Protein and Peptide Separations
J. Giacometti∗ and D. Josić∗, †, ∗Department of Biotechnology, University of Rijeka, Croatia, †Warren Alpert Medical School, Brown University, Providence, Rhode Island,
Outline
7.1. Introduction
7.2. Methods of Protein Liquid Chromatography
7.2.1. Size-Exclusion Chromatography
7.2.2. Ion-Exchange Chromatography
7.2.3. Methods Based on the Hydrophobic Interaction
7.2.4. Affinity Chromatography
7.2.5. Chromatography on Hydroxyapatite
7.2.6. Chromatography on Monolithic Supports
7.2.7. Displacement Chromatography
7.3. Conclusions
7.1 Introduction
The development of techniques and methods for protein purification is very important for bioscience and biotechnology. Various chromatographic techniques with different selectivities are powerful methods for the purification of biomolecules, especially the separation of proteins. New developments in high-resolution liquid chromatography made this method compatible with mass spectrometry and an indispensable tool in proteomic research.
In general, a chromatographic separation depends on the differential partition of proteins between the stationary phase (the chromatographic medium or the adsorbent) and the mobile phase (the buffer or organic solvent). Several liquid chromatographic methods are used for the separation of proteins. They differ mainly in the type of stationary phase employed (Table 7.1).
TABLE 7.1
Methods of Protein Liquid Chromatography
| Type of separation | Type of chromatography |
| Size and shape | Gel filtration (GF)/size-exclusion chromatography (SEC) |
| Net charge | Ion-exchange chromatography (IEX) |
| Hydrophobicity | Hydrophobic-interaction chromatography (HIC) Reversed-phase chromatography (RPC) |
| Biological function | Affinity chromatography (AC) |
| Interaction with ligands Metal binding |
Pseudo-affinity chromatography Immobilized metal ion affinity chromatography (IMAC) |
| Other | Chromatography on hydroxyapatite (HT/HTP) |
A wide variety of materials have been used as chromatographic matrices for protein separations, classified as inorganic supports, synthetic organic polymers, or polysaccharides. Traditional standard chromatography media as well as modern high-performance media are included among these groups, as shown in Figure 7.1 [1].

FIGURE 7.1 Chromatographic materials for protein separation.
All these bulk materials are designed to fulfill a general matrix requirement for protein chromatography—to minimize the interaction between the sample and the surface of the support.
A combination of optimal chemical and physical properties is achieved by the use of hydroxyl or amino groups, and such standard chromatographic media, based on neutral polysaccharides and modified polyamides, are widely used for protein separations [2].
Sample preparation and extraction procedures are an important prepurification step. Different methods are used in the purification of a target protein, such us precipitation, preparative centrifugation, two-phase systems, electrophoresis, or chromatography. In chromatography, proteins are separated on the basis of their different affinity to a stationary or a mobile phase.
Purification of proteins is often associated with a number of steps in order to obtain higher purity, defined as a purification strategy. However, all of these steps are associated with sample loss. On the other hand, a single chromatographic step is most often insufficient, and it is necessary to apply several steps to achieve the desired purity of the target molecule. To meet these criteria, further development of higher selectivity and effectiveness of chromatographic supports is still required. To determine the conditions for best performance, it is also necessary to optimize the chromatographic steps during protein purification [3].
In addition, sometimes, it is necessary to remove solvent from the eluate to achieve higher concentrations of stable proteins in a final form, ready for the intended application. That requires an additional step in the product preparation (e.g., freeze drying, ultrafiltration, or dialysis).
Recombinant DNA techniques are widely used in the production of therapeutic proteins, and the problems associated with solubility, structural integrity and biological activity, and contamination with host proteins have to be reduced to a minimum. Consequently, the development of fast, reliable analytical assays is essential for monitoring the progress of a purification process and assessing its effectiveness (yield, biological activity, recovery) [4].
Until the end of the last century, bulk chromatographic materials containing porous or nonporous particles were used. Monolithic materials made of synthetic or natural polymers and silica-based monoliths with similar surface chemistry have also been used as additional chromatographic supports in the last 15–20 years. Due to the rapid interaction between the sample components and the surface, monolithic materials enable very fast chromatographic separation of large molecules such as proteins and nucleic acids and nanoparticles such as viruses and protein aggregates [5,6].
7.2 Methods of Protein Liquid Chromatography
7.2.1 Size-Exclusion Chromatography
The matrices in size-exclusion chromatography (SEC) consist of porous particles, and the separation of proteins is achieved according to the size and shape of the protein (see Figure 7.2) that passes through a packed column. This technique is sometimes referred as gel-filtration, molecular sieve, or gel-permeation chromatography. SEC can be used in LC for the analysis of sample components if there are sufficient molecular-weight differences among solutes [7].
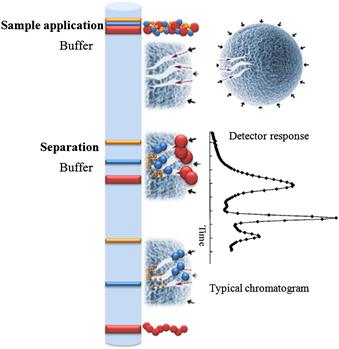
FIGURE 7.2 Principles of size-exclusion chromatography (SEC).
In this chromatographic method, the analyte does not interact with the surface of the stationary phase. Separation is achieved by the differential penetration and exclusion of the sample components in and out of the pores of the packing material. Particles of different sizes elute at different rates. Small molecules, which can penetrate into the pores of the stationary phase, elute later. On the other hand, a very large molecule, which cannot penetrate into the pore system, elutes earlier, in the dead volume of the column. The molecules of intermediate size, which can partially penetrate the pores of the stationary phase elute in the intermediary time, between very large and very small molecules.
The selectivity of porous bulk materials in SEC depends solely on their porosity. Consequently, an appropriate matrix for SEC is a support with reduced adsorptive properties such as natural (mostly agarose or dextran) and synthetic polymers (mostly polyacrylamide). The pore size of chromatographic materials (e.g., Sephadex) depends on the degree of cross-linking in the particular polymer. Predominantly, hydroxyl groups coverage is favorable for use in the fractionation of hydrophilic proteins. After addition of water, some polymers such as agarose form gels spontaneously. To prevent of denaturation of proteins, macroporous silica, used as a support for SEC, must be coated with a protecting hydrophilic layer [7].
To achieve an optimal and reproducible separation, proper selection of a gel with a suitable separation range is necessary. In general, resins are classified based on the different hypothetical sizes of globular proteins. The residence of proteins and their separation in SEC depends on their size and shape. In general, the gels with smaller pores are used for fast desalting procedures (e.g., peptide purification), and the gels with larger ones are used for separation of small- and middle-sized proteins. Stationary phases containing particles with very large pore sizes are used for purification of biological complexes [8]. Uniformity of pore distributions within the gel is also crucial for the optimal separation of proteins [7].
Contrary to other chromatographic media, the selectivity of SEC matrix is not dependent on adjusting the composition of the mobile phase. To preserve the structure and biological activity of proteins, the sample can sometimes require a buffer solution of defined pH and ionic composition. However, high salt concentration in the mobile phase should be avoided to prevent the undesirable interaction of proteins with the support [7].
Compared to other chromatographic methods, both capacity and resolution in SEC are relatively low. An additional problem is the degree of sample dilution. SEC is a useful and simple method for the separation of multimers. An advantage of this approach is the high biological activity of proteins after SEC due to the absence of surface interactions of the analyte with the chromatographic support. Sometimes, this method is also used as the final polishing step in cases of low sample volume. Finally, SEC has a wide range of applicability both in preparative (buffer exchange and protein fractionation) and analytical protein purification [9].
Finally, size-exclusion chromatography coupled with more than one detector, such as laser light-scattering (LS) and refractive-index (RI) detection, provides an excellent approach for determination of the molecular weight (MW) of proteins [10].
7.2.2 Ion-Exchange Chromatography
Ion-exchange chromatography (IEX) has been used for more than 50 years for the separation and purification of proteins. In comparison with other chromatographic methods, 40% of all protein separations are related to IEX, 18% to SEC, and 29% to affinity chromatography (including immobilized-metal-affinity and dye-affinity chromatography) [1].
IEX is a method for purification of proteins based on ionic interactions between proteins and surface charges opposite to those of the charged groups on an ionic resin (see Figure 7.3). In cation-exchange chromatography, positively charged molecules are attracted to a negatively charged solid support. Conversely, in anion-exchange chromatography, negatively charged molecules are attracted to a positively charged solid support. The separation occurs due to competition between proteins with different charges on an ion-exchange resin.
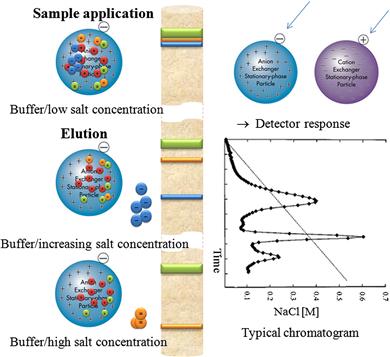
FIGURE 7.3 Principles of ion exchange chromatography (IEX).
Biomolecules exhibit different degrees of interaction with charged chromatography media according to differences in their overall charge, charge density, and surface charge distribution. The separation process is based on the formation of ionic bonds between the charged groups on biomolecules (mostly, –NH3+ , =NH2+, ≡NH+, –COO–, PO43–, SO32–), and an ion-exchange support with the opposite charge. Nonbound biomolecules (i.e., neutral molecules with no electrical charge or molecules with the same charge as the ion-exchange support) are removed by washing, and bound biomolecules are recovered by elution with a buffer of either higher ionic strength or altered pH.
Proteins are complex ampholytes whose charges depend on the proportions of the amino acid residues in their structure as well as the acidity of the aqueous separation media. The isoelectric point (pI) of a protein depends on the structural proportion of ionizable amino acids. Positive charges are typically found when the pH of the protein solution is below 8, due to the N-terminal amine and basic residues such as arginines, lysines, and histidines. Similarly, negative protein charges typically exist above pH 6 and are due to the C-terminal carboxyl group and acidic (e.g., aspartate and glutamate) residues. The charged groups are almost always on the surface of proteins, except in case of metalloproteins, where the metal ion (usually inside the molecule) is usually coordinated by ligands (amino acid residues of the protein) [11].
A stoichiometric model describes the relationship between the charged groups in a protein and the stationary phase. The number of charged groups of the protein binds to the same number of oppositely charged groups of an ionic exchanger, and counterions are released both from the protein and the ion exchanger (see Figure 7.3). Protein retention on an ionic surface depends on the protein charge, surface charge, and the charge characteristics of the surrounding medium. To describe this phenomenon, Kopaciewicz et al. [12,13] developed a nonmechanistic model that shows a positive correlation between protein retention and the number of charges associated with the adsorption-desorption process.
Generally, anion-exchange chromatography (AEX) is used at pH values above the isoelectric point of the protein of interest, while cation-exchange chromatography (CEX) is performed below the isoelectric point. At low pH or even at very high ionic strength, proteins may adsorb very strongly to an ion exchanger. This probably occurs due to an increase in the number of hydrogen bonds [11].
The interaction between a protein and an ion exchanger depends not only on the net charge and the ionic strength but also on the surface charge distribution and conformation of the protein. Some structural changes can affect the separation by IEX. Urea is widely employed to facilitate protein separations in ion exchange chromatography at various scales. Hou et al. [14] indicated that the retention times correlate well with structural changes and they are more sensitive to the change of the tertiary structure.
The properties of the ion exchanger also influence the protein separation. Depending on the functional group, ion exchangers are classified as weak or strong. Use of strong ion exchangers, such as sulfonate (CEX) and quaternary ammonium (AEX), with pKa values outside the pH range for work with proteins (i.e., pH 4–10) result in the charge of the ion exchanger remaining the same despite changes in mobile phase pH. These ion exchangers are applicable in case of weakly ionizable proteins. The benefits of the use of weak ion exchangers are related to a reduced tendency for sample denaturation, less ability to bind impurities, as well as enhanced resolution.
In general, two methods are applicable in the elution strategy in IEX: changing the pH of the eluting buffer, and increasing the ionic strength by addition of salts, mostly NaCl.
The most common active groups related to the ion exchange chromatographic matrices are carboxyl (–COOH), sulfonyl (–SO3H), secondary or tertiary amines (–NH2, –NRH), and a quaternary amino group (-N+R2H). The ion exchangers are classified as weak-cation, strong-cation, weak-anion, and strong-anion exchangers, respectively.
Lendero et al. [15,16] studied a universal nondestructive, noncontaminating method for the characterization of ion-exchange chromatographic columns. This method is based on making a step change in ionic strength of buffer solutions with the same pH in the ion exchange columns and can be used for identification and determination of the type of ion exchange groups on all sorts of ion exchangers (see Figure 7.4). This investigation resulted in the observations that, after the step change from Tris–HCl buffer to Tris–HCl buffer with sodium chloride: (1) the effluent pH for strong-anion-exchange columns remains nearly the same or rises by less than 0.5 pH units in the shape of sharp and relatively short peak; (2) for weak-anion-exchange columns, it raises more than 1 pH unit and this lasts several column volumes (CV); (3) for cation-exchange columns, it drops by more than 1 pH unit.
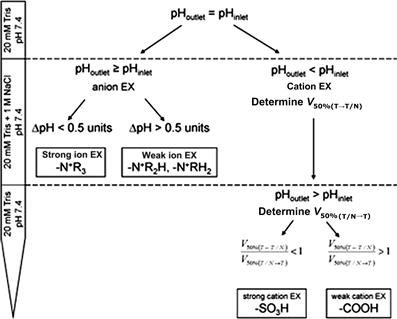
FIGURE 7.4 A scheme to distinguish among different active groups on the most common ion exchangers.
To distinguish among different ion exchange groups on the convection interaction media supports (CIM), Tris–HCl buffer pH 7.4 was pumped through the column. The change in pH values of the solution (Tris–HCl buffer with NaCl) and at the column outlet were measured. The pH profiles versus elution volume normalized to the column volume were compared in three parts of the profile: (a) at low ionic-strength buffer (the effluent pH for the weak-anion exchanger DEAE was lower than for the other ion exchangers), (b) at high ionic-strength buffer (the pH of the cation exchangers reduced, whereas the pH of the strong anion exchanger remained the same), and (c) the two cation exchangers were classified by the duration of the pH drop (for the weak cation exchanger, the pH drop lasted longer than for the strong cation exchanger). Source: Printed from the Ref. [15] with permission.
In practice, the strategy based on the ionic strength by the addition of NaCl is the method of choice. As a rule, more weakly charged proteins are eluted at lower salt concentrations, while the more strongly charged proteins are eluted at higher salt concentrations. Stepwise elution is often used for recovery of a concentrated protein especially in preparative chromatography. In this case, the optimization of gradient conditions is needed [17–19].
Finally, IEX is one of the most used separation techniques in protein purification, for an advantage of this technique is that the elution normally takes place under mild conditions, and the protein can maintain its native conformation during the chromatographic process. Limited selectivity is the major disadvantage of this method [20].
To establish the structural and functional relationships in the characterization of all human proteins, of greatest importance are proteins of the blood. Blood plasma contains an unusually small group of high-abundance proteins, namely, serum albumin, immunoglobulins, transferrin, and α-2-macroglobulin. The amount of these proteins is about 85% of the total serum protein, and they often interfere with the identification of proteins of lower abundance. In proteomics, characterization of the blood proteome requires extensive fractionation prior to mass spectrometry analyses, and the removal of high-abundance proteins and subsequent enrichment of low-abundance proteins is a crucial step. For this purpose, IEX can be combined with other chromatographic methods, mostly with SEC and chromatography on hydrophobic resins [21,22].
As shown in Figure 7.5, CEX and AEX, used after SEC and before hydrophobic interaction chromatography (HIC), were applied in the separation of human plasma proteins. After the initial fractionation steps using ammonium sulfate precipitation and SEC, in the next steps, cation- and anion-exchange chromatography resulted in the highest number of identified proteins in the human plasma proteome [23].
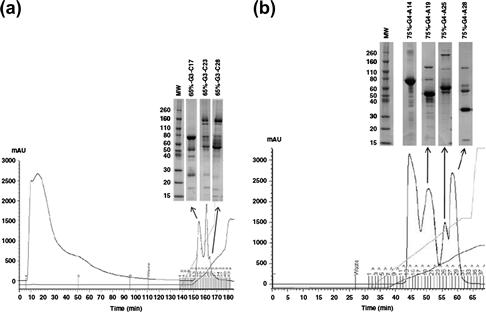
FIGURE 7.5 (a) Cation-exchange chromatography trace (UV 280 nm, black line) of the interim Analyte Library fraction of 65%-G3 (65% ammonium sulfate saturation, third pooled gel-filtration fraction). Conditions: HiTrap SP HP column with 0 to 0.5 M KCl (in 50 mM phosphate buffer, pH 5.5) gradient elution, flow rate: 1 ml/min, 1-ml fractions. (b) Anion-exchange chromatography trace (UV 280 nm, dark line) of the cation exchange chromatography flow-through of fraction 75%-G4 (75% ammonium sulfate saturation, fourth pooled gel filtration fraction). Conditions: HiTrap Q HP column, 0–0.5 M KCl (in 20 mM Tris–HCl, pH 8.5) gradient elution; flow rate, 1 ml/min, 1-ml fractions. Source: Reprinted from Ref. [23] with permission.
In proteomics, multidimensional chromatographic separation and analysis of the proteins are performed at the peptide level, after proteolytic digestion of the entire proteins extracted from tissue samples (the bottom-up approach) [24]. Strong cation-exchange chromatography (SCEX) is one of the frequently used liquid-chromatography strategies, where it has been shown that peptides are eluted according to their charge in a defined process. Compared to the classical strong cation exchange followed by ion-pair reversed-phase (SCX × IP–RPLC) approach, the reversed-phase ion-pair reversed-phase (RP × IP–RPLC) showed a more homogenous distribution of eluted peptides at high pH [25,26].
7.2.3 Methods Based on the Hydrophobic Interaction
As shown in Figure 7.5, proteins can be separated according to differences in their surface hydrophobicity using two methods: hydrophobic-interaction chromatography and reversed-phase chromatography.
Hydrophobic Interaction Chromatography
Hydrophobic-interaction chromatography (HIC) is a powerful method for protein purification based on the reversible interaction between the protein surface and a hydrophobic chromatographic sorbent [27]. This technique is used in protein purification as a complement to other separation techniques and as a next step after ammonium sulfate precipitation of the sample or after IEX separation.
Van der Waals forces are the major contributing factor to the hydrophobic interactions [28]. Using HIC, the structural damage to protein molecules is minimized and its biological activity is maintained, due to a weaker interaction than affinity, ion-exchange, or reversed-phase chromatography (RPC) [29,30].
Hydrophobicity is repulsion between a nonpolar compound and a polar environment, such as water. The distribution and hydrophobicity of hydrophobic amino acids are characteristic of each protein, so a specific separation is possible with use of hydrophobic supports. Different models are used for the description of protein hydrophobicity and hydrophobic interactions, and different theories are proposed for the retention mechanism of proteins in HIC [31]. In addition to the important role of water in the strengthening–weakening of the hydrophobicity, these interactions can be induced by changing the ionic strength, presence of organic solvents, temperature, and the pH value of the chromatographic media. Based on these properties, the HIC separation occurs by the modulation of the frequency and distribution of surface-exposed hydrophobic amino-acid residues, the hydrophobicity of the medium, the nature and composition of the sample, as well as type and concentration of salt in the mobile phase (see Figure 7.6).
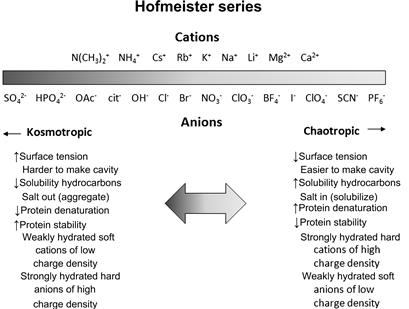
FIGURE 7.6 Typical ordering of cations and anions in a Hofmeister series. Source: Reprinted from Ref. [36] with permission.
HIC is most strongly affected by the specific effects of the salts. To enhance protein binding to the resin at the weakly hydrophobic ligand (such as phenyl group), a salting-out salt is required. Ammonium sulphate ((NH4)2SO4) and sodium sulphate (Na2SO4) are the most-utilized salts in HIC. Since binding of proteins to the HIC resin depends on the hydrophobicity of the proteins and salt concentration, both binding and elution of proteins are modulated by the (NH4)2SO4 concentration. Those salts that interact unfavorably with the proteins or increase the surface tension enhance binding, while those salts that interact favorably with the proteins, enhance elution. The unfavorable interaction increases with salt concentration, meaning that binding to HIC resins occurs at higher salt concentration and lower salt concentration weakens binding, leading to elution of proteins bound at high salt concentration [32]. Amino acids can be hidden into the hydrophobic core of proteins or may be on the surface. Both anions and cations can be sorted, according to the Hofmeister (lyotropic) series in a list of salts that influence the water tension and consequently the interaction between proteins and the HIC adsorbent [33]. Salt effects in protein precipitation and HIC were investigated for a broad combination of proteins, salts, and HIC resins in the study of Nfor et al. [34].
The interaction between hydrophobic proteins and a HIC medium is significantly influenced by the presence of certain salts in the running buffer. The most frequently used salt for the preparation of the mobile phase is (NH4)2SO4. [35]. The high salt concentration enhances the interaction between the hydrophobic components of the sample and the chromatography medium, and the lower salt concentration weakens the interaction. These interactions are a result of the presence of side chains of hydrophobic amino acids. They do not form hydrogen bonds with water and are not surrounded by water molecules. As a result of this phenomenon, the hydrophobic interaction depends on the behavior of the water molecules rather than on direct attraction between the hydrophobic molecules with the support.
To and Lenhoff [37] investigated the contribution of protein solubility to retention in HIC using isocratic elution with four commercially available agarose media and four model proteins (ribonuclease A, lysozyme, myoglobin, and ovalbumin). Various retention trends (type of adsorbents, type and concentration of salt, pH) were tested as a function of the properties of each protein and the importance of protein–surface interaction or conformational change for protein binding was found.
Selectivity of HIC depends on the nature of the ligands and the matrix (the degree of ligand substitution on the matrix), the nature of the target protein, as well as the type and concentration of salt used in the mobile phase [38]. Proteins with the highest degree of hydrophobicity are most strongly retained and are eluted at the end of the chromatogram.
HIC media are composed of resins that contain alkyl or aryl groups coupled to an inert, porous matrix made of spherical particles with a high internal surface area. Agarose is the main matrix suitable for preparing adsorbents for HIC [39]. Silica or organic polymer resins (e.g., phenyl and butyl) are also used extensively [40].
The pH of the buffers used in HIC separations is important because of the adsorption of proteins to the chromatographic support. Increase in the pH value (up to 9–10) of the mobile phase also decreases the hydrophobic interactions between proteins and the hydrophobic ligands, due to the change in charge of the protein. At high pH, silica-based supports are unstable and inadequate for protein purification. Generally, lowering the temperature promotes protein elution. Therefore, labile proteins should be separated at low temperatures [40].
Separations by HIC are often designed using nearly opposite conditions to those used in IEX. The sample is loaded in a buffer containing a high concentration of salt, which makes this method very useful as a subsequent step after proteins are eluted from ion-exchange columns by use of buffers with high salt concentration. The proteins are eluted from the HIC resin as the concentration of the salt in the buffer is decreased. Frequently, they are ready for the next purification step by IEX without further buffer exchange (Figure 7.7).
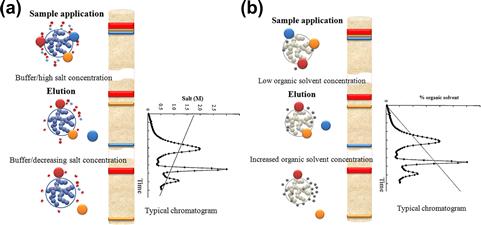
FIGURE 7.7 Methods based on the interaction between surface and proteins. Both methods involve the separation of molecules on the basis of hydrophobicity.
(a) Hydrophobic interaction chromatography (HIC) is based on the adsorption of biomolecules to a weakly hydrophobic surface at high salt concentrations. Elution is carried out with a descending salt gradient. (b) The protein separation in the reversed-phase chromatography (RPLC) depends on the hydrophobic binding of the solute molecule from the mobile phase to the immobilized hydrophobic ligands attached to the stationary phase (i.e., the sorbent).The solutes are eluted in order of increasing molecular hydrophobicity. Elution can carried out by either isocratic conditions (at the constant concentration of organic solvent) or gradient elution (the amount of organic solvent is increased over a period of time).
Reversed-Phase Chromatography
Due to the excellent resolving power, convenience, versatility, stability, and reproducibility, reversed-phase liquid chromatography (RPLC) is one of most important techniques for protein separations and the method of choice for peptide separations. RPLC has been applied on the nano, micro, and analytical scales and can be scaled up for preparative purification at the industrial scale [41,42]. In contrast to reversed phase chromatography, mechanisms of the ion-exchange and hydrophobic-interaction chromatography for peptide and protein separation is based on differences in surface hydrophobicity or surface charge [41]. Because of its compatibility with mass spectrometry (MS), RPLC is an indispensable tool in proteomic research. The increase in resolution offered by LC separation greatly enhances MS detection of sample components, and high-resolution separation reduces ion suppression in MS [42].
Reversed-phase chromatography is a separation method based on the hydrophobicity of the protein. In RPC, the hydrophobic stationary phase is based on silica gel or a synthetic polymer. In recent years, instead of bulk materials for column packing, polymer- or silica gel-based monolithic stationary phases have also been used [43].
In the presence of aqueous buffers, the solute mixture is applied at the start to the sorbent, then the solutes are eluted by the addition of organic solvent to the mobile phase. The stationary phase bears the hydrophobic ligands, mainly C4–, C8–, or C18–alkyl chains. The mobile phase contains water and a water-miscible organic solvent, such as methanol, acetonitrile, or isopropanol. Elution is continued by isocratic conditions at the constant organic solvent concentration or by gradient elution by increasing of the amount of organic solvent over a period of time. Acid (usually, formic, acetic, or trifluoroacetic acid) is added to the mobile phase to render the proteins and peptides positively charged and to reduce undesirable interactions with the stationary phase.
Analytical applications range from the assessment of purity of peptides following solid phase peptide synthesis to the analysis of tryptic maps of proteins, while preparative RPLC is used for the micropurification of protein fragments for sequencing to large-scale purification of synthetic peptides and recombinant proteins [44–46]. The numbers of applications of RPLC in peptide and protein purification continue to expand at an extremely rapid rate.
Mant and Hodges [47] developed more quantitative selectivity parameters for peptide separations using RPLC for control de-novo design of synthetic peptide standards with the same (amino acid) composition and minimal sequence variation (SCMSV). They used a range of stationary phases (C8 and C18 alkyl, polar embedded, polar endcapped, ether-linked phenyl, and phenylhexyl) such as those already available for small molecules, further enhancing the universal value of utilizing peptide standards to compare column performance in the separation of peptide mixtures.
A reversed-phase column was used to purify the ErITC-labeled tryptic fragments, unlabeled peptides, and peptides labeled with erythrosine (see Figure 7.8).
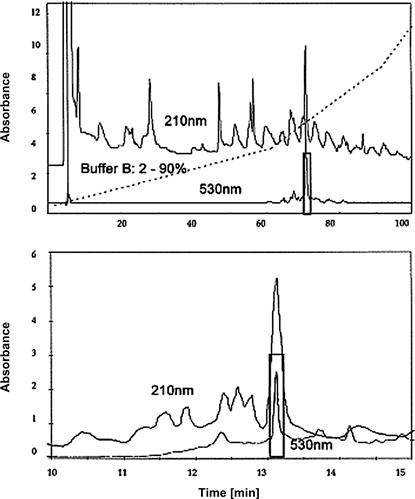
FIGURE 7.8 Separation of tryptic peptides by RPLC on a Merck 250-3 Lichrospher WP300 RP-18 (5-μm) column.
Absorbance of the eluate was recorded at 210 nm to detect all peptides and at 530 nm to detect erythrosin-labeled peptides. The upper chromatogram shows the peptide separation with a gradient of 2–90% acetonitrile in 25 mM KH2PO4 at pH 7.0. The lower chromatogram shows the rechromatography of the fraction with a retention time of 70–73 min (the bar in the upper graph) in the first step under isocratic conditions in a mixture of equal volumes of acetonitrile and 25 mM KH2PO4 pH 7.0. The peak at 13.0–13.3 min (the bar in the lower graph) was used for amino-acid sequence analysis. Source: Reprinted from Linnertz et al. [48], with permission.
Reversed-phase chromatographic separation involves loading of a protein mixture onto a hydrophobic stationary phase in water or a water–solvent mixture, usually containing very dilute acid (usually 0.1% formic or trifluroacetic acid; TFA). The elution of bound components occurs by increasing the organic solvent (usually, acetonitrile or methanol) concentration (see Figure 8.8). According to the theory, HIC and RPC are related techniques, since both are based on interactions between hydrophobic regions (amino acids) in protein molecules and hydrophobic ligands of a chromatographic support. However, experimentally, these techniques are different in the following ways:
1. The surface of the RPC supports is usually more hydrophobic than that of the HIC medium.
2. In HIC, concentrated salt solutions are used for sample application. In RPC, proteins and peptides are dissolved in water or a water–organic solvent mixture, usually containing a dilute acid (see previously).
3. Because of the stronger hydrophobic interaction of proteins with the stationary phase, organic solvents are used for RPC elution. Elution in HIC occurs by decreasing the salt concentration in the mobile phase.
4. The use of organic solvents in RPC frequently leads to denaturation and loss of biological activity of biopolymers, especially of high-molecular weight proteins [49].
5. Temperature, especially elevated temperature, is an important variable in controlling selectivity in RPC separations of proteins. In routine use, most HIC separations are performed at room temperature.
RPC provides excellent resolution of complex protein mixtures at analytical and micro- and nano-analytical scale (see Figure 7.9).
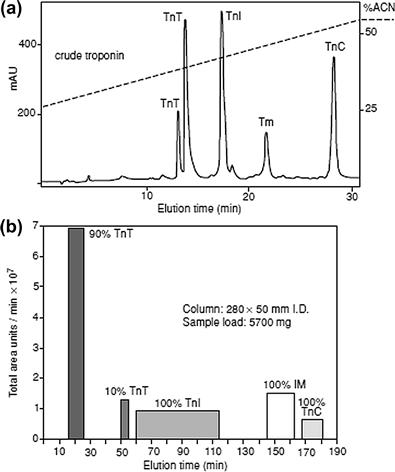
FIGURE 7.9 Analytical RPLC of a crude mixture of proteins from rabbit skeletal muscle: tropomyosin (Tm) and components of troponin (Tn) complex, comprising troponin T (TnT), troponin I (TnI), and troponin C (TnC).
(a) Analytical RPLC. Column, Zorbax SB300 C8 (150 × 4.6 mm i.d., 5-μm particle size, 300 Å pore size; Agilent Technologies). Conditions: linear A–B gradient (1% B/min starting from 25% acetonitrile) at a flow rate of 1 ml/min. Eluent A, water containing 0.05% TFA; Eluent B, acetonitrile containing 0.05% TFA. (b) Preparative RPLC (optimized after scaling-up experiments). Column, μBondapack C8 packing, 5-μm particle size, 300 Å pore size, from Waters, in column of dimensions 280 × 50 mm i.d. Eluent A, water containing 0.05% TFA; Eluent B, acetonitrile containing 0.05% TFA. Sample load, 5700 mg in 440 ml water containing 0.05% TFA (∼13 mg protein/ml); following sample loading at 22 ml/min, a 10-min isocratic hold (at constant solvent concentration) with water containing 0.05% TFA, followed by linear A–B gradient (1.7% acetonitrile/min) up to 25% acetonitrile, then 0.1% acetonitrile/min up to 35% acetonitrile and, finally, 0.5% acetonitrile/min up to 55% acetonitrile. The positions of the individual components identified following fraction analysis are denoted in histograms. Source: Reprinted from Mant and Hodges [50], with permission.
This chromatographic method also has great potential for high-resolution purification as well as for low-resolution desalting steps, especially for sample preparation for proteomics applications [51]. Combined with other chromatographic methods, especially ion-exchange chromatography, and as a component (dimension) in so-called multidimensional liquid chromatography (MDLC), RPC is a key method for separation, identification, and characterization of proteins in very complex mixtures [52].
The performance of a typical RPC separation of proteins is summarized in Figure 7.9. In analytical-scale RPC separations, a continuous linear gradient is often used, as shown in Figure 7.9(a), while preparative-scale separations typically employ an optimized elution scheme combining isocratic elution and gradients with different slopes, see Figure 7.9(b).
A successful RP–HPLC separation is influenced by many parameters. Consequently, to meet the requirements for an application, the RPC separation has to be optimized.
Choosing the optimal support and column dimensions is crucial for a successful chromatographic separation. The selection of an RPC support must be made empirically. The most critical factor in choosing the appropriate stationary phase is sample hydrophobicity. For separation of highly hydrophobic components, a less-hydrophobic stationary phase should be used to facilitate the elution. Proteins that bind strongly to a more hydrophobic support, bind more weakly to a less hydrophobic medium, and also are eluted at lower concentrations of organic solvent. The most important characteristics of the chromatographic support are surface chemistry, alkyl chain density, particle size, pore size, and mechanical stability.
To balance high efficiency and short separation time, spherical nonporous or porous particles with diameters between 1.5 and 5 μm packed in short columns (3 to 5 cm long) are often used. The efficiency, but also the back pressure, of the column rises with smaller particle diameters.
For large-scale preparative separations, porous particles with larger diameters are used. Use of chromatographic supports with a larger particle size allows the use of higher flow rates at lower back pressure, especially at the early stages of protein or peptide purification.
7.2.4 Affinity Chromatography
Affinity chromatography is a specific purification technology based on biological function or individual chemical structure. The application of this technique is in separation of active biomolecules from denaturated or functionally different forms in the isolation of pure proteins present at low concentration and also for removing specific contaminants [53].
Proteins are separated on the basis of reversible specific interactions between proteins and a ligand coupled to a chromatography matrix (see Figure 7.10). The interaction between target molecules and binding sites with complementary surfaces to their ligands can involve different intermolecular chemical bonds, such as a combination of electrostatic or hydrophobic interactions, van der Waals forces, and hydrogen bonds. However, a specific ligand is covalently attached to an inert chromatographic matrix. This allows a high selectivity and high resolution as well as high capacity for the proteins of interest. Recovery of active material is generally very high, which enables the purification of a protein on the order of several thousand-fold. The advantage of this technique compared with others is the shortening of the purification process. On the other hand, the elution of the target molecule that binds to the affinity ligand can sometime be difficult. It can be achieved by addition of a molecule that binds competitively to the ligand (e.g., a sugar in the case of lectin-affinity chromatography) or by a drastic change in the pH value, ionic strength, or polarity of the eluent [54].
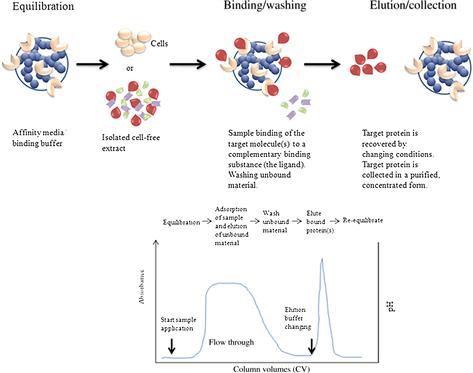
FIGURE 7.10 Typical affinity purification.
Hydrophilic and neutral matrices are favored because they prevent nonspecific interactions between proteins and the gel matrix. The matrix should be macroporous, which means that the protein should be accessible for binding with the immobilized ligand. Agarose is the most-used material for affinity matrices. Specific ligands are covalently bound to the chromatographic matrix. These ligands are selective and bind proteins such as antibodies, protein receptors, and some enzyme inhibitors. Staphylococcal protein A is one of the first discovered IgG binding molecules and has been extensively used as a biospecific ligand. Today, protein A chromatography remains the most common technique for antibody purification on a large scale, and its further application will have great importance for both small- and large-scale purification of native polyclonal and monoclonal and recombinant antibodies [54].
Various methods can be used for activation of an affinity adsorbent. They differ on the binding chemistry of the ligand and adsorbents and on the need for a spacer arm. The activation occurs by the introduction of an electrophilic group into the matrix. In the next step, the ligand reacts with nucleophilic groups, such as amino, thiol, and hydroxyl residues (see Table 7.2). Ligands that contain a coupled amino group, attached by the ester bond, give a very stable amide linkage. The amide bond is stable up to pH 13. That makes the N-hydroxysuccinimide-activated Sepharose (NHS) supports, such as Sepharose, suitable for applications at high pH. Cyanogen bromide reacts with hydroxyl groups on Sepharose to form reactive cyanate ester groups. Under mild conditions, proteins, peptides, amino acids, and nucleic acids can be coupled to a CNBr-activated matrix via primary amino groups or similar nucleophilic groups. In weakly alkaline conditions (pH 9–10), CNBr-activated supports with primary amines give an isourea derivative. The isourea linkage of the ligand causes several problems during the purification procedure, including nonspecific binding, due to the charge and the leakage of ligand from the instability of the isourea bond [55].
TABLE 7.2
Overview of Some Commonly Used Immobilization Procedures
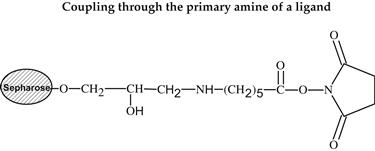
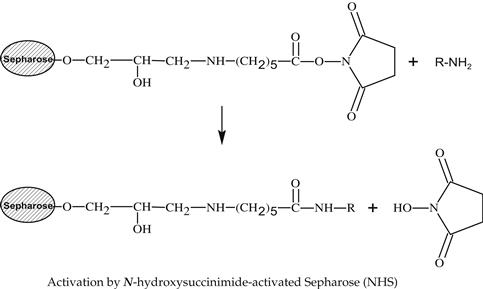
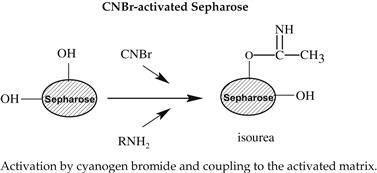
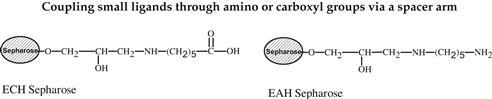
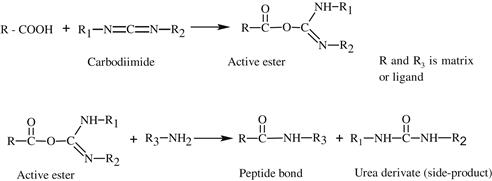

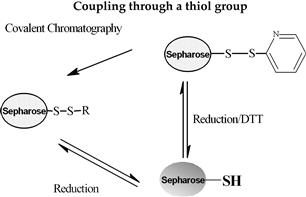
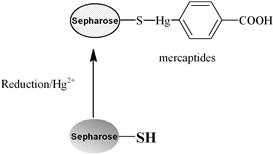
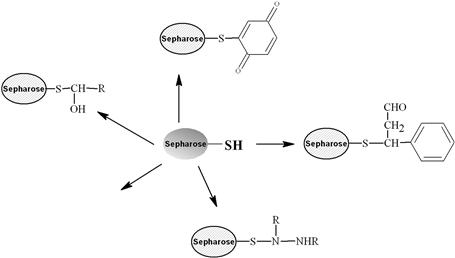
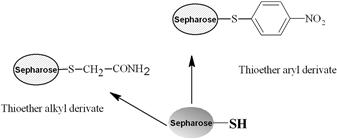
Source: Reproduced with permission from ref [56].
In the presence of a coupling reagent, such as carbodiimide, ligands are coupled in a simple one-step procedure. Epoxy-activated matrices, such as Sepharose 6B, are used for coupling ligands that contain hydroxyl, amino, or thiol groups. Because of the long hydrophilic spacer arm, it is useful for coupling small ligands, such as choline, ethanolamine, and sugars.
Three types of interactions for stability of the bound protein help in choosing the suitable eluant: pH change, adjustments of the ionic strength of the buffer, and addition of a competitor (with a higher concentration and higher affinity to the active groups on the surface of the support).
Affinity chromatography has a wide range of applications for protein purification, such as immunoaffinity, purification of immunoglobulins, purification of glycoproteins, DNA-binding proteins, receptor proteins, enzymes, cell isolation, and nucleotide isolation [53].
Pseudo-Affinity Chromatography
Since no biospecific interactions are characteristic of affinity separations, immobilized metal-affinity chromatography) and chromatography on synthetic dye ligands are usually called pseudo-affinity techniques.
Immobilized Metal-Affinity Chromatography
Immobilized metal-affinity chromatography is a separation technique that has proven to be an efficient and versatile technology for the isolation and purification of industrial enzymes as well as proteins that are of commercial importance or used in research fields, such as genetics, molecular biology, and biochemistry.
IMAC is based on the formation of immobilized metal complexes, formed by the reaction between metal ions with chelating ligands that are attached to a chromatographic support (e.g., Sepharose). The separation of proteins is possible through interaction of specific metal binding sites in the molecule (amino-acid side chains on the protein surface or specifically developed tags, mainly histidine residues) [57].
The selectivity of IMAC depends on the choice of metal ion, chelating ligand, absorption and elution conditions, and the modification of the target protein.
A schematic diagram showing protein adsorption and desorption from the matrix is shown in Figure 7.11.
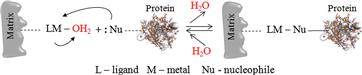
FIGURE 7.11 Protein adsorption and desorption from the matrix.
The adsorption of proteins in IMAC is based on the coordination between an immobilized metal ion and electron donor groups from the protein surface. Most commonly used are the transition-metal ions (Cu2+, Ni2+, Zn2+, Co2+, Fe3+) as electron-pair acceptors (or electrophiles). Electron-donor atoms (or nucleophiles such as N, S, O) present in the chelating compounds attached to the chromatographic support are capable of forming metal chelates. Elution (desorption) of the target protein is achieved by protonation (using elution buffers with lower pH or lowering pH gradients) and ligand exchange of the metal ion by a stronger chelator (such as EDTA).
Histidine is the amino acid that shows the strongest interaction with metals. Electron donor groups on the imidazole ring in histidine form coordination bonds with the immobilized transition metal. Since many proteins contain amino acids, such as Cys, Trp, Phe, and Tyr, located at the surface, that are suitable for interaction with metal ions, it is expected that almost all proteins are capable of binding to metal chelators. The strength of interaction depends on the number coordinative bindings between the metal ion and protein. In general, the supports in IMAC are the same as those applied in affinity chromatography. Beaded agarose is the predominantly used support.
Ligands like iminodiacetic acid (IDA) and nitrilotriacetic acid (NTA) complexed with different metal ions such as Cu(II) or Ni(II) have been used widely, and gels incorporating these ligands are available from commercial suppliers. In general, the supports in IMAC are the same as in affinity chromatography [58].
Divalent ions, such as the transition metals Fe2+, Co2+, Ni2+, Cu2+ and Zn2+, are most commonly used. The affinities of many retained proteins and their respective retention times, such as in the IDA chelator, are in the following order: Cu2+ > Ni2+ > Zn2+ > Co2+. The loss of metal ions at lower pH values leads to reduced adsorption capacity of the sorbent and can also cause damage to the target proteins by metal-catalyzed reactions. Buffers of relatively high ionic strength (containing 0.1 to 1.0 M NaCl) are used to reduce nonspecific ionic adsorption. A preelution step with a low concentration of imidazole or EDTA can be used for the elution of various contaminating proteins that contain histidine residues [59].
The benefits of IMAC separation technologies are related to ligand stability, high protein loading, mild elution conditions, simple column regeneration, and low cost. These are reasons for the wide application of this technology in protein separation. An advantage of this technique is that it is also applicable under denaturing conditions. Such conditions are often necessary when recombinant proteins are highly expressed in the cell culture system in the form of inclusion bodies (e.g., in E. coli). However, IMAC is not the preferred technique for the production of therapeutic proteins in substantial quantities, mostly due to problems with reproducibility and loss of metal ions during protein elution [59].
7.2.5 Chromatography on Hydroxyapatite
Hydroxyapatite (HT), (Ca5(PO4)3OH)2, is a form of calcium phosphate used in the chromatographic separation of biomolecules. Five calcium doublets (Ca sites) and pairs of –OH containing phosphate triplets (P sites) are arranged in a repeating geometric pattern. Hydroxyapatite has unique separation properties, such as high selectivity and resolution, and can be used in separations of proteins by electrophoretic and chromatographic techniques. Applications of hydroxyapatite chromatography include the purification of different subclasses of monoclonal and polyclonal antibodies, antibody subtypes with different chain compositions, antibody fragments, isozymes, supercoiled DNA from linear duplexes, and differentiating single-strand from double-strand DNA [60].
Ceramic hydroxyapatite (CHT) is a spherical, macroporous form of hydroxyapatite. Compared to most other chromatography adsorbents, CHT is unusual in that it is both the ligand and the support matrix. Two types of CHT ceramic hydroxyapatite, Type I and Type II, have elution characteristics similar to crystalline hydroxyapatite but also have some important differences. CHT Type I has a higher protein-binding capacity and better capacity for acidic proteins, while CHT Type II has a lower overall protein-binding capacity but has better resolution for nucleic acids and certain proteins. The Type II material also has a very low affinity for albumin and is especially suitable for the purification of many species and classes of antibodies.
Hydroxyapatite contains two types of binding sites, positively charged calcium (Ca2+) and negatively charged phosphate (PO43-) groups. These sites are distributed into the crystal structure of the matrix. Cation exchange occurs by positively charged proteins with the negatively charged phosphates, and analogously, they are repulsed by the calcium sites (see Figure 7.12).
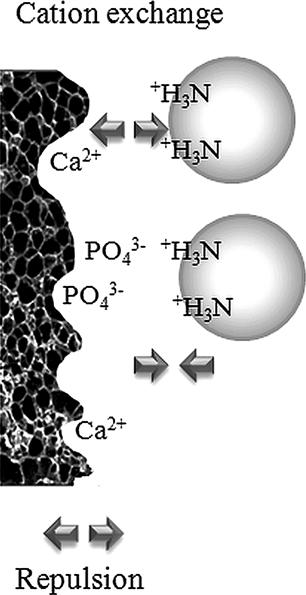
FIGURE 7.12 CHT binding mechanisms.
The ion-exchange interactions are weakened by the addition of neutral salts, such as NaCl or phosphate buffer (from mobile phase), or an increase in pH.
7.2.6 Chromatography on Monolithic Supports
In proteomic applications and for in-process analysis in biotechnology, analytical columns with high efficiency and short analysis times are required. Hence, high-speed packing materials are in demand. If porous materials are used, separation speed is limited by mass transfer between the mobile phase and stationary liquid in the pores of the chromatographic support. For nonporous materials, the binding of sample components and subsequent elution occurs on the surface of the support so separation speed is minimally limited by mass transfer. To increase the performance of nonporous chromatographic material, particles with very small diameter (less than 3 μm, e.g., Tosoh Bioscience, Tokyo, Japan, offers polymer-based nonporous columns and bulk materials) are used, and high column back pressure is often the limiting factor. Frequently, monoliths are an alternative to columns packed with bulk stationary phases. The structure of monolithic materials (see Figure 7.13) allows very fast mass transfer during chromatographic separation that is hardly limited by the flow rate.
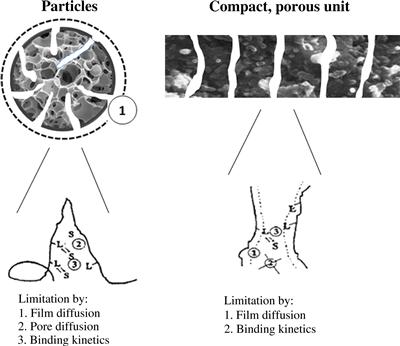
FIGURE 7.13 Comparison of particle-based chromatography and chromatography on compact, porous units. Source: Reprinted from Ref. [61] with permission.
Because of the low black pressure and fast mass transfer, monolithic columns can be run at very high speed (see Figure 7.14). As a result, separation time is up to one order of magnitude faster than that of columns packed with bulk particles [51,62–63]. Similar to columns packed with spherical particles, monolithic columns are silica or polymer based.
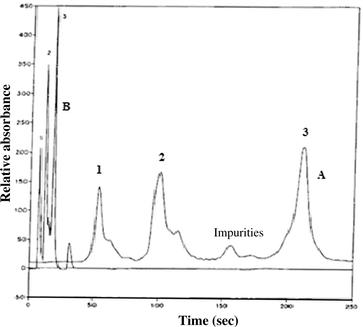
FIGURE 7.14 Comparison of a conventional porous particle medium (Mono Q) with a CIM disk with regard to their separation performance under fast gradient elution conditions: (a) a Mono Q anion exchange column 50 × 5 mm i.d.; (b) a QA anion exchange CIM disk 3 × 10 mm i.d. Conditions: buffer A, 10 mM Tris, pH 7.4; buffer B, buffer A + 0.5 M NaCl. Gradient time: (a) 0–100% buffer B in 4 min; (B) 0–100% buffer B in 6 sec. Flow rate: (a) 1 ml/min; (b) 10 ml/min. Back pressure: (a) 0.9 MPa; (B) 0.8 MPa. Calibration solution: myoglobin (peak 1), conalbumin (peak 2), and soybean trypsin inhibitor (peak 3) in buffer A. Source: Reprinted from Ref. [64] with permission.
In the reversed-phase mode, monolithic columns are mostly used for protein and peptide separation in proteomics applications [51]. In the ion-exchange and hydrophobic-interaction modes, monolithic columns are used for analytical and preparative separation of large biopolymers, such as proteins and nucleic acids, as well as nanoparticles, such as plasmids and viruses [65].
Hennessy et al. [66] describe a general procedure for the generation of peptide maps of proteins with monolithic silica-based columns. The use of reversed-phase monolithic sorbents has been demonstrated to significantly reduce separation times for peptide map analysis, thus enabling increased sample throughput (see Figure 7.15).
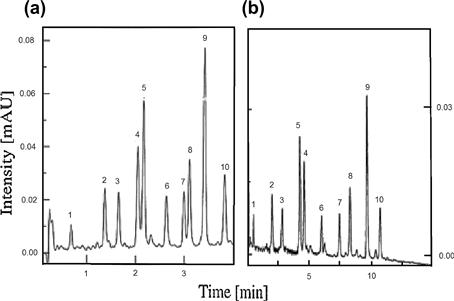
FIGURE 7.15 RPLC separation of tryptic peptide fragments of equine cytochrome c (peaks 1–10) on Chromolith SpeedROD RP-18e (a) 100 mm and (b) 50 mm. Source: Reprinted from Ref. [66] with permission.
The obtained difference between the Chromolith Performance RP-18e column and the SpeedROD 50 mm or SpeedROD 100 mm columns is the pore size. The wide-pore monolith influences the flow rate. Thus, compared to the SpeedROD 50 mm, where total separation time for the 10 tryptic peptides was about 10 min, the longer SpeedROD 100-mm column could be exploited to generate very fast separations; for example, total separation time for the 10 tryptic peptides was less than 4 min, see Figure 7.15(a), at the same conditions (flow rate of 8 ml/min).
7.2.7 Displacement Chromatography
Displacement chromatography was first discovered by Tiselius, who also classified the three modes of chromatography as frontal, elution, and displacement [67]. More than 30 years ago, this technique was employed by Horvath, who first used the modern high-performance columns and equipment [68].
The principle of displacement chromatography for separation is based on the Langmuir isotherm. Only a finite number of sites are on the chromatographic support (stationary phase) for the binding of sample components, and if a site is occupied by one molecule, it is not available to the other sample components. Because the number of binding sites is limited, they are saturated when the concentration of the molecules in the sample is large in comparison to the dissociation constant for the sites. The displacer is a molecule with a high affinity to the given chromatographic support, and it will compete more effectively for binding sites. It causes the displacement of the sample molecules and their enrichment in the lower-affinity solute.
In elution chromatography, the retention of the substances that have to be separated is controlled by adjusting the composition of the mobile phase. The resolution in elution chromatography is generally better if the substance has a higher affinity to the mobile phase and the peaks are strongly retained. On the other hand, the conditions that give good resolution of the early peaks lead to strong retention of the late ones, to long separation times, and to the broadening of later peaks unless gradient elution is employed. The use of a gradient adds to the complexity of the chromatographic process, particularly on a large scale.
Solutes separated in the displacement mode form sharp-edged zones, and peak spreading is less emphasized. The separated zones in this kind of chromatography are self-sharpening. This means that, if a component gets ahead of its band, it enters a zone in which it is more strongly retained and runs more slowly until the original zone catches up. Furthermore, since the loadings in displacement chromatography are deliberately high, more material can be separated on a smaller column, with the purified components recovered at significantly higher concentrations. The displacer is selected to have higher a affinity for the stationary phase than the sample components. The retention conditions in displacement chromatography depend on the displacer concentration. Consequently, the conditions that allow high retention can be employed without a gradient operation, and the displacement pushes all components of the sample out of the column in the designed run time [69].
One of the biggest advantages of displacement chromatography, is that this chromatography mode is well suited for the purification of components from dilute solutions. The disadvantages are the formation of overlapping zones between the separated components, and additionally, the removal of the displacer and column regeneration frequently limits the throughput [70].
Displacement chromatography is a very efficient technique for the purification of proteins from complex mixtures at high column loadings (see Figure 7.16). An important advance in the development of displacement chromatography was the discovery of low molecular-mass displacers such as 4-toluenesulfonic acid salts, streptomycin sulfate A (and similar substances), chloroquine diphosphate, and other small molecules instead of the large polyelectrolyte polymers [69,71,72]. These displacers can be readily separated from the protein after the separation, such as by size-exclusion chromatography or simple ultrafiltration. Their adsorption is also salt dependent, and it significantly facilitates the column regeneration. Displacement chromatography is well suited for the preparative separation of large quantities of proteins from complex mixtures by use of relatively small columns. This mode of chromatography can be performed with a variety of chromatographic resins, such as ion exchange, hydrophobic interaction, reversed phase, hydroxyapatite and pseudo affinity mode phases [71–75], and by use of particle-based as well as monolithic supports [76], for the separation of variety of proteins [77–79].
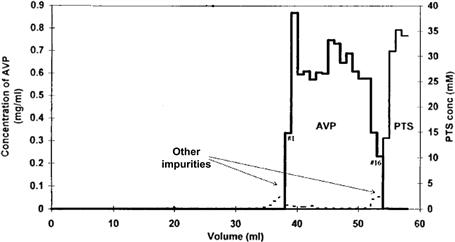
FIGURE 7.16 Selective displacement of the antigen vaccine protein (AVP).
Displacer: 35 mM p-toluenesulfonic acid (PTS). Mobile phase: 20 mM, pH 7.0 Tris buffer. Column: 10 × 290 mm DEAE Fast Flow Sepharose. Loading: ~6.5 ml of AVP feedstock per ml of column volume. Flow rate: 1 ml/min. Fraction size: 1 ml. AVP fractions are numbered from 1 to 16. Source: Reprinted from Ref. [80] with permission.
7.3 Conclusions
• Chromatography is an indispensible method for protein separation on the analytical and preparative scales.
• Ion-exchange, hydrophobic-interaction chromatography; chromatography on hydroxyapatite; and different types of affinity and pseudo-affinity chromatography are the most frequently used methods for isolation of nondenatured, physiologically active proteins.
• Use of the organic solvents necessary in reversed-phase chromatography frequently causes protein denaturation and loss of physiological activity.
• Monoliths are newly introduced chromatographic supports that enable very fast and effective separation of macromolecules, especially large proteins and protein complexes.
• Because of the high grade of the concentrating effects, displacement chromatography is well suited for the purification of proteins from dilute solutions.
Acknowledgments
This work was supported by the Croatian Ministry of Science, Projects No. 335-0000000-0221 and the National Science Foundation of Republic of Croatia. Neither of the authors has conflicts of interest with respect to this work.
References
1. Ersson B, Rydán L, Janson J- C. Introduction to protein purification. Chapter 1 In: Janson J-C, ed. Protein purification: principles, high resolution methods, and applications. 3rd ed Hoboken, NJ: Wiley & Sons; 2011.
2. Biswas MK, DeVido DR, Dorsey JG. Evaluation of methods for measuring amino acid hydrophobicities and interactions. J Chromatogr A. 2003;1000(1–2):637–655.
3. Karlsson D, Jakobsson N, Axelsson A. Model-based optimization of a preparative ion-exchange step for antibody purification. J Chromatogr A. 2004;1055(1–2):29–39.
4. Swartz JR. Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotech. 2001;2(2):195–201.
5. Trauner A, Bennett MH, Williams HD. Isolation of bacterial ribosomes with monolith chromatography. PLoS ONE. 2011;6(2):e16273. doi 10.1371/journal.pone.0016273.
6. Josić D, Štrancar A. Application of membranes and compact, porous units for the separation of biopolymers. Ind Eng Chem Res. 1999;38:333–342.
7. Stanton P. Gel filtration chromatography. Chapter 5 In: Aguilar M-L, ed. Totowa, NJ: Humana Press; 2010; HPLC of peptides and proteins, methods and protocols, methods in molecular biology. vol. 251.
8. Strecker V, Wumaier Z, Wittig I, Schägger H. Large pore gels to separate mega protein complexes larger than 10 MDa by blue native electrophoresis: isolation of putative respiratory strings or patches. Proteomics. 2010;10(18):3379–3387.
9. Azevedo AM, Rosa PAJ, Ferreira LF, Aires-Barros MR. Integrated process for the purification of antibodies combining aqueous two-phase extraction, hydrophobic interaction chromatography and size-exclusion chromatography. J Chromatogr A. 2008;1213(2):154–161.
10. Oliva A, Llabrés M, Fariña JB. Estimation of uncertainty in size-exclusion chromatography with a double detection system (light-scattering and refractive index). Talanta. 2009;78(3):781–789.
11. Stanton P. Ion-exchange chromatography. Chapter 3 In: Aguilar MI, ed. Totowa, NJ: Humana Press; 2010; HPLC of peptides and proteins, methods and protocols, methods in molecular biology. vol. 251.
12. Kopaciewicz W, Rounds MA, Fausnaugh J, Regnier FE. Retention model for high-performance ion-exchange chromatography. J Chromatogr A. 1983;266(26):3–21.
13. Kopaciewicz W, Regnier FE. Nonideal size-exclusion chromatography of proteins: effects of pH at low ionic strength. Anal Biochem. 1982;126(1):8–16.
14. Hou Y, Hansen TB, Staby A, Cramer SM. Effects of urea induced protein conformational changes on ion exchange chromatographic behavior. J Chromatogr A. 2010;1217(47):7393–7400.
15. Lendero N, Vidič J, Brne P, Frankovič V, Štrancar A, Podgornik A. Characterization of ion exchange stationary phases via pH transition profiles. J Chromatogr A. 2008;1185(1):59–70.
16. Podgornik A, Vidič J, Jančar J, Lendero N, Frankovič V, Štrancar A. Noninvasive methods for characterization of large-volume monolithic chromatographic columns. Chem Eng Techn. 2005;28:1435–1441.
17. Kato Y, Nakamura K, Hashimoto T. New ion exchanger for the separation of proteins and nucleic acids. J Chromatogr A. 1983;266:385–394.
18. Harinarayan C, Mueller J, Ljunglöf A, Fahrner R, Van Alstine J, van Reis R. An exclusion mechanism in ion exchange chromatography. Biotech Bioeng. 2006;95(5):775–787.
19. Karlsson E, Hirsch I. Ion exchange chromatography. Chapter 4 In: Janson J-C, ed. Protein purification: principles, high resolution methods, and applications. 3d ed Hoboken, NJ: Wiley & Sons; 2011.
20. Pohl AC, Stillian JR, Jackson PE. Factors controlling ion-exchange selectivity in suppressed ion chromatography. J Chromatogr A. 1997;789(1-2):29–41.
21. Li X, Gong Y, Wang Y, et al. Jiao l, He H, Zhang Z, He F, Zhao X, Qian X Comparison of alternative analytical techniques for the characterisation of the human serum proteome in HUPO Plasma Proteome Project. Proteomics. 2005;5(13):3423–3441.
22. Berglöf JH, Eriksson S, Curling JM. Chromatographic preparation and in vitro properties of albumin from human plasma. J Appl Biochem. 1983;5(4–5):282–292.
23. Kovács A, Sperling E, Lázár J, et al. Fractionation of the human plasma proteome for monoclonal antibody proteomics-based biomarker discovery. Electrophoresis. 2011;32(15):1916–1925.
24. Melchior K, Tholey A, Heisel S, et al. Protein versus peptide fractionation in the first dimension of two-dimensional high-performance liquid chromatography–matrix-assisted laser desorption/ionization tandem mass spectrometry for qualitative proteome analysis of tissue samples. J Chromatogr A. 2010;1217(40):6159–6168.
25. Schley C, Altmeyer MO, Swart R, Müller R, Huber CG. Proteome analysis of Myxococcus xanthus by off-line two-dimensional chromatographic separation using monolithic poly-(styrene-divinylbenzene) columns combined with ion-trap tandem mass spectrometry. J Proteome Res. 2006;5(10):2760–2768.
26. Delmotte N, Lasaosa M, Tholey A, Heinzle E, Huber CG. Two-dimensional reversed-phase x ion-pair reversed-phase HPLC: an alternative approach to high-resolution peptide separation for shotgun proteome analysis. J Proteome Res. 2007;6(11):4363–4373.
27. Jennissen HP. Hydrophobic interaction chromatography: harnessing multivalent protein-surface interactions for purification procedures. Methods Mol Biol. 2005;305:81–99.
28. Van Oss CJ, Good RJ, Chaudhury MK. Nature of the antigen-antibody interaction Primary and secondary bonds: optimal conditions for association and dissociation. J Chromatogr. 1986;376:111–119.
29. Fausnaugh JL, Kennedy LA, Regnier FE. Comparison of hydrophobic-interaction and reversed-phase chromatography of proteins. J Chromatogr. 1984;317:141–155.
30. Regnier FE. The role of protein structure in chromatographic behavior. Science. 1987;238:319–323.
31. Quieroz JA, Tomaz CT, Cabral JMS. Hydrophobic interaction chromatography of proteins. J Biotech. 2001;87:143–159.
32. Tsumoto K, Ejima D, Senczuk AM, Kita Y, Arakawa T. Effects of salts on protein–surface interactions: applications for column chromatography. J Pharm Sci. 2007;96:1677–1690.
33. Gagnon P, Mayes T. Danielsson Å An adaptation of hydrophobic interaction chromatography for estimation of protein solubility optima. J Pharm Biomed Anal. 1997;16(4):587–592.
34. Nfor KB, Hylkema NN, Wiedhaup KR, Verhaert PDEM, van der Wielen LAM, Ottens M. High-throughput protein precipitation and hydrophobic interaction chromatography: salt effects and thermodynamic interrelation. J Chromatogr A. 2011;1218(49):8958–8973.
35. Murphy PJ, Stone OJ, Anderson ME. Automated hydrophobic interaction chromatography column selection for use in protein purification. J Vis Exp (55). pii: 30602011. doi: 10.3791/3060.
36. Kunz W. Specific ion effects in colloidal and biological systems. Curr Opin Colloid Interface Sci. 2010;15(1–2):34–39.
37. To BC, Lenhoff AM. Hydrophobic interaction chromatography of proteins, II Solution thermodynamic properties as a determinant of retention. J Chromatogr A. 2007;1141(2):235–243.
38. Mahn A, Asenjo JA. Prediction of protein retention in hydrophobic interaction chromatography. Biotech Ad. 2005;23(5):359–368.
39. Protein purification. Handbooks from Amersham biosciences, hydrophobic interaction chromatography principles and methods Amersham: Uppsala; 2001; 18-1020-90.
40. Benedek K. High-performance hydrophobic interaction chromatography. In: Aguilar M-I, ed. Chapter 4. Totowa, NJ: Humana Press; 2010; HPLC of peptides and proteins, methods and protocols, methods in molecular biology vol. 251.
41. Aguilar M-I, Hearn MT. High-resolution reversed-phase high-performance liquid chromatography of peptides and proteins. Meth Enzymol. 1996;270:3–26.
42. Shi Y, Xiang R, Horvath CS, Wilkins JA. Role of chromatographic techniques in proteomic analysis. J Chromatogr A. 2004;1053:27–36.
43. Jungbauer A. Chromatographic media for bioseparation. J Chromatogr A. 2005;1065(1):3–12 10.1016/j.chroma.2004.08.162.
44. Evans EW, Beach GG, Wunderlich J, Harmon BG. Isolation of antimicrobial peptides from avian heterophils. J Leukocyte Biol. 1994;56(5):661–665.
45. Jang A, Lee M. Purification and identification of angiotensin converting enzyme inhibitory peptides from beef hydrolysates. Meat Sci. 2005;69(4):653–661.
46. Tripet B, Yu L, Bautista DL, Wong WY, Irvin RT, Hodges RS. Engineering a de novo-designed coiled-coil heterodimerization domain for the rapid detection, purification and characterization of recombinantly expressed peptides and proteins. Protein Eng. 1996;9(11):1029–1042.
47. Mant CT, Hodges RS. Design of peptide standards with the same composition and minimal sequence variation to monitor performance/selectivity of reversed-phase matrices. J Chromatogr A. 2012;1230:30–40.
48. Linnertz H, Kost H, Obsil T, Kotyk A, Amler E, Schoner W. Erythrosin 5′-isothiocyanate labels Cys549 as part of the low-affinity ATP binding site of Na+/K+-ATPase. FEBS Lett. 1998;441(1):103–105.
49. McNay J, Fernandez EJ. How does a protein unfold on a reversed-phase liquid chromatography surface?. J Chromatogr A. 1999;849:135–148.
50. Mant CT, Hodges RS. Reversed-phase liquid chromatography of proteins from rabbit skeletal troponin, a multi-protein complex. J Chromatogr A. 2002;972:101–114.
51. Josic D, Clifton JG. Use of monolithic supports in proteomics technology. J Chromatogr A. 2007;1144:2–13.
52. Wagner K, Miliotis T, Marko-Varga G, Bischoff R, Unger KK. An automated on-line multidimensional HPLC system for protein and peptide mapping with integrated sample preparation. Anal Chem. 2002;74:809–820.
53. Lee W-C, Lee KH. Applications of affinity chromatography in proteomics. Anal Biochem. 2004;324(1):1–10.
54. Batista-Viera F, Janson J-C, Carlsson J. Affinity chromatography. Chapter 9 In: Janson J-C, ed. 3rd ed Hoboken, NJ: Wiley & Sons; 2011; Protein purification: principles, high resolution methods, and applications .
55. Wilchek M, Miron T. Thirty years of affinity chromatography. Reactive Functional Poly. 1999;41:263–268.
56. Affinity chromatography: principles and methods, Handbooks from GE Healthcare, 18-1022-29 AE 10/2007.
57. Zachariou M. Immobilized metal ion affinity chromatography of proteins. Chapter 7 In: Aguilar M-L, ed. Totowa, NJ: Humana Press Inc.; 2010; HPLC of peptides and proteins, methods and protocols, methods in molecular biology. vol. 251.
58. Gaberc-Porekar V, Menart V. Perspectives of immobilized-metal affinity chromatography. J Biochem Biophys Meth. 2001;49:335–360.
59. Block H, Maertens B, Spriestersbach A, et al. Immobilized-metal affinity chromatography (IMAC): a review. Meth Enzymol. 2009;463:439–473.
60. Gagnon P, Ng P, Zhen J, et al. A ceramic hydroxyapatite-based purification platform. BioProcess Int. 2006;4(2):50–60.
61. Thömmes J, Kula M- R. Membrane chromatographys— an integrative concept in the downstream processing of proteins. Biotech Prog. 1995;11:357–367.
62. Josić D, Štrancar A. Application of membranes and compact, porous units for the separation of biopolymers. Indus Eng Chem Res. 1999;38(2):333–342.
63. Svec F, Huber CG. Monolithic materials: promises, challenges, achievements. Anal Chem. 2006;78:2100–2107.
64. Štrancar A. Ph.D. thesis. Separation of Biopolymers with Different Techniques of Liquid Chromatography. University of Ljubljana, Slovenia, 1997.
65. Forcic D, Brgles M, Ivancic-Jelicki J, et al. Concentration and purification of rubella virus using monolithic chromatographic support. J Chromatogr B. 2011;879:981–986.
66. Hennessy T, Boysen RI, Huber MI, Unger KK, Hearn MTW. Peptide mapping by reversed-phase high-performance liquid chromatography employing silica rod monoliths. J Chromatogr A. 2003;1009:15–28.
67. Tiselius A. Displacement chromatography in adsorption analysis. Ark Kemi Mineral Geol. 1943;16A:1–18.
68. Horvath C, Nahum A, Frenz J. High-performance displacement chromatography. J Chromatogr. 1981;218:365–393.
69. Cramer SM, Jayaraman G. Preparative chromatography in biotechnology. Curr Opin Biotech. 1993;4:217–225.
70. Ford JC. Displacement chromatography. In: Cazes J, ed. Encyclopedia of chromatography. New York: Marcel Dekker; 2001.
71. Jayaraman G, Li JA, Moore A, Cramer SM. Ion-exchange displacement chromatography of proteins Dendritic polymers as novel displacements. J Chromatogr A. 1995;702:143–155.
72. Antia FD, Fellegvari I, Horvath C. Displacement of proteins in hydrophobic Interaction chromatography. Ind Eng Chem Res. 1995;34:2796–2804.
73. Kundu A, Barnthouse KA, Cramer SM. Selective displacement chromatography of proteins. Biotech Bioeng. 1997;56:119–129.
74. Morrison HCJ, Gagon P, Cramer SM. Unique selectivity windows using selective displacers/eluents and mobile phase modifiers on hydroxyapatite. J Chromatogr A. 2010;1217:6484–6495.
75. Kim YJ, Cramer SM. Experimental studies in metal affinity displacement chromatography of proteins. J Chromatogr A. 1994;686:193–203.
76. Schmidt B, Wandrey C, Freitag R. Investigation of particle-based and monolithic columns for cation exchange protein displacement chromatography using poly (diallyl-dimethylammonium chloride) as displacer. J Chromatogr A. 2003;1018:155–167.
77. Barnthouse KA, Trompeter W, Jones R, Inampudi P, Rupp R, Cramer SM. Cation-exchange displacement chromatography for the purification of recombinant protein therapeutics from variants. J Biotech. 1998;66:125–136.
78. Vogt S, Freitag R. Comparison of anion-exchange and hydroxyapatite displacement chromatography for the isolation of whey proteins. J Chromatogr A. 1997;760:125–137.
79. Evans ST, Holstein M, Cramer SM. Detection of trace proteins in multicomponent mixtures using displacement chromatography. Anal Chem. 2011;83:4184–4192.
80. Shukla AA, Hopfer RL, Chakravarti DN, Bortell E, Cramer SM. Purification of an antigenic vaccine protein by selective displacement chromatography. Biotech Prog. 1998;14:92–101.