CHAPTER 8
From the One Percent
At the 1993 International Conference on AIDS in Berlin, the most depressing AIDS conference ever, Heiko Jessen was waiting to hear a talk from Bruce Walker. The year before, in 1992, Walker had come across an unusual patient, one who had become infected with HIV in 1978 in San Francisco. But he didn’t know it at the time. Instead, the diagnosis was made later from blood products collected during a hepatitis B vaccine trial. Curiously, despite never having taken antiviral drugs, the man maintained healthy levels of T cells and did not progress to AIDS. Walker remained particularly interested in a special set of immune cells called the killer T cells, the storm troopers of the immune system. These cells are like trained killers. They have a finely tuned mechanism for detecting cells that are cancerous, infected with viruses, or are in some way damaged. Once they identify a cell that needs to be killed, they release cellular toxins, enzymes capable of tearing holes in the cell’s membrane, eventually killing it.
The way these killer cells are able to identify a cancer cell or one that has been infected with HIV depends, in large part, on our own personal genetics. The commanders of the immune system, the helper T cells, have a receptor on their surface that is able to recognize specific pieces of the invader. But the receptors can’t recognize these bits of the virus or invader by themselves. The virus must first be introduced to them. So bits of the invading virus’s proteins, the antigens mentioned in chapter 3, are introduced to the receptors on the T cells by another cell, aptly named an antigen-presenting cell. This antigen-presenting cell encounters a virus and eats it up. It then takes the virus’s antigens and hoists them up on its surface. Similar to how a conqueror would proudly display the head of its defeated enemy on a stake.
Seeing a head on a stake is enough to frighten anyone into action, especially a commander trained to defeat enemies of the immune system. But what if the commander sees a finger on a stake? It’s not quite as scary. It is our individual genetics that determine which part of the virus will be displayed on the surface of the cell. While some people’s display a head, a clear warning sign that something drastic must be done, others encountering the same invader show only a finger. The commander still mobilizes an attack against the invader but does not mobilize the full force of storm troopers, as it would in response to a head. (We biologists can seem macabre about body parts; forgive me.)
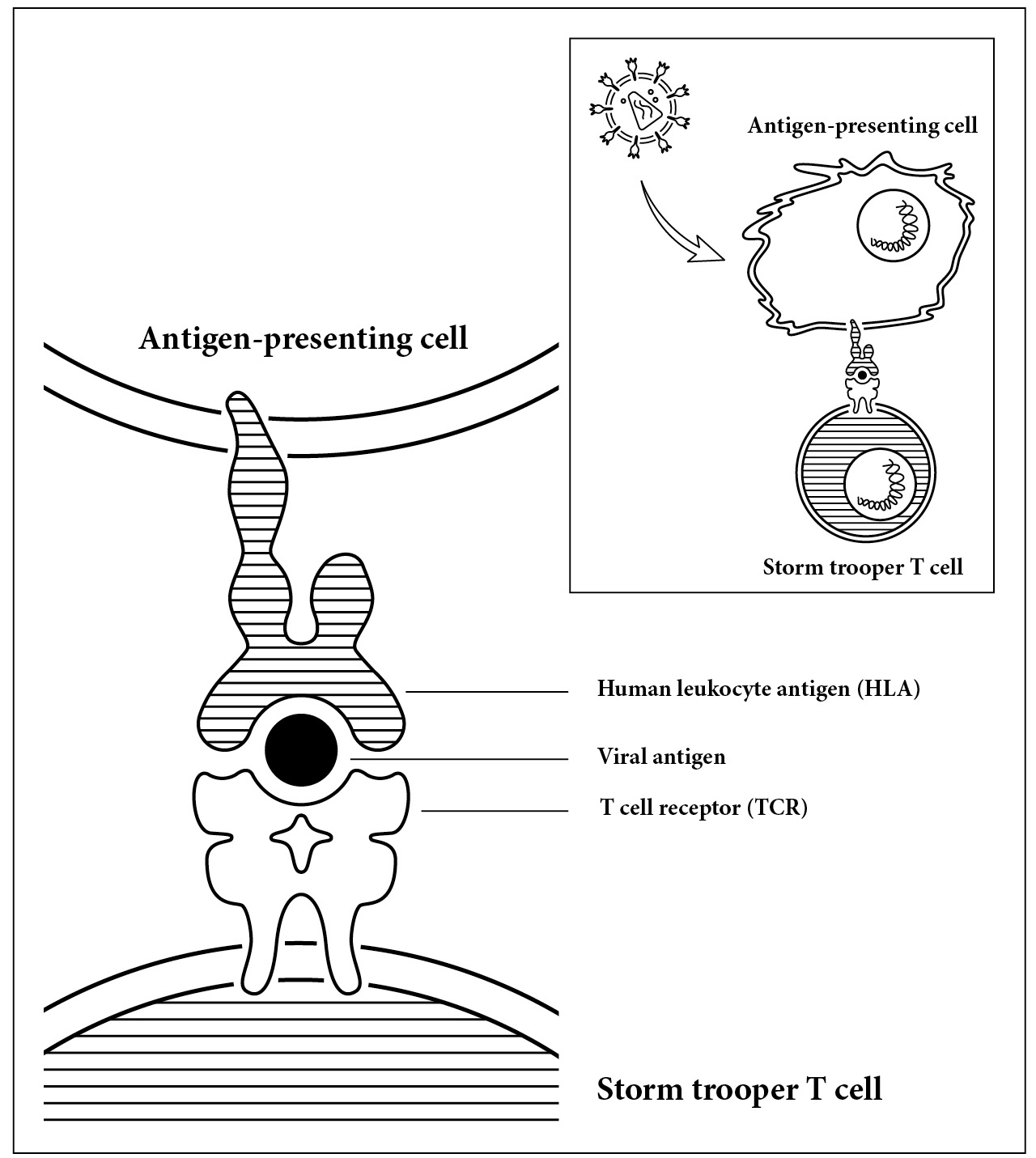
The immune system’s version of a head on a stake. The response to intruders is measured by the interaction between three molecules. First, the human leukocyte antigen (HLA) on an antigen-presenting cell, a small piece of virus called an antigen, and the T cell receptor (TCR) on a T cell. How these three molecules fit together determines the strength of the immune system response.
The response of the immune system is not determined only by the head or finger. Just as important is the stake. What holds the head is the human leukocyte antigen, but let’s just call it the stake. This stake not only holds the head up but also determines which part of the invader is displayed. Depending on the genetics of your stake, you’ll display a different part of your enemy to the immune system. This is key because which part is displayed, the head or the finger, makes all the difference in how the immune system responds.
A critical interaction occurs between these three molecules: the receptor on the commander (TCR), the piece of the invader (antigen), and the stake (HLA). The interaction between these molecules can be seen in the illustration on page 81. The binding of the molecules determines the strength in which the immune system is mobilized. So if we have a special set of genes, when we present the virus to the commander’s receptor it looks like a frightening head on a stake. Our commanders are alerted and our storm troopers are set to high alarm. Alternately, our genes can work against us, showing the receptor what looks like a nonthreatening finger.
Bruce Walker formed the basis of our thinking on killer T cells and how they’re mobilized by our genes against HIV. It’s no wonder that when he met a man who was HIV-positive but did not progress to AIDS, he immediately wondered what the person’s killer T cells were doing and how HIV was being presented to the commander T cells. There’s likely not another HIV physician at that time who would have made the connection. While Walker didn’t quite understand what was happening in his patient, he knew it was significant. As he traveled to Berlin that summer, he was excited to present the results he had, and, even more so, he wanted to learn if other physicians had seen anything like it.
Also attending the conference was David Ho, who had moved to New York City in 1990. Working as an investigator at the New York University School of Medicine, he was desperate to begin clinical trials of new AIDS drugs at Rockefeller University. He recognized the limitations of AZT, but he came to Berlin hoping for good news.
It was a conference of failures. At the top of the list was AZT. The drug was simply not keeping the virus in check; the death rate for AIDS was climbing. Sadly, there were no other options. Drugs from multiple other trials were presented, and nothing worked. A trial called Concorde, which gave AZT to patients early in infection, before symptoms appeared, was worthless. Other, newer drugs, developed to target new parts of the HIV life cycle, were a colossal failure. One study looking at a new combination of drugs appeared to some researchers to have arranged their statistics and study groups in order to show a misleadingly positive result. The subterfuge didn’t work. The response was heated, as attendees accused the group of cheating. One researcher became so upset by the misleading data that, during the discussion section, she angrily asked, “How much is Roche paying you to say this?” By all accounts, the conference was ugly. Eight clinical trials, all of which had been kicked off with high hopes, presented negative results. To top it off, the death rate was the highest yet.
Jessen sat with his brother, sister, and Andrew at the conference. Although neither Jessen’s brother nor his sister worked with AIDS patients yet, they would both find their medical careers shaped by the virus in the years to come. His brother, Arne, would even join Heiko as a physician in his practice. Jessen heard the president of Germany mention AIDS for the first time and was struck by the historical moment. He was even more stunned by what followed. There were no new ideas, no hope. Andrew had been diagnosed only two months previously, and Jessen had been counting on finding some new drug with promise at the conference. He was sure there would be some inkling from an emerging clinical trial. Instead, they were left with nothing. He couldn’t believe that even David Ho, the superstar in the HIV field, had nothing to offer. As he sat at the conference, he began to cry. There was no hope. Andrew would die.
In 1993, the Berlin AIDS conference crushed the hopes of those desperate for a new drug to treat HIV. The drug they were waiting for was still two years away from being approved by the FDA for use. That drug, saquinavir, was designed from the crystal structure of HIV’s protease enzyme. While, in 1989, everyone thought Merck was the first to deduce the crystal structure of the viral enzyme, molecular virologists at Roche knew that the structure was actually quite different. They developed the drug R03I-8959, or saquinavir, based on their modeling of the target.
What no one expected was that the combination of AZT, which targets the reverse transcriptase enzyme, and saquinavir, which targets the protease enzyme, would result in a powerful synergy. By combining AZT and saquinavir, the concentration of each drug increases exponentially inside the cell, making both drugs better at attacking the virus’s replication. This combination therapy, called highly active antiretroviral therapy (HAART), would prove itself capable of knocking out the virus to undetectable levels in the blood (but not the reservoir) in the years ahead.
Fast-forward three years to the AIDS conference in July 1996, where David Ho showed a pivotal slide to the eager audience. The slide presented evidence for what seems obvious now: The average person infected with HIV makes billions of copies of the virus daily. The data marked a turning point. It’s hard to imagine now, but before this conference, physicians were divided on whether the infection even necessitated treatment. After this conference, the picture was clear: Patients needed to be put on antiviral drugs. Even better, new drugs were now available. In 1997, two papers in The New England Journal of Medicine confirmed what had been reported at the 1996 conference. Combination therapy, or HAART, reduces death by 60–80 percent. The 1996 AIDS conference was the opposite of the 1993 conference in Berlin. It was full of optimism. The word cure hovered around the 1996 conference, unspoken by the attendees but present in everyone’s thoughts. The rise of a new class of protease inhibitors was a game changer. Hidden within the optimism of the 1996 conference was the hope that this combination therapy would be powerful enough to end the epidemic. Researchers hoped the drugs could purge the virus from the body. At the forefront of this hope was David Ho, the researcher pioneering the use of protease inhibitors and sparking headlines heralding the end of the AIDS era. But, despite the strides the new combination therapy made, it wasn’t a cure. Not yet.