17
Software Tools for Modeling
- 17.1 13C-Flux2
- 17.2 Antimony
- 17.3 Berkeley Madonna
- 17.4 BIOCHAM
- 17.5 BioNetGen
- 17.6 Biopython
- 17.7 BioTapestry
- 17.8 BioUML
- 17.9 CellDesigner
- 17.10 CellNetAnalyzer
- 17.11 Copasi
- 17.12 CPN Tools
- 17.13 Cytoscape
- 17.14 E-Cell
- 17.15 EvA2
- 17.16 FEniCS Project
- 17.17 Genetic Network Analyzer (GNA)
- 17.18 Jarnac
- 17.19 JDesigner
- 17.20 JSim
- 17.21 KNIME
- 17.22 libSBML
- 17.23 MASON
- 17.24 Mathematica
- 17.25 MathSBML
- 17.26 Matlab
- 17.27 MesoRD
- 17.28 Octave
- 17.29 Omix Visualization
- 17.30 OpenCOR
- 17.31 Oscill
- 17.32 PhysioDesigner
- 17.33 PottersWheel
- 17.34 PyBioS
- 17.35 PySCeS
- 17.36 R
- 17.37 SAAM II
- 17.38 SBMLeditor
- 17.39 SemanticSBML
- 17.40 SBML-PET-MPI
- 17.41 SBMLsimulator
- 17.42 SBMLsqueezer
- 17.43 SBML Toolbox
- 17.44 SBtoolbox2
- 17.45 SBML Validator
- 17.46 SensA
- 17.47 SmartCell
- 17.48 STELLA
- 17.49 STEPS
- 17.50 StochKit2
- 17.51 SystemModeler
- 17.52 Systems Biology Workbench
- 17.53 Taverna
- 17.54 VANTED
- 17.55 Virtual Cell (VCell)
- 17.56 xCellerator
- 17.57 XPPAUT
- Exercises
- References
The databases described in Chapter 16 are huge repositories for the biological data that have been gathered by various techniques. The information in the databases represents raw material for most types of modeling efforts. Modeling tools help us to formulate theoretical ideas and hypothesis and to extract information relevant to these hypotheses from the raw material stored in the databases. Chapter 5 provided a first overview of the most popular modeling tools and data formats. However, there are many more. To provide the reader with a rough overview of the plethora of other tools that exist and that could not be discussed, we have included here a compendium that is based on reviews and surveys from the literature [1,2], as well as various Internet sources. The short description also provides information about the availability, native operating system, and the time when the last update was released (as of May 2015). Tools that have not been updated since 2010 are not included in this list, since that is a strong indication that the active development and maintenance has stopped. Although the compendium contains more than 50 tools, we realize that this cannot be an exhaustive list since the development and usage of software tools is a very dynamic area. Furthermore, because of the large number of programs, we don't claim to be experts for all the programs described here. The description is thus either based on our own experience or paraphrased from the information given at the respective website. We therefore apologize in advance for personal bias and ignorance that entered into the construction of the compendium.
17.1 13C-Flux2
13C-FLUX2 is a high-performance simulator for  -base metabolic flux analysis. All complex interactions between the cellular networks of genes, transcripts, proteins, and metabolites finally result in metabolic fluxes. However, in vivo fluxes are not directly observable and have to be inferred by data from labeling experiments and the use of mathematical models.
-base metabolic flux analysis. All complex interactions between the cellular networks of genes, transcripts, proteins, and metabolites finally result in metabolic fluxes. However, in vivo fluxes are not directly observable and have to be inferred by data from labeling experiments and the use of mathematical models. 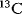 -based metabolic flux analysis emerged as the state-of-the-art technique in the field of fluxomics. 13C-FLUX2 is a software suite of applications for the detailed quantification of intracellular steady-state fluxes.
-based metabolic flux analysis emerged as the state-of-the-art technique in the field of fluxomics. 13C-FLUX2 is a software suite of applications for the detailed quantification of intracellular steady-state fluxes.
Latest version: 13C-FLUX2, January 2013
| Operating system: | Windows □ | Mac OS □ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic ⊠ | Commercial: ⊠ |
17.2 Antimony
Antimony is a text-based model definition language originally based on Jarnac, and extended to be fully modular. Antimony models can be converted to and from SBML, flattening the modularity in the process. Antimony was designed as a successor to Jarnac's model definition language, with some new features that mesh with newer elements of SBML. A programming library, libAntimony, exists in tandem with the language to allow computer translation of Antimony-formatted models into SBML and other formats.
URL: antimony.sourceforge.net/
Latest version: Antimony 2.8, October 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux □ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.3 Berkeley Madonna
Berkeley Madonna is a fast, general-purpose differential and difference equations solver. It is also possible to perform discrete simulations using conveyors, ovens, and queues. Furthermore, (bio)chemical reactions can be entered in a special shorthand syntax (e.g., A + 2B ⇔ C + D) and Berkeley Madonna can automatically find parameter values that minimize the deviation between the model output and a given data set (i.e., parameter fitting). Developed on the Berkeley campus under the sponsorship of NSF and NIH, it is currently used by academic and commercial institutions for constructing mathematical models for research and teaching. The number crunching engine of Berkeley Madonna was originally written in C, and later extended with the Flowchart graphical interface written in Java. Currently, a version of Berkeley Madonna, called JMadonna, is in development. It will have the user interface written in Java, while retaining the simulation engine in C for speed. Also, a Linux version of JMadonna is planned. Student licenses are available at reduced prices as well as a demo version with restricted features (e.g., no saving).
Latest version: Berkeley Madonna 8.3, 2010
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux □ |
| Availability: | Free for all □ | Free for academic □ | Commercial: ⊠ |
17.4 Biocham
BIOCHAM (Biochemical Abstract Machine) is a programming environment for modeling biochemical systems, performing simulations and querying the model in temporal logic. It has a simulator for Boolean, stochastic, and differential models that can be accessed via a GUI. It has its own rule-based modeling language, which is compatible with SBML. The stand-alone versions as well as a Web-based version come with various example models.
URL: contraintes.inria.fr/Biocham
Latest version: BIOCHAM 3.7, July 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.5 BioNetGen
BioNetGen is a tool for automatically generating mathematical models of biological systems from user-specified rules for molecular interactions. Rules are specified in the BioNetGen language (BNGL), which enables precise, visual, and extensible representation of molecular interactions. The language was designed with protein–protein interactions in mind. A user can explicitly indicate the parts of proteins involved in an interaction, the conditions upon which an interaction depends, the connectivity of proteins in a complex, and other aspects of protein–protein interactions. It is one of the few software tools available for generating physicochemical models of systems marked by combinatorial complexity. Apart from the stand-alone versions for the different operating systems, a Web-based version is also available.
URL: www.bionetgen.org
Latest version: BioNetGen 2.2.6, June 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.6 Biopython
Biopython is a Python package that provides a programmatic interface to a large number of bioinformatics databases and tools. Some functions (e.g., downloading and analyzing of nucleotide and protein sequences) can be performed by Biopython alone; for others (“blasting” sequences or calculating phylogenetic trees), the necessary programs have to be installed and Biopython just provides a convenient interface. Reading and writing of DNA, RNA, or protein sequences is, of course, a central feature of Biopython using the subpackages Bio.Seq, Bio.SeqRecord and Bio.SeqIO. Further routines are available to align (Bio.AlignIO) or blast (Bio.Blast.NCBIWWW) sequences. Proteins can be searched in SwissProt (Bio.ExPASy, Bio.SwissProt) and the PDB database for 3D structures (Bio.PDB). It is also possible to cluster sequences (Bio.Cluster) or to construct phylogenetic trees (Bio.Phylo). Finally, the Bio.Entrez module provides routines to search the NCBI sequence databases, but also literature records in PubMed. A detailed description of all the modules is available at http://biopython.org/DIST/docs/tutorial/Tutorial.html.
URL: biopython.org
Latest version: BioPython 1.66, October 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.7 BioTapestry
BioTapestry is an interactive tool for building, visualizing, and simulating genetic regulatory networks. BioTapestry is designed around the concept of a developmental network model, and is intended to deal with large-scale models with consistency and clarity. It is capable of representing systems that exhibit increasing complexity over time, such as the genetic regulatory network controlling endomesoderm development in sea urchin embryos. BioTapestry has the ability to create and handle sets of submodels, which is helpful for structuring large networks in a readable way. These models can be exported in SBML format for dynamic simulation in other tools.
URL: www.biotapestry.org
Latest version: BioTapestry 7, September 2014
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.8 BioUML
BioUML is an open-source integrated Java platform that spans a comprehensive range of capabilities, including access to databases with experimental data, tools for formalized description of biological systems, structure, and functioning, and tools for their visualization, simulation, parameters fitting, and analyses. Due to scripts (R, JavaScript) and workflow support, it provides powerful possibilities for the analyses of high-throughput data. The plug-in-based architecture allows adding new functionality using plug-ins. The system consists of the BioUML server and the BioUML workbench, which can work in a stand-alone mode or in connection with the server. Models written in SBML, CellML, or BioPAX can be used.
URL: www.biouml.org
Latest version: BioUML 0.9.6, November 2013
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.9 CellDesigner
CellDesigner is a structured diagram editor for drawing gene regulatory and biochemical networks. Networks are drawn based on the Systems Biology Graphical Notation (SBGN) and stored using the Systems Biology Markup Language, a standard for representing models of biochemical and gene regulatory networks. CellDesigner can connect to various databases to download models, reaction parameters, and literature information. Models can also be simulated using an internal ODE solver or via a connection to the Copasi solver. Furthermore, models can also be analyzed via a link to the Systems Biology Workbench (SBW). A more in-depth description of CellDesigner is given in Chapter 5.
URL: celldesigner.org
Latest version: CellDesigner 4.4, July 2014
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.10 CellNetAnalyzer
CellNetAnalyzer is a package for Matlab and provides a comprehensive and user-friendly environment for structural and functional analyses of biochemical and cellular networks. CellNetAnalyzer facilitates the analysis of metabolic as well as signaling and regulatory networks solely on their network topology, that is, independent of kinetic mechanisms and parameters. The core concept of visualization and interactivity is realized by interactive network maps where the abstract network model is linked with network graphics. CellNetAnalyzer provides a powerful collection of tools and algorithms for structural network analysis, which can be started in a menu-controlled manner within the interactive network maps. API functionalities have been added to enable interested users to call algorithms of CellNetAnalyzer from external programs.
URL: http://www.mpi-magdeburg.mpg.de/projects/cna/cna.html
Latest version: CellNetAnalyzer 2015.1
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic ⊠ | Commercial: □ |
17.11 Copasi
Copasi (Complex Pathway Simulator) is an application for the simulation and analysis of biochemical networks. It features stochastic and deterministic time course simulation, steady-state analysis, metabolic control analysis (MCA), parameter scans, optimization of arbitrary target functions, and parameter estimation. Copasi provides means to visualize data in customizable plots, histograms, and animations of network diagrams. Models are normally saved in Copasis own format, but import/export of SBML is supported up to Level 3 Version 1. A more detailed description of Copasi is given in Chapter 5.
URL: www.copasi.org
Latest version: Copasi 4.16 (Build 104), August 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.12 CPN Tools
CPN Tools is a tool for editing, simulating, and analyzing Colored Petri Nets as well as noncolored nets. The tool features incremental syntax checking and code generation while a net is being constructed. A fast simulator efficiently handles both untimed and timed nets. Full and partial state spaces can be generated and analyzed, and a standard state space report contains information such as boundedness properties and liveness properties. Models can be saved in the Petri Net Markup Language (PNML).
URL: cpntools.org
Latest version: CPN Tools 4.0.1, February 2015
| Operating system: | Windows ⊠ | Mac OS □ | Linux □ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.13 Cytoscape
Cytoscape is an open-source software platform for visualizing molecular interaction networks and biological pathways as well as integration of these networks with annotations, gene expression profiles, and other state data. Although Cytoscape was originally designed for biological research, now it is a general platform for complex network analysis and visualization. The Cytoscape core distribution provides a basic set of features for data integration, analysis, and visualization. Additional features are available as Apps (formerly called Plugins). Apps are available for network and molecular profiling analyses, new layouts, additional file format support, scripting, and connection with databases. They may be developed by anyone using the Cytoscape open API based on Java and App development is welcomed and encouraged. Most of the Apps are freely available from Cytoscape App Store.
URL: www.cytoscape.org
Latest version: Cytoscape 3.3.0, November 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.14 E-Cell
The E-Cell Project develops general technologies and theoretical support for computational biology with the grand aim to make precise whole-cell simulation at the molecular level possible. Apart from modeling methodologies, formalisms, and techniques, the project developed the E-Cell System, a software platform for modeling, simulation, and analysis of complex, heterogeneous, and multiscale systems like the cell. The source code for E-Cell is available at GitHub (github.com/).
URL: www.e-cell.org
Latest version: E-Cell4, March 2015
| Operating system: | Windows ⊠ | Mac OS □ | Linux □ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.15 EvA2
EvA2 (an Evolutionary Algorithms framework, revised version 2) is a comprehensive heuristic optimization framework with emphasis on Evolutionary Algorithms implemented in Java. EvA2 integrates several derivation-free optimization methods, preferably population based, such as Evolution Strategies (ES), Genetic Algorithms (GA), Differential Evolution (DE), Particle Swarm Optimization (PSO), and classical techniques such as multistart Hill Climbing or Simulated Annealing. Besides typical single-objective problems, multimodal and multiobjective problems are handled directly by the EvA2 framework. Via the Java mechanism of Remote Method Invocation (RMI), the algorithms of EvA2 can be distributed over network nodes based on a client–server architecture. EvA2 aims at two groups of users: first, the end user who does not know much about the theory of evolutionary algorithms but wants to use it to solve an application problem; second, the scientific user who wants to investigate the performance of different optimization algorithms or wants to compare the effect of alternative or specialized evolutionary or heuristic operators. The latter usually knows more about evolutionary algorithms or heuristic optimization and is able to extend EvA2 by adding specific optimization strategies or solution representations. The software consists of a workbench GUI to construct and control the optimization problem and the number crunching core.
URL: http://www.ra.cs.uni-tuebingen.de/software/JavaEvA/
Latest version: EvA 2.1.0, September 2013
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.16 FEniCS Project
The FEniCS Project is a collection of free software tools with an extensive list of features for automated, efficient solution of partial differential equations by finite element methods. FEniCS has an extensive list of features for automated, efficient solution of differential equations, including automated solution of variational problems, automated error control and adaptivity, a comprehensive library of finite elements, high-performance linear algebra, and many more. FEniCS is organized as a collection of interoperable components that together form the FEniCS Project. These components include the problem-solving environment DOLFIN, the form compiler FFC, the finite element tabulator FIAT, the just-in-time compiler Instant, the code generation interface UFC, and the form language UFL. DOLFIN is a c++/Python library that functions as the main user interface of FEniCS. A large part of the functionality of FEniCS is implemented as part of DOLFIN. It provides a problem-solving environment for models based on partial differential equations and implements core parts of the functionality of FEniCS, including data structures and algorithms for computational meshes and finite element assembly. To provide a simple and consistent user interface, DOLFIN wraps the functionality of other FEniCS components and external software, and handles the communication between these components.
URL: fenicsproject.org/
Latest version: FEniCS 1.6.0, July 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.17 Genetic Network Analyzer (GNA)
Genetic Network Analyzer (GNA) is a computer tool for the modeling and simulation of genetic regulatory networks. The aim of GNA is to assist biologists and bioinformaticians in constructing a model of a genetic regulatory network using knowledge about regulatory interactions in combination with gene expression data. Genetic Network Analyzer consists of a simulator of qualitative models of genetic regulatory networks in the form of piecewise linear differential equations. Instead of exact numerical values for the parameters, which are often not available for networks of biological interest, the user of GNA specifies inequality constraints. This information is sufficient to generate a state transition graph that describes the qualitative dynamics of the network. The simulator has been implemented in Java and has been applied to the analysis of various regulatory systems, such as the networks controlling the initiation of sporulation in Bacillus subtilis and the carbon starvation response in Escherichia coli.
URL: ibis.inrialpes.fr/article122.html
Latest version: GNA 8.5.0.1, December 2013
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic ⊠ | Commercial: □ |
17.18 Jarnac
Jarnac is the numerical solver that is part of the Systems Biology Workbench (SBW). There it is used with the network designer JDesigner, but it can also import SBML files produced by other tools. In addition, Jarnac can also be used as a stand-alone tool with its own language for describing reaction networks. This simple control language is similar to the Basic language and supports many of the constructs one would expect, for loops, conditionals, while/do, and repeat/until. It supports several different data types, including integers, floats, Booleans, strings, vectors, matrices, and lists. Jarnac also supports user-defined functions and external modules. There is built-in computational support for dynamic simulation (using LSODA or CVODE integrator), steady-state analysis using the NLEQ solver, simple stability analysis (eigenvalues, using IMSL library), matrix arithmetic (all the main operators, including transpose, det, etc., using IMSL library), metabolic control analysis (all steady-state control coefficients and elasticities), metabolic structural analysis (null space and conservation relation analysis and others), and stochastic simulation (using standard Gillespie method).
URL: http://sbw-app.org/jarnac/
Latest version: Jarnac 3.32a, May 2013
| Operating system: | Windows ⊠ | Mac OS □ | Linux □ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.19 JDesigner
Like Jarnac, JDesigner is a component of the Systems Biology Workbench (SBW). It is a visual design tool for biochemical networks and uses SBML as its native file format. SBW has to be installed before JDesigner can be installed. JDesigner runs under MS-Windows and allows users to draw a biochemical network and save it in SBML. JDesigner has an SBW interface that allows it to be called from other SBW compliant modules. In addition, JDesigner has the ability to use Jarnac as a simulation server (via SBW), thus allowing models to be run from within JDesigner. In this mode, JDesigner is both a network design tool and a simulator.
URL: http://sbw-app.org/jdesigner/
Latest version: JDesigner 2.4.7, May 2013
| Operating system: | Windows ⊠ | Mac OS □ | Linux □ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.20 JSim
JSim is a Java-based simulation system for building quantitative numeric models and analyzing them with respect to experimental reference data. JSim's primary focus is on physiology and biomedicine; however, its computational engine is quite general and applicable to a wide range of scientific domains. JSim models may intermix ODEs, PDEs, implicit equations, integrals, summations, discrete events, and procedural code as appropriate. JSim's model compiler can automatically insert conversion factors for compatible physical units as well as detect and reject unit unbalanced equations. JSim models are normally written using its own Mathematical Modeling Language (MML), but JSim also imports flawlessly models written in SBML or CellML and automatically converts them into MML code. Once loaded, models can be simulated and the results graphically displayed as XY, contour, or surface plots. Models can also be analyzed via a sensitivity analysis and automatic parameter optimization to fit a model to experimental data.
Latest version: JSim 2.16, December 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic ⊠ | Commercial: □ |
17.21 Knime
KNIME (Konstanz Information Miner) is a user-friendly graphical workflow management system for the entire data analysis process, including data access, data transformation, initial investigation, predictive analytics, visualization, and reporting. The open integration platform provides over 1000 modules (nodes), including those of the KNIME community and its extensive partner network. The strength of KNIME is not so much in the area of kinetic modeling, but in the graphical creation of workflows for the analysis of data via clustering, classification, descriptive statistics, and visualization. The workbench itself resembles strongly the Eclipse IDE for program development and is centered around a large canvas on which icons (nodes) are dropped and connected to form the data workflow.
URL: www.knime.org/
Latest version: KNIME 3.1, December 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.22 libSBML
libSBML is a library designed to help you read, write, manipulate, translate, and validate SBML files and data streams. It is not an application itself (although it does come with many example programs), but rather a library you can embed in your own applications. libSBML is written in ISO C and C++, but as a library it may be used from many different programming languages such as C/C++, C#, Java, Python, Perl, Ruby, or Matlab. libSBML offers powerful features such as reading/writing compressed SBML files, support for SBML Level 3 packages, detecting overconstrained models, checking units, an API for SBML <annotation> content, and support for the three most popular XML parser libraries: Xerces, Expat, and libxml2.
URL: http://sbml.org/Software/libSBML
Latest version: libSBML 5.12.0, November 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.23 MASON
MASON is a fast Java library for the construction of multiagent simulations and designed to be the foundation for large custom-purpose Java simulations. MASON contains both a model library and an optional suite of visualization tools in two and three dimensions. Models are completely independent of visualization, which can be added, removed, or changed at any time. This separation makes it possible to run computationally demanding simulations on high-performance servers, while the visualization is performed later on a local computer. MASON can represent continuous, discrete, or hexagonal 2D, 3D, or Network data, and any combination of it. Provided visualization tools can display these environments in two or three dimension, by scaling, scrolling, or rotating them as needed.
URL: http://cs.gmu.edu/∼eclab/projects/mason/
Latest version: MASON 19, June 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.24 Mathematica
Mathematica is a general-purpose tool for the calculation and visualization of any type of mathematical model. It is produced by Wolfram Research (www.wolfram.com) and exists for the operating systems Microsoft Windows, Macintosh, Linux, and several Unix variants. The Mathematica system consists of two components: the kernel that runs in the background performing the calculations and the graphical user interface (GUI) that communicates with the kernel. The GUI has the form of a so-called notebook that contains all the input, output, and graphics. Apart from its numerical calculation and graphics abilities, Mathematica is known for its capability to perform advanced symbolic calculations. Mathematica can be used either by interactively invoking the available functions or by making use of the built-in programming language to write larger routines and programs, which are also stored as or within notebooks. For many specialized topics, Mathematica packages (a special kind of notebook) are available that provide additional functionality. J/Link, .NET/Link, and MathLink, products that ship with Mathematica, enable the two-way communication with Java, .NET, or C/C++ code. This means that Mathematica can access external code written in one of these languages and that the Mathematica kernel can actually be called from other applications. The former is useful if an algorithm has already been implemented in one of these languages or to speed up time-critical calculations that would take too long if implemented in Mathematica itself. In the latter case, other programs can use the Mathematica kernel to perform high-level calculations or render graphic objects. Besides an excellent help-utility, there are also many sites on the Internet that provide additional help and resources. The site mathworld.wolfram.com contains a large repository of contributions from Mathematica user all over the world. If a function or algorithm does not exist in Mathematica, it is worthwhile to check this site before implementing it yourself. If questions and problems arise during the use of Mathematica, a valuable source of help is also the newsgroup: http://news://comp.soft-sys.math.mathematica.
URL: http://www.wolfram.com/mathematica/
Latest version: Mathematica 10.1, March 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic □ | Commercial: ⊠ |
17.25 MathSBML
MathSBML is an open-source package for working with SBML models in Mathematica. It contains three functions that can be invoked directly: SBMLRead, SBMLNDSolve, and SBMLPlot. In addition, the MathSBML Model Builder consists of a suite of functions that can be used to build SBML Models manually. SBMLRead is the primary function provided by this package; it reads a model encoded in SBML into Mathematica, converts the model into a system of differential and possibly algebraic equations, and can generate a formatted listing of the model. SBMLNDSolve is used to solve the system of differential–algebraic equations produced by SBMLRead. SBMLPlot can be used to generate plots of the resulting solutions. Plots can also be generated directly with the Mathematica Plot command. MathSBML supports SBML Level 1 Version 2 as well as Level 2 Version 3.
URL: http://sourceforge.net/projects/xlr8r/files/mathsbml/
Latest version: MathSBML 1203-002-1102, March 2012
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.26 Matlab
Like Mathematica, Matlab is also a general-purpose computation package. In many respects, both products are very similar and it is up to the taste of the user which one to prefer. Matlab is available for the same platforms as Mathematica, has very strong numerical capabilities, and can also produce many different forms of graphics. It too has its own programming language and functions are stored in the so-called M-files. Toolboxes (special M-files) add additional functionality to the core Matlab distribution and like Mathematica, Matlab can be called by external programs to perform high-level computations. A repository exists for user-contributed files (http://www.mathworks.com/matlabcentral/fileexchange and http://www.mathtools.net/MATLAB/toolboxes.html) as well as a newsgroup (http://news://comp.soft-sys.matlab) for getting help. Although still slower than traditional programming languages like C/C++ or Java, Matlab code runs generally faster than Mathematica code.
URL: http://www.mathworks.com/products/matlab
Latest version: Matlab R2015b, 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic □ | Commercial: ⊠ |
17.27 MesoRD
MesoRD (Mesoscopic Reaction Diffusion Simulator) is a tool for stochastic and deterministic simulation of chemical reactions and diffusion in three dimension, and planar 2D spaces. The description of the system that should be simulated is written in the SBML file format. In addition to the SBML file, MesoRD requires information about how the simulation should be executed, such as spatial discretization of the reaction volume, duration of the simulation, visualization, output options, and for deterministic simulations, also choice of integration method. These parameters are given through the MesoRD user interface. The output files from MesoRD are intended for external data analysis and visualization packages, for instance, the freely distributed MesoRD Matlab toolbox available from the MesoRD website. In both the deterministic and the stochastic modes of simulation, the reaction volume is discretized into a large number of small subvolumes and the state of the system is given by the number of molecules per subvolume. In the stochastic simulation, the number of molecules per subvolume is discrete. Furthermore, the reaction and diffusion events that change the number of molecules are probabilistic, in the sense that the next event in the system is sampled from a distribution function. In the deterministic simulation, the state is assumed to be a continuous variable and the change in the number of molecules per time unit is given by the average change as defined from the stochastic model. In addition to precompiled binaries for Windows, the C++ source code is also available.
Latest version: MesoRD 1.1, October 2012
| Operating system: | Windows ⊠ | Mac OS □ | Linux □ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.28 Octave
GNU Octave is a high-level interpreted language, primarily intended for numerical computations. It provides capabilities for the numerical solution of linear and nonlinear problems, and for performing other numerical experiments. It also provides extensive graphics capabilities for data visualization and manipulation. Octave is normally used through its interactive command line interface, but it can also be used to write noninteractive programs. The aim of Octave is that all code that runs in Matlab should also run in Octave. In practice, however, most Matlab programs, except for very simple ones, require modifications. To run under Windows, the Cygwin library (www.cygwin.com) has to be installed.
URL: https://www.gnu.org/software/octave/
Latest version: Octave 4.0.0, May 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.29 Omix Visualization
Omix® is a user-friendly and highly customizable editor and modeling tool for metabolic network diagrams, equipped with extensive data visualization features. Main application fields are the interactive mapping of multi-omics data in the direct context of network drawings, in particular in the fields of transcriptomics, metabolomics, and fluxomics. Omix features the drawing of high-quality network diagrams, interactive visualization of experimental data, analysis of time-dependent data by animations, modeling of 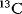 isotope labeling experiments as input for 13C FLUX2, and static network analysis via elementary flux models, network dependencies, and flux balances.
isotope labeling experiments as input for 13C FLUX2, and static network analysis via elementary flux models, network dependencies, and flux balances.
Latest version: Omix 1.8.13, April 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic □ | Commercial: ⊠ |
17.30 OpenCOR
OpenCOR is an open-source modeling environment that is supported on Windows, Linux, and OS X [3]. It relies on a modular approach, which means that all of its features come in the form of plug-ins. These plug-ins can be used to organize, edit, annotate, simulate, and analyze models encoded in the CellML (up to 1.1) format. Together with JSim, OpenCOR is the tool of choice for working with CellML files.
URL: www.opencor.ws
Latest version: OpenCOR 0.4.1, May 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.31 Oscill8
Oscill8 is a suite of tools for analyzing large systems of ODEs, particularly with respect to understanding how the high-dimensional parameter space controls the dynamics of the system. It features time course integration, one- and two-parameter bifurcation diagrams, and bifurcation searches. It also exposes lower level numerical control parameters to the expert user, including a “raw” AUTO interface (see also XPPAUT).
Latest version: Oscill8 2.0.11, February 2011
| Operating system: | Windows ⊠ | Mac OS □ | Linux □ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.32 PhysioDesigner
PhysioDesigner is an open platform that supports multilevel modeling of physiological systems in the field of integrated life sciences and systems biology, including physiology and neuroscience. Users can combine and build mathematical models of biological and physiological functions in PhysioDesigner. Users can also integrate morphometric data into a model, which is used, for example, to define a domain in which partial differential equations are solved. The models developed by PhysioDesigner are stored in PHML (Physiological Hierarchy Markup Language) format, which is an XML-based specification, to describe a wide variety of models of biological and physiological functions with a hierarchical structure. PhysioDesigner also allows the creation of SBML–PHML hybrid models, which is a novel way of creating multilevel physiological systems. Simulation of the models created by PhysioDesigner can be performed using the Flint simulator, which is developed concurrently with PhysioDesigner. Additionally, PhysioDesigner can export C++ and Java source code, including numerical integration solvers. Thus, users can easily perform simulations by compiling them. Finite element simulation for partial differential equations can be done by exporting a model in FreeFem++ format and running the script on FreeFem++.
URL: physiodesigner.org/
Latest version: PhysioDesigner 1.3.1, July 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.33 PottersWheel
PottersWheel is a Matlab toolbox for the construction and analysis of kinetic mathematical models. A model can be entered as a system of ordinary differential equations (ODEs) or loaded from a SBML file. The model is displayed as reaction network, but the real strength of PottersWheel lies in its parameter fitting capabilities. The tool allows the simultaneous fitting of multiple data sets offering a range of different fitting algorithms. In addition to the parameter values, confidence intervals are also calculated and an identifiability analysis is performed. Since fitting problems can be computationally very demanding, the code can be run on a cluster of computers. PottersWheel can be controlled via scripts or a graphical user interface, the use of which is described by several video tutorials that are available on the website. The results are represented by different plots and can also be saved in a report (available as PDF, doc, or html).
URL: www.potterswheel.de/
Latest version: PottersWheel 4.0, 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic □ | Commercial: ⊠ |
17.34 PyBioS
PyBioS is an integrated, Web-based software platform for the design, modeling, and simulation of cellular systems, in particular for human and mouse. Models in PyBioS can be constructed in a Web-based user interface that defines different objects for cellular components, such as genes, mRNAs, proteins, and metabolites as well as cellular compartments. PyBioS keeps track of accession numbers of these entities, such as ENSEMBL gene, transcript or protein identifiers, or ChEBI and KEGG IDs. As each cellular component is defined by a reference entity in the underlying PyBioS database, it allows a modular design of models that can subsequently be merged into larger and more comprehensive models. This supports the easy reuse of models. Models can also be imported from external sources. An interface to the meta-pathway database ConsensusPathDB (see Chapter 16) that is integrating several public pathway resources allows the automatic import of reactions and pathways from databases such as KEGG and Reactome. Models can also be imported from BioModels.org or directly as SBML. PyBioS comes along with several predefined kinetic laws that simplify the definition of new reactions. For quantitative simulation, ODE systems of the models are created by PyBioS on the fly. Simulation results can either be displayed in a diagram or they can be uploaded into the models network graph. This allows the animation of the changes of fluxes and concentrations by appropriate color codes of reaction and species nodes.
Latest version: PyBioS 3, November 2014
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic ⊠ | Commercial: □ |
17.35 PySCeS
PySCeS (Python Simulator for Cellular Systems) is written in Python and provides a variety of tools for the simulation and analysis of cellular systems. The input is via a text-based model description language. Solvers for time course integration (LSODA, CVODE) and steady-state calculations (HYBRD, NLEQ2) exist. Various modules perform metabolic control analysis (i.e., elasticities, flux, and concentration control coefficients) and bifurcation analysis. The package also allows 1D and 2D parameter scans and their visualization via a flexible plotting interface (Matplotlib and/or Gnuplot). PySCeS can import and export SBML and is developed as open-source software. The use of a proper programming language gives PySCeS a great flexibility, but it should be clear that models are not as easy to construct as in GUI-based modeling tools.
Latest version: PySCeS 0.9.1, December 2014
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.36 R
R is a free programming environment that is especially designed and used to provide a wide variety of statistical tests and techniques. It consists of the R language, an integrated development environment (IDE), and a large set of (statistical) packages that can be downloaded from the Comprehensive R Archive Network (CRAN). Especially, two features make R interesting for biostatistical and bioinformatical analyses: first, the rich choice of publication quality plots that can be generated with R; second, the fact that new (bio)statistical analysis methods are often first available as R packages. Of special interest in this respect is the Bioconductor project (www.bioconuctor.org), which consists of a collection of R packages for the analysis of high-throughput genomic data such as sequence, expression, and interaction data. Although R is a complete programming language, it is rarely used for “normal” programming tasks, since it is very slow compared to languages like Java or C++.
URL: www.r-project.org/
Latest version: R 3.2.3, December 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.37 SAAM II
SAAM II (Simulation Analysis and Modeling) is a modeling, simulation, and analysis software package that supports the development and statistical calibration of compartmental models in biological, metabolic, and pharmaceutical systems. Mathematically, these models translate into systems of ordinary linear or nonlinear differential equations. A flexible graphical user interface makes it easy for researchers with diverse backgrounds to use the software. The Epsilon Group, (TEG) acquired all rights of the SAAM II software packages from the University of Washington in early 2011 and added several features since then. During the modeling process, it is sometimes necessary to understand the effect that data collection errors and collection times can have in identifying model parameters. To improve this process, experiments can be simulated using synthetic data with a variety of errors. Users also have the possibility to create sensitivity plots for a better understanding of relationships between the variables and compartments when there is uncertainty. Furthermore, it is also possible to investigate the robustness of models and select the best alternative model among competing alternatives. A variety of outputs can be automatically generated, including parameter values and their confidence intervals, statistical information, or plots (including experiment simulation, sensitivity plots, and batch processing). Support for SBML is in development, but not yet supported.
URL: http://tegvirginia.com/solutions/saam-ii/
Latest version: SAAM II, 2.3, November 2013
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux □ |
| Availability: | Free for all □ | Free for academic □ | Commercial: ⊠ |
17.38 SBMLeditor
The Systems Biology Markup Language is a free and open interchange format for encoding computational models of biological processes. SBML aims to be a systems to systems format, and users are not expected to write SBML files manually. SBMLeditor is a simple, low-level editor for SBML files. It allows you to manipulate SBML elements in a controlled way while maintaining the validity of the final file. SBMLeditor also allows you to create and modify annotations, as defined in the SBML specifications. The editor supports up to SBML Level 3 Version 1 and checks the validity of the SBML code whenever it is saved.
URL: https://www.ebi.ac.uk/compneur-srv/SBMLeditor.html
Latest version: SBMLeditor 2.0-b1, June 2012
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.39 SemanticSBML
SemanticSBML is a suite of software tools for SBML models, supporting users in the annotation, alignment, and merging of models. During model merging, the software detects inconsistencies between model elements and allows users to change the element alignment and to resolve conflicts. Using semantic annotations in SBML models, it also allows for a semantic clustering of models and for a ranked retrieval from models in the BioModels database at www.ebi.ac.uk/biomodels-main. SemanticSBML can be used online at www.semanticsbml.org. Free python code for programmers, including code for element comparison by semantic similarities, is freely available.
URL: www.semanticsbml.org
Latest version: SemanticSBML 2.0, September 2010
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.40 SBML-PET-MPI
SBML-PET-MPI is a parallel parameter estimation tool for SBML-based models. The tool allows the user to perform parameter estimations by fitting multiple data sets. The tool performs an uncertainty and identifiability analysis of the estimated parameters using maximum likelihood and bootstrap methods. Models containing events are supported as well as those with constraints. The software uses the message parsing interface (MPI) protocol for parallelization and achieves good scalability with the number of available processors. To run under Windows, the Cygwin library (www.cygwin.com) has to be installed.
URL: http://www.bioss.uni-freiburg.de/cms/sbml-pet-mpi.html
Latest version: SBML-PET-MPI 1.2, September 2011
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.41 SBMLsimulator
SBMLsimulator is a fast, accurate, and easily usable program for dynamic model simulation and heuristic parameter optimization of models encoded in the Systems Biology Markup Language. In order to ensure a high reliability of this software, it has been benchmarked against the entire SBML Test Suite and all models from the Biomodels.net database. It includes a large collection of nature-inspired heuristic optimization procedures for efficient model calibration. SBMLsimulator provides an intuitive graphical user interface and several command-line options to be suitable for large-scale batch processing and model calibration. The simulation core library of SBMLsimulator can be obtained as a separate application programming library.
URL: http://www.ra.cs.uni-tuebingen.de/software/SBMLsimulator/
Latest version: SBMLsimulator 1.2.1, July 2014
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.42 SBMLsqueezer
SBMLsqueezer generates kinetic equations for biochemical networks according to the context of each reaction. It can be used as a plug-in for CellDesigner, in which case it uses the information contained in the SBGN representation of the network components. Additionally, it can also be used in a stand-alone mode, in which SBMLsqueezer evaluates the Systems Biology Ontology (SBO) annotations to extract the relevant information. Finally, an online version of SBMLsqueezer is available that runs without the need to install any software on the local machine. The rate laws that can be produced by SBMLsqueezer include several types of generalized mass action, detailed and generalized enzyme kinetics, various types of Hill equations, convenience kinetics, and additive models for gene regulation. User-defined settings specify which equation to apply for any type of reaction and how to ensure unit consistency of the model. Equations can be created using contextual menus. All newly created parameters are equipped with the derived unit and annotated with SBO terms (if available) and meaningful textual names. MathML is inserted directly into the SBML file. A LaTeX and text export of the SBML model via the integrated tool SBML2LaTeX (http://www.cogsys.cs.uni-tuebingen.de/software/SBML2LaTeX) is also provided. Finally, a 90-pages user manual is also available on the website.
URL: http://www.cogsys.cs.uni-tuebingen.de/software/SBMLsqueezer/
Latest version: SBMLsqueezer 2.1, August 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.43 SBML Toolbox
SBMLToolbox is built on top of libSBML and provides a set of basic functions allowing SBML models to be used in both Matlab and Octave. SBMLToolbox provides functions for creating and validating models and for manipulating and simulating these models using ordinary differential equation solvers. SBMLToolbox works by translating SBML models to/from a Matlab structure called MATLAB_SBML. It provides facilities for manipulating this and its substructures within the Matlab or the Octave environment. The libSBML binding enables the import and export of these structures to and from SBML files. The toolbox is not intended to be a complete Systems Biology toolbox for Matlab, but rather a platform facilitating getting SBML in and out of Matlab and serving as a starting point from which users can develop their own functionality. The current version of SBMLToolbox supports all releases of SBML up through Level 3 Version 1.
URL: http://sbml.org/Software/SBMLToolbox
Latest version: SBML Toolbox 4.1.0, January 2012
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.44 SBtoolbox2
The Systems Biology Toolbox 2 (SBToolbox2) for Matlab offers systems biologists a powerful, open, and user-extensible environment in which to build models of biological systems. The toolbox features a wide variety of specialized analysis tools and Matlab adds to that with a large number of built-in functions and a high-level scripting language, allowing the user to quickly and efficiently add new functionality. Models can be defined using a simple syntax based on either differential equations or biochemical reaction equations. In addition, the toolbox also supports the import and export of SBML models. Via the toolbox, models can be simulated deterministically or stochastically and can also be analyzed by a variety of methods to study steady states and their stability and perform metabolic control analysis, bifurcation analysis, parameter sensitivity analysis, or a stoichiometric analysis.
URL: www.sbtoolbox2.org/main.php?display=docu mentationSBT&menu=overview
Latest version: SBtoolbox2 1361, July 2014
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.45 SBML Validator
The SBML Validator is an online resource that checks the syntax and internal consistency of SBML code that is uploaded as file, submitted as URL, or pasted directly into the webpage. The validation procedure is built on the libSBML 5.10.1 library and supports SBML code up to Level 3 Version 1. The maximum allowed file size is 32 MByte, which should be sufficient for most purposes. Several validation options, such as checking the MathML syntax and the consistency of identifiers or performing a static analysis to test if the model is overdetermined, can be selected to fine-tune the validation process. If a model gives problems with SBML processing tools, it is definitely a good idea to run it through the validator.
URL: http://sbml.org/Facilities/Validator
Latest version: Support up to SBML Level 3 Version 1
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.46 SensA
Mathematical models (written in SBML) can contain many parameters and it is important to know how sensitive model properties (i.e., concentrations or fluxes) depend on specific parameter values. SensA is a Web-based application for this type of sensitivity analysis [4]. It is based on metabolic control analysis and computes local, global, and time-dependent properties of model components. Models can be uploaded in SBML format and results are represented graphically in diagrams and color-coded tables. Results can be stored online or downloaded as text and graphics. While other tools such as Copasi and JWS online also offer an MCA-based sensitivity analysis for the steady-state case, SensA includes the computation of time-dependent sensitivities.
URL: gofid.biologie.hu-berlin.de/
Latest version: Support up to SBML Level 2 Version 4 (with the exception of events, constraints, algebraic rules, time variable compartment sizes, and initial assignments).
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.47 SmartCell
SmartCell has been developed to be a general framework for modeling and simulating diffusion–reaction networks in a whole-cell context. It supports localization and diffusion by using a mesoscopic stochastic reaction model. The SmartCell package handles any cell geometry, considers different cell compartments, allows localization of species, and supports DNA transcription and translation, membrane diffusion, and multistep reactions, as well as cell growth. Entities are represented by their copy number and location. In order to introduce spatial information, the geometry is divided into smaller volume elements, called voxel, where stochastic events take place. The use of a mesh allows it to consider diffusion as translocation across adjacent volume sites. The user-defined model is translated into an internal core model, where rates are converted into reaction probabilities per unit time. At this stage, reversible processes, diffusion, and complex formation are converted into an equivalent set of unidirectional elementary processes. Finally, each process is translated into as many individual events as volume elements in the region where the process is defined. The core model is subsequently used by the simulation engine itself. The simulation engine then uses different algorithms in order to have exact stochastic result (NREM, NSVM, Hybrid), exact deterministic result (ODE), or approximate stochastic result (Tau leap).
URL: http://software.crg.es/smartcell/
Latest version: SmartCell 4.3b3, March 2011
| Operating system: | Windows ⊠ | Mac OS □ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.48 STELLA
STELLA is a commercial computer modeling package with a graphical user interface that allows users to construct dynamic models that simulate biological systems. Stocks and flows can be placed on the GUI to model continuous processes or discrete events using so-called queues and ovens. Models are saved in a proprietary format. Furthermore, sliders, switches, and buttons can be placed on the model canvas to manipulate model parameters.
URL: http://www.iseesystems.com/softwares/Education/StellaSoftware.aspx
Latest version: STELLA 10.0.4, October 2013
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux □ |
| Availability: | Free for all □ | Free for academic □ | Commercial: ⊠ |
17.49 STEPS
STEPS (Stochastic Engine for Pathway Simulation) is a package for exact stochastic simulation of reaction-diffusion systems in arbitrarily complex 3D geometries. The core simulation algorithm is an implementation of Gillespie's SSA, extended to deal with diffusion of molecules over the elements of a 3D tetrahedral mesh. Since version 2.0, STEPS also supports accurate and efficient computation of local membrane potentials on tetrahedral meshes, with the addition of voltage-gated channels and currents. Tight integration between the reaction–diffusion calculations and the tetrahedral mesh potentials allows detailed coupling between molecular activity and local electrical excitability. The tool has been implemented as a set of Python modules, which means STEPS can be used via Python scripts to control all aspects of constructing the model, generating a mesh, controlling the simulation, and generating as well as analyzing output. The core computational routines are implemented as C/C++ extension modules for maximal speed.
URL: http://steps.sourceforge.net/STEPS/default.php
Latest version: STEPS 2.2.0, April 2014
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.50 StochKit2
StochKit2 is an efficient, extensible stochastic simulation framework developed in the C++ language. StochKit2 provides implementations of several (exact) stochastic algorithms, including the direct method, optimized direct method, and composition–rejection method. These methods generate exact trajectories from the chemical master equation, but use modified underlying algorithms and data structures to achieve different performance and scaling properties. The direct method, which uses simple data structures, tends to be best for relatively small models, while for very large models, the composition–rejection method is the most efficient. The StochKit2 user interface is simple. When a user chooses to run an exact stochastic simulation, the software immediately analyzes the model and automatically chooses the appropriate algorithm. StochKit2 also provides an interface for running stochastic simulations using an adaptive tau-leaping method. The tau-leaping method sacrifices exactness in exchange for taking larger time steps. The software uses an XML-based file format that is similar to, but not identical with, SBML. However, a function is provided to convert StochKit models to and from SBML.
URL: http://www.engineering.ucsb.edu/∼cse/StochKit/
Latest version: StochKit 2.0.11, February 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.51 SystemModeler
SystemModeler is a commercial product of Wolfram Research, the same company that produces Mathematica. The software provides a graphical user interface for creating models based on mechanical, electrical, or magnetic interactions. With the help of the BioChem library, it is also possible to create kinetic models of biochemical reactions. Reaction networks can be created in a drag-and-drop fashion as with CellDesigner and then solved to obtain time course solutions of the model. If desired, the models can be built in a SBML compliant way and exported as an SBML file. The concept of SystemModeler is quite similar to SimuLink for Matlab, but the software can also be used independent of Mathematica. But if Mathematica is installed, the package Wolfram SystemModeler Link (WSMLink) is automatically installed. This enables a tight connection between Mathematica and SystemModeler and allows controlling and analyzing a SystemModeler model from within Mathematica.
URL: http://www.wolfram.com/system-modeler/
Latest version: SystemModeler 4.2, December 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all □ | Free for academic □ | Commercial: ⊠ |
17.52 Systems Biology Workbench
The Systems Biology Workbench (SBW) [5] is a software system that enables different tools to communicate with each other via a fast binary encoded message system. Thus, SBW-enabled tools can use services provided by other modules and in turn advertise their own specialized services. At the center of the system is the SBW broker that receives messages from one module and relays them to other modules. JDesigner and Jarnac are two modules that come with the SBW standard installation. JDesigner is a program for the graphical creation of reaction networks, and Jarnac is a tool for the numerical simulation of such networks (time course and steady state). Jarnac runs in the background and advertises its services to the SBW broker. JDesigner contacts the broker to find out which services are available and displays the found services in a special pull-down menu called SBW. A reaction model that has been created in JDesigner can now be send to the simulation service of Jarnac. A dialog box opens to enter the necessary details for the simulation and then the broker calls the simulation service of Jarnac. After a time course simulation finishes, the result is transmitted back to JDesigner (via the broker) and can be displayed. The list of SBW-enabled programs contains programs specialized in the graphical creation of reaction networks (JDesigner and CellDesigner), simulation tools (Jarnac and TauLeapService), analysis and optimization tools (Metatool, Bifurcation, and Optimization), and utilities such as the Inspector module, which provides information about other modules.
URL: http://sbw.kgi.edu/research/sbwintro.htm
Latest version: SBW 2.9, February 2012
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.53 Taverna
Taverna is an open-source and domain-independent Workflow Management System similar to KNIME. Thus, it provides a suite of tools used to design and execute scientific workflows around the analysis of data. The Taverna suite is written in Java and includes the Taverna Engine (used for enacting workflows) that powers both the Taverna Workbench (the desktop client application) and the Taverna Server, which allows remote execution of the created workflows. Taverna is also available as a Command Line Tool for a quick execution of workflows from a terminal. Finally, Taverna Online lets you edit and run Taverna workflows on the Web. Workflows are created via drag-and-drop method and the resulting workflow diagrams are then displayed using the external tool Graphviz.
URL: www.taverna.org.uk/
Latest version: Taverna 2.5, May 2014
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.54 Vanted
VANTED stands for Visualization and Analysis of Networks containing Experimental Data. This system makes it possible to load and edit graphs, which may represent biological pathways or functional hierarchies. It is possible to map experimental data sets onto the graph elements and visualize time series data or data of different genotypes or environmental conditions in the context of underlying biological processes. Built-in statistic functions allow a fast evaluation of the data (e.g., t-test or correlation analysis). The latest version also contains support for models in the SBML format.
URL: immersive-analytics.infotech.monash.edu/vanted/
Latest version: VANTED 2.6, November 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.55 Virtual Cell (VCell)
Users can build models with a Java interface to specify compartmental topology and geometry, biochemical reactions, and relevant interaction parameters. VCell can handle compartmental and spatial models, which can be simulated deterministically or stochastically. Models are normally stored in VCML (Virtual Cell Markup Language), but deterministic nonspatial (and some spatial) models can also be imported and exported as SMBL. Models are stored in a personal online repository, but they can also be saved on the local computer. Virtual Cell automatically converts the biological description into a corresponding mathematical system of ordinary (compartment model) and/or partial differential equations (2D and 3D spatial models). Mathematically experienced users may directly specify the complete mathematical description of the model, bypassing the schematic interface. Normally the equations are then sent to online servers of the University of Connecticut, where the model is simulated by appropriate solvers and the results are then transferred back for display. But if desired, the model can also be run at the local machine. Finally, VCell can also perform parameter estimations for which it internally uses the Copasi engine (for nonspatial deterministic models). Results can be displayed and analyzed online or downloaded to the user's computer in a variety of formats. A more in-depth description of VCell is given in Chapter 5.
URL: www.vcell.org/
Latest version: VCell 5.3, June 2015
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.56 xCellerator
xCellerator is a Mathematica® package designed to aid biological modeling via the automated conversion of chemical reactions into ODEs. These equations can then be solved via numerical integration and are displayed as time course diagrams. SBML is also supported via plug-ins and MathSBML. The current version generates some warnings with Mathematica version 9, but nevertheless works fine.
URL: www.cellerator.org
Latest version: xCellerator 0.91, November 2012
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
17.57 Xppaut
XPPAUT is a tool for solving differential equations, difference equations, delay equations, functional equations, boundary value problems, and stochastic equations. XPPAUT contains the code for the popular bifurcation program AUTO. Thus, you can switch back and forth between XPPAUT and AUTO, using the values of one program in the other and vice versa.
URL: http://www.math.pitt.edu/∼bard/xpp/xpp.html
Latest version: XPPAUT 7, December 2012
| Operating system: | Windows ⊠ | Mac OS ⊠ | Linux ⊠ |
| Availability: | Free for all ⊠ | Free for academic □ | Commercial: □ |
Exercises
- When and why should a system be modeled stochastically instead of deterministically?
- Which development in the last years is important for the exchange of models between different simulation tools?
- What is the purpose of libSBML?
- Is it possible to develop models in (a) Mathematica or (b) Matlab that support SBML?
- Use CellDesigner to model the irreversible reaction S → P using a Michaelis–Menten kinetics. Draw the diagram, specify the kinetics (for Km = 2 mmol l−1, Vmax = 5 mmol (l*s)−1, St=0 = 100 molecules, and Pt=0 = 0 molecules) and run a time course simulation.
- Export the model as SBML and import it into Copasi. Run a time course simulation to see if it is identical to the one from CellDesigner.
- Use the following three time/substrate concentration data points for model fitting: P1: 5 s60 mmol−1, P2: 10 s 50 mmol−1, P3: 15 s 20 mmol−1. What are the values of Km and Vmax after fitting?
- Import the CellDesigner SBML model into Virtual Cell and run a stochastic simulation. Do you see any differences to the deterministic solution?
References
- 1. Klipp, E., Liebermeister, W., Helbig, A., Kowald, A., and Schaber, J. (2007) Systems biology standards: the community speaks. Nat. Biotechnol., 25 (4), 390–391.
- 2. Ghosh, S., Matsuoka, Y., Asai, Y., Hsin, K.Y., and Kitano, H. (2011) Software for systems biology: from tools to integrated platforms. Nat. Rev. Genet., 12 (12), 821–832.
- 3. Garny, A. and Hunter, P.J. (2015) OpenCOR: a modular and interoperable approach to computational biology. Front. Physiol., 6, 26.
- 4. Floettmann, M., Uhlendorf, J., Scharp, T., Klipp, E., and Spiesser, T.W. (2014) SensA: web-based sensitivity analysis of SBML models. Bioinformatics, 30 (19), 2830–2831.
- 5. Hucka, M., Finney, A., Sauro, H.M., Bolouri, H., Doyle, J., and Kitano, H. (2002) The ERATO Systems Biology Workbench: enabling interaction and exchange between software tools for computational biology. Pac. Symp. Biocomput., 450–461.