Analysis of Vitamins by Liquid Chromatography
A. Gentili and F. Caretti, Department of Chemistry, Faculty of Mathematical, Physical and Natural Sciences, University of Rome La Sapienza, Italy
Outline
18.1. Introduction
18.2. Liquid Chromatographic Determination of Water-Soluble Vitamins
18.2.1. Vitamin B1
18.2.2. Vitamin B2
18.2.3. Vitamin B3
18.2.4. Vitamin B5
18.2.5. Vitamin B6
18.2.6. Vitamin B8
18.2.7. Vitamin B9
18.2.8. Vitamin B12
18.2.9. Vitamin C
18.3. Liquid Chromatographic Determination of Fat-Soluble Vitamins
18.3.1. Vitamin A
18.3.2. Vitamin D
18.3.3. Vitamin E
18.3.4. Vitamin K
18.4. Multivitamin Methods
18.1 Introduction
Vitamins are essential micronutrients, varying widely in chemical structure, biological activity, and physicochemical properties [1]. Owing to this heterogeneity, the classification as water-soluble (B-complex, C) and fat-soluble (A, D, E, K) is based on their solubility characteristics. On the whole, 13 groups are recognized in human nutrition (the B-complex groups together the vitamins B1, B2, B3, B5, B6, B8, B9, and B12) and each of them is composed of several biologically active forms, known as vitamers, which differ in structure, biopotency, and stability.
Due to their relevance in the human physiology, the requirement of accurate data on forms and concentrations of vitamins naturally occurring in foods has become more stringent in recent years. Likewise, the determination of the forms added to fortified foods and supplements needs reliable analytical procedures. Although liquid chromatography (LC) is the ideal technique for vitamin quantitative analysis, many of the current international methods are based on microbiological assays [1–3]; some of them are outdated, time consuming, expensive, and characterized by a high measurement uncertainty. On the contrary, the scientific literature has continuously presented new LC-based procedures suitable for the individual and simultaneous vitamin analysis. At present, also the European Committee for Standardization (CEN) and the Association of Official Analytical Chemists (AOAC) International consider LC as the first choice to determine the vitamins B1, B2, B6, C [1,2] and the fat-soluble vitamins [1,3]. For the other water-soluble vitamins, the published LC methods have to be tested collaboratively before they can be applied as official methods.
Before performing a LC analysis, it is advisable to adopt some preventive measures to restrain losses due to vitamin instability. The most important factors that lead to inactivation are light, air, temperature, pH, trace metals, and ionic strength. Since the majority of the vitamins are photosensitive, the use of low actinic amber glassware and subdued light is recommended for the whole duration of the analysis. Another precaution that cannot be disregarded is the addition of a proper antioxidant to the solvents employed both for the preparation of standard solutions and extraction procedures.
When vitamin determination is applied to food and biological matrices, further problems have to be tackled: (a) (inter- and intragroup) chemical heterogeneity (b) their presence at trace levels in the real samples; (c) matrix complexity; and (d) interactions with other matrix constituents, such as polysaccharides (food), proteins, and lipids (food and biological samples). With regard to the first point, the subtle structural differences among vitamers of a specific group may make their chromatographic separation difficult; another facet pertains to the unavailability and cost of standards for homologs. In consequence, it is preferred to analyze each vitamin singly, applying extraction conditions able to free all bound forms (by means of acidic, alkaline, or enzymatic digestion) or to convert the several forms into a more stable form (see B9, B12 determination as examples). In this way, it is possible to increase the concentration of the single-target homolog in the final extract, to simplify the analysis and hold down expenditure for the purchase of standards.
18.2 Liquid Chromatographic Determination of Water-Soluble Vitamins
The choice of the LC mode for the analysis of water-soluble vitamins depends on the extraction procedure employed and the vitamin form to be quantified (Figure 18.1). The most popular LC modes are normal-phase (NP), reversed-phase (RP), ion-pair RP, ion-suppression RP, and ion-exchange chromatography.
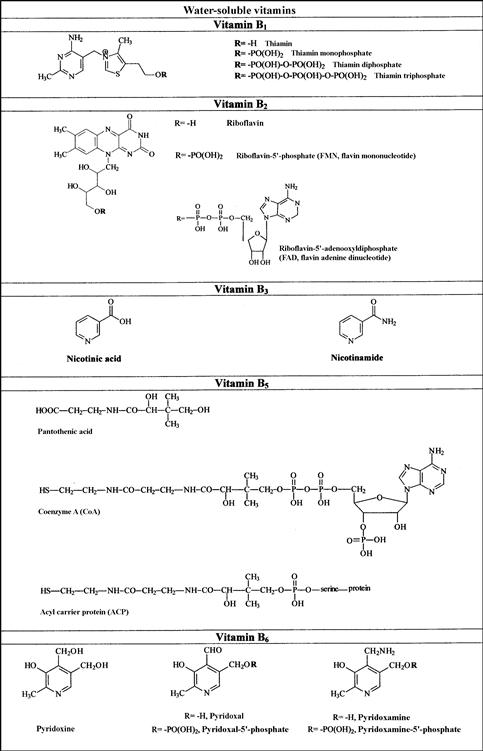
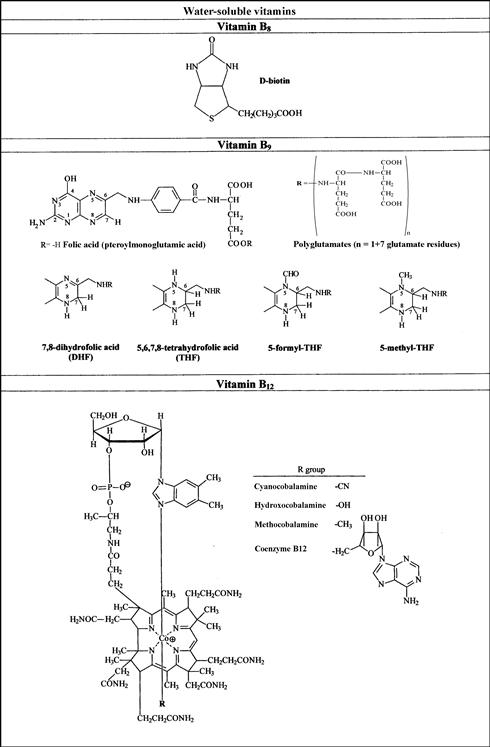
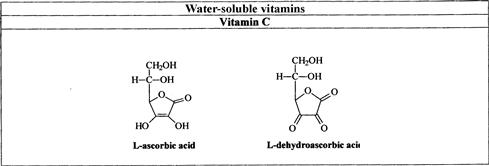
FIGURE 18.1 Names and structures of the water-soluble vitamins naturally occurring in foods.
(a) Vitamins B1, B2, B3, and B5; (b) vitamins B6, B8, sand B9; (c) B12. C.
18.2.1 Vitamin B1
Vitamin B1 [1] exists in nature both in free (thiamin) and esterified form (thiamin monophosphate, diphosphate, and triphosphate), while thiamin hydrochloride is used as a supplement [4]. To evaluate the total content of vitamin B1 in a food, extraction usually consists of an acid hydrolysis (0.1 M HCl in a water bath at 100°C or in an autoclave at 121°C) followed by an enzymatic digestion (diastases possessing a phosphatase activity) [1,2,5,6]. The acid treatment frees protein-bound forms and converts starch into soluble sugars. The enzymatic treatment may require several hours (on average 3 hr) of incubation for complete dephosphorylation of the thiamin esters.
RP and ion-pair RP chromatography are the most common forms of LC used for the free thiamin determination [1,5–10]. Highly deactivated columns contain the phenomenon of peak broadening and tailing, due to the interaction of a vitamin-B1 basic site with silanol groups on the stationary phase. Polarity and low molecular weight are responsible for poor retention on RP columns; mobile phases containing high percentages of water and a suitable ion-pair agent (for example alkyl sulphonates) are expedients that both improve the peak shape and increase retention of the vitamin.
Owing to its low molar absorptivity, the use of UV detection is mainly indicated for the analysis of fortified foods containing high concentrations of thiamin [1,7]. Low content of endogenous vitamin and high quantities of interfering substances in an extract require a more sensitive and selective detector [8–10]. Fluorescence detection can be used, provided the thiamin is oxidased to thiochrome by pre- or postcolumn reaction with alkaline hexacyanoferrate(III). Precolumn derivatization is more often used, but the postcolumn one is convenient for routine analysis and to eliminate the problems of reducing sugars, produced during acid hydrolysis and competing with thiamin for the oxidizing agent [10].
Liquid chromatographic methods for thiamin determination in foodstuffs and other matrices are reviewed by Lynch and Young [11].
18.2.2 Vitamin B2
Vitamin B2 [1] is a generic term indicating a group of compounds characterized by equal biological activity: riboflavin, riboflavin-5’-phosphate (FMN, flavin mononucleotide), and riboflavin-5’-adenosyldisphosphate (FAD, flavin adenine dinucleotide). In animal tissue, FMN and FAD are coenzymes bound tightly but not covalently to the corresponding apoenzymes. Forms for food fortification are FMN and riboflavin hydrochloride [4].
An extraction protocol, analogous to that described for vitamin B1, permits measuring all flavins, both the endogenous ones and those used as supplements [1,6,8,10]: acidic hydrolysis promotes the release of the protein-bound forms and converts FAD to FMN; the succeeding enzymatic digestion (takadiastase, amylase, acid phosphatase, claradiastase) is used for dephosphorylating FMN and hydrolyzing starch.
RP and ion-pair RP chromatography on C18 stationary phases with UV–Vis [12,13] or fluorescence detection [6,8,10] are the LC methods most frequently employed; other works report separations on C8 [14] and amide-C16 columns [15]. Free flavins in aqueous solution exhibit an intense yellowish-green fluorescence (450 nm excitation/522 nm emission) due to their 3-imino group, whereas protein-bound forms do not fluoresce, since their interaction is established through this functional group [1]. The UV–Vis spectrum of riboflavin shows four bands centered at 223, 266, 373, and 445 nm [1]. The use of a 254-nm fixed-wavelength absorbance detector is common [12], but the detection at 446 nm is less susceptible to interferences [13].
18.2.3 Vitamin B3
Two forms of vitamin B3, also known as niacin, are found in food [1,2]: nicotinic acid and nicotinamide. In living tissues, nicotinamide is a moiety of the coenzymes nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP); in meat, it is found free because of the postmortem hydrolysis of NAD [1]. Nicotinamide is also a form used for food fortification [4]. Nicotinic acid is the prevalent vitamer in mature cereal grains; nevertheless, it is unavailable due to its linkage to a number of polysaccharides (niacytin) and polypeptides (niacinogen) [1].
The determination of “total” (free plus bound) or “free” (bioavailable) niacin is based on well established extraction procedures [1,2,16]: Total niacin is released from the food matrix by autoclaving at 121°C with alkali [17] or 1–2.5 N mineral acid [18]; free niacin is isolated by boiling with 0.1 N mineral acid for 1 hr [19] or incubating with NAD glycohydrolase (NADase) at 37°C for 18 hr [20].
Most studies used absorbance detection at 254 to 264 nm for both B3 vitamers [17–19,21]. Their absorptivity is affected by pH: in an acidic solution, it is higher and λmax remains almost unchanged at 261 nm [1]. UV detection is convenient but not very sensitive or selective and the interfering compounds have to be removed by a cleanup step. To improve detection limits and selectivity without having recourse to the purification of hydrolysates, some researchers [20,22,23] propose an LC method based on the UV irradiation of the postcolumn effluent in the presence of H2O2 and Cu2+ to obtain fluorescent compounds (280 nm excitation/380 nm emission).
When the extraction procedure is applied to the determination of total niacin, nicotinic acid is the only B3 vitamer to be monitored by LC, because all nicotinamide is converted into the acid form. Anion exchange chromatography [18] is applied less frequently than RP chromatography under ion-suppression and ion-pairing conditions [17,19]. Van Niekerk et al. [24] assembled a two-dimensional chromatography system, provided by C18 and anion-exchange columns, to increase the selectivity of the UV detection at 254 nm.
Recently, electrospray (ESI) ionization in the positive ion mode was utilized to reduce interference problems due to matrix coextracted materials, detecting icotinic acid in food materials [25] and both B3 vitamers in roasted coffee [26], selectively.
18.2.4 Vitamin B5
Vitamin B5 occurs in three biologically active forms in foods [1]: pantothenic acid, coenzyme A (CoA), and acyl carrier protein (ACP). Calcium or sodium pantothenate are the forms generally used as supplements in infant formula [4]. The total quantification of vitamin B5 requires the release of pantothenic acid from CoA and ACP. Since it consists of pantoic acid linked through an amide linkage to β-alanine, chemical hydrolysis cannot be used. The only alternative to free pantothenic acid from CoA is the digestion with a number of enzymes (pepsin, alkaline phosphatase, pantetheinase); nevertheless, this treatment is unable to release the vitamin from ACP [27,28]. For the extraction of free pantothenic acid from milk and calcium pantothenate from infant formula an acidic deproteination is often used, followed by centrifugation and filtration [29,30].
Ion-suppression RP (TFA, formic acid, phosphate buffer) on C18 [27–29] and C8 columns [30] is the commonly used chromatographic mode. The poor selectivity and sensitivity of UV detection (a very weak absorbance at 204 nm due to the carbonyl group) makes LC–UV unsuitable to determine the low concentration of vitamin B5 in nonformulated foods. Some researchers have overcome these problems using multiwavelength UV detection (at 200, 205, and 240 nm) [30], fluorimetric detection (postcolumn derivatization of β-alanine with ortho-phthaldialdehyde in the presence of 2-mercaptoethanol) [27], and mass spectrometry with electrospray ionization [28,31]. The last solution allows the measurement of endogenous pantothenic acid in starch-containing foods [28], achieving a limit of quantitation (LOQ) adequate to quantify vitamin B5 contents greater than 0.024 mg/100 mg. Also, Pakin et al. [27] suggested a method that is sensitive and applicable to the determination of free and total pantothenic acid in any foodstuff, but it is probably too complex to be utilized for routine analyses.
18.2.5 Vitamin B6
Six vitamers of B6, having equivalent biopotency, are found in nature [1,16]: pyridoxine or pyridoxol, pyridoxal, pyridoxamine, and their 5’-phosphate esters; acid pyridoxic and pyridoxine-glucoside are inactive forms occurring in plant tissues. Regulation 1925/2006/EC cites pyridoxine hydrochloride, pyridoxine 5’-phosphate, and pyridoxine dipalmitate as the utilizable forms to enrich foods [4].
The preferred technique for vitamin B6 assay is LC, due to its high selectivity, which permits the simultaneous separation and quantitation of the several homologs. Different extraction protocols can be applied prior to LC analysis to estimate the total or bioavailable vitamin B6 [1,2,16]. For routine analysis, the approach used is the hydrolysis of the conjugated forms; in this way, chromatography is limited to pyridoxine, pyridoxamine, and pyridoxal, and the quantitation results are comparable to those obtained by microbiological assay. Traditional methods using acidic media and high temperature (0.1 N HCl, 121°C) denature proteins, disintegrate the food matrix, and hydrolyse phosphorylated and glycosilated forms completely [16,32]; the inconvenience may be an overestimation of bioavailable vitamin. A selective extraction, carried out at room temperature with a deproteinizing agent (sulfosalicylic acid, trichloroacetic acid, metaphosphoric acid, and perchloric acid), frees pyridoxal hydrolyzing Schiff bases formed with food proteins, preserves all vitamers (free, phosphorylated, and glycosylated) and allows their individual quantification; in this case, the chromatographic separation of all homologs is more complex [33–35]. The two most common approaches are based on a combination of chemical hydrolysis [36,37] or deproteination [38,39] with enzymatic digestion. The latter is usually performed with acid phosphatase [38] or takadiastase [36]; β-glucosidase is indispensable to determine bioavailable vitamin B6 in plant foods [37]. Ndaw et al. [32] tested a mixture of enzymes (α-amylase, papain, acid phosphatase) to release, in a single step, the bound forms of vitamins B1, B2, and B6; the acid hydrolysis proved to be superfluous, due to the presence of protease.
Usually, the individual and simultaneous separation of free [32,36,38,39] and conjugated B6 [33–35] vitamers is carried out by means of RP chromatography on C18 columns with acidic mobile phases. These methods utilize the native fluorescence of B6 vitamers [1,32–39], increasing the weak intensity of phosphate esters at low pH by means of postcolumn derivatization with sodium hydrogensulphite.
18.2.6 Vitamin B8
The only biologically active form of vitamin B8 is D-(+)-biotin, a unique steroisomer found in nature among the eight isomers theoretically possible [1,40]. In animal and plant tissues, most biotin is covalently bound to a lysine residue, free (D-biocytin) or belonging to biotin-dependent enzymes through an amide attachment. Biotin is also the form used for food enrichment [4].
Extraction of total biotin is obtained by acidic hydrolysis (2–3 M HCl at 100°C or 1–3 M H2SO4 by autoclaving at 121°C) that breaks bonds with proteins and totally converts D-biocytin into D-biotin [41,42]. Possible losses of vitamin depend on both the acid concentration and the duration of autoclaving. Enzymatic digestion with papain for 18 hr leaves biocytin intact and allows the determination of available biotin (biotin plus biocytin); takadiastase is added for starchy foods, such as cereals [42].
Owing to the low concentration and the absence of a strong chromophore, few RP HPLC methods for the endogenous vitamin B8 analysis in foods have been published [1,2]. Most of them [41,42] take advantage of avidin, the egg-white protein, labeled with a fluorescent marker (fluorescein 5-isothiocyanate) as a postcolumn derivatizing agent; its fluorescence is enhanced on binding to biotin and biocytin, its specific ligands. A LC tandem mass spectrometry (MS) method [42], using an ESI source in the positive ion mode and biotin-d6 as internal standard, was able to determine biotin in foods at low concentrations after hydrolysis with sulfuric acid and enzymatic digestion with papain. The whole procedure was reliable and faster than the microbiological assay.
18.2.7 Vitamin B9
Folate and folacin [1] are generic terms referred to a group of compounds with vitamin B9 activity, including folic acid (pteroylglutamic acid), dihydrofolic acid, tetrahydrofolic acid (H4folic acid), 5-formyl-H4folic acid, and 5-methyl-H4folic acid. Folic acid is not considered a natural physiological form, and is the chemical form used as a food supplement [4]. All folates exist in nature at low levels and predominantly as polyglutamates containing no more than seven glutamate residues with a γ-peptide linkage. Even if the theoretical number of folates approaches 150, in nature about 50 are observed.
Multiplicity, instability, and low concentrations of folates in animal and plant tissues have constituted an obstacle to the development of LC methods for their determination [1,2,43]. For the same reasons, sample preparation and purification are key steps carried out according to a well established protocol [2,44]. During extraction, a number of precautions should be taken to avoid the pH-dependent interconversion of some species, oxidative losses (adding antioxidants, such as ascorbate and 2-mercaptoethanol), and thermal degradation [45]. Folates are released from the food matrix by autoclaving the food sample in a buffered aqueous medium: particles are broken up, starch is gelatinized, folate-binding proteins are denaturated as well as enzymes catalyzing folate degradation or interconversion. The autoclaved sample is submitted to a tri-enzyme treatment [44]. The first digestion is performed with protease to free the protein-bound folates definitively. The second uses α-amilase to release the starch-bound folates. Finally, the third treatment, carried out with the folic acid conjugase, deconjugates the polyglutamates to the monoglutamate forms.
A cleanup step on solid-phase extraction (SPE) cartridges is often employed to remove interfering compounds co-extracted with folates from the real matrix. The most used stationary phases include strong anion-exchange (SAX) materials and affinity chromatographic sorbents with immobilized folate-binding protein. Both provide high recoveries, but only the affinity column purifies and concentrates the extracts efficiently.
LC with UV detection (at 280 or 290 nm) is used for the analysis of foods with low concentrations of folates [46,47], after tri-enzyme digestion followed by cleanup on the affinity sorbent to concentrate the extracts by about 10-fold [46]. Nevertheless, fluorimetric detection is more efficient at determining naturally occurring folates [48] as well as ESI–MS that, combined with isotope dilution, allows accurate quantification [49,50]. Ion-suppression RP chromatography is the most suitable mode for coupling with both detectors [48]. In fact, folates show native fluorescence (288 nm excitation/353–356 nm emission) that is enhanced in an acidic medium (mobile phase at pH 2.3 using phosphate buffer) for the reduced forms but not for folic acid; the solution adopted for the latter is to produce a fluorescent pterin fragment through its postcolumn oxidative cleavage [51]. Acidic mobile phases support ESI ionization of folates, but the phosphate buffer has to be substituted by the more volatile formic acid [49,53–55]. Ion-pair RP chromatography is carried out at neutral or basic pH, but it requires acidification of the column effluent to make fluorescence detection possible [52].
18.2.8 Vitamin B12
Vitamin B12 [1] is a term that generically describes a group of cobalt-containing organometallic compounds with antipernicious anaemia activity, known as cobalamins,. The major forms occurring in foods of animal origin include 5’-deoxyadenosylcobalamin (coenzyme B12) and methylcobalamin, two coenzymes covalently bound to their apoenzymes; hydroxocobalamin is their photooxidation product. The general extractive protocol [56] applied for determining total vitamin B12 is used to release the protein-bound vitamers and to convert all natural cobalamins into a single and stable form, known as cyanocobalamin. To this end, Heudi et al. [57] dissolved a food sample in a buffered solution (pH 4) containing sodium cyanide (at 100°C for 35 min) then digested it with pepsin (at 37°C for 3 hr). Determination of vitamin B12 by LC–UV is difficult to perform in nonsupplemented foodstuffs because of the very low concentrations of the vitamin and the poor sensitivity and selectivity of the detection system employed. These problems were overcome using an immunoaffinity column and monitoring the vitamin at 361 nm [58]. The extractive procedure of Liebiedzińska et al. [7] was based on an enzymatic digestion with α-amilase and papain after autoclaving salmon samples at 121°C in the presence of cyanide; cyanocobalamin was monitored by an electrochemical detector.
Fluorescence detection [59] can be used after chemical or enzymatic hydrolysis, indispensable to release α-ribazole, a characteristic fluorescent fragment of vitamin B12. Nevertheless, the latter may occur in foods as a vitamin B12 metabolite; therefore, it is essential to proceed with extensive purification of the extracts before carrying out the hydrolysis. LC–MS using electrospray ionization was successfully applied to the analysis of vitamin B12 in some fortified foods [60] and cultivated mushrooms [61]. It might constitute a promising tool for the cobalamin analysis since it requires neither a derivatizion step nor an accurate cleanup procedure.
18.2.9 Vitamin C
L-ascorbic acid (AA) and L-dehydroascorbic acid (DHAA) are the two main C vitamers occurring in nature [1]. In food analysis, the valuation of the vitamin C total content should account for both forms, since DHAA is readily reduced to AA in the animal body. D-isoascorbic acid (D-IAA), also known as erythorbic acid or D-araboascorbic acid, has analogous reductive properties but only 5% of the antiscorbutic activity of L-AA; this epimer is a by-product of vitamin C, and is approved within the European Community as an antioxidant additive [62]. The capability of LC to distinguish the two ascorbic acid isomers and their primary oxidation products is very useful for analyzing processed foods. Forms used for supplementation are AA, sodium-, calcium-, or potassium-L-ascorbate and L-ascorbyl 6-palmitate [4].
AA is susceptible to both chemical and enzymatic oxidation [1]. Chemical oxidation is catalyzed by minerals, such as Cu(II) and Fe(III), in the presence of oxygen at a pH-dependent rate; enzymatic oxidation by the ascorbate oxidase occurring in plant tissues. Light and heat are other factors that promote its degradation. For these reasons, the extraction procedure should be designed to stabilize the vitamin; for example, metaphosphoric acid [63–66] denatures proteins, inactivates enzymes, provides a medium below pH 4 (degradation rate is minimal at pH 2), and inhibits metal catalysis, whereas EDTA [67,68] chelates the minerals efficiently.
Several LC methods have been proposed for vitamin C analysis [1,62–73]. The good selectivity of silica-based aminopropyl columns in separating homologues of vitamin C is probably due to the hydrogen bonding between hydroxyl protons of vitamin C with the neutral amino group of the stationary phase rather than to differences in pKa values of the vitamers [64,69]. A disadvantage is the short lifetime of this kind of stationary phase because of the possible reaction of the amine group with the carbonyl group of reducing sugars, or other compounds, to form Schiff bases. Other modes are ion exclusion (poorly selective) [70], RP with and without ion suppression [63,65,71], and RP ion-pair chromatography [72,73]. The problem with RP packings is that ascorbic acid is only weakly retained and, therefore, requires the addition of a cationic ion-pair agent to the mobile phase. To this end, several primary, secondary, and tertiary amines have been employed as relatively hydrophobic modifying reagents to obtain sharp, well-defined peaks; tetrabutylammonium has been the most used [1]. The UV (254 nm) [65,66] and amperometric detection (a potential of +0.7 V is often chosen using either a platinum or glassy carbon electrode) [70,71] have been utilized for the direct monitoring of AA, while its fluorimetric detection requires chemical derivatization [64]. DHAA has a weak molar absorptivity and it is electrochemically inactive. A precolumn reduction to AA by using cystein or dithiotreitol makes possible absorbance detection but not electrochemical (high noise) detection [74]. Recently, LC–MS with electrospray ionization in the negative ion mode [75,76] was used for the analysis of vitamin C in several food commodities; the main advantage was the simultaneous determination of AA and DHAA with no need for oxidation–reduction or derivatization steps [76].
18.3 Liquid Chromatographic Determination of Fat-Soluble Vitamins
Fat-soluble vitamins occur in the lipid fraction of foods [1], composed mainly of triglycerides and partly of sterols, phospholipids, and other lipoidal constituents. These substances, having solubility analogous to those of the fat-soluble vitamins, complicate the isolation of fat-soluble vitamins and constitute a source of interference during the following analysis [1,3].
Hot saponification [1, 3,77–79] is the most effective tool for removing the majority of fatty material, hydrolyzing ester linkages of glycerides, phospholipids, esterified sterols, and carotenols. Moreover, alkaline hydrolysis frees bound forms of vitamins (for instance, esterified and protein-bound forms) and degrades chlorophylls in water-soluble products. Also gelatine of the vitamin premix, added to supplemented foods, is dissolved. Starch-containing products, such as breakfast cereals, are digested first with takadiastase and then saponified to avoid the formation of undissolved particles [78]. Normally, saponification is carried out with a mixture of ethanol and 50% (w/v) aqueous KOH solution in the presence of an antioxidant (pyrogallol, ascorbic acid, butylated hydroxytoluene) for 30 min at about 80°C [1,3]. The unsaponifiable fraction (fat-soluble vitamins, carotenoids, sterols, etc.) is extracted from the alkaline digest by liquid–liquid extraction using a water-immiscible organic solvent (hexane, petroleum ether, petroleum ether: diethyl ether at 1:1, v/v). Hexane has the advantage of providing extracts that do not contain soaps and are nearly neutral; nevertheless, when used, it is advisable maintaining the percentage of ethanol below 40% (v/v) to avoid losses of slightly polar vitamins, such as retinols and tocopherols. Saponification can be used for vitamins A, D, and E, while it is not suitable for K vitamers, which are quickly decomposed in alkaline media at high temperature [1,3].
Alcoholysis [1] is milder (ambient temperature) and more rapid (2 min) than saponification. Methanol reacts with KOH to form potassium methoxide, which, in turn, converts glycerides to methyl esters and soaps. It has been used to determine vitamin A palmitate in nonfat milk and vitamin D in whole milk [80].
Enzymatic hydrolysis [81,82] with lipase (from Candida rugosa or from porcine pancrease) is an alternative procedure to remove glycerides in vitamin K determinations. Addition of papain (from Carica Papaya) aids the digestion of meat and foods of animal origin. The hydrolysate is first alkalinized (potassium carbonate in ethanol) to precipitate fatty acids as soaps then extracted with a water immiscible organic solvent (hexane or pentane). It has also been used in combination with supercritical fluid extraction (SFE) [83].
Also, direct solvent extraction [84] is used for the isolation of vitamers susceptible to degradation in alkaline media (retinyl esters, vitamers K), but it is ineffectual for removing interfering fatty substances. In some cases, ultrasonication has been employed to break up the lipoproteic complex encapsulating fat-soluble vitamins [85].
Several LC modes have been used for the separation of fat-soluble vitamins. The choice depends on the vitamin forms to be determined, the nature of the food matrix, and the sample treatment. Adsorption chromatography has two main advantages: (a) geometric and positional isomers are generally resolved on silica stationary phases [86,87]; (b) relatively high loads of lipoidal material can be tolerated by this type of column. The latter feature allows the direct injection of extracts obtained by means of direct liquid extraction [88] or sample dissolution in hexane (e.g., vitamin E from oils) [89], when the recourse to saponification is not essential for the analyte isolation. In these cases, fluorescence detection is preferred to absorption detection, which may reveal intrusive peaks of lipid origin.
The majority of LC separations of fat-soluble vitamins are based on RP chromatography on C18 columns, but the use of a triacontyl stationary phase (C30) is becoming more common, due to its superior shape selectivity [90,91]. Shape discrimination with C30 columns improves at subambient temperature, permitting resolution of geometric and positional isomers partly or fully, depending on the mobile phase composition. Nevertheless, C30 columns are less efficient and the chromatographic peaks broader than those obtained with C18 columns.
Nonaqueous reversed-phase (NARP) chromatography [92] has been employed for the separation of fat-soluble vitamins [93] and carotenoids [94]. This chromatographic technique uses either C18 columns with a high carbon loading (20%) or C30 columns and low polarity mobile phases. A typical NARP mobile phase consists of a polar solvent (e.g., acetonitrile), and a solvent of lower polarity (e.g., dicloromethane) to solubilize analytes and adjust the mobile phase strength. The good chromatographic selectivity is due to the small difference in polarity between the mobile and stationary phases.
18.3.1 Vitamin A
Vitamin A-active compounds [1] are present in foods of animal origin as retinoids (retinol, retinyl esters, retinylaldehyde, retinoic acid) and in those of plant origin as carotenoids (only carotenoids with one unsubstituted β-ionone ring and with an 11-carbon polyene chain at least are provitamins A). Retinyl palmitate is the main form used as a food supplement [4].
Food sample pretreatment may consist of either (a) saponification to quantify the free forms (retinol or xanthophylls may occur free or esterified in foods) [95,96] or (b) direct extraction to determine the unaltered A vitamers [84,88]. Alkaline hydrolysis is also an expedient to simplify the vitamin A analysis, since retinol is the only form to be quantified; nevertheless, due to its sensitivity to light and oxygen, it is important to prevent photo-oxidation by inclusion of a antioxidant (ascorbic acid, hydroquinone, or pyrogallol). A drawback of hot saponification is the generation of artifacts, such as geometric isomers of retinol and carotenoids [97].
Retinol and its esters can be monitored by UV (325 nm) [88,96] and fluorescence (324–328 nm excitation/470 nm emission) detection [98]; the latter has the advantage that β-carotene does not interfere with the vitamin A determination even if there is coelution, while the disadvantage is a more limited linear dynamic range. RP chromatography on a C18 column with semiaqueous mobile phases is the most often used mode for vitamin A analysis [3,100,101], but adsorption chromatography on silica is the more efficient approach for separating geometric isomers of retinol [86,96].
Carotenoids show a characteristic three peak spectrum in the UV–Vis region [99]. The absorption maxima of all-trans β-carotene, main provitamin A, occur at 428, 453, and 478 nm (in hexane or ethanol); its cis isomers may be identified according to the following indications: (a) a small hypsochromic shift of λmax (usually 2–6 nm for mono-cis, 10 nm for di-cis, and 50 nm for poli-cis isomers); (b) an ipochromic effect; (c) a reduction in fine structure; and (d) the appearance of a cis-peak in the near-UV range, between 330 and 350 nm. RP chromatography [102] is preferred to NP for the determination of provitamins A, because many carotenoids can be monitored within the same chromatographic run: the xanthophylls are eluted early, while the carotenes require strong mobile phases (little or no water) for their displacement. Carotenes and their cis isomers are poorly resolved on monomeric C18 phases, while their separation on polymeric C18 or C30 phases depends on the organic modifier and gradient elution conditions [103]. In addition, the C30 sorbent provides the highest selectivity [90,91] at low temperature (19°C) because of the improved alignment with the alkyl chains; only under these conditions can lutein and zeaxanthin (structural isomers) be adequately separated.
Lately, LC–MS analysis of carotenoids and retinoids has been reported for food matrices such as fish eggs [104] and infant formula [105,106]. Atmospheric pressure chemical ionization (APCI) is the method of choice for their detection in the positive ion mode: Retinol gives an intense dehydrated pseudomolecular ion, [MH–H2O]+, at m/z 269; retinyl esters are fragmented in the ion source producing [MH–fatty acid–H2O]+ ions (i.e., retinol dehydrated at m/z 269); carotenoids generate several ions such as [M + H]+, [M]+•, and [M–H]+. The ESI interface is better for the ionization of xanthophylls when semiaqueous mobile phases are used for their separation.
18.3.2 Vitamin D
Vitamin D is the name given to a series of compounds with antirachitic activity [1]: cholecalciferol (vitamin D3) is present in foods of animal origin, whereas ergocalciferol (vitamin D2) is produced by plants, fungi, and yeast (Figure 18.2). In animal organisms, vitamin D is metabolized to its biologically active forms, 25-hydroxy- and 1α,25-dihydroxycholcalciferol.
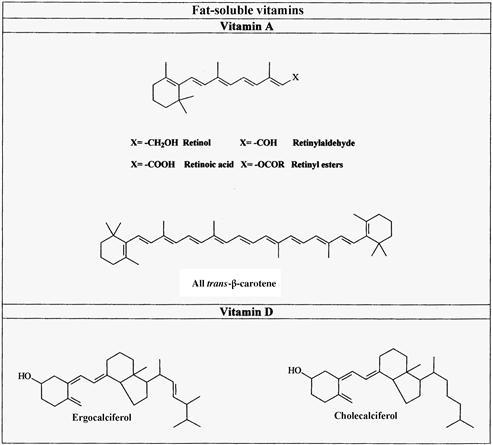
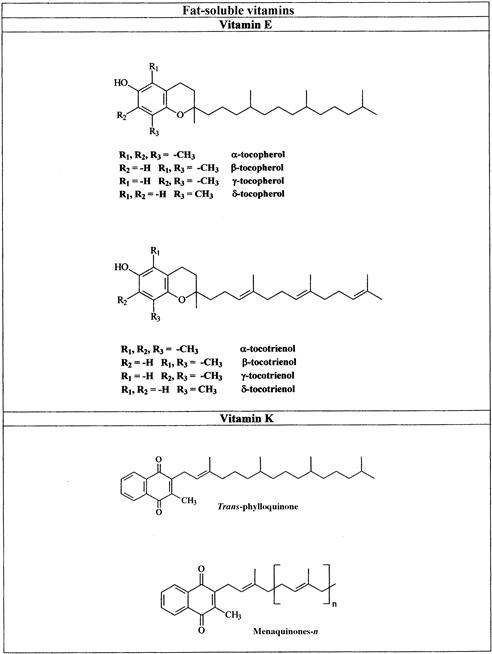
FIGURE 18.2 Names and structures of the fat-soluble vitamins and pro-vitamins A carotenoid naturally occurring in foods.
(a) Vitamins A and D; (b) vitamins E and K.β
The extraction of vitamin D from fatty foods necessitates alkaline hydrolysis [1,3,85]. Thermal isomerization of vitamin D to previtamin D during hot saponification entails losses of 10–20% making its quantification difficult. Overnight cold saponification (prolonged digestion at room temperature) is not affected by this problem and, moreover, demands less operator attention and yields greater recovery than the hot saponification. Cleanup or fractionation procedures [1, 16, 85,106–109] include sterol precipitation in a digitonin solution stored at –20°C overnight, solid-phase extraction, gel permeation chromatography, and LC on a semipreparative scale. These steps are essential (a) to remove the excessive amounts of sterols, which might alter the retention time of the vitamin and interfere with its UV detection (265 nm in ethanol or hexane); (b) to achieve an adequate enrichment factor of the final extract as vitamin D occurs at very low concentrations in nature. SPE is more advantageous than the other methods; the most used sorbent is C18, but polar sorbents, such as silica, aminopropyl silica, and Florisil, are also efficient at removing sterols, carotenoids, vitamin E, and other interfering material. An alternative procedure to perform a reliable quantitative analysis of total vitamin D is the conversion of both D vitamers to isotachisterol [110], a compound more stable to heat and light; moreover, the absorption maximum at 301 nm and the high extinction coefficient contribute to improve selectivity and sensitivity.
Ultraviolet and electrochemical detectors are the most widely used for the quantification of vitamin D. The UV detector has the advantage that all vitamin-D-active compounds absorb within the range of 250–265 nm [1,106,107], the wavelength 280 nm has also been used to reduce interference problems [111]. The highest sensitivity, selectivity, and linear dynamic range of the electrochemical detector [109] allows the analysis of samples containing low concentrations of vitamin D and the simplification of sample treatment. APCI is advantageous for vitamin D detection in milk products [78,112,113], due to its identification power in complex matrices with low concentrations of cholecalciferol. The high sensitivity and selectivity of the LC–MS technique allows simplification and improvement of the reliability of the analysis of endogenous vitamin D contents in foods [114,115].
NP chromatography [1,85] has the advantage of tolerating relatively heavy loads of fatty material and separating vitamin D from its hydroxylated metabolites; nevertheless, it cannot resolve vitamins D2 and D3. Hexane containing a small percentage (less than 5% v/v) of a more polar solvent (isopropanol, dichloromethane, or ethyl acetate) is the most used mobile phase [85].
RP columns having a high carbon loading [107–109] can differentiate vitamin D2 from D3, making possible the use of one homolog as an internal standard for the other [107] as well as facilitating the separation of their hydroxylated metabolites [108].
18.3.3 Vitamin E
Vitamin E has eight biologically active forms [1]: four tocopherols (saturated isoprenoid side chain) and four tocotrienols (unsaturated isoprenoid side chain), designed as α-, β-, γ-, and δ-according to the number and position of methyl groups on the chromanol ring. Of these, α-tocopherol is the most important (plant sources) and active.
During sample treatment, vitamin E must be protected from light and oxygen (flushing with nitrogen and adding an antioxidant) to quantify its actual content. Except for its quantification in oils, which can be performed by direct injection onto a NP column, of a sample diluted with hexane or mobile phase [89] and hot saponification is required for all other foods [77–79]; nevertheless, alkaline conditions (volumes of alkali and ethanol, time, and temperature) must be carefully balanced to avoid losses [77]. The highest recovery is obtained when the digestion is completed under reflux conditions [1].
NP chromatography [87,116] completely resolves the eight homologues, which elute in order of increasing polarity: α-tocopherol, α-tocotrienol, β-tocopherol, γ-tocopherol, β-tocotrienol, γ-tocotrienol, δ-tocopherol, and δ-tocotrienol. The decreasing number of methyl groups and the unsaturation in the side chain make these compounds more polar and therefore more retentive. Separation of β- and γ- positional isomers is due to the diverse interactions that the methyl groups establish with the silanol groups of the silica stationary phase. RP chromatography [116] is less effective for separating the E vitamers; in fact, β- and γ- positional isomers coelute on C18 columns, but a complete separation is achieved on C30 columns [117].
Tocopherols and tocotrienols show a low intensity of absorption between 292 and 298 nm in ethanol [1], and fluorescence detection (290–296 nm excitation/330 nm emission) [89,118] is preferred to analyze vitamin E in complex food matrices. Polar solvents, such as diethyl ether and alcohols, provide a strong flourescence response, and their inclusion in the hexane mobile phase improves the detection limits achievable in NP chromatography [1]. The first description of LC–MS analysis dates back to 1998 [119]; tocopherols and carotenoids were detected by ESI as silver ion adducts, after postcolumn treatment with acetone containing silver perchlorate. In spite of the low detection limits, the poor solubility of this salt in organic solvents causes contamination problems and makes this ionization technique, unsuitable for routine analysis. ESI in the negative ion mode has been used [120] for the quantification of tocopherols and tocotrienols after pressurized liquid (solvent) extraction (PLE) with methanol at 50°C and 110 bar from cereal samples. Nevertheless, Lanina et al. [121], comparing the performances of the ESI and APCI sources in both ionization modes, observed that the positive ion mode is less efficient than the negative ion mode because of the signal dispersion between [M + H]+ and [M]•+; the authors chose APCI in the negative ion mode for the detection of tocopherols in sunflower oil and milk due to the larger dynamic range and lower detection limits. Positive ion APCI was instead the preferred ionization mode of other researchers [122,123].
18.3.4 Vitamin K
The K homologues [1] are a family of 2-methyl-1,4-naphthoquinones possessing cofactor activity for γ-glutamylcarboxylase and differ in the side chain attached at C3. Phylloquinone (vitamin K1) has a phytyl side chain and occurs in green plants. Vitamin K2 includes a group of compounds synthesized by bacteria and characterized by a polyisoprene side chain; each of them is designated menaquinone-n (MK-n, with n from 4 to 13) according to the number of isoprenyl units. Menadione (vitamin K3), menadiol (vitamin K4), and vitamin K1(25) are synthetic forms [1,4].
Owing to their instability to alkali, hot saponification is unworkable. Enzymatic hydrolysis [81,82,124,125] and direct solvent extraction [124–127] are the most-common techniques employed for extraction. Liquid-phase reductive extraction with zinc chloride in an acid medium was tested to extract K vitamers as acetonitrile soluble hydroquinones from dairy products [125,126]. Acid hydrolysis [125] proved advantageous in isolating long chain menaquinones from cheese, provided the digestion time (10 min) was short. Semipreparative LC [125,127,128] and SPE [126] have sometimes been employed as a cleanup and concentration step after solvent extraction. Matrix solid-phase dispersion (MSPD) followed by PLE with ethyl acetate at 50°C and 1500 psi [129] and SFE using carbon dioxide at 8000 psi and 60°C [130] are fast, alternative procedures for extracting phylloquinone.
Phylloquinone and menaquinones exhibit a UV spectrum characteristic of the naphthoquinone ring (five absorption maxima at 242, 248, 260, 269, and 325 nm), but UV detection is seldom used [131], due to its poor sensitivity and selectivity. Most methods employ fluorescence detection (238–244 nm excitation/418–430 nm emission) after a postcolumn reduction with a methanolic solution containing zinc chloride, sodium acetate, and acetic acid in a reactor packed with zinc metal powder [81, 82,124–126]; this LC method is sufficiently sensitive for the determination of low concentrations of menaquinones in animal products [81,125]. Electrochemical detection is an alternative for the determination of phylloquinone in foods of animal and plant origin [127,128].
The K homologs are separated on C18 columns [81, 125–128,] which cannot resolve their geometric isomers; however, this separation is achieved on C30 columns [82,124] and on silica columns [131]. Due to the lipophilic nature of the long chain menaquinones, NARP chromatography is well suited to their separation and determination.
18.4 Multivitamin Methods
Chromatographic techniques are suitable for quantitative multianalyte determinations. In particular, LC is the technique of choice for the direct analysis of polar, nonvolatile, and heat-sensitive compounds, such as water-soluble vitamins (see Figure 18.3 for an example); moreover, having no molecular weight limitations, it can be used for the separation of cobalamins, polyglutamates, FAD, and CoA. LC is also the most common technique used for the concurrent analysis of fat-soluble vitamins and provitamin A carotenoids (see Figure 18.4 for an example).
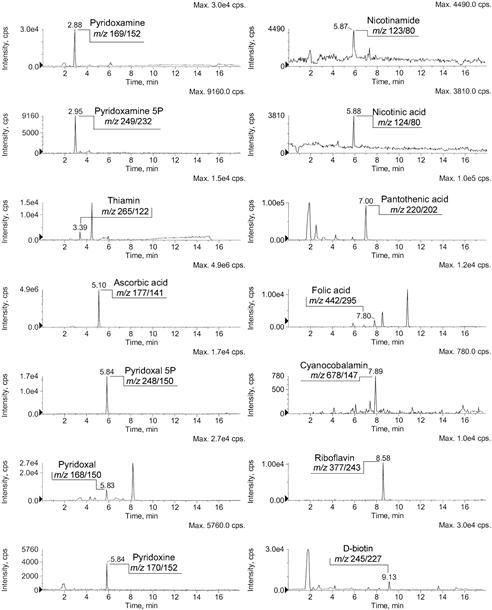
FIGURE 18.3 Extracted ion current profiles of the water-soluble vitamins detected in a green kiwi extract (see reference [138] for the details).
The LC–SRM (standard reference material) chromatogram was acquired by a high-flow ESI source (TurboIonSpray source). Each analyte was identified on the basis of the retention time, the two selected SRM transitions and their relative abundance. Only the most intense SRM ion current is reported in the figure.
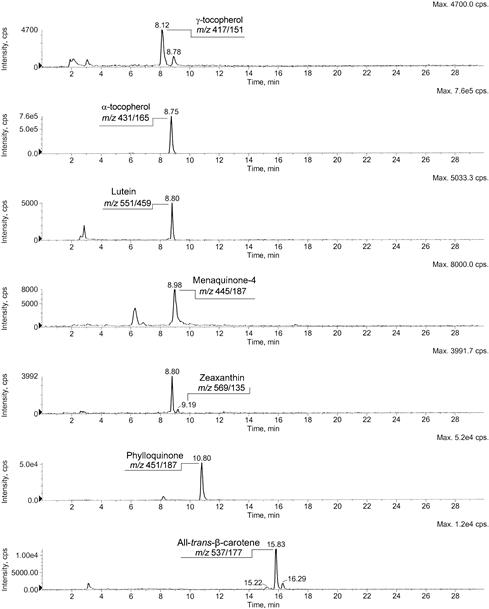
FIGURE 18.4 Extracted ion current profiles of the fat-soluble vitamins detected in a green kiwi extract.
This is an example of NARP chromatography (see Table 18.1, reference [137], for the details): a nonaqueos mobile phase was chosen as the best compromise between chromatographic resolution and support to positive APCI ionization of analytes.
The key step of a multivitamin method is the development of a simultaneous and quantitative extraction procedure. The intra- and intergroup heterogeneity of water-soluble vitamins makes it difficult to realize this goal. The application of an acid treatment, to hydrolyze the bound forms, can be used for simultaneous extraction of B1, B2, B3, B6, B8, B12, and C vitamins [132,133], but it is not appropriate for B5 and B9, which are sensitive to low pH [133]. Since the heterogeneity of the fat-soluble vitamins is less marked, it was simpler to develop an extraction protocol suitable for isolating more than one vitamin from the same matrix. Hot saponification is the most used extraction procedure [78, 107,134–136], especially for foods with a high fat content. It is indicated for the majority of fat-soluble vitamins and carotenoids but causes a rapid decomposition of K vitamers. MSPD was tested as an alternative technique for isolating both fat-soluble [137] and water-soluble vitamins [138] from plant foods.
The UV–diode array [132, 134–136,139,140] and fluorescence detection [141,142] have been used to develop multivitamin LC methods, which, however, remain limited to a few analytes responding to the same detection system and extracted quantitatively with the same procedure. Moreover, by means of a UV detector, other difficulties are represented by the absence of a strong chromophoric group in some vitamins (pantothenic acid and biotin), which absorb with modest sensitivity in the low UV region only, where the selectivity is scarce (absorption of interfering compounds).
A promising detector for the development of multivitamin LC methods is the mass spectrometer, which overcomes problems due to poor chromatographic resolution by extracting the characteristic ions associated with target analytes. The ESI interface is the most efficient at promoting the ionization of polar substances, such as water-soluble vitamins, which, provided with acidic or basic groups, can be deprotonated or protonated. Moreover, ESI can ionize high molecular-weight compounds and produce multicharged pseudomolecular ions, such as [M + nH]n+ or [M–nH]n–. This feature is useful in the detection of cobalamins that give intense mono- and double-charged pseudomolecular ions [138]. Since all water-soluble vitamins respond in the positive ion mode, this has been chosen for the development of multivitamin methods [138,143–145]. Similarly, the APCI interface operating in the positive ion mode represents the best compromise [78,137,145] for the simultaneous detection of fat-soluble vitamins and carotenoids.
The use of a mass spectrometer as chromatographic detector offers a great advantage in vitamin analysis: the possibility of simplifying the extraction procedure. The selectivity of the LC–MS technique reduces problems due to intrusive peaks from matrix components, while its sensitivity (ng or pg injected for real samples) allows the direct injection of an extract, eliminating the concentration step and the exposure to heat (most water-soluble vitamins posses low thermal stability). Sample preparation time is reduced as well as the duration of exposition to air and light (most vitamins and carotenoids are susceptible to these factors).
Few analytical methods have been proposed for multivitamin determination in dietary supplements and food products by LC. The most emblematic multivitamin methods published in the last 10 years are summarized in Table 18.1.
TABLE 18.1
LC Methods for the Simultaneous Determination of Vitamins





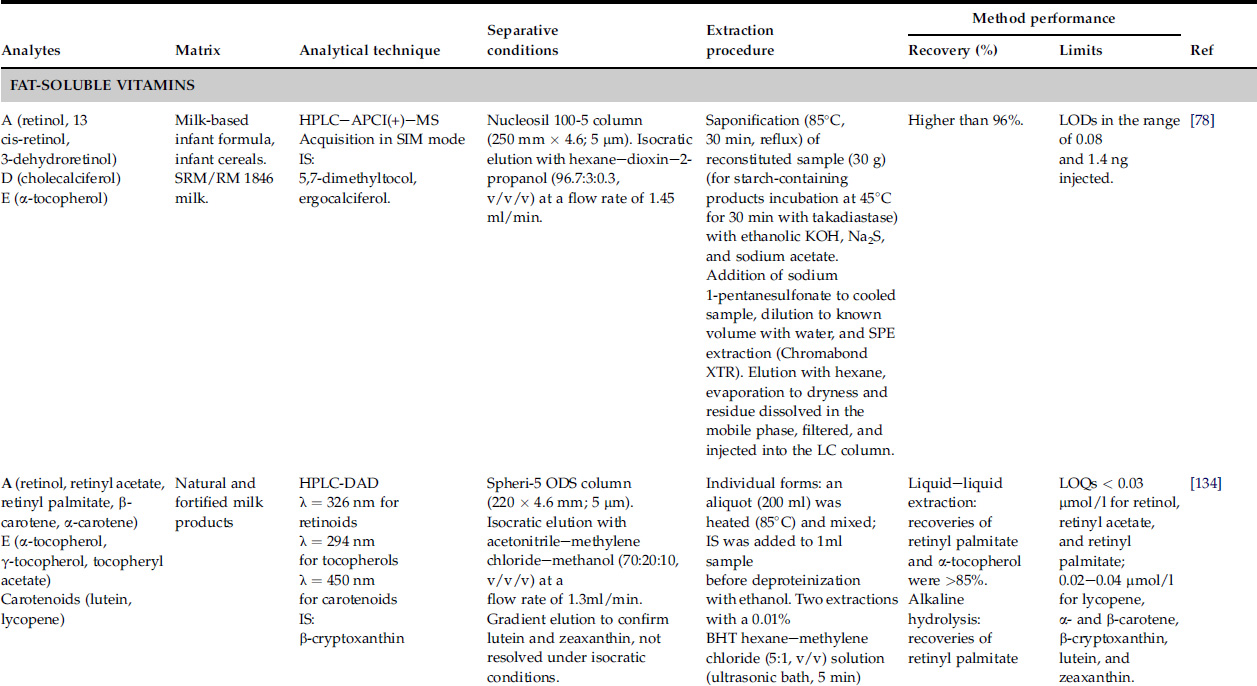



Acronyms: BHT = butylated hydroxytoluene; FL = fluorescence detection; HILIC = hydrophilic-interaction LC; IDMS = isotope-dilution mass spectrometry; IS = internal standard; MSPD = matrix solid-phase dispersion; QqQ = triple quadrupole; SRM = standard reference material; TCA = trichloroacetic acid; TFA = trifluoroacetic acid; UPLC = ultraperformance liquid chromatography.
References
1. Ball GFM, ed. Vitamins in foods Analysis, bioavalability, and stability. Boca Raton, FL: CRC Press; 2006.
2. Blake CJ. Analytical procedures for water-soluble vitamins in foods and dietary supplements: a review. Anal Bioanal Chem. 2007;389:63–76.
3. Blake CJ. Status of methodology for the determination of fat-soluble vitamins in foods, dietary supplements, and vitamin premixes. J AOAC Int. 2007;90(4):897–910.
4. Rychlik M, ed. Fortified foods with vitamins analytical concepts to assure better and safer products. Weinheim, Germany: Wiley-VCH Verlag; 2011.
5. Valls F, Checa MA, Fernández-Muiño MA, Sancho MT. Determination of thiamin in cooked sausages. J Agri Food Chem. 1999;47:170–173.
6. Tang X, Cronin DA, Brunton NP. A simplified approach to the determination of thiamine and riboflavin in meats using reverse phase HPLC. J Food Comp Anal. 2006;19(8):831–837.
7. Lebiedzińska A, Marszall ML, Kuta J, Szefer P. Reversed-phase high-performance liquid chromatography method with coulometric electrochemical and ultraviolet detection for the quantification of vitamins B1 (thiamine), B6 (pyridoxamine, pyridoxal and pyridoxine) and B12 in animal and plant foods. J Chromatogr A. 2007;1173(1–2):71–80.
8. Esteve M-J, Farré R, Frígola A, García-Cantabella J- M. Simultaneous determination of thiamin and riboflavin in mushrooms by liquid chromatography. J Agri Food Chem. 2001;49:1450–1454.
9. Ohta H, Baba T, Suzuki Y, Okada E. High-performance liquid chromatographic analysis of thiamine in rice flour with fluorimetric post-column derivatization. J Chromatogr. 1984;284(1):281–284.
10. Wehling RJ, Wetzel DL. Simultaneous determination of pyridoxine, riboflavin, and thiamin in fortified cereal products by high-performance liquid chromatography. J Agri Food Chem. 1984;32:1326–1331.
11. Lynch PLM, Young IS. Determination of thiamine by high-performance liquid chromatography. J Chromatogr A. 2000;881:267–284.
12. Vidal-Valverde C, Reche A. Reliable system for the analysis of riboflavin in foods by high performance liquid chromatography and UV detection. J Liq Chromatogr. 1990;13(10):2089–2101.
13. Stancher B, Zonta F. High performance liquid chromatographic analysis of riboflavin (vitamin B2) with visible absorbance detection in Italian cheeses. J Food Sci. 1986;51(3):857–858.
14. Reyes ESP, Norris KM, Taylor C, Potts D. Comparison of paired-ion liquid chromatographic method with AOAC fluorometric and microbiological methods for riboflavin determination in selected foods. J AOAC. 1988;71(1):16–19.
15. Viñas P, Balsalobre N, López-Erroz C, Hernández-Córdoba M. Liquid chromatographic analysis of riboflavin vitamers in foods using fluorescence detection. J Agri Food Chem. 2004;52(7):1789–1794.
16. Rizzolo A, Polesello S. Chromatographic determination of vitamins in foods. J Chromatogr. 1992;624(1–2):103–152.
17. Tyler TA, Genzale JA. Liquid chromatographic determination of total niacin in beef, semolina, and cottage cheese. J AOAC. 1990;73(3):467–469.
18. LaCroix DE, Wolf WR, Porter E, Cantellops D, Chase Jr GW, Woollard D. Determination of niacin in infant formula by solid-phase extraction and anion-exchange liquid chromatography. J AOAC Int. 2001;84(3):789–804.
19. Vidal-Valverde C, Reche A. Determination of available niacin in legumes and meat by high performance liquid chromatography. J Agri Food Chem. 1991;39(1):116–121.
20. Ndaw S, Bergaentzlé M, Aoudé-Werner D, Hasselmann C. Enzymatic extraction procedure for the liquid chromatographic determination of niacin in foodstuffs. Food Chem. 2002;78(1):129–134.
21. Valls F, Sancho MT, Fernández-Muiño MA, Checa MA. Simultaneous determination of nicotinic acid and nicotinamide in cooked sausages. J Agri Food Chem. 2000;48(8):3392–3395.
22. Lahély S, Bergaentzlé M, Hasselmann C. Fluorimetric determination of niacin in foods by high-performance liquid chromatography with post-column derivatization. Food Chem. 1999;65(1):129–133.
23. Rose-Sallin C, Blake CJ, Genoud D, Tagliaferri EG. Comparison of microbiological and HPLC–fluorescence detection methods for determination of niacin in fortified food products. Food Chem. 2001;73(4):473–480.
24. van Niekerk PJ, Smit SCC, Strydom ESP, Armbruster G. Comparison of a high-performance liquid chromatographic and microbiological methods for the determination of niacin in foods. J Agri Food Chem 3. 1984;2(2):304–307.
25. Goldschmidt RJ, Wolf WR. Determination of niacin in food materials by liquid chromatography using isotope dilution mass spectrometry. J AOAC Int. 2007;90(4):1084–1089.
26. Lang R, Yagar EF, Eggers R, Hofmann T. Quantitative investigation of trigonelline, nicotinic acid, and nicotinamide in foods, urine, and plasma by means of LC–MS/MS and stable isotope dilution analysis. J Agri Food Chem. 2008;56:11114–11121.
27. Pakin C, Bergaentzlé M, Hubscher V, Aoudé-Werner D, Hasselmann C. Fluorimetric determination of pantothenic acid in foods by liquid chromatography with post-column derivatization. J Chromatogr A. 2004;1035(1):87–95.
28. Rychlik M. Pantothenic acid quantification by a stable isotope dilution assay based on liquid chromatography-tandem mass spectrometry. Analyst. 2003;128:832–837.
29. Romera JM, Ramirez M, A, Gil A. Determination of pantothenic acid in infant milk formulas by high performance liquid chromatography. J Dairy Sci. 1996;79(4):523–526.
30. Woollard DC, Indyk HE, Christiansen SK. The analysis of pantothenic acid in milk and infant formulas by HPLC. Food Chem. 2000;69(2):201–208.
31. Mittermayr R, Kalman A, Trisconi MJ, Heudi O. Determination of vitamin B5 in a range of fortified food products by reversed-phase liquid chromatography–mass spectrometry with electrospray ionisation. J Chromatogr A. 2004;1032(1–2):1–6.
32. Ndaw S, Bergaentzlé M, Aoudé-Werner D, Hasselman C. Extraction procedures for the liquid chromatographic determination of thiamin, riboflavin and vitamin B6 in foodstuffs. Food Chem. 2000;71:129–138.
33. Addo C, Augustin J. Changes in the vitamin B6 content in potatoes during storage. J Food Sci. 1988;53(3):749–752.
34. Sampson DA, Eoff LA, Yan XL, Lorenz K. Analysis of free and glycosylated vitamin B6 in wheat by high-performance liquid chromatography. Cereal Chem. 1995;72(2):217–221.
35. Argoudelis CJ. Simple high-performance liquid chromatographic method for the determination of all seven vitamin B6-related compounds. J Chromatogr A. 1997;790:83–91.
36. Esteve MJ, Farré R, Frígola A, García-Cantabella JM. Determination of vitamin B6 (pyridoxamine, pyridoxal and pyridoxine) in pork meat and pork meat products by liquid chromatography. J Chromatogr A. 1998;795(2):383–387.
37. Sierra I, Vidal-Valverde C. A simple method to determine free and glycosilated vitamin B6 in legumes. J Liq Chromatogr Rel Tech. 1997;20(6):957–969.
38. Kall MA. Determination of total vitamin B6 in foods by isocratic HPLC: a comparison with microbiological analysis. Food Chem. 2003;82:315–327.
39. van Schoonhoven J, Schrijver J, van den Berg H, Haenen GRMM. Reliable and sensitive high-performance liquid chromatographic method with fluorometric detection for the analysis of vitamin B6 in foods and feeds. J Agri Food Chem. 1994;42:1475–1480.
40. Livaniou E, Costopoulou D, Vassiliadou I, et al. Analytical techniques for determining biotin. J Chromatogr A. 2000;881:331–343.
41. Staggs CG, Sealey WM, McCabe BJ, Teague AM, Mock DM. Determination of the biotin content of select foods using accurate and sensitive HPLC/avidin binding. J Food Comp Anal. 2004;17:767–776.
42. Höller U, Wachter F, Wehrli C, Fizet C. Quantification of biotin in feed, food, tablets, and premixes using HPLC–MS/MS. J Chromatogr B. 2006;831:8–16.
43. Arcot J, Shrestha A. Folate: methods of analysis. Trends Food Sci Tech. 2005;16:253–266.
44. Hyun TH, Tamura T. Trienzyme extraction in combination with microbiologic assay in food folate analysis: an updated review. Exp Biol Med. 2005;230:444–454.
45. Quinlan EP, Hanson AD, Gregory JF. The analysis of folate and its metabolic precursors in biological samples. Anal Biochem. 2006;348:163–184.
46. Pfeiffer CM, Rogers LM, Gregory III JF. Determination of folate in cereal-grain food products using trienzyme extraction and combined affinity and reversed-phase chromatography. J Agri Food Chem 4. 1997;5:407–413.
47. Ruggeri S, Vahteristo LT, Aguzzi A, Finglas P, Carnovale E. Determination of folate vitamers in food and in Italian reference diet by high-performance liquid chromatography. J Chromatogr A. 1999;855:237–245.
48. Vahteristo LT, Ollilainen V, Koivistoinen PE, Varo P. Improvements in the analysis of reduced folate monoglutamates and folic acid in food by high-performance liquid chromatography. J Agri Food Chem. 1996;44:477–482.
49. Rychlik M, Englert K, Kapfer S, Kirchhoff E. Folate contents of legumes by optimized enzyme treatment and stable isotope dilution analysis. J Food Comp Anal. 2007;20:411–419.
50. Thomas PM, Flanagan VP, Pawlosky RJ. Determination of 5-methyltetrahydrofolic acid and folic acid in citrus juices using stable isotope dilution–mass spectrometry. J Agri Food Chem. 2003;51:1293–1296.
51. Doherty RF, Beecher GR. A method for the analysis of natural and synthetic folate in foods. J Agri Food Chem. 2003;51:354–361.
52. Osseyi ES, Wehling RL, Albrecht JA. HPLC determination of stability and distribution of added folic acid and some endogenous folates during breadmaking. Cereal Chem. 2001;78(4):375–378.
53. De Brouwer V, Storozhenko S, Van De Steene JC, et al. Optimisation and validation of a liquid chromatography–tandem mass spectrometry method for folates in rice. J Chromatogr A. 2008;1215:125–132.
54. Vishnumohan S, Arcot J, Pickford R. Naturally-occurring folates in foods: method development and analysis using liquid chromatography–tandem mass spectrometry. Food Chem. 2011;125:736–742.
55. Zhang G-F, Storozhenko S, Van Der Straeten D, Lambert WE. Investigation of the extraction behavior of the main monoglutamate folates from spinach by liquid chromatography–electrospray tandem mass spectrometry. J Chromatogr A. 2005;1078:59–66.
56. Kumar SS, Chouhan RS, Thakur MS. Trends in analysis of vitamin B12. Anal Biochem. 2010;398:139–149.
57. Heudi O, Kilinç T, Fontannaz P, Marley E. Determination of vitamin B12 in food products and in premixes by reversed-phase high performance liquid chromatography and immunoaffinity extraction. J Chromatogr A. 2006;1101:63–68.
58. Campos-Giménez E, Fontannaz P, Trisconi MJ, Kilinc T, Gimenez C, Andrieux P. Determination of vitamin B12 in food products by liquid chromatography/UV detection with immunoaffinity extraction: single-laboratory validation. J AOAC Int. 2008;91(4):786–793.
59. Pakin C, Bergaentzlé M, Aoudé-Werner D, Hasselmann C. α-Ribazole, a fluorescent marker for the liquid chromatographic determination of vitamin B12 in foodstuffs. J Chromatogr A. 2005;1081:182–189.
60. Luo X, Chen B, Ding L, Tang F, Yao S. HPLC–ESI–MS analysis of vitamin B12 in food products and in multivitamins-multimineral tablets. Anal Chim Acta. 2006;562:185–189.
61. Koyyalamudi SR, Jeong S-C, Cho KY, Pang G. Vitamin B12 is the active corrinoid produced in cultivated white button mushrooms (Agaricus bisporus). J Agri Food Chem. 2009;57:6327–6333.
62. Kall MA, Andersen C. Improved method for simultaneous determination of ascorbic acid and dehydroascorbic acid, isoascorbic acid and dehydroisoascorbic acid in food and biological samples. J Chromatogr B. 1999;730:101–111.
63. Nisperos-Carriedo MO, Buslig BS, Shaw PE. Simultaneous detection of dehydroascorbic, ascorbic, and some organic acids in fruits and vegetables by HPLC. J Agri Food Chem. 1992;40(7):1127–1130.
64. Bognár A, Daood HG. Simple in-line postcolumn oxidation and derivatization for the simultaneous analysis of ascorbic and dehydroascorbic acids in foods. J Chromatogr Sci. 2000;38(4):162–168.
65. Castro RN, Azeredo LC, Azeredo MAA, de Sampaio CST. HPLC assay for the determination of ascorbic acid in honey samples. J Liq Chromatogr Rel Tech. 2001;24(7):1015–1020.
66. Rizzolo A, Brambilla A, Valsecchi S, Eccher-Zerbini P. Evaluation of sampling and extraction procedures for the analysis of ascorbic acid from pear fruit tissue. Food Chem. 2002;77(2):257–262.
67. Pérez AG, Olías R, Ezpada J, Olías JM, Sanz C. Rapid determination of sugars, non-volatile acids, and ascorbic acid in strawberry and other fruits. J Agri Food Chem. 1997;45(9):3545–3549.
68. Barros AIRNA, Silva AP, Gonçalves B, Nunes FM. A fast, simple, and reliable hydrophilic interaction liquid chromatography method for the determination of ascorbic and isoascorbic acids. Anal Bioanal Chem. 2010;396(5):1863–1875.
69. Doner LW, Hicks KB. High-performance liquid chromatographic separation of ascorbic acid, erythorbic acid, dehydroascorbic acid, dehydroerythorbic acid, diketogulonic acid, and diketogluconic acid. Anal Biochem. 1981;115(1):225–230.
70. Wagner HP, McGarrity MJ. The use of pulsed amperometry combined with ion-exclusion chromatography for the simultaneous analysis of ascorbic acid and sulfite. J Chromatogr A. 1991;546:119–124.
71. Behrens WA, Madére R. A procedure for the separation and quantitative analysis of ascorbic acid, dehydroascorbic acid, isoascorbic acid, and dehydroisoascorbic acid in food and animal tissue. J Liq Chromatogr. 1994;17(11):2445–2455.
72. Fontannaz P, Kilinç T, Heudi O. HPLC-UV determination of total vitamin C in a wide range of fortified food products. Food Chem. 2006;94(4):626–631.
73. Daood HG, Biacs PA, Dakar MA, Hajdu F. Ion-pair chromatography and photodiode-array detection of vitamin C and organic acids. J Chromatogr Sci. 1994;32(11):481–487.
74. Ziegler SJ, Meier B, Sticher O. Rapid and sensitive determination of dehydroascorbic acid in addition to ascorbic acid by reversed-phase high performance liquid chromatography using a post-column reduction system. J Chromatogr A. 1987;391:419–426.
75. Garrido Frenich A, Hernández Torres ME, Belmonte Vega A, Martínez Vidal JL, Plaza Bolaños P. Determination of ascorbic acid and carotenoids in food commodities by liquid chromatography with mass spectrometry detection. J Agri Food Chem. 2005;53:7371–7376.
76. Fenoll J, Martínez A, Hellín P, Flores P. Simultaneous determination of ascorbic and dehydroascorbic acids in vegetables and fruits by liquid chromatography with tandem-mass spectrometry. Food Chem. 2011;127:340–344.
77. Lee J, Ye L, Landen Jr WO, Eitenmiller RR. Optimization of an extraction procedure for the quantification of vitamin E in tomato and broccoli using response surface methodology. J Food Comp Anal. 2000;13(1):45–57.
78. Heudi O, Trisconi M-J, Blake C- J. Simultaneous quantification of vitamins A, D3 and E in fortified infant formulae by liquid chromatography–mass spectrometry. J Chromatogr A. 2004;1022(1–2):115–123.
79. Indyk HE. Simplified saponification procedure for the routine determination of total vitamin E in dairy products, foods and tissues by high-performance liquid chromatography. Analyst. 1988;113:1217–1221.
80. Grace ML, Bernhard RA. Measuring vitamins A and D in milk. J Dairy Sci. 1984;67(8):1646–1654.
81. Indyk HE, Woollard DC. Vitamin K in milk and infant formulas: determination and distribution of phylloquinone and menaquinone-4. Analyst. 1997;122:465–469.
82. Woolard DC, Indyk HE, Fong BY, Cook KK. Determination of vitamin K1 isomers in foods by liquid chromatography with C30 bonded-phase column. J AOAC Int. 2002;85(3):682–691.
83. Turner C, Persson M, Mathiasson L, Adlercreutz P, King JW. Lipase-catalyzed reactions in organic and supercritical solvents: application to fat-soluble vitamin determination in milk powder and infant formula. Enzyme Microb Tech. 2001;29(2–3):111–121.
84. Blanco D, Fernández MP, Gutiérrez MD. Simultaneous determination of fat-soluble vitamins and provitamins in dairy products by liquid chromatography with a narrow-bore column. Analyst. 2000;125:427–431.
85. Perales S, Alegría A, Barberá R, Farré R. Review: determination of vitamin D in dairy products by high performance liquid chromatography. Food Sci Tech Int. 2005;11(6):451–462.
86. Stancher B, Zonta F. High-performance liquid chromatography of the unsaponifiable from samples of marine and freshwater fish: fractionation and identification of retinol (vitamin A1) and dehydroretinol (vitamin A2) isomers. J Chromatogr. 1984;287(2):353–364.
87. Kamal-Eldin A, Görgen S, Pettersson J, Lampi AM. Normal-phase high-performance liquid chromatography of tocopherols and tocotrienols: comparison of different chromatographic columns. J Chromatogr A. 2000;881(1–2):217–227.
88. Thompson JN, Hatina G, Maxwell WB. High-performance liquid chromatographic determination of vitamin A in margarine, milk, partially skimmed milk, and skimmed milk. J AOAC. 1980;63(4):894–898.
89. Gama P, Casal S, Oliveira B, Ferreira MA. Development of an HPLC/diode-array/fluorimetric detector method for monitoring tocopherols and tocotrienols in edible oils. J Liq Chromatogr Rel Tech. 2000;23(19):3011–3022.
90. Sander LC, Sharpless KE, Pursch M. C30 Stationary phases for the analysis of food by liquid chromatography. J Chromatogr A. 2000;880(1–2):189–202.
91. Rimmer CA, Sander LC, Wise SA. Selectivity of long chain stationary phases in reversed phase liquid chromatography. Anal Bioanal Chem. 2005;382:698–707.
92. Parris NA. Non-aqueous reversed-phase liquid chromatography: a neglected approach to the analysis of low polarity samples. J Chromatogr A. 1978;157:161–170.
93. Barnett SA, Frick LW, Baine HM. Simultaneous determination of vitamins A, D2 or D3, E and K1 in infant formulas and dairy products by reversed-phase liquid chromatography. Anal Chem. 1980;52(4):610–614.
94. Nelis HJCF, De Leenheer AP. Isocratic nonaqueous reversed-phase liquid chromatography of carotenoids. Anal Chem. 1983;55(2):270–275.
95. Hulshof PJM, van Roekel-Jansen T, van de Bovenkamp P, West CE. Variation in retinol and carotenoid content of milk and milk products in the Netherlands. J Food Comp Anal. 2006;19(1):67–75.
96. Heinonen M, Ollilainen V, Linkola E, Varo P, Koivistoinen P. Carotenoids and retinoids in Finnish foods: cereal and bakery products. Cereal Chem. 1989;66(4):270–273.
97. Lietz G, Henry CJK. A modified method to minimise losses of carotenoids and tocopherols during HPLC analysis of red palm oil. Food Chem. 1997;60(1):109–117.
98. Jensen SK. Retinol determination in milk by HPLC and fluorescence detection. J Diary Res. 1994;61(2):233–240.
99. Britton G, Liaaen-Jensen S, eds. Carotenoids handbook. Basel: Switzerland; 2008; H. Pfander.
100. Thompson JN, Maxwell WH. Reversed-phase high-pressure liquid chromatography of vitamin A in margarine, infant formula, and fortified milk. J AOAC. 1977;60(4):766–771.
101. Zahar M, Smith DE. Vitamin A quantification in fluid dairy products: rapid method for vitamin A extraction for high performance liquid chromatography. J Dairy Sci. 1990;73(12):3402–3407.
102. Epler KS, Sander LC, Ziegler RG, Wise SA, Craft NE. Evaluation of reversed-phase liquid-chromatographic columns for recovery and selectivity of selected carotenoids. J Chromatogr. 1992;595:89–101 199.
103. Emenhiser C, Sander LC, Schwartz SJ. Capability of a polymeric C30 stationary phase to resolve cis-trans carotenoid isomers in reversed-phase liquid chromatography. J Chromatogr A. 1995;707:205–216.
104. Li H, Tyndale ST, Heath DD, Letcher RJ. Determination of carotenoids and all-trans-retinol in fish eggs by liquid chromatography–electrospray ionization–tandem mass spectrometry. J Chromatogr B. 2005;816:49–56.
105. Huang T, Zhang W, Liu J, Tian Y, Yang G. Determination of vitamin A in infant/adult formula by HPLC–isotope dilution mass spectrometry. J Liq Chromatogr Rel Tech. 2009;32(5):712–722.
106. Lim YO, Kim B, Ahn S, Kim J. Improvement of accuracy for the determination of vitamin A in infant formula by isotope dilution–liquid chromatography/tandem mass spectrometry. Food Chem. 2011;126:1393–1398.
107. Salo-Väänänen P, Ollilainen V, Mattila P, Lehikoinen K, Salmela-Mölsä E, Piironen V. Simultaneous HPLC analysis of fat-soluble vitamins in selected animal products after small-scale extraction. Food Chem. 2000;71(4):535–543.
108. Mattila PH, Piironen VI, Uusi-Rauva EJ, Koivistoinen PE. Contents of cholecalciferol, ergocalciferol, and their 25-hydroxylated metabolites in milk products and raw meat and liver as determined by HPLC. J Agri Food Chem. 1995;43(9):2394–2399.
109. Hasegawa H. Vitamin D determination using high-performance liquid chromatography with internal standard-redox mode electrochemical detection and its application to medical nutritional products. J Chromatogr A. 1992;605(2):215–220.
110. Argawal VK. Liquid chromatographic determination of vitamin D in infant formula. J Assoc Off Anal Chem. 1989;72(6):1007–1009.
111. Indyk H, Woollard DC. Determination of vitamin D [cholecalciferol] in fortified milk powders and infant formulae by HPLC. J Micronut Anal. 1985;1(2):121–141.
112. Craige Trenerry V, Plozza T, Caridi D, Murphy S. The determination of vitamin D3 in bovine milk by liquid chromatography mass spectrometry. Food Chem. 2011;125:1314–1319.
113. Dimartino G. Convenient analysis of vitamin D in cheese and other food matrixes by liquid chromatography/mass spectrometry. J AOAC Int. 2007;90(5):1340–1345.
114. Min H, LaLuzerne P, Winters D, Sullivan D. Measurement of vitamin D in foods and nutritional supplements by liquid chromatography/tandem mass spectrometry. J AOAC Int. 2009;92(5):1327–1335.
115. Byrdwell WC. Comparison of analysis of vitamin D3 in foods using ultraviolet and mass spectrometric detection. J Agri Food Chem. 2009;57(6):2135–2146.
116. Tan B, Brzuskiewicz L. Separation of tocopherol and tocotrienol isomers using normal- and reverse-phase liquid chromatography. Anal Biochem. 1989;180(2):368–373.
117. Strohschein S, Pursch M, Lubda D, Albert K. Shape selectivity of C30 phases for RP-HPLC separation of tocopherol isomers and correlation with MAS NMR data from suspended stationary phases. Anal Chem. 1998;70:13–18.
118. Hewavitharana AK, Lanari MC, C, Becu C. Simultaneous determination of vitamin E homologs in chicken meat by liquid chromatography with fluorescence detection. J Chromatogr A. 2004;1025:313–317.
119. Rentel C, Strohschein S, Albert K, Bayer E. Silver-plated vitamins: a method of detecting tocopherols and carotenoids in LC/ESI–MS coupling. Anal Chem. 1998;70:4394–4400.
120. Bustamante-Rangel M, Delgado-Zamarreno MM, Sanchez-Perez A, Carabias-Martınez R. Determination of tocopherols and tocotrienols in cereals by pressurized liquid extraction–liquid chromatography–mass spectrometry. Anal Chim Acta. 2007;587:216–221.
121. Lanina SA, Toledo P, Sampels S, Kamal-Eldin A, Jastrebova JA. Comparison of reversed-phase liquid chromatography–mass spectrometry with electrospray and atmospheric pressure chemical ionization for analysis of dietary tocopherols. J Chromatogr A. 2007;1157:159–170.
122. Stöggl WM, Huck CW, Scherz H, Popp M, Bonn GK. Analysis of vitamin E in food and phytopharmaceutical preparations by HPLC and HPLC–APCI–MS–MS. Chromatographia. 2001;54:179–185.
123. Kalman A, Mujahid C, Mottier P, Heudi O. Determination of α-tocopherol in infant foods by liquid chromatography combined with atmospheric pressure chemical ionisation mass spectrometry. Rapid Comm Mass Spec. 2003;17:723–727.
124. Cook KK, Mitchell GV, Grundel E, Rader JI. HPLC analysis for trans-vitamin K1 and dihydro-vitamin K1 in margarines and margarine-like products using C30 stationary phase. Food Chem. 1999;67:79–88.
125. Koivu-Tikkanen TJ, Ollilainen V, Piironen VI. Determination of phylloquinone and menaquinones in animal products with fluorescence detection after postcolumn reduction with metallic zinc. J Agri Food Chem. 2000;48:6325–6331.
126. Booth SL, Davidson KW, Sadowski JA. Evaluation of an HPLC method for the determination of phylloquinone (vitamin K1) in various food matrices. J Agri Food Chem. 1994;42:295–300.
127. Koivu TJ, Piironen VI, Henttonen SK, Mattila PH. Determination of phylloquinone in vegetables, fruits, and berries by high-performance liquid chromatography with electrochemical detection. J Agri Food Chem. 1997;45:4644–4649.
128. Piironen V, Koivu T, Tammisalo O, Mattila P. Determination of phylloquinone in oils, margarines and butter by high-performance liquid chromatography with electrochemical detection. Food Chem. 1997;59(3):473–480.
129. Chase Jr GW, Thompson B. Accelerated solvent extraction of vitamin K1 in medical foods in conjunction with matrix solid-phase dispersion. J AOAC Int. 2000;83(2):407–410.
130. Schneiderman MA, Sharma AK, Mahanama KRR, Locke Dc. Determination of vitamin K1 in powdered infant formulas, using supercritical fluid extraction and liquid chromatography with electrochemical detection. J AOAC. 1988;71(4):815–817.
131. Hwang S-M. Liquid chromatographic determination of vitamin K1 trans- and cis-isomers in infant formula. J AOAC. 1985;68(4):684–689.
132. Viñas P, López-Erroz C, Balsalobre N, Hernández-Córdoba M. Reversed-phase liquid chromatography on an amide stationary phase for the determination of the B group vitamins in baby foods. J Chromatogr A. 2003;1007:77–84.
133. Leporati A, Catellani D, Suman M, Andreoli R, Manini P, Niessen WMA. Application of a liquid chromatography tandem mass spectrometry method to the analysis of water-soluble vitamins in Italian pasta. Anal Chim Acta. 2005;531:87–95.
134. Herrero-Barbudo MC, Granado-Lorencio F, Blanco-Navarro I, Olmedilla-Alonso B. Retinol, α- and γ-tocopherol and carotenoids in natural and vitamin A- and E-fortified dairy products commercialized in Spain. Int Dairy J. 2005;15:521–526.
135. Kienen V, Costa WF, Visentainer JV, Souza NE, Oliveira CC. Development of a green chromatographic method for determination of fat-soluble vitamins in food and pharmaceutical supplement. Talanta. 2008;75:141–146.
136. Chauveau-Duriot B, Doreau M, Noziere P, Grailet B. Simultaneous quantification of carotenoids, retinol, and tocopherols in forages, bovine plasma, and milk: validation of a novel UPLC method. Anal Bioanal Chem. 2010;397:777–790.
137. Gentili A, Caretti F. Evaluation of a method based on liquid chromatography–diode array detector–tandem mass spectrometry for a rapid and comprehensive characterization of the fat-soluble vitamin and carotenoid profile of selected plant foods. J Chromatogr A. 2011;1218:684–697.
138. Gentili A, Caretti F, D’Ascenzo G, et al. Simultaneous determination of water-soluble vitamins in selected food matrices by liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Comm Mass Spec. 2008;22:2029–2043.
139. Albalá-Hurtado S, Veciana-Nogués MT, Izquierdo-Pulido M, Mariné-Font A. Determination of water-soluble vitamins in infant milk by high-performance liquid chromatography. J Chromatogr A. 1997;778:247–253.
140. Ciulu M, Solinas S, Floris L, et al. RP–HPLC determination of water-soluble vitamins in honey. Talanta. 2011;83:924–929.
141. Woollard DC, ndyk HE. Rapid determination of thiamine, riboflavin, pyridoxine, and niacinamide in infant formulas by liquid chromatography. J AOAC Int. 2002;85(4):945–951.
142. Zafra-Gómez A, Garballo A, Morales JC, García-Ayuso LE. Simultaneous determination of eight water-soluble vitamins in supplemented foods by liquid chromatography. J Agri Food Chem. 2006;54:4531–4536.
143. Lu B, Ren Y, Huang B, Liao W, Cai Z, Tie X. Simultaneous determination of four water-soluble vitamins in fortified infant foods by ultra-performance liquid chromatography coupled with triple quadrupole mass spectrometry. J Chromatogr Sci. 2008;46:225–232.
144. Goldschmidt RJ, Wolf WR. Simultaneous determination of water-soluble vitamins in SRM 1849 infant/adult nutritional formula powder by liquid chromatography-isotope dilution mass spectrometry. Anal Bioanal Chem. 2010;397:471–481.
145. Phinney KW, Rimmer CA, Thomas JB, Sander LC, Sharpless KE, Wise SA. Isotope dilution liquid chromatography–mass spectrometry methods for fat- and water-soluble vitamins in nutritional formulations. Anal Chem. 2001;83:92–98.