Environmental Analysis
Emerging Pollutants
M. Petrovic∗, †, M. Farré†, E. Eljarrat†, M.S. Díaz-Cruz† and D. Barceló∗∗, ∗Catalan Institute of Water Research (ICRA), Girona, Spain, †Department of Environmental Chemistry, Institute of Environmental Assessment and Water Research (IDAEA-CSIC), Barcelona, Spain, ∗∗King Saud University, Riyadh, Saudi Arabia
Outline
14.1. Introduction
14.2. General Trends
14.2.1. Fast Chromatography
14.2.2. On-Line SPE–LC–MS Coupling
14.2.3. Multiresidue Methods
14.3. Target Analysis of Specific Contaminant Groups Using LC–MS
14.3.1. Brominated Flame Retardants
14.3.2. Drugs of Abuse
14.3.3. Hormones and Other Endocrine-Disrupting Compounds
14.3.4. Nanomaterials
14.3.5. Perfluorinated Compounds
14.3.6. Pharmaceuticals
14.3.7. Sunscreen Agents
14.4. Conclusions
14.1 Introduction
Impressive improvements in detection limits for organic contaminants, mostly due to the development of hyphenated chromatography–mass spectrometry (MS) techniques, have pushed the target concentrations from the microgram to the nanograms or picograms per liter range [1]. With the progresses in analytical instrumentation, extraction techniques have also become more simple, fast, and inexpensive, providing the enrichment of analytes of interest from complex environmental matrices [2]. All these improvements led to the detection of many harmful compounds at the levels at which they have a biological effect in the environment and several new or previously ignored or unrecognized contaminants have come under scrutiny.
Several groups of organic compounds have emerged as particularly relevant:
• Brominated flame retardants, new classes other than polybrominated diphenyl ethers.
• Drugs of abuse and their metabolites.
• Hormones and other endocrine-disrupting compounds.
• Organophosphate flame retardants and plasticizers.
• Pharmaceuticals and personal care products.
• Polar pesticides and their degradation–transformation products.
All these compound classes are referred to as emerging contaminants, which does not necessarily mean “new substances,” that is, newly introduced chemicals and their degradation products, metabolites, or by-products but also refers to compounds, including naturally occurring compounds, with previously unrecognized adverse effects on ecosystems. The study of their occurrence, fate, and behavior in the environment has been the focus of attention by the scientific community during recent years and concern is raised regarding the risks posed by these emerging contaminants to humans.
This chapter attempts to survey the current state of the art in the application of modern LC–MS techniques to environmental analysis of emerging contaminants, which is a very broad, very active field. Consequently, a large number of papers are published every year and comprehensive coverage of all contaminant classes and trends is an impossible task. Therefore, we focus on recent trends in the application of LC–MS, including sample preparation, in the environmental analysis of several important classes of emerging contaminants, such as pharmaceuticals and personal care products, drugs of abuse, endocrine-disrupting compounds, perfluorinated compounds, brominated flame retardants, and nanoparticles.
14.2 General Trends
Currently, the main breakthrough in the analysis of emerging environmental contaminants is observed in the application of tandem and hybrid LC–MS techniques with several notable trends [3]:
• Shift toward high-throughput methods involving fast chromatography or on-line coupling of sample preparation.
• Shift toward multiresidue methods vs. compound–class-specific methods.
• Shift toward polar contaminants.
• Shift from parent compound analysis to the analysis of metabolites and transformation product.
14.2.1 Fast Chromatography
Among the different modern approaches to achieving fast separation without compromising resolution and separation efficiency, two approaches are frequently used in environmental analysis of emerging contaminants: liquid chromatography at ultra high pressures using sub-2-μm-particle packed columns (ultra high-pressure liquid chromatography, UHPLC) and chromatography using fused-core columns.
Rapid multiresidue screening of organic contaminants in environmental samples is one of the most promising fields of application of UHPLC technology. Recently, UHPLC–MS multiresidue methods were developed for the analysis of pharmaceuticals and steroid hormones in river and wastewater [4,5], pharmaceuticals and endocrine-disrupting compounds (EDCs) in sewage sludge and sediment [6], drugs of abuse in river and wastewater [7,8], UV filters in water [9,10], perfluorinated compounds in river water and fish [11], and polar pesticides in wastewater [12].
Several successful applications of fused-core columns for the analysis of emerging contaminants in environmental samples are described in the literature, including the analysis of pharmaceuticals and their metabolites in wastewater [13], personal care products (PCPs; antimicrobials, preservatives), benzotriazole UV stabilizers and organophosphorus compounds in fish tissue [14], sulphonamide antibiotics in fish [15], bisphenol A and its chlorinated metabolites in wastewater and drinking water [16], and drugs of abuse and metabolites in wastewater [17].
14.2.2 On-Line SPE–LC–MS Coupling
Over the past 10 years, there has been an increase in the use of automated instruments that integrate the extraction, purification, and detection steps. Several generic approaches have been developed for on-line sample extraction coupled to LC–MS using different extraction supports or sorbents, such as disposable solid–phase extraction (SPE) cartridges, restricted access materials (RAM), large-size particles, or monolithic materials [18]. The most often used automated commercial systems include Symbiosis and Prospekt-2 systems manufactured by Spark Holland, using disposable SPE cartridges. Recently, applications have been developed for the analysis of illicit drugs [19,20], multiclass pharmaceuticals [21,22], biocides and pesticides [23], pesticides [24], PFOS (perfluorooctanesulfonic acid) and PFOA (perfluorooctanoic acid) [25]. Another approach for automated and integrated extraction, preconcentration and cleanup is based on turbulent-flow chromatography (TFC), using large diameter particles and has shown great potential for on-line sample pretreatment, but applications in environmental analysis are still scarce [26].
14.2.3 Multiresidue Methods
Due to their comprehensive approach, multiresidue analytical methods have become preferred tools for tracking down different compound classes at low nanogram per liter levels. They allow determination of a large number of micropollutants in a single analysis, thus reducing its time and cost. In an ideal case, a multiclass–multiresidue analytical method for determination of trace organic pollutants in environmental matrices should fulfill several criteria, such as
1. Simultaneous extraction and preconcentration of all target analytes in a single step.
2. Limits of quantification (LOQs) low enough for each analyte.
3. Substance-specific detection.
4. Applicability to various matrices (e.g., natural water, drinking water, wastewater).
Some of the recent examples of multiresidue methods for emerging contaminants in environmental samples are a method for the analysis of 34 polar organic compounds (pharmaceutical compounds, pesticides and their degradation products, perfluorinated acids, and endocrine disrupting compounds) in river water [27]; a method for 46 basic, neutral, and acidic compounds, including selected iodinated contrast media, analgesics, anti-inflammatories, stimulants, beta blockers, antibiotics, lipid regulators, antihistamines, psychiatric drugs, herbicides, corrosion inhibitors, and the gastric acid-regulator pantoprazole [28]; a method for 60 pharmaceuticals and illicit drugs in suspended particulate matter [29]; a method for 65 stimulants, opiod and morphine derivatives, benzodiazepines, antidepressants, dissociative anaesthetics, drug precursors, human urine indicators, and their metabolites in wastewater and surface water [7]; a method for 70 high-priority pharmaceuticals (of the U.S. EPA) in water [30]; a method for 23 emerging pollutants belonging to personal care products (antimicrobials, preservatives), benzotriazole UV stabilizers, and organophosphorus compounds in fish using high-speed solvent extraction (HSSE), followed by silica gel cleanup and ultra fast liquid chromatography coupled with tandem mass spectrometry (UFLC–MS/MS) analysis [31].
However, it is important to mention that simultaneous analysis of multiclass compounds with quite different physicochemical characteristics often imposes compromises between the performance parameters and extraction, separation, and detection conditions acceptable for the majority of compounds but not optimal for all of them. Another potential pitfall of multiresidue determination methods is an enhanced matrix effect, particularly when studying complex samples, such as wastewater. In multiresidue methods, usually simple sample pretreatment is used to reduce the analysis time. This simplification of the sample cleanup step can result in dirty extracts with high coextractive substance content, which may lead to significant ion suppression (or enhancement) when using LC–MS/MS.
14.3 Target Analysis of Specific Contaminant Groups Using LC–MS
14.3.1 Brominated Flame Retardants
Flame retardant (FR) compounds are a structurally diverse group of chemicals added to or reacted with polymers, and they are used in plastics, textiles, electronic circuitry, and other materials to reduce the risk of fire. One of these groups of compounds is brominated FRs (BFRs), some of which are ubiquitous, and many of which have been detected in biota, sediments, air, water, marine mammals, and even human milk.
Brominated flame retardants have typically been analyzed using gas chromatography (GC)–MS but the high injection port temperatures required to transfer the analytes to the GC column can result in degradation of some desired compounds. This could occur in the case of highly polybrominated diphenyl ethers (PBDEs) and other BFRs (e.g., hexabromocyclododecane, HBCD). Moreover, in the case of tetrabromobisphenol A (TBBPA) or PBDE metabolites, a derivatization step is needed, giving more possibilities for error, because derivatization is not always reproducible and quantitative. LC–MS/MS appears to be the methodology of choice to analyze TBBPA, HBCD diastereoisomers, and HBCD enantiomers because no derivatization is needed and isomeric separation is obtained [32]. Also important is the recent development of methods to identify metabolites of PBDEs and HBCD by LC–MS/MS, thereby avoiding the problems of derivatization with diazomethane.
LC–MS/MS appears to be the method of choice for TBBPA analysis because no derivatization of the phenolic group is required [33–35]. To obtain a good chromatographic separation of TBBPA, it is important to take into account its great dependence on the mobile phase composition. Chu, Haffner, and Letcher [34] found that, using methanol as the mobile phase, the response of the target compound was about one third greater than when using acetonitrile. The most suitable LC–MS interface for TBBPA analysis is electrospray ionization (ESI) operating in the negative ion mode. Tollbäck, Crescenzi, and Dyremark [36] reported limits of detection (LODs) 30–40 times lower than with atmospheric pressure photoionization (APCI). In terms of sensitivity, LC–ESI–MS can be competitive with published GC–EI (electron ionization)–MS techniques with LODs in the ppt range [37].
To obtain HBCD diastereoisomer-specific data, it is essential to use LC, since, using GC, the diastereoisomers interconvert at temperatures above 160°C. Therefore, LC–MS/MS using ESI or APCI are versatile tools for the isomer-specific determination of trace levels of HBCDs, monitoring the specific transitions m/z 640.6 to m/z 78.9 and 80.9. An example of separation using permethylated β-cyclodextrin column is shown in Figure 14.1.
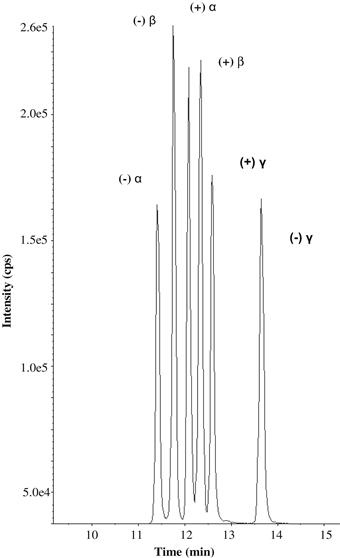
FIGURE 14.1 Enantiomeric HBCD separation by LC-ESI-MS/MS for a standard solution, using a Nucleodex β-PM (200 mm × 4.0 mm, 5 μm) chiral column.
LC–MS/MS is also used to analyze metabolites and transformation products of BFRs. Abdallah et al. [38] describe HBCD transformation-product detection in dust samples by LC–MS/MS analysis. These products were identified as four pentabromocyclododecene (PBCDe) isomers and two congeners of tetrabromocyclododecadienes (TBCDe). Brandsma et al. [39] studied the presence of hydroxylated metabolites of HBCD in three wildlife species and rats exposed to HBCD for 28 days. Four types of metabolites were found using LC–MS/MS, the monohydroxy metabolites of TBCDe, PBCDe, and HBCD and a dihydroxy-HBCD. The analysis of OH-PBDE metabolites by LC–MS seems interesting. However, these metabolites are poorly ionized by LC–ESI–MS, so they are not easily detected at low levels [40]. In 2007, Mas et al. [41] proposed the analysis of eight underivatized OH-PBDEs (OH-di- to OH-tetra-) by negative ion-spray ionization (ISP) –MS/MS. This method was shown to be efficient, robust, sensitive, and selective with LOQs at the high pg/g dry weight level.
14.3.2 Drugs of Abuse
Almost all the published methods to determine the drugs of abuse in water include a sample preparation based on either off-line SPE using Oasis HLB, Strata-X, Oasis MCX, or Strata-XC sorbents [42,43] or on-line SPE coupled directly to LC–MS [19,20]. Reversed-phase LC is the most used chromatographic technique for drugs of abuse [44], but recently, some methods were reported using hydrophilic interaction liquid chromatography (HILIC) [19,45]. Some studies have also been reported with a new particle column based on Fused-Core™ technology which has been developed to provide similar high speed and high efficiency as sub-2-μm particles but at half the back pressure. For example, Fontanals, Marcι, and Borrull [19] successfully used on-line SPE–HILIC–MS with a Fused-Core particle column to achieve a rapid and sensitive (low ng/l) analytical method for polar drugs in water, while Pedrouzo et al. [17] developed a method based on SPE–LC–MS/MS for the simultaneous analysis of six drugs and four metabolites in water using Ascentis Express C18. As already mentioned, UHPLC is being increasingly used in environmental analysis [7,8,42], because it offers advantages over conventional LC, such as improved separation efficiency and faster separations.
14.3.3 Hormones and Other Endocrine-Disrupting Compounds
The number of analytical methods described in the literature for the determination of endocrine-disrupting compounds (EDCs), such as natural and synthetic hormones (estrogens and progestogens), alkylphenolic compounds, and bisphenol A, has grown considerably. Because of the very low environmental levels (especially of hormones) and the complexity of some matrices, very efficient extraction–purification methods, in addition to selective and sensitive analytical techniques, are required. Due to its sensitivity and selectivity and because of its suitability for identification purposes in nontarget analysis, LC–MS/MS, preceded by SPE, is the method of choice for EDCs. For natural and synthetic hormones, which require especially sensitive detection, several on-line SPE–LC–MS/MS methods were developed [46–48], generally yielding LODs in the sub-ng/l range. Because of their low estrogenic potency, estrogen conjugates and progestogens have received comparatively less attention than estrogens. Liu et al. [49] used off-line SPE ultrasonic extraction and silica gel cartridge cleanup followed by rapid-resolution (RR) LC–MS/MS for the analysis of 28 steroids including 4 estrogens, 14 androgens, 5 progestagens, and 5 glucocorticoids in surface water, wastewater, and sludge samples. Several LC–MS/MS based methods were developed for the simultaneous analysis of EDCs belonging to different compound classes (hormones, nonylphenol, BPA, phthalates, etc.) in mixtures with pharmaceutical and other emerging contaminants, such as in the method for the simultaneous analysis of free and conjugated estrogens and antibiotics in runoff water and soil [50]; a method for the analysis of steroid hormones, pharmaceuticals and hormone-like personal care products (parabens, triclosan) in sewage sludge [6]; method for human pharmaceuticals and synthetic hormones in wastewater and river water [4,51].
14.3.4 Nanomaterials
In spite of the dynamic growth of manufacturing and use of nanomaterials (NMs), there are still very few analytical methods for their quantification in environmental matrices. Most quantitative procedures for NMs in environmental samples are devoted to carbon based NMs. For the quantification of fullerenes at low concentration, LC combined with MS [52], TOF MS, and TOF/TOF is mostly used [53]. To date, no measurements of the exposure or accumulation have been made in wild aquatic organisms, and few works have developed methods for the analysis of complex biological samples. Xia, Monteiro-Riviere, and Riviere [54] reported a trace analytical method for fullerenes from a sample containing a large amount of proteins. After the optimization of a liquid–liquid extraction LC method, a limit of detection of 0.34 μg/l was achieved. In another work, quantitative liquid–liquid extraction followed LC–MS was reported for the determination of fullerenes in water samples with a limit of detection of 0.4 μg/l [55]. The method was applied to determine the uptake of C60 by embryonic zebra fish. Recently, the same group presented a new analytical method for the analysis of aqueous fullerene C60 aggregates by asymmetric FFF with size determination by dynamic light scattering and quantification by LC–APPI–MS [56]. A method based on ultrasonic extraction followed by LC–MS/MS was developed for the quantification of fullerenes absorbed to suspended material in wastewater by Farré et al. [57], and this has given the first evidence of fullerenes in real environmental samples. In this case, the sensitivity was adequate (0.2–1 ng/l) for the analysis of real environmental samples. Recently, a group evaluated the occurrence of aerosol bound fullerenes in the Mediterranean Sea atmosphere [58].
14.3.5 Perfluorinated Compounds
Perfluorinated compounds (PFCs) constitute a large group of compounds characterized by a fully fluorinated hydrophobic linear carbon chain attached to one or more hydrophilic head groups. PFCs repel both water and oil and are therefore ideal chemicals for surface treatments. These compounds have been used for many industrial applications, including stain repellents (such as Teflon), textile, paints, waxes, polishes, electronics, adhesives, and food packaging [59].
In recent years a significant effort has been devoted to the detection of PFCs in environmental samples and biota, and several reviews have been published [60]. One of the major problems encountered in PFCs analysis is cross-contamination during the analytical process [61]. The major source of contamination of PFCs in laboratories is contact with laboratory materials made of or containing fluoropolymers, such as polytetrafluoroethylene or perfluoroalkoxy compounds [61]. On the other hand, different causes of losses have been identified associated with adsorption to sample containers, such as glass [62], or polymeric containers, such as polypropylene (PP), and high-density ethylene (HDPE) container surfaces. Biodegradation and biotransformations also should be prevented. Whereas good results were obtained for samples stored in a freezer or using combinations of solvents, like acetonitrile, and freezing [63] among others.
For the extraction of complex matrices, such as food, procedures based on ion-pair extraction using a tetrabutylammonium (TBA) hydroxide solution are widely employed [64,65]. However, this method has some disadvantages, such as (a) coextraction of lipids and other matrix constituents and (b) the wide variety of recoveries observed, which are related to the matrix effects mentioned previously. Liquid–solid extraction (LSE) is also applied to the analysis of biota and food samples [66]. During recent years, KOH digestion, to release covalently-bound fluoride ions followed by extraction using solid-phase extraction has been used. This method was initially reported by Yamashita et al. [67] and later applied in different studies [68]. For liquid samples, analysis protocols based on filtration followed by SPE are widely used.
For quantitative aspects, LC is the separation technique of choice. LC separation of PFCs has been carried out mainly with C18 and C8 columns. Taniyasu et al. [69] explored the chromatographic properties and separation of short-chain PFAs on RP C18 and ion-exchange columns. The results suggest that RP columns are not suitable for the analysis of short-chain PFAs, and ion-exchange columns are a suitable alternative, and have superior retention properties for more hydrophilic substances.
ESI operating in the negative ion mode has been the interface most widely used for the analysis of anionic PFCs. In addition, ESI has been optimized for the determination of neutral compounds, such as the sulfonamides PFOSA, Et-PFOSA, and t-Bu-PFOS. The use of atmospheric pressure photoionization was explored by Takino, Daishima, and Nakahara [70]. The authors found the main advantage of this technology to be the absence of matrix effects, but the limits of detection were considerably higher than those obtained by LC–ESI–MS/MS.
LC–MS/MS performed using triple quadrupole (QqQ) combined with multiple reaction monitoring (MRM) is one of the more widely applied detectors as well as one of the better-suited detectors for quantification of PFCs. Nowadays, the performance of ion trap (IT) and TOF also are considered suitable for trace quantification of PFCs. Berger and Haukas present a comparison between IT, QqQ, and time-of-flight (TOF) instruments [71]. Tandem mass spectrometry showed excellent specificity, as the matrix background is eliminated by the instrument. Applying TOF–MS gives an estimation of the amount of matrix left in the extract, which could impair the ionization performance, and the high mass resolution of the TOF–MS instrument offers excellent specificity for PFC identification after a crude sample injection.
14.3.6 Pharmaceuticals
The need to monitor pharmaceutical residues in the environment resulted in numerous analytical methods developed for their determination and the determination of their metabolites. Today, multiresidue analytical methods have become preferred tools for tracking down different therapeutic categories, and the pharmaceuticals more frequently included in such multiresidue methods are analgesics and anti-inflammatory drugs, antibiotics, lipid regulators, psychiatric drugs, and ß blockers. These drugs have very high consumption worldwide and are the most ubiquitous in both surface and wastewaters. SPE followed by LC coupled to tandem MS (QqQ) is the usual method of choice for quantitative determinations. Table 14.1 gives several examples of current methods used to simultaneously analyze pharmaceuticals belonging to different therapeutical classes. Furthermore, in the last few years, state-of-the-art analytical methodologies using hybrid MS detection systems, such as QqTOF, QqLIT, linear ion trap–Fourier transform–ion-cyclotron resonance (LIT–FT–ICR) and LTQ Orbitrap [3] have been applied to the analysis of pharmaceuticals in environmental and wastewaters. Schriks et al. [72] used LC–high resolution Orbitrap MS/MS to identify and quantify various glucocorticoids in wastewaters in the Netherlands. Figure 14.2 shows the high resolution MS spectra and low resolution product ion spectra for a selection of five glucocorticoids. Herrera-Rivera at al. [73] used an Orbitrap to study the influence of natural organic matter on the screening of pharmaceuticals in water, while in another study, an Orbitrap was used for the high-throughput identification of microbial transformation products of pharmaceuticals [74].
TABLE 14-1
Overview of Most Representative LC-MS Methods for Quantitative Determination of Pharmaceuticals in Aqueous and Solid Environmental Samples

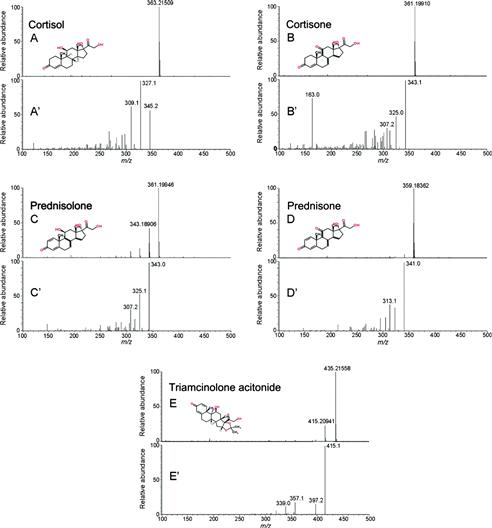
FIGURE 14.2 High-resolution full scan MS spectra (A−E) and low resolution MS product ion spectra (A′−E′) for a selection of 5 glucocorticoid standards. Reprinted with permission from [72]. Copyright © 2010 American Chemical Society
14.3.7 Sunscreen Agents
Sunscreen agents, also known as UV filters, are used extensively in personal care products and also are present in a wide variety of industrial goods, such as textiles, paints, and plastics to prevent photodegradation of polymers and pigments [75]. However, recent concern has risen due to their potential for endocrine disruption and developmental toxicity [76,77].
A number of LC–MS/MS based methods have been developed for their trace analysis in environmental samples. For water analysis, LC–ESI–MS/MS is the technique of choice; however, in recent studies, UHPLC separation and APCI ionization has become increasingly popular. Pedrouzo et al. [78] developed a new UHPLC–MS/MS method using solventless stir-bar sorptive extraction to investigate four UV filters, dihydroxy methoxybenzophenone (DHMB), benzophenone-3 (BP-3), octocrylene (OC), and ethylhexyl dimethyl-aminobenzoate (OD-PABA) in surface water and wastewater. Detection limits of 2.5 ng/l and 10 ng/l, respectively, were achieved. Recently, Wick, Fink, and Ternes combined LC–ESI–MS/MS and LC–APCI–MS/MS after SPE extraction for the determination of five UV filters in wastewater and surface water [79]. Quantification limits achieved ranged from 0.5 to 5 ng/l and 2.5 to 50 ng/l in surface water and wastewater, respectively. An innovative method, more simple and rapid, based on the direct analysis of the surface of a polydimethylsiloxane-coated stir bar previously used to extract the UV filters from water was developed by Haunschmidt et al. [80]. With this direct analysis in real time (DART) –MS method, seven UV filters were determined in different environmental waters with detection limits below 40 ng/l in all cases.
Several methods have been developed for the analysis of sludge and sediment samples. For sewage sludge, PLE combining extraction and cleanup in a single step, followed by UHPLC–ESI–MS/MS was used by Nieto et al. [81] to measure four UV filters with limits of detection below 8 μg/kg dry weight. Using this method, OD-PABA and BP-3 were detected for the first time in sewage sludge. Gago-Ferrero, Díaz-Cruz, and Barcelo [82] employed a similar method to determine eight UV filters and degradation products in sewage sludge. The determination was fast and sensitive, affording limits of detection lower than 19 ng/g dry weight. Good recovery rates, especially given the high complexity of the sludge matrix (between 70% and 102% were afforded. Rodil, Schrader, and Moeder [83] described a method based on LC–APPI–MS/MS for the determination of 11 UV filters in sludge with limits of detection in the range 0.3–25 ng/g. With this method, UV filters were found at levels ranging from 1.4 to 2479 ng/g, and seven of them were detected for the first time. In sediments, existing data on UV filters is scarce. The extraction methods used were based on solid–liquid extraction of the freeze dried sediments with different organic solvents, and analyses were performed by gas chromatography–mass spectrometry, with separation times longer than 60 min and requiring a derivatization step for the most polar compounds. Based on LC, Gago-Ferrero, Díaz-Cruz, and Barcelo [84] created a fast, selective and sensitive multiresidue analytical method involving the concentration and purification in a single step by placing aluminum oxide as cleanup sorbent in the PLE extraction cell. PLE extraction and purification in connection with UHPLC enabled all compounds to be separated in less than 9 min and with a total chromatographic analysis time of 18 min. This method significantly decreased the overall time of analysis as compared to those of previously developed methods. Quantitative recoveries (>80%), satisfactory precision (RSD, 5–15%), and low limits of detection (0.5–15 ng/g dry weight) were achieved. By using this method, UV filters could be detected in 95% of the sediment samples analyzed. Results revealed a widespread presence of OC, reaching concentrations up to 2400 ng/g dry weight, the highest reported so far. OD-PABA and BP-3 were also frequently detected (60–65%) but at lower concentrations (4.4–27 ng/g dry weight).
14.4 Conclusions
The application of advanced tandem and hybrid LC–MS instruments in the field of environmental analysis has allowed the determination of a broader range of compounds and, thus, permitted more comprehensive assessment of environmental contaminants. However, one of the drawbacks of current analytical methods is that the majority of them focus on only parent target compounds and rarely include metabolites and transformation products, which sometimes can be more toxic and persistent than the original compounds. Moreover, identification of reaction pathways and identification of transformation products is of crucial importance in understanding the fate or organic contaminants in the environment. On the other hand, the high complexity of environmental samples sometimes requires the application of high resolution techniques to provide additional structure information needed for unequivocal identification of contaminants and confirmation of positive findings. The introduction of Orbitrap, TOF, QqTOF, and QqLIT instruments, which allow the simultaneous determination of both parent and transformation products within a single analytical run, overcomes this drawback and provides a higher degree of certainty in compound identification. In comparison to triple quadrupole mass spectrometers, which operate at unit resolution and generally in the SRM mode for specific target analytes, TOF, QqTOF, and Orbitrap mass spectrometers are capable of acquiring full-scan mass spectra at high resolution for all analytes without loss in sensitivity. Since these instruments have high resolution (at least 10,000 and higher) at full-width–half-maximum (fwhm) peak height, isotopic patterns are evident and chemical structures can be proposed for unknowns or confirmed for target analytes [3].
Therefore, a gradual shift from parent compound analysis to the analysis of metabolites and transformation product is expected. But the question is whether chemical analysis of specific compounds is sufficient to assess contaminants present in the environment. The main drawback of the conventional approach is target compound monitoring, which is often insufficient to assess the environmental relevance of emerging contaminants. The real challenge is to decide on the significance of the chemical data, and the discussion of the risks posed is still open. General screening for unknown substances is time consuming and expensive and is often shattered by problems, such as lack of standards and mass spectral libraries. Therefore, effect-related analysis, focused on relevant compounds, nowadays, seems to be more appropriate to assess and study environmental contamination problems.
References
1. Barcelo D, Petrovic M. Challenges and achievements of LC–MS in environmental analysis: 25 years on. Trac Trend Anal Chem. 2007;26:2–11.
2. Runnqvist H, Bak SA, Hansen M, Styrishave B, Halling-Sørensen B, Björklund E. Determination of pharmaceuticals in environmental and biological matrices using pressurised liquid extraction–are we developing sound extraction methods?. J Chromatogr A. 2010;1217:2447–2470.
3. Petrovic M, Farré M, Lopez de Alda MJ, et al. Recent trends in the liquid chromatography-mass spectrometry analysis of organic contaminants in environmental samples. J Chromatogr A. 2010;1217:4004–4017.
4. Huerta-Fontela M, Galceran MT, Ventura F. Fast liquid chromatography–quadrupole-linear ion trap mass spectrometry for the analysis of pharmaceuticals and hormones in water resources. J Chromatogr A. 2010;1217(25):4212–4222.
5. López-Roldán R, de Alda ML, et al. Advanced monitoring of pharmaceuticals and estrogens in the Llobregat River basin (Spain) by liquid chromatography–triple quadrupole–tandem mass spectrometry in combination with ultra performance liquid chromatography–time of flight-mass spectrometry. Chemosphere. 2010;80(11):1337–1344.
6. Yu Y, Huang Q, et al. Determination of pharmaceuticals, steroid hormones, and endocrine-disrupting personal care products in sewage sludge by ultra-high-performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2011;399(2):891–902.
7. Baker DR, Kasprzyk-Hordern R. Multi-residue analysis of drugs of abuse in wastewater and surface water by solid-phase extraction and liquid chromatography–positive electrospray ionisation tandem mass spectrometry. J Chromatogr A. 2011;1218(12):1620–1631.
8. Hernández F, Bijlsma L, et al. Rapid wide-scope screening of drugs of abuse, prescription drugs with potential for abuse and their metabolites in influent and effluent urban wastewater by ultrahigh pressure liquid chromatography–quadrupole-time-of-flight-mass spectrometry. Anal Chim Acta. 2011;684(1–2):87–97.
9. Gago-Ferrero P, Díaz-Cruz MS, et al. Fast pressurized liquid extraction with in-cell purification and analysis by liquid chromatography tandem mass spectrometry for the determination of UV filters and their degradation products in sediments. Anal Bioanal Chem 2011;1–10.
10. Pedrouzo M, Borrull F, et al. Stir-bar-sorptive extraction and ultra-high-performance liquid chromatography–andem mass spectrometry for simultaneous analysis of UV filters and antimicrobial agents in water samples. Anal Bioanal Chem. 2010;397(7):2833–2839.
11. Gosetti F, Chiuminatto U, et al. Determination of perfluorochemicals in biological, environmental and food samples by an automated on-line solid phase extraction ultra high performance liquid chromatography tandem mass spectrometry method. Chromatogr A. 2010;1217(50):7864–7872.
12. Barco-Bonilla N, Romero-González R, et al. Analysis and study of the distribution of polar and non-polar pesticides in wastewater effluents from modern and conventional treatments. J Chromatogr A. 2010;1217(50):7817–7825.
13. arcomnicu I, Van Nuijs ALN, et al. Simultaneous determination of 15 top-prescribed pharmaceuticals and their metabolites in influent wastewater by reversed-phase liquid chromatography coupled to tandem mass spectrometry. Talanta. 2011;83(3):795–803.
14. Kim JW, Ramaswamy BR, et al. Multiresidue analytical method for the determination of antimicrobials, preservatives, benzotriazole UV stabilizers, flame retardants and plasticizers in fish using ultra high performance liquid chromatography coupled with tandem mass spectrometry. J Chromatogr A. 2011;1218(22):3511–3520.
15. Lu Y, Shen Q, et al. Development of an on-line matrix solid-phase dispersion/fast liquid chromatography/tandem mass spectrometry system for the rapid and simultaneous determination of 13 sulfonamides in grass carp tissues. J Chromatogr A. 2011;1218(7):929–937.
16. Gallart-Ayala H, Moyano E, et al. On-line solid phase extraction fast liquid chromatography-tandem mass spectrometry for the analysis of bisphenol A and its chlorinated derivatives in water samples. J Chromatogr A. 2010;1217(21):3511–3518.
17. Pedrouzo M, Borrull F, Pocurull E, Marce RM. Drugs of abuse and their metabolites in waste and surface waters by liquid chromatography–tandem mass spectrometry. J Sep Sci. 2011;34(10):1091–1101.
18. Rodriguez-Mozaz S, de Alda MJL, Barcelo D. Advantages and limitations of on-line solid phase extraction coupled to liquid chromatography–mass spectrometry technologies versus biosensors for monitoring of emerging contaminants in water. J Chromatogr A 2007;1152.
19. Fontanals N, Marcé RM, Borrull F. On-line solid-phase extraction coupled to hydrophilic interaction chromatography–mass spectrometry for the determination of polar drugs. J Chromatogr A. 2011;1218(35):5975–5980.
20. Postigo C, Lopez de Alda arceló MJD, arceló D. Fully automated determination in the low nanogram per liter level of different classes of drugs of abuse in sewage water by on-line solid-phase extraction–liquid chromatography–electrospray-tandem mass spectrometry. Anal Chem. 2008;80(9):3123–3134.
21. Trenholm RA, Vanderford BJ, Snyder SA. On-line solid phase extraction LC–MS/MS analysis of pharmaceutical indicators in water: A green alternative to conventional methods. Talanta. 2009;79:1425–1432.
22. López-Serna R, Pérez S, Ginebreda A, Petrović M, Barceló D. Fully automated determination of 74 pharmaceuticals in environmental and waste waters by online solid phase extraction–liquid chromatography–electrospray–tandem mass spectrometry. Talanta. 2010;83(2):410–424.
23. Singer H, Jaus S, Hanke I, Lück A, Hollender J, Alder AC. Determination of biocides and pesticides by on-line solid phase extraction coupled with mass spectrometry and their behaviour in wastewater and surface water. Environ Pollut. 2010;158(10):3054–3064.
24. Jansson C, Kreuger J. Multiresidue analysis of 95 pesticides at low nanogram/liter levels in surface waters using online preconcentration and high performance liquid chromatography/tandem mass spectrometry. J AOAC Int. 2010;93(6):1732–1747.
25. Wilson SR, Malerod H, Holm A, Molander P, Lundanes E, Greibrokk T. On-line SPE-nano-LC-nanospray-MS for rapid and sensitive determination of perfluorooctanoic acid and perfluorooctane sulfonate in river water. J Chromatogr Sci. 2007;45:146–152.
26. Mottier P, Hammel YA, Gremaud E, Guy PA. Quantitative high-throughput analysis of 16 (fluoro)quinolones in honey using automated extraction by turbulent flow chromatography coupled to liquid chromatography-tandem mass spectrometry. J Agri Food Chem. 2008;56(1):35–43.
27. Loos R, Locoro G, Contini S. Occurrence of polar organic contaminants in the dissolved water phase of the Danube River and its major tributaries using SPE-LC-MS2 analysis. Water Res. 2010;44(7):2325–2335.
28. Nödler K, Licha T, Bester K, Sauter M. Development of a multi-residue analytical method, based on liquid chromatography–tandem mass spectrometry, for the simultaneous determination of 46 micro-contaminants in aqueous samples. J Chromatogr A. 2010;1217(42):6511–6521.
29. Baker DR, Kasprzyk-Hordern B. Multi-residue determination of the sorption of illicit drugs and pharmaceuticals to wastewater suspended particulate matter using pressurised liquid extraction, solid phase extraction and liquid chromatography coupled with tandem mass spectrometry. J Chromatogr A. 2011;1218(44):7901–7913.
30. Ferrer I, Zweigenbaum JA, Thurman EM. Analysis of 70 Environmental Protection Agency priority pharmaceuticals in water by EPA Method 1694. J Chromatogr A. 2010;1217:5674–5686.
31. Joon-Woo K, Babu Rajendran R, Kwang-Hyeon C, Tomohiko I, Shinsuke T. Multiresidue analytical method for the determination of antimicrobials, preservatives, benzotriazole UV stabilizers, flame retardants and plasticizers in fish using ultra high performance liquid chromatography coupled with tandem mass spectrometry. J Chromatogr A. 2011;1218:3511–3520.
32. Guerra P, Eljarrat E. Determination of halogenated flame retardants by liquid chromatography coupled to mass spectrometry. Trac Trends Anal Chem. 2011;30:842–855.
33. Covaci A, Voorspoels S, Abdallah MAE, Geens T, Harrad S, Law RJ. Analytical and environmental aspects of the flame retardant tetrabromobisphenol-A and its derivatives. J Chromatogr A. 2009;1216:346–363.
34. Chu S, Haffner GD, Letcher RJ. Simultaneous determination of tetrabromobisphenol A, tetrachlorobisphenol A, bisphenol A and other halogenated analogues in sediment and sludge by high performance liquid chromatography–electrospray tandem mass spectrometry. J Chromatogr A. 2005;1097:25–32.
35. Covaci A, Gerecke AC, Law RJ, et al. Hexabromocyclododecanes (HBCDs) in the environment and humans: a review. Environ Sci Tech. 2006;40:3679–3688.
36. Tollback J, Crescenzi C, Dyremark E. Determination of the flame retardant tetrabromobisphenol A in air samples by liquid chromatography–mass spectrometry. J Chromatogr A. 2006;1104:106–112.
37. Hayama T, Yoshida H, Onimaru S, et al. Determination of tetrabromobisphenol A in human serum by liquid chromatography-electrospray ionization tandem mass spectrometry. J Chromatogr B. 2004;809:131–136.
38. Abdallah MAE, Ibarra C, Neels H, Harrad S, Covaci A. Comparative evaluation of liquid chromatography-mass spectrometry versus gas chromatography–mass spectrometry for the determination of hexabromocyclododecanes and their degradation products in indoor dust. J Chromatogr A. 2008;1190:333–341.
39. Brandsma SH, Van Der Ven LTM, De Boer J, Leonards PEG. Identification of hydroxylated metabolites of hexabromocyclododecane in wildlife and 28-days exposed wistar rats. Environ Sci Tech. 2009;43:6058–6063.
40. Díaz-Cruz MS, García-Galán MJ, Guerra P, et al. Analysis of selected emerging contaminants in sewage sludge. TRAC Trends Anal Chem. 2009;28:1263–1975.
41. Mas S, Jauregui O, Rubio F, De Jua A, Tauler R, Lacorte S. Comprehensive liquid chromatography–ion-spray tandem mass spectrometry method for the identification and quantification of eight hydroxylated brominated diphenyl ethers in environmental matrices. J Mass Spec. 2007;42:890–899.
42. Bijlsma L, Sancho JV, Pitarch E, Ibáñe M, Hernández F. Simultaneous ultra-high-pressure liquid chromatography–tandem mass spectrometry determination of amphetamine and amphetamine-like stimulants, cocaine and its metabolites, and a cannabis metabolite in surface water and urban wastewater. J Chromatogr A. 2009;1216(15):3078–3089.
43. Vazquez-Roig P, Andreu V, Blasco C, Picó Y. SPE and LC-MS/MS determination of 14 illicit drugs in surface waters from the Natural Park of L’Albufera (València, Spain). Anal Bioanal Chem. 2010;397(7):2851–2864.
44. Castiglioni S, Zuccato E, Chiabrando C, Fanelli R, Bagnati R. Mass spectrometric analysis of illicit drugs in wastewater and surface water. Mass Spec Rev. 2008;27:378–394.
45. van Nuijs ALN, Pecceu B, Theunis L, et al. Spatial and temporal variations in the occurrence of cocaine and benzoylecgonine in waste- and surface water from Belgium and removal during wastewater treatment. Water Res. 2009;43:1341–1349.
46. Salvador A, Moretton C, Piram A, Faure R. On-line solid-phase extraction with on-support derivatization for high-sensitivity liquid chromatography tandem mass spectrometry of estrogens in influent/effluent of wastewater treatment plants. J Chromatogr A. 2007;1145:102–109.
47. López-Roldán R, de Alda ML, Gros M, Petrovic M, Martín-Alonso J, Barceló D. Advanced monitoring of pharmaceuticals and estrogens in the Llobregat River basin (Spain) by liquid chromatography–triple quadrupole–tandem mass spectrometry in combination with ultra performance liquid chromatography–time of flight-mass spectrometry. Chemosphere. 2010;80(11):1337–1344.
48. Viglino L, Aboulfadl K, Prévost M, Sauvé S. Analysis of natural and synthetic estrogenic endocrine disruptors in environmental waters using online preconcentration coupled with LC–APPI–MS/MS. Talanta. 2008;76(5):1088–1096.
49. Liu S, Ying GG, Zhao JL, et al. Trace analysis of 28 steroids in surface water, wastewater and sludge samples by rapid resolution liquid chromatography–electrospray ionization tandem mass spectrometry. J Chromatogr A. 2011;1218(10):1367–1378.
50. Tso J, Dutta S, Inamdar S, Aga DS. Simultaneous analysis of free and conjugated estrogens, sulfonamides, and tetracyclines in runoff water and soils using solid-phase extraction and liquid chromatography–-tandem mass spectrometry. J Agri Food Chem. 2011;59(6):2213–2222.
51. Al-Odaini NA, Zakaria MP, Yaziz MI, Surif S. Multi-residue analytical method for human pharmaceuticals and synthetic hormones in river water and sewage effluents by solid-phase extraction and liquid chromatography–tandem mass spectrometry. J Chromatogr A. 2010;1217(44):6791–6806.
52. Deye JR, Shiveley AN, Oehrle SA, Walters KA. Separation of substituted fullerenes using non-aqueous reversed-phase liquid chromatography–mass spectrometry. J Chromatogr A. 2008;1181(1–2):159–161.
53. Ilchenko S, Cotter RJ. Collision energetics in a tandem time-of-flight (TOF/TOF) mass spectrometer with a curved-field reflectron. Int J Mass Spec. 2007;265(2–3):372–381.
54. Xia XR, Monteiro-Riviere NA, Riviere JE. Trace analysis of fullerenes in biological samples by simplified liquid–liquid extraction and high-performance liquid chromatography. J Chromatogr A. 2006;1129(2):216–222.
55. Isaacson CW, Usenko CY, Tanguay RL, Field JA. Quantification of fullerenes by LC/ESI–MS and its application to in vivo toxicity assays. Anal Chem. 2007;79(23):9091–9097.
56. Isaacson CW, Bouchard D. Asymmetric flow field flow fractionation of aqueous C60 nanoparticles with size determination by dynamic light scattering and quantification by liquid chromatography atmospheric pressure photo-ionization mass spectrometry. J Chromatogr A. 2010;1217(9):1506–1512.
57. Farré M, Gajda-Schrantz K, Kantiani L, Barceló D. Ecotoxicity and analysis of nanomaterials in the aquatic environment. Anal Bioanal Chem. 2009;393(1):81–95.
58. Sanchís J, Berrojalbiz N, Caballero G, Dachs J, Farré M, Barceló D. Occurrence of aerosol-bound fullerenes in the Mediterranean Sea atmosphere. Environ Sci Tech 2012; 46(3):1335–43.
59. Clara M, Scheffknecht C, Schar S, Weiss S, Gans O. Emissions of perfluorinated alkylated substances (PFAS) from point sources—identification of relevant branches. Wat Sci Tech. 2008;58:59–66.
60. Schulte C. Perfluorinated compounds—environmental findings and distribution. PFT–Nachweise Umwelt Theor Verbreit. 2007;12(2):67–71.
61. Taniyasu S, Kannan K, Man KS, et al. Analysis of fluorotelomer alcohols, fluorotelomer acids, and short- and long-chain perfluorinated acids in water and biota. J Chromatogr A. 2005;1093(1–2):89–97.
62. Martin JW, Kannan K, Berger U, et al. Analytical challenges hamper perfluoroalkyl research. Environ Sci Tech. 2004;38(13):248A–255A.
63. Wang N, Szostek B, Buck RC, et al. Fluorotelomer alcohol biodegradation—direct evidence that perfluorinated carbon chains breakdown. Environ Sci Tech. 2005;39(19):7516–7528.
64. Taniyasu S, Kannan K, Horii Y, Hanari N, Yamashita N. A survey of perfluorooctane sulfonate and related perfluorinated organic compounds in water, fish, birds, and humans from Japan. Environ Sci Tech. 2003;37(12):2634–2639.
65. Guruge KS, Manage PM, Yamanaka N, Miyazaki S, Taniyasu S, Yamashita N. Species-specific concentrations of perfluoroalkyl contaminants in farm and pet animals in Japan. Chemosphere. 2008;73(Suppl. 1):S210–S215.
66. Powley CR, George SW, Ryan TW, Buck RC. Matrix effect-free analytical methods for determination of perfluorinated carboxylic acids in environmental matrixes. Anal Chem. 2005;77(19):6353–6358.
67. Yamashita N, Kannan K, Taniyasu S, et al. Analysis of perfluorinated acids at parts-per-quadrillion levels in seawater using liquid chromatography-tandem mass spectrometry. Environ Sci Tech. 2004;38(21):5522–5528.
68. So MK, Taniyasu S, Lam PKS, Zheng GJ, Giesy JP, Yamashita N. Alkaline digestion and solid phase extraction method for perfluorinated compounds in mussels and oysters from south China and Japan. Arch Environ Contam Toxicol. 2006;50(2):240–248.
69. Taniyasu S, Kannan K, Yeung LWY, Kwok KY, Lam PKS, Yamashita N. Analysis of trifluoroacetic acid and other short-chain perfluorinated acids (C2–C4) in precipitation by liquid chromatography–tandem mass spectrometry: comparison to patterns of long-chain perfluorinated acids (C5–C18). Anal Chim Acta. 2008;619(2):221–230.
70. Takino M, Daishima S, Nakahara T. Determination of perfluorooctane sulfonate in river water by liquid chromatography/atmospheric pressure photoionization mass spectrometry by automated on-line extraction using turbulent flow chromatography. Rapid Comm Mass Spec. 2003;17(5):383–390.
71. Berger U, Haukas M. Validation of a screening method based on liquid chromatography coupled to high-resolution mass spectrometry for analysis of perfluoroalkylated substances in biota. J Chromatogr A. 2005;1081(2):210–217.
72. Schriks M, Van Leerdam JA, Van Der Linden SC, Van Der Burg B, Van Wezel AP, De Voogt P. High-resolution mass spectrometric identification and quantification of glucocorticoid compounds in various wastewaters in the Netherlands. Environ Sci Tech. 2010;44(12):4766–4774.
73. Herrera Rivera Z, Oosterink E, Rietveld L, Schoutsen F, Stolker L. Influence of natural organic matter on the screening of pharmaceuticals in water by using liquid chromatography with full scan mass spectrometry. Anal Chim Acta. 2011;700(1–2):114–125.
74. Helbling DE, Hollender J. E.Kohler H-P, Singer H, Fenner K High-throughput identification of microbial transformation products of organic micropollutants. Environ Sci Tech. 2010;44(17):6621–6627.
75. Lowe NJ, Shaatg NA, Pathak MA. Sunscreens: development, evaluation and regulatory aspects New York: Marcel Dekker; 1997.
76. Kunz PY, Fent K. Multiple hormonal activities of UV filters and comparison of in vivo and in vitro estrogenic activity of ethyl-4-aminobenzoate in fish. Aquat Toxicol. 2006;79:305–324.
77. Schreurs RHMM, Sonneveld E, Jansen JHJ, Seinen W, van der Burg B. Interaction of polycyclic musks and UV filters with the estrogen receptor (ER), androgen receptor (AR), and progesterone receptor (PR) in reporter gene bioassays. Toxicol Sci. 2005;83:264–272.
78. Pedrouzo M, Borrull F, Marce RM, Pocurull E. Stir-bar-sorptive extraction and ultra-high-performance liquid chromatography-tandem mass spectrometry for simultaneous analysis of UV filters and antimicrobial agents in water samples. Anal Bioanal Chem. 2010;397:2833–2839.
79. Wick A, Fink G, Ternes TA. Comparison of electrospray ionization and atmospheric pressure chemical ionization for multi-residue analysis of biocides, UV-filters and benzothiazoles in aqueous matrices and activated sludge by liquid chromatography–tandem mass spectrometry. J Chromatogr A. 2010;1217:2088–2103.
80. Haunschmidt M, Klampfl CW, Buchberger W, Hertsens R. Determination of organic UV filters in water by stir bar sorptive extraction and direct analysis in real-time mass spectrometry. Anal Bioanal Chem. 2010;397:269–275.
81. Nieto A, Borrull F, Marcé RM, Pocurull E. Determination of personal care products in sewage sludge by pressurized liquid extraction and ultra high performance liquid chromatography–tandem mass spectrometry. J Chromatogr A. 2009;1216:5619–5625.
82. Gago-Ferrero P, Díaz-Cruz MS, Barcelo D. Occurrence of multiclass UV filters in treated sewage sludge from wastewater treatment plants. Chemosphere. 2011;84:1158–1165.
83. Rodil R, Schrader S, Moeder M. Pressurised membrane-assisted liquid extraction of UV filters from sludge. J Chromatogr A. 2009;1216:8851–8858.
84. Gago-Ferrero P, Díaz-Cruz MS, Barcelo D. Fast pressurized liquid extraction with in-cell purification and analysis by liquid chromatography tandem mass spectrometry for the determination of UV filters and their degradation products in sediments. Anal Bioanal Chem. 2011;400:2195–2204.
85. López-Serna R, Pérez S, Ginebreda A, Petrovic M, Barceló D. Fully automated determination of 74 pharmaceuticals in environmental and waste waters by online solid phase extraction-liquid chromatography–electrospray-tandem mass spectrometry. Talanta. 2010;83:410–424.
86. López-Serna R, Petrovic M, Barceló D. Development of a fast instrumental method for the analysis of pharmaceuticals in environmental and wastewaters based on ultra high performance liquid chromatography (UHPLC)–tandem mass spectrometry (MS/MS). Chemosphere. 2011;85:1390–1399.
87. Gros M, Petrovic M, Barceló D. Tracing pharmaceutical residues of different therapeutic classes in environmental waters by using liquid chromatography/quadrupole-linear ion trap mass spectrometry and automated library searching. Anal Chem. 2009;81:898–912.
88. Grujic S, Vasiljevic T, Lausevic M. Determination of multiple pharmaceutical classes in surface and ground waters by liquid chromatography–ion trap–tandem mass spectrometry. J Chromatogr A. 2009;1216:4989–5000.
89. Wu C, Spongberg AL, Witter JD. Use of solid phase extraction and liquid chromatography-tandem mass spectrometry for simultaneous determination of various pharmaceuticals in surface water. Int J Environ Anal Chem. 2008;88:1033–1048.
90. Busetti F, Linge KL, Heitz A. Analysis of pharmaceuticals in indirect potable reuse systems using solid-phase extraction and liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2009;1216:5807–5818.
91. Kasprzyk-Hordern B, Dinsdale RM, Guwy AJ. Multi-residue method for the determination of basic/neutral pharmaceuticals and illicit drugs in surface water by solid-phase extraction and ultra performance liquid chromatography–positive electrospray ionisation tandem mass spectrometry. J Chromatog A. 2007;1161:132–145.
92. Baker DR, Kasprzyk-Hordern B. Multi-residue determination of the sorption of illicit drugs and pharmaceuticals to wastewater suspended particulate matter using pressurised liquid extraction, solid phase extraction and liquid chromatography coupled with tandem mass spectrometry. J Chromatogr A. 2011;1218:7901–7913.
93. Jelic A, Petrovic M, Barceló D. Multi-residue method for trace level determination of pharmaceuticals in solid samples using pressurized liquid extraction followed by liquid chromatography/quadrupole-linear ion trap mass spectrometry. Talanta. 2009;80:363–371.
94. Radjenovic J, Jelic A, Petrovic M, Barceló D. Determination of pharmaceuticals in sewage sludge by pressurized liquid extraction (PLE) coupled to liquid chromatography–tandem mass spectrometry (LC–MS/MS). Anal Bioanal Chem. 2009;393:1685–1695.
95. Barron L, Tobin J, Paull B. Multi-residue determination of pharmaceuticals in sludge and sludge enriched soils using pressurized liquid extraction, solid phase extraction and liquid chromatography with tandem mass spectrometry. J Environ Monitor. 2008;10:353–361.
96. Spongberg AL, Witter JD. Pharmaceutical compounds in the wastewater process stream in Northwest Ohio. Sci Total Environ. 2008;397:148–157.
97. Yu Y, Huang Q, Cui J, Zhang K, Tang C, Peng X. Determination of pharmaceuticals, steroid hormones, and endocrine-disrupting personal care products in sewage sludge by ultra-high-performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2011;399:891–902.