Appendix
Neuroscience 101
Consider two different scenarios.
First: Think back to when you hit puberty. You’d been primed by a parent or teacher about what to expect. You woke up with a funny feeling, found your jammies alarmingly soiled. You excitedly woke up your parents, who, along with my family, got tearful; they took embarrassing pictures, a sheep was slaughtered in your honor, you were carried through town in a sedan chair while neighbors chanted in an ancient language. This was a big deal.
But be honest—would your life be so different if those endocrine changes had instead occurred twenty-four hours later?
Second scenario: Emerging from a store, you are unexpectedly chased by a lion. As part of the stress response, your brain increases your heart rate and blood pressure, dilates blood vessels in your leg muscles, which are now frantically working, sharpens sensory processing to produce a tunnel vision of concentration.
And how would things have turned out if your brain took twenty-four hours to send those commands? Dead meat.
That’s what makes the brain special. Hit puberty tomorrow instead of today? So what? Make some antibodies in an hour instead of now? Rarely fatal. Same for delaying depositing calcium in your bones. But much of what the nervous system is about is encapsulated in the frequent question in this book: What happened one second before? Incredible speed.
The nervous system is about contrasts, unambiguous extremes, having something or having nothing to say, maximizing signal-to-noise ratios. And this is demanding and expensive.[*]
One Neuron at a Time
The basic cell type of the nervous system, what we typically call a “brain cell,” is the neuron. The hundred billion or so in our brains communicate with each other, forming complex circuits. In addition, there are “glia” cells, which do a lot of gofering—providing structural support and insulation for neurons, storing energy for them, helping to mop up neuronal damage.
Naturally, this neuron/glia comparison is all wrong. There are about ten glial cells for every neuron, coming in various subtypes. They greatly influence how neurons speak to each other, and also form glial networks that communicate completely differently from neurons. So glia are important. Nonetheless, to make this primer more manageable, I’m going to be very neuron-centric.
Part of what makes the nervous system so distinctive is how distinctive neurons are as cells. Cells are usually small, self-contained entities—consider red blood cells, which are round little discs.
Neurons, in contrast, are highly asymmetrical, elongated beasts, typically with processes sticking out all over the place. Consider this drawing of a single neuron seen under a microscope in the early twentieth century by one of the patriarchs of the field, Santiago Ramón y Cajal:

It’s like the branches of a manic tree, explaining the jargon that this is a highly “arborized” neuron (a point explored at length in chapter 7, concerning how those arbors form in the first place).
Many neurons are also outlandishly large. A zillion red blood cells fit on the proverbial period at the end of this sentence. In contrast, there are single neurons in the spinal cord that send out projection cables that are many feet long. There are spinal cord neurons in blue whales that are half the length of a basketball court.
Now for the subparts of a neuron, the key to understanding its function.

What neurons do is talk to each other, cause each other to get excited. At one end of a neuron are its metaphorical ears, specialized processes that receive information from another neuron. At the other end are the processes that are the mouth, that communicate with the next neuron in line.
The ears, the inputs, are called dendrites. The output begins with a single long cable called an axon, which then ramifies into axonal endings—these axon terminals are the mouths (ignore the myelin sheath for the moment). The axon terminals connect to the spines on the branches of dendrites of the next neuron in line. Thus, a neuron’s dendritic ears are informed that the neuron behind it is excited. The flow of information then sweeps from the dendrites to the cell body to the axon to the axon terminals, and is then passed to the next neuron.
Let’s translate “flow of information” into quasi chemistry. What actually goes from the dendrites to the axon terminals? A wave of electrical excitation. Inside the neuron are various positively and negatively charged ions. Just outside the neuron’s membrane are other positively and negatively charged ions. When a neuron has gotten an exciting signal from the previous neuron at a spine on a dendritic branch, channels in the membrane in that spine open, allowing various ions to flow in, others to flow out, and the net result is that the inside of the end of that dendrite becomes more positively charged. The charge spreads toward the axon terminal, where it is passed to the next neuron. That’s it for the chemistry.
Two gigantically important details:
The Resting Potential
So when a neuron has gotten a hugely excitatory message from the previous neuron in line, its insides can become positively charged, relative to the extracellular space around it. When a neuron has something to say, it screams its head off. What might things look like then when the neuron has nothing to say, has not been stimulated? Maybe a state of equilibrium, where the inside and outside have equal, neutral charges.[*] No, never, impossible. That’s good enough for some cell in your spleen or your big toe. But back to that critical issue, that neurons are all about contrasts. When a neuron has nothing to say, it isn’t some passive state of things just trickling down to zero. Instead, it’s an active process. An active, intentional, forceful, muscular, sweaty process. Instead of the “I have nothing to say” state being one of default charge neutrality, the inside of the neuron is negatively charged.
You couldn’t ask for a more dramatic contrast: I have nothing to say = inside of the neuron is negatively charged. I have something to say = inside is positive. No neuron ever confuses the two. The internally negative state is called the resting potential. The excited state is called the action potential. And why is generating this dramatic resting potential such an active process? Because neurons have to work like crazy, using various pumps in their membranes, to push some positively charged ions out, to keep some negatively charged ones in, all in order to generate that negative internal resting state. Along comes an excitatory signal; channels open, and oceans of ions rush this way and that to generate the excitatory positive internal charge. And when that wave of excitation has passed, the channels close and the pumps have to get everything back to where they started, regenerating that negative resting potential. Remarkably, neurons spend nearly half their energy on the pumps that generate the resting potential. It doesn’t come cheap to generate dramatic contrasts between having nothing to say and having some exciting news.
Now that we understand resting potentials and action potentials, on to the other gigantically important detail.
That’s Not What Action Potentials Really Are
What I’ve just outlined is that a single dendritic spine receives an excitatory signal from the previous neuron (i.e., the previous neuron has had an action potential); this generates an action potential in that spine, which spreads the axonal branch that it is on, on toward the cell body, over it, and on to the axon and the axon terminals, and passes the signal to the next neuron in line. Not true.
Instead: The neuron is sitting there with nothing to say, which is to say that it’s displaying a resting potential; all of its insides are negatively charged. Along comes an excitatory signal at one dendritic spine on one dendritic branch, emanating from the axon terminal of the previous neuron in line. As a result, channels open and ions flow in and out in that one spine. But not enough to make the entire insides of the neuron positively charged. Simply a little less negative just inside that spine. Just to attach some numbers here that don’t matter in the slightest, things shift from the resting potential charge being around −70 millivolts (mV) to around −60 mV. Then the channels close. That little hiccup of becoming less negative[*] spreads farther to nearby spines on that branch of dendrite. The pumps have started working, pumping ions back to where they were in the first place. So at that dendritic spine, the charge went from −70 mV to −60 mV. But a little bit down the branch, things then go from −70 to −65 mV. Farther down, −70 to −69. In other words, that excitatory signal dissipates. You’ve taken a nice smooth, calm lake, in its resting state, and tossed a little pebble in. It causes a bit of a ripple right there, which spreads outward, getting smaller in its magnitude, until it dissipates not far from where the pebble hit. And miles away, at the lake’s axonal end, that ripple of excitation has had no effect whatsoever.
In other words, if a single dendritic spine is excited, that’s not enough to pass the excitation down to the axonal end and on to the next neuron. How does a message ever get passed on? Back to that wonderful drawing of a neuron by Cajal.
All those bifurcating dendritic branches are dotted with spines. And in order to get sufficient excitation to sweep from the dendritic end of the neuron to the axonal end, you have to have summation—the same spine must be stimulated repeatedly and rapidly and/or, more commonly, a bunch of the spines must be stimulated at once. You can’t get a wave, rather than just a ripple, unless you’ve thrown in a lot of pebbles.
At the base of the axon, where it emerges from the cell body, is a specialized part (called the axon hillock). If all those summated dendritic inputs produce enough of a ripple to move the resting potential around the hillock from −70 mV to about −40 mV, a threshold is passed. And once that happens, all hell breaks loose. A different class of channels opens in the membrane of the hillock, which allows a massive migration of ions, producing, finally, a positive charge (about +30 mV). In other words, an action potential. Which then opens up those same types of channels in the next smidgen of axonal membrane, regenerating the action potential there, and then the next, and the next, all the way down to the axon terminals.
From an informational standpoint, a neuron has two different types of signaling systems. From the dendritic spines to the axon hillock, it’s an analog signal, with gradations of signals that dissipate over space and time. And from the axon hillock on to the axon terminals, it’s a digital system with all-or-none signaling that regenerates down the length of the axon.
Let’s throw in some imaginary numbers, in order to appreciate the significance of this. Let’s suppose an average neuron has about one hundred dendritic spines and about one hundred axon terminals. What are the implications of this in the context of that analog/digital feature of neurons?
Sometimes nothing interesting. Consider neuron A, which, as just introduced, has one hundred axon terminals. Each one of those connects to one of the one hundred dendritic spines of the next neuron in line, neuron B. Neuron A has an action potential, which propagates down to all of its hundred axon terminals, which excites all one hundred dendritic spines in neuron B. The threshold at the axon hillock of neuron B requires fifty of the spines to get excited around the same time in order to generate an action potential; thus, with all one hundred of the spines firing, neuron B is guaranteed to get an action potential and is going to pass on neuron A’s message.
Now instead, neuron A projects half of its axon terminals to neuron B and half to neuron C. It has an action potential; does that guarantee one in neurons B and C? Each of those neurons’ axon hillocks has that threshold of needing a signal from fifty pebbles at once, in which case they have action potentials—neuron A has caused action potentials in two downstream neurons, has dramatically influenced the function of two neurons.
Now instead, neuron A evenly distributes its axon terminals among ten different target neurons, neurons B through K. Is its action potential going to produce action potentials in the target neurons? No way—continuing our example, the ten dendritic spines’ worth of pebbles in each target neuron is way below the threshold of fifty pebbles.
So what will ever cause an action potential in, say, neuron K, which only has ten of its dendritic spines getting excitatory signals from neuron A? Well, what’s going on with its other ninety dendritic spines? In this scenario, they’re getting inputs from other neurons—nine of them, with ten inputs from each. In other words, any given neuron integrates the inputs from all the neurons projecting to it. And out of this comes a rule: the more neurons that neuron A projects to, by definition, the more neurons it can influence; however, the more neurons it projects to, the smaller its average influence will be upon each of those target neurons. There’s a trade-off.
This doesn’t matter in the spinal cord, where one neuron typically sends all its projections to the next one in line. But in the brain, one neuron will disperse its projections to scads of other ones and receive inputs from scads of other ones, with each neuron’s axon hillock determining whether its threshold is reached and an action potential generated. The brain is wired in these networks of divergent and convergent signaling.
Now to put in a flabbergasting real number: your average neuron has about ten thousand to fifty thousand dendritic spines and about the same number of axon terminals. Factor in a hundred billion neurons, and you see why brains, rather than kidneys, write good poetry.
Just for completeness, here are a couple of final facts that should be ignored if this has already been more than you wanted. Neurons have some additional tricks, at the end of an action potential, to enhance the contrast between nothing-to-say and something-to-say even more, a means of ending the action potential really fast and dramatically—something called delayed rectification and another thing called the hyperpolarized refractory period. Another minor detail from that diagram above: a type of glial cell wraps around an axon, forming a layer of insulation called a myelin sheath; this “myelination” causes the action potential to shoot down the axon faster.
And one final detail of great future importance: the threshold of the axon hillock can change over time, thus changing the neuron’s excitability. What things change thresholds? Hormones, nutritional state, experience, and other factors that fill this book.
We’ve now made it from one end of a neuron to the other. How exactly does a neuron with an action potential communicate its excitation to the next neuron in line?
Two Neurons at a Time: Synaptic Communication
Suppose an action potential triggered in neuron A has swept down to all those tens of thousands of axon terminals. How is this excitation passed on to the next neuron(s)?
The Defeat of the Synctitium-ites
If you were your average nineteenth-century neuroscientist, the answer was easy. Their explanation would be that a fetal brain is made up of huge numbers of separate neurons that slowly grow their dendritic and axonal processes. And eventually, the axon terminals of one neuron reach and touch the dendritic spines of the next neuron(s), and they merge, forming a continuous membrane between the two cells. From all those separate fetal neurons, the mature brain forms this continuous, vastly complex net of one single superneuron, called a “synctitium.” Thus, excitation readily flows from one neuron to the next because they aren’t really separate neurons.
Late in the nineteenth century, an alternative view emerged, namely that each neuron remained an independent unit, and that the axon terminals of one neuron didn’t actually touch the dendritic spines of the next. Instead, there’s a tiny gap between the two. This notion was called the neuron doctrine.
The adherents to the synctitium school were arrogant as hell and even knew how to spell “synctitium,” so they weren’t shy in saying that they thought that the neuron doctrine was asinine. Show me the gaps between axon terminals and dendritic spines, they demanded of these heretics, and tell me how excitation jumps from one neuron to the next.
And then in 1873, it all got solved by the Italian neuroscientist Camillo Golgi, who invented a technique for staining brain tissue in a novel fashion. And the aforementioned Cajal used this “Golgi stain” to stain all the processes, all the branches and branchlets and twigs of the dendrites and axon terminals of single neurons. Crucially, the stain didn’t spread from one neuron to the next. There wasn’t a continuous merged net of a single superneuron. Individual neurons are discrete entities. The neuron doctrine–ers vanquished the synctitium-ites.[*]
Hooray, case closed; there are indeed micro-microscopic gaps between axon terminals and dendritic spines; these gaps are called synapses (which weren’t directly visualized until the invention of electron microscopy in the 1950s, putting the last nail in the synctitial coffin). But there’s still that problem of how excitation propagates from one neuron to the next, leaping across the synapse.
The answer, whose pursuit dominated neuroscience in the middle half of the twentieth century, is that the electrical excitation doesn’t leap across the synapse. Instead, it gets translated into a different type of signal.
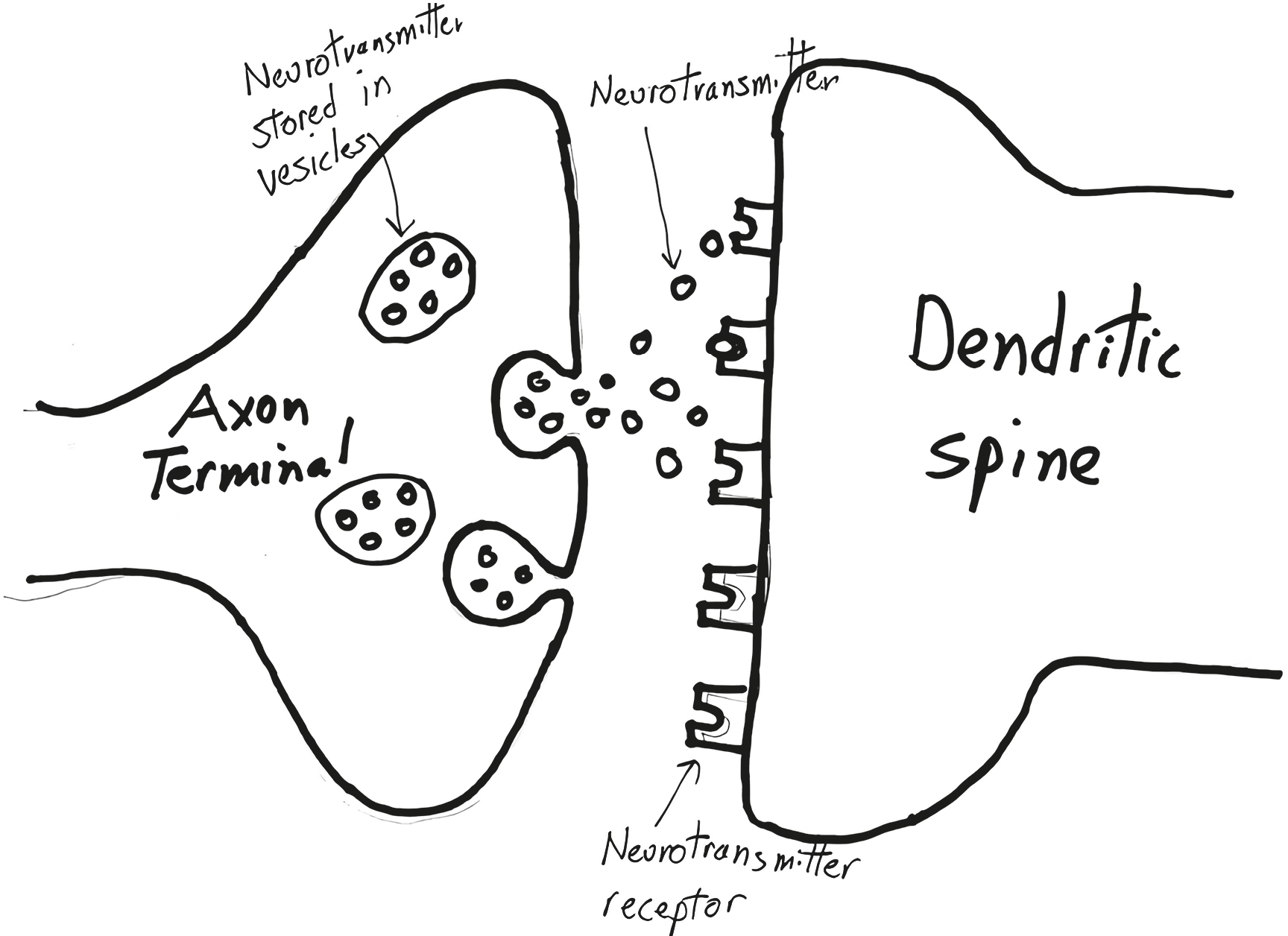
Neurotransmitters
Sitting inside each axon terminal, tethered to the membrane, are little balloons, called vesicles, filled with many copies of a chemical messenger. Along comes the action potential that initiated at the very start of the axon, at that neuron’s axon hillock. It sweeps over the terminal and triggers the release of those chemical messengers into the synapse. Which they float across, reaching the dendritic spine on the other side, where they excite the neuron. These chemical messengers are called neurotransmitters.
How do neurotransmitters, released from the “presynaptic” side of the synapse, cause excitation in the “postsynaptic” dendritic spine? Sitting on the membrane of the spine are receptors for the neurotransmitter. Time to introduce one of the great clichés of biology. The neurotransmitter molecule has a distinctive shape (with each copy of the molecule having the same). The receptor has a binding pocket of a distinctive shape that is perfectly complementary to the shape of the neurotransmitter. And thus the neurotransmitter—cliché time—fits into the receptor like a key into a lock. No other molecule fits snugly into that receptor; the neurotransmitter molecule won’t fit snugly into any other type of receptor. Neurotransmitter binds to receptor, which triggers those channels to open, and the currents of ionic excitation begin in the dendritic spine.
This describes “transsynaptic” communication with neurotransmitters. Except for one detail: What happens to the neurotransmitter molecules after they bind to the receptors? They don’t bind forever—remember that action potentials occur on the order of a few thousandths of a second. Instead, they float off the receptors, at which point the neurotransmitters have to be cleaned up. This occurs in one of two ways. First, for the ecologically minded synapse, there are “reuptake pumps” in the membrane of the axon terminal. They take up the neurotransmitters and recycle them, putting them back into those secretory vesicles to be used again.[*] The second option is for the neurotransmitter to be degraded in the synapse by an enzyme, with the breakdown products flushed out to sea (i.e., the extracellular environment, and from there on to the cerebrospinal fluid, the bloodstream, and eventually the bladder).
These housekeeping steps are hugely important. Suppose you want to increase the amount of neurotransmitter signaling across a synapse. Let’s translate that into the excitation terms of the previous section—you want to increase excitability across the synapse, such that an action potential in the presynaptic neuron has more of an oomph in the postsynaptic neuron, which is to say it has an increased likelihood of causing an action potential in that second neuron. You could increase the amount of neurotransmitter released—the presynaptic neuron yells louder. Or you could increase the amount of receptor on the dendritic spine—the postsynaptic neuron is listening more acutely.
But as another possibility, you could decrease the activity of the reuptake pump. As a result, less of the neurotransmitter is removed from the synapse. Thus, it sticks around longer and binds to the receptors repeatedly, amplifying the signal. Or as the conceptual equivalent, you could decrease the activity of the degradative enzyme; less neurotransmitter is broken down, and more sticks around longer in the synapse, having an enhanced effect. As we’ll see, some of the most interesting findings that help explain individual differences in the behaviors that concern us in this book relate to the amounts of neurotransmitter made and released, and the amounts and functioning of the receptors, reuptake pumps, and degradative enzymes.
Types of Neurotransmitters
What is this mythic neurotransmitter molecule, released by action potentials from the axon terminals of all of the hundred billion neurons? Here’s where things get complicated, because there is more than one type of neurotransmitter.
Why more than one? The same thing happens in every synapse, which is that the neurotransmitter binds to its key-in-a-lock receptor and triggers the opening of various channels that allow the ions to flow and makes the inside of the spine a bit less negatively charged.
One reason is that different neurotransmitters depolarize to different extents—in other words, some have more excitatory effects than others—and for different durations. This allows for a lot more complexity in information being passed from one neuron to the next.
And now to double the size of our palette, there are some neurotransmitters that don’t depolarize, don’t increase the likelihood of the next neuron in line having an action potential. They do the opposite—they “hyperpolarize” the spine, opening different types of channels that make the resting potential even more negative (e.g., shifting from −70 mV to −80 mV). In other words, there are such things as inhibitory neurotransmitters. You can see how that has just made things more complicated—a neuron with its ten thousand to fifty thousand dendritic spines is getting excitatory inputs of differing magnitudes from various neurons, getting inhibitory ones from other neurons, integrating all of this at the axon hillock.
Thus, there are lots of different classes of neurotransmitters, each binding to a unique receptor site that is complementary to its shape. Are there a bunch of different types of neurotransmitters in each axon terminal, so that an action potential triggers the release of a whole orchestration of signaling? Here is where we invoke Dale’s principle, named for Henry Dale, one of the grand pooh-bahs of the field, who in the 1930s proposed a rule whose veracity forms the very core of each neuroscientist’s sense of well-being: an action potential releases the same type of neurotransmitter from all of the axon terminals of a neuron. As such, there will be a distinctive neurochemical profile to a particular neuron: Oh, that neuron is a neurotransmitter A–type neuron. And what that also means is that the neurons that it talks to have neurotransmitter A receptors on their dendritic spines.[*]
There are dozens of neurotransmitters that have been identified. Some of the most renowned: serotonin, norepinephrine, dopamine, acetylcholine, glutamate (the most excitatory neurotransmitter in the brain), and GABA (the most inhibitory). It’s at this point that medical students are tortured with all the multisyllabic details of how each neurotransmitter is synthesized—its precursor, the intermediate forms the precursor is converted to until finally arriving at the real thing, the painfully long names of the various enzymes that catalyze the syntheses. Amid that, there are some pretty simple rules built around three points:
-
You do not ever want to find yourself running for your life from a lion and, oopsies, the neurons that tell your muscles to run fast go off-line because they’ve run out of neurotransmitter. Commensurate with that, neurotransmitters are made from precursors that are plentiful; often, they are simple dietary constituents. Serotonin and dopamine, for example, are made from the dietary amino acids tryptophan and tyrosine, respectively. Acetylcholine is made from dietary choline and lecithin.[*]
-
A neuron can potentially have dozens of action potentials a second. Each involves restocking of the vesicles with more neurotransmitter, releasing it, mopping up afterward. Given that, you do not want your neurotransmitters to be huge, complex, ornate molecules, each of which requires generations of stonemasons to construct. Instead, they are all made in a small number of steps from their precursors. They’re cheap and easy to make. For example, it only takes two simple synthetic steps to turn tyrosine into dopamine.
-
Finally, to complete this pattern of neurotransmitter synthesis as cheap and easy, generate multiple neurotransmitters from the same precursor. In neurons that use dopamine as the neurotransmitter, for example, there are two enzymes that do those two construction steps. Meanwhile, in norepinephrine-releasing neurons, there’s an additional enzyme that converts the dopamine to norepinephrine.
Cheap, cheap, cheap. Which makes sense. Nothing becomes obsolete faster than a neurotransmitter after it has done its postsynaptic thing. Yesterday’s newspaper is useful today only for house-training puppies. A final point that will be of huge relevance to come: just as the threshold of the axon hillock can change over time in response to experience, nearly every facet of the nuts and bolts of neurotransmitter-ology can be changed by experience as well.
Neuropharmacology
As these neurotransmitterology insights emerged, this allowed scientists to begin to understand how various “neuroactive” and “psychoactive” drugs and medicines work.
Broadly, such drugs fall into two categories: those that increase signaling across a particular type of synapse and those that decrease it. We already saw some of the strategies for increasing signaling: (a) administer a drug that stimulates more synthesis of the neurotransmitter (for example, by administering the precursor or using a drug that increases the activity of the enzymes that synthesize the neurotransmitter; as an example, Parkinson’s disease involves a loss of dopamine in one brain region, and a bulwark of treatment is to boost dopamine levels by administering the drug L-DOPA, which is the immediate precursor of dopamine); (b) administer a synthetic version of the neurotransmitter or a drug that is structurally close enough to the real thing to fool the receptors (psilocybin, for example, is structurally similar to serotonin and activates a subtype of its receptors); (c) stimulate the postsynaptic neuron to make more receptors (fine in theory, not easily done); (d) inhibit degradative enzymes so that more of the neurotransmitter sticks around in the synapse; (e) inhibit the reuptake of the neurotransmitter, prolonging its effects in the synapse (the modern antidepressant of choice, Prozac, does exactly that in serotonin synapses and thus is often referred to as an “SSRI,” a selective serotonin reuptake inhibitor).[*]
Meanwhile, a pharmacopeia of drugs is available to decrease signaling across synapses, and you can see what their underlying mechanisms are going to include—blocking the synthesis of a neurotransmitter, blocking its release, blocking its access to its receptor, and so on. Fun example: Acetylcholine stimulates your diaphragm to contract. Curare, the poison used in darts by indigenous people in the Amazon, blocks acetylcholine receptors. You stop breathing.
More Than Two Neurons at a Time
We have now triumphantly reached the point of thinking about three neurons at a time. And within not too many pages, we will have gone wild and considered even more than three. The purpose of this section is to see how circuits of neurons work, the intermediate step before examining what entire regions of the brain have to do with our behaviors. As such, the examples here were chosen merely to give a flavor of how things work at this level. Having some understanding of the building blocks of circuits like these is enormously important for chapter 12’s focus on how circuits in the brain can change in response to experience.
Neuromodulation
Consider the following diagram below:
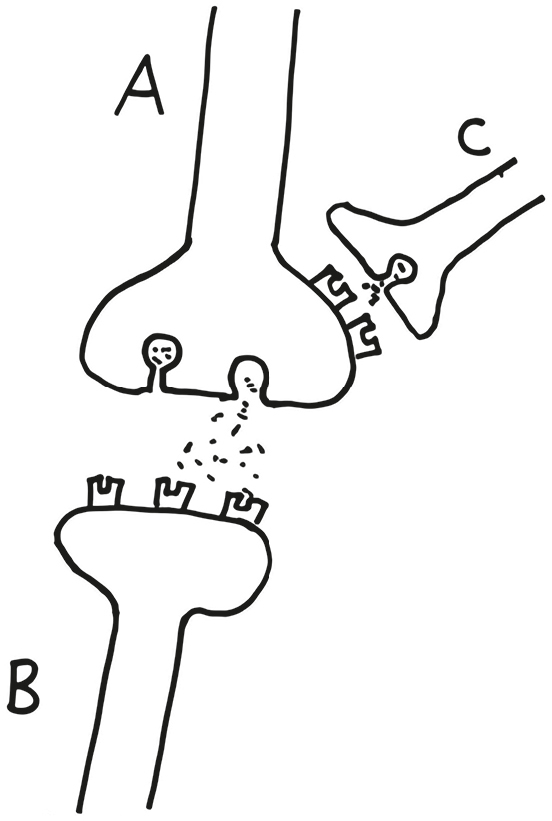
The axon terminal of neuron A forms a synapse with the dendritic spine of postsynaptic neuron B and releases an excitatory neurotransmitter. The usual. Meanwhile, neuron C sends an axon terminal projection on to neuron A. But not to a normal place, a dendritic spine. Instead, its axon terminal synapses onto the axon terminal of neuron A.
What’s up with this? Neuron C releases the inhibitory neurotransmitter GABA, which floats across that “axo-axonic” synapse and binds to receptors on that side of neuron A’s axon terminal. And its inhibitory effect (i.e., making that −70 mV resting potential even more negative) snuffs out any action potential hurtling down that branch of the axon, keeps it from getting to the very end and releasing neurotransmitter; thus, rather than directly influencing neuron B, neuron C is altering the ability of neuron A to influence B. In the jargon of the field, neuron C is playing a “neuromodulatory” role in this circuit.
Sharpening a Signal over Time and Space
Now for a new type of circuitry. To accommodate this, I’m using a simpler way of representing neurons. As diagrammed, neuron A sends all of its ten thousand to fifty thousand axonal projections to neuron B and releases an excitatory neurotransmitter, symbolized by the plus sign. The circle in neuron B represents the cell body plus all the dendritic branches that contain ten thousand to fifty thousand spines:

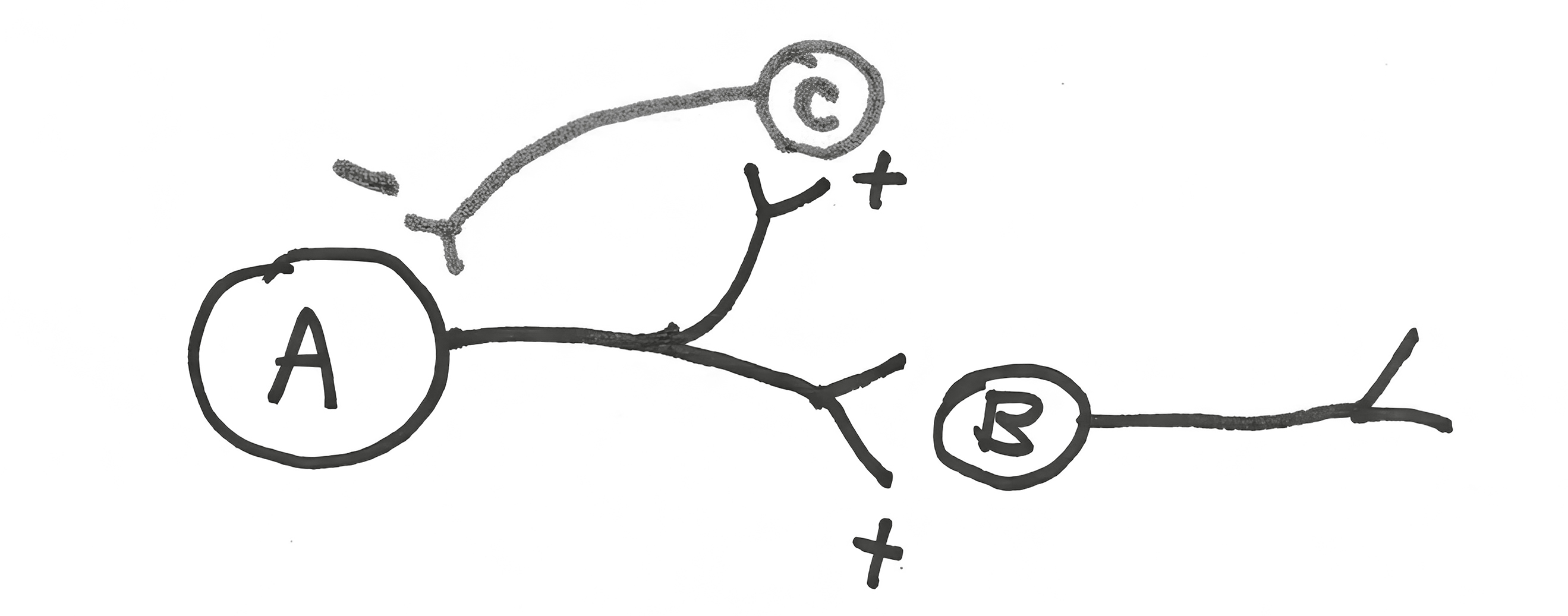
Now consider the next circuit. Neuron A stimulates neuron B, the usual. In addition, it also stimulates neuron C. This is routine, with neuron A splitting its axonal projections between the two target cells, exciting both. And what does neuron C do? It sends an inhibitory projection back on to neuron A, forming a negative feedback loop. Back to the brain loving contrasts, energetically screaming its head off when it has something to say, and energetically being silent otherwise. This is a more macro level of the same. Neuron A fires off a series of action potentials. What better way to energetically communicate when it’s all over than for it to become majorly silent, thanks to the inhibitory feedback loop? It’s a means of sharpening a signal over time.[*] And note that neuron A can “determine” how powerful that negative feedback signal will be by how many of the ten thousand axon terminals it shunts toward neuron C instead of B.
Such “temporal sharpening” of a signal can be accomplished in another way:
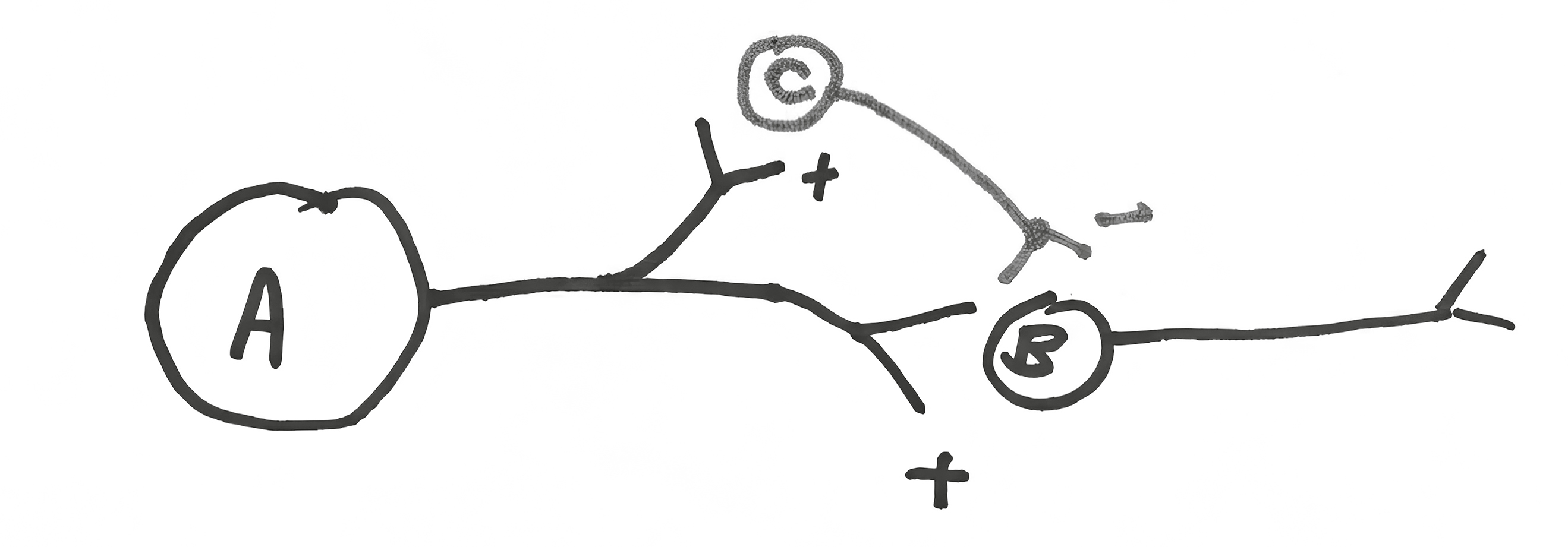
Neuron A stimulates B and C. Neuron C sends an inhibitory signal on to neuron B that will arrive after B starts getting stimulated (since the A/C/B loop is two synaptic steps, versus one for A/B). Result? Sharpening a signal with “feed-forward inhibition.”
Now for the other type of sharpening of a signal, of increasing the signal-to-noise ratio. Consider this six-neuron circuit, where neuron A stimulates B, C stimulates D, and E stimulates F:

Neuron C sends an excitatory projection on to neuron D. But in addition, neuron C’s axon sends collateral inhibitory projections on to neurons A and E.[*] Thus, if neuron C is stimulated, it both stimulates neuron D and silences neurons A and E. With such “lateral inhibition,” C screams its head off while A and E become especially silent. It’s a means of sharpening a spatial signal (and note that the diagram is simplified, in that I’ve omitted something obvious—neurons A and E also send inhibitory collateral projections on to neuron C, as well as the neurons on the other sides of them in this imaginary two-dimensional network).
Lateral inhibition like this is ubiquitous in sensory systems. Shine a tiny dot of light onto an eye. Wait, was that photoreceptor neuron A, C, or E that just got stimulated? Thanks to lateral inhibition, it is clearer that it was C. Ditto in tactile systems, allowing you to tell that it was this smidgen of skin that was just touched, not a little this way or that. Or in the ears, telling you that the tone you are hearing is an A, not an A-sharp or A-flat.[*]
Thus, what we’ve seen is another example of contrast enhancement in the nervous system. What is the significance of the fact that the silent state of a neuron is negatively charged, rather than a neutral zero millivolts? A way of sharpening a signal within a neuron. Feedback, feed-forward, and lateral inhibition with these sorts of collateral projections? A way of sharpening a signal over space and time within a circuit.
Two Different Types of Pain
This next circuit encompasses some of the elements just introduced and explains why there are, broadly, two different types of pain. I love this circuit because it is just so elegant:
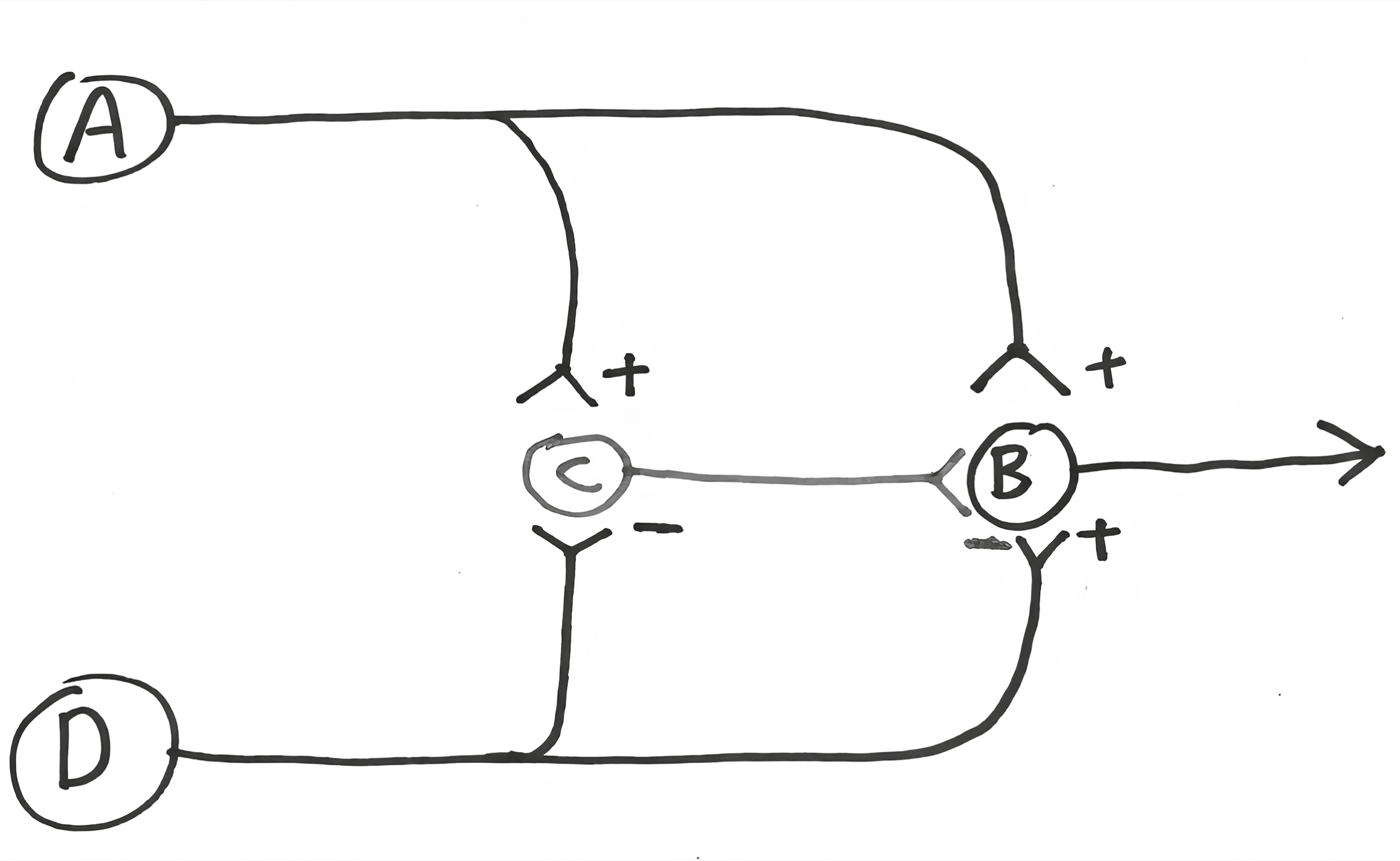
Neuron A’s dendrites sit just below the surface of the skin, and the neuron has an action potential in response to a painful stimulus. Neuron A then stimulates neuron B, which projects up the spinal cord, letting you know that something painful just happened. But neuron A also stimulates neuron C, which inhibits B. This is one of our feed-forward inhibitory circuits. Result? Neuron B fires briefly and then is silenced, and you perceive this as a sharp pain—you’ve been poked with a needle.
Meanwhile, there’s neuron D, whose dendrites are in the same general area of the skin and respond to a different type of painful stimulus. As before, neuron D excites neuron B, message is sent up to the brain. But it also sends projections to neuron C, where it inhibits it. Result? When neuron D is activated by a pain signal, it inhibits the ability of neuron C to inhibit neuron B. And you perceive it as a throbbing, continuous pain, like a burn or abrasion. Importantly, this is reinforced further by the fact that action potentials travel down the axon of neuron D much slower than in neuron A (having to do with that myelin that I mentioned earlier—details aren’t important). So the pain in neuron A’s world is not only transient but immediate. Pain in the neuron D branch is not only long-lasting but has a slower onset.
The two types of fibers can interact, and we often intentionally force them to. Suppose that you have some sort of continuous, throbbing pain—say, an insect bite. How can you stop the throbbing? Briefly stimulate the fast fiber. This adds to the pain for an instant, but by stimulating neuron C, you shut the system down for a while. And that is precisely what we often do in circumstances. An insect bite throbs unbearably, and we scratch hard right around it to dull the pain. And the slow-chronic-pain pathway is shut down for up to a few minutes.
The fact that pain works this way has important clinical implications. For one thing, it has allowed scientists to design treatments for people with severe chronic pain syndromes (for example, certain types of back injury). Implant a little electrode into the fast-pain pathway and attach it to a stimulator on the person’s hip; too much throbbing pain, buzz the stimulator, and after a brief, sharp pain, the chronic throbbing is turned off for a while; works wonders in many cases.
Thus we have a circuit that encompasses a temporal sharpening mechanism, introduces the double negative of inhibiting inhibitors, and is just all-around cool. And one of the biggest reasons why I love it is that it was first proposed in 1965 by these great neurobiologists Ronald Melzack and Patrick Wall. It was merely proposed as a theoretical model (“No one has ever seen this sort of wiring, but we propose that it’s got to look something like this, given how pain works”). And subsequent work showed that’s exactly how this part of the nervous system is wired.
Circuitry built on these sorts of elements is extremely important as well in chapter 12 in explaining how we generalize, form categories—where you look at a picture and say, “I can’t tell you who the artist is, but this is by one of those Impressionist painters,” or where you’re thinking of “one of those” presidents between Lincoln and Teddy Roosevelt, or “one of those” dogs that herd sheep.
One More Round of Scaling Up
A neuron, two neurons, a neuronal circuit. We’re ready now, as a last step, to scale up to the level of thousands, hundreds of thousands, of neurons at once. Look up an image of a liver sliced through in cross section and viewed through a microscope. It’s just a homogeneous field of cells, an undifferentiated carpet; if you’ve seen one part, you’ve seen it all. Boring.
In contrast, the brain is anything but that, showing a huge amount of internal organization.
In other words, the cell bodies of neurons that have related functions are clumped together in particular regions of the brain, and the axons that they send to other parts of the brain are organized into these projection cables. What all this means, crucially, is that different parts of the brain do different things. All the regions of the brain have names (usually multisyllabic and derived from Greek or Latin), as do the subregions and the sub-subregions. Moreover, each talks to a consistent collection of other regions (i.e., sends axons to them) and is talked to by a consistent collection (i.e., receives axonal projections from them). Which part of the brain is talking to which other part tells you a lot about function. For example, neurons that receive information that your body temperature has risen send projections to neurons that regulate sweating and activate them at such times. And just to show how complicated this all gets, if you’re around someone who is, well, sufficiently hot that your body feels warmer, those same neurons will activate projections that they have to neurons that cause your gonads to get all giggly and tongue-tied.
You can go crazy studying all the details of connections between different brain regions, as I’ve seen tragically in the case of many a neuroanatomist who relishes all these details. For our purposes, there are some key points:
—Each particular region contains millions of neurons. Some familiar names on this level of analysis: hypothalamus, cerebellum, cortex, hippocampus.
—Some regions have very distinct and compact subregions, and each is referred to as a “nucleus.” (This is confusing, as the part of every cell that contains the DNA is also called the nucleus. What can you do?) Some probably totally unfamiliar names, just as examples: the basal nucleus of Meynert, the supraoptic nucleus of the hypothalamus, the charmingly named inferior olive nucleus.
—As described, the cell bodies of the neurons with related functions are clumped together in their particular region or nucleus and send their axonal projections off in the same direction, merging together into a cable (aka a “fiber tract”).
—Back to that myelin wrapping around axons that helps action potentials propagate faster. Myelin tends to be white, sufficiently so that the fiber tract cables in the brain look white. Thus, they’re generically referred to as “white matter.” The clusters where the (unmyelinated) neuronal cell bodies are clumped together are “gray matter.”
Enough with the primer. Back to the book.