
I.Introduction. Tachyarrhythmias have been classically categorized by their location and mechanism. Tachyarrhythmias can originate from ventricular tissue (ventricular tachycardia [VT]) or, alternatively, can originate from or involve supraventricular tissue (supraventricular tachycardia [SVT]). The three mechanisms of tachyarrhythmias include abnormal automaticity, triggered activity, and reentry.
A.Mechanisms
1.Abnormal automaticity. Automaticity refers to the ability of cardiac tissue to spontaneously generate pacemaker activity. There are both normal and abnormal sources of automaticity.
An example of normal accelerated automaticity is the rapid firing rates of a normal pacemaker focus, such as the sinus node (SN), atrioventricular (AV) node, or Purkinje system because of ischemia, metabolic disturbance, exercise, or pharmacologic manipulation. A clinical example would be accelerated sinus tachycardia or junctional rhythm.
Abnormal automaticity refers to tissues that under normal circumstances do not demonstrate automaticity, but can become automatic in the setting of ischemia, metabolic disturbance, or pharmacologic manipulation. Overall, abnormal automaticity is responsible for <10% of tachyarrhythmias. These latent or ectopic loci of cells generate automatic, spontaneous impulses that usurp control of the cardiac rhythm. These usually have a warm-up and cool-down period and cannot be induced by programmed electrical stimulation. A clinical example would be accelerated idioventricular rhythm (see Section V.B.3.b) or multifocal atrial tachycardia (see Section II.E.3.c).
2.Triggered activity refers to pacemaker activity that is dependent on afterdepolarizations from a prior impulse or series of impulses. Afterdepolarizations are oscillations in the membrane potential. If these reach the critical threshold for depolarization of the surrounding cardiac tissue, they may trigger an action potential, thereby precipitating further afterdepolarizations and perpetuating the pacemaker activity. The two categories of afterdepolarizations are early and delayed.
Early afterdepolarizations (EADs) occur before repolarization of the cardiac tissue is completed (during phase 3 of the action potential) and may be the mechanism responsible for the ventricular arrhythmias of the long QT syndromes (LQTSs), as well as torsade de pointes (“twisting of the points”) produced by class I and class III antiarrhythmics, sympathetic discharge, and hypoxia. Antibiotics such as macrolides, certain azole antifungal agents, some psychotropic medications such as haloperidol, and several nonsedating antihistamines have been shown to produce EADs. Rapid heart rates and the administration of magnesium have been shown to suppress EADs.
Delayed afterdepolarizations (DADs) occur after the repolarization of the surrounding tissue is complete (during phase 4 of the action potential) and are thought to be the mechanism of triggered atrial tachycardia, arrhythmias of digitalis toxicity, and rare VTs responsive to calcium channel blockers. These have been demonstrated in various cardiac issues, including parts of the conducting system, myocardial cells, and valve tissues. Increases in intracellular calcium are associated with DADs, such as those caused by digitalis preparations or excessive sympathetic stimulation. Drugs that block the influx of calcium (such as calcium channel blockers and β-blockers) and drugs that decrease the sodium current (such as lidocaine and phenytoin) suppress the occurrence of DADs, whereas rapid heart rates augment DADs.
3.Reentry. Reentry is the most common cause of tachyarrhythmias. In order for reentry to occur, three conditions must be met:
•Two functionally distinct conducting pathways must connect to form a circuit.
•Unidirectional conduction block occurs in one of the pathways because of differences in refractory periods (block occurs in pathway with the longer refractory period).
•Slow conduction occurs down the unblocked pathway (which has the shorter refractory period), allowing the blocked pathway time to recover excitability and sustain the arrhythmia.
Reentrant circuits can occur in the SN, the atrium, the AV node, between the atrium and ventricle via bypass tracts, and within the ventricle itself. The typical substrate for malignant reentry in the ventricle is scar or ischemia, which can produce regions in the heart that depolarize and repolarize heterogenously. Therefore, the impulse can spread to an area that has already repolarized after being previously depolarized. This can set up a circular movement of the impulse resulting in sustained tachyarrhythmias such as VT. Reentry can typically be induced by premature electrical stimulation during electrophysiologic (EP) testing.
Elucidation of the mechanisms of tachyarrhythmias has led to the development of catheter-based treatment strategies and more advanced medical therapy.
II.Supraventricular tachyarrhythmias
A.Sinus tachycardia
1.Clinical presentation. Sinus tachycardia manifests as sinus rhythm with a rate above 100 beats/min. Although the rate may be as high as 200 beats/min in younger individuals, it is generally 150 beats/min or less in older individuals.
2.Pathophysiology. The SN is an epicardial structure that is located laterally near the junction between the superior vena cava and the right atrium. Under normal circumstances, the rate of SN discharge is governed by sympathetic and vagal stimulation. Sinus tachycardia generally reflects an underlying process, metabolic state, or effect of medication. Fever, hypovolemia, shock, congestive heart failure (CHF), anxiety, pulmonary disease including pulmonary embolism, anemia, thyrotoxicosis, caffeine, nicotine, atropine, catecholamines, or withdrawal from alcohol or drugs (both therapeutic and illicit) can cause sinus tachycardia. Sinus tachycardia can be appropriate, where it represents a normal physiologic response, or inappropriate, as in defects in vagal or sympathetic tone or an intrinsic problem with the SN itself.
The clinical consequences of sinus tachycardia vary based on the presence or absence of underlying heart disease. Patients with significant coronary artery disease (CAD), left-ventricular (LV) dysfunction, or valve disease may not tolerate sinus tachycardia. Patients with inappropriate sinus tachycardia may experience significant symptoms such as palpitations, dyspnea, and/or chest pain.
3.Diagnostic testing. Electrocardiography is the primary diagnostic test. The main differential is between sinus tachycardia, sinus node reentry tachycardia (SNRT) (see Section II.B), and inappropriate sinus tachycardia. Inappropriate sinus tachycardia is characterized by the following features: (a) heart rate > 100 beats/min, (b) P-wave axis and morphology during tachycardia similar or identical to that during sinus rhythm, (c) exclusion of secondary causes of sinus tachycardia, (d) exclusion of atrial tachycardias, and (e) symptoms clearly documented to be related to resting or easily provoked sinus tachycardia.
Therapy is generally directed at the elimination of the underlying cause whenever possible. If withdrawal from a therapeutic medication is suspected, then reinstitution or slow tapering of this medication can be attempted, if clinically appropriate. In the case of inappropriate sinus tachycardia, β-blockers and calcium channel blockers may be necessary to control the heart rate. Ivabradine, a new agent that affects the If channel, can also be considered. In medically refractory cases, catheter ablation for sinoatrial nodal modification may be considered.
B.SNRT accounts for 2% to 5% of all supraventricular tachyarrhythmias.
1.Clinical presentation. SNRT is most frequently seen in patients with structural heart disease or CAD, especially in inferior myocardial infarctions (MIs). The rate varies from 80 to 200 beats/min. SNRT’s characteristic abrupt onset and termination (paroxysmal nature) along with its ability to be induced and terminated by pacing imply that the underlying mechanism is reentry and distinguish it from sinus tachycardia and inappropriate sinus tachycardia.
2.Pathophysiology. Reentry occurs within or adjacent to the SN and then conducts via the normal conduction pathway to the rest of the heart. The morphology of the P-wave is identical to the underlying sinus morphology. Block at the AV node may occur, but it does not slow the tachycardia. In fact, a Wenckebach-type block often occurs with this rhythm. The development of a bundle branch block does not affect the cycle length or the PR interval.
3.Therapy. Vagal maneuvers or adenosine may successfully terminate this arrhythmia. Rapid atrial pacing can be used to induce and terminate this tachycardia. Various agents such as β-blockers, calcium channel blockers, digoxin, or amiodarone may help prevent recurrences. SN ablation or modification is rarely necessary.
C.Atrial fibrillation (AF) is the most common sustained arrhythmia, occurring in up to 1% of the general population. The prevalence of AF increases with age, affecting up to 10% of the population older than 80 years (see Chapter 24).
D.Atrial flutter. Atrial flutter is the second most common of the atrial tachyarrhythmias. Its reported incidence varies from 0.4% to 1.2% in hospital reports of electrocardiogram (ECG). The clinical significance of atrial flutter is generally due to its association with AF (with all of the attendant risks of AF) and/or its association with rapid rates of ventricular response.
1.Clinical presentation. The clinical presentation may vary widely depending on the presence of underlying heart disease, the ventricular rate, and the overall condition of the patient. It is occasionally reported to persist for days and, less commonly, for weeks or longer. Careful examination of the jugular venous pulse may reveal frequent, regular a-waves that correspond to the atrial flutter rate. Like AF, it is commonly seen after open heart surgery, as well as with other conditions commonly associated with AF, such as pulmonary disease, thyrotoxicosis, atrial enlargement because of any cause including mitral/tricuspid valve disease, and SN dysfunction.
2.Pathophysiology. “Typical” atrial flutter is the result of a macroreentrant circuit in the right atrium. Atypical atrial flutter generally involves other macroreentrant circuits around scar tissue or surgical incisions.
In typical atrial flutter, the reentrant circuit most commonly travels in a counterclockwise rotation down the right atrial anterolateral free wall across the cavotricuspid isthmus (area of slow conduction) and up the interatrial septum. Clockwise rotation of this circuit may also be seen.
Atrial flutter has been classified into type I and type II based on the following characteristics:
Type I atrial flutter can be terminated with rapid atrial pacing and typically has an atrial rate in the range of 240 to 340 beats/min in the absence of drug therapy.
Type II atrial flutter cannot be terminated with rapid atrial pacing and typically has an atrial rate in the range of 340 to 440 beats/min in the absence of drug therapy.
Types I and II are not synonymous with typical and atypical atrial flutters. Type I atrial flutter can include typical and atypical atrial flutters. Type II atrial flutter is less well characterized than type I with respect to etiology and therapy; therefore, we refer to type I atrial flutter throughout this discussion.
Vagal maneuvers include carotid sinus massage and Valsalva maneuver. Caution must be exercised when attempting carotid sinus massage in patients with known or suspected carotid disease or vagal maneuvers in patients with CAD who are at risk for ischemia.
Agents for rate control include the intravenous calcium channel blocking agents verapamil and diltiazem and the intravenous β-blockers esmolol and metoprolol. Adenosine can be administered if the diagnosis is in question: 6 mg rapid intravenous push, followed by 12 mg if there is no response (a second 12-mg dose can be given if there is no response). The half-life of this medication is very short, approximately 9 seconds. This causes transient (lasting seconds), complete AV block. Patients should be connected to a transcutaneous pacing device during the administration of this medication for reasons of safety. The clinician can also record from a temporary atrial epicardial pacing wire (placed at open heart surgery). This results in an ECG with clearer atrial complexes and thus simplifies diagnosis. This strategy also allows a method of delivering rapid atrial pacing in an attempt to terminate the atrial flutter.
On the surface ECG, typical counterclockwise atrial flutter shows the classic negatively directed “sawtooth” waveform in the inferior leads (II, III, and aVF) (Fig. 21.1). Conversely, the atrial depolarizations are positive in these leads in clockwise atrial flutter (Fig. 21.2).
FIGURE 21.1 “Typical” atrial flutter, leads II and III.
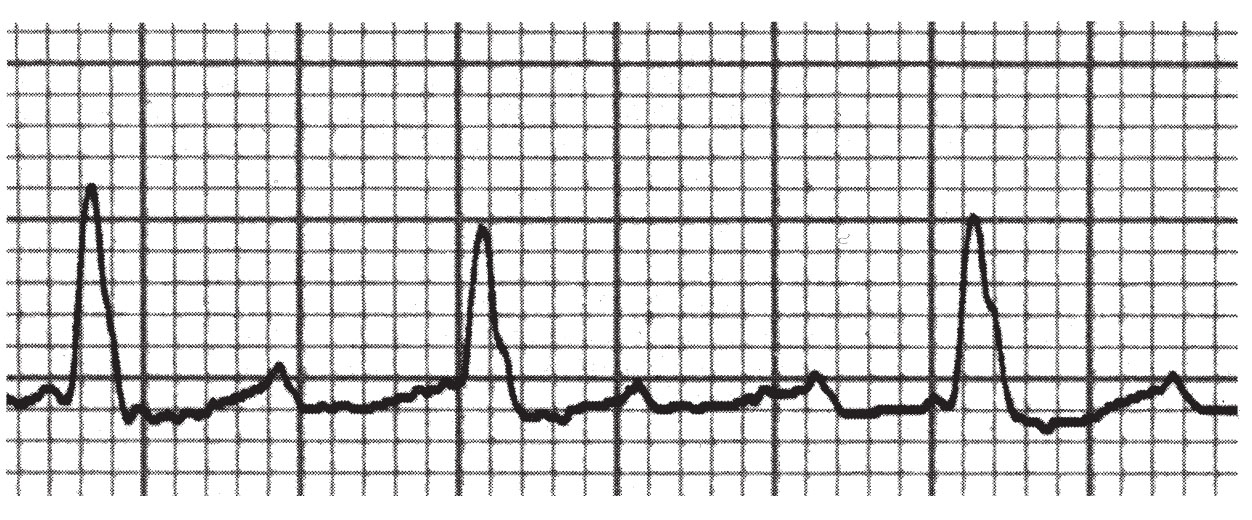
FIGURE 21.2 “Atypical” atrial flutter, lead II.
The atrial rate in the absence of drug therapy is 240 to 340 beats/min. The QRS complex should be the same as that seen during sinus rhythm although aberrant conduction may occur, and the QRS may be slightly distorted by the atrial flutter waves. The ventricular response can be irregularly irregular, because of varying degrees of block (2:1, 4:1, and so on), but is more typically regular as a fixed ratio of the flutter rate.
4.Therapy. Medical therapy differs very little from that for AF (see Chapter 24). Control of the ventricular response rate with a β-blocker, a calcium channel blocker, or digoxin is critical prior to initiating therapy with agents such as the class IA or IC agents. The class IA or IC agents may either enhance AV nodal conduction through their vagolytic effects, thereby enabling 1:1 (AV) conduction, or slow the atrial rate to a point where 1:1 conduction is facilitated. The conversion from atrial flutter to AF after cardioversion is substantially reduced by the administration of antiarrhythmic drugs prior to direct current cardioversion (DCC), thereby increasing the chance of converting to sinus rhythm.
a.Anticoagulation. There are no prospective data looking at the incidence of thromboembolic events with atrial flutter. However, retrospective data suggest an increased incidence of thromboembolic events. The American College of Cardiology/American Heart Association/Heart Rhythm Society 2014 guidelines recommend managing anticoagulation in atrial flutter in a manner similar to that for AF, including cardioversions. Optimal management needs to be individualized with the patients’ profile for thromboembolic risk, dictating the type and duration of therapy using the CHA2DS2-VASc score. There is also recent evidence to support the use of the novel anticoagulants for use as anticoagulation and their safety for use in DCC.
b.Direct current cardioversion. DCC is the preferred and most effective therapy for most patients. A starting energy as low as 25 to 50 J is often effective. Because DCC may result in conversion from atrial flutter to AF, a second shock is sometimes necessary to convert AF to sinus rhythm. Rapid atrial pacing should be considered as the first line of therapy for all patients who have epicardial atrial pacing wires in place after open heart surgery. It may be considered via a transesophageal pacing lead or via a transvenously placed pacing lead in patients for whom DCC fails or who are not candidates for DCC. Before attempting to rapidly pace the atria, one should confirm absence of ventricular capture by first pacing at a relatively slow rate while observing for such a phenomenon. Once this is confirmed, the atrium is paced at a rate of 10 to 20 beats/min faster than the underlying atrial flutter rate. Once atrial capture is attained, the rate is increased steadily until the hallmark negative-sawtooth waveform converts to a positive waveform. The pacing is then either halted abruptly or slowed rapidly to an acceptable atrial pacing rate. In cases that require extremely rapid rates of pacing (>400 beats/min) or high amplitudes of pacing stimulus strength (>20 mA), there is an increased tendency for the atrial flutter to convert to AF. When pacing via a transesophageal lead, a higher stimulus strength (up to 30 mA) may be necessary. Because this type of pacing can be quite painful, a sufficient energy to convert the atrial flutter should be used initially to minimize the conversion attempts.
c.Percutaneous therapy. Radiofrequency ablation (RFA) of the cavotricuspid isthmus is often curative, with an efficacy >90% for the long-term elimination of atrial flutter. Despite the high success rate of catheter-based therapy, a significant number of patients may subsequently develop AF (>80% at 5 years).
E.Atrial tachycardias. This term encompasses a number of different types of tachycardias that originate in the atria. These tachycardias account for between 10% and 15% of the tachycardias seen in older patients, usually in the setting of structural or ischemic heart disease, chronic obstructive pulmonary disease, electrolyte imbalances, or drug toxicity (particularly digitalis).
2.Diagnostic testing
a.ECG. The P-wave axis or morphology is usually different from that of sinus rhythm. One exception is atrial tachycardias originating from the right superior pulmonary vein, which is anatomically close to SN. The axis can be used to predict the origin of the atrial tachycardia. Atrial rhythm is regular, except with automatic atrial tachycardia, which displays a warm-up period (see Section II.E.3.a). A QRS complex that is generally identical to sinus rhythm (QRS can be wide if aberrant conduction occurs) follows each P-wave. PR interval is within normal limits or prolonged. Nonspecific ST-T–wave changes may be present. When an AV block is present, there is an isoelectric baseline between P-waves in all leads. EP study has become critical in determining the underlying mechanism of these tachycardias, because the clinical differences are subtle and overlapping.
3.Subclassifications. The current subclassifications are based on mechanisms and include automatic atrial tachycardia, triggered atrial tachycardia, and intra-atrial reentry.
Intra-atrial reentry is usually a disorder seen in those with underlying heart disease or history of atrial arrhythmia, such as AF or atrial flutter. The mechanism is not well understood. The ventricular rate is typically 90 to 120 beats/min because of the frequent occurrence of 2:1 AV block, such that hemodynamic effects are generally minimal. This rhythm can be difficult to distinguish from other supraventricular tachyarrhythmias. One clue is that despite any AV conduction block, the rhythm continues. The ability to terminate with adenosine and β-blockers is variable. RFA may be effective, with success rates >75%. Antiarrhythmics (the same drugs as for AF and atrial flutter) have been disappointing in the prevention of recurrence.
a.Automatic atrial tachycardia appears to be generated by an ectopic atrial focus, which usually arises from regions around the crista terminalis in the right atrium and around the base of the pulmonary veins in the left atrium. The mechanism is not well understood. Automatic atrial tachycardia is seen more often in younger patients, displays a warm-up phenomenon (the supraventricular tachyarrhythmia accelerates after its initiation), does not respond to vagal maneuvers, and is more likely to be incessant. Automatic atrial tachycardia can be induced with treadmill testing or with administration of isoproterenol. Atrial stimulation during EP has no effect on either initiating or terminating this arrhythmia. Propranolol has been used successfully to suppress automatic atrial tachycardia. Catheter ablation is the preferred therapy when the tachycardia is incessant or associated with a cardiomyopathy. Although adenosine may transiently slow automatic atrial tachycardia, it is unlikely to terminate it. Likewise, verapamil has been used without success.
b.Triggered atrial tachycardia is the least common of the atrial tachycardias and is virtually never incessant. It is more likely to appear in older individuals. It can be induced with rapid atrial pacing and is cycle length–dependent. The mechanism of triggered atrial tachycardia is thought to be due to DADs (see Section I.A.2) secondary to digitalis toxicity or sympathetic discharge. Catecholamines may play a role in the initiation of this arrhythmia, and thus exercise testing and isoproterenol may provoke it. Verapamil and adenosine have been shown to terminate triggered atrial tachycardia. β-Blockers have been less effective. RFA is preferred when the tachycardia is very symptomatic and not responsive to medication.
c.Multifocal atrial tachycardia
(1)Clinical presentation. This atrial arrhythmia is uncommon and estimated to occur in 0.37% of hospitalized patients. The atrial rate is generally 100 to 130 beats/min. It occurs most often in elderly, critically ill patients and is frequently associated with concurrent pulmonary disease, particularly chronic obstructive pulmonary disease. It may also be seen in CHF and can degenerate into AF.
(2)Pathogenesis and diagnostic tests. The mechanism appears to be abnormal automaticity or triggered activity arising from distinct atrial sites. The diagnosis requires the following criteria: (1) atrial rate > 100 beats/min, (2) P-waves with three or more different morphologies, (3) varying P–P, P–R, and R–R intervals, and (4) the P-waves separated by isoelectric intervals. Loss of AV conduction of each P-wave is uncommon, making it possible to distinguish multifocal atrial tachycardia from AF.
Therapy is directed at the underlying illness, with little role for antiarrhythmics. Calcium channel blockers in high doses may be useful, or amiodarone when antiarrhythmic therapy is deemed necessary. Maintenance of electrolyte balance, particularly potassium and magnesium, may suppress the occurrence of multifocal atrial tachycardia.
d.Atrioventricular nodal reentrant tachycardia (AVNRT)
(1)Clinical presentation. AVNRT usually has a narrow QRS complex with a ventricular rate typically in the range of 150 to 250 beats/min, although faster rates are infrequently observed. AVNRT is generally seen in patients without underlying heart disease. Palpitations and dyspnea are common presenting complaints. Angina, CHF, and rarely shock may be seen in those with a history of underlying heart disease. Syncope may occur due to rapid ventricular rates or due to a prolonged pause or bradycardia seen occasionally when this tachycardia terminates.
(2)Pathophysiology. The mechanism in AVNRT is a reentrant circuit composed of separate fast and slow atrial pathways involving the AV node. In 50% to 90% of patients with “typical” AVNRT, the antegrade conduction to the ventricles travels over the slow pathway and the retrograde conduction to the atria occurs over the fast pathway. The initiating event may be either a premature atrial complex (PAC) or a premature ventricular complex (PVC). The PAC blocks the fast pathway antegradely and conducts down the slow pathway, then backs up the fast pathway after it has repolarized. Less commonly, a PVC conducts retrogradely to the atria via the fast pathway and then returns to the ventricles via the slow pathway. In the remaining 5% to 10% of patients, with atypical AVNRT, the antegrade conduction is down the fast pathway and retrograde via the slow pathway. The cycle length is thus dependent on the conduction velocity of the slow pathway, because the fast pathway generally has rapid conduction. Termination of the tachycardia is often the result of a block in the slow pathway. AV dissociation may develop during the tachycardia because the ventricles are not involved in the reentry circuit. This does not affect the rate of tachycardia nor does the development of bundle branch block.
(3)Laboratory features and diagnosis. P-waves are generally hidden within the QRS complex or at the terminal portion of the QRS in typical AVNRT. This may be visible as a small pseudo-R′ in lead V1 or small negative deflections in the inferior leads, as depolarization of the atria occurs simultaneously with ventricular depolarization. The RP segment is generally <100 ms. AVNRT is often induced abruptly by a PAC, and its termination, which also tends to be abrupt, is often followed by a retrograde P-wave. The termination may be followed by a brief period of asystole or bradycardia before the SN recovers from its tachycardia-induced suppression. The cycle length may vary, especially at the beginning and at the end of the tachycardia. This variation reflects the variable antegrade AV nodal conduction time. Vagal maneuvers may slow or terminate this tachycardia.
(4)Therapy. Presently, the success and safety of percutaneous catheter ablation have allowed this approach to be considered equally with medical therapy as first-line therapy for long-term management of AVNRT. The decision about treatment approach should be individualized according to the characteristics of each patient and his or her arrhythmic patterns.
RFA has the advantage of curing the arrhythmia in the majority of instances and eliminating the need for long-term suppressive therapy with medications. Cure rates with catheter ablation for AVNRT are in excess of 95%.
Medical therapy. Medications that suppress AV nodal conduction such as β-blockers, calcium channel blockers, digoxin, and adenosine all slow or block conduction in the antegrade slow pathway, whereas class IA and class IC antiarrhythmic drugs slow the conduction in the retrograde fast pathway. Adenosine may be considered as first-line drug therapy for acute termination of AVNRT. This medication is available in an intravenous form only and has a very short half-life of about 9 seconds. The use of intravenous or oral β-blockers or calcium channel blockers is an alternative if adenosine is unsuccessful. The onset of action of digoxin limits its usefulness in terminating these arrhythmias, although it may be useful to prevent recurrences. Recurrences may be prevented in patients with frequent sustained episodes with any of the above-mentioned agents except adenosine. Antiarrhythmic drug therapy is not routinely necessary or desirable for AVNRT, given the high success rates and low complication rates for catheter ablation. DCC should be considered for patients whose disease is unstable or highly symptomatic. Low energies of 10 to 50 J are usually sufficient to terminate AVNRT.
e.Atrioventricular reentrant tachycardia (AVRT)
(1)Clinical presentation. Similar to AVNRT, this is another example of an AV nodal–dependent SVT. AVRT usually has a narrow QRS with ventricular rates similar to those of AVNRT, although it more often tends to have a ventricular rate >200 beats/min. The clinical features are very similar to those of AVNRT but are distinct on an EP basis.
(2)Pathophysiology. The mechanism in AVRT relies on the presence of an accessory pathway as one portion of the circuit and the AV node as the other portion. The atrium and the ventricle on the same side as the accessory pathway are necessary components of the circuit. AVRT may be orthodromic or antidromic. Orthodromic AVRT usually has a narrow complex that uses the AV node as the antegrade limb and the accessory pathway as the retrograde limb of the circuit. Antidromic AVRT has a wide complex that is the opposite of the orthodromic variety, such that the accessory pathway serves as the antegrade limb and the AV node as the retrograde limb of the circuit. AVRT is most often of the orthodromic type. Accessory pathways may be “concealed” (inapparent by ECG) because of having only retrograde (V to A) conduction properties or “manifest” (apparent on ECG as delta waves, i.e., Wolff–Parkinson–White [WPW] pattern). Unlike AVNRT, the AVRT circuit must involve one of the ventricles; therefore, the development of bundle branch block on the side ipsilateral to the accessory pathway can prolong the ventricular to atrial conduction time and often the cycle length of the tachycardia. Bundle branch block, particularly left bundle branch block (LBBB), occurs more commonly in AVRT than in AVNRT. AVRT can be distinguished from AVNRT by EP study. The presence of AV or ventriculoatrial (VA) block with continuation of the tachycardia should exclude the presence of an accessory AV pathway.
(3)Laboratory features and diagnosis. The P-waves of AVRT are frequently inscribed on the ST-segment or T-wave, because the atrial depolarization and ventricular depolarization are in series rather than in parallel. The RP segment is generally >100 ms. Orthodromic AVRT is more common, accounting for about 95% of all AVRTs, whereas antidromic AVRT accounts for only about 5%. Orthodromic AVRT is usually characterized by a narrow QRS complex as opposed to antidromic AVRT, which is characterized by a wide QRS complex.
(4)Therapy. See the discussion of therapy for WPW syndrome.
III.Preexcitation syndromes. Preexcitation was originally used to describe the premature activation of ventricle in patients with WPW. The term has broadened to include all conditions in which antegrade ventricular activation or retrograde atrial activation occurs partially or totally via an anomalous pathway distinct from the normal cardiac conduction system. The incidence of preexcitation on ECG is approximately 1.5 per 1,000 cases, most of which occur in otherwise healthy subjects without organic heart disease. About 7% to 10% of these patients have associated Ebstein anomaly and are thus more likely to have multiple accessory pathways. There is a higher rate of preexcitation in males, with the prevalence decreasing with age, although the frequency of paroxysmal tachycardia increases with age.
A.Clinical presentation. Approximately 50% to 60% of patients with preexcitation report symptoms such as palpitations, anxiety, dyspnea, chest pain or tightness, and syncope. In approximately 25% of the cases, the disease will become asymptomatic over time. Those patients older than 40 years whose disease has been asymptomatic are likely to remain symptom free. The absence of preexcitation on ECG despite the discovery of accessory pathways in patients with asymptomatic disease likely identifies a group of patients at low risk for developing symptoms.
B.Pathophysiology. Patients with preexcitation generally have an accessory pathway(s) that alters the conduction between the atria and the ventricles. These accessory pathways are likely congenital, because relatives of subjects with preexcitation have an increased incidence of preexcitation. AVRT is the most common mechanism associated with preexcitation (80% to 85%), with permanent junctional reciprocating tachycardia, Mahaim fiber tachycardia, and Lown–Ganong–Levine (LGL) syndrome accounting for the remainder.
C.WPW syndrome. The basic abnormality lies in the existence of an accessory pathway of conducting tissue, outside of the normal conducting system, which connects the atria and the ventricles. This accessory pathway permits the atrial impulse to bypass the normal pathway through the AV node to the ventricles. In the past, these accessory pathways have been referred to as “bundles of Kent.” An impulse from the atria can be conducted down both the accessory pathway and the AV node, arriving at the ventricle at nearly the same time. This results in preexcitation of the ventricle, which is really a fusion beat, as a portion of the ventricle is activated via the accessory pathway (giving rise to the delta wave; Fig. 21.3) and the remainder of the ventricle is activated by the normal activation pathway. If antegrade conduction occurs exclusively via the accessory pathway, the resultant QRS is maximally preexcited and is a wide complex. These accessory pathways may conduct rapidly, but frequently have longer refractory periods than the AV node. The inciting event for AVRT is frequently a PAC that is blocked in the accessory pathway and that conducts to the ventricles via the AV node, which has recovered more rapidly. The resultant QRS complex in this instance is normal in appearance. After the QRS complex, the accessory pathway has had sufficient time to recover excitability, and the impulse thus conducts retrogradely to the atria. A small but significant percentage (5% to 10%) of patients have multiple accessory pathways.
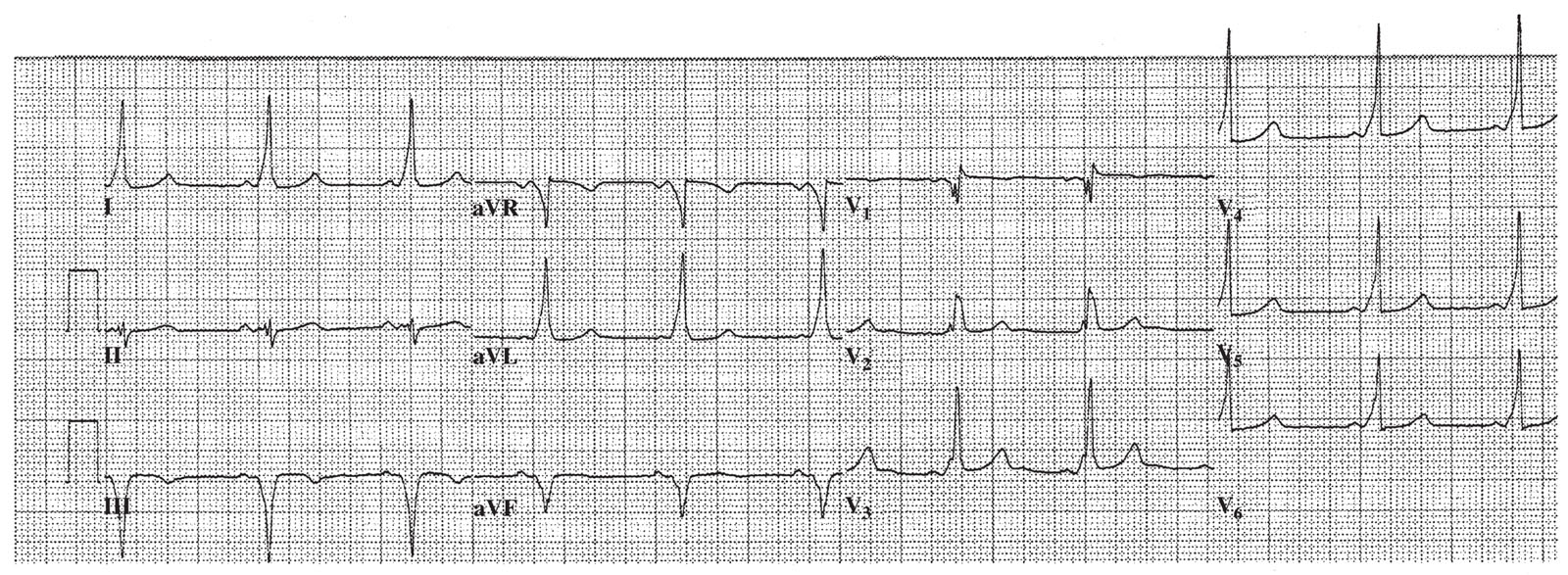
FIGURE 21.3 Wolff–Parkinson–White syndrome, with widespread delta waves seen at the upstroke of the QRS complexes.
D.Permanent junctional reciprocating tachycardia is a variant of AVRT. It is often an incessant supraventricular tachyarrhythmia with an unusual accessory pathway. Here, the accessory pathway behaves like the AV node in that it displays decremental retrograde conduction properties. Thus, the faster the stimulation of such an accessory pathway, the slower the conduction through the pathway. The accessory pathway is most often located in the posteroseptal region and acts as the retrograde limb of the reentrant circuit. The VA conduction is slowed by the decremental nature of the accessory pathway. Because of the incessant nature of this tachycardia, a tachycardia-induced cardiomyopathy may result and ablation is the therapy of choice when this occurs.
E.Mahaim fiber tachycardias are another variant of reentrant tachycardia. The two most common varieties that are recognized are atriofascicular and fasciculoventricular. In the former, the accessory pathway is located within the right atrium and inserts into the right bundle branch. The reentrant tachycardia conducts antegrade via the accessory pathway, resulting in a typical LBBB morphology with left-axis deviation. The retrograde circuit is via the AV node. In the second form of Mahaim reentry, the accessory pathway arises in the His-Purkinje fibers and allows bypass of the distal conducting system. This second type is not associated with a clinical tachycardia syndrome and further therapy is not needed.
LGL syndrome is diagnosed by the presence of a short PR interval and a normal QRS complex on the surface ECG. LGL syndrome likely represents one end (enhanced) of the normal spectrum of AV nodal conduction properties, but in some cases it is impossible to exclude a distinct perinodal accessory pathway or an abnormality in conduction characteristics of the AV node. It is uncertain if this abnormality in AV conduction is itself associated with arrhythmias.
1.EKG. The following electrocardiographic criteria are suggestive of an accessory pathway consistent with a WPW pattern. The WPW syndrome occurs in the setting of the WPW pattern and SVT.
The PR interval is short, typically <120 ms.
The QRS complex exceeds 120 ms, with some leads showing the characteristic slurred upstroke known as a delta wave (Fig. 21.3) and a normal terminal QRS portion.
The ST-T–segment is directed opposite to the major delta and QRS vectors. The most commonly seen tachycardia in WPW syndrome is characterized by a normal QRS with a regular rate of 150 to 250 beats/min. Onset and termination are abrupt.
2.Localization of accessory pathway. The surface ECG may provide information that allows localization of the accessory pathway. Using the initial 20 ms of the delta wave in leads I, II, aVF, and V1 [classified as positive (+), negative (–), or isoelectric (±)] and the ratio of R- and S-wave amplitudes in leads III and V1 (classified as R ≥ S or R < S) is used in the Arruda criteria. Step 1 uses leads I and V1. If lead I is isoelectric or negative or if R > S in lead V1, it is a left-sided pathway. Using aVF will then help determine if the pathway is anterolateral, lateral, or posterior. Step 2 uses lead II. If lead II is negative, the pathway is located in a branch of the coronary sinus. Step 3 uses lead V1. If V1 is isoelectric or negative, the pathway is septal. Again using lead aVF and QRS vector will help determine if the pathway is anterior, middle, or posteroseptal. Step 4 locates all others on the right side. Leads aVF and II will then help to differentiate anterior, lateral, or posterior locations. The most precise localization method is EP study with ventricular pacing or during orthodromic AVRT (the latter condition is especially helpful because there is VA conduction purely through the accessory pathway, and fusion with VA conduction through the AV node is, therefore, avoided).
3.Risk stratification should be considered for patients with WPW pattern or ventricular preexcitation according to ECG findings. The appearance or disappearance of preexcitation on serial ECGs is of no predictive value. However, the intermittent loss or appearance of preexcitation on a beat-to-beat basis is indicative of lower risk. This may be assessed with ambulatory Holter monitoring during usual activities or with formal exercise stress testing. Such intermittent preexcitation suggests a pathway without the ability for rapid AV conduction and, therefore, lower risk of sudden cardiac death (SCD). However, the reverse is not necessarily true in that most patients with persistent preexcitation may still be at low risk for SCD, but these patients cannot be distinguished from those at risk. Because the greatest danger to patients with preexcitation may be the development of AF, the induction of AF may be most useful in risk stratification. This can be done via transesophageal pacing; however, EP study is the procedure of choice for risk stratification in patients with persistent ventricular preexcitation.
4.Therapy
a.Emergency management of acute tachycardia episodes. A patient demonstrating hemodynamic instability or extreme symptomatology should be cardioverted rapidly. Stable patients may be treated medically.
b.Normal QRS width. Both types of AVRT (orthodromic and antidromic) are AV node–dependent and thus respond to AV nodal blocking therapies. Although it is reasonable to use vagal maneuvers and AV nodal blocking medications acutely in patients presenting with a narrow QRS (immediate synchronized DCC should be available should the rhythm degenerate), it is not safe in patients when they present with a wide QRS. Atrial pacing, either transvenous or transesophageal, is also quite efficacious for terminating these types of tachycardias. Adenosine, although effective in treating orthodromic and antidromic AVRTs, may induce AF in up to 15% of cases and should, therefore, be used with caution. In patients with WPW syndrome, AF is a potentially life-threatening arrhythmia, especially when the accessory pathway has a short antegrade refractory period capable of rapid ventricular conduction.
If the patient develops AF, it has been observed that definitive therapy for the AV reentrant circuit, such as ablation of the accessory pathway, often results in the decrease or even prevention of future episodes of AF.
5.Long-term management
a.Priority of therapy. Patients whose disease is asymptomatic at diagnosis are at low risk for sudden death. As such, it may not be justified to pursue medical or ablative therapy in these patients unless there is a family history of sudden death or the patients are competitive athletes or are in a high-risk occupation. Patients whose disease is symptomatic or who have a history of AF or aborted sudden death may be at higher risk, and such patients warrant further study.
b.Medical therapy. Medical therapy may be appropriate for those with increased risk but no prior symptoms, those with accessory pathways located near the normal conduction pathway that might develop AV block with RFA, or those at increased risk from invasive procedures. Single-drug therapy may be attempted with amiodarone, sotalol, flecainide, or propafenone. These drugs work to slow conduction in both the accessory pathway and the AV node.
c.Combination therapy can be accomplished with drugs that work on the AV node (calcium channel blockers, β-blockers) and with drugs that work exclusively on the accessory pathway (class IA antiarrhythmics).
d.Percutaneous therapy. RFA is effective 85% to 98% of the time, depending on the location of the accessory pathway. Recurrence rates are approximately 5% to 8%. Catheter ablation should be considered for any patient at high risk, patients with symptoms or tachycardias refractory to medical therapy, those who have intolerance to medical therapy, and those with high-risk occupations such as pilots.
IV.Atrial premature depolarizations (APDs). APDs are premature depolarizations that arise from a region other than the SN. The P-wave morphology and PR interval may be different from the sinus P-wave and normal P interval, depending on the location and timing of the APD.
A.Clinical presentation. APDs are usually asymptomatic and in isolation are considered to be benign. Some patients may feel palpitations or skipped beats. If there is atrial bigeminy with each APD causing AV block, patients may develop symptoms of bradycardia. APDs may trigger SVT (AVNRT, AVRT, and atrial tachycardia) or AF in patients with the electrical and structural substrate for these arrhythmias. APDs increase in frequency as patients age and may be more frequent in patients with mitral valve disease, LV dysfunction, hypertrophic cardiomyopathy (HCM), mitral stenosis, pulmonary disease, and renal failure. Stress, alcohol and caffeine consumption, and smoking can promote APDs. However, APDs also occur in healthy individuals with structurally normal hearts and without significant external exposures (caffeine, alcohol, and stress). Although one study showed a correlation between the frequency of APDs within a 24-hour period and the risk of stroke in men, it is unclear what percentage of these men developed AF. Furthermore, in the Atherosclerosis Risk in Communities (ARIC) study which followed patients with APDs and ventricular premature depolarizations (VPDs), only patients with APDs did not have increased incidence of SCD.
B.Pathophysiology. APDs may be caused by a variety of mechanisms, including reentry, triggered activity, and increased automaticity. Reentry is thought to be the most common mechanism.
C.Therapy. Asymptomatic individuals do not need treatment for APDs. For symptomatic patients, β-blockers and class IA, class IC, and class III antiarrhythmic drugs may be considered, although no randomized controlled trials have been performed in this patient population.
V.Ventricular tachyarrhythmias. Ventricular tachyarrhythmias, including monomorphic VT, polymorphic VT, and VF, account for up to 80% of SCD.
A.Ventricular tachycardia. VT is defined as three or more consecutive QRS complexes of ventricular origin at a rate exceeding 100 beats/min.
1.Clinical presentation. The presentation is variable and depends on the clinical setting, the heart rate, the presence of underlying heart disease, and other medical conditions. Some patients have no or minimal symptoms, whereas others may present with syncope or sudden death. The loss of normal AV synchrony may cause symptoms in patients with decreased cardiac function at baseline. Heart rates <150 beats/min can be surprisingly well tolerated in the short term, even in the most compromised individuals. Exposure to these rates for more than a few hours is likely to be associated with heart failure in patients with poor ventricular function, whereas those with normal ventricular function may tolerate prolonged periods at such rates. The range of 150 to 200 beats/min is tolerated variably, according to the factors noted previously. Once the rate reaches and exceeds 200 beats/min, there are symptoms in virtually all patients. Nonsustained ventricular tachycardia (NSVT) is generally defined as a VT of duration <30 seconds. VT is generally regular in rate and appearance, although it can be polymorphic in appearance, slightly irregular with respect to rate, and may have capture and/or fusion beats within it.
2.Differential diagnosis. VT needs to be distinguished from supraventricular tachyarrhythmia with aberrant intraventricular conduction, bundle branch block, and morphologic changes of the QRS complex secondary to metabolic derangement or pacing.
Brugada criteria. Distinguishing VT from supraventricular tachyarrhythmia with aberrancy can be challenging. Various criteria have been proposed. A good rule of thumb is that any WCT in a patient with ischemic heart disease is VT until proven otherwise. Some have reported that >80% of WCTs in such patients are VTs. The algorithm proposed by Brugada may be helpful in making this distinction, and the algorithm is both sensitive (99%) and specific (96.5%) in patients without a preexisting bundle branch block. As shown in Figure 21.4, a stepwise approach is applied. In the first step, the precordial leads are examined for the presence or absence of an RS complex. If an RS is uniformly absent, VT is established. If an RS is present in at least one precordial lead, one moves to the second step, which is measuring the interval from the onset of the QRS complex to the nadir of the S-wave. If this distance is >100 ms in at least one precordial lead, then the diagnosis of VT is made. If there is no RS interval >100 ms, the third step is used. In the third step, one looks for evidence of AV dissociation. If there are more QRS complexes than P-waves, then the diagnosis is VT. If not, then one moves to the fourth step, which involves examining the morphology of the QRS in the precordial leads V1 and V6. If the morphology criteria for VT (Fig. 21.5) are present in these leads, then the diagnosis of VT is established. If not, the diagnosis is supraventricular tachyarrhythmia with aberrant intraventricular conduction.
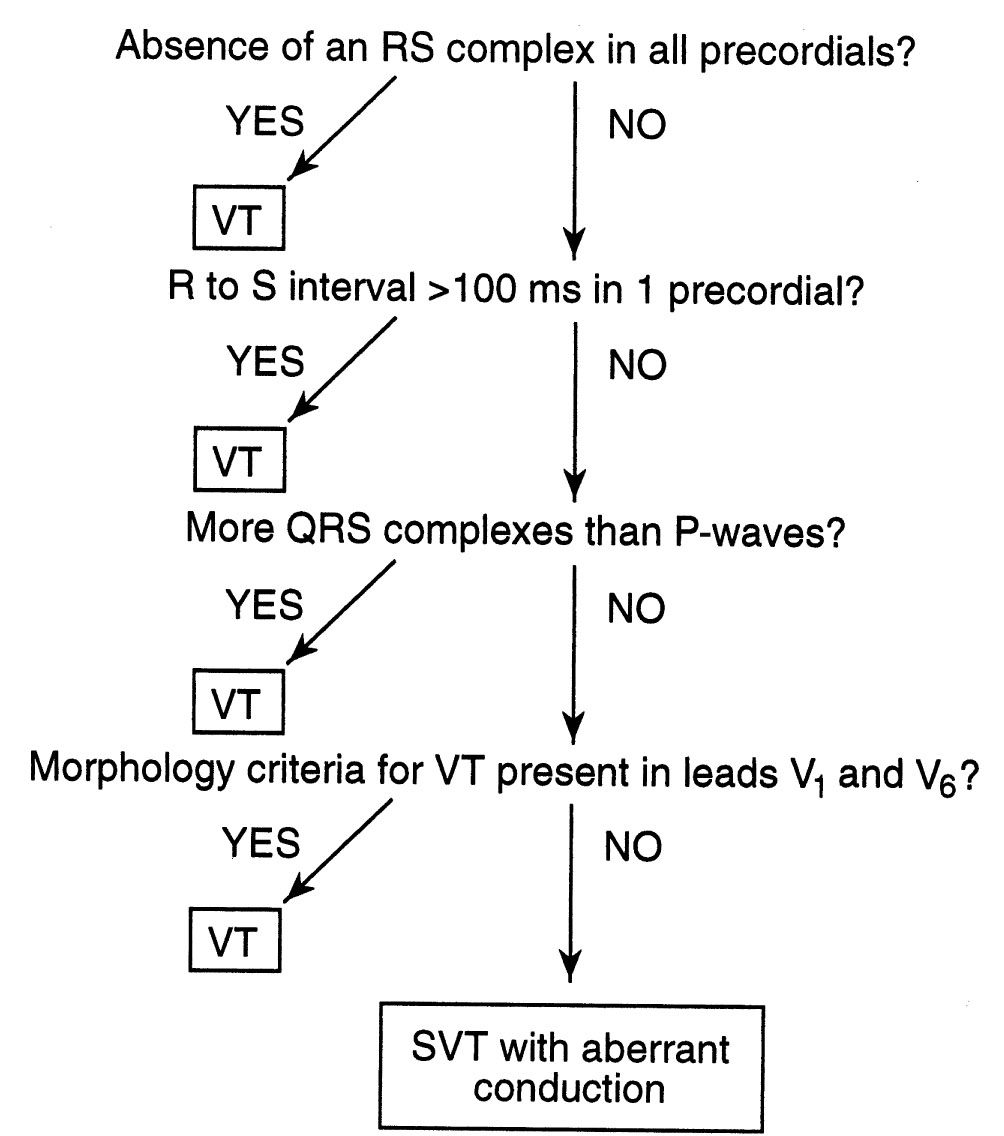
FIGURE 21.4 Brugada criteria for differentiating ventricular tachycardia from supraventricular tachycardia with aberrant intraventricular conduction. VT, ventricular tachycardia; SVT, supraventricular tachycardia.
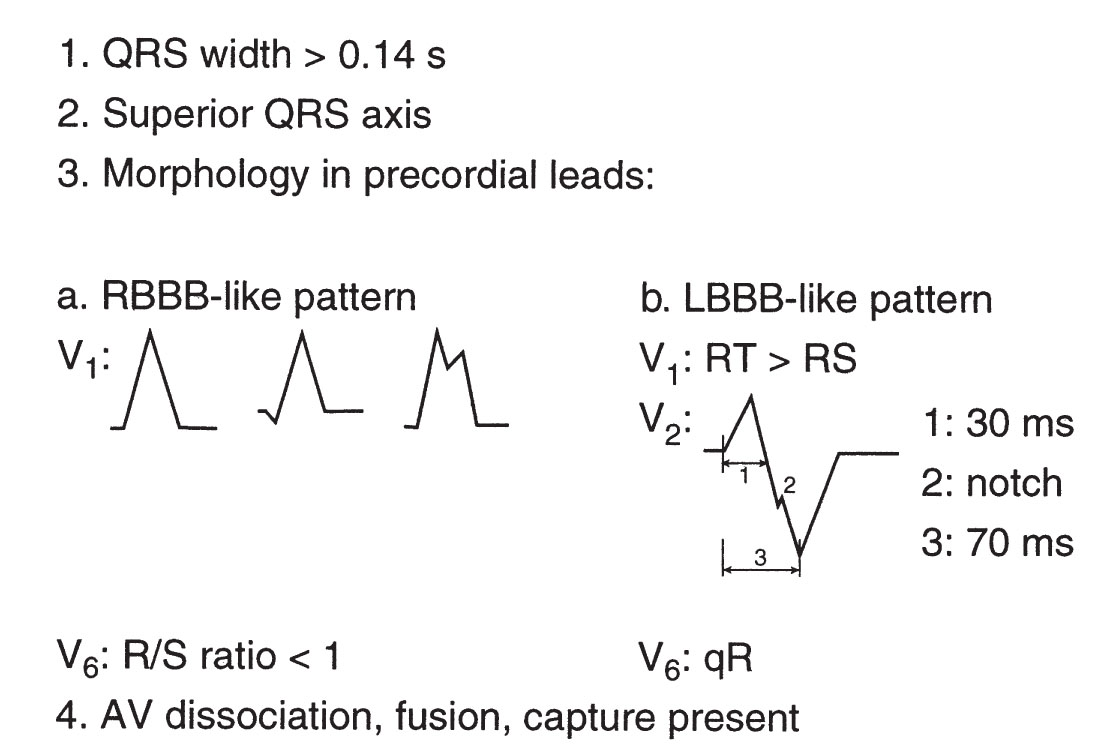
FIGURE 21.5 Classic morphology criteria for ventricular tachycardia. AV, atrioventricular; LBBB, left bundle branch block; RBBB, right bundle branch block.
The Brugada criteria have been further refined to distinguish between VT and supraventricular tachyarrhythmia with antegrade conduction over an accessory pathway. After applying the preceding criteria, a second stepwise algorithm is applied (Fig. 21.6). This second algorithm has a sensitivity of 75% and a specificity of 100% to diagnose VT and exclude preexcited tachycardia. In the first step, leads V4 to V6 are examined to see if the QRS is predominantly negative. If so, then VT is favored. If not, then the second step, examining leads V2 to V6 for the presence of a QR complex in one or more of these leads, is applied. If there is a QR complex in any of these leads, then the diagnosis is VT. The third criterion, presence of AV dissociation, is 100% specific for VT. If there is no AV dissociation, then supraventricular tachyarrhythmia with antegrade accessory pathway conduction is favored.
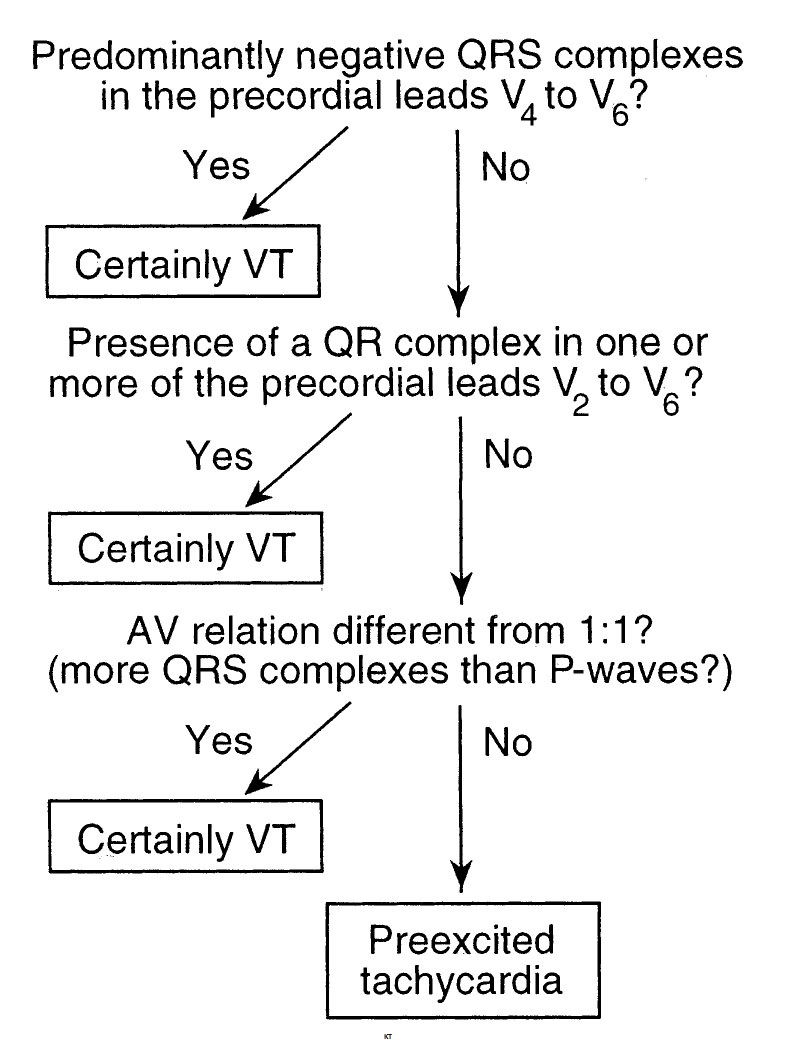
FIGURE 21.6 Brugada criteria for differentiating ventricular tachycardia from antidromic tachycardia over an accessory pathway. AV, atrioventricular; VT, ventricular tachycardia.
A new criterion for differentiating VT from SVT was published in 2008 by Vereckei et al., which boasts a >90% accuracy in their cohort (Fig. 21.7). The rationale to use the new method was to simplify the approach by using one electrocardiographic lead (aVR) in a four-step, tree-like model. The method starts with identifying the presence of an initial R-wave in aVR. If present, VT is diagnosed. If not, then the next step is to assess the presence of an initial R- or Q-wave >40 ms, and if present, VT is present. If this criterion is not satisfied, then the presence of a notch on the descending limb of a negative onset and predominantly negative QRS gives the diagnosis of VT. If this is not present, then one should compare the voltage of the initial 40 ms (Vi) with the voltage of the terminal 40 ms (Vt) of the QRS complex. If Vi/Vt ≤ 1, then it is VT. If none of these criteria are satisfied, then SVT is diagnosed.
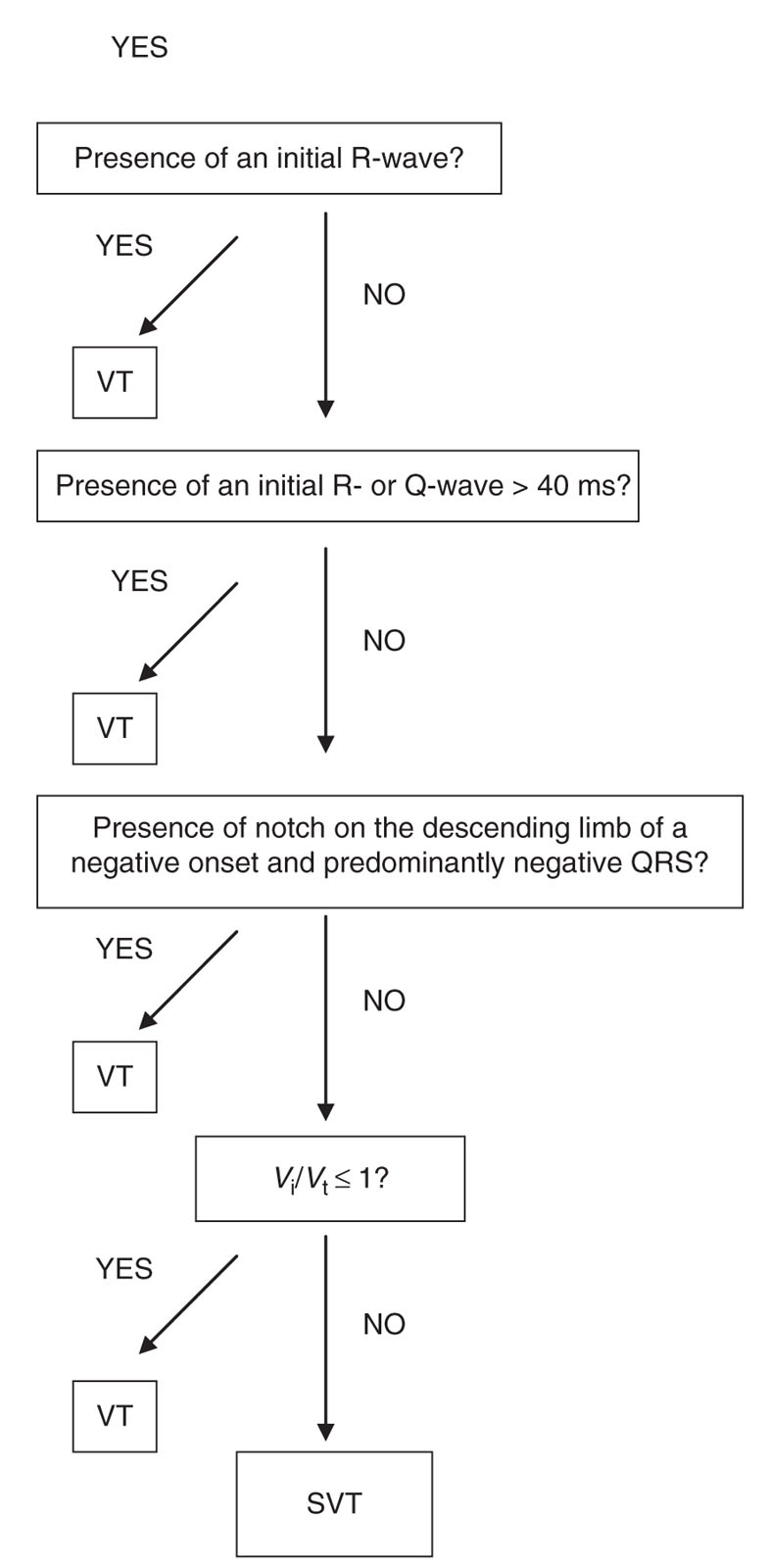
FIGURE 21.7 aVR criteria for diagnosing ventricular tachycardia. Vi, vertical excursion (mV) during initial (Vi) and terminal (Vt) 40 ms of the QRS complex. SVT, supraventricular tachycardia; VT, ventricular tachycardia.
3.Therapy
a.General management. The treatment of VT may involve DCC, discontinuation of offending proarrhythmic drugs, specific antiarrhythmic therapy with drugs, correction of electrolyte imbalances, implantable devices, ablation, revascularization, and surgery. The appropriate selection of the preceding therapies is aided by the assessment of the patient, an understanding of the etiology and mechanism of the VT, knowledge of any exacerbating medical conditions contributing to the VT, and the risk-to-benefit ratio of the available therapies.
b.Priority of therapy. A patient who has no hemodynamic compromise can be treated medically, at least initially. As with most types of tachyarrhythmias, the treatment of any unstable patient with VT is rapid DCC. The treatment for pulseless VT is asynchronous DCC with a starting energy of 200 to 360 J. If the patient is conscious but has unstable vital signs or is extremely symptomatic, synchronized DCC is recommended. The most recent Advanced Cardiac Life Support guidelines (AHA) currently emphasize the delivery of high-quality cardiopulmonary resuscitation (CPR): effective chest compressions (at least 100/min and compression depth of 2 in.) with minimal interruptions, rescue breaths given over 1 second with visible chest rise while avoiding hyperventilation (30:2 ratio before an advanced airway and 10 asynchronous breaths/min after airway is secured), and a single shock to attempt to defibrillate pulseless VT patients followed by immediate continuation of CPR.
Magnesium (2 g over 5 minutes) should be administered for torsades de pointes. Whenever possible, a reversible cause for VT should be sought. Elimination of ischemia and correction of electrolyte abnormalities are recommended. Bradycardia may cause frequent premature ventricular contractions or VT. Maneuvers including temporary pacing and agents that increase heart rate should be used. Hypotension should be promptly corrected. Therapy for CHF should be optimized with the agents known to promote survival in this disorder. Offending agents should be stopped whenever possible, and antidotes should be administered in the case of overdosage and poisoning.
d.Prevention and prophylactic treatment. All antiarrhythmic agents to date, except β-blockers, have not been shown in randomized clinical trials to be effective as the primary management of patients with life-threatening ventricular arrhythmias or in the prevention of SCD. Since the Cardiac Arrhythmia Suppression Trial (CAST) data have become available, there has been a shift away from the use of class I agents and toward the use of class III agents and β-blockers for prophylactic maintenance therapy of VT. The development of curative catheter-based therapies and surgical procedures has somewhat reduced the role of antiarrhythmics in the prevention of recurrence, especially for VT occurring in normal hearts, which has very high cure rates with catheter ablation. However, antiarrhythmic drug therapy remains the first-line treatment for VT, particularly for patients with cardiomyopathy. The greatest impact on survival in sudden death has been made by the implantable cardioverter–defibrillator (ICD). Data from the Multicenter Unsustained Tachycardia Trial investigations have shown that patients with CAD, an ejection fraction (EF) <40%, and NSVT who have inducible sustained VT on testing are at substantially increased risk over those who do not have inducible VT.
e.Medical therapy. Although drug therapy continues to have a role in the prevention of VT and sudden death, this role has become more limited because there has been no decrease in mortality with the use of antiarrhythmic drugs. The Electrophysiologic Studies Versus Electrocardiographic Monitoring trial studied the efficacy of seven antiarrhythmics (imipramine, mexiletine, pirmenol, procainamide, propafenone, quinidine, and sotalol) in preventing the recurrence of sustained VT. Sotalol was seen to be the most effective, although even with sotalol, the recurrence rate was disappointing. The European Myocardial Infarct Amiodarone Trial and the Canadian Amiodarone Myocardial Infarction Arrhythmia Trial (CAMIAT) investigations were designed to study the effectiveness of empiric amiodarone for the prevention of VT after MI. Although both of these trials showed a decrease in arrhythmic deaths, no survival benefit was recorded.
f.Combination therapy. Drug therapy is becoming an adjunct to ICD therapy in this high-risk population. At present, fully half of those with ICDs remain on antiarrhythmic therapy. The rationale for this combined therapy includes preventing atrial tachyarrhythmias and reducing the frequency of VT and thus the frequency of ICD discharge.
Calcium channel blockers are used primarily in the management of supraventricular tachyarrhythmia. However, some of the idiopathic monomorphic VTs, (the VTs originating in the right ventricular outflow tract [RVOT]), fascicular VT, and the VTs of digitalis toxicity are responsive to calcium channel blocking agents such as verapamil and diltiazem (because of the underlying mechanism of calcium-dependent triggered activity). RFA is potentially curative for idiopathic VTs and should be considered despite effective termination with calcium channel blockers.
β-Blockers may be effective, particularly for outflow tract VT. Idiopathic left VT may respond to calcium channel blockers.
g.Percutaneous therapy
(1)ICDs. Two large trials comparing ICDs with amiodarone in high-risk patients with prior infarction, the Multicenter Automatic Defibrillator Implantation Trial (MADIT) and the Antiarrhythmics Versus Implantable Defibrillator (AVID) trial, have been completed. High risk implies either an EF of 35% or less or the presence of inducible sustained VT at EP study. Both trials showed a decided advantage for ICDs, with 30% to 50% reductions in mortality with ICDs. In fact, the AVID trial found no survival benefit from amiodarone, β-blockers, or any other antiarrhythmic agent. Newer ICDs often have antitachycardia pacing (ATP) capabilities, can recognize monomorphic ventricular rhythms with rates <200 beats/min, and can rapidly pace the ventricles to restore sinus rhythm, aborting the need for countershock. In the Primary Prevention Parameters Evaluation study which evaluated the effects of ICD programming in a patient population receiving ICDs for primary prevention, using ATP as first-line therapy for fast VT (≥182 and <250 beats/min), including a monitoring zone at 167 beats/min, and applying SVT versus VT discriminators for rates <200 beats/min helped in reducing shocks without negatively affecting the mortality. Data from MADIT II have shown that in patients with a prior MI and an EF < 30%, the implantation of a defibrillator is associated with a significant improvement in survival. Data from the MADIT-RIT trial showed decreased inappropriate therapy and all-cause mortality with higher rate or longer detection intervals as compared with conventional programming in primary prevention patients. Programmed high-rate therapy (with a 2.5-second delay before the initiation of therapy at a heart rate of ≥200 beats/min) or delayed therapy (with a 60-second delay at 170 to 199 beats/min, a 12-second delay at 200 to 249 beats/min, and a 2.5-second delay at ≥250 beats/min) was associated with a decrease in the number of patients with a first occurrence of inappropriate ATP or shocks, and all-cause mortality, as compared with conventional programming (with a 2.5-second delay at 170 to 199 beats/min and a 1.0-second delay at ≥200 beats/min).
(2)Catheter-based therapy. RFA may be effective for reducing the incidence of VT. The success rate depends on the type of VT, with the highest success rates (>90%) in structurally normal hearts. VT associated with underlying cardiomyopathy has lower success rates with ablation, particularly those with arrhythmogenic right ventricular (RV) cardiomyopathy and ischemic cardiomyopathy. However, catheter ablation still remains an effective and feasible approach, even for these types of VTs. Presently, catheter ablation of VT does not obviate the need for an ICD in a patient with an indication for one. The role of ablation is to target the source in focal VT or the critical isthmus is reentrant VT. The VISTA trial compared targeted ablation (ablation of the critical isthmus of the reentrant circuit) versus a substrate-based ablation (ablation of all abnormal electrograms located within scar) in patients with ischemic cardiomyopathy and stable VT. The substrate-based approach resulted in fewer VT recurrences and less antiarrhythmic drug use at 1 year.
B.Diagnostic evaluation of a patient with VT. Once the diagnosis of VT has been established and the patient has been acutely managed with either DCC or medical therapy, further management depends on the underlying cardiac pathology. In broad terms, the substrate can be divided into two categories: the structurally normal heart and the structurally abnormal heart. Various modalities are available to determine the cardiac structure and function, which include electrocardiography, cardiac catheterization, echocardiography, nuclear imaging, and magnetic resonance imaging.
1.VT in a structurally normal heart. About 10% of VT in the United States occurs in structurally normal hearts, the so-called idiopathic VT. These patients have no significant CAD, no family history of arrhythmia or sudden death, and normal surface ECGs. They can be focal VTs or reentry VTs. Focal VTs are a result of triggered activity, abnormal automaticity, or reentry within the Purkinje fibers.
a.Focal VT
(1)Mechanism. Focal VTs most commonly arise from the RVOT and account for up to 70% of idiopathic VTs. They may be caused by cyclic adenosine monophosphate (cAMP)-mediated EADs. Of particular importance in the diagnosis of a patient who presents with LBBB VT is to be cognizant of the possibility of arrhythmogenic right ventricular dysplasia (ARVD), which falls into the category of VT/PVCs in the structurally abnormal heart. The clinician should investigate for RV structural abnormalities (fatty infiltration), ask about a family history of ARVD, and review the electrogram for the presence of T-wave inversion across the right precordial leads, and/or epsilon waves (Fig. 21.10). A cardiac magnetic resonance imaging or cardiac positron emission tomography scan may also be useful to rule out the presence of cardiac sarcoidosis, which also would fall into the category of the structurally abnormal heart.
(2)ECG. The surface ECG usually demonstrates an LBBB and inferior axis with very positive QRS voltage in inferior leads. Other locations of focal VTs may include the LV outflow tract, aortic cusps, pulmonary artery, mitral and tricuspid annuli, papillary muscles, and epicardium.
(3)Treatment. In general focal VTs are benign, carrying a very low risk of SCD. Therefore, the treatment is predominantly guided by symptoms. Given the role of cAMP in inducing this form of VT, adenosine may be effective at acute termination. For longer term therapy in the symptomatic patient, β-blockers are typically the first-line agents and can be effective in up to 50% of patients. The nondihydropyridine calcium channel blockers may also be effective in 25% to 50% of patients. Very effective medications are sotalol and amiodarone, with up to 90% success rate in eliminating symptoms, but potential side effects may limit their use. Patients who wish to potentially avoid lifelong medications or who are refractory to medical therapy can be considered for ablation, which has variable success rate depending on the location. RVOT VT ablation success rate may be as high as 90%. Ablation procedures, although generally safe, may be associated with infrequent but life-threatening complications, including cardiac perforation and tamponade.
b.Fascicular VTs. Fascicular VT involves reentry using the tissue of the LV septum as the antegrade limb and usually the posterior fascicle in the retrograde limb.
(1)ECG. This typically produced a right bundle branch block (RBBB) with left-axis deviation pattern. Less commonly, the QRS pattern is an RBBB with right-axis deviation (left anterior fascicular VT).
(2)Treatment. This subtype of VT may be verapamil-sensitive, but catheter ablation can be considered in patients who want to avoid long-term medication or in whom medical therapy is ineffective.
2.VT/VF associated with channelopathies. Various cardiac ion channel disturbances can predispose to ventricular arrhythmias. Patients with these channelopathies have no overt structural heart disease. They are genetically heterogeneous and have variable penetrance. They include LQTS, short QT syndrome, Brugada syndrome, and catecholaminergic polymorphic VT.
a.Long QT syndrome
(1)Pathology and presentation. This channelopathy is characterized by prolonged cellular repolarization resulting in an increase of the QT interval. Clinical presentation includes syncope or sudden death as a result of torsade de pointes, and usually an autosomal dominant transmission pattern.
LQT1 patients typically have broad-based T-waves and exercise-induced arrhythmias, especially during swimming. LQT2 syndrome is characterized by low-amplitude or notched T-waves and auditory triggers such as sudden loud sounds like alarm clocks or strong emotion, and LQT3 is characterized by a long isoelectric ST-segment and arrhythmias during sleep.
(2)Treatment. In patients with LQTS, risk stratification involves assessment of age, gender, clinical history, and possibly the QT interval and genetic mutation. Whereas β-blockers have a variable efficacy depending on the type of LQT mutation, typically high doses of propranolol or nadolol are used to prevent clinical symptoms. β-Blockers are recommended as first-line therapy for all LQTS patients. For the LQT3 patients, there may be a role for sodium channel blockers (flecainide) as add-on therapy if the QTc is >500 ms and acute dosing results in a >40 ms shortening of the QTc. Patients should be advised against high-intensity sports and should be educated regarding avoidance of QT-prolonging drugs. Patient with syncope despite β-blocker therapy or history of aborted sudden death should undergo ICD implantation. Left cardiac sympathetic denervation can be used as an adjunctive therapy to reduce recurrence of arrhythmias.
b.Short QT syndrome. This syndrome is characterized by gain-of-function mutations in the IKs, IKr, and IK1 potassium channels or CACNA1 and CACNB2 L-type calcium channel mutations. ICD therapy is the primary treatment modality (class I) in patients with aborted SCD or VT. However, particular attention needs to be given to the prevention of inappropriate shocks, because patients may have T-wave oversensing (tall T-waves) and a high incidence of AF. Quinidine can be considered (class IIb) in asymptomatic patients with a family history of SCD.
c.Brugada syndrome. Brugada syndrome is a condition associated with SCD in the setting of a structurally normal heart, characterized by an electrocardiographic pattern of RBBB and ST-segment elevation in leads V1 to V3 (Fig. 21.8). It is inherited in an autosomal dominant pattern with a male predominance. It is a genetically heterogeneous disease with many mutations linked to the gene SCN5A, which encodes for a cardiac sodium channel, leading to unopposed Ito potassium current in the RV epicardium. The diagnosis can be difficult because of the variable expression of the ECGs at baseline, changes in the ECG over time induced by a host of factors (fever, heart rate, autonomic tone, and medications), and the wide range of clinical manifestations. The diagnosis should be considered in patients who have documented VF, self-terminating polymorphic VT, family members with ST-segment elevation, syncope, or family history of sudden death in the setting of the electrocardiographic findings noted previously. Currently, no medication has proved effective in preventing SCD in these patients, but quinidine, which blocks the Ito channel, may be used as an adjunctive therapy to reduce the likelihood of arrhythmias. ICDs are currently the only available treatment and are recommended in patients with previous cardiac arrest or VT (class I), and syncope felt to be from arrhythmia with spontaneous ECG pattern (class IIa), or induced VF during EP study (IIb). It is generally recommended to implant an ICD in symptomatic patients and clinically follow asymptomatic patients with an abnormal ECG only on pharmacologic provocation and no inducible ventricular arrhythmias.
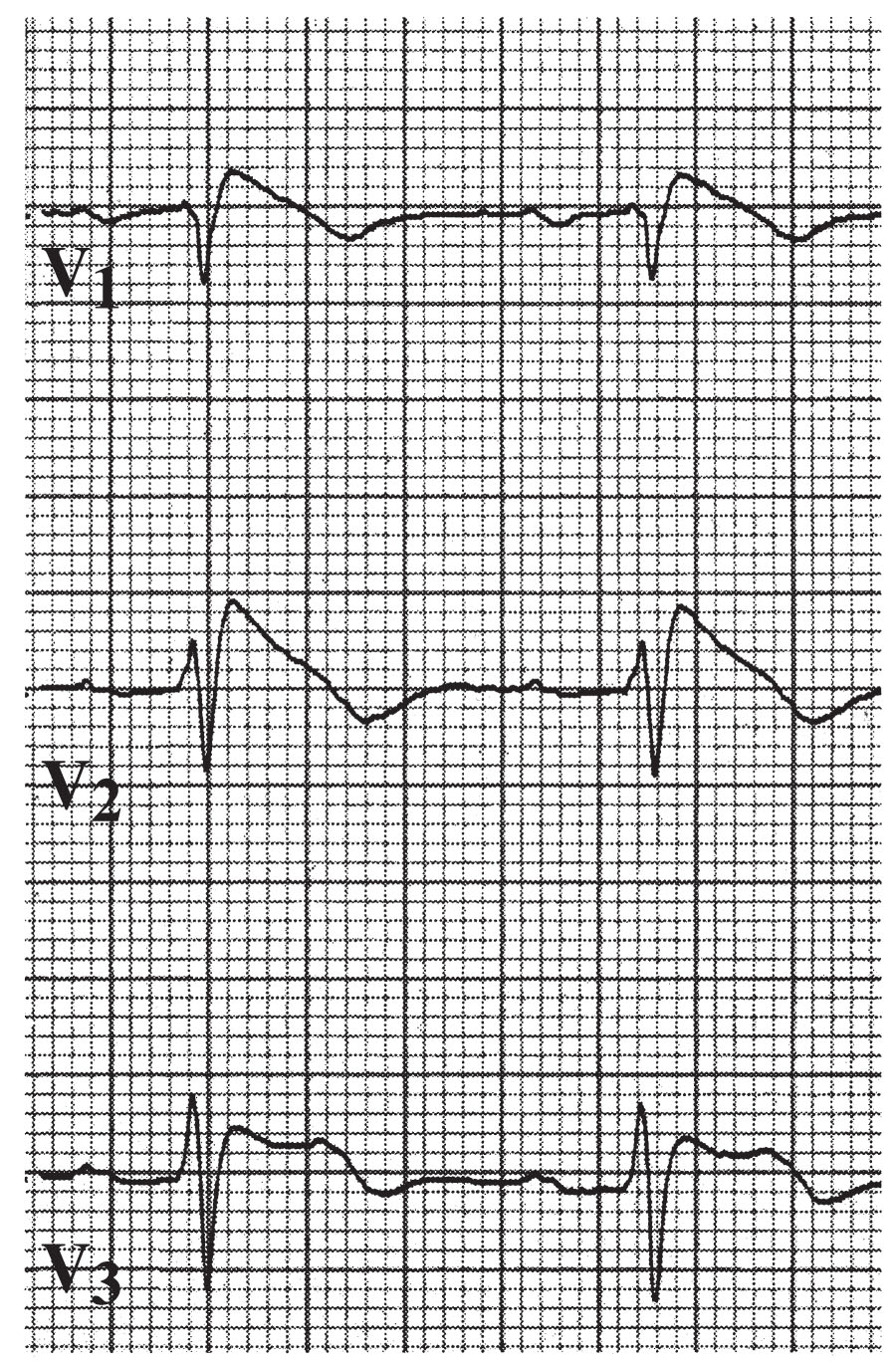
FIGURE 21.8 Leads V1 through V3, demonstrating type 1 Brugada pattern.
d.Catecholaminergic polymorphic VT. This arrhythmia is more common in adolescents and children and may present with SCD or stress-induced syncope. Although usually familial, it can also occur because of de novo mutations. Triggers often include emotional or physical stress, and the arrhythmia can be polymorphic, bidirectional, and less commonly, VF. Two culprit genes have been identified thus far: calsequestrin 2 (autosomal recessive pattern) and cardiac ryanodine receptor (autosomal dominant pattern). ICDs are indicated in patients with this syndrome and syncope and/or VT. β-Blockers can reduce the incidence of arrhythmias as well. Flecainide may be useful (class IIa), and sympathetic denervation can be considered (class IIb) in patients who have recurrent arrhythmia despite β-blocker therapy.
3.VT in the structurally abnormal heart
a.Ischemic VT. Patients with ischemic VT may have acute ischemia leading to MI or a history of ischemic heart disease with scar. Patients who have VF/VT within 48 hours of an acute MI have a relatively high in hospital and 30-day mortality compared with patients who do not have VF/VT.
(1)Etiology and pathophysiology. At the cellular level, ischemia may alter action potentials, prolong refractoriness of cells, and uncouple the cell-to-cell propagation of depolarization. The biochemical milieu in which the cells exist with respect to ion concentrations, acid–base balance, and so forth can be altered. Also, the myocardial damage as a result of infarction is structurally heterogeneous. Therefore, scar tissue and healthy tissue are admixed in the region of the infarction. As described before, a reentrant circuit requires two functionally distinct pathways with unidirectional block in one pathway and slowed conduction down a second pathway. The changes associated with ischemia provide the anatomic substrate for reentry. The VT in the setting of ischemia tends to be polymorphic, whereas VT in the setting of established myocardial scar tends to be monomorphic. Ischemia has been shown to prolong the QT interval in some subjects, often with associated T-wave inversion. The QT interval in ischemic-mediated polymorphic VT is not as prolonged as that in torsade de pointes, another polymorphic VT. Ischemia is by far the most common cause of polymorphic VT with normal QT interval.
(2)Predictors of VT. As might be expected, larger infarcts with greater resultant impairment of LV systolic function are more likely to be associated with VT. In fact, LV systolic function is the single most important predictor of sudden death because of arrhythmia. Similarly, the presence of an open artery appears to reduce the occurrence of VT and other arrhythmias. Other proposed predictors include syncope, abnormal signal-averaged electrocardiogram (SAECG) result, NSVT, absence of heart rate variability, abnormal EP study outcome, and T-wave alternans (TWA); however, currently, the LVEF remains the most accurate predictor of sudden death.
(3)Laboratory examination and diagnostic testing. The various tests for risk stratification (EP study, SAECG, heart rate variability, TWA, and so forth) have shown poor specificity and positive predictive value for VT and thus should not be used alone to guide therapy but in combination with the rest of the clinical information.
(4)Role for ICD. In patients who present with a VF/VT arrest in the setting of an acute MI (within 24 to 48 hours of infarction), revascularization should be the primary initial treatment. Given the fact that acute ischemia is considered a “transient or potentially correctable cause” of VF/VT, and such patients were excluded from the AVID trial, based on current guidelines, an ICD would be indicated 90 days after revascularization if the LVEF is ≤35% or after 40 days if no revascularization was performed, but treatment should be guided on a patient-to-patient basis. Of note, patients in the AVID registry who were excluded from the AVID trial because of a “transient or potentially correctable cause” had a high mortality risk in follow-up. Of the 278 patients studied, 183 patients were determined to have ischemic causes. Of these, 161 were categorized as new MI and 22 were categorized as transient ischemia. Other causes included electrolyte abnormalities, antiarrhythmic drug interaction, and “other (illicit drug use, sepsis, hypoxia, electrocution, drowning).” For patients who are post-MI and are deemed to be at high risk for SCD during the waiting period of 40 to 90 days, a wearable cardioverter–defibrillator (WCD) may provide protection against cardiac arrest, but no randomized clinical trials comparing WCDs with medical therapy in this post-MI period have been completed to date. Patients with late VT/VF (i.e., >48 hours after acute MI) are deemed to be particularly high risk for recurrent VT/VF and therefore typically receive ICDs before hospital discharge. These patients are considered to meet secondary prevention indications for ICDs.
b.Accelerated idioventricular rhythm (Fig. 21.9) is a form of VT seen almost exclusively in ischemic heart disease, particularly during an MI and especially after reperfusion of an occluded territory. It may be seen with digitalis toxicity, but can also be present in healthy adults and children with no structural heart disease.
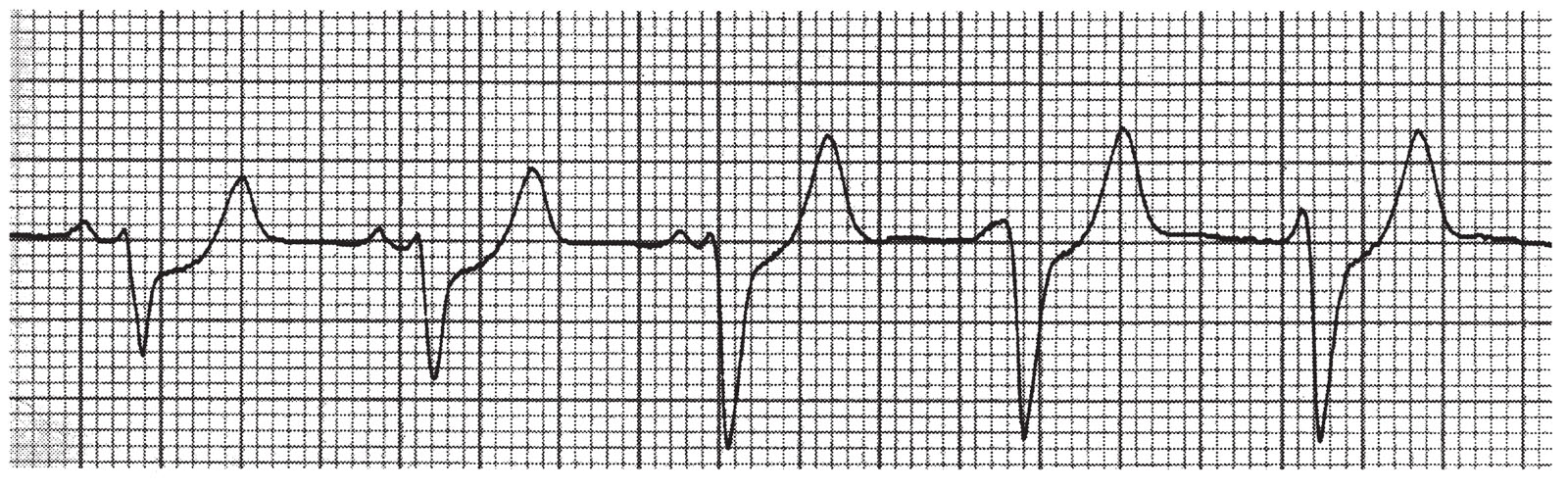
FIGURE 21.9 Accelerated idioventricular rhythm (beats 3 through 5), interspersed with normal sinus rhythm (beats 1 and 2), lead IV.
(1)The EKG features include regular or slightly irregular ventricular rhythm, a rate of 60 to 110 beats/min, a QRS morphology resembling that of PVCs, and, often, AV dissociation as well as fusion beats and capture beats.
Accelerating the sinus rhythm with atropine or atrial pacing can be useful to suppress the accelerated idioventricular rhythm.
(3)Therapy is rarely necessary, unless the loss of AV synchrony results in hemodynamic compromise, a more rapid VT intervenes, the accelerated idioventricular rhythm falling on the T-wave of the preceding beat (R-on-T phenomenon), the ventricular rate being rapid enough to produce symptoms, or occurrence of VF.
c.Dilated cardiomyopathy (DCM). Risk stratification is particularly difficult in patients with DCM because SAECG, microvolt TWA, and an EP study are not reliable predictors in this population, and asymptomatic ventricular arrhythmias are common. The Defibrillators in Nonischemic Cardiomyopathy Treatment Evaluation and Sudden Cardiac Death in Heart Failure Trial have influenced the current guidelines for implanting ICDs in patients with DCM. ICDs are recommended for patients who manifest life-threatening arrhythmias or syncope and for primary prevention in patients who have an LVEF <35% and are New York Heart Association classes I to III (less evidence exists for class I). All patients should be receiving chronic optimal medical therapy and have a life expectancy >1 year. Bundle branch reentrant tachycardia occurs most commonly in patients with DCM for which EP testing is helpful in diagnosing and guiding ablative treatment. Although ablation may be curative if bundle branch reentry is the mechanism, such patients should still be considered for ICD implantation.
d.Hypertrophic cardiomyopathy. Supraventricular tachyarrhythmia and AF are particularly poorly tolerated by these patients, as is ischemia, and may lead to VT. No prospective randomized trials regarding ICD therapy have been carried out to date in this patient population. Consequently, the precise risk stratification is debated. ICDs are recommended for patients who have sustained VT or VF, or both, and for primary prevention in patients who have either one of the preceding life-threatening arrhythmias or one or more of other major risk factors for SCD (nonsustained spontaneous VT, family history of premature SCD, unexplained syncope, LV thickness ≥30 mm, or abnormal exercise blood pressure). Again, all patients should be receiving chronic optimal medical therapy and have a life expectancy >1 year. EP study may be helpful in stratifying risk for VT and sudden death. Patients at low risk for HCM include those with infrequent or brief episodes that are asymptomatic or mildly symptomatic. Although amiodarone may be beneficial in this population, an ICD is increasingly used in those considered to be at high risk.
e.Muscular dystrophies, particularly Duchenne muscular dystrophy and myotonic dystrophy, have been associated with frequent defects in the conduction system. Heart block and bundle branch block as well as sudden death because of ventricular tachyarrhythmias are well-recognized complications of these muscular disorders.
f.Congenital heart disease. Structural abnormalities such as repaired tetralogy of Fallot and mitral valve prolapse have been associated with increased risk of VT and sudden death. In tetralogy of Fallot, the VT often originates in the RVOT, at the site of a previous repair. Risk of VT and sudden death in this population has been associated with QRS width (and rate of QRS width increase) as well as severity of pulmonary insufficiency. Mitral valve prolapse has been uncommonly linked to sudden death, although ventricular arrhythmias are not uncommon. The prognosis with respect to VT is quite good in mitral valve prolapse.
g.Arrhythmogenic right ventricular cardiomyopathy is a cardiomyopathy that begins in the right ventricle and often progresses to involve the left ventricle. It results in RV dilation with resultant poor contractile function. The RV muscle becomes increasingly replaced by adipose and fibrous tissues as the disease progresses. VT arising in the right ventricle is often an early manifestation of this disorder. The VT is a reentrant type and has an LBBB morphology, although in sinus rhythm, there is often inversion of the T-waves in the anterior precordial leads and a slurring of the terminal portion of the QRS complex, known as an epsilon wave (Fig. 21.10). These patients frequently have a positive SAECG for late potentials. The combination of the scarring and the late potentials provides the anatomic substrate for reentry. During EP study, it may be possible to elicit VT of varying morphologies, because of the prolific scarring of the myocardium. The risk of VT correlates with the extent of myocardial involvement. Therapy with sotalol or high-dose amiodarone may be somewhat successful. Ablation via catheters is often successful, but only temporizing, because the generalized involvement tends to give rise to arrhythmias at a different locus later in the disease course. ICDs are often the only reliable therapy to prevent sudden death in this disorder. Patients are advised against intense exercise, because this may promote the incidence and progression of arrhythmias.
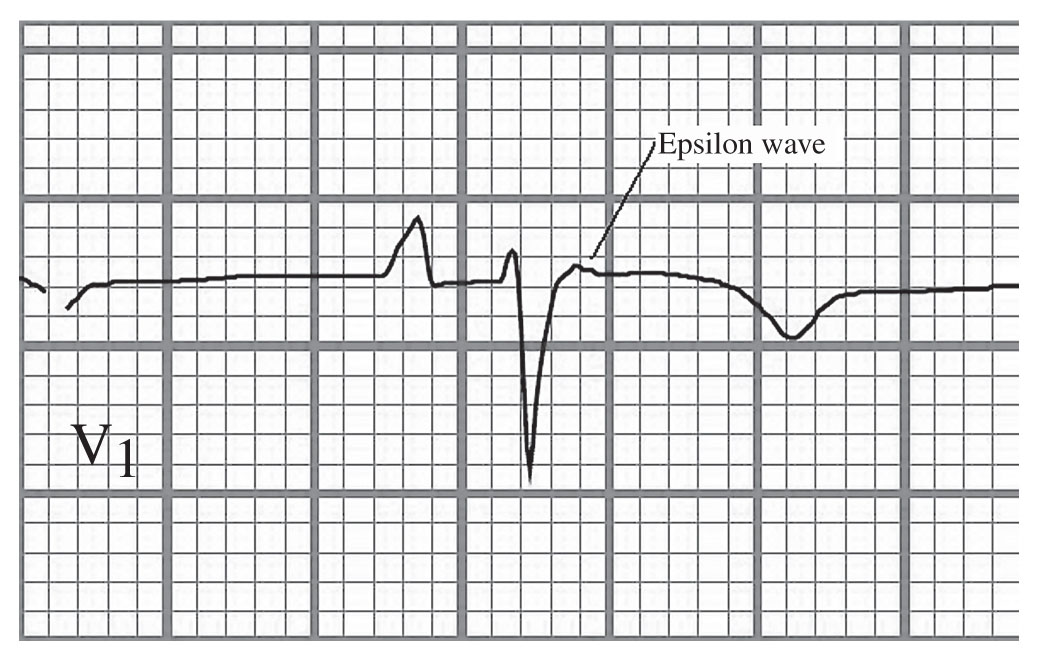
FIGURE 21.10 Epsilon wave in a patient with arrhythmogenic right ventricular dysplasia.
h.Several inflammatory or infectious conditions have been associated with VT.
Sarcoidosis is frequently cited as a cause of heart block and may also cause VT and VF. Amiodarone and sotalol are the most efficacious agents in this disorder, although an ICD may be necessary in addition to the drug therapy. Catheter ablation may be considered for recurrent arrhythmia.
Acute myocarditis has been associated with both polymorphic and monomorphic VTs. It may be intractable in giant cell myocarditis. Antiarrhythmic therapy and anti-inflammatory therapy are generally combined in the treatment of these patients.
Chagas disease, caused by the parasite Trypanosoma cruzi, is a well-known cause of cardiomyopathy, particularly in South and Central America. VT and other arrhythmias because of conduction system involvement are common complications. Therapy involves antiparasitic treatment, standard therapy for CHF, antiarrhythmics, and pacemaker or ICD implantation, as appropriate. Some patients require catheter ablation of refractory VT, which sometimes must be performed epicardially.
i.Coronary anomalies. Anomalous aortic origin of the coronary artery is recognized as a cause of sudden death and/or exercise-induced death in young individuals. In an autopsy study of over 200 patients conducted by the Armed Forces Institute of Pathology, the most common coronary anomalies included the right coronary artery and left main coronary artery arising from the left sinus, the left main and right coronary arteries arising from the right sinus, single coronary artery from the aorta, and the left main or left anterior descending artery arising from the pulmonary artery. Patients whose coronary arteries take an interarterial course (between the pulmonary artery and the aorta) may develop exercise-induced ischemia and/or sudden death. Surgical revascularization in patients with symptomatic coronary anomalies has been well described. Surgical treatment for patients with high-risk coronary anomalies who are asymptomatic is controversial.
Digitalis toxicity can propagate DADs, which generate action potentials, leading to VT. The VT of digitalis toxicity is typically monomorphic and often responds to calcium channel blockers. Rarely, digitalis toxicity manifests as a bidirectional VT, meaning that it has a regular rhythm with an axis that alternates from −60° to −90° to +120° to +130°, with a ventricular rate from 140 to 200 beats/min. Because digitalis toxicity may have a narrow QRS complex and may respond to calcium channel blockers, it may be confused with supraventricular tachyarrhythmia. This type of VT is best managed by removing the offending agent, digoxin, with its binding antibody. The treatment for digitalis toxicity is the same in the face of bidirectional VT.
k.Torsade de pointes is a type of polymorphic VT associated with delayed myocardial repolarization, most often manifested as a prolonged QT interval. Although the duration of torsade de pointes is typically brief (<20 seconds), it can be sustained and can degenerate into VF. It generally has an irregular ventricular rate (>200 beats/min) and displays a polymorphic structure with an undulating appearance. The QRS complexes appear to twist around an isoelectric axis. Characteristics that distinguish torsade de pointes from other forms of VT include (1) prolonged QT interval, (2) initiation with a short–long–short sequence, and (3) typical “twisting of the points” appearance of the VT.
(1)Etiology. QT prolongation can be congenital or acquired.
The congenital forms are seen in the LQTS, discussed in Chapter 23.
The acquired forms are most often drug induced, although polymorphic VT with a prolonged QT can be caused by electrolyte abnormalities, hypothyroidism, cerebrovascular events, MI or ischemia, starvation diets, organophosphate poisoning, myocarditis, severe CHF, and mitral valve prolapse.
The most commonly implicated drugs have been the class IA drugs, although less frequent occurrences have been reported with all subclasses of class I antiarrhythmics. The class III drugs, such as sotalol, dofetilide, and, less commonly, amiodarone, have been implicated. The incidence of torsade de pointes with sotalol is in the range of 2% to 5% and with dofetilide it is ~1%. Ibutilide is an antiarrhythmic agent for supraventricular tachyarrhythmias that is associated with an incidence of torsade de pointes at least as high as that of sotalol. Other drugs implicated include the phenothiazines, haloperidol, and the tricyclic antidepressants. Antibiotics, including erythromycin and other macrolides, as well as trimethoprim–sulfamethoxazole combinations, have been implicated. The macrolides are particularly prone to cause torsade de pointes when combined with certain antihistamines such as astemizole and terfenadine. These antihistamines have also been found to cause torsade de pointes when combined with certain azole antifungal agents such as ketoconazole. Ionic contrast and promotility agents such as cisapride have also been associated with torsade de pointes. Medications associated with increasing QT interval are listed on the following website: www.torsades.org. It is maintained by the University of Arizona Center for Education and Research on Therapeutics.
Bradycardia can promote torsade de pointes in patients with prolonged QT intervals, although it is not clear if bradycardia by itself predisposes to torsade de pointes. Specifically, pause-dependent VT occurs in the setting of bradycardia and a prolonged QT interval. Usually a long RR interval followed by a short RR interval followed by another long RR interval initiates the VT.
Electrolyte disorders. Hypokalemia is the electrolyte disorder most reliably linked to torsade de pointes. Hypomagnesemia has been proposed as a logical cause, because the administration of magnesium frequently terminates torsade de pointes. However, there is scant evidence to confirm this. Likewise, although hypocalcemia is associated with prolongation of the QT interval, there are only rare reports of torsade de pointes associated with hypocalcemia.
Short coupled VT. Polymorphic VT is initiated <400 ms following the preceding QRS complex.
R-on-T phenomenon occurs when a defibrillation or pacing current or spike is delivered simultaneously with occurrence of the electrocardiographic T-wave resulting in polymorphic VT.
A variety of cerebrovascular events have been associated with torsade de pointes, most notably subarachnoid hemorrhage. The prolongation of the QT interval sometimes seen with intracranial bleeding is usually transient, resolving within weeks.
(2)Therapy. Acute management is aimed at terminating the arrhythmia.
If torsade de pointes is sustained or associated with hemodynamic compromise, prompt DCC should be carried out. Starting voltages are generally 50 to 100 J and can be advanced to 360 J if necessary.
Correction of hypokalemia, hypomagnesemia, and hypocalcemia should be undertaken promptly. Magnesium can be given in a bolus form at a dose of 1 to 2 g, with a total dose of 2 to 4 g given over 10 to 15 minutes. This successfully terminates torsade de pointes within 5 minutes in up to 75% of patients and within 15 minutes in virtually all patients.
Bradycardia can be corrected with either isoproterenol infusion or temporary transvenous pacing. Pacing may be preferable when readily available, because of the potential complications of isoproterenol therapy (worsened ischemia and hypertension). Offending agents should be discontinued.
l.Miscellaneous
Commotio cordis. Commotio cordis is the sudden ventricular arrhythmia occurring as a result of a blunt, nonpenetrating impact to the precordial region, which is most commonly observed in young healthy persons during participation in sports. The blow likely falls within a small 10- to 30-ms window of ventricular vulnerability just prior to the peak of the T-wave that results in polymorphic VT and sudden death. A 2002 case series of 128 individuals showed that only 16% of patients survived an episode of commotio cordis, with most returning to a baseline level of function. Prompt CPR/defibrillation was the only identifiable factor associated with a favorable outcome.
C.Ventricular fibrillation. VF is a chaotic ventricular rhythm that reflects no organized electrical activity and hence no cardiac output from the ventricle. It is devoid of the distinct elements that make up the usual electrical complex of ventricular activity. It is a rapidly fatal rhythm, and if resuscitation is not begun within 5 to 7 minutes, death is virtually certain. VF is often preceded by VT. Virtually all of the risk factors and conditions discussed for VT are applicable to VF. It may arise without any inciting cardiac rhythm or event.
1.Course of disease. Of patients who experience an out-of-hospital cardiac arrest, 75% have VF as their initial cardiac rhythm. Of those successfully resuscitated, 75% have significant CAD and 20% to 30% have a transmural infarction. Patients without an ischemic etiology have an increased risk of further episodes of sudden death, whereas those who have an MI associated with sudden death have a 1-year recurrence rate of 2%. Anterior MI complicated by VF represents a subgroup at high risk for recurrence of sudden death. Predictors of SCD include evidence of ischemia, decreased LV systolic function, 10 or more PVCs per hour on telemetry, inducible or spontaneous VT, hypertension and LV hypertrophy, smoking, male sex, obesity, elevated cholesterol, advanced age, and excessive alcohol use.
2.Therapy. As noted previously, VF is a rapidly fatal rhythm, which virtually never terminates spontaneously. CPR must be initiated promptly and rapid, asynchronous DCC performed as soon as possible. A single shock of 200 to 360 J (biphasic devices, 200 J; monophasic devices, 360 J) should be given initially followed by immediate resumption of CPR for 2 minutes before checking for a pulse. If VF/pulseless VT persists, an immediate second shock (biphasic devices, ≥200 J; monophasic devices, 360 J) should be given followed by a vasopressor (1 mg of epinephrine every 3 to 5 minutes; single dose of 40 units of vasopressin may replace first or second dose of epinephrine). If VF/pulseless VT persists after two or three shocks, CPR, and a vasopressor, administration of an antiarrhythmic should be considered (amiodarone is preferred and lidocaine as an alternative). The emphasis should be on performing high-quality CPR with interruptions in chest compressions only for ventilation (until an advanced airway is established), rhythm checks (pulse checks only if an organized rhythm is observed), and shocks.
See Chapter 23 for a discussion about the long-term treatment of survivors of VF.
D.Ventricular premature depolarizations and NSVT. VPDs are common in patients with both structurally normal and structurally abnormal hearts. They are usually not hemodynamically significant, except in patients with depressed EF or in patients with frequent VPDs and/or bradycardia. Whether or not VPDs increase risk of subsequent cardiovascular events depends on the study. In the ARIC study, after controlling for cardiovascular risk factors, patients with a single VPD on a 2-minute Holter had over a twofold incidence of dying of CAD over a 10-year follow-up period compared with patients who had no VPDs. However, in the Baltimore Longitudinal Study on Aging which evaluated ambulatory ECGs on apparently healthy subjects ≥60 years of age, VPDs on ambulatory ECG monitoring did not predict the development of coronary events. One main difference between the ARIC study and the Baltimore Longitudinal Study was that in the latter, inclusion criteria were more strict, requiring a normal exercise stress test, and therefore, likely had less patients with subclinical CAD.
1.Therapy
a.VPDs. Although patients post-MI with VPDs have a higher mortality than patients post-MI without VPDs, suppression of VPDs with antiarrhythmic medication is associated with increased mortality. For patients with symptomatic VPDs, β-blockers or calcium channel blockers should be the first-line agents. If this fails and the patient has no structural heart disease, class IC agents may be effective; however, potential risks and benefits of the antiarrhythmic drug should be explained to the patient. Sotalol has been shown to reduce the frequency of VPDs by 70% to 80%. In CAMIAT, amiodarone reduced VPDs and arrhythmic deaths, but did not reduce overall mortality. A very high burden of VPDs (>20,000/d) may be associated with reduced EF, although it may be difficult to determine if the VPDs caused the cardiomyopathy or vice versa. However, in some patients with a high burden of VPDs and idiopathic DCM, treating the VPDs either with medication or with catheter ablation may improve the LVEF. Ablation can be an alternative approach to treatment in patients with very symptomatic VPDs refractory to medical therapy and/or patients with reduced EF thought to be a result of the VPDs.
b.NSVT is defined as VT of duration <30 seconds. It can occur in up to 4% of healthy adults and increases in frequency as people age, and NSVT during exercise is not associated with a poor cardiovascular prognosis. The approach to NSVT is based on the cardiac substrate. However, patients with frequent polymorphic NSVT should undergo an evaluation for LQTS and catecholaminergic VT. One should also be mindful of repetitive monomorphic NSVT that may be of LBBB morphology in patients who have a family history of arrhythmias or sudden death, which may suggest ARVD. Monomorphic NSVT may also be a part of the spectrum of the outflow tract VTs.
In patients in whom structural heart disease has been excluded, NSVT does not carry prognostic significance. Treatment should therefore only be guided by symptoms. A similar approach to NSVT with regard to medication choices is used as with symptomatic VPDs.
c.NSVT in structural heart disease. In patients with MI, NSVT within the first 24 to 48 hours has little prognostic significance. However, NSVT >48 hours after acute MI may increase the risk of sudden death by almost twofold, and the risk is even higher in patients with reduced LV function. Patients with CAD, an EF <40%, and NSVT who have inducible sustained VT on testing are at substantially increased risk over those who do not have inducible VT. In patients with HCM, NSVT on Holter monitoring is one of the major risk factors of SCD. Patients with mitral valve prolapse and aortic stenosis who have NSVT are not at increased risk for SCD compared with those without NSVT. In patients with nonischemic DCM, NSVT is not an independent predictor of sudden death.
ACKNOWLEDGMENTS: The author thanks Drs. Omeed Zhardkoohi, Ross Downey, Keith Ellis, and Thomas Dresing for their contributions to earlier editions of this chapter.
KEY REVIEWS AND REFERENCES
Affirm Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002;347:1825–1833.
Antiarrhythmics Versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576–1583.
Antunes E, Brugada J, Steurer G, et al. The differential diagnosis of a regular tachycardia with a wide QRS complex on the 12-lead ECG: ventricular tachycardia, supraventricular tachycardia with aberrant intraventricular conduction, and supraventricular tachycardia with anterograde conduction over an accessory pathway. Pacing Clin Electrophysiol. 1994;17:1515–1524.
Arruda MS, McClelland JH, Wang X, et al. Development and validation of an ECG algorithm for identifying accessory pathway ablation site in Wolff–Parkinson–White syndrome. J Cardiovasc Electrophysiol. 1998;9:2–12.
Biase L, Burkhardt J, Lakkireddy D, et al. Procedural benefit of substrate base ablation versus conventional mapping and ablation of clinical stable ventricular tachycardia: results from the vista randomized trial. J Am Coll Cardiol. 2015;65(10 suppl):A412. doi:10.1016/S0735-1097(15)60412-0.
Bigger JT Jr, Fleiss JL, Rolnitzky LM. Prevalence, characteristics and significance of ventricular tachycardia detected by 24-hour continuous electrocardiographic recordings in the late hospital phase of acute myocardial infarction. Am J Cardiol. 1986;58(13):1151.
Buxton AE, Lee KL, DiCarlo L, et al. Electrophysiology testing to identify patients with coronary artery disease who are at risk for sudden death. N Engl J Med. 2000;342:1937–1945.
Cairns JA, Connolly SJ, Roberts R, et al. Randomised trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarisations: CAMIAT. Canadian Amiodarone Myocardial Infarction Arrhythmia Trial Investigators. Lancet. 1997;349:675–682.
Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406–412.
Cardiac Arrhythmia Suppression Trial II Investigators. Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. N Engl J Med. 1992;327:207–233.
Chung MK, Martin DO, Sprecher D, et al. C-reactive protein elevation in patients with atrial arrhythmias. Inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104:2886–2891.
Duffee DF, Shen WK, Smith HC. Suppression of frequent premature ventricular contractions and improvement of left ventricular function in patients with presumed idiopathic dilated cardiomyopathy. Mayo Clin Proc. 1998;73(5):430.
Engstrom G, Hedblad B, Juul-Moller S, et al. Cardiac arrhythmias and stroke: increased risk in men with high frequency of atrial ectopic beats. Stroke. 2001;32:1443.
Fontaine G, Fontaliran F, Lascault G, et al. In: Zipes DP, Jalife J, eds. Cardiac Electrophysiology: From Cell to Bedside. 2nd ed. Philadelphia, PA: WB Saunders; 2009:745–751.
Julian DG, Camm AJ, Frangin G, et al. Randomised trial of effect of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. European Myocardial Infarct Amiodarone Trial Investigators. Lancet. 1997;349:667–674.
Kaufman ES. Mechanisms and clinical management of inherited channelopathies: long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. Heart Rhythm. 2009;6(suppl 8):S51–S55.
Kennedy HL, Fleg JL. Long-term prognostic significance of ambulatory electrocardiographic findings in apparently health subjects ≥ 60 years of age. Am J Cardiol. 1992;70:748–751.
Klein AL, Grimm RA, Black IW, et al. Cardioversion guided by transesophageal echocardiography: the ACUTE pilot study. Ann Intern Med. 1997;126:200–209.
Klein AL, Grimm RA, Murray DR, et al. Use of transesophageal echocardiography to guide cardioversion in patients with atrial fibrillation. N Engl J Med. 2001;344:1411–1420.
Lerman BB, Stein KM, Markowitz SM. Mechanism of idiopathic ventricular tachycardia. J Cardiovasc Electrophysiol. 1997;8:571–583.
Mason JW. A comparison of seven antiarrhythmic drugs in patients with ventricular tachyarrhythmias. Electrophysiologic Study versus Electrocardiographic Monitoring Investigators. N Engl J Med. 1993;329:452–458.
Massing MW, Simpson RJ Jr, Rautaharju PM, et al. Usefulness of ventricular premature complexes to predict coronary heart disease events and mortality (from the Atherosclerosis Risk in Communities Cohort). Am J Cardiol. 2006;98(12):1609–1612.
Moss AJ, Hall WJ, Cannom DS, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. Multicenter Automatic Defibrillator Implantation Trial Investigators. N Engl J Med. 1996;335:1933–1940.
Moss AJ, Schuger C, Beck CA, et al; MADIT-RIT Trial Investigators. Reduction in inappropriate therapy and mortality through ICD programming. N Engl J Med. 2012;367:2275–2283.
Mumtaz MA, Lorber RE, Arruda J, et al. Surgery for anomalous aortic origin of the coronary artery. Ann Thorac Surg. 2011;91(3):811–814.
Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74.
Prystowsky EN. Management of patients with atrial fibrillation: a statement for healthcare professionals from the Subcommittee on Electrocardiography and Electrophysiology, American Heart Association. Circulation. 1996;93:1262–1277.
Roberts-Thomson KC, Lau H, Sanders P. Diagnosis and management of ventricular arrhythmias. Nat Rev Cardiol. 2011;8:311–318.
Taylor AJ, Rogan KM, Virmani R. Sudden cardiac death associated with isolated congenital coronary artery anomalies. J Am Coll Cardiol. 1992;20:640–647.
Teerlink JR, Jalaluddin M, Anderson S, et al. Ambulatory ventricular arrhythmias in patients with heart failure do not specifically predict an increased risk of sudden death. PROMISE (Prospective Randomized Milrinone Survival Evaluation) Investigators. Circulation. 2000;101(1):40.
Vereckei A, Duray G, Szénási G, et al. New algorithm using only lead aVR for differential diagnosis of wide QRS complex tachycardia. Heart Rhythm. 2008;5(1):89–98.
Wellens HJ. Contemporary management of atrial flutter. Circulation. 2002;106:649–652.
Wilkoff BL, Williamson BD, Stern RS, et al; for the PREPARE Study Investigators. Strategic programming of detection and therapy parameters in implantable cardioverter-defibrillators reduces shocks in primary prevention patients. Results from the PREPARE (Primary Prevention Parameters Evaluation) Study. J Am Coll Cardiol. 2008;52(7):541–550.
Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–988.
Wyse DG, Friedman PL, Brodsky MA, et al; for the AVID Investigators. Life-threatening ventricular arrhythmias due to transient or correctable causes: high risk for death in follow-up. J Am Coll Cardiol. 2001;38:1718–1724.