3
A Medicine for Memory
All life is chemistry.
J. B. van Helmont, Ortas Medicinae, 1648
ON 5 NOVEMBER 1986, in a proclamation to raise awareness for Alzheimer’s, President Ronald Reagan addressed the crowd: ‘No cure or treatments yet exist… but through research we hope to overcome what we now know is a disease…’1 For the scientists who had worked for so long to prove that Alzheimer’s is not a normal part of ageing, this public recognition of the Alzheimer’s epidemic was a landmark moment. Three years earlier, President Reagan had declared November as America’s National Alzheimer’s Disease Month. Reagan would himself later succumb to Alzheimer’s.
On the same November day in 1986, the office of William Summers, a neuroscientist at the University of Southern California, Los Angeles, was flooded with telephone calls from the press. They had in their possession an advance copy of the New England Journal of Medicine article that Summers was about to publish, which, he claimed, demonstrated a treatment for Alzheimer’s disease.
Recent attempts to understand the brain have mainly involved breaking it down into its constituent parts, examining those parts, and then fitting them into a larger theoretical framework–a philosophy known as reductionism. Though many now believe it’s time to move on from this way of thinking because the brain is proving more complicated than the sum of its parts, reductionism has provided us with an enormous amount of knowledge, upon which much of the research into a cure for Alzheimer’s has relied.
Broadly speaking, the brain is composed of two main cell types–neurons and glia. Neurons, the nerve cells of the brain, are electrical cells that send chemical messages to one another at specialised contact points called synapses. They are often compared to trees in a dense forest or wires in a telecommunications network. You could also think of them as the masters of social media: each neuron is like a person that has around 85 billion ‘friends’, and they are part of a ‘network’ of synaptic connections that is 100 trillion strong. This means that every second, billions of neurons are sending trillions of synaptic messages in the deepest recesses of your mind.
Glia (Greek for ‘glue’) are non-electrical cells that protect and support neurons. It was thought they did little else–hence the disparaging Greek translation. But there is now good evidence that glia command far more illustrious roles in the brain, and swathes of neuroscientists on the front lines of Alzheimer’s research are busy deciphering those roles in a bid to exploit them therapeutically.
According to the British biologist Lewis Wolpert, the best way to appreciate a neuron’s complexity is to imagine each is the size of a human. At this scale the whole brain would cover an area of ten kilometres, nearing the size of Manhattan–and ten kilometres into the sky. The population of Manhattan is around 1.6 million people, but this space would be occupied by billions of ‘neuron-people’ piled high on top of one another, each one talking to between 100 and 1,000 of its neighbours.2 If you can imagine that, then you can get a sense of how sophisticated the neuron is.
A typical neuron is composed of a cell body, numerous fine projections called dendrites, and one long projection called the axon. Several ‘internal organs’, or organelles, exist inside the cell body–such as the nucleus, which houses the neuron’s DNA, the mitochondria, which provide the neuron with energy, and the ribosomes, which act as microscopic protein factories. Closely spaced along the length of the dendrites are the synapses, each one making contact with another neuron by almost touching the terminal of its axon. In this way neurons form a fixed but highly dynamic web of interactions.
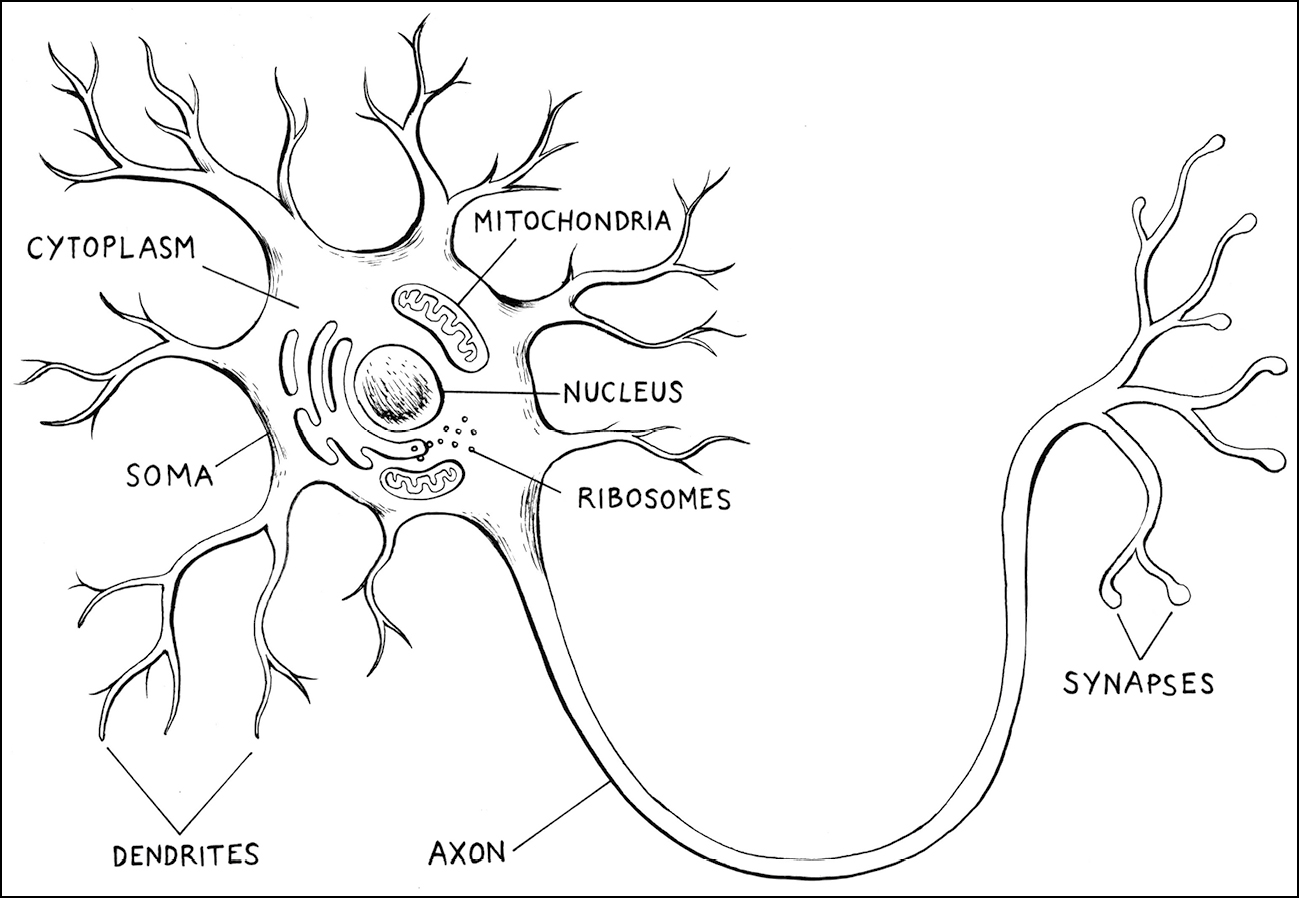
Zooming out from this level, the brain is held together in distinct anatomical units, like pieces of glass in a stained-glass window. Each unit takes on the duty of controlling different functions.3 The medulla oblongata, for instance, is located at the base of the brainstem and performs the onerous task of regulating heart rate, blood pressure and breathing. The cerebellum, just above the brain stem, helps coordinate movement. The thalamus, buried deep within the centre of the brain, controls sleep and wakefulness. The cerebral cortex, that eye-catching folded outer layer of the brain, gives rise to the higher human faculties, such as language, emotion and consciousness. And the hippocampus (Greek for ‘seahorse’), one of the very first regions to succumb to Alzheimer’s, plays a pivotal role in converting short-term memory to long-term memory.


The brain works by constantly transmitting chemical messages across synapses. When such a message is delivered, the neuron is said to have ‘fired’, resulting in countless different processes–from making sure you continue to breathe to ensuring your fingers do what you tell them to do. We call these messages neurotransmitters and most come in the form of chemical compounds. Glutamate, for instance, is a major neurotransmitter. Acetylcholine is another.
The signals these molecules convey form the roots of many aspects of normal brain function: emotion, learning, memory. While pinpointing a thought’s origin in the brain is like deciding where a forest begins, thoughts are essentially generated by neurons triggering the release of neurotransmitters. It comes as no surprise, then, that scientists in the 1970s turned their heads when a striking loss of the neurotransmitter acetylcholine was seen in the brains of Alzheimer’s patients.
It was 1978 and, almost simultaneously, three groundbreaking studies by separate teams of British biochemists were changing the face of Alzheimer’s research: one led by Peter Davies at Edinburgh University;4 the second by Elaine and Robert Perry at Newcastle University;5 the third by David Bowen at the London Institute of Neurology.6
The 1970s were a fertile decade for dementia awareness. In America, Florence Mahoney, a prominent health activist, began lobbying politicians to create a new institute that specialised in age-related disorders to complement the already existing National Institute of Health (NIH). With her help, Congress convinced President Nixon to pass the Research on Aging Act of 1974, and a National Institute on Aging (NIA) was established. In the UK, health activist Peter Campbell founded the Mental Patients’ Union (MPU), which campaigned against the then asylum-based psychiatric system that had been repeatedly exposed as a source of neglect and abuse for people with dementia,7 and in 1979 a small group of patient relatives and prudent medical practitioners formed the Alzheimer’s Disease Society (known today as the Alzheimer’s Society). Across Europe, prestigious academic institutes–in Berlin, Paris, Rome and Stockholm–began to bring together scientists from diverse backgrounds and establish university departments with the sole purpose of unearthing the disorder’s unknown origins.
In Britain, the biochemists had noticed a mysterious link between the effects of a childbirth anaesthetic and memory formation. From the turn of the century until the 1960s, a drug called scopolamine was used to spare mothers the pain of childbirth. Before that, chloroform had been the only option, but it was widely rebuked in the medical establishment as the source of life-threatening complications such as heart failure. Scopolamine, derived from the Asian flowering plant Scopolia tangutica, signified progress for its capacity to induce a ‘Twilight Sleep’: a state where the patient felt no pain while simultaneously remaining completely awake. More striking, though, was the intriguing observation that mothers often emerged from the treatment with no memory of their birthing experience whatsoever. No one could explain it. But what scientists did know was that scopolamine disrupted acetylcholine signalling in the nervous system.
Pinning down the neurochemistry of how memories are formed was, and remains, a Holy Grail for neuroscience. We still don’t know how memory works. In the 1970s the Norwegian scientists Per Anderson and Terje Lømo had offered the most convincing theory to date. They argued that memories are made and lost by a respective strengthening and weakening of neuronal synapses. They called their model ‘long-term potentiation’ (LTP).8 The phenomenon occurs, they said, after a synapse receives a high-frequency electrical stimulation. This causes a long-lasting increase in the strength of connections between neurons. Like almost everything in science, the truth (or in this case, the hypothesis) is almost too strange to comprehend, but Anderson and Lømo suggested that those connections–simply put–are our memories. They are nothing like how we perceive them to be–images and feelings passing through our minds. Memories are physically encoded.
So when a memory is born, say, from meeting someone for the first time, the information is first sent to the hippocampus to be encoded in a network of synapses. Some of this information will linger here as short-term memory, which lasts around thirty seconds, but if something about the meeting is important or has a strong emotional element, then the information is channelled to synapses in the cortex where it resides as long-term memory. If the science here sounds imprecise and unsatisfactory, that’s because it is. We know that long-term memory can be divided broadly into declarative and procedural. Declarative memory refers to knowledge gathered over a lifetime, like the name of your dog, or how many children you have. Procedural memory involves remembering how to do certain things, like tying shoelaces, or driving a car. But in terms of memory’s underlying neurophysiology, Anderson and Lømo’s theory of LTP, combined with the observation that neurotransmitter signalling is somehow involved, was (and still is) the best description of memory in neuroscience.
With that, the British biochemists immediately asked the obvious question: could acetylcholine loss be the key to the decline in memory seen in Alzheimer’s disease? It was a highly attractive theory. If proven true it could collapse the entire puzzle into a single piece: scientists would be able to develop a drug that simply replaces the acetylcholine. Indeed, Parkinson’s research had triumphed this way in the 1960s, when a loss of the neurotransmitter dopamine was identified and scientists discovered a way to replace it using the drug Levodopa. Though not curative, the therapeutic gain for Parkinson’s patients has been remarkable.
But in the case of Alzheimer’s the answer was not so straightforward, and the success of the idea depended upon a number of questions. The first: was there a reduction in acetylcholine found in brain samples from deceased Alzheimer’s patients? After an extensive search of post-mortem tissue the British groups were in unanimous agreement, and by 1978 all had published their findings. ‘Yes’, it appeared.
The next question: could artificially blocking acetylcholine in young healthy people trigger the same kind of memory loss seen in the elderly? As luck would have it this question had already been answered. In 1974 David Drachman and Janet Leavitt at Northwestern University, Chicago, took a group of young student volunteers, gave them a dose of ‘Twilight Sleep’ using scopolamine, and tested their ability to store and retrieve new memories.9 Could they, for example, remember and repeat a random sequence of numbers after listening to them on a tape recorder? And how many nouns could they list that were categorised as animals, fruits and girls’ names? They then gave the same tests to healthy volunteers aged between fifty-nine and eighty-nine who had not been given the drug. Astonishingly, while the untreated students far outperformed the elderly subjects in every test, the students under the influence of scopolamine performed just as poorly as their elderly counterparts. Here again, the answer was an affirmative.
The final question–and indeed the most crucial–was: does boosting acetylcholine release in the brains of living Alzheimer’s patients improve their memory? The simplest way to test this was dietary. To make acetylcholine, neurons first need choline, a vitamin found circulating in the blood. The vitamin is provided in significant amounts from the food we eat–eggs, beef and fish, for instance, are plentiful in choline.
From 1978 to 1982 a number of European and American clinical trials tested the effect of dietary choline supplements on Alzheimer’s patients.10 They used doses up to fifty times the average human intake, gave it every day for months at a time, tested hundreds of patients of different ages, and implemented dozens of new methods for assessing memory and cognitive ability. The trials were the culmination of a collective effort of more than a dozen studies performed by a total of nearly 100 of the world’s leading scientists. It hailed the first milestone in translating the findings of basic research into a real-world therapy for Alzheimer’s. And it marked a new precedent in the history of humanity’s response to the disease.
But it failed. The results of almost every study reported no effects on memory and no improvement on any tests of cognition. While a few groups declared some benefit, the data backing up such claims was inadequate. Whatever the reason, neurons had given up making acetylcholine, and giving them copious amounts of choline in the hope they would kick-start the mechanism back into action wasn’t enough.
But all was not lost. ‘Truth in science,’ the Austrian zoologist Konrad Lorenz said, ‘can be defined as the working hypothesis best suited to open the way to the next better one.’ What if, some scientists asked, instead of trying to make new neurotransmitter from scratch, we simply kept the acetylcholine that was already present in the brain around for longer?
On the evening of 16 March 1988 huge clouds of yellow smoke began to rise into the air in the small city of Halabja, in the foothills of the Hawraman Mountains in Iraqi Kurdistan. Confused families rushed indoors and into basements; others ran into their cars and closed the windows. What they saw defied belief. In the streets among the smoke, crowds of people were uncontrollably vomiting, urinating and defecating, before violently convulsing and falling to the ground in seizures. The attack left an estimated 5,000 people, mostly civilians, dead, massacred by Saddam Hussein’s forces in the final days of the Iran–Iraq war. The weapon of choice was a deadly chemical nerve gas known as sarin.
Sarin is twenty times more toxic than cyanide. It works by meddling with the neurotransmitter acetylcholine. Specifically, it binds to and paralyses another protein, called acetylcholinesterase, responsible for degrading acetylcholine. This causes a build-up of excess neurotransmitter, which wreaks havoc on the nervous system because acetylcholine signalling is also responsible for controlling muscle contraction. As a result the victim experiences a grotesque and undignified purge from every orifice before the muscles controlling their lungs fail, their chest tightens, and they eventually stop breathing altogether. Depending on the dose, death can occur within minutes.
The grim and lethal effect of sarin gas on humans was known long before the Iraqi massacre brought this horrifying reality to the world stage. As early as the 1950s its reputation as the deadliest nerve agent made the USSR and America begin stockpiling it for military purposes. So in 1981 when a neuroscientist named William Summers proposed using a drug that also works by binding and paralysing acetylcholinesterase to treat Alzheimer’s patients, he trod carefully.
Summers was interested in a drug called tacrine (otherwise known as 1,2,3,4-tetrahydroacridin-9-amine). Synthesised by an Australian chemist during the Second World War in a hunt to develop antiseptics for treating wounded infantry, tacrine was effectively shelved and forgotten to make way for penicillin. But animal tests using tacrine during the war effort revealed a curious property: it always counteracted the anaesthetic that scientists administered to an animal (usually a mouse) to put it to sleep. This intrigued another Australian, a psychiatrist named Sam Gershon, in the late 1950s. The arousing effect of tacrine appeared to stem from its ability to block acetylcholinesterase–making its behaviour similar to that of sarin gas but with a much less dangerous effect.
Raised on the wooded outskirts of a small town in central Missouri, Summers is a very different breed of scientist. His pragmatic yet unorthodox approach seems to have run in his family: both his father and grandfather were physicians and the young Summers, who was expected to follow in the family tradition, would often accompany them to the local hog-rendering plant to collect pig thyroids. The family would hang and dry the thyroids in their garden and then harvest the extract to treat patients with hypothyroidism–an effective method that was eventually replaced by synthetic means. Summers first became interested in Alzheimer’s while training in psychiatry at Washington University in St Louis, when the idea of Alzheimer’s as a form of dementia was beginning to take hold.
However, Summers remains unconvinced by the popular idea that plaques and tangles cause the disease. ‘I think they just mark where the dead neurons are,’ he told me, during a long conversation in the office of his private practice in Albuquerque, New Mexico. ‘They might cause some neuronal death, but I think for the most part they’re actually protective mechanisms against other things. There are probably more than fifty different targets and Alzheimer’s is just the result of a final common pathway.’ For Summers, acetylcholinesterase is the target–and tacrine the ideal weapon.
In February 1981 he administered the drug to twelve Alzheimer’s patients intravenously.11 The doses varied–after all, no one had any idea what amount would be effective, detrimental, or even fatal. But for all the suspense and trepidation the results were remarkably positive. Nine patients showed significant improvements in cognition only a few hours following treatment. And the side effects were mercifully limited to mild nausea and excessive sweating. ‘It impressed me,’ said Summers, ‘that a toxin can, in the proper circumstances, become a medicine.’
Summers was eager to push his study further, but others in the hospital remained sceptical. As a full-time clinician at the University of Southern California the majority of Summers’ working day was spent seeing patients, which left little time for research. Determined to pursue his research, Summers left the clinic and went into private practice: ‘I thought what the hell, I would go earn money in the private sector, pay for my own research, and who gave a damn what they thought at the NIH!’
A straight-talking and resolute man by nature, Summers spent $90,000 out of his own pocket–$300,000 by today’s standards–to develop a tacrine pill with the help of a few supporters at the University of California, Los Angeles (UCLA) and the Aldrich Chemical Company in Milwaukee. Mice and primates underwent testing, the US Food and Drug Administration (FDA) approved human trials, and by 1986 Summers published the results of seventeen patients given the drug orally in the New England Journal of Medicine (NEJM).12
His findings were an instant sensation: the drug did appear to temporarily improve memory and cognition in Alzheimer’s patients. It wasn’t a cure, but for the millions watching loved ones slipping away, the beacon of light it offered was blinding. ‘The genie was out of the bottle,’ said Summers. ‘There was a Saudi prince who had Alzheimer’s and they were going to send a jet to LAX to pick me up and take me to Saudi Arabia to treat the prince. I had no idea what I was playing with to be honest.’
For all the excitement, though, there were many doubters. One troubling issue for the FDA and fellow neuroscientists was that Summers did the study out of his private psychiatric practice, under his own steam, out of sight of the academic establishment. This raised suspicion. Indeed, other researchers failed to replicate the result. Before long, Summers found himself embroiled in a year-long federal investigation.
‘There’s a rule of invention,’ he told me, ‘the lion’s share of invention takes place in the wrong place, by the wrong people, at the wrong time, for the wrong reasons. I fit all of those. The discovery of the first treatment for Alzheimer’s should certainly have come from the NIH, not from an individual in private practice in LA, and it should’ve come from big government grants, not from somebody who’s self-funded on a shoestring.’ The NEJM article, said the FDA, was overstated and misleading. According to them, there were gaping holes in the techniques and conclusions, reflecting an additional failure on the part of the NEJM reviewers.
Eventually, Summers was vindicated. A special committee of faculty at UCLA, charged with scrutinising his work with a fine-tooth comb, told the FDA that it checked out and on 10 September 1993 tacrine became the first FDA-approved treatment for Alzheimer’s disease. As more patients started enrolling in the treatment it became clear that the effects–though positive–were modest. But it was enough to bolster hope in the idea that the disease would, and could, yield to treatment–something many in the broader scientific community never thought possible.
Today there are four internationally approved drugs for the treatment of Alzheimer’s: Donepezil (AriceptTM), Rivastigmine (ExelonTM), Galantamine (RazadyneTM), and Memantine (NamendaTM). The first three work using the same scientific principle as tacrine. Patients who take them usually show a delay in the worsening of their symptoms for about six to twelve months–allowing them to cope better with simple daily tasks like dressing, shopping and personal hygiene. Whether you agree with Summers’ somewhat renegade methods or not, he was a trailblazer for modern Alzheimer’s research. His work created a desperately needed inroad of hope in a world with Alzheimer’s disease rapidly on the rise. More importantly, his efforts signalled the first major assault on the disease that was based on a scientific, evidence-based hypothesis, giving other scientists the confidence to assert their own mechanistic theories. Would he do it again? ‘Yeah, I’d do it in a heartbeat. Look at the people who have hope and are getting benefit. So it was worth every battle.’
We have entered a very different world from the one Summers and his predecessors entered. Their study of Alzheimer’s taught us that memory is a material phenomenon, a subtle and exquisite product of healthy brain cells. They taught us that if memory resides anywhere, it resides in a network of durable connections and neurotransmitter systems between those brain cells. And if the rhythms of a damaged heart could be exposed and repaired, then so could memory. They had demonstrated that Alzheimer’s could be abolished.