TWO
Are You Experienced?

THE PROBLEM, I’M CONVINCED, IS THAT IT FALLS trippingly off the tongue: “Nature versus nurture.” With that great alliteration, rhyme, and snare drum beat, it’s just fun to say, like “Might makes right” or “If the glove doesn’t fit, you must acquit.” The British polymath Francis Galton didn’t invent this catchy expression,1 but he popularized it starting in 1869, and it’s been messing things up ever since. First of all, why say “nature” to mean “heredity”? The word nature typically means the entirety of the natural world, as in “the wonders of nature,” or the essence or moral character of something, as in “the better angels of our nature.” But it never means heredity, except in this one idiosyncratic phrase.
Then there’s “versus.” The idea that nature and nurture must be in opposition to explain human traits is silly. We know (although Galton and his contemporaries did not) that a few traits (like earwax type) are entirely hereditary, while others (like speech accent) are entirely nonhereditary, but that most traits fall somewhere in between. Even more crucially, we know that nature and nurture interact in various ways to determine traits: To have the symptoms of PKU, you need to both inherit two broken copies of the relevant gene and eat foods rich in phenylalanine. Similarly, you won’t be able to reach your full genetic potential for height if you are malnourished or chronically infected. If you’re born with athletic talent, you’re more likely to seek out opportunities to play sports and improve with practice. The oppositional construction of “nature versus nurture” is just wrong.
But the part of this horrid expression that really chaps my ass is “nurture.” The word means how your parents raised you—how they cared for and protected you (or failed to do so) when you were a child. But, of course, that’s only one small part of the nonhereditary determination of traits. As we will explore in this chapter, a more correct term would be “experience,” which I mean in the broadest sense. Not just social experience and not just the experience of events that you have stored as memories, but rather every single factor that impinges upon you, from the moment that the sperm fertilizes the egg to your last breath. These experiences start even before the embryo implants in the womb and encompass everything from the foods your mother ate while she was carrying you in utero to the waves of stress hormones you secreted on the first day of your first real job.
And there’s another important factor that is neither heredity nor experience. That’s the random nature of development, particularly the self-assembly of the brain and its five hundred trillion connections. As I mentioned earlier, developmental randomness is a large part of what we measure in twin studies in the category of non-shared environment. This self-assembly is guided by the genome, but it is not precisely specified at the finest levels of anatomy and function. The genome is not a detailed cell-by-cell blueprint for the development of the body and brain, but rather a vague recipe jotted down on the back on an envelope. The genome doesn’t say, “Hey you, glutamate-using neuron #12,345,763! Grow your axon in the dorsal direction for 123 microns and then make a sharp left turn to cross to the other side of the brain.” Rather, the instruction is more like, “Hey, you bunch of glutamate-using neurons over there! Grow your axons in the dorsal direction for a bit and then about 50 percent of you make a sharp left to cross the midline to reach the other side of the brain. The rest of you, turn your axons to the right.” The key point is that the genetic instructions for development are not precise. In one growing identical twin, 40 percent of the axons in this area will make the left turn; in an another, 60 percent will. The example here is from the brain, but the principle applies to all the organs. That’s the main reason why identical twins, who share the same DNA sequence and nearly the same uterine environment, are not born with wholly identical bodies, brains, or temperaments.
This means that your individuality is not a matter of “nature versus nurture” but rather “heredity interacting with experience, filtered through the inherent randomness of development.” It’s not nearly as fun to say but, unlike the former expression, it’s true. The exciting part is that we now have a general understanding of the molecular mechanisms by which heredity, experience, and developmental randomness interact to make you unique. Let me tell you about them.

NEARLY EVERY CELL IN your body contains your entire genome—all nineteen thousand or so genes and the vast stretches of DNA in between them.2 Yet, in a given cell, only some of these genes will ever be activated to instruct the production of a protein, a process called gene expression. When you think about it, this makes sense. You don’t want the cells that form the hair follicles in your scalp to be turning on the genes to make insulin, and you don’t want the cells of your pancreas to be growing hair. For example, most of the electrically excitable cells of the nervous system, the neurons, express about thirteen thousand genes. Of those, about seven thousand perform general cellular housekeeping functions, and so most other cells in the body express them too. There are about four hundred genes that tend to be expressed in much higher levels in neurons than in other cell types. Some specialized genes are shared between tissues. For example, both neurons and heart muscle cells are electrically active, so they share expression of certain genes that are required to generate electrical activity.
Doing the arithmetic, we can calculate that there are about six thousand genes that are never expressed in neurons.3 There are several ways to shut genes off so that they cannot be used to instruct the production of proteins. The longest-lasting way involves attaching small, globular chemical structures called methyl groups (—CH3) along the length of the gene’s DNA sequence.4 That blocks the information in the gene from being read out. The genes that are never expressed in a given cell type are usually shut off by this methylation of DNA.
In addition to those genes that are always shut off in a particular cell type, there are others that might be turned on or off at various times. For example, during childhood, certain growth-related genes are activated in tissues like muscle, bone, and cartilage, but those genes are turned off once a child stops growing. Other genes are turned on or off on a more rapid time scale. There are many genes in neurons and other tissues that turn on every night and are shut off during the day (or vice versa) and still more that are activated within minutes in response to a particular pattern of electrical activity in the nervous system or rising levels of a hormone.
These transient cycles of gene expression are controlled by different mechanisms. One involves modifying ball-shaped proteins called histones, around which the strands of DNA are wound. Attaching various chemical groups to histones can allow the DNA to unwind, which is a first, necessary step for gene expression. Other chemical groups can prevent this unwinding and therefore block gene expression. Another regulatory step involves proteins called transcription factors, which bind to a section of DNA near the start site of a gene and, in so doing, turn on expression of that gene. In many cases, genes need several transcription factors, all working together, for expression to start and the protein to be made.5 The regulation of gene expression—by the action of transcription factors, or the attaching of various chemical groups to DNA or histone proteins—is epigenetics. The crucial point here is that none of these mechanisms alter the underlying sequence of As, Cs, Ts, and Gs. That’s why it’s called epigenetics, rather than genetics.
Gene expression is exquisitely regulated. Genes can be turned on and off in different cell types, at different times, in response to all forms of experience—from hormonal fluctuations to infection to electrical activity from the sense organs. The regulation of gene expression, over both the short and long term, is the crucial place where genes and experience interact to forge human individuality.6
IN DECEMBER 1941, THE Imperial Japanese Army invaded the tropics, rapidly overrunning opposition in the steamy colonial enclaves of British Malaya and Burma, Dutch Indonesia, French Indochina, and the American Philippines, as well as the Kingdom of Thailand. Those were heady days for the Japanese military, as they routed, among others, the vaunted British Army. The Japanese enjoyed rapid and decisive military victories, and by March 1942 they stood at the frontier of India. However, not all was well among this tropical fighting force. One serious problem was that many Japanese soldiers were succumbing to heatstroke, rendering them temporarily unable to fight. When army doctors investigated, they found that soldiers from the colder northern Japanese island of Hokkaido had a much higher incidence of heatstroke then their comrades in arms from the subtropical southern island of Kyushu. The reason was that the northern soldiers sweated less and so had reduced evaporative cooling, resulting in dangerously elevated core body temperatures in hot climates. Skin biopsies revealed that northern and southern soldiers had the same total number of sweat glands. These are the eccrine sweat glands, which cover most of the body and secrete saltwater—not the apocrine sweat glands of the armpits and crotch that secrete oily, protein-laden sweat, which were discussed earlier in relation to the dry earwax gene, ABCC11. Upon more detailed inspection, doctors discovered that the southern soldiers had more eccrine sweat glands that received nerve fibers carrying sweat-activating electrical signals from the temperature-regulating region of the brain. These are the sweat glands that matter most for keeping your body’s core cool on a hot day.
The classic genetic explanation for how this difference came about would be that, over many generations, people living in Kyushu developed differences in their genes compared to those in Hokkaido. These genetic differences would give rise to more innervated sweat glands and better tolerance of hot climates and would be passed down to the offspring of Kyushu parents. If that were true, then you would imagine that children who were born in Hokkaido but whose parents were from established Kyushu families would inherit Kyushu-typical gene variants and so have larger numbers of activated sweat glands. And, conversely, you’d expect that Kyushu-born and -raised babies of parents from long-established Hokkaido families would have fewer activated sweat glands.
That explanation turned out to be utterly wrong. Instead, the degree of sweat gland innervation is determined by the ambient temperature experienced in your first year and is then locked in for the rest of your life. If you’re born in a cold place and you move to a hot place later in life, you’re just out of luck—you’ll carry your cold-appropriate, reduced-sweating skin with you. However, if you stay in the tropics and have and raise children there, they will have more activated sweat glands and improved thermal regulation.7
This potential mismatch between adaptation to the environment of early life and the experience of a different environment in later life seems like a problem for people who move from one place to another, but it may actually be beneficial. Genetic changes in response to the environment are often slow, requiring many generations to emerge. But adaptations determined by early life experience can appear in the very same generation. You and your mate, as northern-born people, may be prone to heatstroke after moving to the tropics; but your child, who carries northern genes, will sweat more extensively and fare better in the heat. This kind of experience-driven developmental plasticity may be part of what has allowed humans to rapidly migrate over long distances. For example, after the first humans crossed the land bridge from Siberia to Alaska, some of them settled all the way down to the tip of South America, spanning many climate zones, within less than one thousand years.
The story of the sweating Japanese soldiers shows us that we can be influenced by early life experiences, like temperature, that are not social in nature. In fact, those experiences can even start in the womb and, in other animals, can be quite dramatic. Some reptiles and amphibians, for example, have temperature-dependent sex determination. Males and females have identical chromosomes, but the pattern of gene expression that determines sex is set by the temperature of the incubating egg during the middle third of development, when the gonads differentiate.8 When American alligators lay their eggs, those embryos that experience a middle range of temperatures (from 32 to 34 degrees Celsius) will become male, while those either above or below this range will be female. It’s not clear if the adult female alligator, when burying her clutch of eggs, is choosing a nest location to influence the sex of her offspring—or if she will be able to modify that choice to keep her offspring from becoming all female as the climate warms.
This process, by which the external physical environment influences the traits of developing animals, can be found in mammals as well. It may sound suspiciously like a validation of astrology, but there’s good evidence from several mammals that birth season can influence development. For example, meadow voles born in the fall arrive with thicker fur than those born in the spring, even if both litters have the same parents. This trait is not influenced by the ambient temperature, which is similar in fall and spring. Instead, coat thickness is determined by the changes in day length experienced by the pregnant mother. When meadow voles are brought to the lab, day length can be manipulated with artificial lights. Mothers who experience lengthening days over the course of their twenty-one-day pregnancy, thereby mimicking springtime, give birth to pups with thinner fur. When these same mothers are bred to the same males and subjected to artificial shortening days over the course of their next pregnancy, simulating fall, they give birth to pups with thicker fur.9
There are some tantalizing hints from epidemiological studies that birth-season effects are present in humans too. Nicholas Tatonetti and his coworkers at Columbia University analyzed a huge data set: the medical records of over 1.7 million people treated at NewYork-Presbyterian/Columbia University Medical Center who were born between 1900 and 2000. They were looking for statistical associations between a patient’s birth month and lifetime incidence of 1,688 different medical conditions, covering the breadth of medicine from middle-ear infections to schizophrenia. Of those 1,688 conditions, only 55 were significantly influenced by birth month, including acute bronchiolitis, which is more prevalent among fall births, and angina (cardiac chest pain) which is overrepresented in those born in early spring (figure 3).10
There were some nice things about the design of this study. First, there were no decisions made about which conditions to test or report (which can lead to a bias for reporting positive associations and ignoring negative ones). Second, the population of patients in the database was quite diverse in terms of ancestry and affluence, so the statistics don’t just apply to affluent white people, who have been historically overrepresented in the pool of biomedical research subjects. However, there are some important limitations as well. The most obvious is that the patients were drawn from the New York City area, with its particular seasons, range of foods, weather, types of pollution, etc.
More importantly, birth month can reflect different types of influence, both prenatal and postnatal. For example, babies born in the late spring were carried in the later stages of pregnancy during the winter and spring months, when sunlight-driven vitamin D production is weakest. Low maternal vitamin D is thought to be a risk factor for certain autoimmune diseases, such as rheumatoid arthritis and systemic lupus.11 Babies born in the summer and fall arrived into peak indoor dust mite season, which has been suggested to underlie their higher rates of asthma and rhinitis as adults. And of course, some infectious diseases, like influenza, have varying seasonal incidence.
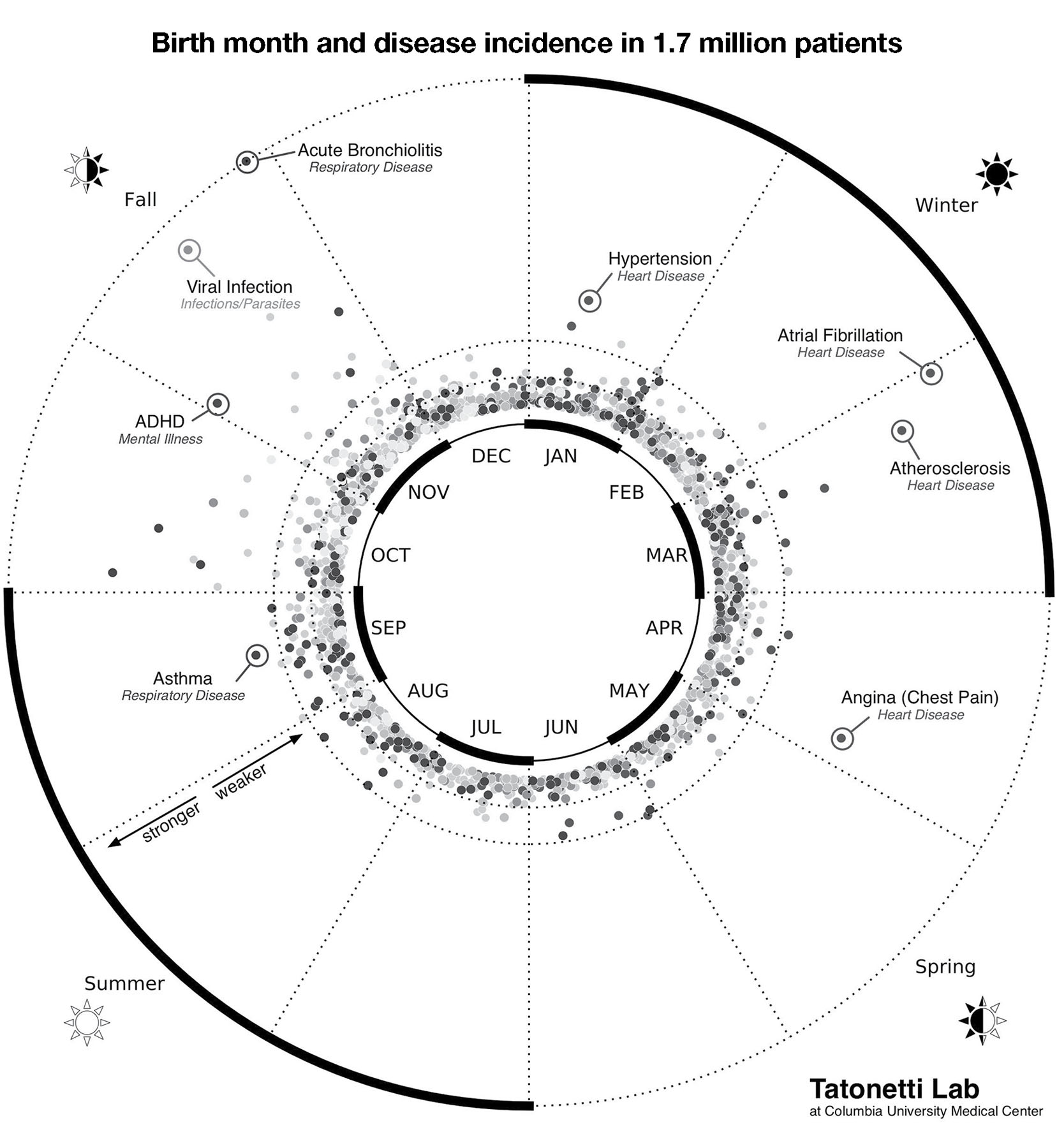
FIGURE 3. Some diseases are significantly more prevalent in people born in particular seasons. In this polar plot, greater distance from the center indicates a stronger statistical association between the disease incidence and birth month. For example, ADHD and acute bronchiolitis are more prevalent in people born in the fall, while atrial fibrillation (a heart problem) is found at higher rates in those born in the winter. This graph applies to the temperate latitudes of the Northern Hemisphere. Figure by Dr. Nicholas Tatonetti. Used with permission.
In addition to physical effects, birth month can also have social influences based on the cutoff date for entering school. If the cutoff day is October 1, then children born in October or November will be among the eldest in their school year, and those born in August and September will be among the youngest. Relative age in school will tend to give a child an advantage in sports. This can also affect medical conditions, as kids who participate in sports tend to have more injuries. Conversely, children younger than their peers are more likely to experience bullying, which can then impact neurological development.
To study the potential effects of age relative to peers,
Tatonetti, together with an international cast of collaborators, compiled medical data from 10.5 million patients across six locations in three countries (Taiwan, South Korea, and the United States) with varying latitudes (and therefore seasons), local weather, customs, and school cutoff dates. They calculated the incidence of 133 diseases, chosen so that there would be at least 1,000 patients with that disease at each of the six locations. Of those 133 diseases, only one showed a positive association with age relative to peers: attention deficit hyperactivity disorder (ADHD). Children who are younger relative to their peers in school had an 18 percent higher risk.12 Why? We don’t know. Maybe bullying is a risk factor for ADHD. Maybe it’s something else, social or biological. This uncertainty shows us an inherent limitation: epidemiological studies, no matter how carefully designed, cannot prove causality; they can only point us in interesting and useful directions. To go further, we’ll need experiments.
THE INFLUENZA PANDEMIC OF 1918 was the deadliest mass infection in modern history. The H1N1 flu strain originated in birds, moved to pigs, and then to humans. The first cases, in the spring of 1918, were reported at Fort Riley, a huge army base in Kansas. The virus spread east through the United States, leaving death and panic in its wake, before hopping the Atlantic to Europe and then Asia during the final months of World War I. The countries fighting on both sides of the war had strong press censorship, which suppressed reporting of the pandemic. Spain, which was neutral, had no such restrictions, and so the Spanish press spread the word. This is why the 1918 strain became known as the Spanish flu, even though it probably originated in North America.13
The 1918 pandemic flu was unusual: it had a high mortality rate, typically from secondary bacterial infections like pneumonia, and it was particularly fatal for those in the prime of life (people older than 40 probably had some degree of immunity from exposure to a milder, related flu strain that appeared in 1889). Worldwide, about one in three people were infected and over fifty million people died, including about 675,000 in the United States. To put this in perspective, more US soldiers died from the flu than from combat in World War I. The 1918 flu killed more people in twenty-four weeks than AIDS did in its first twenty-four years in North America.14
Many women were pregnant during the 1918 flu season, and about one third of them were infected but survived and gave birth in 1919. The echoes of this pandemic are seen in their children. Caleb Finch of the University of Southern California examined the medical records of soldiers who enlisted in World War II in 1941 and 1942. This sample included 2.7 million men who were born between 1915 and 1922. His team found that men whose mothers had carried them through the time of the 1918 pandemic flu were, on average, about one millimeter shorter than one would expect from comparing them to their fellow soldiers born just before the onset or conceived just after the end of the 1918 flu season.15 Now, one millimeter of height seems trivial, but in a huge sample like that it’s highly statistically significant.
Height is just the tip of the iceberg. The babies born in 1919 grew up to have higher rates of cardiovascular disease (about 20 percent more), perform slightly worse on standardized cognitive tests, and even earn somewhat less money. Perhaps most striking is that the incidence of schizophrenia in this population increased from about 1 percent to about 4 percent. Subsequent studies of other populations with maternal viral exposure in utero have confirmed a similar increased rate of schizophrenia,16 and have extended it to encompass a higher rate of autism as well.17
There are at least two different potential explanations for these findings. One hypothesis is that gene variants that allow the mother (or perhaps the fetus) to survive an influenza infection have other effects, including a reduction in average height and a higher incidence of heart disease, schizophrenia, and autism. Alternatively, we know that viral infection produces activation of the immune system, so perhaps virus-fighting immune cells from the maternal bloodstream or the chemical signals they secrete cross the placenta, enter the umbilical cord, and then affect development of the brain and other organs of the fetus.
IF YOU IMAGINE A young, brilliant, scientist power couple, you may well see Gloria Choi and Jun Huh, of MIT and Harvard Medical School, respectively. He’s an immunologist and she’s a neuroscientist. In the evening, when their children are put to bed and the dinner dishes are clean, sometimes they talk shop. Choi and Huh had read the scientific literature showing a higher rate of autism in those whose mothers had fought off a viral infection during pregnancy. And they had read a report from Paul Patterson’s lab at Caltech showing that, in mice, maternal infection can produce autism-like behaviors, and this process could be blocked by interfering with the action of an immune system signaling molecule called IL-6 in the mother.18 IL-6 is well known to trigger the production of yet another immune signaling molecule, IL-17a, which can pass from the mother into the developing fetus.
So, Choi and Huh thought that by performing experiments in mice to measure and manipulate IL-17a, they might reveal how maternal infection could change the brain of the developing fetus to produce behaviors associated with autism. To mimic viral infection, they used a well-established method. They injected pregnant mice with synthetic double-stranded RNA about midway through pregnancy, at a time when the neocortex is forming. Then they waited for the pups to be born and grow into adults before subjecting them to analysis. There were two exciting findings. First, the outermost portion of the neocortex was malformed in the mice whose mothers had been infected. Normally, the neocortex looks like a cake with six layers of varying thickness. Now, at various locations in the late-fetal brain, these regular layers were disrupted by protrusions, where blobs of neurons stuck out. When the fetuses from infected mothers grew to adulthood, a different pattern of cortical disruption emerged, which altered local electrical activity and was concentrated in a region called S1DZ. Second, the mice displayed behaviors roughly consistent with autism, including social interaction deficits and repetitive, compulsive behavior (in mice, this behavior is compulsive marble burying). Importantly, when the mothers were infected a few days later in pregnancy, by which time the layered structure of the neocortex had been established, neither the disrupted brain structure nor the autism-like behaviors were produced.
The next step in this inquiry is to understand the cellular and molecular steps that link maternal infection to altered brain development. I apologize in advance for bombarding you with a bunch of names for biomolecules. It’s not important that you memorize them. The crucial point is that there is a detailed, specific, and testable hypothesis for maternal-infection-triggered autism here, not just a bunch of hand-waving generalities.
The double-stranded RNA injected into the mother can’t cross the placenta into the fetal mice, but it can trigger immune cells in the mother’s body, called dendritic cells, to secrete signaling molecules called pro-inflammatory cytokines (please see figure 4 to follow along). These molecules (which have mind-numbing names like IL-6, IL-1beta, and IL-23) stimulate yet another type of immune cell (T helper 17 cell) to secrete IL-17a, a cytokine previously found to be elevated in the blood of autistic kids. IL-17a produced in the mother’s body crosses the placenta, flows through the umbilical cord, and binds receptors for IL-17a on developing neurons in the fetal neocortex. Crucially, when molecular or genetic tricks were used to interfere with maternal IL-17a production or signaling, the ability of maternal infection to produce disrupted cortical patches and autism-like behaviors in the pups was abolished. And, to put the icing on the cake, when IL-17a was injected directly into the developing fetal brain, this also produced both neocortical malformation and autism-like behaviors when the pups grew up.19
Presumably, when maternal IL-17a binds receptors on the developing fetal brain cells, it causes changes in gene expression in those cells, and those changes lead to the emergence of cortical patches and autism-like behavior. These results are consistent with reports in humans showing that, when autopsies are performed on autistic adults, malformations are sometimes found in the neocortex and that IL-17a is present at higher levels in the blood of some autistic children.20
These findings are super-exciting. They describe a molecular pathway to potentially explain the well-established link between maternal infection and the increased incidence of autism. And they point to potential therapies—perhaps interfering with IL-17a or the changes it evokes in the fetal brain could prevent autism from maternal infection. So, it was interesting when the basic finding could not be replicated in Choi and Huh’s labs using genetically identical lab mice that came from a different breeder. In mice supplied by Jackson Laboratory, maternal infection produced no elevation in IL-17a and no cortical patches or autism-like behaviors in the adult offspring. Then it was noticed that the original mice, which came from Taconic Biosciences, had a common, innocuous type of bacteria in their gut (called segmented filamentous bacteria, or SFB), while the Jackson mice did not. Sure enough, when Taconic mice were treated with an antibiotic to wipe out SFB, the maternal infection autism effect was abolished, and when Jackson mice had SFB introduced into their guts, the effect was restored.21
It turns out that SFB, through a process that is not yet understood, allows T helper 17 cells to differentiate and thereby become competent to secrete IL-17a. The essential point here is that, in order to produce the surge of IL-17a that causes fetal brain trouble, several things all have to happen: the female mouse must be pregnant, she must be carrying the right bacteria in her gut, and she must become infected with a virus. To produce autism in the pups, all of this must happen just as the fetal neocortex is developing, around day twelve of mouse pregnancy. If it happens a bit too early or too late, then the IL-17a surge produced by the infection will have no effect.22
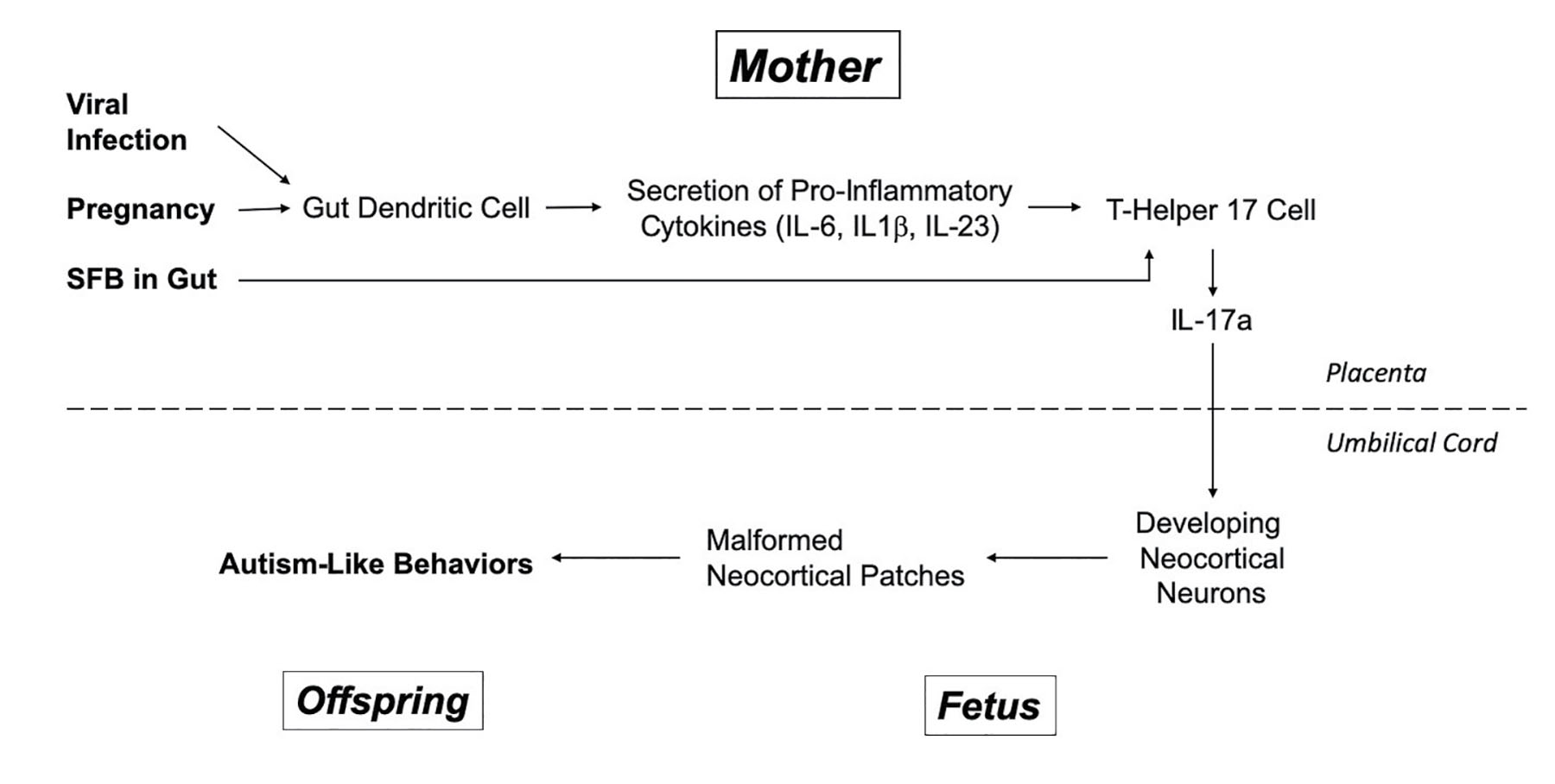
FIGURE 4. A molecular model for the contribution of maternal viral infection to fetal autism risk based on the work of Drs. Choi and Huh and their coworkers, as well as some other labs.
Of course, there are some caveats to sound. The cortical patches produced in Huh and Choi’s fetal mice are not exactly like the ones in humans with autism. And not all autopsy tissue from humans with autism reveals these cortical patches. And plenty of people have autism even though their mothers did not have a viral infection during pregnancy, so this IL-17a pathway is not the whole story for autism. Conversely, plenty of pregnant women get the flu and their children do not all have autism or schizophrenia. Nonetheless, these results make us think about individuality in a new way: both our mother’s experience with infection during pregnancy and her complement of gut microbiota (and the way these factors interact over time) can potently influence our neuropsychiatric development.
IT’S EASY TO IMAGINE that the effects of social experience on the development of individuality somehow occur in a different realm than those produced by the formative physical experiences we’ve discussed, like maternal viral infections. When we talk about early social experience we use words—like attachment, bonding, emotional warmth, and neglect—that are different from the biological terms, like IL-17a and dendritic cell. Let’s be clear: these behavioral terms are important and useful, but they should not suggest to us that social experience operates in some special, woo-woo space where biology doesn’t apply. When social experience—like parental neglect or bullying or nurturing—affects individuality in adulthood, it does so through biological effects on the brain. And when talk therapy works to ameliorate negative behavioral effects later on in life, it does so by changing the brain as well.
Here’s an example. We know that children who don’t receive regular loving touch in the first two years of life tend to have a broad array of lifelong neuropsychiatric problems, such as anxiety, depression, and intellectual disability. They also have a higher incidence of somatic (non-neuropsychiatric) illnesses, including persistent diseases of the gastrointestinal and immune systems. In recent years, a group of studies has shown that many different forms of early life social adversity—from a lack of parental loving touch to harsh, inconsistent discipline—produce exaggerated reactions to stress that persist through adult life and can contribute to these neuropsychiatric and somatic conditions. At least a portion of this enhanced stress reactivity comes from methylation in a region of DNA that suppresses the expression of the glucocorticoid receptor gene in certain brain regions.23 Through a hormonal feedback loop, this increases the secretion of a key stress hormone, CRH, by neurons in the hypothalamus brain region, producing widespread biological effects. While methylation of the glucocorticoid receptor gene is only a part of the story of how social adversity in early life affects individual traits in adulthood, it is an important example. It shows that we can potentially understand the molding effects of early social experience in terms of identified molecular and cellular signals.
WHEN BARBRA STREISAND’S ADORED Coton de Tuléar dog, Samantha, was near death in 2017, the famous singer was devastated at the impending loss of her loyal and loving companion. So, as befits a wealthy star, she had her vet take tiny biopsy samples from Samantha’s belly skin and cheek and sent them off, together with $50,000, to a company in Texas called ViaGen Pets. Using techniques originally developed at Seoul National University in South Korea, the ViaGen scientists were able to produce puppies from those cells that were genetic clones of Samantha. Streisand is now raising two of these puppies, which she has named Miss Violet and Miss Scarlett. While Miss Violet, Miss Scarlett, and Samantha are all genetically identical, they are not perfectly identical in terms of either looks or temperament. “They have different personalities,” Streisand said. “I’m waiting for them to get older so I can see if they have her brown eyes and her seriousness.”24
That Streisand’s two cloned puppies are not precise copies of either her original dog or each other should be no surprise. After all, human identical twins share the same sequence of DNA, and yet, even when raised together, they have certain differences in appearance and even more noticeable differences in personality. These differences have been codified in guidelines for forensic science. While the total number of ridges in a person’s fingerprints is about 90 percent heritable,25 when the exact pattern of ridges and whorls is examined, it is revealed that identical twins do not have identical fingerprints. Furthermore, identical twins don’t smell exactly the same. Well-trained sniffer dogs can reliably distinguish between the body odor of identical twins, even if they live in the same house and eat mostly the same foods.26 This is a general finding. Genetically identical twins, raised in the same household, will still show differences in both physical and behavioral traits. Perhaps the best illustration of this is that people married to one member of an identical twin pair rarely find themselves romantically attracted to their spouse’s twin. And this lack of spark is mutual: few identical twins are attracted to their co-twin’s spouse.27
So why aren’t identical twin people (or dogs) raised together more similar than they appear to be? Recall that twin studies suggest three general factors that influence traits: heritability, shared environment, and non-shared environment. In the case of identical twins raised together, differences in the first two factors are near zero. Does that mean that non-shared experiences account for all of the differences in traits? Not really. The truth is that “non-shared environment” is a garbage bag of a term that includes factors most of us would not regard as experience at all.
One of these important factors is the inherent randomness in the development of the body, particularly the nervous system. This is not “experience” or “environment” as we typically think of it—as something that impinges on an individual from the outside, like social experience or a viral infection. Rather, developmental randomness is intrinsic to the individual. During development, the human brain is estimated to produce about two hundred billion neurons, of which about one hundred billion survive competitive pruning during early life. In the adult brain, each of these surviving one hundred billion neurons makes about five thousand synaptic connections with other neurons. Those five hundred trillion synapses are not made randomly. The signals from the retina must be conveyed to the visual processing regions of the brain, and the signals from the parts of the brain that initiate movement must find their way to the appropriate muscles, and so on. The biological challenge is that the wiring diagram of the human brain is so enormous and complicated that it cannot be specified exactly in the sequence of an individual’s DNA.28 Subtle, random changes in the number, position, biochemical activity, or movement of cells within the developing nervous system can cascade through time to produce important differences in neural wiring and function between genetically identical twins raised together. Neurogeneticist Kevin Mitchell nicely sums this situation up by noting, “If you or I were cloned 100 times, the result would be 100 new individuals, each one of a kind.”29
One might imagine that the differences between genetically identical people come about as a result of non-shared experience or developmental randomness affecting the timing or pattern of gene expression in various cells of the body. Indeed, this is sometimes true. For example, one of the chemical processes that regulates gene expression is the pattern of DNA methylation and the transfer of chemical acetyl groups (C2H3O) to histone proteins (histone acetylation). When we compare these processes in identical twin pairs, we find that the twins are very similar in early life, but that older identical twins accumulate more and more of these epigenetic differences as they age, causing their gene expression profiles to slowly drift apart.30 That’s really compelling, and so one might be tempted to imagine that regulation of gene expression is the whole story when it comes to understanding how experience drives individuality. But it’s not. There are other important aspects of individual experience that are completely independent of regulated gene expression.
LIKE ALL BIOLOGISTS OF my era, I was taught that somatic cells (all cells in the body except eggs and sperm) are genetically identical. In this way, the differences between cell types arise from varying patterns of gene expression, as determined by the process of development and by experience. That’s what makes a liver cell different from a skin cell, even though they presumably both have the very same sequence of DNA.
Until recently, reading someone’s DNA required a goodly amount of it: you’d take a blood draw or a cheek swab and pool the DNA from many cells before loading it into the sequencing machine. However, in recent years it has become possible to read the complete sequence of DNA, all three billion or so nucleotides, from individual cells, such as a single skin cell or neuron. With this technique in hand, Christopher Walsh and his coworkers at Boston Children’s Hospital and Harvard Medical School isolated thirty-six individual neurons from three healthy postmortem human brains and then determined the complete genetic sequence for each of them.31 This revealed that no two neurons had exactly the same DNA sequence. In fact, each neuron harbored, on average, about 1,500 single-nucleotide mutations. That’s 1,500 nucleotides out of a total of three billion in the entire genome—a very low rate, but those mutations can have important consequences. For example, one was in a gene that instructs the production of an ion channel protein that’s crucial for electrical signaling in neurons. If this mutation were present in a group of neurons, instead of just one, it could cause epilepsy. Another was in a gene linked to higher incidence of schizophrenia. There’s nothing special about the brain in this regard. Every cell in your body has accumulated mutations, and therefore every cell has a slightly different genome. This phenomenon is called mosaicism, and when it occurs in cells other than sperm and eggs it is called somatic mosaicism. Sometimes somatic mosaicism is obvious. For example, the famous port-wine mark that adorned the head of Soviet leader Mikhail Gorbachev resulted from a spontaneous somatic mutation in a single progenitor cell, which then divided to give rise to a patch of cells forming enlarged blood vessels, thereby darkening that bit of skin.
Life begins as a single cell: a newly fertilized egg with a single genome. During development, both in utero and in early life, cells divide and divide again (figure 5). Early cells are multipotent: a single cell in a sixteen-cell embryo will have its progeny contribute to many different tissues of the body. As time goes on, cells and their offspring can become more restricted in their fate, giving rise to just skin cells or just brain cells, for example. In the end, the body is composed of about thirty-seven trillion cells, all originating from that single fertilized egg. Some cell types, like skin cells, keep dividing throughout your life to replace ones that die. Others, like most neurons, reach a point in early postnatal life where they stop dividing.32 Most somatic mutations that change single nucleotides occur when a cell is not dividing.33 The mutations that happen during cell division tend to be more drastic, involving loss, duplication, or inversion of big chunks of chromosomes or even entire chromosomes.34
When Walsh and his collaborators looked at neurons from the same person, they sometimes found the very same mutation in several neurons. In some cases, that group of neurons was clustered together in the same part of the brain. In other cases, they were spread widely across brain regions. Mutations found in brain cells were also found in single cells from the heart, liver, and pancreas. It is likely that these situations result from early mutations. Later mutations were shared by a few neighboring cells and were less likely to change bodily functions (unless the mutations activated the cell-division pathway that causes cancer), while earlier mutations were shared by many more cells, distributed more widely in the body.
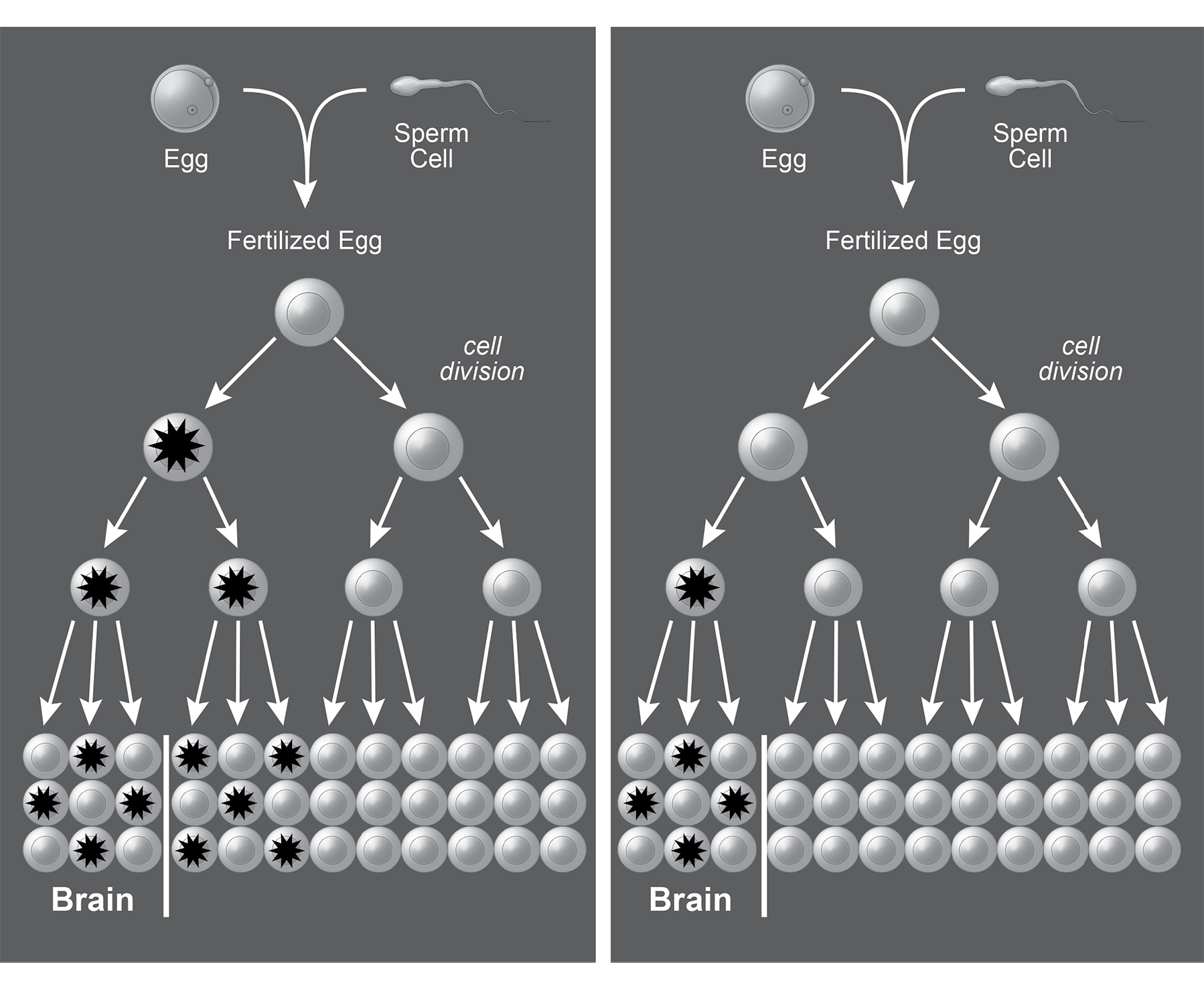
FIGURE 5. A spontaneous random mutation occurring in one cell can be passed to all of its progeny through cell division, resulting in a somatic mosaic. The left panel shows a spontaneous mutation (asterisk) occurring early in development that is passed to cells in various tissues. The right panel shows a mutation occurring somewhat later in development, which is passed to fewer cells and restricted to one organ—in this case, the brain. © 2019 Joan M. K. Tycko. Adapted with permission from Poduri, A. et al. (2013). Somatic mutation, genomic variation, and neurological disease. Science 341, 1237758.
At present, we don’t have a very big data set of fully sequenced individual human neurons, and the ones we have are mostly from autopsy tissue. We know of a few cases where spontaneous somatic mutations have given rise to severe neurological diseases—such as an overgrowth of one cerebral hemisphere, called hemimegalencephaly.35 It’s almost certain that a fraction of heretofore mysterious neurological diseases, like epilepsy of unknown origin, results from spontaneous somatic mutations affecting the electrical function of a group of neurons. It’s also likely that somatic mosaicism contributes to individual differences in cognition or personality that do not rise to the level of disease. Stated another way, a portion of your individuality results from each cell in your body rolling the dice over and over again as you develop, grow, and age. These random changes, because they are in your somatic cells, not your eggs or sperm, will be unique to you and not passed down to your children. This distinction underlies an important point of terminology. The terms “genetic” and “hereditary” are often used interchangeably, but this is incorrect. Somatic mutations are genetic changes but—because they are neither inherited nor passed down to offspring—they are not hereditary.
So, we’re actually a collection of thirty-seven trillion cells, each with a somewhat different genome. That’s pretty hard to imagine. It’s not just that it takes a village to raise a child. Each child is a village—or rather, a huge metropolis—of related but genetically unique individual cells. But it gets even more complicated. Sometimes the metropolis admits immigrants.
IN 1953, LONG BEFORE analysis of DNA was possible, a curious report was published in the British Medical Journal.36
Mrs. McK., a donor aged 25, gave her first pint of blood in March of this year. When the blood came to be grouped, it seemed to be a mixture of A and O cells, for anti-A serum caused large agglutinates to appear to the naked eye, but the microscope showed these agglutinates to be set in a background of unagglutinated cells. The appearance was such as might be seen for a time after a large transfusion of O blood into an A recipient: but Mrs. McK. had never been transfused.
Now here was a mystery. How could Mrs. McK. have two different blood types running through her veins? Careful replication of the result showed that it wasn’t due to simple contamination in the lab. One possible explanation was that Mrs. McK.’s conception resulted from a very rare situation in which a single egg is fertilized by two different sperm cells and develops into a single individual. But such dispermic people always have some degree of asymmetry in their bodies, like one ear noticeably larger than the other or different colored eyes. Mrs. McK. was normally symmetrical. Then, one of the doctors thought to pose an important question. “When asked if she were a twin, Mrs. McK., somewhat surprised, answered that her twin brother had died of pneumonia, 25 years ago, at the age of 3 months.”
The explanation for Mrs. McK.’s double blood type is that the placenta is not a perfect barrier to the flow of cells. Some of her brother’s type-A cells had passed into her body in utero and had replicated and survived for twenty-five years. At that age, about one-third of her blood cells were derived from her twin. When her blood was analyzed in the years to follow, the fraction gradually decreased but was never entirely eliminated—an odd kind of immortality.37
When cells from two different individuals become mixed, it is called chimerism. Recent work has shown chimerism to be widespread.38 In fact, we’re all chimeras, because cells readily pass from mother to fetus. In some cases, the maternal cells are cleared during childhood, but in others, the cells take up residence in a wide variety of organs and can persist for decades. Moving in the other direction, every woman has fetal cells in her body during late pregnancy, and about 75 percent of women still have fetal cells distributed throughout their bodies many years later.39 In one recent study of autopsy tissue, 63 percent of women who had given birth decades earlier (their median age was seventy-five) had fetal cells in their brains.40 It’s worthwhile to note that fetal-to-maternal cell transfer can happen even in the case of miscarriage or abortion. There are women who don’t even realized that they miscarried early in pregnancy, yet are still chimeras from invasion by those early fetal cells.41
Cell transfer across the placenta is a potential source of individuality, but we know very little about how nonself cells function in the body. Those fetal cells in the maternal brain can become electrically active neurons embedded within larger circuits. But it remains unclear if they matter for mental function and behavior. We don’t know whether those invading fetal cells change a woman’s experience of the world. When I was growing up in the 1970s, the mother of a friend liked to drink her coffee out of a mug emblazoned with the phrase “Insanity Is Hereditary: You Get It from Your Kids.” Maybe she was right, but in a different way than she imagined.
Fetal-to-maternal cell transfer can be both harmful and beneficial.42 At least some of the fetal cells that invade the mother’s body are stem cells—undifferentiated cells that can ultimately become any type of cell. In some cases, the mother’s immune system attacks these cells, producing autoimmune diseases like systemic sclerosis, which can damage the mother’s skin, heart, lungs, and kidneys. In other women, the fetal stem cells can sometimes produce miraculous repair.43 In one case study, a mother with a failing thyroid saw spontaneously renewed thyroid function. When a biopsy was performed, the cells of the regenerated thyroid were male, presumably seeded by a stem cell transferred from her son in utero. A similar case was reported for a mother’s spontaneously remitting liver disease, but in that case the regenerating liver cells were from a pregnancy that was terminated.
WE HAVE DISCUSSED SEVERAL ways in which experience, broadly considered, drives individual traits: First, experience-driven regulated gene expression, in which stimuli such as temperature, social interaction, and birth season—acting epigenetically through transcription factors, DNA methylation, and histone modifications—determine which genes are turned on or off at various times in various cells. Second, somatic mosaicism, where random mutations accrue in the (non-sperm, non-egg) cells of your body to change their individual DNA sequence. Third, chimerism, when cells from another person invade your body. Finally, we have discussed the randomness of the development of the body and the brain, from the moment of conception to adulthood, that produces individual variation but is not really experience, in the sense that it is not a process that impinges upon your body from the outside.
Importantly, these are all mechanisms by which genetically identical twins can have divergent traits. Even side by side in the womb, twins do not have identical histories of developmental randomness, somatic mutation, experience, or chimerism. And, of course, the experiences, development, and accumulated somatic mutations of identical twins will continue to diverge after birth.
IN RECENT YEARS, THERE’S been a lot of media attention about a phenomenon that scientists call transgenerational epigenetic inheritance, and which the popular press has mostly called “you can inherit your grandmother’s trauma.” The idea is that if your grandmother (or grandfather) lived through some physically or emotionally traumatic experience, like the 1918 pandemic flu, her trauma could be passed down through epigenetic changes (such as DNA methylation or histone acetylation) to her offspring. These changes would cause her offspring to experience some consequences of trauma (for example, anxiety, overeating, or high blood pressure) and the epigenetic changes could then be passed through the same mechanism down to you. Just to be clear, the mode of transmission here is thought to be epigenetic (modification of the patterns of and timing of gene expression), not genetic (modification of the DNA sequence itself as happens in the mutation and selection of conventional evolutionary change). And the mode of transmission is not merely intergenerational, passing from parent to offspring and then ending, but rather transgenerational, passing through at least two generations.
As of this writing, there are over fifty scientific papers claiming transgenerational epigenetic inheritance in humans. Some of the most cited are a group of reports from the rural Swedish region of Överkalix, which, over the years, has suffered from poor harvest and intermittent famine. These reports found that Överkalix grandsons lived longer if their grandfathers lived through famine in their prepubescent years. But the granddaughters of women who had survived famine had lower life expectancy.44 The authors write, “We conclude that sex-specific, male-line transgenerational responses exist in humans and hypothesize that these transmissions are mediated by the sex chromosomes, X and Y.”
I won’t burden you with the details, but I’m sad to say that there’s not a single one of these fifty-plus epidemiological studies that I find convincing. They tend to suffer from inadequate sample size, poor statistics (which do not correct for multiple comparisons), and hypothesizing only after the results are known.45 The few studies in humans that have sought to actually measure epigenetic marks across generations—for example, in human sperm cells—have suffered from many of the same methodological problems.
For transgenerational epigenetic inheritance to work, the epigenetic modifications of your grandmother’s DNA that occur in her brain to produce anxiety must also be transmitted to her eggs so that they can be passed to the next generation. Then, these marks must somehow act on the brain and body to change expression in specific target cells to reproduce the same behavioral and somatic traits in the next generation. Then, of course, this whole process must happen a second time—from your mother or father to you.
There is no evidence to show that any of these steps occur in humans. The long-standing dogma in developmental biology has been that epigenetic marks on DNA and histone proteins are removed very early in development, at a point where any given cell in the developing embryo has the potential to become any type of cell in the body. Recently, it has been shown that there are a very small number of sites in the mouse genome where these epigenetic marks are not completely erased, and so these could potentially serve as a substrate for transgenerational epigenetic inheritance.46 There are some other non-DNA inheritance mechanisms, involving RNA interference with gene expression, that operate in plants and worms, but they have yet to be demonstrated in mammals, much less in humans.47 At present, I remain unconvinced by claims for human transgenerational epigenetic inheritance. As they say, “Extraordinary claims require extraordinary evidence,” and such extraordinary evidence has not emerged. However, I’m not willing to slam the door on the possibility that such a mode of human inheritance might be convincingly and mechanistically demonstrated in some limited way in the future.
THERE’S SOMETHING IN NATURE that loves individuality, even in a situation that’s designed to squash it. This was demonstrated when Benjamin de Bivort and his colleagues at Harvard University took genetically identical fruit flies and raised them in the lab to have as similar experiences as possible. Then, they placed individual flies in tiny Y-shaped mazes and made videos as they explored.48 Some flies had a noted preference for turning left and others for turning right. On average, there were about the same number of righties as lefties. This wasn’t just a spur-of-the-moment thing. Righty flies preferred to turn right day after day, and the same was true for lefty flies. It wasn’t an artifact of lingering odors, as mutant flies that couldn’t smell also had consistently behaving righties and lefties. When the scientists bred righty flies together, their offspring had, on average, an equal preference for left and right turns. The same thing happened with the progeny of lefty fly pairs. These results indicate that the trait of turn preference is not heritable.49
Then the scientists looked at several different strains of fly, where each individual fly of a single strain was genetically identical but there were genetic differences among strains. All of the strains had nearly identical average turning bias: about 50 percent righty. But some strains of fly had a greater number of individuals with an extreme preference: they would nearly always turn right or nearly always turn left. This means that, while the bias of an individual fly to turn left or right is not heritable, the total amount of variability across the population is determined by genetics. One way to think about this is that there are genes in flies that don’t determine right or left preference, but rather influence something like decisiveness (or maybe, at the risk of anthropomorphizing, stubbornness). The specification of a fly to be a righty or lefty is random, but once that has been set, whether that fly will display a mild or extreme preference is genetically influenced.
This is important because it implies that behavioral individuality is itself a trait that is subject to evolutionary forces. Genes that drive a wide range of individual behaviors can be nature’s way of ensuring that there’s enough variation that a population won’t be entirely wiped out by a catastrophic event.50 If, say, only extreme left turners or extreme shade seekers would survive some drastic perturbation of the environment, then complete annihilation of the group would be avoided if there were a few of these kooky eccentrics left to live and breed another day.