Derivatization in Liquid Chromatography
C.F. Poole, Department of Chemistry, Wayne State University, Detroit, Michigan
Outline
2.1. Introduction
2.2. Reagent Selection
2.2.1. Reagents for UV–Visible Detection
2.2.2. Reagents for Fluorescence and Chemiluminescence Detection
2.2.3. Reagents for Electrochemical Detection
2.2.4. Reagents for Mass-Spectrometric Detection
2.2.5. Reagents for the Formation of Diastereomers
2.2.6. Multifunctional Reagents for the Formation of Cyclic Derivatives
2.2.7. Solid-Phase Analytical Derivatization
2.3. Postcolumn Reaction Detectors
2.3.1. Photoreactors
2.4. Conclusions
2.1 Introduction
Derivatization represents an added step in the analysis of a sample and is justified only when it facilitates the isolation, separation, or detection of a compound or leads to more-robust results by enhancing the stability of the compound, reduces matrix interferences, improves the reproducibility of the method, or simplifies the operational steps in the method. Many compounds lack a suitable chromophore (UV-Visible detection), fluorophore (fluorescence and chemiluminescence detection), electrophore (electrochemical detection) or possess low ionization efficiently (mass spectrometric detection) for detection at anticipated sample concentrations. For compounds with reactive functional groups, simple chemical reactions allow the required detection characteristics to be acquired by the compound by modifying its chemical structure to that required for facile detection or serves to minimize matrix interference by moving the compound to a position in the chromatogram where interference in the detector response is minimal. Replacing a reactive functional group (or groups) with a substituent of different polarity, and perhaps, a relatively complex structure, alters the separation characteristics of the compound, and if favorable, might lead to an improvement in the separation, but just as easily, might make the compounds more difficult to separate or extend the separation time. Individual reagents behave differently in this respect, and a suitable reagent to enhance sample detection must also not lead to an inferior or unacceptable separation. Since derivatizing reactions can be performed either precolumn, and affect the separation of the compounds, or postcolumn, and affect only the detection step, there is some flexibility in the selected approach. Occasionally, the derivatization reaction is performed close to the start of an isolation procedure to increase the recovery of the analytes, to ensure their stability to the conditions employed in the isolation procedure, or to increase the selectivity of the method for the target compounds.
For precolumn derivatization, the selected reaction must be quantitative, or nearly so, and free from by-products. Reaction conditions can usually be optimized free of time constraints. When possible, samples are processed in batches with a high level of automation and control of the reaction conditions but can also be performed manually or for individual samples. In general, a simple method must be available to separate excess reagent and other products from the derivatives, if these interfere in the separation or detection of the derivatives. This is quite likely if the reagent and derivative share a common core structure responsible for the detector response.
Postcolumn reactions occur in a continuous-flow reactor and need not be quantitative, so long as they are reproducible. Reaction times are usually constrained by the design of the reactor and should be sufficiently fast that the column resolution is not destroyed by diffusion in the reactor device. Although artifact formation is rarely a problem, both the reagent and by-products (if any) must either not respond to the detector under the same conditions used to detect the analytes or must be easily separated from the analytes after derivatization and before detection. The latter operation can be performed by extraction in a solvent-segmented stream but adds to the complexity of the reactor design. Although postcolumn derivatization allows independent optimization of the conditions for separation and detection, it is not uncommon for the optimum solvent composition for the separation to be incompatible with the requirements of the derivatization reaction. This may limit some applications of postcolumn detection.
2.2 Reagent Selection
A derivatizing reagent can be considered to comprise two parts: a reactive functional group that controls the rate, extent, and selectivity of the chemical reaction (derivative formation) and a structural unit that provides a favorable detector response to the derivative and, in precolumn reactions, modifies the isolation and separation properties of the analytes. Some typical reagent functional groups and the complementary analyte functional groups they react with are summarized in Table 2.1 [1–4]. In selecting a reagent based on one of these groups, important considerations are the reaction time, the completeness of the reaction, the conditions required for the reaction and whether these affect the stability of the analytes, the stability of the derivatives (storage properties), and the ease with which excess reagent can be separated from the derivatives. Depending on the sample type, whether the reaction requires completely anhydrous conditions or occurs in aqueous solution can be an important factor. The desire to use fast and quantitative reactions places a restriction on the number of reagents that are commonly used and the types of analyte functional groups that can be easily derivatized.
TABLE 2.1
Classification of Reactions Based on the Reactive Functional Group of the Reagent
Note: R is any part of the reagent or analyte not directly involved in the reaction and A is an aromatic ring containing the reactive functional group for the reagent.
2.2.1 Reagents for UV–Visible Detection
The majority of reagents used to introduce a chromophore contain an aromatic ring with different substituents to optimize the absorption wavelength or the molar absorption coefficient. Ideally, the chromophoric group should have a large molar absorption coefficient (>10,000) at some convenient wavelength for detection limits at the low nanogram level. Some representative chromophores are summarized in Table 2.2. There is a preference for reagents that absorb at relatively long wavelengths, since this minimizes interferences from the sample matrix and solvents used in the separation. Since most solvents absorb strongly at wavelengths <210 nm, reagents that form derivatives that can be detected at wavelengths >240 nm are preferred. Many reagents forming colored derivatives possess a higher molar absorption in the UV region, and, in general visible detection is less common. However, visible detection can provide higher selectivity for analytes with a high matrix burden, since most organic compounds are transparent in the visible region.
TABLE 2.2
Some Common Chromophores and Their Properties
| Chromophore | Wavelength for maximum absorption (nm) | Molar absorption coefficient at 254 nm |
| 4-Nitrophenyl | 265 | 5,200 |
| 3,5-Dinitrobenzyl | >10,000 | |
| 4-Chlorobenzoate | 236 | 6,300 |
| 4-Nitrobenzoate | 254 | >10,000 |
| 2,4-Dinirophenyl | >10,000 | |
| Toluoyl | 236 | 5,400 |
| Anisyl | 262 | 16,000 |
| Phenacyl | 250 | 10,000 |
| 4-Bromophenacyl | 260 | 18,000 |
| 2-Naphthacyl | 248 | 12,000 |
Some common reagents for derivatizing specific analyte functional groups are summarized in Table 2.3 [1–12]. Alcohols are usually converted to esters by reaction with an acid chloride in the presence of a base catalyst. For compounds with labile functional groups, reaction with a phenyl isocyante to form phenylurethanes can be performed under mild conditions. Alcohols in aqueous solution can be derivatized with 3,5-dinitrobenzoyl chloride. Amines are usually derivatized with an acid chloride to form phenyl substituted amides or sulfonamides. Sanger’s reagent, 2,4-dinitro-1-fluorobenzene, originally introduced for the identification of N-terminal amino acid residues in proteins, has been widely used for the derivatization of amines. Carbamate and phenylacetate reagents can be used to derivatize amines under mild conditions without a catalyst. Phenyl isothiocyanate is commonly used for the precolumn derivatization of amino acids (phenylthiocarbamyl derivatives) for separation by reversed-phase liquid chromatography [13]. Alkylation of carboxylic acids with haloalkyl reagents in the presence of a catalyst in aqueous or anhydrous conditions is widely used [6]. O-(4-nitrobenzyl)-N,N’-diisopropylurea facilitates the derivatization of carboxylic acids under mild conditions without addition of a catalyst. Aldehydes and ketones can be selectively derivatized in the presence of other functional groups using reagents with a hydroxylamine or hydrazine reactive group [9]. Aldehyde groups in monosaccharides and more complex glycogens are detected after derivatization with a number of reagents containing a reactive amine group [10].
TABLE 2.3
Reagents for the Introduction of a Chromophore into Compounds with Complementary Reactive Functional Groups
| Analyte functional group | Derivatizing reagent | Reference |
| Hydroxyl | 3,5-Dinitrobenzyl chloride | [1,2] |
| Benzoyl chloride | [1,2] | |
| 4-Nitrobenzoyl chloride | [1,2] | |
| 4-Nitrophenyl chloroformate | [3] | |
| Phenyl isocyanate | [1,2] | |
| 4-Dimethylaminophenyl isocyanate | [3] | |
| Phenol | 3,5-Dinitrobenzyl chloride | [1,2] |
| 4-Toluenesulfonyl chloride | [3] | |
| Diazo-4-aminobenzonitrile | [3] | |
| Carboxylic acid | 4-Bromophenacyl bromide | [6] |
| O-(4-nitrobenzyl)-N,N’-diisopropylisourea | [1,2] | |
| N-Chloromethyl-4-nitrophthalimide | [3] | |
| Methylphthalimide | [3] | |
| Naphthyldiazomethane | [6] | |
| 4-Nitrobenzyl bromide | [6] | |
| 4-Methoxyaniline | [6] | |
| 1-Naphthylamine | [6] | |
| Ketones and aldehydes | 2,4-Dinitrophenylhydrazine | [9] |
| 4-Nitrobenzylhydroxylamine | [1,2] | |
| Isocyanates | N-4-Nitrobenzyl-N-n-propylamine | [1,3] |
| Thiols | 4-Dimethylaminoazobenzene-4’-sulfonyl chloride | [9,11,12] |
| 1-Benzyl-2-chloropyridinium bromide | [10] | |
| N-Ethylmaleimide | [10] | |
| 2-Chloro-1-methylquinolinium tetrafluoroborate | [10] | |
| Amines | 3,5-Dinitrobenzoyl chloride | [1,2] |
| 4-Methoxybenzoyl chloride | [1,2] | |
| n-Succinimidyl-4-nitrophenylacetate | [3] | |
| 4-Nitrobenzoyl chloride | [1,2] | |
| 4-Toluenesulfonyl chloride | [1,2] | |
| 2-Naphthacyl bromide | [1,2] | |
| 2,4-Dinitro-1-fluorobenzene | [1–3] | |
| 2-Naphthalenesulfonyl chloride | [1–3] | |
| phenyl isocyanate | [8] | |
| 4-(Dimethylamino)benzaldehyde | [8] |
2.2.2 Reagents for Fluorescence and Chemiluminescence Detection
Fluorescence detection is used for those methods that require either low detection limits or higher selectivity. Few organic compounds are naturally fluorescent and nonspecifc interferences in the determination of fluorescent compounds are reduced compared with UV–Visible absorption methods. In addition, the fluorescence process requires excitation at one wavelength and emission at a different (longer) wavelength, which further enhances both selectivity and detectability. The choice of excitation and emission wavelengths provides a possible mechanism to distinguish between fluorescent compounds in a mixture improving selectivity. The longer wavelength of the emission signal minimizes the contribution of scattered light from the excitation wavelength, resulting in low background noise and the possibility of using higher amplification to maximize the signal. This, combined with the higher excitation energy of sources used for fluorescence, is largely responsible for the two or more orders of magnitude increase in signal that allows the detection of fluorescent compounds at low pictogram amounts. The fluorescence signal, however, is subject to a number of matrix interferences that can affect both the signal intensity and the emission wavelength [14]. Impurities in the mobile phase, particularly oxygen, may quench the signal from low concentrations of fluorescent compounds, even after solvent degassing. Provided that the mobile phase contains a minimum of 1% (v/v) methanol, oxygen can be efficiently removed by a short precolumn containing an immobilized catalyst located between the pump and the injector [15]. Both the emission wavelength and fluorescence intensity of ionizable fluorophores are critically dependent on pH and solvent hydrogen-bonding interactions. Intensity changes on an order of magnitude and large shifts in the emission wavelength are possible for neutral fluorophores that undergo strong solvent interactions. Many fluorophores exhibit significant temperature dependence with an increase in temperature of 1°C causing a 1–2% decrease in fluorescence intensity and short-term temperature fluctuations increasing the background detector noise. Coeluting components of the sample matrix can also modify the observed response compared with standards prepared in neat solvent.
Reagents containing fluorophores can have complex structures and some representative reagents are shown in Figure 2.1 [1–6,16–20]. A number of reagents have a common fluorophore and different reactive functional groups. This is because the number of stable and intense fluorophores is not that large, while the number of potential applications can be significantly increased by variation in the reactive group. When selecting a reagent, those forming derivatives with a relatively high quantum efficiency and long-wavelength fluorescence are the most useful. Fluorophores with a large Stoke’s shift (difference between the excitation and emission maxima) are usually favored and provide higher selectivity when analyzing complex mixtures.
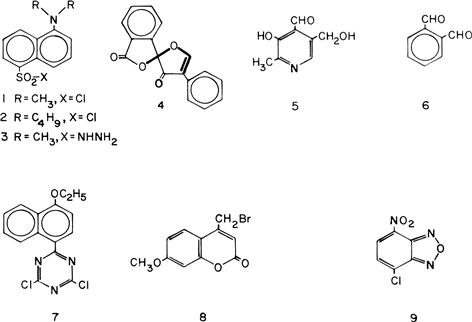
FIGURE 2.1 Reagents for introducing a fluorophore into compounds with a complementary reactive functional group.
Reagents: 1 = dansyl chloride; 2 = dabsyl chloride; 3 = dansyl hydrazine; 4 = fluorescamine; 5 = 2,4-dihydro-6,7-dimethoxy-4-methyl-3-oxo-quinoxaline-2-carbonyl azide; 6 = o-phthalaldehyde; 7 = fluorenylmethyloxycarbonyl chloride; 8 = 4-bromomethyl-7-methoxycoumarin; and 9 = 4-chloro-7-nitrobenzofurazan.
Some common reagents for derivatizing compounds with reactive functional groups are summarized in Table 2.4 [1–6,16–27]. Reagents containing a methoxycoumarin, dimethylaminonaphthalene, or benzoxadiazole fluorophore usually provide lower detection limits than those containing polycyclic aromatic hydrocarbon or acridine fluorophores. Dansyl chloride (and its analogs) is a popular multipurpose reagent [1–3]. Dansyl chloride forms derivatives with primary and secondary amines readily, less rapidly with phenols and imidazoles, and very slowly with alcohols. The reaction media is usually an aqueous organic solution (e.g., 1:1 acetone–water) buffered to pH 9.5–10. Several analogs of dansyl chloride are also in use. 5-Dibutylaminonaphthalenesulfonyl chloride forms derivatives with better storage capability and facilitates the extraction of hydrophilic compounds. Dansyl hydrazine is a selective reagent for the detection of aldehydes and ketones (it will also form derivatives with carboxylic acids) and dansyl aziridine is a selective reagent for the detection of thiols. Several reagent-containing maleimide or iodoacetamide groups that form adducts with thiols are also widely used [12].
TABLE 2.4
Reagents for the Introduction of a Fluorophore into Compounds with Complementary Reactive Functional Groups
| Analyte functional group | Derivatizing reagent | Reference |
| Hydroxyl | 4-Dimethylamino-1-naphthoyl nitrile | [1–3] |
| 9-Anthronyl nitrile | [1–3,22] | |
| 7-[(Chlorocarbonyl)methoxy]-4-methylcoumarin | [1,5] | |
| 3,4-Dihydro-6,7-dimethoxy-4-methyl-3-oxo-quinoaline-2-carbonyl azide | [3–5] | |
| 1-Naphthyl isocyanate | [23] | |
| 4-(7-diethylaminocoumarin-3-yl)benzoyl cyanide | [24] | |
| Phenol | 5-Dimethylaminonaphthalene-1-sulfonyl chloride | [1,2] |
| 4-Bromomethyl-7-methoxycoumarin | [1,2] | |
| Carboxylic acids | 4-Bromomethyl-7-methoxycoumarin | [1–3,6] |
| 9-(Chloromethyl)anthracene | [6] | |
| 9-Anthradiazomethane | [6] | |
| 9-(Hydroxymethyl)anthracene | [3,6] | |
| 2-Bromoacetyl-6-methoxynaphthalene | [25] | |
| Ketones and aldehydes | 5-Dimethylaminonaphthalene-1-sulfonylhydrazine | [1–3] |
| 2-Aminooxy-N-[3-(5-dimethylaminonaphthalene)-1-sulfonylaminoprpyl]acetamide | [26] | |
| 4-Hydrazino-7-nitrobenzofurazan | [19] | |
| Thiols | N-(9-Acridinyl)maleimide | [12] |
| N-[4-(2-benzoxazoyl)phenyl]maleimide | [12] | |
| 4-(Aminosulfonyl)-7-fluorobenzo-2-oxa-1,3-diazole | [12] | |
| Amines | 5-Dimethylaminonaphthalene-1-sulfonyl chloride | [1–3] |
| 1-Pyrenesulfonyl chloride | [27] | |
| Fluorescamine | [1–3] | |
| o-Phthalaldehyde | [17] | |
| Naphthalene-2,3-dicarboxaldehyde | [20] | |
| 4-Fluoro-7-nitrobenzofurazan | [19] | |
| Fluorenylmethyloxycarbonyl chloride | [17] | |
| 3-Chlorocarbonyl-6,7-dimethoxy-1-methyl-2(1H)- quinoxalinone | [27] |
Numerous reagents have been described for the detection of amines, and in particular, for the on-line detection of amino acids separated by liquid chromatography [17,18,27]. Fluorescamine reacts with water, alcohols, and amines but forms only stable fluorescent products with primary amines. Quantitative reaction in an aqueous–organic solvent mixture buffered at pH 8–9.5 occurs in less than a minute, facilitating postcolumn detection strategies; hydrolysis of the reagent occurring concurrently. o-Phthalaldehyde reacts rapidly, 1–2 min, with amines in an aqueous solution buffered to pH 10 in the presence of a thiol (2-mercaptoethanol, ethanthiol, etc.) to form a highly fluorescent 1-(alkylthio)-2-alkylisoindole, Figure 2.2. This reaction can be used for both precolumn and postcolumn derivatization of amino acids. Isoindole derivatives are unstable and cannot be stored [20]. In some sources it is claimed that the isoindole derivatives formed by naphthalene-2,3-dicarboxaldehde in the presence of the cyanide ion are more stable and a better choice as a precolumn derivatizing reagent for amines [20]. When stability of the derivatives is important, amide derivatives containing an acridine or carbazole fluorophore provide a further alternative [21]. These are conveniently prepared using reagents with a reactive acetyl chloride group. Fluorenylmethyloxycarbonyl chloride reacts with amines in basic solution in less than one minute. The derivatives can be stored but should be isolated from excess reagent, since the derivatives and reagent have similar fluorescent properties. Alternatively, the reaction can be quenched by adding an excess of an amine, easily separated from the derivatives of interest. 4-Chloro-7-nitrobenzofurazan forms strongly fluorescent derivates with amines but only weakly or nonfluorescent products with anilines, phenols, and thiols, often in poor yield [19]. Although the reagent is nonfluorescent, it interferes in the fluorescence of the derivatives and excess reagent should be removed prior to separation. The 4-fluoro-7-nitrobenzofurazan analog is more reactive and has replaced 4-chloro-7-nitrobenzofurazan in many of its conventional applications.
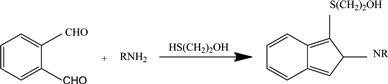
FIGURE 2.2 Reaction of o-phthalaldehyde in the presence of 2-mercaptoethanol to form a highly fluorescent 1-(alkylthio)-2-alkylisoindole.
Only reagents with a hydroxyl group can be considered chemically selective for the derivatization of carboxylic acids. These reactions usually require an activating agent as a catalyst. Reagents containing the diazoalkane group react rapidly and smoothly with carboxylic acids in aqueous or anhydrous solvents to form esters [6]. Diazoalkane reagents do not store well and may require purification before use [16]. Other common reagents for derivatizing carboxylic acids are alkylating reagents which simultaneously react to various extents with phenols, thiols, and amides. 4-Bromomethyl-7-methoxycoumarin in anhydrous acetone smoothly alkylates carboxylic acids in the presence of potassium carbonate solvolysed by a crown ether catalyst [6,16]. Coumaric acid salts formed by the base catalysed solvolysis of the lactone ring of the reagent are potential interfering fluorescent by-products.
Reagents for the derivatization of alcohols vary significantly in their reaction rates. Reagents with acid chloride or nitrile groups require anhydrous conditions. 3,4-Dihydro-6,7-dimethoxy-4-methyl-3-oxo-quinoxaline-2-carbonyl azide is the only choice for tertiary alcohols [3–5]. Many of the reagents used to derivatize alcohols also react with other functional groups, and this can be a problem when specificity is desired or chromatograms are complicated by a large number of unrelated compounds [5]. Detection limits for alcohol derivatives are also frequently no more than modest, leaving some scope for the development of new reagents.
The limiting factor for the measurement of fluorescence is background noise resulting from stray light and instability of the source. A detection method that does not require optical excitation, such as chemiluminescence, should be capable of lower detection limits, perhaps by one to three orders of magnitude, as well as affording a wider linear response range [5,28–30]. Chemiluminescence is the emission of light observed when a chemical reaction yields an electronically excited intermediate or product, which either luminesces (direct chemiluminescence) or donates its energy to a compound containing a fluorophore responsible for the emission (indirect or sensitized chemiluminescence). Except for the excitation process, the production of luminescence is the same as for fluorescence. The compounds of interest are either derivatized with a reagent containing a chemiluminergenic group (direct method) or reacted with a conventional fluorogenic reagent (indirect method), which after separation, generates chemiluminescence in a postcolumn reaction. The first approach could be considered general, while for the second approach, only specific fluorophores generate meaningful chemiluminescence. The most popular of the chemiluminergenic reagents are based on luminol (5-amino-2,3-dihydro-1,4-phthlazine-1,4-dione) or isoluminol (6-amino analog), which react with an oxidizing agent, such as hydrogen peroxide and potassium hexacyanoferrate (III), in an alkaline medium, in a post-olumn reactor, to produce chemiluminescence from the resultant excited aminophthalate dianion. For the indirect approach, the peroxyoxalte chemiluminescence reaction is the most widely used. This reaction involves the oxidation of an aryl oxalate ester with hydrogen peroxide, leading to the formation of an energy-rich intermediate(s) capable of exciting a large number of fluorophores. The intermediate forms a charge transfer complex with the fluorophore, donating one electron to the intermediate, which is transferred back to the fluorophore, raising it to an excited state, which relaxes by emission (and other processes). The process is efficient for fluorophores with high quantum efficiency, a low oxidation potential, and low singlet energy. As a consequence, it is not effective for all fluorophores. Electrochemically produced chemiluminescence employing the oxidation of the tris(2,2’-bipyridyl)ruthenium (II) complex as the reagent species provides an alternative excitation mechanism but has found few applications in liquid chromatography. Chemiluminescence applications are not as common as those based on conventional fluorescence. The much lower detection limits are not always needed, and the greater complexity of the postcolumn reaction conditions detracts from its use for routine methods. The chemiluminescence emission is strongly dependent on the reaction conditions (reagent concentration, pH, solvent composition, temperature, time, etc.), and fluctuations in these conditions have a major effect on precision.
2.2.3 Reagents for Electrochemical Detection
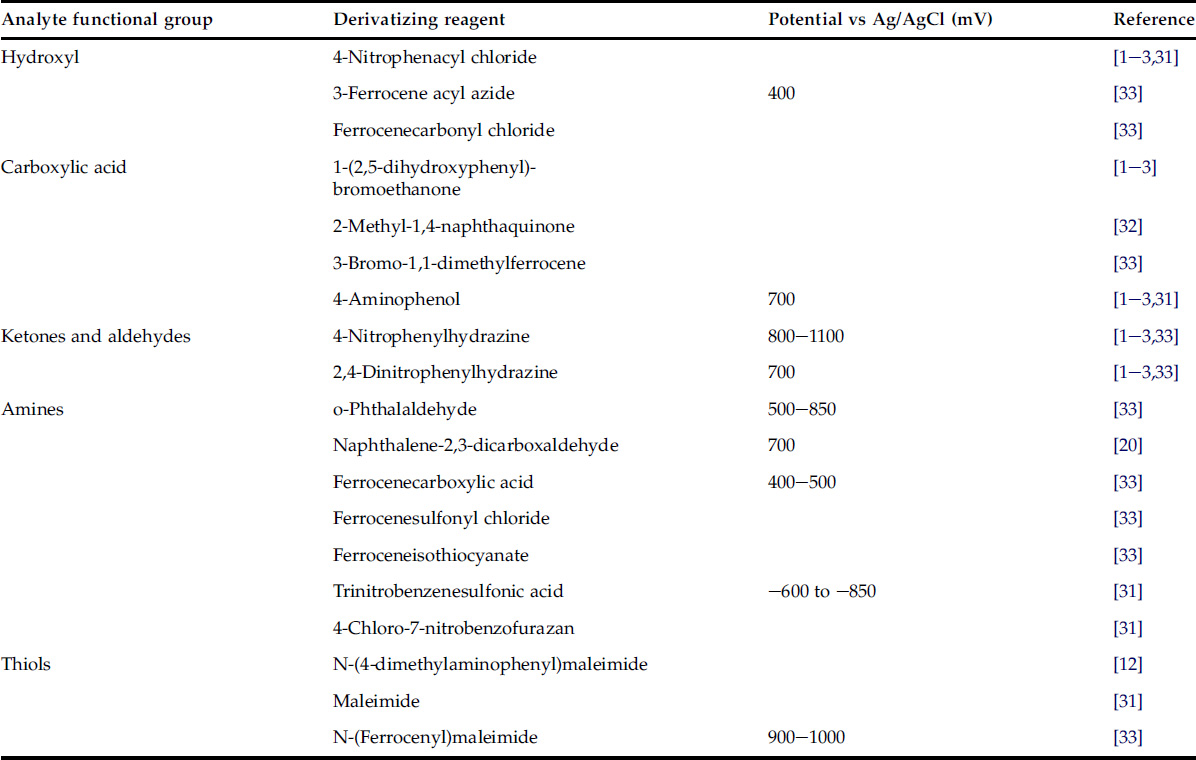
Electrochemical detection complements fluorescence detection in providing an alternative approach to obtain low detection limits (nanomolar) and improved selectivity compared with UV–Visible detection. The common reagents used to install an electrophore into compounds with a reactive functional group are the same as those used to introduce a chromophore or fluorophore, Table 2.5 [31–33]. The isoindole derivatives of amines and amino acids formed by reaction with o-phthalaldehyde (postcolumn) or naphthalene-2,3-dicarboxaldehyde (precolumn) can be determined at low levels with an aperometric detector as an alternative to fluorescence detection [31]. A useful feature of this reaction is that the reagents are not electrochemically active at the potential used to detect the derivatives. A number of reagents containing ferrocene with different reactive groups facilitate reactions with a wide range of compounds containing complementary reactive functional groups forming derivatives with a relatively low oxidation potential (200–600 mV) [33]. This allows selective detection of the ferrocene derivatives, even in the presence of other aromatic compounds. In general, reagents for oxidative electrochemical detection are preferred, due to operational problems in the reduction mode resulting from the difficulty of excluding oxygen from the solvents used to prepare samples and mobile phases. Electrochemical detection is best performed in a buffered aqueous solution, and the presence of organic solvent and variations in pH and ionic strength can degrade the detector response. To minimize changes in the detector response accompanying changes in the mobile phase composition, separations are usually limited to the isocratic mode. Excess reagent and reaction by-products are a possible source of interference that may require isolation of the derivatives prior to detection. From the perspective of typical applications, electrochemical detection of compounds in their native form (when they contain an electrophore) is more common than the detection of compounds containing an electrophore introduced by derivatization. Also, the number of applications in which the electrochemical detector was used for the detection of electrophoric derivatives is only a small fraction of those employing fluorescence detection and fluorophore-containing derivatives.
TABLE 2.5
Reagents for the Introduction of an Electrophore into Compounds with Complementary Reactive Functional Groups
2.2.4 Reagents for Mass-Spectrometric Detection
Many laboratories currently use mass spectrometry for detection based on one or more characteristic mass ions or for identification by fragmentation of one or more characteristic mass ions [14]. When used in combination with liquid chromatography the most important ionization methods are electrospray ionization (ESI) and atmospheric-pressure chemical ionization (APCI). Both are soft ionization methods that produce molecular ions, protonated or deprotonated molecular ions, or catonized molecular ion adducts with very little excess energy and rather limited fragmentation. ESI and APCI differ in several ways, but of note for developing a role for derivatization for mass-spectrometric detection is that ionization in ESI takes place in solution, with ions being expelled into the gas phase, while APCI ionization employs reactions with a plasma of thermalized electrons and solvent-derived ions occurring in the gas phase. In terms of application, the two techniques are complementary rather than competitors. ESI is capable of generating multiply charged ions that allow the identification of large molecules on mass spectrometers of limited scan range. This is of value for the analysis of biopolymers and similar compounds but is not important (or useful) for the analysis of compounds of low mass. In the majority of cases, ESI is selected for the analysis of compounds that exist in an ionic form in solution (this includes compounds that can be easily ionized by buffering the mobile phase or readily form ion adducts with additives added to the mobile phase). APCI is selected for compounds of low to medium polarity and, in particular, for compounds containing elements or groups with a high proton or electron affinity. Derivatization to enhance ionization efficiency is far less common in the case of APCI for small molecules than with ESI. In both cases, derivatization is employed primarily to enhance the ionization efficiency of compounds that are otherwise difficult to detect, to minimize interference in the ionization process by matrix components (largely ion suppression problems), and to produce stable product ions in tandem mass spectrometry for compound class identification or low-level detection using selected reaction monitoring.
A selection of derivatizing reagents for mass-spectrometric detection is summarized in Table 2.6 [34–44]. Some compounds of modest polarity containing hydroxyl, aldehyde, and ketone functional groups are usually poorly ionized by ESI, and derivatives with a charged or chargeable group can dramatically improve detection limits. Low-mass carboxylic acids can be determined as negative ions, but sensitivity is usually low due to high background noise. Lower detection limits are possible after derivatization by moving the characteristic mass ions to regions were background contributions are reduced. Compounds having an amino group are easily protonated under acidic conditions and suitable for ESI mass spectrometry. Derivatization can improve their separation and lower detection limits by minimizing matrix interferences.
TABLE 2.6
Reagents for the Introduction of Ionic or Ionizable Groups into Compounds with Complementary Reactive Functional Groups for Detection by Mass Spectrometry
| Analyte functional group | Derivatizing reagent | Reference |
| Hydroxyl or phenol | 4-(4-Methyl-1-piperazyl)-3-nitrobenzoyl azide | [42] |
| Dansyl chloride | [37,39] | |
| Ferrocenecarbonyl chloride | [33] | |
| Carboxylic acid | Tris(2,4,6-trimethoxyphenyl)phosphonium propylamine bromide | [40] |
| Aminoarenesulfonic acid | [34] | |
| 3-Hydroxy-1-methylpiperidine | [37] | |
| 2-Hydrazinopyridine | [44] | |
| Ketones and aldehydes | 2,4-dinitrophenylhydrazine | [39] |
| 2-Hydrazino-1-methylpyridine | [41] | |
| Girard P and T reagents | [37] | |
| Amines | 5-N-(succinimidoxy-5-oxopentyl)triphenylphosphonium bromide | [38] |
| p-N,N,N-trimethylammonioanilyl N’-hydroxysuccinimidylcarbamate iodide | [43] | |
| 9-Fluorenylmethoxycarbonyl chloride | [34,37] | |
| Dansyl chloride | [34,37] | |
| Thiols | 2-Fluoro-1-methylpyridinium 4-toluenesulfonate | [34] |
To enhance the ionization efficiency in ESI, the derivative should contain either an ionic group or functional group that is easily charged under ESI conditions. In addition, either the reagent or analyte should contain a low polarity region, since during the ESI process, compounds of low polarity accumulate at the surface of the drop and transfer to the gas phase more readily than those in the drop interior. For low mass compounds, derivatives of higher molecular mass are preferred, as they produce characteristic ions in the higher mass range, where noise caused by matrix ions is lower and a cleaner signal is obtained. For high-mass compounds, low-mass derivatives are generally preferred. The size and hydrophobicity of the reagent also influences the chromatography. For almost all applications described so far, reversed-phase liquid chromatography was utilized for the separation. The derivative employed should be tailored to provide suitable separation and mass-spectrometry properties, and for compounds with the same reactive functional groups but different size, different derivatizing reagents may be required. The ESI process is generally more efficient for mobile-phase compositions containing a high volume fraction of organic solvent and the optimum separation conditions by liquid chromatography–mass spectrometry (LC–MS) may be different to those selected for optical detection. High-mass derivatizing reagents may react either slowly or incompletely with some compounds, due to steric hindrance. This can be a limitation for some applications or require more complex protocols using internal standards to improve precision.
Tandem mass spectrometry is being increasingly used for trace analysis and identification. Derivatives can assist in this process by fragmenting into a small number of stable and intense product ions [37–39,44]. If the product ion is characteristic of the derivative, then the parent ions for all compounds that form a similar derivative can be identified. Alternatively, selected reaction monitoring of characteristic product ions is used to facilitate the detection of target compounds at low levels in complex mixtures only partially separated by liquid chromatography. For derivatives with an ionic group, the fixed charge may be responsible for directing fragmentation, producing intense characteristic ions suitable for low-level detection.
2.2.5 Reagents for the Formation of Diastereomers
Enantiomers are stereoisomers that behave identically in an achiral environment but may exhibit different properties in a chiral environment. These differences in a chiral environment explain the general interest in enantiomers in biology and the environment. They also provide the mechanism that allows their separation by liquid chromatography. The interaction or reaction between single enantiomers to form diasteromers is the basis of their separation by liquid chromatography. In practice, two complementary strategies based on this mechanism are used. The most popular strategy, the direct method, involves the formation of transient diastereomer association complexes between a mixture of enantiomers and a chiral stationary phase or chiral additive in the mobile phase (see Chapter 4 for details). The alternative approach, the indirect method, involves formation of diasteromers by a chemical reaction employing a single enantiomer reagent. The last two decades have witnessed enormous advances in the direct approach, which tends to dominate enantiomer separations today. However, the formation of covalent diastereomer derivatives remains a viable option for many compounds, and sometimes it is the only option available.
The formation of diasteriomers by itself does not guarantee that a separation can be obtained; it merely establishes that a separation may be possible using a conventional (achiral) separation system. A useful separation depends on the differences in physical properties of the diastereomers, the extent to which these differences affect the relative distribution of the diasteromers in a biphasic separation system, and the chromatographic efficiency of the separation system.
Several factors need to be considered in the selection of a suitable single enantiomer reagent for a particular application [2,45,46]. The analyte must contain at least one functional group capable of reacting with the derivatizing reagent. Several hundred single enantiomer reagents have been described for reaction with amine, alcohol, phenol, and carboxylic acid functional groups, mainly, as well as a smaller number for reaction with aldehydes, ketones, thiols, epoxides, and the like, Table 2.7 [2,45–50]. Some representative structures are shown in Figure 2.3. The most widely used reactions employ relatively mild conditions and short reaction times, to minimize racemization. The diastereomer transition states should have similar conversion rates for the enantiomers; otherwise, differences in reaction rates may result in kinetic resolution. The diastereomers should also have similar stability in the reaction media. Diasteromers can have different spectrophotometric properties, and calibration of the individual diastereomers may be required for quantitative analysis.
TABLE 2.7
Single Enantiomer Reagents for the Formation of Diastereomers with Enantiomers Containing a Complementary Reactive Functional Group
| Analyte functional group | Derivatizing reagent | Reference |
| Hydroxyl | α-Methoxy-α-(trifluoromethyl)phenylacetic acid | [2,46] |
| 2-t-Butyl-2-methyl-1,3-benzodioxazolo-4-carboxylic acid | [48] | |
| 2-Phenylproionyl chloride | [2,46] | |
| 1-Phenylethyl isocyanate | [46,47] | |
| 1-(1-Naphthylethyl) isocyanate | [48] | |
| Menthyl chloroformate | [47] | |
| 1-(9-Fluorenyl)ethyl chloroformate | [48] | |
| Carboxylic acid | 2-Butanol | [2,46] |
| Menthyl alcohol | [2,46] | |
| 1-Phenylethanol | [46,47] | |
| α-Methyl-4-nitrobenzylamine | [45] | |
| 1-(4-Dansylaminophenyl)ethylamine | [2] | |
| 2-[4-(1-Aminoethyl)phenyl]-6-methoxybenzoate | [45] | |
| Amines | 1-(4-Nitrophenylsulfonyl)propionyl chloride | [50] |
| α-Methoxy-α-(trifluoromethyl)phenylacetyl chloride | [45] | |
| (2,3,4,6-Tetra-O-acetyl)-β-glucopyranosyl isothiocyanate | [2,45,46] | |
| 1,3-Diacetoxy-1-(4-nitrophenyl)-2-propyl isothiocyanate | [47] | |
| 1-Phenylethyl isocyanate | [46,47] | |
| 4-(3-Isothiocyanatopyrrolidin-1-yl)-7-(N,N-dimethylaminosulfonyl)- | ||
| 2,1,3-benzoxadiazole | [47] | |
| 1-Fluoro-2,4-dinitrophenyl-5-alaninamide | [49] | |
| 1-(Fluorenyl)methyl chloroformate | [45] | |
| Menthyl chloroformate | [45] |
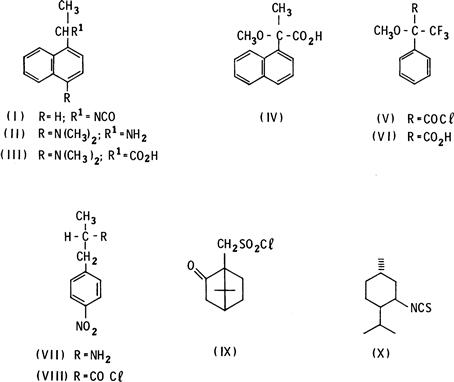
FIGURE 2.3 Reagents for the separation of enantiomers by forming diasereomeric derivatives with compounds containing complementary reactive functional groups.
Reagents: (I) 1-(1-naphthylethyl) isocyanate; (II) 1-(4-dimethylamino-1-naphthyl)ethylamine; (III) 1-(4-dimethylamino-1-naphthyl)acetic acid; (IV) α-methoxy-α-methyl-1-naphthalenecarboxylic acid; (V) α-methoxy-α-trifluoromethylphenacetyl chloride; (VI) carboxylic acid analog of (V); (VII) α-methoxy-4-nitrobenzylamine; (VIII) acid chloride analog of (VII); (IX) camphor-10-sulfonyl chloride; and (X) neomenthyl isothiocyanate.
For amine-activated carboxylic acids (e.g., acid chlorides), chlorofrormates (forming carbonate derivatives), isocyantes (forming urea derivatives), and isothiocyantes (forming thiourea derivatives) are popular general choices. The isocyanate group is selective toward primary amines and secondary amines under mild conditions, no other free functional groups need be protected, and the thiourea derivatives produced allow sensitive UV detection [47]. Marfey’s reagent (1-fluoro-2,4-dinitrophenyl-5-L-alanineamide) is popular for the separation of peptides and related compounds, forming aniline derivatives with good UV detection properties [50]. A number of reagents are used for the introduction of fluorophores into enantiomers combining separation with low-level detection [48–50]. The reaction of amino acids with o-phthalaldehyde and a chiral thiol (e.g., N-butyrylcysteine) results in the formation of highly fluorescent diasteromeric isoindole derivatives [45].
For alcohols, activated carboxylic acids or acyl nitrile reagents (forming ester derivatives), chloroformates (forming carbonate derivatives) and isocyanates (forming carbamate derivatives) are widely used. Reagents containing an acid chloride, anhydride, or acyl cyanide groups are suitable for the derivatization of hydroxyl groups under anhydrous conditions with a catalytic amount of organic base. Because of the lower reactivity of hydroxyl groups with isocyanates and chloroformates, a catalyst (organic base) is usually required.
The most frequently used approaches for derivatizing carboxylic acids are esterification with a number of single enantiomer alcohols or formation of amides with single enantiomer amines. Esterification reactions generally require harsh conditions, and this needs to be considered if either the analyte or diasteromeric derivative is conformationally labile or unstable.
The selection of the reactive functional group of the reagent is quite straightforward but the choice of the correct chiral substituent to facilitate the separation of the diesteromers is often arrived at empirically. Separation is usually favored if the diasteromers have conformationally rigid groups attached to the asymmetric centers and a large size difference between the groups. The distance between the asymmetric centers should be short and ideally less than three bonds. Both reversed-phase and liquid–solid chromatography are used for the separation of diastereomers, with liquid–solid chromatography favored for moderately polar diastereomers, lacking strong hydrogen-bonding functional groups. This takes advantage of the greater stereoselectivity of the adsorption mechanism on surfaces with immobilized active sites. From a practical point of view, single enantiomer reagents of high purity (to detect the presence of 0.5% of a minor enantiomer in a mixture of enantiomers requires a reagent with at least 99.9% enantiomer purity) and with a reasonable shelf life is required. It is advantageous if the derivatizing reagent is available in both the R and S form, to allow elution order reversal, to facilitate the quantification of the minor enantiomer by eluting it before the major enantiomer.
2.2.6 Multifunctional Reagents for the Formation of Cyclic Derivatives
Liquid chromatography is well suited to the separation of cations directly or after formation of stable metal chelates with reasonable solubility in organic solvents [51]. The most widely used chelating reagents are the dithiocarbamates, dithionates, ketoamines, salicylaldimines, dialkyl thiophosphonates, 8-hydroxyquinolines, and tetradentate Schiff bases. The stability of the metal chelates cover a wide range, and some are kinetically labile under typical separation condition used for liquid chromatography. Others may undergo ligand substitution reactions with metal components of the separation system. These considerations tend to define the chelating reagent chosen for the separation of a particular series of cations. Most metal chelates also exhibit strong UV–Visible absorption facilitating their detection. The separation conditions for many metals by ion-exchange chromatography are well known. In this case, chelation reactions are often employed in a postcolumn reactor for multielement detection with detection limits at the nanogram level [51,52]. The reagent is usually a chelating dye, producing strongly absorbing metal complexes that absorb at a different wavelength to the reagent. For a series of metals, it is advantageous if the wavelength maxima for the metal complexes are reasonably close together, so that a single compromise detection wavelength can be used. Common reagents for multielement detection are 4-(2-pyridyl)-azoresorcinol, arsenazo dyes, xylenol organge, dithizone, and dithiocarbamates. Most of the metals that can be separated by ion chromatography can be determined with one or more of these reagents.
Bifunctional organic compounds are characterized by the presence of at least two reactive functional groups on a molecular framework that places these groups into close proximity to one another. As such they do not constitute a defined chemical class but examples are found within almost all of the common classes of functionalized organic compounds, some of which are of biomedical or environmental interest. Typically, these are compounds containing reactive functional groups on 1,2-, 1,3- or 1,4-carbon atoms in an alkyl chain or 1,2-disubstituted aromatic rings. Specific reagents have been developed to react with these functional groups, forming cyclic derivatives [2, 3,53–55]. Reagents used to form cyclic derivatives can be organized into two classes: reagents that are highly selective for specific functional groups and general reagents that react with a wider range of compounds. The specific reagents, although very useful, is not discussed further in this section, because each reaction is its own story and there is little to generalize. The reaction of o-phthalaldehyde in the presence of a thiol with primary amines to form a cyclic isoindole derivative (see Section 2.2.2) is an example of this reagent type. The formation of acetals and ketals by the reaction of aldehydes and ketones with diols, quinoxalinols from the reaction of diamines with α-keto acids, and thiohydantoins formed by amino acids and phenyl isocyanate are further examples.
The most versatile of the general reagents are the boronic acids, which react with hydroxyl, amine, thiol, and carboxylic acid functional groups, as shown in Figure 2.4. Their dominant position as a derivatizing reagent for bifunctional compounds is a consequence of their versatility, ease of reaction under mild conditions, and the possibility of varying the substituent group on the boronic acid to facilitate a wide range of detection options. Some derivatives are labile in aqueous solution, which can be a problem for their separation by liquid chromatography [56].
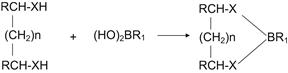
FIGURE 2.4 Formation of cyclic boronates by reaction of a boronic acid with a bifunctional compound (X = O, N, S, or CO2 and n = 0, 1, or 2).
Varying the structure of R1 allows different detection options for the derivatives.
2.2.7 Solid-Phase Analytical Derivatization
Derivatization reactions can be used at an early stage in sample preparation to improve the efficiency or selectivity of the isolation step when using solid-phase extraction, solid-phase microextraction, liquid–liquid extraction, or liquid–phase microextration [57,58]. When a solid phase is used for extraction the derivatization reaction can be performed after elution of the analytes from the extraction column and occurs in solution (conventional reaction) or in the presence of the sorbent prior to elution of the derivatives (heterogeneous reaction). The latter approach called solid–phase analytical derivatization, combines the extraction and derivatization steps into a single procedure, in one of several ways [59,60]. The sorbent could be coated with the derivatizing reagent and the reagent-coated sorbent used to enhance the extraction process as well as simultaneously (or subsequently if a change of conditions is required) perform the derivatization reaction [61]. A typical example is the determination of volatile aldehydes and ketones by gas-phase trapping of the analytes on a sorbent coated with 2,4-dinitrophenylhydazine and subsequent elution of the hydrazone derivatives [62]. Alternatively, the analytes could be extracted by the sorbent and the sorbent immobilized analytes reacted with the derivatizing reagent in a two-step process. This is the most common arrangement for solid-phase extraction and solid-phase microextraction procedures [63,64]. To be effective, it is generally required that, after the reaction, the derivatives can be isolated by a sequence of solvent rinses with minimal contamination by reagents and by-products of the reaction. Full automation of the extraction, derivatization, and separation by liquid chromatography has been demonstrated using a short precolumn connected to the separation column by a multifunctional valve, together with associated pumps and other equipment to introduce the reagent to the precolumn, transfer the derivatives to the analytical column, and clean and prepare the precolumn for the next extraction and reaction [64]. Reactions employing physically adsorbed reagents are most often used. In earlier times, solid-phase reagents utilizing ionic or covalently attached reagents to a polymeric support were used for precolumn and postcolumn reactions, but these are little used today, although solid-phase reagents continue to be used in synthetic organic chemistry [65].
In spite of its attractive features solid-phase analytical derivatization is not widely used at present. It is most useful for trace analysis but requires careful optimization. Reaction yields are rarely quantitative (although reproducible) and background contamination from by-products derived from the extraction sorbent can be a problem. Most applications so far have been set up with gas chromatography as the determinant step rather than liquid chromatography.
2.3 Postcolumn Reaction Detectors
Postcolumn reaction systems are convenient for on-line derivatization of target compounds to facilitate their detection, often at low concentrations [14]. Reactions must be reasonably fast (<20 min) for convenient real-time detection and to maintain the separation integrity. Efficient mixing of the column effluent and reagents is required to form a homogeneous solution. A significant source of detector noise arises from the auxiliary equipment required to provide reagents to the reactor [51]. Although many suitable reaction chemistries have been described [2–4], the general complexity of postcolumn reaction systems and the need for lengthy optimization mean that they are rarely selected for short-term applications. Principal applications are for the detection of amino acids and amines [17], carbohydrates [66,67], pesticides and herbicides [68], and metal ions [51] in clinical, environmental, and food samples. To some extent the use of postcolumn reaction detection has declined in recent years, with the wider availability of affordable mass-spectrometric detectors, and this trend will likely continue.
Some typical postcolumn reaction configurations are shown in Figure 2.5 [14]. The postcolumn reaction system must provide for the continuous addition of one or more reagent solutions to the column effluent, homogeneous mixing of the effluent–reagent streams, and incubation for some time and temperature to complete the reaction. Detection of the reaction products is usually by UV–Visible, fluorescence, or electrochemical detection [51,69]. Pneumatic and syringe pumps are often preferred for reagent delivery as these provide a stable low flow with minimal pulsation (a significant source of detector noise). Peristaltic pumps are suitable for segmented flow systems that generally operate at lower back pressures. Nearly all reaction detectors contain a T or Y piece or cyclone mixer. The mixing device should have a small volume to minimize band broadening and reduce peak dilution. The reaction vessel is usually an open tube or packed bed of appropriate dimensions to store the combined effluent and reagent volumes for the time required for the reaction. If the segmentation principle is employed, a device for introducing a bubble of liquid or gas into the column effluent upstream of the reaction vessel and a phase separator after the reaction vessel but before the detector to remove the segmentation agent are required. The segmentation principle is used for reactions that require a relatively long time for completion or to incorporate an extraction step to remove excess reagent prior to detection. For reactions using solid-phase reagents or catalysts (e.g., immobilized enzymes), a packed=bed reactor is used. Reactions that are too slow at room temperature are accelerated by thermostatting the reaction vessel to a higher temperature. A cooling coil may be required before the detector to minimize thermal noise or bubble formation.
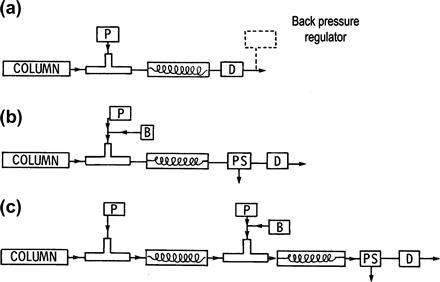
FIGURE 2.5 Some possible postcolumn reaction configurations for liquid chromatography: (a) nonsegmented tubular reactor; (b) segmented tubular reactor; (c) extraction segmented reaction detector.
P = pump, PS = phase separator, B = device for introducing bubbles, and D = detector. Source: Used with permission from [14].
The parabolic-low profile in an open-tubular reactor restricts applications to fast reactions if extensive band broadening is to be avoided [70,71]. The reaction time can be extended by using reactors prepared from optimally deformed capillary tubes. The introduction of secondary flow, even at low flow rates, breaks up the parabolic profile and minimizes band broadening. These reactors are now commercially available in a wide variety of dimensions as knitted open-tubular (KOT) reactors or stitched open-tubular (SOT) reactors. KOT reactors are generally fabricated from PTFE capillary tubes, deformed in such a way as to produce alternating left and right loops with small coiling diameters. SOT reactors are usually prepared from stainless steel capillary tubes woven through a steel mesh in serpentine fashion, with alternate loops displaced to the right and left [51]. For fast reactions (i.e., <1 min), open-tubular or KOT reactors are commonly used. Reagents such as fluorescamine and o-phthalaldehyde are frequently used in this type of reactor to determine primary amines, amino acids and indoles in biological and environmental samples with fluorescence detection [17]. Residues of N-methylcarbamate pesticides in environmental and food extracts can be determined in a two-stage reaction [72]. In the first reactor, the N-methylcarbamates are rapidly hydrolyzed to methylamine, which is then converted to a highly fluorescent isoindole by reaction with o-phthalaldehyde and 2-mercaptoethanol in the second reactor. For reactions of intermediate kinetics (i.e., reaction times <5 and >1 min), a KOT or packed-bed reactor is used. A packed-bed reactor is constructed from a length of column tubing packed with an inert material of small diameter, such as glass beads [73]. The pressure drop across the packed-bed reactor limits the length and smallest particle size that can be used. The inner diameter can be varied to adjust the reactor volume to the required reaction time. For slow reactions (reaction times >5 min), an air or liquid segmented reactor is typically used, although the complexity of these systems means that contemporary applications are few.
2.3.1 Photoreactors
Photolysis can be used to convert some compounds into easier-to-detect products for subsequent on-line derivatization [29,31,68,69,73]. Photolysis causes degradation into smaller molecules (photolysis) and possibly their further reaction with water (photohydrolysis), intramolecular rearrangements, photodimerization, or photoionization as well as possible electron transfer. These photolysis products can be detected at low concentrations directly or after conversion to suitable derivatives by UV–Visible, fluorescence, or electrochemical detection. The primary reagent for photolysis reactions are photons, and their reactivity can be controlled to some extent by varying the excitation wavelength and intensity. However, the main limitation remains the limited number of compounds that yield useful detection products by photolysis.
Photoreactors are simple in design, consisting of one or more high-intensity discharge lamps with a PTFE KOT tube reactor wound around one of the lamps. The PTFE tube creates a light-tube effect, which increases the effective photon flux overcoming the inefficiency of direct transmission of UV light by PTFE. The KOT reactor is housed in a reflective housing to maximize the photon flux and usually has some provision to remove excess heat. A typical application is the photodecomposition of N-nitrosocompounds releasing nitrite, which can be determined calorimetrically by the Greiss reaction. Nitrites are also released by photolysis of organonitro compounds (explosives), which can be conveniently detected with an electrochemical detector. Phenyl urea herbicides yield alkylamines by photolysis that can be detected by reaction with one of the standard amine reagents for fluorescence detection.
2.4 Conclusions
A wide variety of chemical reactions can be employed to transform target compounds into new compounds that are easier to separate by liquid chromatography or to detect with common detectors. With such a wealth of possible reagents, selection can seem more confusing than it need be and several reagents may be equally suitable for a particular problem. In other cases, where either chromatographic or detector selectivity is important, identifying a suitable reagent is more critical; and to be able to start from several possible options more often leads to success. One aspect of reagent selection that could be improved is in the area of comparative studies, so that the reagent used for a particular application could be identified more easily as the preferred choice for compounds of that type or for matrices of a certain type. Derivatives are not prepared without a need, and the buoyant literature on this topic is an indication that chemistry still plays a significant role in instrumental analysis.
References
1. Knapp DR. Handbook of analytical derivatization reactions. New York: John Wiley & Sons; 1979.
2. Blau K, Halket J, eds. Handbook of derivatives for chromatography. Chichester, England: John Wiley; 1993.
3. Lunn G, Hellwig LC. Handbook of derivatization reactions for HPLC. New York: John Wiley & Sons; 1998.
4. Toyo’oka T. Modern derivatization methods for separation sciences. Chichester, England: Wiley; 1999.
5. Fukushima T, Usui N, Santa T, Imai K. Recent progress in derivatization methods for LC and CE analysis. J Pharm Biomed Anal. 2003;30:1655–1687.
6. Mukherjee PS, Karnes HT. Ultraviolet and fluorescence derivatization reagents for carboxylic acids suitable for high performance liquid chromatography: a review. Biomed Chromatogr. 1996;10:193–204.
7. Rosenfeld JM. Derivatization in the current practice of analytical chemistry. Trends Anal Chem. 2003;22:785–798.
8. Medvedovici A, Farca A, David V. Derivatization reactions in liquid chromatography for drug assaying in biological fluids. Adv Chromatogr. 2009;47:283–322.
9. Uchiyama S, Inaba Y, Kunugita N. Derivatization of carbonyl compounds with 2,4-dinitrophenylhydrazine and their subsequent determination by high-performance liquid chromatography. J Chromatogr B. 2011;879:1282–1289.
10. Lamari FN, Kuhn R, Karamanos N. Derivatization of carbohydrates for chromatographic, electrophoretic and mass spectrometric structure analysis. J Chromatogr B. 2003;793:15–36.
11. Kusmiere K, Chwatko G, Glowacki R, Kubalczyk P, Bald E. Ultraviolet derivatization of low-molecular-mass thiols for high performance liquid chromatography and capillary electrophoresis analysis. J Chromatogr B. 2011;879:1209–1307.
12. Toyo’oka T. Review recent advances in separation and detection methods for thiol compounds in biological samples. J Chromatogr B. 2009;877:3318–3330.
13. Palego L, Giannaccini G, Saccomanni G, et al. Modified RP-LC of phenylthiocarbamyl amino acid adducts in plasma acetonitrile extracts using multiple internal standards and phot-diode UV detection. Chromatographia. 2010;71:291–297.
14. Poole CF. The essence of chromatography. Amsterdam: Elsevier; 2003.
15. MacCrehan WA, Schonberger F. Determination of vitamin K1 inserum using catalytic-reduction liquid chromatography with fluorescence detection. J Chromatogr B. 1995;670:209–217.
16. Toyo’oka T. Fluorescent tagging of physiologically important carboxylic acids, including fatty acids, for their detection in liquid chromatography. Anal Chim Acta. 2002;465:111–130.
17. Molnar-Perl I. Advancement in the derivatization of the amino groups with o-phthalaldehyde-thiol and with 9-fluorenylmethyloxycarbonyl chloride reagent. J Chromatogr B. 2011;87:1241–1269.
18. Hernandez-Cassou S, Saurina J. Derivatization strategies for the determination of biogenic amines in wines by chromatographic and electrophoretic techniques. J Chromatogr B. 2011;879:1270–1281.
19. Santa T, Fukushima T, Ichibangase T, Imai K. Recent progress in the development of derivatization reagents having a benzofurazan structure. Biomed Chromatogr. 2008;22:343–353.
20. Rammouz G, Lacroix M, Garrigues JC, Poinsot V, Coudere FO. The use of naphthalene-2,3-dicarboxaldehyde for the analysis of primary amines using high-performance liquid chromatography and capillary electrophoresis. Biomed Chromatogr. 2007;21:1223–1239.
21. Fan X, You J, Kang Q, Ou Q, Zhu Q. New reagents for the determination of amino acids by liquid chromatography with pre-column fluorescence derivatization. Anal Chim Acta. 1998;367:81–91.
22. Glowka FK, Karazniewicz M, Lipnicka E. RP-HPLC with fluorescence detection for the determination of small quantities of triamcinolone in plasma in the presence of endogeneous steroids after derivatization with 9-anthronyl nitrile, pharmacokinetic studies. J Chromatogr B. 2006;839:54–61.
23. McEntyre CJ, Slow S, Lever M. Measurement of plasma free choline by high performance liquid chromatography with fluorescence detection following derivatization with 1-naphthyl isocyanate. Anal Chim Acta. 2009;644:90–94.
24. Takechi H, Goto Y, Machida M. 4-(7-diethylaminocoumarin-3-yl)benzoyl cyanide (DBCB-CN): a highly sensitive fluorescent derivatization reagent for alcohols in high performance liquid chromatography. Chem Pharm Bull. 1998;46:159–162.
25. Gotti R, Bousquet E, Bonazzi D, Cavrini V. Determination of carboxylic acid salts in pharmaceuticals by high performance liquid chromatography after pre-column fluorogenic labeling. Biomed Chromatogr. 1996;60:9–24.
26. Houdier S, Perrier S, Defrancq E, Legrand M. A new fluorescent probe for sensitive detection of carbonyl compounds: sensitivity improvement and application to environmental water samples. Anal Chim Acta. 2000;412:221–233.
27. Kurihara T, Min JZ, Toyo’oka T, Fukushima T, Inagaki S. Determination of fluorescence-labeled asparaginyl-oligosaccharide in glycoprotein by reversed-phase ultraperformance liquid chromatography and time-of-flight mass spectrometry. Anal Chem. 2007;79:8694–8698.
28. Ohba Y, Kuroda N, Nakashima K. Liquid chromatography of fatty acids with chemiluminescence detection. Anal Chim Acta. 2002;465:101–109.
29. Lara FJ, Garcia-Campana AM, Aaron J- J. Analytical applications of photoinduced chemiluminescence in flow systems—a review. Anal Chim Acta. 2010;679:17–30.
30. Gamiz-Gracia L, Garcia-Campana AM, Huertas-Perez JF, Lara FJ. Chemiluminescence detection in liquid chromatography: applications to clinical, pharmaceutical, environmental and food analysis—a review. Anal Chim Acta. 2009;640:7–28.
31. Rose MJ, Lunte SM, Carlson RG, Stobaugh JF. Transformation of analytes for electrochemical detection: a review of chemical and physical approaches. Adv Chromatogr. 2001;41:203–248.
32. Trojanowicz M. Recent developments in electrochemical flow detections—a review, Part II Liquid chromatography. Anal Chim Acta. 2011;688:8–35.
33. Seiwert B, Karst U. Ferrocene-based derivatization in analytical chemistry. Anal Bioanal Chem. 2008;390:181–200.
34. Zaikin VG, Halket JM. Derivatization in mass spectrometry—8 Soft ionization mass spectrometry of small molecules. Eur J Mass Spectrom. 2006;12:79–115.
35. Zaikin V, Halket JM. A handbook of derivatives for mass spectrometery. Chichester: England; 2009; I.M. Publications.
36. Xu FG, Zou L, Liu Y, Zhang ZJ, Ong CN. Enhancement of the capabilities of liquid chromatography–mass spectrometry with derivatization: general principles and applications. Mass Spectrom Rev. 2011;30:1143–1172.
37. Santa T. Derivatization reagents in liquid chromatography/electrospray ionization tandem mass spectrometry. Biomed Chromatogr. 2010;25:1–10.
38. Inagaki S, Tano Y, Yamakata Y, Higashi T, Min JZ, Toyo’oka T. Highly sensitive and positively charged precolumn derivatization reagents for amines and amino acids in liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:1358–1364.
39. Iwasaki Y, Nakano Y, Mochizuki K, et al. A new strategy for ionization enhancement by derivatization for mass spectrometry. J Chromatogr B. 2011;879:159–169.
40. Cartwright AJ, Jones P, Wolf JC, Evans EH. Derivatization of carboxylic acid groups in pharmaceuticals for enhanced detection using liquid chromatography with electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:1058–1062.
41. Higashi T, Yamauchi A, Shimada K. 2-Hydrazino-1-methylpyridine a highly sensitive derivatization reagent for oxosteroids in liquid chromatography-electrospray ionization–mass spectrometry. J Chromatogr B. 2005;825:214–222.
42. Nishio T, Higashi T, Funaishi A, Tanaka J, Shimada K. Development and application of electrospray–active derivatization reagents for hydroxysteroids. J Pharm Biomed Anal. 2007;44:786–795.
43. Shimbo K, Yahashi A, Hivayama K, Nakazawa M, Miyano H. Multifunctional and highly sensitive precolumn reagents for amino acids in liquid chromatography/tandem mass spectrometry. Anal Chem. 2009;81:5172–5179.
44. Higashi T, Ichikawa I, Inagaki S, Min JZ, Fukushima T, Toyo’oka T. Simple and practical derivatization procedure for the enhanced detection of carboxylic acids in liquid chromatography–electrospray ionization-tandem mass spectrometry. J Phar Biomed Anal. 2010;52:809–818.
45. Srinivas NR. Evaluation of experimental strategies for the development of chiral chromatographic methods based on diastereomer formation. Biomed Chromatogr. 2004;8:207–233 1.
46. Gorog S, Gazdag M. Enantiomeric derivatization for biomedical chromatography. J Chromatogr B. 1994;659:51–84.
47. Peter M, Peter A, Fulop J. Application of (1S,2S)- and (1R,2R)-1,3-diacetoxy-1-(4-nitrophenyl)-2-propylisothiocyanate to the indirect enantioseparation or racemic proteinogenic amino acids. J Chromatogr A. 2000;871:115–126.
48. Toyo’oka T. Fluorescent chiral derivatization reagents possessing benzofurazan structure for the resolution of optical isomers in HPLC: the synthesis, characteristics and application. Current Pharm Anal. 2005;1:57–64.
49. Toyo’oka T. Recent progress in liquid chromatographic enantioseparaton based upon diastereomer formation with fluorescent chiral derivatization reagents. Biomed Chromatogr. 1996;10:265–277.
50. Ilisz I, Berkeez R, Peter A. Application of chiral derivatizing agents in the high-performance liquid chromatographic separation of amino acid enantiomers: a review. J Pharm Biomed Anal. 2007;47:1–15.
51. Nesterenko PN, Jones P, Paull B. High performance chelation ion chromatography. Cambridge, England: Royal Society of Chemistry; 2011.
52. Ueno K, Imamura T, Cheng KL. Handbook of organic analytical reagents. Boca Raton, FL: CRC Press; 2000.
53. Poole CF, Zlatkis A. Cyclic derivatives for the selective chromatographic analysis of bifunctional compounds. J Chromatogr A. 1980;184:99–183.
54. Zalkin VG, Halket JM. Derivatization in mass spectrometry, 4 Formation of cyclic derivatives. Eur J Mass Spectrom. 2004;10:555–568.
55. Hammad SF, Mabrouk MM, Habib A, et al. Precolumn fluorescence labeling method for simultaneous determination of hydroxyzine and cetirizing in human serum. Biomed Chromatogr. 2007;21:1030–1050.
56. Zhong QQ, Ngim KK, Sun M, Li J, Deese A, Chetwyn NP. Strategies for the analysis of highly reactive pinacolboronate esters. J Chromatogr A. 2012;1229:216–222.
57. Nerin C, Salafranea J, Aznar M, Bartlle R. Critical review on recent developments in solventless techniques for extraction of analytes. Anal Bioanal Chem. 2009;393:809–833.
58. Xu L, Basheer C, Lee H- K. Chemical reactions in liquid-phase microextraction. J Chromatogr A. 2009;1216:701–707.
59. Rosenfeld JM. Solid-phase analytical derivatization: enhancement of sensitivity and selectivity of analysis. J Chromatogr A. 1999;843:19–27.
60. Johnson ME, Carpenter TS. The use of solid-phase supports for derivatization in chromatography and spectroscopy. Appl Spectrosc Rev. 2005;40:391–412.
61. Atapattu SN, Rosenfeld JM. Solid-phase analytical derivatization of anthropogenic and natural phenolic estrogen mimics with pentafluoropyridine for gas chromatography–mass spectrometry. J Chromatogr A. 2011;1218:9135–9141.
62. Prokai L, Szarka S, Wang X, Prokai-Tatrai K. Capture of the volatile carbonyl metabolites of flecainide on 2,4-dinitrophenylhydrazine cartridge for quantitative stable isotope dilution mass spectrometry coupled with chromatography. J Chromatogr A. 2012;1232:281–287.
63. Yu LZ, Wells MJM. Establishing the feasibility of coupled solid-phase extraction–solid-phase derivatization for acidic herbicides. J Chromatogr A. 2007;1143:16–25.
64. Lord HL, Rosenfeld J, Volovich V, Kumbhare D, Parkinson B. Determination of malondialdehyde in human plasma by fully automated solid-phase analytical derivatization. J Chromatogr B. 2008;877:1292–1298.
65. Krull IS, Szulc ME, Bourque AJ, Zhou FX, Yu J, Strong R. Solid-phase derivatization reactions for biomedical liquid chromatography. J Chromatogr B. 1994;659:19–50.
66. El Rassi Z, ed. Carbohydrate analysis High performance liquid chromatography and capillary electrophoresis. Amsterdam: Elsevier; 1995.
67. Harvey DJ. Derivatization of carbohydrates for analysis by chromatography, electrophoresis and mass spectrometry. J Chromatogr B. 2011;879:1196–1225.
68. Ruberu SR, Draper WM, Perera SK. Multiresidue HPLC method for phenyl urea herbicides in water. J Agric Food Chem. 2000;48:4109–4115.
69. Lunte SM. Pre- and post-column derivatization reactions for liquid chromatography– electrochemical detection. Trends Anal Chem. 1991;10:92–97.
70. Waiz S, Cedillo BM, Jambunathan S, Holinbolt SG, Dasgupta PK, Wolcott DK. Dispersion in open tubular reactors of various geometries. Anal Chim Acta. 2010;428:163–171.
71. Neue UD. Geometrically deformed tubes as efficient vessels for post-column derivatization in high-performance liquid chromatography. Chem Eng Process. 2010;49:662–671.
72. McGarvey BD. Derivatization reactions applicable to pesticide determination by high performance liquid chromatography. J Chromatogr B. 1994;659:243–257.
73. Fedorowski J, LaCourse WR. A review of post-column photochemical reaction systems coupled to electrochemical detection in HPLC. Anal Chim Acta. 2010;657:1–8.