Canine distemper virus (CDV), a member of the genus Morbillivirus, which also includes the human pathogen measles virus (MeV) as well as several animal viruses, is one of the most severe viral diseases in ferrets. The infection is invariably lethal, and even certain live-attenuated vaccine strains used safely in dogs can cause severe disease or death in ferrets. This high sensitivity makes ferrets an attractive animal model not only for the characterization of morbillivirus pathogenesis mechanisms but also for the evaluation of morbilliviral vaccines and treatments, and the development of morbilliviruses as gene therapy vectors and recombinant vaccine platform.
Morbillivirus Biology
Morbilliviruses are enveloped single-stranded RNA viruses with a genome of negative polarity. The genome includes six genes that encode eight proteins, and each gene is flanked by untranslated regions that carry transcription signals recognized by the viral polymerase. Morbilliviruses enter target cells after binding of the viral attachment (H) protein to the respective cellular receptor via pH-independent membrane fusion mediated by the fusion (F) protein. The viral ribonucleoprotein complex comprised of the nucleoprotein (N)-encapsidated RNA genome with the associated phosphoprotein (P) and the RNA-dependent RNA polymerase (L) protein is then released in the cytoplasm and initiates the replication cycle. At early infection stages, the polymerase preferentially transcribes viral messenger RNAs (mRNAs) and then switches to genome replications once sufficient N protein has accumulated. The matrix (M) protein interacts with the newly formed ribonucleoprotein complexes as well as the cytoplasmic tails of the viral glycoproteins, mediating particle assembly and release. In addition to the six structural proteins, morbilliviruses produce two nonessential proteins V and C, which are both synthesized from the P gene open reading frame, either by RNA editing or by use of an alternate open reading frame [1]. While the role of the V protein in the interference with innate immune activation is well characterized, the role of the C protein is less clear.
Course of CDV Disease in Ferrets
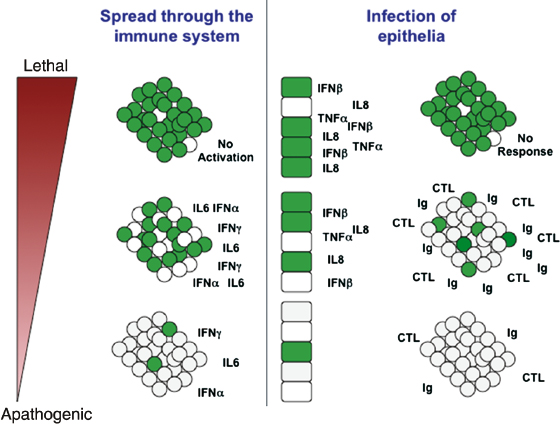
Morbilliviruses are transmitted by aerosol, not only via respiratory droplets but also urine [2], which may play an important role in the transmission among carnivores, which use urine to mark their territory. The virus initially infects immune cells in the upper and lower respiratory tracts [3,4]. In ferrets, the prodromal period lasts around 7–10 days and is characterized by dissemination of the virus through the lymphatic system (Fig. 26.1) [5,6]. Virus can be detected in peripheral blood mononuclear cells (PBMC) and lymphatic organs as early as 2–3 days after infection, and infection rates of above 60% in circulating T and B lymphocytes can be seen in the context of lethal disease [4]. Aside from a short spike in body temperature 2–3 days after infection, no clinical signs are observed, and lethal infections are characterized by a complete lack of innate immune activation. In contrast, immune cells of surviving animals rapidly induce the expression of type I and II interferons (IFNs) and proinflammatory cytokines (Fig. 26.2) [7].
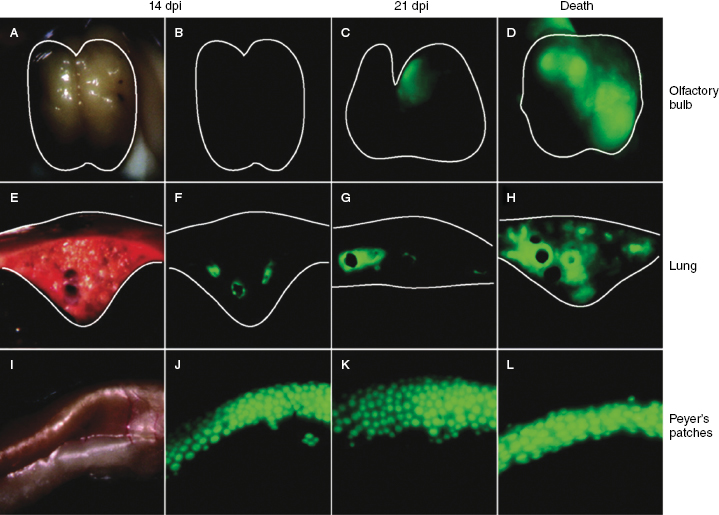
Fig. 26.1. Overview of CDV pathogenesis.Fig. 26.2. Time course of CDV dissemination in different tissues. Shown are a macroscopic visualization of infection in the olfactory bulb (A–D), a transverse section of a lung lobe (E–H), and Peyer's patches (panels I–L). The contours of the organs are outlined by a white line. (A, E, and I) Normal light photographs of the different tissues from an animal killed at 14 d.p.i. (B, F, and J) Same organs and time point as above but photographed after eGFP fluorescence excitation. Also shown are the same organs as above from an animal killed at 21 d.p.i. (C, G, and K) and at the time of euthanasia (D, H, and L), photographed after eGFP fluorescence excitation. (Rudd PA et al. (2006) J Virol, 80: 9361–9370).
The onset of clinical signs coincides with the spread to epithelia throughout the body (Fig. 26.3). Animals develop a sustained fever that can reach temperatures above 40°C/104°F, and an erythematous rash on lips and chin as one of the first notable signs. As the disease progresses, a severe rash covering the entire body and an increasing inflammation of the mucous membranes in the upper respiratory tract, the ocular conjunctiva, and the rectal mucosa are observed [6,8,9]. This inflammation results in reduced uptake and absorption of nutrients by the intestinal muscosa, and facilitates the establishment of secondary gastrointestinal and respiratory infections. If the immune system is unable to control the infection by mounting a humoral and cellular response, epithelial barriers are increasingly compromised, leading to septicemia and death. If the animal survives, clinical signs gradually resolve, leading to complete recovery within 3–5 weeks after infection. The increase in neutralizing antibodies correlates with the decline of the cell-associated viremia. Replicating virus can be isolated from conjunctival and nasal exsudates, and tracheal swabs as well as lymphatic and epithelial tissues throughout the symptomatic phase and has been found in PBMCs of recovering animals until day 21, and in individual cases even until day 28 [6,10]. In ferrets, infection of the central nervous system (CNS) is common [11], but cases of persistent CNS infections are rare due to the high mortality during the acute disease phase.
Fig. 26.3. Model of correlation between interference with innate immune activation and disease severity.
Innate and Adaptive Host Responses
In the context of lethal disease outcome, the virus completely inhibits the induction of type I and type II IFNs and proinflammatory cytokines in immune cells and tissues, whereas sublethal infections are characterized by robust upregulation of cytokine mRNAs as early as 3 days after infection [7]. Expression of proinflammatory as well as Th1- and Th2-type cytokines increases over the first week and then gradually subsides with the onset of the adaptive immune response characterized by the detection of neutralizing antibodies around 10 days after infection. This increase in CDV-specific antibodies and the development of a cell-mediated antiviral response correlate with virus clearance, and antibody titers above the protective level are usually detected within 3–4 weeks after infection [6,12]. While the postinfection titer may drop over time, values well above the protective threshold of 100 are detected even years after infection [13].
Transmission Studies
Because of their high susceptibility, ferrets have been used frequently to determine the particle stability and modes of transmission of CDV, and the results can probably be applied to all morbilliviruses. Even though most of the recent infection studies use intranasal inoculation, contact transmission has also been reported [14]. In addition, an early study demonstrated the possibility of transmission via contaminated surfaces for short periods of time [15]. The relative ease of housing larger numbers of animals in a laboratory setting has led to the use of ferrets to study CDV population dynamics. This work revealed that postexposure vaccination has little benefit for the vaccinee or outbreak control, and that herd immunity above 70% prevents an epizootic [16].
Vaccines and Immunization Schedules
The uniform and severe course of CDV disease in ferrets has led to their extensive use for vaccine development, including safety and efficacy studies in naïve animals of different ages, and comparison of the efficacy of vaccination when different administration routes are used. Early studies demonstrated that subcutaneous, intramuscular, or intraperitoneal immunization with live-attenuated vaccines induced protection from challenge with a virulent strain within 1–2 days [17], and immunization via aerosol protected within 5 days [18]. However, the mechanisms underlying this rapid protection remain to be characterized. Different studies provide compelling evidence that a single immunization of a naïve with a live-attenuated vaccine induces long-term and likely even lifelong protection [13,19].
While the generation of live-attenuated vaccines through repeated passages of wild-type strains in different mammalian cell types or embryonated chicken eggs [20] was a great success, subsequent experiments revealed that passaging of these attenuated viruses in vivo could result in reversion to virulence [21]. The reattenuation observed after cell culture passages of the reverted virus [22] and the differences in virulence observed after inoculation of ferrets with cell lysates from different persistently CDV-infected cell lines [23] illustrate the difficulties associated with predicting and assuring the attenuation level of live-attenuated vaccines.
Vaccination Strategies for Highly Susceptible Species
While currently available live-attenuated CDV vaccines have proven safe and effective for dogs, most retain residual immunosuppressive properties and can cause severe or even lethal disease in more sensitive species [24]. The uniform and severe course of disease in ferrets has led to their extensive use as animal model for the development of vaccines targeting these susceptible species, especially in the context of zoos or wildlife conservation programs. After the near extinction of the black-footed ferret due to a CDV vaccination campaign [25], an experimental beta-propiolactone-inactivated vaccine was developed. This vaccine induced lower antibody levels and was slightly less protective than the original live-attenuated vaccine, but was no longer immunosuppressive [26]. The use of this inactivated vaccine or the live-attenuated measles vaccine followed by immunization with one of the live-attenuated CDV vaccines upon verification of antibody titers was subsequently recommended for the protection of domestic ferrets and other highly sensitive species [27,28]. This immunization scheme has since been replaced by the recombinant canarypox vaccine expressing the CDV glycoproteins F and H [29], which received regulatory approval in North America in 1996 after thorough dog and ferret efficacy studies [8,30].
Vaccination in the Context of Pregnancy and Maternal Immunity
Immunization of pregnant jills at different gestation times did not result in infection of the fetuses or malformations or abnormalities in the offspring [31], demonstrating an additional level of safety of the live-attenuated vaccine. In offspring of dams previously immunized with a modified-live CDV vaccine, maternal antibodies had a half-life of 9 days and were below the limit of detection within 8–12 weeks [32]. Consistent with these kinetics, a separate study reported that kits from CDV-immunized dams had to be at least 7 weeks old before a live-attenuated vaccine induced protective immunity, regardless of the route of immunization [33]. In contrast, offspring of CDV-naïve females responded to immunization from 8 days old onward [34].
More recently, ferrets have been used to evaluate different strategies to overcome maternal antibody interference with vaccination, which also plays an important role in the MeV eradication effort. Even though parenteral immunization with recombinant vaccinia or canarypox viruses expressing the CDV envelope glycoproteins F and H induced a better antibody response than the live-attenuated vaccine in ferrets with residual maternal antibodies, they did not improve survival after challenge with wild-type virus. However, one-third of the animals survived when the poxviral vaccines were given simultaneously via the intranasal and parenteral routes [35], indicating that mucosal immunization may partially circumvent preexisting maternal immunity.
Vaccine Development
In addition to the CDV glycoprotein-expressing poxvirus vaccine vectors [8] mentioned earlier, poxviruses expressing the envelope glycoproteins of the closely related bovine morbillivirus rinderpest [36] or MeV [37] were shown to protect up to 60% of immunized ferrets and all dogs, respectively, from CDV challenge. The lack of CDV-specific neutralizing antibodies in some of these animals suggests that the observed protection was due to cross-reactive cellular immunity. However, the duration and protective capacity of the cellular immune response remains poorly characterized. Other recent experimental vaccine approaches include canine adenovirus-based vaccine expressing the CDV F and H glycoproteins, which conferred protection from challenge in dogs [38], and studies in mink and dogs have illustrated the potential of DNA-based vaccines [39,40]. Even though immunized animals were protected from challenge with wild-type virus, DNA vaccination alone or in combination with a cationic lipid-based transfection agent did not reliably induce CDV-specific antibodies [41], making it difficult to identify immunized animals or to predict if the vaccination conferred protection.
The potential of specifically attenuated vaccines through targeted alteration of the CDV genome has also been explored. Insertion of the green fluorescent protein gene into the CDV polymerase gene resulted in complete attenuation of an otherwise lethal wild-type strain, and animals infected with this virus were protected from subsequent challenge with a different wild-type virus [42]. A chimeric virus consisting of the MeV polymerase complex combined with the envelope proteins of a CDV wild-type strain was also completely attenuated, induced a robust antibody response after intramuscular or intranasal inoculation, and protected animals from lethal challenge [43]. Together, these studies demonstrate not only the safety and efficacy of these attenuated recombinant viruses for highly sensitive species but also the feasibility of approaches to develop new live-attenuated vaccines by introducing specific attenuating changes into their genome. As more tools and reagents become available to characterize host immune responses in ferrets, this model will likely be used even more frequently to explore the potential of new vaccination strategies.
Antiviral Therapies
At this time, no specific drugs are available to treat morbillivirus infections. High doses of vitamin A are recommended for children with MeV based on epidemiological studies, and the relationship between hypovitaminosis A and enhanced CDV-induced disease severity has been shown in ferrets [44]. This study revealed that ferrets are profoundly sensitive to deficiencies of vitamin A, developing severe corneal lesions, profuse diarrhea, weight loss, oily skin, and are becoming moribund within 4–6 weeks on a vitamin A-deficient diet, indicating that this animal may be particularly well suited to investigate the relationship between vitamin A and susceptibility to infectious diseases.
Ribavirin, 5-ethynyl-1-beta-d-ribofuranosylimidazole-4-carboxamide (EICAR), and proanthocyanidin A2 all exhibit an inhibitory effect on CDV replication in vitro [45,46], but controlled studies or even case reports of treated pet ferrets or dogs are not available. Similarly, a therapeutic effect of the commercially available recombinant feline IFN omega, which is approved for the treatment of canine parvovirus and feline calicivirus [47,48], has been suggested, but clinical efficacy data are still outstanding. Several proof-of-concept studies for new antiviral drugs targeting MeV are currently ongoing using CDV in ferrets as model system, and with the worldwide effort to eradicate MeV, these studies are likely to increase.
Pathogenesis
Since MeV and CDV share high genetic and functional similarities, the study of CDV in ferrets is increasingly used to elucidate morbillivirus pathogenetic mechanisms. Infection of ferrets with genetically modified CDVs unable to bind to the immune cell receptor signaling lymphocytic activation molecule (SLAM/CD150) confirmed that immune and not epithelial cell infection is essential for the establishment of a productive morbillivirus infection [49]. However, the animals still developed a strong antibody response, likely due to localized replication in epithelial cells close to the inoculation site in the upper respiratory tract, making it an attractive vaccine candidate. A similar study using an epithelial cell receptor-blind virus highlighted the importance of epithelial cell infection not only for shedding but also for the development of clinical disease [50]. Animals infected with this virus still experienced severe leukopenia and inhibition of lymphocyte proliferation, thereby disconnecting morbillivirus-induced immunosuppression and clinical signs.
Within the infected cell, the virus modulates the innate immune response to prevent immune recognition. The accessory V protein and, to a lesser extent, the C and P proteins inhibit cellular signaling pathways involved in innate immune activation. The V protein prevents the nuclear translocation of signal transducers and activators of transcription (STAT) 1 and 2 transcription factors, and interferes with the IFN-induced helicase melanoma differentiation-associated gene (mda) 5 mediated signaling, thereby inhibiting cellular responses to IFN stimulation and induction of an antiviral state [51]. In ferrets, viruses that lack the V protein caused only a mild disease and were no longer immunosuppressive, while the absence of the C protein alone had no effect. However, a virus lacking both V and C proteins was completely attenuated, indicating that C contributes to virulence [49]. Ongoing studies with viruses carrying V proteins that are unable to interfere with STAT1-, STAT2-, or mda5-mediated signaling revealed that STAT2 and mda5 signaling inhibition is essential for virulence, while interaction with STAT1 is less important. These specifically attenuated viruses are promising candidates for the next generation of live-attenuated vaccines, since the attenuating mutations are known and can thus be confirmed for each batch as part of the quality control process.
Several studies with viruses carrying modifications or deletion in different genes or untranslated regions have illustrated the intricate balance between efficient replication and avoidance of immune recognition [52–54]. Delayed as well as increased replication efficacy has proven detrimental for virulence, leading to a vigorous immune response and rapid control of the infection.
Morbillivirus Immunosuppression
Even though CDV infection or vaccination results in long-term and possibly life-long protection, the acute infection is associated with severe immunosuppression characterized by leukopenia and an inability of PBMCs to proliferate upon specific or nonspecific stimulation [55,56]. The up to 80% infection levels observed in immune cells of ferrets infected with lethal CDV strains partially explains the extent of leukopenia observed [4], but apoptosis of noninfected bystander lymphocytes also contributes to the dramatic loss of circulating immune cells [57,58]. Interestingly, infected immune cells were found to be more resistant than noninfected cells to exogenous induction of apoptosis, indicating that coexisting pro- and antiapoptotic processes contribute to immunosuppression.
The mechanisms underlying the infection-associated inhibition of lymphocyte proliferation remain poorly understood. Consistent with reports from MeV patients, lymphocytes from CDV-infected ferrets rapidly lose their ability to proliferate, and recovery to preinfection levels can take several weeks after the infection is cleared [6]. The observed correlation between infection level and inhibition of proliferation points toward a direct role of infected immune cells during the cell-associated viremia, and studies with MeV indicate that contact with infected cells expressing the viral envelope glycoproteins on their surface is sufficient to induce an anergic state in noninfected immune cells [59]. However, this does not explain the inhibition observed after the virus is cleared.
Neuroinvasion and CNS Persistence
Among morbilliviruses, CDV is associated with the highest incidence of neurological involvement, with up to 30% of dogs and most ferrets and wild carnivores exhibiting some evidence of infection in the CNS [60,61]. Similar to MeV, CDV-mediated neurological complications are categorized as acute encephalopathy, inclusion body polionecephalitis, and subacute to chronic demyelinating encephalitis [62]. Consequently, CDV has been extensively used to study morbillivirus neuropathology. In experimentally infected dogs, the onset and magnitude of the immune response determine if the animal recovers without CNS involvement, develops persistent CNS infection after recovery from the acute disease, or dies during the acute disease phase involving CNS infection [12]. Similar results were obtained when ferrets were infected with a recombinant non-neurovirulent virus that carried the H protein of a neurovirulent strain and the inverse virus. This study revealed that the extent of immunosuppression determined CNS invasion [63]. Early histopathological analyses of CDV time courses in dogs revealed the presence of infected lymphocytes in the white matter and the choroid plexus during the acute disease phase, indicating neuroinvasion via the hematogenous route [64].
More recently, a similar study in ferrets using an enhanced green fluorescent protein (eGFP)-expressing derivative of a neurovirulent CDV strain confirmed these findings and illustrated the contribution of anterograde neuroinvasion via the olfactory bulb for CDV CNS infection [11]. Virus is first detected in the olfactory bulb during the second week after infection (Fig. 26.3), and then spreads into the brainstem and the frontal lobes, while less virus is found in the caudal areas of the brain and the cerebellum. Immunohistochemical staining of sections of the olfactory mucosa indicate that the virus spreads from infected epithelial cells to dendrites of the olfactory receptor neurons [11]. Similar transmission may also occur at other sites of close proximity between epithelial and other cranial nerves. Thus, neuroinvasion occurred only after widespread infection of epithelial tissues, which may explain the observed lack of neurological involvement in uncomplicated rapidly controlled cases or after vaccination.
Even though most of the CDV positive cells in tissues from clinical cases or experimentally infected dogs are neurons and astrocytes, virus antigen has been found in all cell types present in the CNS, including microglia and oligodendrocytes [65–67]. While the characterization in ferrets is less advanced, infection of neurons and astrocytes cells has also been reported [11], indicating that the CDV CNS tropism is broad and species independent.
In dogs, the most common chronic neuropathological changes are first observed at the end of the acute disease around 3 weeks after infection and continue to progress after virus clearance from the periphery and recovery from clinical disease [68]. The intracerebral infiltration of circulating immune cells, perivascular cuffing of lymphocytes and monocytes, MHC upregulation, and high levels of CDV-specific antibodies in the cerebrospinal fluid observed in these animals are also characteristics of subacute sclerosing panencephalitis (SSPE) [69–71], a devastating late-onset complication of MeV infection. In ferrets, clinical signs of neurological involvement including retching, vomiting, seizures, and paralysis are usually observed during the acute phase, when the animal is severely immunosuppressed. While little lymphocyte infiltration is seen, the observed activation of microglia and the expression of proinflammatory cytokines in infected and neighboring cells are indicative of an infection-associated innate immune activation, which may contribute to the clinical presentation [72]. Since ferrets rarely survive, little is known about degenerative changes and host responses in the context of persistent CNS in this model.
Ferrets as Model for SSPE Pathogenesis
Among the MeV-associated neurological complications, SSPE has the lowest incidence with around one in a million cases. It occurs mainly in individuals who contracted the virus before 2 years of age and manifests months to years after the initial infection. The disease is characterized by a persistent CNS infection and low-level replication of a highly cell-associated derivative of the originally infecting virus, which has acquired multiple mutations [1]. To improve the understanding of the underlying mechanisms, ferrets were inoculated intracerebrally with brain cell cultures from SSPE patients. While wild-type MeV strains and SSPE isolates that efficiently replicated in cell culture did not induce any disease or histopathological changes, typical cell-associated nonproductive SSPE isolates produced an acute encephalitis in young ferrets [73]. Follow-up studies revealed that the course of disease was strongly dependent on the SSPE strain and the cell type used for its amplification, but the route of inoculation was less important, since intracardiac injection of cell-associated nonproductive SSPE strains induced the same disease and pathological changes [74]. However, despite severe clinical signs and considerable infection levels, only discreet CNS lesions and little to no inflammatory changes were found in histopathological analyses, and no antiviral antibodies were detected in the cerebrospinal fluid (CSF) [75], all of which are hallmarks of SSPE.
To more accurately reproduce the SSPE disease phenotype, ferrets were immunized with the live-attenuated MeV vaccine prior to intracerebral inoculation with a nonproductive SSPE strain. While around half of the animals succumbed to the disease, several of the survivors developed subacute encephalitis within weeks or months after infection [76]. These ferrets fell into a coma and ultimately succumbed to the disease, similar to the end stage of SSPE. Virus was found in the brain and, in some cases, the spinal cord, and inflammatory lesions were found in the white and gray matters. In addition, increased antiviral immunoglobulin G (IgG) concentrations were detected in the CSF of animals that developed subacute encephalitis [76,77], with M-specific antibodies being underrepresented or even absent compared with antibodies against other viral proteins [74], all of which is typical for SSPE. Even though the intracerebral route of inoculation limits the value of this model for pathogenesis studies, its ability to reproduce most of the aspects of SSPE makes it an attractive system to study the role of the host response in the development of this unique MeV complication. With the increasing availability of reagents and modern imaging tools, this model will likely be revisited and further developed.
Perspectives
The similarities in biology and pathogenesis observed among different members of the genus Morbillivirus have led to an increased use of the study of host–CDV interactions as a model for the different aspects of MeV vaccination, treatment, disease manifestation, and complications. Ferrets are particularly attractive for these studies, since they recapitulate the clinical signs of measles and are commonly used laboratory animals that can be accommodated by most animal facilities. The consistent disease phenotype observed and the availability of different strains that reproducibly result in defined neurological complications have resulted in a resurgence of the use of this model in recent years. It can be expected that the development of tools and reagents for this model in the context of the ongoing influenza research effort will further increase its application in the development of new morbillivirus vaccines and antiviral drugs.
References
1. Griffin DE (2007) Measles virus. In: Knipe DM, Howley PM, eds. Fields virology, 5th ed. Philadelphia: Lippincott, Williams & Wilkins, pp. 1551–1585.
2. Shen DT, Gorham JR, Pedersen V (1981) Viruria in dogs infected with canine distemper. Vet Med Small Anim Clin 76: 1175–1177.
3. Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yuksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL (2011) Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog 7: e1001263.
4. von Messling V, Milosevic D, Cattaneo R (2004) Tropism illuminated: lymphocyte-based pathways blazed by lethal morbillivirus through the host immune system. Proc Natl Acad Sci U S A 101: 14216–14221.
5. Crook E, Gorham JR, McNutt SH (1958) Experimental distemper in mink and ferrets. I. Pathogenesis. Am J Vet Res 19: 955–977.
6. von Messling V, Springfeld C, Devaux P, Cattaneo R (2003) A ferret model of canine distemper virus virulence and immunosuppression. J Virol 77: 12579–12591.
7. Svitek N, von Messling V (2007) Early cytokine mRNA expression profiles predict Morbillivirus disease outcome in ferrets. Virology 362: 404–410.
8. Stephensen CB, Welter J, Thaker SR, Taylor J, Tartaglia J, Paoletti E (1997) Canine distemper virus (CDV) infection of ferrets as a model for testing Morbillivirus vaccine strategies: NYVAC- and ALVAC-based CDV recombinants protect against symptomatic infection. J Virol 71: 1506–1513.
9. Liu C, Coffin DL (1957) Studies of canine distemper infection by means of fluorescein-labeled antibody. I. The pathogenesis, pathology, and diagnosis of the disease in experimentally infected ferrets. Virology 3: 115–131.
10. Appel MJ (1970) Distemper pathogenesis in dogs. J Am Vet Med Assoc 156: 1681–1684.
11. Rudd PA, Cattaneo R, von Messling V (2006) Canine distemper virus uses both the anterograde and the hematogenous pathway for neuroinvasion. J Virol 80: 9361–9370.
12. Appel MJ, Shek WR, Summers BA (1982) Lymphocyte-mediated immune cytotoxicity in dogs infected with virulent canine distemper virus. Infect Immun 37: 592–600.
13. Cabasso VJ, Stebbins MR, Cox HR (1953) Onset of resistance and duration of immunity to distemper in ferrets following a single injection of avianized distemper vaccine. Vet Med 48: 147.
14. Shen DT, Gorham JR (1978) Contact transmission of distemper virus in ferrets. Res Vet Sci 24: 118–119.
15. Shen DT, Gorham JR (1980) Survival of pathogenic distemper virus at 5C and 25C. Vet Med Small Anim Clin 75: 69–72.
16. Kelker D (1980) The effect of immunes on the spread of distemper in small ferret populations. Comput Biol Med 10: 53–60.
17. Shen DT, Gorham JR, Evermann JF, McKeirnan AJ (1984) Comparison of subcutaneous and intramuscular administration of a live attenuated distemper virus vaccine in ferrets. Vet Rec 114: 42–43.
18. Gorham JR, Leader RW, Gutierrez JC (1954) Distemper immunization of ferrets by nebulization with egg adapted virus. Science 119: 125–126.
19. Schultz RD (2006) Duration of immunity for canine and feline vaccines: a review. Vet Microbiol 117: 75–79.
20. Rockborn G, Norrby E, Lannek N (1965) Comparison between the immunizing effect in dogs and ferrets of living distemper vaccines, attenuated in dog tissue cultures and embryonated eggs. Res Vet Sci 6: 423–427.
21. Appel M (1978) Reversion to virulence of attenuated canine distemper virus in vivo and vitro. J Gen Virol 41: 385–393.
22. Goto H, Shen DT, Gorham JR (1976) Reversion to virulence of an attenuated distemper virus vaccine strain induced by rapid serial passage in ferrets. Fed Proc 35: 391.
23. Axthelm MK, Krakowka S, Gorham JR (1987) Canine distemper virus: in vivo virulence of in vitro-passaged persistent virus strains. Am J Vet Res 48: 227–234.
24. Bush M, Montali RJ, Brownstein D, James AEJ, Appel MJ (1976) Vaccine-induced canine distemper in a lesser panda. J Am Vet Med Assoc 169: 959–960.
25. Carpenter JW, Appel MJ, Erickson RC, Novilla MN (1976) Fatal vaccine-induced canine distemper virus infection in black-footed ferrets. J Am Vet Med Assoc 169: 961–964.
26. Williams ES, Anderson SL, Cavender J, Lynn C, List K, Hearn C, Appel MJ (1996) Vaccination of black-footed ferret (Mustela nigripes) x Siberian polecat (M. eversmanni) hybrids and domestic ferrets (M. putorius furo) against canine distemper. J Wildl Dis 32: 417–423.
27. Montali RJ, Bartz CR, Teare JA, Allen JT, Appel MJ, Bush M (1983) Clinical trials with canine distemper vaccines in exotic carnivores. J Am Vet Med Assoc 183: 1163–1167.
28. Qin Q, Wei F, Li M, Dubovi EJ, Loeffler IK (2007) Serosurvey of infectious disease agents of carnivores in captive red pandas (Ailurus fulgens) in China. J Zoo Wildl Med 38: 42–50.
29. Wimsatt J, Biggins D, Innes K, Taylor B, Garell D (2003) Evaluation of oral and subcutaneous delivery of an experimental canarypox recombinant canine distemper vaccine in the Siberian polecat (Mustela eversmanni). J Zoo Wildl Med 34: 25–35.
30. Pardo MC, Bauman JE, Mackowiak M (1997) Protection of dogs against canine distemper by vaccination with a canarypox virus recombinant expressing canine distemper virus fusion and hemagglutinin glycoproteins. Am J Vet Res 58: 833–836.
31. Hagen KW, Goto H, Gorham JR (1970) Distemper vaccine in pregnant ferrets and mink. Res Vet Sci 11: 458–460.
32. Appel MJ, Harris WV (1988) Antibody titers in domestic ferret jills and their kits to canine distemper virus vaccine. J Am Vet Med Assoc 193: 332–333.
33. Farrell RK, Skinner SF, Gorham JR, Lauerman LH (1971) The aerosol and subcutaneous administration of attenuated egg adapted distemper vaccine to ferret kits from distemper immune females. Res Vet Sci 12: 392–393.
34. Ott RL, Gorham JR (1955) The response of newborn and young ferrets to intranasal administration with egg-adapted distemper virus. Am J Vet Res 16: 571–572.
35. Welter J, Taylor J, Tartaglia J, Paoletti E, Stephensen CB (2000) Vaccination against canine distemper virus infection in infant ferrets with and without maternal antibody protection, using recombinant attenuated poxvirus vaccines. J Virol 74: 6358–6367.
36. Jones L, Tenorio E, Gorham J, Yilma T (1997) Protective vaccination of ferrets against canine distemper with recombinant pox virus vaccines expressing the H or F genes of rinderpest virus. Am J Vet Res 58: 590–593.
37. Taylor J, Pincus S, Tartaglia J, Richardson C, Alkhatib G, Briedis D, Appel M, Norton E, Paoletti E (1991) Vaccinia virus recombinants expressing either the measles virus fusion or hemagglutinin glycoprotein protect dogs against canine distemper virus challenge. J Virol 65: 4263–4274.
38. Fischer L, Tronel JP, Pardo-David C, Tanner P, Colombet G, Minke J, Audonnet JC (2002) Vaccination of puppies born to immune dams with a canine adenovirus-based vaccine protects against a canine distemper virus challenge. Vaccine 20: 3485–3497.
39. Dahl L, Jensen TH, Gottschalck E, Karlskov-Mortensen P, Jensen TD, Nielsen L, Andersen MK, Buckland R, Wild TF, Blixenkrone-Moller M (2004) Immunization with plasmid DNA encoding the hemagglutinin and the nucleoprotein confers robust protection against a lethal canine distemper virus challenge. Vaccine 22: 3642–3648.
40. Cherpillod P, Tipold A, Griot-Wenk M, Cardozo C, Schmid I, Fatzer R, Schobesberger M, Zurbriggen R, Bruckner L, Roch F, Vandevelde M, Wittek R, Zurbriggen A (2000) DNA vaccine encoding nucleocapsid and surface proteins of wild type canine distemper virus protects its natural host against distemper. Vaccine 18: 2927–2936.
41. Fischer L, Tronel JP, Minke J, Barzu S, Baudu P, Audonnet JC (2003) Vaccination of puppies with a lipid-formulated plasmid vaccine protects against a severe canine distemper virus challenge. Vaccine 21: 1099–1102.
42. Silin D, Lyubomska O, Ludlow M, Duprex WP, Rima BK (2007) Development of a challenge-protective vaccine concept by modification of the viral RNA-dependent RNA polymerase of canine distemper virus. J Virol 81: 13649–13658.
43. Rouxel RN, Svitek N, von Messling V (2009) A chimeric measles virus with canine distemper envelope protects ferrets from lethal distemper challenge. Vaccine 27: 4961–4966.
44. Rodeheffer C, von Messling V, Milot S, Lepine F, Manges AR, Ward BJ (2007) Disease manifestations of canine distemper virus infection in ferrets are modulated by vitamin A status. J Nutr 137: 1916–1922.
45. Dal Pozzo F, Galligioni V, Vaccari F, Gallina L, Battilani M, Scagliarini A (2010) Antiviral efficacy of EICAR against canine distemper virus (CDV) in vitro. Res Vet Sci 88: 339–344.
46. Gallina L, Dal Pozzo F, Galligioni V, Bombardelli E, Scagliarini A (2011) Inhibition of viral RNA synthesis in canine distemper virus infection by proanthocyanidin A2. Antiviral Res 92: 447–452.
47. de Mari K, Maynard L, Eun HM, Lebreux B (2003) Treatment of canine parvoviral enteritis with interferon-omega in a placebo-controlled field trial. Vet Rec 152: 105–108.
48. Hennet PR, Camy GA, McGahie DM, Albouy MV (2011) Comparative efficacy of a recombinant feline interferon omega in refractory cases of calicivirus-positive cats with caudal stomatitis: a randomised, multi-centre, controlled, double-blind study in 39 cats. J Feline Med Surg 13: 577–587.
49. von Messling V, Svitek N, Cattaneo R (2006) Receptor (SLAM [CD150]) recognition and the V protein sustain swift lymphocyte-based invasion of mucosal tissue and lymphatic organs by a morbillivirus. J Virol 80: 6084–6092.
50. Sawatsky B, Wong XX, Hinkelmann S, Cattaneo R, von Messling V (2012) Canine distemper virus epithelial cell infection is required for clinical disease but not for immunosuppression. J Virol 86: 3658–3666.
51. Rothlisberger A, Wiener D, Schweizer M, Peterhans E, Zurbriggen A, Plattet P (2010) Two domains of the V protein of virulent canine distemper virus selectively inhibit STAT1 and STAT2 nuclear import. J Virol 84: 6328–6343.
52. Anderson DE, von Messling V (2008) Region between the canine distemper virus M and F genes modulates virulence by controlling fusion protein expression. J Virol 82: 10510–10518.
53. Dietzel E, Anderson DE, Castan A, von Messling V, Maisner A (2011) Canine distemper virus matrix protein influences particle infectivity, particle composition, and envelope distribution in polarized epithelial cells and modulates virulence. J Virol 85: 7162–7168.
54. Anderson DE, Castan A, Bisaillon M, von Messling V (2012) Elements in the canine distemper virus M 3′ UTR contribute to control of replication efficiency and virulence. PLoS ONE 7: e31561.
55. Kauffman CA, Bergman AG, O'Connor RP (1982) Distemper virus infection in ferrets: an animal model of measles-induced immunosuppression. Clin Exp Immunol 47: 617–625.
56. Krakowka S, Cockerell G, Koestner A (1975) Effects of canine distemper virus infection on lymphoid function in vitro and in vivo. Infect Immun 11: 1069–1078.
57. Pillet S, von Messling V (2009) Canine distemper virus selectively inhibits apoptosis progression in infected immune cells. J Virol 83: 6279–6287.
58. Schobesberger M, Summerfield A, Doherr MG, Zurbriggen A, Griot C (2005) Canine distemper virus-induced depletion of uninfected lymphocytes is associated with apoptosis. Vet Immunol Immunopathol 104: 33–44.
59. Schlender J, Schnorr JJ, Spielhoffer P, Cathomen T, Cattaneo R, Billeter MA, ter Meulen V, Schneider-Schaulies S (1996) Interaction of measles virus glycoproteins with the surface of uninfected peripheral blood lymphocytes induces immunosuppression in vitro. Proc Natl Acad Sci U S A 93: 13194–13199.
60. Summers BA, Greisen HA, Appel MJ (1984) Canine distemper encephalomyelitis: variation with virus strain. J Comp Pathol 94: 65–75.
61. Krakowka S, Axthelm MK, Gorham JR (1987) Effects of induced thrombocytopenia on viral invasion of the central nervous system in canine distemper virus infection. J Comp Pathol 97: 441–450.
63. Bonami F, Rudd PA, von Messling V (2007) Disease duration determines canine distemper virus neurovirulence. J Virol 81: 12066–12070.
64. Summers BA, Greisen HA, Appel MJ (1979) Early events in canine distemper demyelinating encephalomyelitis. Acta Neuropathol 46: 1–10.
65. Stein VM, Czub M, Schreiner N, Moore PF, Vandevelde M, Zurbriggen A, Tipold A (2004) Microglial cell activation in demyelinating canine distemper lesions. J Neuroimmunol 153: 122–131.
66. Zurbriggen A, Schmid I, Graber HU, Vandevelde M (1998) Oligodendroglial pathology in canine distemper. Acta Neuropathol 95: 71–77.
67. Wyss-Fluehmann G, Zurbriggen A, Vandevelde M, Plattet P (2010) Canine distemper virus persistence in demyelinating encephalitis by swift intracellular cell-to-cell spread in astrocytes is controlled by the viral attachment protein. Acta Neuropathol 119: 617–630.
68. Vandevelde M, Higgins RJ, Kristensen B, Kristensen F, Steck AJ, Kihm U (1982) Demyelination in experimental canine distemper virus infection: immunological, pathologic, and immunohistological studies. Acta Neuropathol 56: 285–293.
69. Vandevelde M, Zurbriggen A, Steck A, Bichsel P (1986) Studies on the intrathecal humoral immune response in canine distemper encephalitis. J Neuroimmunol 11: 41–51.
70. Alldinger S, Wunschmann A, Baumgartner W, Voss C, Kremmer E (1996) Up-regulation of major histocompatibility complex class II antigen expression in the central nervous system of dogs with spontaneous canine distemper virus encephalitis. Acta Neuropathol 92: 273–280.
71. Schneider-Schaulies J, Niewiesk S, Schneider-Schaulies S, ter Meulen V (1999) Measles virus in the CNS: the role of viral and host factors for the establishment and maintenance of a persistent infection. J Neurovirol 5: 613–622.
72. Rudd PA, Bastien-Hamel LE, von Messling V (2010) Acute canine distemper encephalitis is associated with rapid neuronal loss and local immune activation. J Gen Virol 91: 980–989.
73. Katz M, Rorke LB, Masland WS, Koprowski H, Tucker SH (1968) Transmission of an encephalitogenic agent from brains of patients with subacute sclerosing panencephalitis to ferrets. Preliminary report. N Engl J Med 279: 793–798.
74. Thormar H, Mehta PD, Barshatzky MR, Brown HR (1985) Measles virus encephalitis in ferrets as a model for subacute sclerosing panencephalitis. Lab Anim Sci 35: 229–232.
75. Brown HR, Thormar H, Barshatzky M, Wisniewski HM (1985) Localization of measles virus antigens in subacute sclerosing panencephalitis in ferrets. Lab Anim Sci 35: 233–237.
76. Mehta PD, Thormar H (1979) Immunological studies of subacute measles encephalitis in ferrets: similarities to human subacute sclerosing panencephalitis. J Clin Microbiol 9: 601–604.
77. Thormar H, Mehta PD, Lin FH, Brown HR, Wisniewski HM (1983) Presence of oligoclonal immunoglobulin G bands and lack of matrix protein antibodies in cerebrospinal fluids and sera of ferrets with measles virus encephalitis. Infect Immun 41: 1205–1211.