1.1 DNA-Binding Molecules
1.1.1 Naturally Occurring Molecules

B-form double helical DNA and Watson–Crick base pairs

Naturally occurring products that bind to DNAs in a sequence-dependent manner
1.1.2 Pyrrole–Imidazole Polyamides

Chemical structure of pyrrole–imidazole polyamide (PIP) and the crystal structure of the complex of a PIP and a duplex DNA (PDB code: 3omj)

Base-pair discrimination by a P-P or I-P pair of PIPs
A crucial advance in the issues of PIP synthesis was reported in 1996, describing a method for the solid-phase synthesis (SPS) of PIPs using Boc chemistry and the gram-scale synthesis of monomer units without chromatographic purification [19]. Therefore, the synthetic timescale was shortened for one polyamide from months to days. The subsequent progress of the solid-phase approach using an Fmoc chemistry has been reported, showing optimized yields and purities [20]. Nowadays, the Fmoc SPS approach has been adopted for rapid and facile PIP synthesis.
The sequence-specific binding of PIPs to the predetermined sequences permits the transcriptional regulation of intended genes. There have been numerous successful examples to date [21]. Gottesfeld, Dervan, and colleagues first reported a prominent work, where a PIP designed to target TFIIIA binding site (5′-ATGACT-3′) interfered with 5S RNA gene expression in a kidney cell [22]. Continuously, the Dervan group has been a leading research group in this field of targeted gene regulation by PIP molecules and keeping offering the invaluable insight into PIP design for enhanced specificity and affinity, methodologies of how to target a specific gene of interest, the molecular mechanism of the action of PIPs, and in vivo application (see Dervan’s group website http://dervan.caltech.edu/).

A PIP-indole-seco-CBI conjugate to target the oncogenic mutant codon 12 in KRAS gene

SAHA-PIP conjugates that activate a particular gene network

CTB-PIP conjugate (I) that exhibits a similar gene regulation profile compared to the SAHA-PIP conjugate (I) as indicated by a heatmap obtained from DNA microarray analysis

Bromodomain inhibitor-PIP conjugates; a Bromodomain inhibitors, b Syn-TEH1, c Bi-PIP
1.1.3 Trinucleotide Repeat-Targeting Molecules
In the human genome, a vast number of microsatellite repeat sequences were prevalent and some of them have profound implications in biological and neuropathological contexts [37, 38]. In this section, we highlight the trinucleotide microsatellite sequence and its implications in hereditary diseases from the viewpoint of DNA conformations and their targeting molecules as potential therapeutic agents and diagnosis tools.

Genomic locations and expansion thresholds of triplet repeats associated with trinucleotide repeat diseases. The repeat expansion exceeding defined thresholds is a pathogenic origin. UTR; untranslated regon,. SCA; spinocerebellat ataxia
Mutant RNAs and proteins derived from the expanded repeat DNA regions are directly associated with the pathogenesis of each hereditary disorder [41]. For example, the expansion of repetitive CAG trinucleotides (>36 repeats) within the first exon of the Huntingtin (HTT) gene causes Huntington’s disease [42]. The expanded repeat-derived polyglutamine (PolyQ) tracts tethered to a full-length HTT protein and toxic long hairpin RNA structures formed by the repetitive region of HTT transcripts are primary pathogenesis substances [43–45]. In myotonic dystrophy type 1 (DM1), transcripts harboring excessive CUG repeats that are transcribed from CTG repeat sequences situated at the 3′ UTR of DMPK gene, form aggregative RNA foci within the nucleus by assembling multiple RNA-binding proteins [46, 47]. Those recruited proteins such as splicing regulators are unable to function properly any longer, ultimately leading to the pathogenesis of DM1. The CGG repeats located in the 5′-UTR of FMR1 gene is associated with Fragile X syndrome (FXS, > 200 repeats) and FXTAS (55–200 repeats). While the pathogenesis of FXTAS is thought to be attributable to the capability of extended CGG repeated RNAs to form multiple high-order structures [48], more frequent CGG repeats in FXS cause epigenetic repression of the gene transcription by DNA methylation within the CGG repeat and 5′-UTR region [49]. The repetitive GAA runs observed in Friedreich’s ataxia (FRDA) also have a similar situation, where repressive chromatin across these repetitive genome regions hinders the RNA polymerase initiation and elongation, thus silencing the gene expression [50, 51]. Recently, it has been found that repeat-associated non-ATG translation (RAN translation) is also the pathogenic source of (CAG)n, (CGG)n, and other repeat-associated disorders [52, 53].

Non-canonical DNA structures formed by expandable repeats; a hairpin formed by CNG repeats, b G-quadruplex (G4) formed by GGGGCC or CGG repeats, c slipped hairpin formed by CTG/CAG repeats, d triple helix formed by GAA/CTT repeats

(Left) crystal structure of a 2:1 actinomycin-d(ATGCTGCAT)2 complex (PDB code: 1mnv); (Right) crystal structure of a 2:1 actinomycin-d(ATGCGGCAT)2 complex (PDB code: 4hiv)

a Solution structure of a 2:1 NA-d(CTAACAGAATG•CATTCAGTTAG)2 complex (PDB code: 1 × 26). b Intermolecular hydrogen bonds between NA and guanine or adenine bases

Designer molecules that dually target d(CTG/GTC)n and the derived r(CUG/GUC)n transcripts; (left) compound 5; (center) compound 6; (right) compound 9 in Ref. [57]

Three-way junction formed by CAG and CTG repeat DNAs and triptycene molecules that bind to the cavity of the junction
1.2 G-Quadruplex
1.2.1 Structures and Biological Significance of G-Quadruplexes

a Structure and schematic illustration of a G-tetrad. b Schematic illustrations of typical intramolecular G-quadruplex (G4) structures: (left) crystal structure of parallel-type telomere G4 (PDB code: 1kf1); (center) solution structure of antiparallel-type telomere G4 (PDB code: 143d); (right) solution structure of hybrid-type telomere G4 (PDB code: 2gku)

Large variety of biological functions of G4 DNA. a The interferene of transcription factor binding or nucleosome occupancy by G4s can regulate the downstream gene expression. b The arrest of DNA polymerase progression occasionally induces strand breakage. c A series of actions of G4-binding proteins leads to the recruitment of histone H3.3 variants to reconstitute correctly the structure of the nucleosome. G4s play an important role in initiating replication. d Stalling the duplication process temporarily at G4s results in irreversible changes in histone modification patterns. Recycled and newly recruited histones are colored in green and red, respectively
Historically, the G4 structures observed in a telomere tandem repeat (GGGTTA)n region at the end of the chromosome initially attracted a great deal of attention, as a single-stranded background offers a greater likelihood of G4 formation. The group of Neidle and Hurley first identified that a telomere G4 DNA-interactive molecule is a telomerase inhibitor, using a series of 2,6-diamidoanthraquinone derivatives [78]. The discovery of telomestatin, a naturally occurring macrocycle compound that exhibits telomerase-inhibiting activity by binding to telomeric G4 structures, suggested the existence of G4s in vivo [79, 80]. The creation of extremely high affinity and specific antibodies to G4s has enabled the visualization of G4s by immunofluorescence, which demonstrated the existence of G4s not only in the single-stranded telomere region but also in duplex regions in nuclei [81, 82]. Regarding G4s in duplex regions, structural analysis initially focused on one that was observed in the nuclease hypersensitive element (NHE) III1 located in the promoter region of a c-myc oncogene [83], the biological significance of which Hurley demonstrated by showing that it was associated with control of gene expression [69]. At a relatively early stage of G4 studies, other biologically important genes, such as hTERT [84], c-kit [85], KRAS [86, 87], BCL2 [88, 89], and VEGF [90], were also identified as genes in which the formation of a G4 was involved in transcriptional regulation. In parallel, many high-resolution G4 structures were elucidated at the atomic level using nuclear magnetic resonance (NMR) and X-ray crystallography, opening a new avenue for the rational design of G4 ligands [91–96]. Recently, G4 ChIP seq analysis revealed that approximately 10,000 actual G4 structures form in the human genome [97]. In addition, the growing number of reports of G4-interacting proteins and their relevant functions supports the biological functions of G4s [98–102]. For instance, some of the RNA helicases recognize G4 structures and manifest their unfolding activity so that DNAs are correctly replicated by DNA polymerases [101]. When such helicases were mutated so as to be devoid of unfolding ability, replication forks stalled in the genome at folded G4s, which resulted in genome instability. These mechanisms have been shown in some cases to be associated with genetic diseases [101, 102].
1.2.2 G-Quadruplexes and Cancers
As mentioned earlier, the formation of G4 structures in human telomeric DNA was first assumed, due to the characteristic guanine-rich sequence (TTAGGG)n and a single-stranded context of human telomere. Abundant evidence has accumulated during the past two decades that the G4 structures are truly formed in the telomere region and has an important role in telomere-end processing in cells [81, 103]. More importantly, stabilization of telomere G4s and blockage of telomerase activities by small molecules, exemplified by telomestatin, is a new strategy for antitumor therapy [79, 80].
G4-forming sequences observed in the promoter of cancer-related genes have also received a great deal of attention as potential biomedical targets for antitumor therapy [104]. Generally, targeting the promoter region rather than expressed proteins has several advantages, including the lower likelihood of point mutations and the development of drug resistance. Quarfloxin, a G4-interacting ligand, had completed Phase II trials as a candidate therapeutic agent candidate against several tumors, including neuroendocrine tumors, carcinoid tumors, and lymphomas [105]. Quarfloxin disrupts the G4–nucleolin complexes of ribosomal DNA in the nucleolus, which in turn redistributes nucleolin into the nucleoplasm where it binds specifically to a G4 in the promoter region of c-myc proto-oncogene to inhibit its gene expression. Although the Phase III trials for Quarfloxin are currently not proceeding due to high albumin binding, several tumor-related genes were identified as genes in which the formation of a G4 was involved in transcriptional regulation, showing that G4s are potential molecular targets for cancer therapy. In this section, we would like to discuss the detailed exposition of the telomere and G4-driven oncogenes in terms of the direct targetability by synthetic ligands.
1.2.2.1 Telomere
A telomere is a structure of the ends of the chromosome, in which a repeated microsatellite sequence and its specifically interacting components (called a shelterin complex) protect the DNAs from DNA repair mechanisms [106]. The human telomeric DNA comprises a single microsatellite repeat sequence, (GGGTTA)n, with a 3′ overhang at its terminus (200 ± 75 nucleotides). In normal somatic cells, the length of the telomere sequence gradually shortens with DNA replication, which limits cell growth and proliferation, as the expression of telomerase is almost entirely silenced. Telomerase is a reverse transcriptase enzyme that adds a repeated DNA sequence to the 3′ end of telomeres. It consists of a catalytic subunit, hTERT, and TR/TERC, which is an RNA template that is used during the elongation of telomeres by hTERT. Although TR/TERC is globally expressed (regardless of cell type), hTERT is silenced in somatic cells and is reactivated in nearly 90% of human cancers. Aberrant telomerase activity disturbs the balance of the normal telomere maintenance mechanisms, contributing to the acquisition of immortality. Hence, inhibition of telomerase has long been considered as a potential therapeutic strategy for human cancers, and several telomerase inhibitors have entered preclinical or clinical trials. However, no clinically important benefits of these drugs have been reported to date. Recently, quadruplex-binding telomerase inhibitors have been considered as an alternative strategy for curing telomerase-positive cancers, as they exhibit high antitumor activity while minimally affecting normal somatic cells in vivo.
As mentioned before, 2,6-diamidoanthraquinone derivatives and telomestatin were first found to be telomerase inhibitors through their binding to telomere G4s. Similarly, RHPS4 was shown to induce telomere dysfunction by disturbing the integrity of the shelterin complex in mammal cancer cells [107]. The later relevant studies found that a large repertoire of alternative higher-order structures derived from the canonical telomere G4 have been thought to be adopted at 3′ overhang region [65, 108–110]. Those structures and their specific motifs are amenable to a gain of specificity for telomere G4s.
1.2.2.2 c-myc
c-myc encodes a multifunctional transcription factor that can act as a transcription activator of some genes involving the cell proliferation, while acting as a transcription repressor of other genes involving the growth arrest [111, 112]. There are a broad variety of c-myc-responsive genes that engage in the important cellular functions in concert, such as cell proliferation, metabolic transformation, and metastatic capacity [113]. In tumor cells, MYC protein function is almost always activated primarily through upstream oncogenic pathways. As the overexpression of the MYC protein is observed in various human malignancies (particularly in 80% of solid tumors), the downregulation of the gene may be an effective way toward cancer therapy. However, it is generally considered to be an undruggable target at the protein level because of its short half-life and unstructured nature [104].

a c-myc promoter has one putative G4-forming sequence (PQS). b Solution structure of G4 from a NHE III1 region in the vicinity of the P1 promoter (PDB code: 1xav)
1.2.2.3 VEGF

a VEGF promoter has one PQS located close to the transcription start site (TSS) and hormone response element (HRE) that regulate the transcription. b Solution structure of G4 from the vicinity of the promoter (PDB code: 2m27)
Initially, the interaction of TMPyP4 and telomestatin with G4 oligonucleotides proved to unwind the duplex DNA oligomer into ssDNA oligomer and stabilize the G4 structure [90], and Se2SAP, a global G4-interacting ligand, efficiently suppressed VEGF expression in two adenocarcinoma cell lines (HEC1A and MDA-MB-231) [125]. These data offer the possibility that the transcription regulation of VEGF is controllable by ligand-mediated G4 stabilization and lead to the application of G4-interacting ligand to cancer therapy. Similarly, a perylene monoimide derivative, PM2, was found to be a VEGF downregulator likely by direct interaction with the G4 structure [126]. A quindoline derivative, SYUIQ-FM05 also demonstrated strong interactions with a VEGF G4 and exhibited potential antiangiogenic and antitumor activities [127]. On the basis of these successful reports, several VEGF G4-preferred ligands have been developed, through small screening using docking and/or spectroscopic approaches [128, 129]. Biological activities of these ligands have never examined thus far, and therefore, a future study is awaited.
1.2.2.4 BCL2
BCL2 (B-cell lymphoma 2) is recognized as an apoptosis-related gene whose translated product resides on the cytoplasmic face of the mitochondrial outer membrane and acts to suppress the movability of apoptosis-induced proteins by controlling mitochondrial membrane permeability [130]. Overexpressed BCL2 protein expression is associated with aberrant carcinoma growth in various human diseases, particularly solid tumors such as lymphomas, non-small cell lung cancer, myeloma, and melanoma, being recognized as a target for cancer therapy in the past three decades [131]. Several approaches have been made to downregulate of the BCL2 expression in cancer cells by small molecule to disrupt protein–protein interactions [132], antisense oligonucleotides [133], and peptidomimetics [134] toward cancer therapy. Overexpression of BCL2 is also indicated to be a principal element of chemoresistance, particularly for lymphocytic cancers [135, 136]. For instance, transfection of BCL2 into A549 cells induced resistance to the apoptotic effect triggered by triazine derivative 12459, a G4-interacting ligand that inhibits telomerase activity [137]. As another approach, the molecular decay effect by guanine-rich AS1411 aptamer that can be stably folded into a G4 structure causes the destabilization of BCL2 mRNA and degradation with RNase by interfering with the binding of nucleolin to the AU-rich element of BCL2 mRNA, eventually inducing apoptosis [138]. This approach is reminiscent of the involvement of G4 formation in the gene expression.

a BCL2 promoter has two G4-forming elements that were shown to attenuate the BCL2 promoter activity. b Solution structure of G4 from the vicinity of the P1 promoter (PDB code: 2f8u)
In addition to the G4s, i-motif, another form of DNA that forms in cytosine-rich sequences is involved in transcriptional regulation, in which the binding of hnRNP LL to the i-motif structure likely activates the BCL2 gene expression [145]. Moreover, an i-motif-interacting molecule, IM-48, was identified to modulate the BCL2 gene expression by affecting the dynamic equilibrium of the i-motif and the flexible hairpin form [145], opening a new avenue to more precisely modulate the gene expression of BCL2. Targeting such canonical DNAs formed in the regulatory element of the promoter may be an effective way to specifically target a particular target to combat the tumor.
1.2.2.5 c-kit
The c-kit proto-oncogene encodes a receptor tyrosine kinase that is bridged and activated by the binding of dimerized stem cell factors (SCF), and in turn stimulate proliferation, differentiation, and survival in hemopoietic precursor cells [146–148] Malfunctions of the KIT protein acquired by overexpression or mutations have been associated with several diseases including gastrointestinal stromal tumors (GIST), mastocytosis, and acute myelogenous leukemia (AML). Although the kinase inhibitor Imatinib (Glivec) has been successfully developed as an FDA approved drug for GIST, the long-term exposure often causes secondary mutations at exon 13, 14 or 17 that encodes tyrosine kinase domains [149]. Notably, drug resistance derived from mutations at exon 17 is found to severely attenuate the therapeutic effect by imatinib [150]. A compelling approach to fundamentally suppress c-kit expression would be highly desirable.

a c-kit promoter has two PQSs, where several transcription factors are likely involved. b Solution structure of G4 from the proximal PQS in the vicinity of the promoter (PDB code: 2o3m)
1.2.2.6 hTERT
hTERT (human telomerase reverse transcriptase, TERT), which encodes the catalytic subunit of telomerase, has considerable attention as a compelling biomedical target particularly for cancers, since elevated TERT expression was often observed in ~90% of human cancer cells, whereas it is normally silenced in most of the normal cells [157, 158]. Aberrantly expressed TERT accelerates telomerase activity to irregularly maintain the telomere length [159]. Other than the canonical role as the maintenance of telomere length, TERT has been considered to suppress BCL2-dependent apoptosis [160] to regulate chromatin state [161, 162] and DNA damage responses [163], and to promote MYC and Wnt-driven cellular proliferation [163, 164].
The mutations that were identified in >70% of melanomas partially account for the elevated level of TERT expression [165]. The recent studies demonstrated that C to T mutations in the sense strand (G to A mutations in the antisense strand) in the TERT promoter highly activated transcription through creating a new consensus sequence for the binding of ETS/TCF (E-twenty six/ternary complex factor) [166]. Patients who have tumors expressing elevated levels of TERT exhibit even worse entire survival rates compared to those who do them expressing relatively lower levels of it [167]. These observations clearly indicate that a TERT promoter targeting based on the mutations might have a great impact on tumor therapeutics covering a wide range of tumors.
1.2.2.7 KRAS
The RAS gene family including HRAS, NRAS, and KRAS was first discovered in human tumors as driver oncogenes and has long been recognized as important therapeutic targets. Mutation of the KRAS gene is one of the most oncogenic driver mutations in pancreatic, colorectal, and lung cancers and plays a role in acquiring and increasing the drug resistance [168, 169]. Hence, the direct targeting for active KRAS by small molecules was considered to be a compelling strategy to combat the KRAS mutant tumors, yet it remains at an unsuccessful stage. Recently, our group has developed a novel approach that directly targets the mutant DNA using an alkylating pyrrole–imidazole polyamide (PIP) molecule, where it is capable for selectively alkylating oncogenic codon 12 mutant DNA and causing strand cleavage and consequent tumor growth suppression in tumor xenograft model of cancer in mice [26].
G4-mediated promoter targeting is also reported. The NHE in the KRAS proximal promoter is highly abundant in G-rich sequences, and several transcription factors interact with a G4 structure formed in this region [86, 87, 170–172]. A polypurine G-rich element located in approximately −300 to −100 nucleotides upstream of the exon 0/intron 1 boundary in a murine genome, or human genome was likely to be a component of the promoter activity and the PQS [86, 87, 170–174]. Importantly, pyrene-modified oligonucleotides that were devised to be a more stable form of the KRAS G4 was able to attract the transcription factors essential for transcription and to exhibit a strong antiproliferative activity through a G4-decoy effect in pancreatic cancer cells [175].
1.2.2.8 c-myb
c-myb is largely expressed in an early stage of the differentiation of hematopoietic cells, and its expression is gradually decreased toward the end of the differentiation [176]. It encodes a transcription factor that plays a critical role in the proliferation, differentiation, and survival of haematopoietic progenitor cells. c-myb was identified by the discovery of v-myb oncogene found in avian myeloblastosis virus and E26 [177]. This gene is also recognized as a proto-oncogene, high expression of which is related to promoting the development of hematologic cancers and adenocarcinomas by a mechanism based on its canonical proliferative property [178–182].
The regulation of c-myb expression at a transcription level relies on multiple activating and repressing transcription factors in a cell-type-dependent fashion [183–188]. Notably, a region in the promoter with three (GGA)4 triplet repeats beginning 17 nucleotides downstream of the transcription initiation site on the antisense strand was implicated in the promoter activity by forming very thermally stable higher-order parallel G4structures [189–191]. Partial deletion of the (GGA)4 triplet repeats not to be capable of forming the dimerized G4 enhances the promoter activity, suggesting that the G4 structures formed by utilizing together the three (GGA)4 triplet repeats should function as a negative regulator of the c-myb promoter activity [189]. Additionally, MAZ protein may bind to the c-myb G4 structure and negatively regulate the promoter activity.

a c-myb promoter has multiple PQSs. b Chemical structure of topotecan that efficiently represses the MYB protein expression
When the story moves more specifically to human diseases, c-myb proto-oncogene is identified as a target in glioma stem cells for glioblastoma multiforme (GBM) therapy, in which expression was considerably elevated in GBM tissues relative to normal tissues [193]. Interestingly, telomestatin, a global G4-interacting ligand, cause the impairment of the maintenance of GSC stem cell state through an apoptotic pathway largely by reducing a c-myb expression in vitro and in vivo. Although the direct interplay of telomestatin and c-myb G4s in the promoter has not examined, these observations offer the possibility that direct c-myb G4 DNA targeting might be a compelling therapeutic approach to GBM treatment.
1.2.2.9 Others (PDGFR-β, PDGF-A, STAT3, FGFR2)
Other G4s formed in putative regulatory elements in the promoters of cancer-related genes have been reported and are proposed as targetable by G4-interacting ligands (in promoters in genes for PDGFR-β [194], PDGF-A [195], STAT3 [196], FGFR2 [197]). For instance, GSA11129, which can interact with a G4 in the gene for PDGFR-β promoter to shift the equilibrium to a G4 species, was demonstrated to reduce the transcription level and to inhibit PDGF-β-driven cell proliferation and migration [194]. The G-rich element of the proximal promoter in the gene for PDGF-A also forms a stable G4 structure even in the duplex context, and TMPyP4 reduced the basal promoter activity of PDGF-A, suggesting that targeting the PDGF-A G4 by the ligand specific for this G4 may be feasible as cancer therapy for gliomas, sarcomas, and astrocytomas [195, 198–202].
1.2.3 G-Quadruplex-Interacting Ligands
Studies on the G4s from extensive aspects render researches led to a belief in the notion that G4s can form in the guanine-rich region in the human genome and are regarded as biologically and pharmaceutically important. In this context, numerous researchers have made tremendous efforts to get highly active G4 ligands and some of them attained great success in the development of drugs in vivo [203]. However, these drugs are still only midway toward approval for clinical use.
One conceivable obstacle to impede the clinical application of G4-interacting molecules seems to rest with selectivity, although the global or multiple G4 targeting approaches may in some cases be effective [204–207]. As mentioned earlier, approximately 10,000 G4 structures exist in the human chromatin [97]. A growing number of G4-driven genes have also been reported, suggesting the high importance of the expanded varieties of G4-interacting ligands that possess differential binding profiles [208, 209]. However, poor ligand designability originating from the topological similarity of the skeleton of diverse G4s has remained a bottleneck for gaining specificity toward the individuals. Very recently, researchers came to enter the new phase of the development of next-generation G4-interacting ligands in which they consider the ligand selectivity to a particular G4 to be targeted, not only leading to developing highly antitumor and bioactive molecules with minimized side effects toward antitumor therapy, but also creating chemical biology tools for the detailed investigation of the functions of individual G4s in the genome [209]. In the next section, we address the recent progress of G4-interacting molecules that can discriminate particular G4 structures from the others.
1.2.4 Addressing the Specificity of Ligands to Particular G-Quadruplexes
1.2.4.1 Global G-Quadruplex-Selective Ligands
Since G4-interacting molecules were developed based on duplex DNA-binding molecules, researchers initially endeavored the development of G4 ligands that have a clear selectivity to G4 structures over the duplex DNA [210, 211]. A telomere G4-interacting molecule, 2,6-diamidoanthraquinone derivatives, was first found to act as a telomerase inhibitor by the group of Neidle and Hurley, as discussed before. Cationic porphyrin, TMPyP4, was also identified to be a G4 binder, whose planar skeleton and cationic propensity would be preferable for G4 binding [212]. Moreover, several commercially available G4 ligands such as BRACO19 [213], Pyridostatin [214], Phen-DC3 [215], L2H2-6OTD [216], and L1H1-7OTD [217] that have negligible binding affinities to duplex DNAs is dispensable to biochemical, biophysical, and chemical biology studies on G4s.
1.2.4.2 Flat-Shaped Compounds that Were Originally Developed in Different Fields

DNA G4 ligands with a preference toward particular topologies or G4s. a, b Studies in the field of G4s shed light on NMM IX and crystal violet as topology-preferred ligands. c–m Synthetic ligands likely to interact with loops and grooves that offer distinct environments as scaffolds for specific molecular recognition
1.2.4.3 Loops and Grooves that Offer Distinct Environments for Specific Molecular Recognition
In the past three decades, a series of intensive studies using NMR techniques and X-ray crystallography for the atomic-level elucidation of a library of G4 structures has facilitated and rationalized the design of G4 ligands that exhibit specificity between different G4s. One approach to gain specificity among many types of G4s without reducing the binding affinity is the use of loops and grooves that offer distinct environments for specific molecular recognition. For instance, the core G-tetrad layers of three types of the telomere G4 structures are centered with loops differently positioned (Fig. 1.15b). Based on this principle, several successful attempts were made to achieve preference toward a particular G4 over other quadruplexes. CPT2 can visually discern antiparallel G4s from parallel ones by fitting into the shape of the groove (Fig. 1.22c) [222]. ThT-HE, a thioflavin T analogue that was modified by the addition of a hydroxyethyl group at the N3 position of the benzothiazole ring, exhibits a clear preference toward a c-myc parallel G4 relative to other parallel structures (c-kit DNA, c-src DNA, and NRAS RNA) in a sodium-dominant buffer using fluorescence detection (Fig. 1.22d) [223]. Acridine–peptide conjugates were developed to discriminate between distinct G4s. Two peptide sequences (with substituents at different sites to contact distinctly the loops and grooves) were attached to an acridine core moiety that targets the planar surface of a G-tetrad. SPR-binding assays showed that Compounds 10, 14, 19, and 21 could distinguish specific G4s (Fig. 1.22e) with very high binding affinities (KD = 4–25 × 10−9 M) [224]. Molecular modeling suggested that the spatial allowance of the rectangular acridine moiety upon occupying the wider square shape of a G-tetrad would facilitate the correct positioning of substituents and their distinct interaction with the loops and grooves. GQR was identified among several BODIPY derivatives as a particular parallel G4-preferred light-up probe (93del: 5′-G4TG3AG2AG3T-3′ over c-myc and c-kit parallel G4s) (Fig. 1.22f) [225]. NDI 3 was developed as a ligand with specificity for a c-kit G4, in which a planar core naphthalenediimide was functionalized with two lysines with boc-protected side chains (Fig. 1.22g) [226]. The preference for this interaction possibly relies on the specific contact with the loops or grooves. Phen-Et, a phenanthroline-bisbenzimidazole caroboxyamide molecule, shows a preference for c-myc and c-kit parallel quadruplexes over any topology of telomere G4s (parallel, antiparallel, hybrid, or higher-ordered topologies), albeit with a moderate binding affinity (KD ~ 1.6 × 10−5 M) (Fig. 1.22h) [153]. Computer-aided modeling studies underscored the significance of the optimal projection of N,N-dimethylaminoethyl side chains at the N position of the benzimidazole moiety for recognizing the propeller loops of promoter G4s. Guanosine moiety can be used for specific recognition when attached to a dansyl moiety to yield DDG, in which two azide-labeled dinucleosides are linked across a dansyl dialkyneamide through click chemistry (Fig. 1.22i) [227]. This conjugate is capable of recognizing specifically a c-myc parallel G4 against a c-kit parallel one. TOxaPy, a crescent-shaped molecule that is alternately made up of pyrimidine and oxazole rings, shows preferential binding to a telomere with antiparallel topology over a telomere with parallel topology with a high binding affinity (KD = 2 × 10−7 M) (Fig. 1.22j) [228]. Specific groove binding of ToxaPy to the antiparallel topology has been predicted by a docking analysis, but this has not been confirmed. It is worth describing the last three molecules, BTC-f [229], TH3 [230], and IZCZ-3 [231], because these molecules have been shown to reduce off-target effects in biological experiments (Fig. 1.22k–m). Therefore, information about their in vitro preference is biologically confirmed.
1.2.4.4 Template-Guided Component Assembly and Linkage Through Click Chemistry

Template-guided component assembly and linkage through click chemistry gave the highly effective ligand, pyridostatin-based adduct 10, which is a more potent TRF1 competitor than PDS
1.2.4.5 Targeting of Non-canonical Higher-Order G-Quadruplex Structures

a GTC365 is a highly specific to hTERT G4 containing a mismatched hairpin stem loop, which is recognized by a guanidine moiety. b Coaddition of netropsin and Phen-DC3 is able to simultaneously recognize both duplex and G4 segments on duplex stem-loop-containing G4 motifs, respectively. N-methylpyrrole is highlighted in blue
1.2.4.6 Cell-Based Screening of G-Quadruplex Ligands

Hit compounds by cell-based screenings. a, b The c-kit-targeting ligands that were identified. The different parts of chemical structures are highlighted in purple, for clarity. c PDC12 was identified as the most potent candidate with the ability to induce G4-dependent transcriptional reprogramming by stabilizing a single G4 located at the BU-1 locus
1.2.4.7 Specific Targeting of Telomere G-Quadruplexes

Specific telomere G4 targeting by ligands. a Telomere G-stretch sequences potentially adopt non-orthodox G4s that offer specific binding motifs. b Several telomere G4-preferred binders based on the specific-motif recognition
1.2.4.8 RNA G-Quadruplex-Interactive Molecules
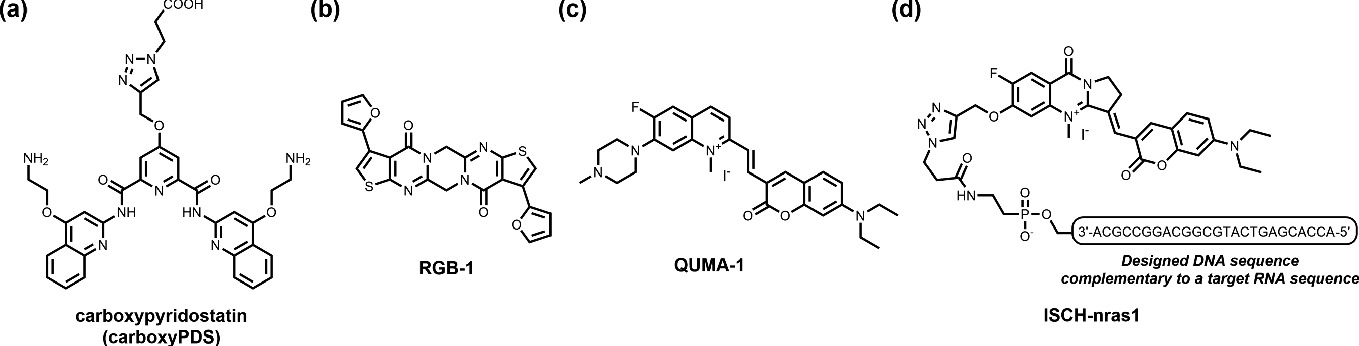
a–d RNA G4-targeting ligands. d It is noteworthy that ISCH-nras1 was able selectively to target and detect an NRAS RNA G4 in a cellular context
1.2.4.9 Specific Localization for the Selective Targeting of Particular G-Quadruplexes; the Mitochondrial G-Quadruplex

Representative mitochondria-localized compounds that are likely to bind to G4 structures; a ZnPC1, b TP2py
1.2.4.10 Alternative Nucleic Acid form as a Biomedical Target, G-Triplex

a–c G-triplex-targeting ligands. b A platform for their evaluation constructed by DNA origami
Our group has devised a nanoplatform constructed by DNA origami for studying such intermediates of G4 such as G-triplex and G-hairpin and found that PDC, a well-known G4-interacting ligand, unexpectedly recognized the G-triplex and G-hairpin structures (Fig. 1.29c) [271]. Considering this, the ability to recognize the intermediates of G4 might be an essential component for the high binding affinity, selectivity, or inducing ability of the G4 structures from the stable duplex or single-stranded DNA. The platform manifests the power to assess an unprecedented G4-binding property of a ligand.
1.3 Conclusion and Future Prospects
I have discussed expandable trinucleotide repeats and G-quadruplexes (G4s) in terms of molecular targets by synthetic ligands toward creating potential drugs or chemical tools. From the therapeutic aspect of G4 ligands, the G4 is relatively recently considered to be a potential biomedical target particularly for tumor or neurologic disease therapy, and a considerable body of evidence has been accumulating that G4-interacting drugs exhibit good antitumor activities. However, limited fruits remain. As protein-targeting drugs face the same situations, G4-interacting drugs displayed low selectivities to the targeted G4 structure, mainly due to the similar skeleton among different G4 forms prevalent in the genome. In this chapter, I have introduced G4-interacting ligands that were devised to gain selectivity to a particular G4 structure. The selectivity issues remain incompletely solved but, if accomplished, would substantially impact cancer therapy. Besides, the G4-driven oncogenes introduced here are known to usually well correlated and concertedly influence tumorigenesis, tumor growth, and malignant transition [160, 163, 183, 272, 273]. Although this relationship is not fully elucidated, combinatorial approaches may be a good option for further therapeutic advancements [273].
Collectively, the abovementioned non-canonical DNA conformations have profound implications in various biological, neurological, pharmacological events, primarily based on human diseases. The subsequent chapters include my Ph.D. study addressing the development of DNA-sequence and DNA-form selective ligands toward elucidating the function of non-canonical DNA structures relevant to human diseases.