CHAPTER 25
Mood Disorders
Sarah E. Dreyer-Oren, B.A.
Larry D. Mitnaul Jr., M.D., M.P.H., M.S.
Paul E. Holtzheimer III, M.D., M.S.
Mood disorders are characterized by abnormalities of mood and affect regulation, cognitive changes, motor activity alterations, sleep abnormalities, appetite changes, and other disturbances of homeostatic/drive states (e.g., libido, motivation). Although the etiology of mood disorders in most patients is “idiopathic,” neuropsychiatric disorders are commonly associated with disturbances of mood and affect, especially depressive syndromes. A growing literature supports a similar neurobiological basis for mood disorders, regardless of etiology. However, rather than defining a single causative “lesion” or neurochemical abnormality, current models propose an integrated set of distinct but interconnected neural systems underlying the phenomenological features of mood disorders. Each of these systems may be more or less disturbed, resulting in variable symptomatic presentation and treatment response. Mood disorders (major depressive disorder, persistent depressive disorder [dysthymia], and bipolar disorder) are a pervasive and costly public health concern. Mood disorders have a 12-month prevalence rate of about 10% and a lifetime prevalence rate of about 20%. Mental and behavioral disorders, especially major depressive disorder, are the leading cause of disability in the United States and present a significant economic burden nationally and globally.
Current and developing mood disorder treatments are based on the complex interplay of anatomy, biochemistry, and phenomenology of mood disorders. By analogy, the treatments for many neurological disorders, such as Parkinson’s disease (PD), vary depending on the paradigm on which an intervention is based, such as biochemistry (e.g., dopamine agonists, enzyme inhibitors, anticholinergic medication), anatomy, or neurocircuitry (e.g., ablation, deep brain stimulation [DBS]). Similarly, greater acknowledgment of the intricate neurobiology of mood disorders will likely generate many diverse and hopefully effective treatments.
Clinical Features of Mood Disorders
On the basis of the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5; American Psychiatric Association 2013), mood disorders are characterized by the types of mood episodes that occur over the course of the illness. Thus, major depressive disorder (MDD) is categorized by one or more major depressive episodes (MDEs), where as bipolar I disorder is categorized by at least one manic episode and the possible occurrence of depressive or hypomanic episodes.
In DSM-5, prior diagnoses of dysthymia and chronic MDD are now subsumed under persistent depressive disorder. Although criteria for MDD have remained unchanged from DSM-IV (American Psychiatric Association 1994), a specifier, “with mixed features,” has been added to indicate the presence of an MDE with at least three manic symptoms (inadequate to fulfill criteria for a manic episode). Similarly, in bipolar I disorder, the specifier “mixed episode” was discarded in lieu of a new specifier, “with mixed features,” to describe individuals with episodes of hypomania/mania in the presence of depression. Bipolar I disorder no longer requires that individuals’ symptoms simultaneously meet full criteria for both mania and an MDE to be considered “mixed.” For both bipolar and depressive disorders, specifiers were added for “anxious distress.”
This classification of mood disorders is useful for both clinical and research purposes but belies the phenomenological complexity of mood disorders. Two patients with MDD, for example, may present with very different symptoms of an MDE. One may have decreased sleep with early morning awakening, severe psychomotor retardation, profound anhedonia, absence of mood reactivity, and a distinct quality of “depressed” mood that differs from normal sadness (e.g., melancholic features). The other may present with sad mood and atypical features consisting of increased sleep, appetite, and mood reactivity. Although both patients’ symptoms meet criteria for an MDD diagnosis, the manifestations of the two illnesses appear quite distinct and reflect a biological difference between subtypes of depression. When mood disorders manifest in the context of neuropsychiatric conditions (such as PD), they can have even greater variability in presentation, given the overlap between psychiatric and neurological symptoms.
Mood Disorders Associated With Neuropsychiatric Conditions
Correctly diagnosing a mood disorder in the neuropsychiatric patient can be difficult because symptoms of the neurological illness may mimic or mask the symptoms of mood disturbance. Thus, when the clinician is evaluating patients with neuropsychiatric disease, it is important to be vigilant for any and all symptoms of depression, paying particular attention to symptoms less likely to be independently associated with the underlying neurological illness, including mood disturbance, anhedonia, excessive guilt, and suicidality.
In patients with neurological illness, depression may result from the pathophysiology or treatment of the underlying neurological illness, reaction to the psychosocial stress of having the underlying illness, recurrence of a premorbid depressive disorder, or a combination of all of these. Table 25–1 lists common associations of depression with various neuropsychiatric illnesses and treatments. In many patients, the etiology of depression will be multifactorial.
Psychiatric (idiopathic) |
Major depressive disorder |
Bipolar disorder |
Persistent depressive disorder (dysthymia) |
Cyclothymic disorder |
Pharmacological/iatrogenic |
Corticosteroids |
Thyroid ablation |
α-Interferon |
Deep brain stimulation (especially of subthalamic nucleus) |
Substance intoxication/withdrawal |
Neurological |
Basal ganglia disease, especially, |
Parkinson’s disease |
Huntington’s disease |
Wilson’s disease |
Cerebrovascular disease, especially, |
Frontal cortical/subcortical stroke |
Basal ganglia stroke |
Multiple sclerosis |
Infectious encephalitis |
Neoplasm |
Traumatic brain injury |
Dementia |
Epilepsy |
Other |
Hypothyroidism |
Cushing’s syndrome |
Vitamin deficiency (e.g., B12) |
Autoimmune disease |
Neurochemical abnormalities in patients with neurological disease mimic those in patients with idiopathic depression. All three monoamines are disrupted in PD, which is highly associated with depression. Serotonergic dysfunction has also been linked to poststroke depression. Huntington’s disease has been less clearly associated with monoaminergic abnormalities; however, abnormalities of corticotropin-releasing factor and glutamate function have been suggested.
The structural and functional neuroanatomical changes resulting from depression associated with neurological disease also share similarities with those of idiopathic depression. Computed tomography and magnetic resonance imaging (MRI) studies in stroke patients with and without mood disorders have demonstrated a high association of mood changes with infarctions of the left frontal lobe and basal ganglia, the severities of which correlated with lesion proximity to the left frontal pole (Starkstein et al. 1987). Studies of individuals with head trauma, brain tumors, or ablative neurosurgery further suggest that dorsolateral prefrontal lesions, particularly on the left, are associated with depression and depressive-like symptoms.
A number of functional imaging studies of depressed patients with neurological disease have been conducted in order to evaluate depression-associated functional abnormalities without the confound of gross cortical lesions. These studies have typically investigated PD, Huntington’s disease, and lacunar strokes of the basal ganglia—disorders with known or identifiable neurochemical, neurodegenerative, or focal changes that spare frontal cortex (the region repeatedly implicated in idiopathic depression studies). These data have demonstrated that depressed patients with PD have selective hypometabolism involving the caudate nucleus and prefrontal and orbitofrontal cortices. Depressed patients with Huntington’s disease show decreases in paralimbic orbitofrontal and inferior prefrontal cortices (as well as caudate abnormalities inherent to the disease). Patients with depression following stroke also show cortical hypometabolism (Mayberg 1994), suggesting that subcortical lesions can affect function throughout a network of brain regions involved in mood regulation. These data suggest that the depressive syndrome, regardless of etiology, is associated with similar regional brain changes (Figure 25–1).

FIGURE 25–1. Common glucose metabolic positron emission tomography findings in neurological and idiopathic depression.
Decreased prefrontal, dorsal cingulate, and temporal cortical metabolism is a common finding across different depressive syndromes, including patients with Parkinson’s disease, Huntington’s disease, and idiopathic unipolar depression.
Together, imaging data for depressed patients with neurological disease support that cortical, subcortical, and limbic-paralimbic networks are involved in mood regulation. These data also support the notion that disturbance of the network at any critical node can result in behavioral effects and “downstream” activity changes consistent with idiopathic depression.
Neurobiology of Mood Disorders
Neurochemical Findings
Depression has been associated with dysfunction of the monoamine neurotransmitter systems (i.e., serotonin, norepinephrine, dopamine). Growing evidence suggests that the glutamatergic system also plays an important role (Caddy et al. 2014). Of the monoamine neurotransmitter systems, serotonin has received the greatest attention, and there is strong evidence that serotonergic dysfunction plays a major role in the pathophysiology of depression.
Data supporting norepinephrine and dopamine dysfunction in the pathophysiology of depression are more limited but suggest a significant role for these neurotransmitters (Morilak and Frazer 2004). Medications that selectively block norepinephrine reuptake are effective in treating depression, as are bupropion and mirtazapine, which also act on the noradrenergic system. In depressed patients taking noradrenergic antidepressants and euthymic patients with a history of depression, catecholamine depletion can result in depressive relapse. Various studies suggest dopamine transporter activity may be reduced in patients with depression. Medications targeting the dopamine system, such as monoamine oxidase inhibitors (MAOIs), have shown antidepressant efficacy.
Glutamate, an excitatory neurotransmitter, has been implicated in mood disorders, memory, and cognition (Sanacora et al. 2012). A proliferation of studies have explored medication that targets a specific glutamate receptor type, the N-methyl-D-aspartate (NMDA) receptor. In individuals with depression, changes in glutamate level have been identified in brain tissue, cerebrospinal fluid, and plasma concentration. In addition, decreased glutamate and glutamine have been noted in the hippocampus, amygdala, anterior cingulate cortex, left dorsolateral prefrontal cortex, dorsomedial prefrontal cortex, and ventromedial prefrontal cortex of patients with major depression.
Neuroendocrine systems have been implicated in the pathophysiology of mood disorders. The hypothalamic-pituitary-adrenal (HPA) axis is clearly dysfunctional in at least some patients with depression (Pariante and Lightman 2008). Severely depressed patients with prior unsuccessful medication trials may have a hyperactive HPA axis, evidenced by cortisol levels and impaired feedback response using the prednisolone suppression test. Recent reports of alterations in cortisol regulation associated with transient stress in patients with a history of early-life trauma or abuse further suggest that HPA axis dysregulation may be an important marker of vulnerability to various types of affective disorders in later life (Heim and Binder 2012); these data also suggest that HPA axis abnormalities may be causal for certain types of depression rather than the reverse. Corticotropin-releasing factor—the hormone responsible for adrenocorticotropin hormone release—has been shown to be an important modulator of monoaminergic activity.
A growing database supports a role for inflammatory processes in mood disorders, especially depression (Rosenblat et al. 2014). Certain immune mediators and inflammatory markers have been clearly associated with depressive and other mood disorder symptoms. Inflammatory pathways may interact with stress-response systems (i.e., HPA axis) to mediate effects on mood and behavior. Investigations of these targets continue, with many studies exploring the use of anti-inflammatory medications such as acetylsalicylic acid, celecoxib, minocycline, antitumor necrosis factor α agents, curcumin, and omega-3 polyunsaturated fatty acids for mood disorders.
An increasing number of studies have focused on dysregulation of second messenger systems, gene transcription, various neurotrophic factors, and cell turnover in mood disorders. Such pathways have been more extensively studied in bipolar disorder, where medications (e.g., lithium) are known to have effects on these cell-signaling systems (Einat and Manji 2006).
Genetics
The heritability of depression is 33%–50% (Levinson 2006), and the heritability of bipolar disorder may be as high as 80%–90% (McGuffin et al. 2003). When the variability of mood disorders and their complex patterns of inheritance are taken into account, these illnesses likely involve multiple genes and important genetic-environmental interactions.
The genetic literature suggests that depression variability might best be explained by a three-factor model, parsing out the psychomotor/cognitive, mood, and neurovegetative symptom clusters of depression. Data also suggest that specific polymorphisms are associated with distinct personality traits and symptoms of mood disorders, including neuroticism (Heim and Binder 2012) and suicidality (Levinson 2006). Consistent with this concept, several genes involved in monoamine function have been implicated in vulnerability to depression and bipolar disorders. Genetic polymorphisms associated with serotonin transporter inefficiency may contribute to genetic depression vulnerability (see, e.g., Caspi et al. 2003).
With the recognition that monoamine dysfunction cannot fully explain the neurobiology of mood disorders, genes for other neuromodulators are another focus of investigation (Levinson 2006). For example, a functional polymorphism of the promoter region for the brain-derived neurotrophic factor gene may cause susceptibility to mood disorders. Thus, consideration has been given to conceptualizing mood and genetic risk in the context of developmental vulnerability, gene-environment interactions, and epigenetic mechanism (Heim and Binder 2012).
Neuroanatomical Findings
The neuroanatomy of mood disorders has been of interest for more than a century. Advances in structural and functional neuroimaging have allowed increasingly detailed investigation of brain anatomy and have greatly advanced our understanding of how parts of the brain are involved in the pathophysiology of depression.
The most common structural abnormalities associated with depression include decreased volumes of the prefrontal cortex, hippocampus, amygdala, and various basal ganglia structures, although data are inconsistent (Wise et al. 2014). Most studies investigating hippocampal abnormalities have shown that patients with unipolar but not bipolar depression tend to have reduced hippocampal volume. Some research has also shown decreased volume of various systems in the prefrontal cortex. Investigations of the anterior cingulate cortex have likewise been tentatively implicated; although some meta-analyses using region-of-interest measurements show no significant change, voxel-based morphometry meta-analyses show significant volume reduction. Also, variability in the structure of brain regions involved in mood regulation may be related to genetic factors—with polymorphisms of the promoter region for the serotonin transporter gene associated with differences in volume of the subgenual cingulate cortex and amygdala in healthy subjects (Rodríguez-Cano et al. 2014).
Functional neuroimaging research has emphasized the role of a network of brain regions in the pathophysiology of depression. The “default mode network” refers to a set of interconnected neural regions that remain active when subjects are awake and not engaging with any tasks or stimuli (Raichle and Snyder 2007). Several functional MRI (fMRI) studies have supported the idea that depression is associated with increased functional connectivity between the anterior cingulate cortex, as well as other prefrontal cortex structures, and the default mode network (Greicius et al. 2007). Validating these findings, successful treatment for depressive symptoms has been shown to normalize default mode network activity (Dichter et al. 2015). The most common functional neuroanatomical abnormality that is associated with depression is resting state hypometabolism and reduced connectivity between the cortical and limbic systems (Wang et al. 2012). A recent meta-analysis posits that depression is associated with hypometabolism in the superior temporal gyrus and the insula (Chen et al. 2015). However, hyperactivity of the prefrontal cortex has also been reported (Brody et al. 2001). Depression has also been associated with the dysfunction of various subcortical limbic systems (Wang et al. 2012).
Taken together, these data suggest that depression (as a syndrome) is best characterized not by any single functional neuroanatomic abnormality but rather by a pattern of brain activity changes that includes decreased activity in dorsal regions and increased activity in ventral and limbic-paralimbic regions of a mood regulation network. Even in subjects who fail to show this typical pattern, abnormal activity is seen in similar frontal cortical-subcortical brain regions.
Observations of patients undergoing surgery to alleviate treatment-refractory depression provide complementary evidence for this neural systems conceptualization of the depression syndrome (Mayberg et al. 2005). These studies suggest that surgical modulation of a putative mood regulation network (e.g., through tractotomy, vagus nerve stimulation [VNS], or DBS) can alleviate depression—perhaps through “downstream” effects throughout this network.
This multidirectional mood network model has been well supported in recent investigations of emerging treatments for depression. Neuroanatomical targets for mood disorder treatments were determined empirically based on brain imaging data and have been validated by the success of focal stimulation treatment. For example, on the basis of this model, DBS of the subgenual cingulate cortex (Brodmann area 25) was developed and has shown promising antidepressant effects in patients with treatment-resistant depression; these antidepressant effects were associated with systemwide changes in regional brain activity consistent with the effects of combinations of multiple treatments (Holtzheimer et al. 2012). These treatment effects were well maintained in long-term (3-year) follow-up research. From imaging data, researchers hypothesize that DBS’s efficacy lies in the subgenual region’s strong links to structures implicated in mood disorders, including the nucleus accumbens, amygdala, hypothalamus, and prefrontal cortex. This supposition might explain behavioral and neurological evidence that DBS reduces patients’ negative self-bias (Hilimire et al. 2015). In some other, albeit smaller, trials of DBS, stimulation of the ventral capsule/ventral striatum, nucleus accumbens, and medial forebrain bundle/nucleus accumbens have demonstrated antidepressant effect. Repetitive transcranial magnetic stimulation has demonstrated utility in providing antidepressant effect, with the left dorsolateral prefrontal cortex (DLPFC) as the most common target. The DLPFC is implicated in regulating blood-flow response in the anterior cingulate cortex based on prior transcranial magnetic stimulation/positron emission tomography studies (Barrett et al. 2004). Investigations of VNS provide similar evidence that abnormal cerebral blood flow is associated with depression; several studies show that the action of VNS mimics cerebral blood flow effects of antidepressant medications (Conway et al. 2012).
Generally, brain regions implicated in bipolar depression significantly overlap with those identified in unipolar depression (Strakowski et al. 2012); this finding is supported by evidence that patients with unipolar and bipolar depression respond favorably to DBS of the subgenual cingulate cortex and to right-side prefrontal transcranial magnetic stimulation. Interestingly, during mania, activity of prefrontal cortical regions may decrease (as often seen in depression), perhaps suggesting a valence-independent change in cortical activity during mood episodes, although lateralization of decreased activity is dependent on mood state (either manic or depressed). Decrease in subgenual cingulate cortical volume, seen after onset of mania, has also been implicated in bipolar disorder. Abnormalities in limbic structure have also been observed. Some research suggests that abnormally large prefrontal and parahippocampal volumes might predict onset of bipolar disorder, and reduced amygdala volume is consistently associated with bipolar diagnosis.
From these data, it can be concluded that mood disorders cannot be simply explained by a “single lesion” model of regional brain dysfunction (just as no single neurotransmitter abnormality can explain all depressive syndromes). Rather, the functional neuroanatomy of mood disorders involves a diverse set of brain structures, including the prefrontal cortex, anterior cingulate cortex, subgenual cingulate cortex, medial temporal cortex, parietal cortex, hippocampus, and amygdala, as well as subcortical structures, including the ventral striatum, thalamus, hypothalamus, and brain stem. Additionally, there is notable variability in neuroanatomical findings reported to date. Although some of this variability might be explained by inconsistencies in imaging technique and analysis, it is likely that this discordance results from underlying biological heterogeneity across mood disorder patients.
Neurobiology of Symptom Domains
Another approach to the study of mood disorders has been to focus on the neurobiology of specific symptom clusters—based on the presumption that there will be greater homogeneity in the neural basis for a specific symptom than for the larger syndrome (in which symptom manifestation may differ between subjects with the same disorder). In this section, we review symptom clusters relevant to mood disorders and neuropsychiatric conditions, followed by the neurobiological bases of each cluster.
Mood and Affect
Disturbances of mood and affect are fundamental to mood disorder diagnoses. Essentially, every mood disorder requires a specific alteration of mood to justify the diagnosis (with the notable exception of MDD, which allows for no alteration of mood as long as anhedonia is present). Although the terms mood and affect are often used interchangeably, it is useful to make a semantic distinction between the two. Mood is sometimes used to refer to the subjective emotional state experienced by an individual (e.g., the subject feels happy, sad, anxious, numb). Affect is more objective and refers to the individual’s emotional state as it appears to an outside observer (e.g., the subject looks happy, sad, anxious, numb). Importantly, affect can also be defined by the range of emotional states a subject demonstrates (e.g., during an interview), its stability and consistency over time, and how appropriate affect is, given the conversation, stated mood, and so forth. Alternatively, mood may be defined as the sustained, pervasive emotional baseline (the “emotional climate”) within which moment-to-moment changes in emotion, or affect, occur (the “emotional weather”) (American Psychiatric Association 2013; Arciniegas 2013). Within this conceptualization, both mood and affect have objective and subjective components—namely, the expression of emotion and the experience of emotional feeling. This is a critical point discussed in more detail below.
Mood and affective states associated with specific mood episodes often are clinically straightforward to assess, and, typically, mood and affect correspond. Thus, patients diagnosed with depression often describe their mood as “sad,” “blue,” or simply “depressed,” and their affect expresses sadness, sometimes accompanied by restricted emotional range. Similarly, patients with mania or hypomania usually describe elevated moods and often appear excited and euphoric. Patients in mixed episodes often describe their moods as “depressed” or “irritable” but rarely describe elevated moods. However, affect in a mixed episode is often more labile than in depressed patients and may be inappropriate for the situation or stated mood.
Patients with neuropsychiatric disease often present in a manner in which the expression and the experience of emotion appear to be disconnected. For example, a PD patient may deny experiencing persistent and excessive sadness or other symptoms of depression but present with severely restricted range of emotional expression that resembles that often associated with depression. Other neurological patients may present with prominent affective instability, during which their emotional experience is not proportional to the extreme character of their emotional expression (i.e., laughing with little or no feeling of mirth, crying with little or no feeling of sadness) or may even be of a valence contrary to their emotional expression (i.e., laughing while feeling sad, crying while feeling mirth). Such episodes, when uncontrollable, stereotyped, and provoked by sentimentally trivial stimuli, define pathological laughing and crying, also known as pseudobulbar affect or emotional incontinence (Arciniegas 2013). These observed dissociations between mood and affect suggest that mood and affect are controlled by related but distinct neural systems. For a more complete list of the idiopathic and neuropsychiatric etiologies of manic symptoms, see Table 25–2.
Psychiatric (idiopathic) |
Bipolar disorder |
Cyclothymia |
Pharmacological/iatrogenic |
Dopaminergic agents |
Corticosteroids |
Substance intoxication/withdrawal |
Surgical treatments, especially, |
Pallidotomy |
Deep brain stimulation |
Antidepressant medications |
Neurological |
Basal ganglia disease |
Huntington’s disease |
Wilson’s disease |
Cerebrovascular disease |
Frontal cortical/subcortical stroke |
Basal ganglia stroke |
Multiple sclerosis |
Infectious encephalitis |
Neoplasm |
Paraneoplastic syndrome |
HIV encephalopathy |
Epilepsy |
Other |
Cushing’s syndrome |
Vitamin deficiencies (e.g., B12 or niacin) |
Hyperthyroidism |
Systemic infections |
Uremia |
Electrolyte abnormality (e.g., hypocalcemia) |
Neuroanatomical studies have played an important role in elucidating the neurobiology of emotions and emotional regulation. Broadly speaking, it has been shown that emotional processing occurs within neural networks that include predominantly ventral frontal brain structures. In particular, sad mood has been correlated with increased activity in the ventral medial frontal cortex, with several studies implicating Brodmann area 25 of the subgenual cingulate cortex as a critical node in this network. Changes in this region have been associated with antidepressant treatments (Mayberg 2003; Mayberg et al. 2005) (Figure 25–2).

FIGURE 25–2. Imaging data supporting role for Brodmann area (BA) 25 in depression.
(top row) Decreased activity in the subgenual cingulate (BA25; Cg25) is a consistent finding across numerous and diverse treatment studies. (bottom row) Increased subgenual cingulate activity is associated with increased sadness, and functional connectivity of this region during processing of emotional stimuli may be mediated by genetics.
DBS=deep brain stimulation; ECT=electroconvulsive therapy; SERT=serotonin transporter; SSRI=selective serotonin reuptake inhibitor; TMS=transcranial magnetic stimulation.
Source. Adapted from Mayberg 2003.
Suppression of sadness in healthy subjects has been associated with the DLPFC, possibly suggesting a compensatory response in healthy subjects that may be abnormal in depressed patients (where abnormal DLPFC activity is typically observed). Positive emotion is associated with similar frontal, especially prefrontal, brain structures. Temporal lobe structures, especially the amygdala, have also been implicated in emotional processing. Further, cortical-amygdala interactions may be involved in emotional reactivity, with variability between persons explained in part by genetic differences (Hariri et al. 2005). Corroborating evidence that the amygdala is implicated in dysphoria, studies have shown that with successful depression treatment, amygdala hyperactivity associated with depression subsides (Sheline et al. 2001).
Some investigators have suggested the brain has lateralized networks for emotional regulation, such that processing of positive emotions is more strongly associated with left-side brain function and processing of negative emotions is more associated with right-side neural systems. Hemispheric asymmetry is associated with emotion-processing differences in patients with depression: electroencephalogram (EEG) studies show that those with typical (melancholic) depression fail to show right hemispheric dominance (which aids in processing emotionally salient visual stimuli), whereas individuals without depression and those with atypical depression typically have a right hemispheric dominance when processing visual stimuli and linguistic stimuli (Miller et al. 1995). Successful selective serotonin reuptake inhibitor treatment for depression is associated with decreased activation of specific brain regions in response to sad stimuli (Fu et al. 2004). Emotion regulation might also be associated with lateralization; some EEG studies suggest that depressed patients who respond to selective serotonin reuptake inhibitors tend to show greater right hemispheric activation compared with depressed nonresponders (Bruder et al. 2001).
Patients with bipolar disorder tend to be emotionally dysregulated and hyperreactive (Townsend and Altshuler 2012) and show correspondingly higher activity in response to both positive and negative emotional stimuli. Their current mood state further influences emotion regulation; investigators have found that for bipolar patients in depressive episodes, exposure to happy faces induced hyperactivity in the frontal lobe, striatum, and thalamus; for those in manic episodes, exposure to sad faces induced hyperactivity in the fusiform gyrus (Chen et al. 2006).
Disturbance of affect has been associated with dysfunction within the basal ganglia and related structures. PD patients, for example, may demonstrate “depressive” affect in the absence of other depressive symptoms; further, PD patients may have greater difficulty demonstrating and recognizing facial expressions of emotion (Péron et al. 2012).
The neurochemical bases of mood and affect are not clear, although the involvement of monoaminergic systems has been suggested. Acute depletion of tryptophan (resulting in decreased available serotonin) can lead to a recurrence of “depressive symptoms” in vulnerable patients (Faulkner and Deakin 2014), although it is not clear from the literature which depressive symptoms recur. Dopamine function has been strongly associated with positive emotional states.
Interest and Motivation
Mood episodes are commonly associated with disturbances of interest and motivation (i.e., perceived importance of and internal drive to engage with the external world). In fact, a major depressive episode may be diagnosed in the absence of expressed depressed mood if significant anhedonia is present. Individuals with depression, especially melancholic depression, often present with severe anhedonia such that they typically report no pleasure from even highly positive stimuli. Conversely, patients with mania and hypomania often demonstrate an exaggerated level of interest and increased responsiveness to positive stimuli—clinically, this may present as increased goal-directed activity (e.g., vigorous writing, cleaning) or engagement in pleasurable activity regardless of risks (e.g., promiscuity, substance abuse).
In the absence of a mood disorder, however, interest and motivation may also be disturbed. For example, apathy without associated depression is a common symptom associated with PD, Alzheimer’s dementia, and some cases of traumatic brain injury (Cipriani et al. 2014). Further, some neurological patients (often traumatic brain injury, including stroke, patients) demonstrate increased pleasure seeking or disinhibition without having other symptoms associated with mania or hypomania (Starkstein et al. 2004).
Along these lines, it is important to recognize a distinction between anhedonia and apathy. Anhedonia is defined as a decreased ability to experience pleasure, whereas apathy is defined as primarily a diminished drive to engage in goal-directed thought and activity as well as diminished emotion and emotional reactivity.
Studies of interest and motivation strongly implicate function within ventral striatal and cortical systems that are involved in dopamine metabolism. Ventral striatal dopaminergic pathways appear to play a critical role in motivation (Der-Avakian and Markou 2012). Although the constructs of motivation, reward-seeking behavior, and hedonic response are inherently intertwined, studies suggest that dopamine might be more strongly related to motivation-seeking behavior than to hedonic response and reward (Robinson et al. 2005). Compared with healthy control subjects, depressed patients show decreased resting state functional connectivity between the cingulate cortex and striatum, including the caudate nucleus—structures critical to the reward processing system (Bluhm et al. 2009).
In depressed patients, anhedonia has been associated with decreased activity in ventral basal ganglia and ventral prefrontal cortical regions (Keedwell et al. 2005), perhaps related to depressed patients’ attenuated emotional and neural response to positive emotional stimuli.
Studies of apathy (separate from depression and anhedonia) have implicated similar, but somewhat more dorsal prefrontal and subcortical, brain regions, with studies showing decreased activity in the left anterior cingulate and orbitofrontal gyrus and the right inferior and medial frontal gyrus. In the context of neurodegenerative disorders, subtype of dementia influences apathy symptoms: patients with Alzheimer’s disease tend to have apathy in conjunction with dysphoric mood symptoms, but patients with frontotemporal dementia do not (Chow et al. 2009).
In summary, anhedonia and apathy appear to be mediated by overlapping brain regions that primarily include ventral cortical and subcortical areas. These regions largely overlap those involved in mood regulation described above (Der-Avakian and Markou 2012). However, studies show that specific regions may be involved in decreased interest and motivation in the absence of the full depressive syndrome.
Sleep
Sleep is frequently abnormal in patients with mood disorders. Depressed patients often complain of decreased sleep due to difficulty falling asleep (early insomnia), frequent awakenings during the sleep cycle (middle insomnia), or early morning awakening (late insomnia). Other patients describe hypersomnia (common in atypical depression). Manic and hypomanic patients typically report decreased need for sleep—that is, they feel capable of functioning “normally” on little or no sleep at all—in addition to an overall decreased amount of sleep, a measure that correlates with symptom severity. Sleep disturbance in bipolar disorder is common regardless of mood state, although it worsens preceding and during episodic periods. As with disturbances of affect, interest, and motivation, sleep abnormalities in the absence of a mood disorder are commonly found in neuropsychiatric patients, such as those with PD, Huntington’s disease, and dementia (Gagnon et al. 2008).
Sleep physiology has been extensively studied in depressed patients. Sleep EEG abnormalities in depression include prolonged sleep latency, decreased slow-wave sleep, and reduced rapid eye movement (REM) latency with disturbances in the relative time spent in both REM and non-REM sleep. Reduced REM latency is the best studied and most reproducible sleep-related EEG finding in depressed patients, and this abnormality is reversed by most antidepressants. Particularly for those with more variable mood states, sleep deprivation has an effect similar to antidepressant medication, although the rapid, dramatic improvement in depressive symptoms is short-lived. Imaging data have suggested that increased pretreatment activity in the ventral anterior cingulate cortex and ventromedial prefrontal cortex may predict antidepressant response to sleep deprivation (Wu et al. 1999). Electroencephalographic research also suggests that the ventromedial prefrontal cortex in depressed patients is hyperactive during sleep; activity in this region correlates with poor response to treatment and the likelihood of relapse (Broadway et al. 2012). Changes in nocturnal body temperature and attenuation of the normal fluctuations in core body temperature during sleep further suggest a more generalized dysregulation of normal circadian rhythms in patients with depression. To date, however, none of these markers has proven to be specific to depressive disorders, suggesting a neural system underlying sleep and circadian rhythms that is involved in but not specific to mood disorder syndromes. Interestingly, findings in depressed patients with sleep dysregulation suggest genetic vulnerability.
The physiology of sleep disturbances in patients with mania and bipolar depression is less well characterized. Clinically, sleep deprivation is a common precipitant of manic episodes, again suggesting an important biological link between sleep and affective symptoms, with effects on REM measures and sleep continuity similar to those implicated in unipolar depression. Patients with bipolar depression presenting with hypersomnia, however, do not show a consistent reduction in REM latency.
Appetite
In patients with depression, appetite may be decreased, increased, or unchanged. The most common abnormality is a decrease in appetite with corresponding weight loss. However, some patients report increased appetite and weight gain during depressive episodes. In mania, appetite change is not a specific criterion for the disorder, although decreased appetite (or decreased intake) is commonly observed. As with sleep, appetite abnormalities are common in neuropsychiatric disease.
The neurobiology of appetite disturbance in patients with mood disorder and/or neuropsychiatric disease is not well understood. As described above, appetite and weight changes are common in these patients. Also, medications used to treat these conditions (e.g., anticonvulsants, lithium, neuroleptics) have clear effects on appetite, body weight, and metabolism. The regulation of appetite and feeding involves brain regions, including the hypothalamus and amygdala, and neuromodulatory systems, including leptin (a peripheral hormone in central nervous system activity), melanocortin, neuropeptide Y, the HPA axis, and the monoamines (especially dopamine) (Kishi and Elmquist 2005). In particular, depression is hypothesized to disrupt HPA function, particularly the regulation of corticotropin-releasing hormone, which, in turn, reduces appetite.
Psychomotor Activity
Motor and psychomotor deficits in depression include changes in motility, mental activity, and speech. Depressed patients typically report a subjective sense of fatigue and a perceived and observed slowing of thought processes and physical activity. Taken to the extreme, a depressed patient may present with catatonia. Conversely, mania is almost always associated with a dramatic increase in psychomotor speed (often reported as “racing” thoughts and associated with a corresponding increase in activity level [e.g., pressured speech, agitation]). These changes in psychomotor activity (including speech) tend to be state related. Spontaneous motor activity is significantly lower when patients are depressed and not euthymic.
As with emotional state, psychomotor activity has a subjective and objective component. Thus, a depressed patient may “feel” as if he or she has no energy but appear agitated and demonstrate increased physical activity—such a disconnect may be indicative of a mixed mood episode or anxiety. Many neuropsychiatric illnesses are associated with a slowing of thought and motor activity without other symptoms of depression (e.g., PD). Agitation is also a common but nonspecific symptom in neuropsychiatric patients. Although agitation may be indicative of a manic episode or anxiety, it may also arise as a response to pain or as a medication side effect (e.g., akathisia from antipsychotic medications).
Psychomotor abnormalities have largely been linked to monoaminergic neurotransmission and dorsal cortical and subcortical brain activity. Dopamine has been clearly implicated in the neurobiology of psychomotor activity. PD patients have decreased psychomotor activity (in the absence of depression) that clearly improves with dopaminergic therapies, and stimulant medications affecting dopaminergic function are associated with increased psychomotor activity. Decreased dopamine in the basal ganglia has been associated with psychomotor retardation in depressed patients. Dorsolateral prefrontal cortex activity correlates with psychomotor activity in depressed patients, such that decreased blood flow or metabolism in the dorsal prefrontal cortex is associated with psychomotor retardation (Buyukdura et al. 2011).
Emotional Bias
Patients with mood disorders commonly demonstrate mood-congruent emotional bias in cognitive processing. For example, depressed subjects show better recall for negative words and are faster than nondepressed individuals at identifying negative adjectives as self-descriptive (Stuhrmann et al. 2011). In mania, subjects can demonstrate a strong positive emotional bias that presents as grandiosity and overfriendliness.
Neuroticism involves temperamental hypersensitivity to negative stimuli and the tendency to experience exaggerated negative mood states in situations of emotional instability or dissonance. High levels of neuroticism (especially when combined with low levels of extroversion) may indicate a predisposition to developing depression. Using a different model of personality, Cloninger et al. (2006) suggest that personality traits involving negative bias (such as high “harm avoidance” and low “self-directedness”) predict development of depression.
Emotional bias refers to the distorted processing of emotional stimuli. Processing of positive and negative information (such as rewards and punishments) has been linked to the ventral prefrontal cortex, ventral striatum, amygdala, and hippocampus (Stuhrmann et al. 2011) and to dopamine function. Depressed patients have shown abnormal activity in ventral cortical and subcortical brain regions associated with processing of feedback and negative emotional stimuli (George et al. 1997).
In this review, emotional bias is treated as a separate symptom. However, it may be better described as the cognitive processing of emotional stimuli. As such, it is not unreasonable to expect that emotional bias may reflect an interaction of neural systems involved in mood and cognition. For example, older patients with depression have shown slower performance on the emotional Stroop Test (compared with matched control subjects) as well as slower response to negative versus neutral/positive words (a pattern not seen in matched control subjects) (Dudley et al. 2002). Depressed patients, compared with healthy control subjects, show a different functional pattern of frontal-limbic brain activity during the standard and emotional Stroop tasks (George et al. 1997). Negative and positive emotional processing are both implicated in depression, with fMRI data showing that depressed participants, when compared with control subjects, had less activation of the frontotemporal and limbic regions in response to happy words. In response to sad words, depressed participants showed increased activation in the inferior parietal lobe and less activation in the superior temporal gyrus and cerebellum (Canli et al. 2004). However, some research suggests that patients with depression have attenuated neural responses to almost all emotional stimuli, which might explain depressed patients’ inaccuracy in assessing subtle facial expressions and impaired emotional functioning (Stuhrmann et al. 2011).
Suicidal ideation is a type of extreme negative emotional bias. Postmortem brain studies of depressed people who died by suicide report changes in a number of additional serotonin markers, including regional transmitter and metabolite levels, neurotransmitter receptor density, and second messenger and transcription proteins (Arango et al. 2003).
Cognition
The cognitive abnormalities typically seen in depressed patients include slowed thought processes and impaired attention and concentration. Depressed patients also demonstrate impaired executive functioning (e.g., planning, organization, short-term memory). Manic patients show impaired memory encoding, poor concentration and attention, and compromised executive functioning skills in category fluency and mental manipulation and behavioral inhibition (Robinson et al. 2006). Even in the absence of mood disorders, neurological patients commonly show cognitive impairment such that these symptoms may be nonspecific in neuropsychiatric conditions. However, in contrast to deficits associated with many structural neurological disorders, specific impairments in language, perception, and spatial abilities are not usually seen in patients with idiopathic mood disorders (except as a secondary consequence of poor attention, motivation, or organizational abilities). Cognitive deficits in mood disorders are typically of mild to moderate severity but can become quite severe in prolonged or intractable depression—some patients, especially patients with late-life depression, may develop “pseudodementia” (Raskind 1998). Finally, cognitive disturbances may be exacerbated in neurological patients with co-occurring mood disorders.
The neurobiology of cognition has been extensively investigated. Mood disorders primarily disturb cognitive functioning in the dorsal frontal and subcortical brain regions. Executive function, including information organization, strategy planning, and problem solving, as well as executive control of other cognitive functions (e.g., attention, working memory, declarative memory, language), is clearly linked with dorsolateral prefrontal cortical function and tends to be impaired in depression. Depressed patients have shown blunting of an expected left anterior cingulate increase during performance of a cognitive interference task (tests of the Stroop effect). These patients have also shown a corresponding increase in function within the DLPFC (a region not normally recruited during this task) (George et al. 1997), suggesting altered compensatory activity.
Bipolar patients in manic episodes show poor activation in the orbitofrontal cortex during a response inhibition task that reliably increases orbitofrontal activity in nonbipolar control subjects (Altshuler et al. 2005). Disinhibition, a common feature of mania and dementia, has been associated with ventral cortical structures (Starkstein et al. 2004).
Conclusion: A Neural Circuitry Model of Mood Disorders
Considering mood disorders within a neuropsychiatric context highlights that these conditions represent dysfunction within several distinct but interconnected neural circuits (see, e.g., Figure 25–3). An extrapolation is that phenomenological differences between patients with mood disorders might best be explained by differential dysfunction within these circuits. Key features of a neural circuitry model of mood disorders are as follows: 1) network dysfunction (i.e., mood disturbance) may be precipitated by dysfunction at any “critical node” within the network; 2) dysfunction at a critical node will have “upstream” and “downstream” effects that may result in further symptoms; 3) compensatory alterations within the network (in response to dysfunction) may lead to further alterations throughout the network, resulting in further symptomatic manifestation; and 4) successful treatment of network dysfunction may occur through modulation of function at other critical nodes, presuming compensatory responses are intact.
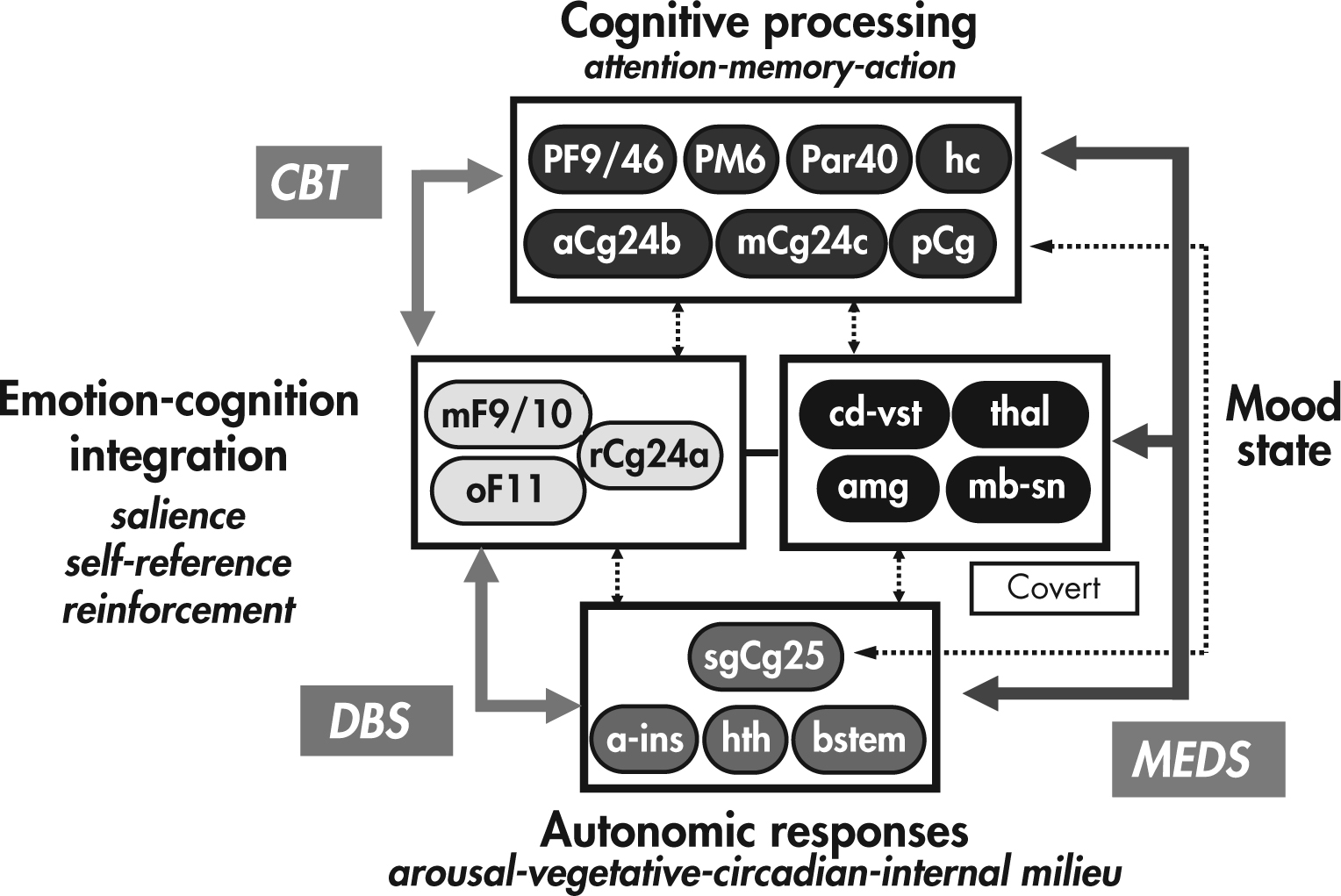
FIGURE 25–3. A proposed model of mood regulation.
Different sets of brain regions are involved in different aspects of mood experience and modulation. Numerous interconnections exist between these different regions, and the system is recognized to be dynamic and potentially modulated at any critical node. Different treatments for mood disorder syndromes may act primarily at different nodes within the system with therapeutic downstream effects.
a-ins=anterior insula; amg=amygdala; aCg24b=Brodmann area 24b/dorsal-perigenual anterior cingulate cortex; bstem=brain stem; CBT=cognitive-behavioral therapy; cd-vst=ventral caudate–ventral striatum; DBS=deep brain stimulation of Brodmann area 25; hc=hippocampus; hth=hypothalamus; mb-sn=midbrain/subthalamic nuclei; mCg24c=Brodmann area 24c/dorsal anterior cingulate cortex; MEDS=antidepressant medications; mF9/10=medial frontal cortex; oF11=orbitofrontal cortex; Par40=dorsal parietal; pCg=posterior cingulate gyrus; PF9/46=dorsolateral prefrontal cortex; PM6=premotor area; rCg24a=Brodmann area 24a/perigenual-subgenual cingulate cortex; sgCg25=Brodmann area 25/subgenual cingulate cortex; thal=thalamus.
Within this model, distinct neural systems are associated with specific functions (and therefore associated with underlying symptom clusters). Cognitive, psychomotor, and sensorimotor processing is associated with dorsal prefrontal, dorsal anterior cingulate, and parietal and posterior cingulate cortices, as well as hippocampus. Medial frontal, orbitofrontal, and perigenual anterior cingulate cortices are associated with overt cognitive processing of emotional stimuli, including salience, reward value, and self-relevance. More covert/masked cognitive-emotional processing is associated with medial temporal and subcortical regions, including the amygdala, ventral basal ganglia, and midbrain structures/nuclei. The brain regions involved in homeostatic/drive processes (e.g., sleep, appetite), as well as body state representation (i.e., the physical aspects of emotional experience), include the subcallosal anterior cingulate cortex, anterior insula, and hypothalamus. Brain stem nuclei are also included in these regions, although it is recognized that monoaminergic projections from these nuclei influence function throughout the entire network.
Emotional-behavioral states (e.g., depression) are then understood to be associated with alteration within several distinct but overlapping neural circuits, with symptomatic presentation corresponding to the direction and degree of dysfunction within each subnetwork. The source of dysfunction may vary (e.g., between idiopathic and neurologically related depression), but the neural systems involved are the same. It is further hypothesized that treatments with different primary mechanisms of action directly alter network activity at distinct nodes. Efficacy is then determined by how adequately the treatment site of action matches the source of dysfunction and/or the compensatory activity of the network. For example, certain “first-line” treatments, such as serotonergic antidepressant medications and cognitive-behavioral therapy (CBT), may have different primary sites of action within the network (frontal cortex for CBT and midbrain-subcortical regions for medications) but rely on intact connections between various regions of the circuit and the ability of these connected regions to respond appropriately (i.e., changes in midbrain-subcortical regions with medications must be able to result in downstream functional changes in frontal cortex, and vice versa for CBT). Similarly, poor adaptive capacity within the network may underlie lack of response to common treatments and explain why progressively more aggressive treatments (such as electroconvulsive therapy and surgery) are needed to ameliorate symptoms.
References
Altshuler LL, Bookheimer SY, Townsend J, et al: Blunted activation in orbitofrontal cortex during mania: a functional magnetic resonance imaging study. Biol Psychiatry 58(10):763–769, 2005 16310510
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th Edition. Washington, DC, American Psychiatric Association, 1994
American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 5th Edition. Arlington, VA, American Psychiatric Association, 2013
Arango V, Huang YY, Underwood MD, et al: Genetics of the serotonergic system in suicidal behavior. J Psychiatr Res 37(5):375–386, 2003 12849930
Arciniegas DB: Emotion, in Behavioral Neurology and Neuropsychiatry. Edited by Arciniegas DB, Anderson CA, Filley CM. Cambridge, UK, Cambridge University Press, 2013, pp 266–298
Barrett J, Della-Maggiore V, Chouinard PA, et al: Mechanisms of action underlying the effect of repetitive transcranial magnetic stimulation on mood: behavioral and brain imaging studies. Neuropsychopharmacology 29(6):1172–1189, 2004 15029151
Bluhm R, Williamson P, Lanius R, et al: Resting state default-mode network connectivity in early depression using a seed region-of-interest analysis: decreased connectivity with caudate nucleus. Psychiatry Clin Neurosci 63(6):754–761, 2009 20021629
Broadway JM, Holtzheimer PE, Hilimire MR, et al: Frontal theta cordance predicts 6-month antidepressant response to subcallosal cingulate deep brain stimulation for treatment-resistant depression: a pilot study. Neuropsychopharmacology 37(7):1764–1772, 2012 22414813
Brody AL, Saxena S, Mandelkern MA, et al: Brain metabolic changes associated with symptom factor improvement in major depressive disorder. Biol Psychiatry 50(3):171–178, 2001 11513815
Bruder GE, Stewart JW, Tenke CE, et al: Electroencephalographic and perceptual asymmetry differences between responders and nonresponders to an SSRI antidepressant. Biol Psychiatry 49(5):416–425, 2001 11274653
Buyukdura JS, McClintock SM, Croarkin PE: Psychomotor retardation in depression: biological underpinnings, measurement, and treatment. Prog Neuropsychopharmacol Biol Psychiatry 35(2):395–409, 2011 21044654
Caddy C, Giaroli G, White TP, et al: Ketamine as the prototype glutamatergic antidepressant: pharmacodynamic actions, and a systematic review and meta-analysis of efficacy. Ther Adv Psychopharmacol 4(2):75–99, 2014 24688759
Canli T, Sivers H, Thomason ME, et al: Brain activation to emotional words in depressed vs healthy subjects. Neuroreport 15(17):2585–2588, 2004 15570157
Caspi A, Sugden K, Moffitt TE, et al: Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301(5631):386–389, 2003 12869766
Chen CH, Lennox B, Jacob R, et al: Explicit and implicit facial affect recognition in manic and depressed States of bipolar disorder: a functional magnetic resonance imaging study. Biol Psychiatry 59(1):31–39, 2006 16112653
Chen ZQ, Du MY, Zhao YJ, et al: Voxel-wise meta-analyses of brain blood flow and local synchrony abnormalities in medication-free patients with major depressive disorder. J Psychiatry Neurosci 40(6):401–411, 2015 25853283
Chow TW, Binns MA, Cummings JL, et al: Apathy symptom profile and behavioral associations in frontotemporal dementia vs dementia of Alzheimer type. Arch Neurol 66(7):888–893, 2009 19597092
Cipriani G, Lucetti C, Danti S, et al: Apathy and dementia: nosology, assessment and management. J Nerv Ment Dis 202(10):718–724, 2014 25265266
Cloninger CR, Svrakic DM, Przybeck TR: Can personality assessment predict future depression? a twelve-month follow-up of 631 subjects. J Affect Disord 92(1):35–44, 2006 16442638
Conway CR, Sheline YI, Chibnall JT, et al: Brain blood-flow change with acute vagus nerve stimulation in treatment-refractory major depressive disorder. Brain Stimulat 5(2):163–171, 2012 22037127
Der-Avakian A, Markou A: The neurobiology of anhedonia and other reward-related deficits. Trends Neurosci 35(1):68–77, 2012 22177980
Dichter GS, Gibbs D, Smoski MJ: A systematic review of relations between resting-state functional-MRI and treatment response in major depressive disorder. J Affect Disord 172:8–17, 2015 25451389
Dudley R, O’Brien J, Barnett N, et al: Distinguishing depression from dementia in later life: a pilot study employing the Emotional Stroop task. Int J Geriatr Psychiatry 17(1):48–53, 2002 11802230
Einat H, Manji HK: Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry 59(12):1160–1171, 2006 16457783
Faulkner P, Deakin JF: The role of serotonin in reward, punishment and behavioural inhibition in humans: insights from studies with acute tryptophan depletion. Neurosci Biobehav Rev 46(Pt 3):365–378, 2014 25195164
Fu CH, Williams SC, Cleare AJ, et al: Attenuation of the neural response to sad faces in major depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imaging study. Arch Gen Psychiatry 61(9):877–889, 2004 15351766
Gagnon JF, Petit D, Latreille V, et al: Neurobiology of sleep disturbances in neurodegenerative disorders. Curr Pharm Des 14(32):3430–3445, 2008 19075719
George MS, Ketter TA, Parekh PI, et al: Blunted left cingulate activation in mood disorder subjects during a response interference task (the Stroop). J Neuropsychiatry Clin Neurosci 9(1):55–63, 1997 9017529
Greicius MD, Flores BH, Menon V, et al: Resting-state functional connectivity in major depression: abnormally increased contributions from subgenual cingulate cortex and thalamus. Biol Psychiatry 62(5):429–437, 2007 17210143
Hariri AR, Drabant EM, Munoz KE, et al: A susceptibility gene for affective disorders and the response of the human amygdala. Arch Gen Psychiatry 62(2):146–152, 2005 15699291
Heim C, Binder EB: Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp Neurol 233(1):102–111, 2012 22101006
Hilimire MR, Mayberg HS, Holtzheimer PE, et al: Effects of subcallosal cingulate deep brain stimulation on negative self-bias in patients with treatment-resistant depression. Brain Stimulat 8(2):185–191, 2015 25499035
Holtzheimer PE, Kelley ME, Gross RE, et al: Subcallosal cingulate deep brain stimulation for treatment-resistant unipolar and bipolar depression. Arch Gen Psychiatry 69(2):150–158, 2012 22213770
Keedwell PA, Andrew C, Williams SC, et al: The neural correlates of anhedonia in major depressive disorder. Biol Psychiatry 58(11):843–853, 2005 16043128
Kishi T, Elmquist JK: Body weight is regulated by the brain: a link between feeding and emotion. Mol Psychiatry 10(2):132–146, 2005 15630408
Levinson DF: The genetics of depression: a review. Biol Psychiatry 60(2):84–92, 2006 16300747
Mayberg HS: Frontal lobe dysfunction in secondary depression. J Neuropsychiatry Clin Neurosci 6(4):428–442, 1994 7841814
Mayberg HS: Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull 65:193–207, 2003 12697626
Mayberg HS, Lozano AM, Voon V, et al: Deep brain stimulation for treatment-resistant depression. Neuron 45(5):651–660, 2005 15748841
McGuffin P, Rijsdijk F, Andrew M, et al: The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry 60(5):497–502, 2003 12742871
Miller EN, Fujioka TA, Chapman LJ, et al: Psychometrically matched tasks for assessment of hemispheric asymmetries of function. Brain Cogn 28(1):1–13, 1995 7546665
Morilak DA, Frazer A: Antidepressants and brain monoaminergic systems: a dimensional approach to understanding their behavioural effects in depression and anxiety disorders. Int J Neuropsychopharmacol 7(2):193–218, 2004 15003145
Pariante CM, Lightman SL: The HPA axis in major depression: classical theories and new developments. Trends Neurosci 31(9):464–468, 2008 18675469
Péron J, Dondaine T, Le Jeune F, et al: Emotional processing in Parkinson’s disease: a systematic review. Mov Disord 27(2):186–199, 2012 22162004
Raichle ME, Snyder AZ: A default mode of brain function: a brief history of an evolving idea. Neuroimage 37(4):1083–1090; discussion 1097–1089, 2007 17719799
Raskind MA: The clinical interface of depression and dementia. J Clin Psychiatry 59 (suppl 10):9–12, 1998 9720476
Robinson LJ, Thompson JM, Gallagher P, et al: A meta-analysis of cognitive deficits in euthymic patients with bipolar disorder. J Affect Disord 93(1–3):105–115, 2006 16677713
Robinson S, Sandstrom SM, Denenberg VH, et al: Distinguishing whether dopamine regulates liking, wanting, and/or learning about rewards. Behav Neurosci 119(1):5–15, 2005 15727507
Rodríguez-Cano E, Sarró S, Monté GC, et al: Evidence for structural and functional abnormality in the subgenual anterior cingulate cortex in major depressive disorder. Psychol Med 44(15):3263–3273, 2014 25066663
Rosenblat JD, Cha DS, Mansur RB, et al: Inflamed moods: a review of the interactions between inflammation and mood disorders. Prog Neuropsychopharmacol Biol Psychiatry 53:23–34, 2014 24468642
Sanacora G, Treccani G, Popoli M: Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62(1):63–77, 2012 21827775
Sheline YI, Barch DM, Donnelly JM, et al: Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry 50(9):651–658, 2001 11704071
Starkstein SE, Robinson RG, Price TR: Comparison of cortical and subcortical lesions in the production of poststroke mood disorders. Brain 110(Pt 4):1045–1059, 1987 3651794
Starkstein SE, Garau ML, Cao A: Prevalence and clinical correlates of disinhibition in dementia. Cogn Behav Neurol 17(3):139–147, 2004 15536301
Strakowski SM, Adler CM, Almeida J, et al: The functional neuroanatomy of bipolar disorder: a consensus model. Bipolar Disord 14(4):313–325, 2012 22631617
Stuhrmann A, Suslow T, Dannlowski U: Facial emotion processing in major depression: a systematic review of neuroimaging findings. Biol Mood Anxiety Disord 1(1):10, 2011 22738433
Townsend J, Altshuler LL: Emotion processing and regulation in bipolar disorder: a review. Bipolar Disord 14(4):326–339, 2012 22631618
Wang L, Hermens DF, Hickie IB, et al: A systematic review of resting-state functional-MRI studies in major depression. J Affect Disord 142(1–3):6–12, 2012 22858266
Wise T, Cleare AJ, Herane A, et al: Diagnostic and therapeutic utility of neuroimaging in depression: an overview. Neuropsychiatr Dis Treat 10:1509–1522, 2014 25187715
Wu J, Buchsbaum MS, Gillin JC, et al: Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulate and medial prefrontal cortex. Am J Psychiatry 156(8):1149–1158, 1999 10450253