8.1 Introduction
The dried stigma of Crocus sativus (L.) i.e., “Saffron” is taken into account as the utmost valuable flavoring herb in the world. It is an autumn-flowering perennial plant with underground storage organ (in the form of corm) (Fernández 2004). The major saffron-producing countries are Iran, India, Greece, Morocco, and Spain, wherein Iran ranks first in terms of the worldwide production (Molina et al. 2005; Carmona et al. 2006). Saffron cultivation earns priority mainly for its red stigmas (style branches) that hold the unique flavoring and coloring attributes (Melnyk et al. 2010). Saffron flower comprises of over 150 aroma and volatile compounds. Significant amounts of apocarotenoids like crocin, picrocrocin, and safranal are produced by saffron stigma. These apocarotenoids exhibit an array of anticancer, neuroprotective, anti-inflammatory, and cardioprotective activities (Baba et al. 2015a).
Naturally, saffron, a renowned member of Iridaceae is a non-fertile herb owing to the triploid nature (2n = 3x = 24) of its genome (Harpke et al. 2013). Its triploid condition is attributed to an irregular meiosis process and allows vegetative propagation via corms that limits its genetic base and hinders its genetic enhancement (Renau-Morata et al. 2012). The data on nuclear DNA content and other karyological features in the genus Crocus present a large and complex genome of about 10 Gb (for C. sativus) (Busconi et al. 2015). Such genome size and ploidy level eventually restrict saffron breeding and genetic improvement. The recent advances in genomic approaches provide new techniques that allowed investigation of the evolutionary origins of saffron. A number of research studies were carried out on the molecular basis of Crocus, but the genetic origin of C. sativus is not clear yet. Phylogenetic analysis based on chloroplast, ribosomal, and nuclear single copy genes sequence could not find the origin of saffron (Petersen et al. 2008; Harpke et al. 2013). Despite these intensive studies, the questions on the ancestor species and the allied evolutionary processes still remain unresolved. The whole genome sequencing of the Crocus provides a powerful tool to reveal diversity, relationships between species, and the origin of saffron, but till today it persists as a challenging problem for cultivated saffron carrying the intricate genome of considerable size. However, a number of sequencing projects are still under way mainly funded by the European Commission Brussels, Belgium. Therefore, it is expected that the whole genomic sequences of diploids and polyploid species of the genus Crocus would provide a fundamental knowledge for understanding the evolution and domestication of saffron.
In addition, not much is understood regarding the synthesis and accumulation of apocarotenoid compounds in the course of stigma growth and development. Numerous researches were carried out to study the transcriptome sequence data for the identification of structural and functional organization of the saffron genome. The same was also used for putative gene identification and networks that are involved in the production of biologically active plant compounds. Expressed sequence tags (ESTs) provide information about the genes expressed in a specific tissue or organ. However, limited EST collections from saffron corms (Álvarez-Ortí et al. 2004a) and mature stigmas (D’Agostino et al. 2007) are available till date. Recently, next-generation transcriptome sequencing efforts were performed for the stigma and flower tissues by Baba et al. (2015a) and Jain et al. (2016) to elucidate the molecular basis of apocarotenoid biosynthesis and its accumulation. A number of MADS-box and MYB-transcription factors that are involved in the flower development were cloned, and their expression were characterized (Tsaftaris et al. 2007; Gómez-Gómez et al. 2012). The surge in expression frequencies of apocarotenogenic genes suggested that the apocarotenoid accumulation might be regulated by gene expression during the stigma and tepals development (Ahrazem et al. 2015). Bioinformatics approaches offer the essential techniques for the identification of responsible genes and pathways of medicinal plants; in addition such approaches analyze the bulk amount of information, generated from high-throughput techniques (Sharma and Sarkar 2012). Bioinformatics studies can contribute in all stages of genotyping experiments in saffron such as structural genomics, comparative genomics, transcriptomics, proteomics, phylogenetic analysis, and system biology (Husaini et al. 2009). In recent years, the development of genomic tools and techniques, such as ESTs, genome and transcriptome sequencing, and bioinformatics, facilitated the research on genetic enhancement of saffron. This chapter provides an outline of the recent developments in genomics- and transcriptomics-based researches of saffron and also summarizes these omics approaches to identify molecular mechanisms of apocarotenoid biosynthesis.
8.2 Saffron Apocarotenoids and Their Use
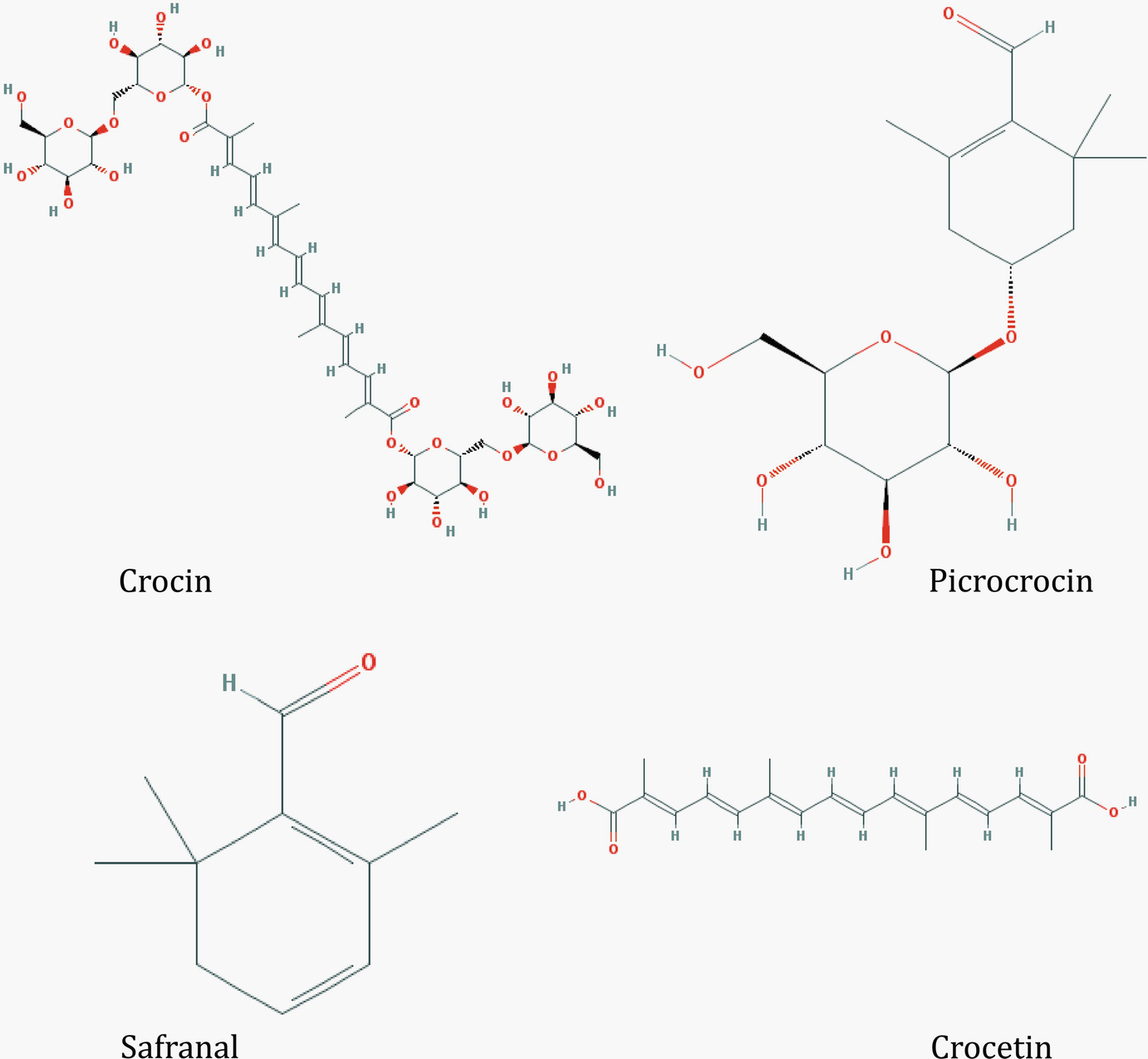
Structures of some key saffron apocarotenoids
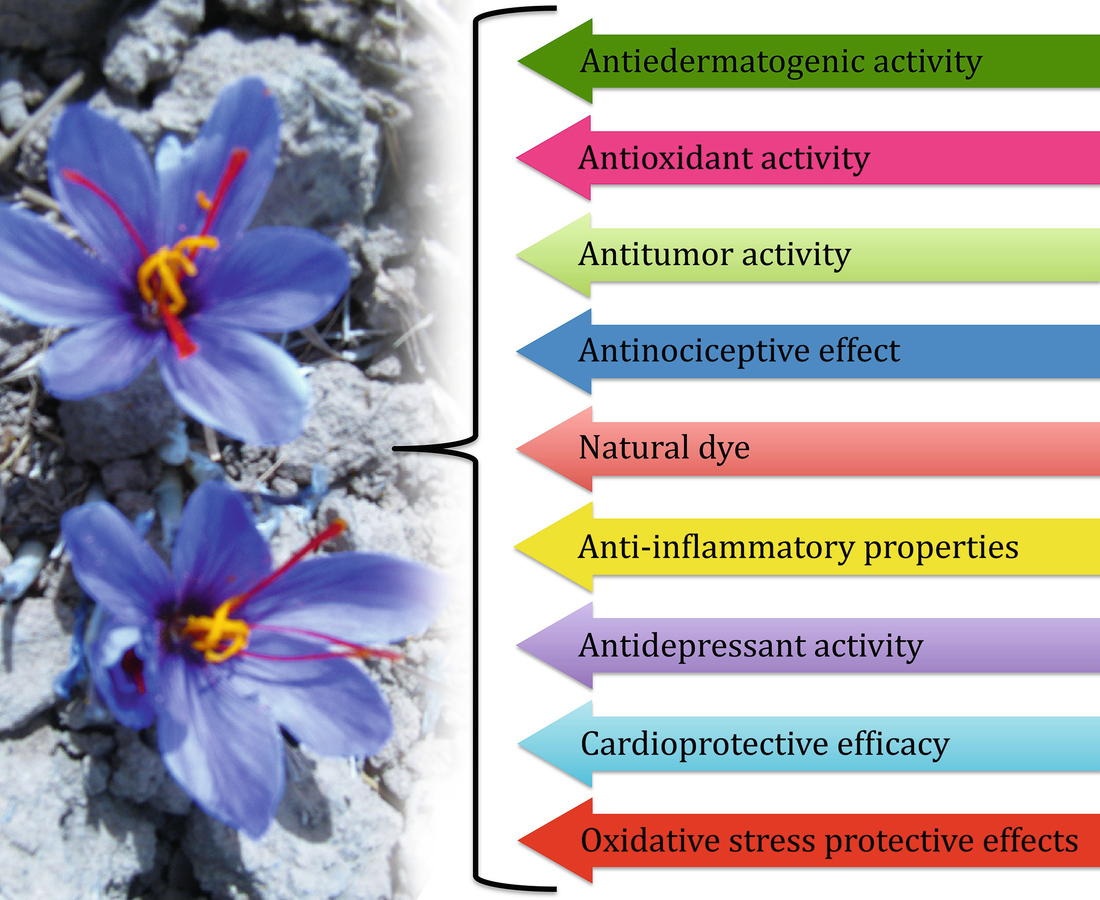
Pharmacological activities and other uses of saffron apocarotenoids
Saffron tablet was referred without much clinical importance based on some biochemical parameters and hematological changes (Mahamadpour et al. 2013). A safety evaluation of saffron tablets in healthy volunteers had shown a certain change in hematological and biochemical parameters. However, these changes were not in abnormal values and not so critically important (Modaghegh et al. 2008). Saffron (200 mg tablets) was observed to show a positive response on a sexual function by enhancing duration of some erectile events in patients with erectile dysfunction after ingesting for a period of 10 days (Hosseinzadeh 2009). One of the reports proposed that crocins may play a critical role in controlling the obsessive-compulsive disorder, a type of psychiatric disorder. It showed a functional interaction among crocin and the serotonergic system (Georgiadou et al. 2012). Several such clinical trials were executed on saffron to evaluate its pharmacological activities (Moshiri et al. 2014; Bhandari 2015). Rajaei et al. (2013) confirmed the hypoglycemic and antioxidative properties of crocin wherein a dose of 60 mg/kg was observed to substantially decline the blood glucose level in diabetic animals. In addition, crocin was reported to suppress the proliferation of K-562 human chronic myelogenous leukemia cells expressing Bcr-Abl protein tyrosine kinase activity (Geromichalos et al. 2014). Crocin supplements were found to be beneficial that enhanced the serum cholesteryl ester transfer protein in patients with metabolic syndrome (Javandoost et al. 2017). As reviewed by Moradzadeh et al. (2018), crocetin has the ability to inhibit cancer cell proliferation via preventing nucleic acid synthesis, improving antioxidative system, as well as stimulating apoptosis and differentiation pathways. A number of preclinical researches have exhibited that dietary intake of some carotenoids has potent antitumor effects both in vitro and in vivo, suggesting their potential preventive and/or therapeutic roles in several tissues (Bolhassani et al. 2014). Pitsikas (2015) critically reviewed the advancements in research on the influence of crocin to deal with memory disorders and explain its benefit over currently commonly used cognitive enhancers. However, these reports seldom considered the clinical safely issue emerged from usage of saffron tablets. On the contrary, Mahamadpour et al. (2013) evaluated the clinical safety of crocin and reported a comparatively safe and standard profile for crocin (in form of tablet) intake at a dose of 20 mg/day for 30 days for healthy human volunteers.
8.3 Biosynthesis of Saffron Apocarotenoids
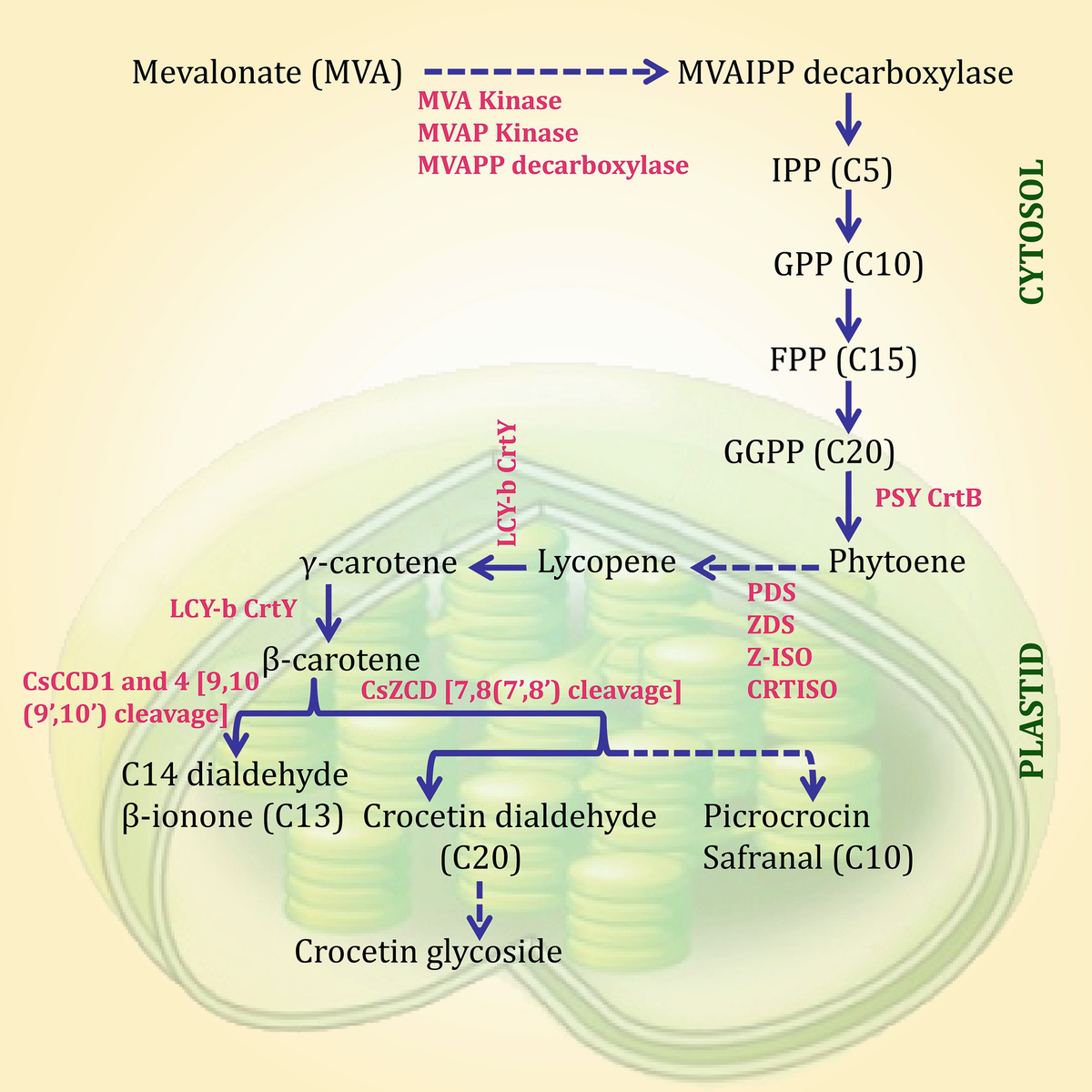
Elementary pathways for synthesis of saffron apocarotenoids. The enzymes involved in these pathways are in red color. The steps that involve multiple enzymes are specified with dashed arrows. (Adapted and redrawn from Rosati et al. 2009)
8.4 Omics Approaches
The latest advancement in molecular genetics has accelerated the identification of apocarotenoid biosynthesis pathway. The sequences of MADS-box genes expressed in saffron flower have been cloned to appraise the molecular events governing the growth and development of flower in the associated wild progenitor species of Crocus as well as cultivated saffron. Comparative structural and phylogenetic analysis of these proteins may be helpful to solve the origin of the cultivated triploid C. sativus (Tsaftaris et al. 2007). The genetic origin or ancestral species of saffron is not yet clearly disclosed (Petersen et al. 2008; Harpke et al. 2013). The current advances in genome sequencing technology of diploids and polyploid species of the series Crocus may clarify some important details of the genus Crocus. Chloroplast genome sequencing of seven Crocus species was performed using illumina platform, which provided genetic information on the phylogeny, species identification, and population genetics of this valuable spice. A new and simple technique of genome walking relying on the initiation of circular genomic DNA fragments (cgDNA) and rolling circle amplification of the circular genomic DNA has been reported to isolate the promoter regions of numerous genes from C. sativus (Tsaftaris et al. 2010). Apart from the biodiversity array in medicinal plants, DNA barcoding can also be applicable for the identification/authentication of such commercially important plants in the postgenomic era (Gantait et al. 2014; Mishra et al. 2016). Three DNA barcodes such as trnH-psbA intergenic spacer (trnH-psbA), a sizable subset of ribulose-bisphosphate carboxylase (rbcL-a), and nuclear internal transcribed spacer 2 (ITS2) were employed to discriminate saffron from its adulterants by sequences diversity assessment (Huang et al. 2015). The barcoding melting curve analysis approach (Bar-MCA) that utilizes the unanimous chloroplast plant DNA barcoding zone trnH-psbA could be a faster method to authenticate saffron and detect its adulterants (Jiang et al. 2014). The sequence of the plastid genes matK and rbcL was applied to construct the new marker for saffron and the adulterant species reorganization (Soffritti et al. 2016). A practical standard operating procedure (SOP) has been introduced by Zhao et al. (2016) for the authentication of saffron. For SOP, loop-mediated isothermal amplification (LAMP) method is used, and it requires four to six different primers that are projected depending on the nucleotide sequence of the internal transcribed spacer 2 (ITS2) nuclear ribosomal DNA of C. sativus to distinguish saffron from its adulterants. A sequence characterized amplified region (SCAR) technique also can be applied for the validation of a wide variety of dried food products containing saffron (Torelli et al. 2014). SCAR markers as a quick, profound, and inexpensive screening approach were developed for identifying dehydrated commercial saffron stigmas often mixed with seven common bulking components (saffron adulterants) (A. montana, B. orellana, C. officinalis, C. tinctorius, C. vernus, C. longa, and Hemerocallis sp.). SCAR markers are effective in identifying these adulterants substantiately (Marieschi et al. 2012). This approach facilitated the recognition of even a very low quantity of each adulterant. Apart from the sequence-related amplified polymorphism (SRAP), the new SCAR combined with ITS maker-based multiplex PCR analysis developed for the rapid identification of substitutes in saffron at molecular level (Babaei et al. 2014a, b). This approach could identify the occurrence of any anticipated plant material and adulterant materials in a single sample. DNA fingerprints are the barcode-like patterns that can be used for authentication of herbal medicines (Ganie et al. 2015).
The transcriptome is the study of all RNA transcripts in one cell at the specific developmental stage that focuses on the gene expression. The C. sativus transcriptome provides insights for understanding the molecular basis of flavor and color biogenesis. The first genomic characterization of a mature saffron stigmas has revealed the occurrence of 6603 high-quality ESTs across the Saffron Genes database (http://www.saffrongenes.org) that categorized into 1893 clusters, each related to a differently expressed gene, and interpreted. Homology analysis by blastX showed the high expression level of some transcripts contigs (TCs) (D’Agostino et al. 2007). A computational analysis was used to identify miRNAs, and their targets using this EST library from mature saffron stigmas, two putative miRNAs (miR414 and miR837-5p), and co-expressed genes including transcription factors and protein kinase which may play roles in apocarotenoid biosynthetic pathways have been characterized (Zinati et al. 2016). Three novel miRNAs, csa-miR1, csa-miR2, and csa-miR3, were forecasted by computational approaches. These objects ensure a function in biotic and abiotic stress resistance, senescence, as well as growth and development of plant. Furthermore, certain objects are engaged in mRNA transfer, translation, and posttranslational amendments (Guleria et al. 2012).
Some of the genes and enzymes that are involved in the abovementioned steps are studied and characterized. Likewise, the Crocus carotenoid cleavage dioxygenase gene (CsCCD) was also cloned (Bouvier et al. 2003). According to Rubio-Moraga et al. (2008), the Crocus CCDs, characterized till date, are analogous to CCD1 and CCD4 enzymes and are differentially expressed in flower organs, and CCD4s solely carry predicted transit peptides for plastid localization. The CCD1-like generic CsCCDs possess 9,10 (9′,10′) cleavage activity on various carotenoid substrates (Bouvier et al. 2003). CCD4-like CsCCD4a and CsCCD4b proteins (Rubio-Moraga et al. 2008) are 98–100% comparable to the CsZCD enzyme, earlier reported to cleave zeaxanthin at the 7,8(7′,8′) positions, resulting in synthesis of crocetin dialdehyde (Bouvier et al. 2003). CsCCD4 enzymes are longer than CsZCD and contain a plastid transit peptide. They perform a 9,10(9′,10′) cleavage and are also able to cleave zeaxanthin, although the expected apocarotenoids could not be detected by neither LC nor GC (Rubio-Moraga et al. 2008). However, advanced experimentations are necessary to explain the enzymatic activity, protein structure, and a precise number of Crocus CCD4/ZCD enzymes. Furthermore, apocarotenoid volatiles and water-soluble crocetin glycosides are collected in vacuoles to express pigmentation. As reviewed by Rosati et al. (2009), an UDP-glucose crocetin 8-8′-glycosyltransferase enzyme was purified from cell suspensions and characterized (Côté et al. 2000), and the product of the stigma-expressed UGTCs2 gene was shown to glucosylate crocetin aglycones and glycosides in vitro (Rubio-Moraga et al. 2004). Although the recent efforts have been focused on the identification of genes that are involved in the apocarotenoid biosynthesis, there are certain genes absent in the entire apocarotenoid biosynthetic pathway. Several enzymes that were identified to catalyze apocarotenoid biosynthesis pathway are the product of the crucial genes, such as PSY, LCY, CCD, BCH, and ZCD, which control the biosynthesis of apocarotenoids during the course of multiple phases of stigma development (Gómez-Gómez et al. 2010; Mir et al. 2015a). The molecular functions of two Crocus carotenoid cleavage dioxygenases, namely, CsCCD and CsZCD, have been detected by Bouvier et al. (2003). CsZCD precisely catalyzes the synthesis of crocetin dialdehyde from zeaxanthin, and CsZCD is responsible for the pigment and aroma synthesis in saffron. The expression patterns of CsPSY, CsPDS, CsLYCb, and CsBCH genes were investigated throughout the growth of stigma. By the modification of immature yellow to completely matured red stigmas, an accumulation of zeaxanthin was detected, supplementing with the expression of CsPSY, phytoene desaturase, and CsLYCb, besides the substantial collection of CsBCH and CsZCD transcripts (Castillo et al. 2005). The garnering of apocarotenoids and expression framework of apocarotenoid biosynthesis genes were researched on focusing on three particular phases of stigma growth (yellow, orange, and scarlet). Reverse transcription (RT)-PCR analysis revealed a distinct association amid apocarotenoid gene expression and apocarotenoid content throughout developmental period (IqbaLMzr et al. 2013). Maximum apocarotenoid biosynthesis and highest levels of CsZCD gene expression occurred during the fully developed scarlet stage of stigma development (Mir et al. 2012). CCD2 was identified during the first steps of stigma development using the 454-based transcriptome sequencing. The expression of CsCCD2 was correlated with the accumulation of crocin since it catalyzes the first step in crocetin biosynthesis (Frusciante et al. 2014). The model of crocin accumulation and the expression of apocarotenoid-related genes were investigated to find the agents affecting the garnering of such bio-active compounds and to recognize the main stages of their biosynthetic pathway. The results showed that the expression of the carotenogenic genes PSY, ZDS-V, BCH, and LCY-II was associated with the accumulation of crocins and increases the transcript levels of CCD2 genes during stigma and tepal development (Ahrazem et al. 2015). Four CCD genes, namely, CsCCD1a, CsCCD1b, CsCCD4a, and CsCCD4b, were identified from C. sativus. The four CCDs are divided into two phylogenetically dioxygenase categories with the same enzymatic activity even though their expression and localization were different (Rubio et al. 2008). In a study on the expression of three isoforms of CCD4 gene (CsCCD4a, CsCCD4b, and CsCCD4c) in response to different stresses, the results indicated that CsCCD4a and CsCCD4b showed enhanced expression in response to dehydration, salinity, and methylviologen, but CsCCD4c did not show any change in expression (Baba et al. 2015a). Functional characterization of CsBGlu12, a β-glucosidase from C. sativus, has shown its role in abiotic stress through reactive oxygen species (ROS) scavenging (Baba et al. 2017). The association between expression of CstNCED and the endogenous ABA quantity was studied in corms and stigma; the results showed the participation of CstNCED in the modulation of ABA-associated activities, for example, corm dormancy and flower senescence of saffron (Ahrazem et al. 2011). CCD7 and CCD8 genes that control the branching of shoots through apical dominance were required for strigolactones (SL) biosynthesis and were first isolated by Rubio-Moraga et al. (2014a, b). The expression patterns of two lycopene-b-cyclase genes, CstLcyB1 and CstLcyB2a, were explored in multiple saffron tissues; CstLcyB1 was substantially expressed in stigma and leaf tissue, and at lesser levels in tepals, contrastingly, CstLcyB2a was characterized only in the stigma tissue (Ahrazem et al. 2010). The spatial and temporal expression array of CsGT45 was investigated by RT-PCR during stigma development. The results showed that CsGT45 expression is developmentally controlled. The CsGT45 expression level in the yellow and orange phases was low, but enhanced since the red phase, and touched its ultimate state during anthesis. CsGT45 is an effective enzyme that performs a major responsibility in the synthesis of flavonoid glucosides in the stigma of saffron (Rubio-Moraga et al. 2009).
Three distinctive homologous CsAP1 genes, viz., CsAP1a, CsAP1b, and CsAPc, are the originally described MADS-box genes that were characterized from leaves and flowers of saffron. The expression pattern genes showed that the transcripts of each of these genes exist in leaves, together with the flowers of C. sativus (Tsaftaris et al. 2004). The expression of a family of five PISTILLATA/GLOBOSA-like (PI/GLO-like) MADS-box genes have been studied in the saffron flower, recognized to produce heterodimers for stamens and petals (Kalivas et al. 2007). SEP3-like cDNAs, transcribed from three genes, were isolated and their expression configurations and prospective protein interactions with other saffron MADS-box proteins investigated (Tsaftaris et al. 2011). The isolated MYB gene from C. sativus when expressed displayed an enhanced expression in the red stigmas of saffron, but a comparatively reduced expression was detected in tepals, alongside no transcripts identified in anthers and leaves (Gómez-Gómez et al. 2012). The first analysis of a comparative expression analysis of floral homeotic genes in relation with senescence was performed at different stages of flower development, identifying the pathway can make last longer flowering of saffron by activation of particular key genes (Wafai et al. 2015). Later, Ashraf et al. (2015) reported the modulatory role of CsULT1 in biosynthesis of Crocus apocarotenoid for the first time; it suggested a potential function in controlling the biosynthetic pathway of crocin. Differentially expressed genes, early inducible proteins (ELIP) and SOUL heme-binding proteins, engaged in the response of saffron stigmas against light, were recognized in saffron stigma (Ahrazem et al. 2016).
There is lack of study about gene expression pattern in the corm of saffron. At a stage characterized by storage accumulation and corm growth, a remarkable amount of sequences with similarity to genes related to cell growth, protein synthesis, folding and degradation, transcription factors, and proteins related to the formation and maintenance of cell wall and other cellular structures were identified (Alvarez-Orti et al. 2004a). The expression profile of the key storage protein, mannose-binding lectin, of saffron corm was greater throughout summer season before sprouting and then declined immediately after sprouting of corm (Álvarez-Ortí et al. 2004b).
The first study on transcriptome sequencing of saffron stigma and flower tissues was carried out using illumina platform that generated 64,604,402 flower and 51,350,714 stigma reads, and 64,438 de novo assembled sequences were categorized into 32,204 unigenes comprising of 9853 clusters and 22,351 singletons. The database provides a basis to identify the regulatory pathway of C. sativus flower development and biosynthesis of apocarotenoids (Baba et al. 2015c). Furthermore, differential gene expression (DGE) in saffron stigma against the rest of the flower indicated that biosynthesis of carotenoids and their subsequent degradation into apocarotenoids occur mainly in stigma. Eighty-one zinc-finger genes were detected in stigma divided into eight subfamilies (Malik and Asharaf 2017). Expression patterns indicated a probable role for CsSAP09 in apocarotenoid metabolism regulation that found to be highly expressed in stigma at anthesis stage corroborating with the accumulation pattern of apocarotenoids. From 206 million high-quality paired-end studies, following the standardization of de novo transcriptome organization, as many as 105,269 distinctive transcripts were attained. Functional annotation helped the discovery of genes involved in flavor and color biogenesis in spice; 54% of C. sativus transcripts could effectively be interpreted with the aid of public databases (Jain et al. 2016). Comprehensive databases in the Yet Another Tool Suite for analyzing RNA-seq derived transcriptome (YeATS) suite from the NCBI and Ensembl databases were established to accelerate the characterization of the saffron metagenome from the transcriptome obtained by Jain et al. (2016). Soybean mosaic virus was detected to be abundantly expressed in all five tissues analyzed; several putative pathogen bacterial and fungal genera transcripts were identified according to the factors based on the homology comparison (Chakraborty 2016).
8.5 Genetic Modifications
Genetic modifications with the aid of biotechnological tools and techniques could be a source for bringing variations in saffron. In fact, genetically transformed saffron could be evolved as a source of new and desirable traits with high economic value and wider adaptability. Such an avenue of research could only be taken up when there are established in vitro protocols for direct and/or indirect regeneration of saffron. As reviewed by Gantait and Vahedi (2015), there are an ample number of in vitro protocols reported by several researchers, and these can pave the way forward for genetic engineering in saffron. The other aspect for genetic modification in saffron is the identification of desirable genes and their regulatory behavior that can fulfill the demand of the breeder or consumer (Mir et al. 2015b). Since recent past, genetic modification through Agrobacterium-mediated gene transfer technology attained significant progress in the regulated genetic enhancement of traits in demand for several other plants in Iridaceae family where this technology has emerged out to be the key approach in modern molecular breeding. Several research achievements have been reported on gene manipulations and modifications, for instance, genes responsible for abiotic stress tolerance and insect resistance, regulation of genes involved in the biosynthetic pathways of secondary metabolites, etc. However, such reports on the genetic modification in saffron are scanty until now. Instead, there are multiple attempts that have been reported on genetic information related to synthesis of aroma compounds during the development of saffron stigma. Naturally, the young stigma has almost no odor, but at pre-anthesis, the aromatic compound β-ionone turns out to be the volatile norisoprenoid in the stigma. Rubio-Moraga et al. (2008) isolated four CCD genes (namely, CsCCD1a, CsCCD1b, CsCCD4a, and CsCCD4b) from saffron. Subsequently, they observed the expression pattern wherein CsCCD1a displayed an incessant expression and CsCCD1b was expressed exclusively in stigma tissue; however, during the stigma development, only CsCCD4a and CsCCD4b expressed harmoniously with the maximum levels of carotene and ionone release. Similarly, Ahrazem et al. (2010) isolated and analyzed the CCD4 genomic DNA regions in saffron. They also recognized multiple alleles, such as CsCCD4a (that includes or excludes an intron) and CsCCD4b (that includes an atypical intron). In addition, they confirmed the occurrence of individual gain or loss based on the relationship of the locations of CCD4 introns within the coding region with CCD4 genes from other plant species. CCD4a promoter sequence was found appropriate to initiative GUS expression in the saffron flower specifically in pollen. This was a functional characterization of CCD4a promoter, was carried out via stable transformation of Arabidopsis plants with a 1400 bp DNA fragment (P-CsCCD4a) integrated to the β-glucuronidase (GUS) reporter gene. Following the isolation of CCD4 genes (CsCCD4a and CsCCD4b) from the saffron stigma tissue and the establishment of their relation to the synthesis of some distinct volatile compounds to attract the pollinators, Rubio-Moraga et al. (2014a, b) confirmed other CCD4 individuals that are linked with carotenoid-derived volatile synthesis during stigma growth. They observed the expression of CsCCD4c confined within the saffron stigma tissue, and it was found to be associated with the synthesis of megastigma-4,6,8-triene.
Additionally, upregulation of CsCCD4c was induced following any external injury or environmental stress that eventually suggests that the apocarotenoid product of this gene is involved during adapting with abiotic stress. Lately, Baba et al. (2015b) studied the substrate specificity of three isoforms of CsCCD4 based on their molecular modeling and docking analysis. High substrate specificity for β-carotene was exhibited by all the three isoforms. Furthermore, they have exposed the three CsCCD4 isoforms to variable stresses and analyzed their expression pattern, which confirmed that CsCCD4a and CsCCD4b showed amplified expression toward water stress, salinity stress, and methylviologen. Such finding supports the earlier observation of Rubio-Moraga et al. (2014a, b) on the function of CsCCD4 isoforms facilitating the defense response of plants against environmental stress. An overexpression of CsCCD4b in genetically transformed Arabidopsis confirmed this attribute of CsCCD4 isoforms. The transgenic Arabidopsis displayed comparatively long roots and more lateral roots in comparison to wild type/non-transformed plants. Additionally, the genetically transformed Arabidopsis exhibited increased performance of reactive oxygen species metabolizing enzymes signifying that CsCCD4b generated β-ionone and β-cyclocitral which could function as stress signals and intervene in the rearrangement of stress-responsive genes that eventually results in plant defense.
8.6 Conclusions and Future Prospects
There is a huge prospect, opportunities, as well as bottlenecks in the concept, implementation, and biotechnological improvement of saffron. The ever-increasing scientific progress and information updates offer remarkable innovative prospective to explain genetic relationships, genomic evolution, and biotechnological improvement of saffron and to use these scientific techniques and database for the persistent advancement of this important medicinal and aromatic plant. Nevertheless, at the same time, if we are unable to utilize these information and technologies at an optimal level, it could be obvious that we ascertain the safeguarding of crucial germplasms; increase storage, manipulation, and access to enormously accumulating genomic data; and establish upgraded functional genomic technologies for phenotyping and genetic management of saffron.
Acknowledgments
The authors are thankful to the anonymous reviewers and the editor of this chapter for their critical comments and suggestions on the manuscript.