving them intact and functional.
For this and other reasons, not many of the events that at first look like deletions actually seem likely to have much more effect on actual genetic integrity than simple point mutations.
When we look at epimutations, the evidence available so far leads me to the same conclusion. The most well-studied kind of epimutation is changes in methylation—a chemical alteration of genes that prevents them from being expressed. Middle-aged mice11 and humans12 have indeed been found to have more alterations in their methylation patterns than immature ones do, but it’s not clear that that trend continues further as the organism actually ages (as would be required of a true, stable form of molecular aging damage). Instead, as they go from adulthood into true old age, the change in methylation epimutation may stop accelerating and may even slow down. This could mean, for instance, that the observed methylation changes occur in the early life of the organism, but that their frequency is subsequently held to tolerable levels by repair mechanisms or even by the elimination of unacceptably aberrant cells, rather than accumulating with age the way that AGE cross-links or mitochondrial mutations do. Just as important, from our perspective as would-be interveners, there’s no evidence that the observed methylation changes cause any actual functional problem over the life span.
Add it all up, and there seems to be remarkably little increase in mutations happening in aging cells—and of what there is, again, very few will actually affect the cell’s functionality.
![]() The Times, Not the Genes, They Are A-Changin’
The Times, Not the Genes, They Are A-Changin’
Vijg’s studies of that period, and those of a few of his forerunners, assessed the impact of mutations on aging by measuring the actual frequency at which genes are structurally altered during aging. But there’s another kind of evidence that’s often invoked to support the role of mutations in aging: gene expression studies. Starting in the late 1990s, scientists were able to use a new technology informally known as “gene chips” to measure the activity, rather than the structure, of nearly all the genes in the cells of tissues. This allowed for comparison studies of aging and younger animals—including humans.13
The results clearly showed that there are pretty substantial changes in gene expression in the cells of aging animals. This result has often been mistakenly thought to prove the importance of mutations in aging, because the gene expression shifts were assumed to be the result of mutations in the genes themselves. This idea was given some seeming support by evidence that the changes in gene expression happened alongside markers of “pre-mutagenic” free radical damage to genes—damage that can still be repaired as it stands, but that can go on to become full-blown mutations if not repaired properly.
But there’s a much easier explanation for these shifts, and that is the very fact that gene expression does change—not just with aging, but all the time. Your cells are in a state of continuous dynamic adaptation to their environment, changing gene expression in response to new conditions at every moment. Every time your body needs to respond to its environment, the expression of genes involved in those responses is altered.
So as cells and tissues acquire aging damage that impairs their normal, youthful function, the body adapts to the changed circumstances as best it can. As oxidative stress climbs with the accumulation of cells that have been taken over by mutant mitochondria, cells ramp up the activity of genes that produce protective antioxidants and “heat shock” factors that help to repair some of the ensuing damage to proteins. When the increase in oxidative stress leads your arteries to become infiltrated with free-radical-damaged LDL, the surrounding cells produce more inflammatory factors to attract macrophages to clear the toxic stuff out. When your heart stiffens as it becomes riddled with cross-linked proteins, the body produces more “remodeling” enzymes that help to degrade the old tissue in hopes of clearing the way for fresh, undamaged replacement material.
Additionally, changes in the cellular environment imposed by aging can also interfere with normal gene expression. In some cases, such effects are easily observed: for example, oxidative stress directly inhibits the expression of some genes. But there are also more subtle effects afoot. For instance, you’ll recall that free radicals, in addition to being produced as simple side effects of metabolic forces, are also produced intentionally as signaling molecules in various systems inside and outside cells. As free radical levels climb in the aging organism’s cells, the excessive oxidative stress distorts these same signalling pathways, introducing “noise” into them. This can in turn result in inappropriate metabolic and gene-expression shifts as the cell responds wrongly to misheard, drowned-out, or counterfeit messages from (or superimposed on) the system.
And so on. The point is that the age-related shifts in gene expression seen in studies of tissues are not the cause of aging, but are an adaptive (and sometimes maladaptive) response to it. Changes in gene expression in cells subjected to free radical attack can occur not because that attack has damaged the genes whose expression levels change, but because an increase in oxidative stress requires the cell to change its metabolism to keep going in face of the challenge.
Now, you may be thinking that I’ve jumped the gun here. So far, I’ve explained how gene expression changes can be a compensatory response to aging rather than a cause of aging—but surely I have not shown that those changes are, overwhelmingly, responses and not causes. But it’s easy to see that they must be responses, because they’re coordinated. The only reason the studies I’m referring to were able to detect a change in expression of a given gene was because it was occurring in the bulk of the cells within the tissue being analyzed. Mutations and epimutations, by contrast, would affect one gene in one cell and a different gene in the next cell.
This coordinated change of gene expression in response to aging is shown most clearly in gene-chip studies that compare normally aging animals to those undergoing calorie restriction (CR),14,15,16 which is, again, the only intervention now at our disposal that slows down aging in mammals, short of tinkering with their genes. These studies show that the greatest number of changes in gene expression that happen with aging occur in the blueprints for antioxidant, inflammatory, remodelling, and heat shock proteins—exactly the ones needed to respond to the damage inflicted by aging. They also show that CR not only slows down, but reverses, many of these changes when it’s implemented late in life. This shows that the changes in gene expression can’t be the result of mutations, because while CR can slow down the accumulation of mutations and other aging damage, it cannot undo damage that has already been done. Rather, it reduces the sources of damage that are leading to the changes in gene expression, cutting down free radical production in the mitochondria, reducing the accumulation of mitochondrially mutant cells, lowering blood glucose to prevent glycation cross-links, and so forth. Change the cells’ pro-aging environment, and they scale back their gene-expression adaptations to that environment.
This sort of logic has led some, among whom Vijg’s team are again prominent, to conduct much trickier studies assessing the variation of gene expression from one cell to the next in old animals as compared with young ones It was found that there is a dramatic increase with age in that variation.17 On the face of it, this increased variability doesn’t seem likely to be the result of the kinds of adaptation that are responsible for the dominant pattern of age-related gene expression shifts that we were talking about a moment ago, because the conditions that create them—and the adaptive responses needed to keep going under their influence—are more or less the same from one cell to the next in a given tissue.
But there are a lot of alternatives to explore before we jump to the conclusion that this increase in the amount of difference in gene expression between a given aging cell and its neighbors is the result of mutations (or epimutations). It may be due to some other age-related stressor, for instance. Vijg’s team found that they could reproduce the increase in variability by exposing cells in culture to oxidative stress—and again, oxidative stress disrupts the ability of cells to relay signals within themselves. Such disruptions could lead to a failure to respond to signals coming in from the cell’s environment—either local signaling molecules from its neighbors, or more “broadcast” signals like hormones and other factors. The effect of this would naturally vary from one cell to the next, because it results from noise in intra-and intercellular signaling, but if the oxidative stress were then removed, the variability might return to its original level.
Variability from one cell to the next can also result from other kinds of damage that have accumulated, randomly, to different degrees from one cell to another: telomere loss in one cell, high levels of AGEs in another, and the presence of mitochondrial mutations (without nuclear mutations) in still a third. Intuitively, the gene expression shifts required to increase the degradation of AGE-ridden proteins on the surface of heart muscle cells are quite distinct from those that occur in response to accumulated junk within them. When neighboring cells suffer different levels of different kinds of molecular damage, they mount different adaptive gene expression responses, both to counterbalance the effect of the damage on their ongoing metabolic processes and in many cases to attempt to repair it. Such effects have been observed at the cellular level in roundworms,18 and preliminary studies suggest that similar factors could be at play in increases in gene expression variability in rats and humans, too.19
Also, such damage can send shock waves outside the cell in which it occurs, which will elicit changes in gene expression in nearby cells trying to cope with its impact. Consider cell senescence, which I discussed in Chapter 10. The activation of the senescence program will certainly change the expression of genes in the senescent cell as compared to its neighbors, but it will also change the expression profile of those neighbors, as they respond to the flood of growth factors, inflammatory signals, and remodeling proteins that it produces. Cells closer to the senescent one will be more affected than cells further away.
Importantly, these changes would distinguish the neighbors of a senescent cell both from the original culprit and from other cells not directly affected by its output. To pick the most obvious example: growth factors secreted by a senescent cell will trigger cell-replication programs in nearby cells. These programs, executed by gene expression changes, are ones that have been permanently shut off in the senescent cell itself, and that are quiescent in cells far enough removed from the senescent cell to evade its influence.
The same would be true of cells that had suffered any number of problems. And the variation can flow in the opposite direction, too. That is, in addition to damaged cells playing havoc with their neighbors by exporting their own internal woes, healthy cells can communicate with damaged ones to keep them functioning more normally. The best-researched case of this is cancer.20,21 A single cell can harbor mutations that would by default lead to malignancy, but be held in check by its neighbors, which can do everything from inhibiting its proliferation (so-called contact inhibition) to inducing apoptosis. As with all other adaptive changes, the output required to impose this control requires shifts in gene expression that will distinguish cells working to keep their antisocial neighbor under control from the patterns of both the would-be cancer cell and noncancerous cells elsewhere in the tissue.
Hence, even the fact that there are more pronounced differences in the activity of genes from one cell to the next in the tissue of old animals (as compared with young) doesn’t necessarily pin the blame on nuclear mutations. Moreover, much of the variability observed is a good thing—the distinct efforts, futile though they may ultimately be, of each cell to preserve its integrity under the unique burden of aging damage that each of them faces.
If, then, age-related gene expression changes don’t flow out of a significant increase in nuclear mutations, but are instead the result of other kinds of damage (and cells’ attempts, successful and otherwise, to carry on in the face of it), the proper engineering response is to leave nuclear mutations alone, and focus our attention on the real culprit in age-related gene dysregulation: the aging damage that forces our cells to flail about in increasingly desperate, disorderly, and panicked attempts to keep their heads above the waters of the aging process. Once our cells are no longer suffering under the onslaught of problems ranging from mitochondrial mutations to AGE cross-linking to visceral fat accumulation, their gene expression profiles can be expected to normalize, because both they and their environment will be normalized—returned to a youthful state of functionality.
Ironically, it may be precisely after we have cleaned up all of these sources of damage that nuclear DNA mutations may, eventually, begin to contribute to age-related death. The evidence shows only that cells accumulate relatively few nuclear DNA mutations, and those mutations do relatively little to impede cell function within a normal lifetime. But what happens when that life span is extended to centuries, with the same amount of metabolic damage to our DNA occurring, and with our DNA-replicating and DNA-repairing machinery no more perfect than it is today? It may well be that effects that are too subtle ever to reach a pathological stage within nine decades or so could build up into a real threat over the longer term.
However, we don’t need to worry about that now. As I explained in respect of AGE cross-links in Chapter 9, one cornerstone of the engineering approach to developing anti-aging biotechnology is that we don’t need to fix all possible forms of damage at once—we only need to do a good enough job of cleaning up those insults that meaningfully contribute to age-related frailty within today’s life expectancies. Once this is accomplished, our bodies will remain youthful during the years in which they are now undergoing a slow descent into decrepitude. Some forms of molecular damage that aren’t causing us problems now will then begin to reach the threshold beyond which we begin to meaningfully, functionally decay.
At that point, the next generation of SENS therapies will need to be brought in, to repair that newly pathological (and in some cases even newly identified) damage to the same standard. Fortunately, our extended lives will buy us the time to observe such damage accumulating in our bodies, and in those of shorter-lived animals whose lives have been extended in the lab, and whose youths renewed, using the same treatment—and as time goes by, we’ll build better and better tools for identifying that new damage and for developing weapons to fight it. The key is to get past the barriers that hold us back today, and then be ready to break new ones as they approach. From all available evidence, nuclear mutations aren’t yet such a limit—but they may well be one in the future.
![]() The Exception That Creates the Rule
The Exception That Creates the Rule
So far in this chapter, I’ve explained that one of the reasons not to worry too much about nuclear DNA mutations is that even the ones that are actually harmful to the cell are usually minor and, even when they are major, the damage that they inflict is mostly constrained to one or a few cells, so that any slack that their dysfunction creates can be picked up by other cells in the tissue. But there is, of course, one screamingly important exception to this rule: cancer. Cancer is famously a disease of nuclear DNA mutations (though, as we’ve seen, it usually takes more than just mutations to turn an aberrant cell into full-blown cancer). And the incidence of cancer clearly goes up with age.
Shortly, I’ll discuss how I think we can really defeat cancer. But first, I’m going to explain why cancer is the reason for our apparently “unnecessarily” good defenses against the accumulation of nuclear mutations.
First, let’s recall that one of the reasons cancer is so formidable an adversary is that any number of different mutations can contribute to the cancer process. A breakdown in any one of the many systems that protect against cancer—mutations that lead to the formation of a defective senescence or apoptosis “tumor suppressor” protein, or to the excessive production of a cell receptor for growth signals, or to the reactivation of a suppressed telomerase-coding gene—can be a key step along the broad and winding road that leads a healthy cell to become a renegade.
As with all other causes of age-related death, the evolved level of protection against potentially cancer-causing mutations is limited by the cost of creating and maintaining the machinery to achieve that protection. On the one hand, there’s no sense in putting all of an organism’s eggs into the basket of being so resistant to aging processes that it can stay young and healthy for two hundred years, if there are strong odds that it will freeze, starve, sicken, or become something’s lunch in its third decade; those resources would be better spent on warmer fur, sharper claws, or simply a shorter gestational period. But on the other hand, the animal does need to remain internally intact for as long as it can reasonably be expected to survive those threats from the external environment, because every year of youth is another opportunity to spread one’s genes around. If you’re unsure about this, go back and reread Chapter 3.
Given those opposing priorities, natural selection will push hard to create machinery that protects against potentially cancerous mutations rigorously enough to keep cancer at bay for at least as long as the organism would likely get through winters, wars, and attacks from predators. But, precisely because of the nature of the cancer threat—that so many alternative mutations can contribute to the cancer—the body can’t afford to cherry-pick the genes that it’s going to protect. The only effective defense against cancer is to safeguard the integrity of every single gene we possess.
So this is how it comes to be that new, age-related nuclear mutations are so rare—why we possess such an elaborate system of oversight of DNA synthesis repair guarding our every gene, despite the fact that the great majority of mutations have negligible effect on the overall economy of the body. To protect the organism adequately against cancer, evolution gets the best bang for its buck from a system in which every gene gets the kind of gold-plated antimutation defense that you might think it would reserve for a privileged few, mission-critical, cancer-avoidance genes.22,23
![]() A 2015 Challenge
A 2015 Challenge
This analysis shows that we don’t need to address all mutations in order to develop a panel of interventions comprehensive enough to result in the first dramatic extensions of human life span. My realization of this was crucial to the nascent development of the SENS platform back in 2000, because it showed me that the sheer scope of the nuclear mutation problem was in one sense much smaller than I had at first feared. It had become clear to me that, for the most part, age-related accumulation of nuclear mutations are basically harmless over the course of a currently normal lifespan: the rate of their accumulation is quite insufficient to contribute significantly to age-related decline. But we emphatically do need to confront the one enormous exception to that rule: cancer. In principle, we can simply ignore nuclear mutations, if and only if we can find a truly effective way to protect ourselves against this one fatal disease.
The stakes here are high. Cancer is a deal-breaker for building an ageless organism. We can shatter the cellular manacles of AGE, free our brains and hearts of the webs of amyloid, clean out the dirty depths of our lysosomes, and all the rest—but if we fail to make a breakthrough against this one disease, we can still expect to be dead in our mid-eighties.
If you’ve been listening to the popular press accounts of progress in the War on Cancer, you may now be feeling much less alarmed than you should. The media, and also the scientists and bureaucrats on whom they report, love to trumpet every advance (indeed, every hint of an advance) in the treatment of cancer. There are so many reports of potential new cancer treatments that one might well think we were already far down the path to the day when we will have cancer mastered. This is especially so in light of the targeted cancer therapies that I reviewed in Chapter 10—therapies that generally are, or can be predicted to be, far safer and more effective than the knives, poisons, and radiation that have been the staples of cancer management for decades.
And you wouldn’t be alone, nor even outside the scientific mainstream, in thinking this. In 2003, none other than Dr. Andrew von Eschenbach, the Director of the National Cancer Institute, famously put forward an ambitious but (so he claimed) realistic vision for his organization: to eliminate suffering and death from cancer by the year 2015. Dr. von Eschenbach wasn’t just whimsically putting his dreams into words: he was putting forward his sober assessment of what the world scientific community, spearheaded by the NCI, could achieve in little over a decade. It became the formal agenda of the Institute: “Challenge Goal 2015.” The timetable is now so embedded in the organization that it is routinely alluded to as simply “2015,” with no further elaboration necessary—in the same way that we once talked about “Y2K.” 24
I think that this goal is utterly unrealistic—and that it only arose because of a failure to appreciate the flaws in the assumptions that are built into it. First, and quite explicitly, “it does not mean ‘curing’ cancer but, rather, it means that we will eliminate many cancers and control the others, so that people can live with—not die from—cancer.”25 If feasible, this is a perfectly legitimate medical goal: to have cancer under the same level of control that we today have over adult-onset diabetes or AIDS—in which the disease is still present but is so well managed that patients can lead nearly normal lives—would represent an enormous alleviation of human suffering and death from a terrible disease.
But cancer is fundamentally different from these diseases in a way that precludes its chronic “management.” Diabetes and hypertension can be held at a safe, manageable level precisely because they are essentially stable diseases. By contrast, what makes cancer so fearsome a foe is that it’s a constantly evolving disease, a hive of genetic inventiveness that continuously finds new and better ways to outwit our attempts to control it. Relegating cancer to the level of a chronic disease is an idea that could only ever be entertained by completely ignoring the basics of natural selection.
Cancer cells are characterized by an immense genetic instability, which results in large part from the fact that they nearly all get started from a mutation in one or more of the “guardians of the genome”—the genes that police mutations and direct either repair of DNA damage or the activation of senescence and apoptosis programs. Without this constant surveillance and maintenance, the random damage that cells suffer every day is allowed to develop into full-blown mutations, and the process feeds on itself as more regulatory genes are lost.
Many of these mutations are fatal to the cancer cell, but a few of them result in viable progeny that are just different from their parents and half-siblings. And that’s where natural selection comes into play. Cancer cells, by definition, are reproducing themselves at an astonishing pace. They throw their bastard children out into the world and let survival of the fittest reign. The immune system or oncologists soon take their best shots at the tumor, exploiting the soft spots in the cancer cells’ metabolism: their reliance on particular growth factors, for example, or their need for a reactivated telomerase gene, or their hunger for folic acid. But within a single tumor exists such an astonishingly varied population of cells, each with its own combination of normal and abnormal genes, that at least some of those cells nearly always have a way to survive any particular attack: a greater ability to detoxify a particular toxin, or an alternative way to keep their growth fueled even when a particular signal transduction pathway is shut down.
As a result, it ultimately just doesn’t matter if a given therapy kills 99 percent of the cells in a tumor. Somewhere within its heart lurks the dark father of a “strain” of the cancer with a novel mutation that allows it to survive the drug that destroyed its cousins. This founding cell’s furious growth continues even as we decimate its cousins, or resumes when the patient can no longer tolerate the stresses of the treatment. Its descendents remain standing after the assault, and are thereby selected for survival by the very thing that killed their cousins. When the ensuing tumor becomes large enough for us to detect, we attack what seems to be the same cancer in the same patient using the same treatment, but this time, the old tricks don’t work. There really is plenty of truth in the saying that you can’t outsmart evolution.
As I sat reflecting on all of this in the wake of my original SENS “Eureka!” moment at the turn of the millennium, a grim formulation of the problem crystallized in my mind. It is not my goal, I thought, to buy time for cancer.
![]() Making Cancer Wilt Away
Making Cancer Wilt Away
Soberly, I contemplated my own cancer challenge: to develop a cancer treatment that would keep us free of clinical cancer for just as long as the other SENS therapies would keep us free of other age-related maladies. The reasoning outlined above immediately ruled out all existing approaches, which leave us fighting one battle after another against an enemy with the implacable force of evolution on its side—a war in which we can win individual campaigns but must ultimately be defeated.
The answer came to me in March 2002 while nursing a beer in a café in Italy. As my mind had done late that night in California in 2000, it now leapt upon an insight that was in some ways completely obvious, and yet led inexorably to revolutionary conclusions. To defeat cancer, I saw, we would need a therapy that does not depend on anything that a cancer could escape through a mutation-driven change in gene expression. So any solution would have to have three key characteristics to be viable. First, it would necessarily involve denying cancerous cells access to some tool that is absolutely indispensable to any cancer’s survival, so that they couldn’t just make up for its loss by tweaking some other gene expression pathway through mutating its other genes. Second, we would have to take away that tool in such a way that no mutation could restore it, either. And third, this tool would have to be one that our normal, noncancer tissues could do without.
I quickly saw the tool that I wanted to lock up: telomerase. I mentioned this enzyme back in Chapter 10, when discussing senescent cells; now’s the time to explore it in more detail. Our DNA comes equipped with a stretch of nonsense or “noise” DNA called the telomere. Telomeres are to our genes as the brief, silent stretch of “leader tape” at the beginning of a music cassette is to the songs on the tape: they give the “cassette player” (the DNA-replicating machinery) something to hold on to and advance over, so that it won’t skip over the essential information at the beginning of the very first “song” (gene) on the tape.
One key difference between telomeres and cassette leaders is that leaders stay intact as long as the tape does, whereas telomeres become ever-so-slightly shorter every time the cell replicates itself or is hit by damaging agents like free radicals. If it weren’t for telomerase, this gradual shortening would eventually lead to the complete loss of the telomeres in cells that replicate frequently during the lifespan, and thus the gradual erosion of the genes themselves. Telomerase periodically relengthens the telomere before it becomes critically short.
As with all of our other genes, the DNA that encodes the telomerase enzyme is present in all of our cells—but, because it’s only needed after quite a few cell divisions have occurred, it’s not needed in most cells for most or all of the time, so it’s turned off. This widespread lack of the need for telomerase is used by evolution as a key component of our defense against cancer, because having a limit to the size and renewal of our telomeres prevents our cells from replicating themselves indefinitely—the crucial hallmark of cancer.
To become a full-blown cancer (as opposed to a cell with a single, potentially threatening mutation—a genetic risk factor for becoming a cancer) requires the accumulation of five to ten mutations, and statistically that requires multiple rounds of cell division and selection. The arithmetic is complex, but the consensus is that, to pose a health threat, cancers have to replicate at least two to three hundred times, even though a clinically relevant tumor contains “only” a million million (a “1” with twelve zeroes after it) cells, which could be achieved by “only” forty or so divisions if the originating cell had all the necessary mutations from the outset. And to be genuinely malignant (i.e., to be the founder of a colony of cancer cells that spreads its way throughout the body, as opposed to a localised tumor that could be simply removed with surgery and forgotten about), cancers must then be able to keep up the feverish pace of their replication even longer. The frenzied reproduction of cancer cells is also a key part of their ability to evade our assaults, because it is essential to their capacity to evolve new solutions to the challenges that we throw up against them.
It’s no surprise, then, that mutations that unleash telomerase from the repressive strictures imposed on it in normal cells are found in over 90 percent of cancers. The remaining 10 percent also have a way to renew their telomeres—a little-understood mechanism called the “Alternative Lengthening of Telomeres” (ALT) pathway, which I’ll discuss a little later on. Either way, without a way to renew their telomeres, the single-minded multiplication of potential cancer cells rapidly grinds to a halt as it reaches the end of its telomere “rope,” and we wind up with a tiny (and generally short-lived) lump in our bodies instead of a life-threatening, malignant disease.
If, then, we could snatch this one tool out of the hands of cancers, we would cause any and all the aspiring cancers we developed to fizzle out before they became life-threatening—indeed, before many of them even became actual cancers, because they wouldn’t get the opportunity to undergo the full spectrum of mutational events needed to give rise to the kind of renegade cell that can truly pose a threat to the body.
Of course, I was hardly the first to think the problem this far through. Several biotech companies—most prominently Geron, which first made a name for itself in telomere research—are working to develop anticancer drugs that would work by deactivating telomerase. But these pharmaceuticals suffer the same problem as all other approaches based on drugs that affect gene expression: they act as a force of natural selection against a disease with evolution at its disposal. A telomerase inhibitor would kill off those cancer cells in which it effectively turns off the enzyme (and in which ALT didn’t take telomerase’s place), but it would leave behind any cells that harbored mutations allowing them to keep on renewing their telomeres in the face of it. Different cancer cells might bear any number of variations that let them escape the drug’s effects. Some would simply crank their telomerase activity up even further; some would enhance the activity of drug-metabolizing enzymes that degrade the inhibitor; still others would change their cell surface proteins in ways that would make it harder for the drug to penetrate into the cell. Whatever the mechanism, if even one cancer cell can evade the effects of such a drug, it can act as the seed for the tumor’s renewed blossoming in a dark spring.
So, again, there was no sense in doing the job only halfway. If we’re going to snatch telomerase out of the hands of cancer cells, I thought, we must really take it away. And there was only one way that I could think to do that reliably: by deleting the gene that encodes it.
Evolution can, of course, create whole new genes—but it takes a very, very long time to do it. Indeed, very little evolutionary change actually involves the creation of new genes, or even the removal of old ones, precisely because it’s so hard to do: instead, evolution finds new ways to regulate old genes, or new functions for gene products other than the ones for which they originally evolved.26 Thus, for example, the lens of the eye is made up of clear, flexible proteins called crystallins, which would seem to have no purpose other than to be used to focus light. Yet, there it is in the nervous system of the sea squirt, where it is part of an organ that keeps track of “down” by sensing gravity. The gene is of course present in every cell in its body, and a mutation in a proto-eye cell of one of our ancestors that carried this gene may have caused it to be expressed there, where previously it would have been turned off, making the protein available to let the light shine on in. And our genome contains no genes similar to the ones for telomerase, ready to mutate to replace it on cancer’s demand.
So deleting the telomerase gene, unlike trying to inhibit it somehow, would be an almost sure-fire way to shut down cancer cells permanently. (Again: this logic ignores Alternative Lengthening of Telomeres but fear not—I will get to ALT shortly.) I had hoped back in 2000, and even speculated in the paper arising from the first SENS workshop,27 that there might be some way that we could do this only in cancer cells. I became increasingly aware, however, that while it might well be possible to do this for most cancer cells, we’d never be able to do it for all, for the same evolutionary reasons that I keep hammering on: any mechanism that targeted cancer cells exclusively would have to have some mechanism for selecting a difference between them and normal cells—and of course, those differences would have a genetic basis, leaving the flap of the “tent” open for a mutant subpopulation of cancer cells to stick its evolutionary “nose” under.
Finally, fully eighteen months later in that Italian café, I stopped trying to run from this conclusion. The only way to be sure that we were denying telomerase to cancer cells would be to deny it to all cells. What we needed, I realized, was to take the telomerase gene out of every cell in the body, along with the ALT mechanism whereby a small minority of cancer cells manage to lengthen their telomeres without relying on telomerase itself. I would soon term this therapeutic target the “Whole-body Interdiction of Lengthening of Telomeres” (WILT).
Removing telomerase from every cell in our body would preempt cancer before it got a chance to get started. But you can surely see why I took so long to explore this option—and why no one else had explored it before me. Deleting telomere elongation capacity throughout the body would also be life-threatening, because it would mean that our regular, proliferating cells (like those in the skin or the lining of the gut) would suddenly have iron limits on their ability to reproduce themselves and thus replenish tissue. From the moment that we denuded our cells of telomerase, a clock would be ticking. With each division the telomere would shorten by a notch from whatever it had been when we took telomerase out. We would be under the specter of a rather horrible death, as our stem cells went off-line one by one under replicative senescence (see Chapter 10): with each failure of a stem cell responsible for supplying key functions, the tissue would fail to be renewed and would slowly degenerate.
So, the effect of telomerase deletion on frequently dividing cells would indeed be very serious indeed—fatal, in fact, in what I calculated to be around a decade from the point when telomerase was deleted.
But hang on, I immediately thought, SENS already has a proposed solution to “normal,” age-related cell loss: stem cells. So we might just be able to deal with cell loss if we had a sufficiently sophisticated program of stem-cell replenishment—using cells engineered to lack the one linchpin function for cancer, namely telomere elongation.
Of course, these stem cells would eventually peter out, too, as their telomeres were worn down—but this is just the same situation that we face with all aging damage. The engineer knows that we don’t have to root every last trace of cellular and molecular injury out of our systems in order to build a body that will not suffer age-related degeneration and death. The tissues of a twenty-year-old are already riddled with aging damage, and the level climbs every day, but you’d be hard-pressed to find much of a health difference between a basically clean-living person at twenty-five and the same person at thirty-five, because their level of damage at thirty-five is still beneath the threshold at which it causes functional deficits. As long as we keep it there, we will remain biologically young.
So if we introduced stem cells with nice, long telomeres in the first place, we could let them wind down and eventually be lost to apoptosis, senescence, or other sources of damage—and just top our tissues up with more stem cells before enough of those cells were lost to begin to impair tissue function. The need for regular treatments in this case would, ultimately, be no different from the need for regular rounds of AGE-breakers or of purges of anergic T-cell clones. Neglect your medicine and you will eventually suffer the consequences; keep up with your schedule, and stay young and healthy into a boundless future.
In this case, the engineer’s logic is even stronger, because the same “damage” that might eventually kill us (in this case, the running down of our telomeres) is simultaneously the very thing that we need to ensure does happen, or else we will be killed by another means (the unchecked cell division at the heart of cancer). Putting an expiry date on all of our cells, but ensuring that they are regularly replenished with new ones, erases both problems at once.
In fact, the case for deleting telomerase was even stronger than this, because placing an absolute limit on the number of cell divisions that our stem cells (and the mature cells derived from them) could undergo would actually bring us an additional anticancer and anti-aging benefit. While we often are given the impression that most age-related nuclear DNA mutations are the result of damaging agents like free radicals, radiation, and mutagenic chemicals, the reality is that most nuclear mutations are the result of errors made in copying the DNA during cell division. And while it’s almost never mentioned in the popular press, most cancers arise not in the mature cells of our bodies, but in our stem cells, where regular cell division and an active telomerase gene makes it relatively easy to take the brakes off cell growth.28 (It was, in fact, precisely my increasing appreciation of this fact during 2001 that forced me along the line of thinking that led to the WILT concept.) By cutting down on the number of divisions that our stem cells can undergo before they die, we would simultaneously reduce the number of mutations that they would ever accumulate—and thus, the risk that they might suffer the combination of mutations that would turn them cancerous.
At that point, we’d have cancer licked. No cancer could reach a clinically significant stage. At worst, we would end up with a few little pebble-tumors, small balls of abnormal cells that have exhausted their ability to grow, no more life-threatening than a mole or a small cyst. And our normal tissues would be preserved intact, provided that we underwent regular rounds of replacement of stem cells. See Figure 1.
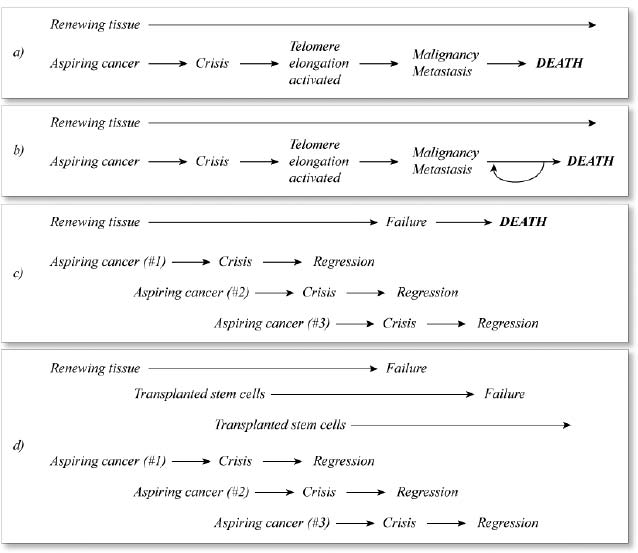
Figure 1. The effects of traditional (late-acting) cancer therapies (b), telomerase deletion (c), and telomerase deletion plus stem cell reseeding (d) on the prognosis for cancer (a).
![]() So Crazy, It Just Might Work
So Crazy, It Just Might Work
I will not conceal that, when they first hear about it, virtually all my colleagues think the WILT proposal is utterly mad. Indeed, I myself, while not doubting my own sanity, initially worried that I must surely be missing some life-threatening side effect of the two-in-one intervention that was beginning to firm itself up in my mind. So I began consulting experts in all of the relevant fields—telomere biology, mutant humans, mice lacking functional telomerase genes, the ALT mechanism, stem cells, bone marrow transplantation, and of course cancer as a disease above and beyond its characteristic preservation of its telomeres—to confirm that I had my facts straight and hadn’t neglected any actual showstoppers, and to ask them what they thought of the technical challenges facing the development of each of the biotechnologies that would be required to implement it.
Their reaction was interesting, and typical of my experience in networking with experts from different fields on interdisciplinary projects. Presented with the whole scheme, each of these experts thought the project as a whole was audacious at best, and something straight out of “soft” science fiction at worst. But to my moderate surprise, when I asked each of them to assess the feasibility and timescales for the development of the individual components that WILT would take from their own discipline, each and every one thought them achievable (albeit ambitious), and felt that nothing in the subfield of biomedicine in which they worked every day would pose an insurmountable challenge. It was in the areas that they didn’t have intimate, working familiarity with the science or the biotechnology involved that they made assumptions of intractability.
Encouraged by these discussions, I held the third SENS workshop specifically on WILT, inviting many of the experts I’d already been consulting about the field. As with the 2000 meeting, my purpose was to put the participants in a room together and simultaneously put them to work fleshing out the way forward (or else the proof that none existed) to the complete, integrated intervention, thus showing them (I hoped) that the plan was sound in its elements and as a whole.
To my enormous satisfaction, it achieved both ends, and we published the results together in the Annals of the New York Academy of Sciences.29 And of all the experts that were involved in the roundtable, only one scientist objected to being given the credit (and blame) for authorship of the paper—and her reasons were extremely revealing, as the Acknowledgements section of the paper, written with her approval, indicates. Dr. Nicola Royle, senior lecturer at the University of Leicester’s Department of Genetics and an expert on telomeres (and especially the ALT mechanism), insisted that her name be withdrawn from its author list, not because she didn’t think that WILT would work, but because she very much feared that it would. Her concern was that, as far as she could see, there was nothing stopping us from developing WILT as a final cure for cancer, and especially as part of a complete panel of SENS interventions that would finally free humanity from age-related degeneration, leading to indefinite youthful, healthy lifespans. But she was not ready to embrace that future: like so many others, her principled (but, I of course maintain, misplaced) fear about the potential drawbacks of unbounded human lifespans on the environment and on existing social structures was so great that she wanted no further part in promoting our progress toward any part of the SENS agenda.30
Let’s look at some of the technical challenges and concerns that were discussed at this SENS roundtable—and, of course, in follow-ups with these scientists and other colleagues at other venues ever since.31
![]() What Happens When We Take Telomerase Away?
What Happens When We Take Telomerase Away?
This is an obvious one. We’re talking about taking away a gene that is at least present in all of our cells, though it’s permanently turned off in tissues where the cells never have to divide, like the muscles, the heart, and the brain. At least on the face of it, we wouldn’t think that taking the gene (either or both of two genes in fact, as telomerase has two subunits) right out of the cell would cause these cells any problems. On the other hand, the enzyme is expressed and routinely used (under strict controls) by cells that need to undergo regular division—most notably stem cells. What would be the effects of taking it away from every last one of them?
Fortunately, we have some pretty reliable evidence on this point, thanks to two models: mice that have been genetically engineered to lack one or other of the two subcomponents of the telomerase enzyme, and a human inherited disease called dyskeratosis congenita (DKC). The picture here, overall, is a pretty optimistic one.
Unlike humans, mice are born with telomeres long enough to last them their whole lives without telomerase. (This makes mice a rather tricky species to use as models for human cancer, in fact.) Therefore, mice with their telomerase genes deleted have to pass their progressively shortening telomeres on through several successive generations before much of anything bad occurs. At that point, they develop the symptoms that you’d expect from a lack of stem cells, which appear first in the tissues that multiply the most. They become sterile as their sperm-forming cells run out of steam, and their guts and skin start to become depleted of cells and fragile.
They also, ironically, start developing high levels of cancer, which might at first seem to fly in the face of the entire program—but this is just one of the ways in which mice are a fraught model for human cancer. First, even though these mice telomeres are short enough to mess up stem cell proliferation, they’re still long enough to let cancers grow to a size that’s dangerous for a mouse, simply because mice’s small size allows tumors that would be harmlessly small in a human to impede organ function and siphon off a fatally high percentage of their tiny bodies’ resources. Second, mouse cells find it relatively easy to activate ALT, so the fact that these mice lack functional telomerase doesn’t guarantee that they can’t lengthen those telomeres anyway. Clever combinations of telomerase deletion with other mutations can largely sidestep these differences between mice and humans, though, and when this has been done,32,33 cancer risk went down dramatically—in one case, to such low levels that none of the telomerase-lacking mice had died of the disease at a point when all of the animals from the same strain but with functional telomerase had been consumed.
DKC patients also give us reason for hope in the midst of their despair. They have a variety of mutations that prevent the effective functioning of their telomerase enzymes—either in telomerase itself, or in genes encoding proteins needed for its normal working. Patients do hold on to some limited telomerase activity, however. The likely reason we never see people completely lacking in the gene is that humans have to undergo a lot more cell division in the womb than mice do (and have shorter telomeres than mice at conception), so fetuses with a more severe telomerase mutation are probably aborted.
But telomerase activity in DKC patients is certainly very low. As a result, their telomeres are shorter than normal folks’, and they develop predictable symptoms similar to those suffered by telomeraseless mice: mottled or web-patterned skin; patches of abnormal, thickened white cells in the mucous membranes, similar to those often seen in lifetime smokers; weak, thin nails with ridges and fissures; hair loss and lung problems; and bone marrow failure, causing problems with immunity, blood clotting, and delivery of oxygen and iron to their tissues.34
It used to be thought that the worst symptoms of DKC usually occur in people in their teens and twenties, but we now know that it isn’t quite that simple—and the reason why that’s so turns out to be very important. Dr. Inderjeet Dokal, who works extensively with DKC patients at the Department of Haematology at Imperial College in London, told me and the other participants back in 2002 at the WILT summit that he had noted that first-generation DKC patients of a particular type, the ones that have a mutation in one copy of a telomerase gene, don’t develop symptoms until they’re in their forties—but that, as they pass on their preshortened telomeres to their children and grandchildren, those that inherit the disease develop symptoms earlier and earlier. He and colleagues later confirmed this preliminary observation in rigorous studies of several families.35,36
This fact reinforces the principle that it is not the lack of telomerase per se, but the reaching of a threshold length by the telomeres themselves, that causes symptoms. This is an optimistic finding, because it implies exactly what we would expect (and hope) would be the case: that if we can periodically replenish the bone marrow and other stem cell pools with new stem cells whose telomeres are well above the critical length, we should be able both to cure DKC and also to avoid all the problems of the disease in people with intentionally extinguished telomerase. Indeed, the best treatment for DKC today is a bone marrow transplant, introducing new stem cells taken from people without DKC to replace the ones that are being depleted.
![]() What About This Periodic Stem Cell Replenishment?
What About This Periodic Stem Cell Replenishment?
The Bone Marrow
Bone marrow transplants are, of course, already a common and nearly routine procedure, not only for DKC sufferers, but also for patients with a range of blood disorders, cancer patients who have lost their bone marrow to radiation therapy, and many others. Still, there are many complications in recipients today, and many technologies that we will absolutely have to master if we are to use bone marrow transplants for WILT.
One reason why bone marrow transplants often don’t “take” is that they are not robustly incorporated into their niche in the bone while the original stem cells are still there. For the first round of WILT bone marrow replacement, we may have to perform chemotherapy to wipe out the native cells—but we’d want to do this anyway to minimize the cancer risk of having those old, telomerase-competent cells left behind. In subsequent rounds, the process will be easier, because we will intentionally wait to replace the cells from the first round of transplantation until the cells introduced in the previous round are beginning to die as their nonrenewing telomeres wear down.
How often will this stem cell replenishment have to be performed? It’s looking good. People have done clever experiments to measure the average time between divisions of blood stem cells, and it’s at least a couple of months in humans. Because it takes around fifty divisions before human cells not expressing telomerase start to feel the shortness of their telomeres, this rate of division should be slow enough to enable us to function just fine for about a decade between successive rounds of bone marrow replacement.
The Skin
Because of the pressure to supply skin grafts for burn patients, disfigured children, and cosmetic surgery, we’ve made remarkably fast progress in mastering the art of making new skin from stem cells. In mice, we can now peel the skin right down to the dermis (the layer of tissue beneath what we normally think of as “skin,” which houses hair follicles, sweat and skin oil-secreting glands, and blood vessels), and fully reconstitute the old skin that we’ve removed. The cells of the dermis do not divide regularly, so we don’t need to worry about their telomeres running out. Remarkably, the epidermis (the layer on top of the dermis, which is where we do need to replace stem cells) can be replaced using stem cells derived even from such different locations as the cornea of the eye, and the dermis orchestrates their rapid transformation into hair follicle stem cells that then expand outward, renewing the tissue.
Again based on how often skin stem cells divide under the status quo, a round of skin stem cell replacement should last us about ten years. Because the skin is so easy to access, it should be among the easiest and least invasive of WILT stem cell replacement routines.
The Lungs
The innermost layer of the lung, like the skin and the gut, is continuously sloughed off and is thus in continuous need of renewal. Unsurprisingly, lung complications are a major cause of death in DKC patients. Because the lung is in important ways similar to the skin, and is relatively easy to access, there is no reason to think that we won’t make quick and relatively painless progress on this front once we put our mind to it. Indeed, some scientists are doing this already, mostly in hopes of treating cystic fibrosis patients. Better yet, the latest estimates are that lung stem cells divide considerably less often than even those in the skin.
Work to date has gone forward using similar approaches to those used in skin, though not yet from stem cells. Even so, progress is being made. In two different models of immunodeficient mice, scientists have purged the lung of its “skin” (epithelium), “scraping” it down to the underlying basement membrane, and then replaced the lost tissue using restructured cells taken from the innermost layer of the human lung. The next step will be to do it with stem cells.
The Gut
So far, there are still significant challenges facing the replacement of stem cells in the gastrointestinal tract in humans. Several years ago, Dr. F. Charles Campbell, now Professor of Surgery at the Queen’s University Belfast’s School of Medicine, made the first major advance. His team extracted stem cells from mouse intestinal tissue, wiped out the cells from small stretches of the colon, and then repopulated the tissue with stem cells, which differentiated into all the appropriate cell types and made fully functional new tissue. Progress has not been rapid since then, however. At the WILT summit Campbell explained that, in studies he has never reported in print, his team tried the same approach in pigs, but the result was a mass of dysfunctional scar tissue. Since then, however, considerable progress has been made by another group working in dogs,37 and the same group has further advanced the technique in rats and mice.
Much work remains to be done here: not least, the opening-up of sections of the gut to remove the existing cells would be far too invasive for human use in WILT. Endoscopy, similar to what’s currently in use to remove potentially cancerous colon polyps, may provide us with a more tolerable solution, and should be much more advanced in a few decades, when we’ll actually need it.
A further question is how often we will need to replace stem cells in the gut. Previous estimates suggested that it would be much more frequently than the decade or so needed in these other tissues—more like a few times a year. Luckily, however, there is an easy way to see that those numbers must be wrong. If gut stem cells divided so much faster than those in other tissues, DKC sufferers and telomerase-lacking mice would suffer gut failure at a younger age than they suffer failure of the bone marrow or skin—but they generally don’t. Rather, all these tissues fail at about the same age in any given patient; if anything, the blood is most often the first to go in humans, which is why bone marrow transplants are temporarily helpful. But still, the replacement of the stem cells of the gut seems likely to be a more invasive procedure than that in bone.
The Rest of Us
The rest of our bodies is, in the relevant respect, like the dermis: composed of cells that don’t divide regularly. Some of these cell types (including the dermal cells, which are called fibroblasts) are quiescent: they can divide but they only do so when called upon to do so, such as to close a wound. Others are postmitotic: totally unable to divide, and instead renewed by incoming progenitor cells, if at all.
Postmitotic cell types are obviously no problem in cancer terms, but quiescent cells require a little more thought. The fact that they are not significantly affected in DKC or telomerase-knockout mice, and the relative rarity of cancers derived from them, gives us a bit more time to work on the job—and, once we have mastered it, probably a lot more time between each round of stem cell reseeding (as opposed to the more targeted replacement of “normal” and pathological cell loss to aging damage, which we will be tackling earlier on using ESCs, as outlined in Chapter 11).
But, precisely because they don’t divide, these cells aren’t going anywhere for a while either—and they do become cancerous occasionally. Thus, we do want to protect ourselves from cancer in them as much as possible. Removing senescent cells will reduce the already-low risk in these tissues considerably, but we will still want to cut it down much further.
The likely solution to this problem is targeted gene deletion. This is again a major challenge, because even in mice (where gene therapy is now routine) it’s very difficult to target genes for insertion or deletion in cells that are already inside the organism (as opposed to putting genes into sperm or egg, or embryos, or into cells that have been taken out of the animal, modified, and replaced). We are, however, getting better at this. Hopes are high for using this technique for gene therapy generally, and it could potentially be used to remove telomere-renewing potential from tissues that aren’t maintained by stem cells.
Over time, however, between replacing age-related cell loss using stem cells engineered to lack telomerase, and eventually replacing the original stem cells for these tissues with new ones engineered the same way, we will slowly “WILT-ify” these tissues a few cells at a time, progressively lowering the cancer risk they may pose.
![]() Does WILT Underestimate Cancer’s Evolutionary Ingenuity?
Does WILT Underestimate Cancer’s Evolutionary Ingenuity?
WILT is a highly complex, multifaceted proposal—but it rests, as I’ve explained, on one absolutely essential assumption. WILT will fail if cancers can figure out a way to grow indefinitely without the genes that we delete. Let’s look at that eventuality in detail.
One formal possibility is that cells could develop a way to replicate their chromosomes without telomere shortening, and thereby avoid the need for any enzyme to reverse that shortening. It’s been hypothesized that some stem cells effectively do just this. When stem cells divide, they typically produce one daughter that’s still a stem cell and one “amplifying” cell that is set on the path to differentiation into its required function. (Indeed, the ability to do this is an essential feature of most people’s definition of a stem cell.) It’s possible that, by a clever system of controlling which DNA strands stay in the daughter stem cell and which ones go into the amplifying cell, stem cells could stop their DNA from shortening. But we don’t need to worry about this possibility in respect of cancer, because it can only confer linear growth rates, not the exponential growth that is seen in cancers. With linear growth, a cancer would take thousands of years to grow large enough to kill us.
Another way to avoid telomere shortening is the way that bacteria, and indeed our own mitochondria, do it: not to have any telomeres! Bacterial and mitochondrial DNA are circles, so there’s no end-replication problem. But it seems that there’s no meaningful risk of this happening in humans. When telomeres get really short, the cell’s DNA-repair machinery mistakes the raw ends of the exposed chromosomes for the result of a chromosome break. That causes the cell to make a misguided attempt at repair, joining the chromosomes together, end to end—which is rapidly fatal for a dividing cell, because the cell division machinery snaps the double chromosome apart again, not at the original join but at some random place, scrambling the genes into a dysfunctional mess.
The final possibility is altogether more real, unfortunately. I’ve mentioned it a few times in passing during this chapter: it is that cancers can occasionally relengthen their telomeres using enzymes other than telomerase. These enzymes have not yet been identified, so the system is given the uninformative name ALT, or Alternative Lengthening of Telomeres. The good news here is that ALT is quite rare—in some tissues it’s almost never seen, and in those few tissues where it does appear it occurs in no more than about half of all tumors. This means that at least as much mutation is needed to activate it as to activate telomerase—and that tells us that ALT is almost certainly dependent on the turning-on of a gene that’s usually very firmly off, rather than only the loss of activity of genes that are normally on. This is great news for WILT, because, if ALT is indeed dependent on the activation of some gene or other, rather than just on the inactivation of genes, we will be able to do the same for that gene (once we identify it) as WILT proposes to do with telomerase. There may be side effects, just as there are with deleting telomerase, but they should be addressable with stem cell or other regenerative therapies, just as telomerase deletion seems likely to be.
A SIDE EFFECT: WILTING FERTILITY?
One potential side effect of the loss of telomerase from all of our cells might be eventual sterility for men. If having children is still a priority in a post-rejuvenation world, then men may choose to freeze their sperm in advance, as is presently done for sperm donors, for IVF. The sole sexual issue will be the actual making of babies, of course: nothing else will wilt from WILT.