CHAPTER 25
The Promise Kept
As the video opens, we see Emily Whitehead, only six years old, lying in a hospital bed. Emma, as she likes to be called, is wearing purple, her favorite color. Her head is bare, the multiple rounds of chemotherapy having stripped her of her once thick, brown hair. She sits emotionless, watching calmly as doctors quietly explain what each tube being placed in her body is for. In 2010, when Emma was five years old, she was diagnosed with acute lymphoblastic leukemia, or ALL. This kind of leukemia is a close cousin to the one Timothy had. Just as the storm trooper T cells in a person with HIV have trouble hunting all the cells infected with the virus, in leukemia, the storm troopers have trouble hunting all the cancerous cells.
Emma went through a year of chemotherapy before learning that her leukemia had reappeared. The relapse was a terrible sign, reducing her chances of defeating the cancer from 80 to 90 percent to less than 30 percent. She started aggressive chemotherapy treatments and was scheduled for a bone marrow transplant in February 2012. Exactly like Timothy’s procedure, the bone marrow transplant would boost her immune system, giving her all the precious immune cells the cancer was killing off. But just two weeks before the transplant, Emma learned she couldn’t receive it. She had again relapsed. Emma went back on chemotherapy as her parents hoped for a miracle. The options for her were shrinking. Cells taken from her bone marrow with a big needle placed in Emma’s spine, showed that 7 percent of the cells were cancerous. The chemo wasn’t working. Emma and her parents were left with one option: a highly experimental clinical trial at the Children’s Hospital of Philadelphia. They had rejected the idea of this clinical trial once already. It was a frightening procedure, and Emma would be the very first child to participate in it.
The clinical trial, called CART-19, would isolate the T cells from Emma’s blood and then genetically engineer them to specifically recognize and kill the cancer cells lurking in her body. How can we transform T cells into cancer-killing machines? The trick, as mentioned above, involves HIV. Carl June, the lead investigator of the trial, knew that HIV is a master at getting into cells. In order to safely exploit this feature of the virus, June used a version of HIV that had been spliced apart, removing all the parts of the virus that make it dangerous. It’s the same vehicle, or vector, that John Zaia, working three thousand miles away in California, was using for his gene therapy. June then stuck the cancer-targeting information the cell needed into the empty shell of the virus. HIV worked as the packaging for the information the cell needed to target cancer. Like a sheep in wolf’s clothing, the virus was able to invade the T cells. Once inside, instead of taking over the cell’s machinery to replicate itself, the virus delivered the blueprint for destroying cancer cells. That blueprint was a chimeric antigen receptor, or CAR. The CAR is a modified version of the T cell receptor, or TCR, the molecule that lies on the surface of T cells and is a key part of how we control our immune system. By modifying the TCR, researchers were changing how the immune system responded to invaders. The CAR that Carl June is using is composed of signaling molecules that lie on the surface of B cells. The gene therapy replaces how T cells decide which target to kill, so that they now put all their energy toward attacking the B cell and B-cell progenitors in the bone marrow (and the rest of the body) that are harboring the cancer.
On April 17, 2012, Emily became the first pediatric patient to receive CART-19. Her T cells were isolated from her blood and then treated with the tempered HIV containing the cancer-killing blueprint. Over the course of three days, those T cells were reinfused into her body. It would take ten long days to know if the T cells were doing their job and killing off the cancer. However, after only three days, Emma became very sick. She was running a fever of 104.9°F, and became delirious. She was rushed to the pediatric ICU, where her breathing tanked and her blood pressure became dangerously low. In a last-ditch effort, she was given steroids, a move that physicians knew could kill off the genetically engineered T cells. The gene therapy was now unimportant as Emma’s life hung in the balance.
In Ross Kauffman’s short video “Fire with Fire,” Carl June describes what happened next: “It was like the calm after the storm. The clouds went away. And she woke up and there was no leukemia.” With tears flooding his eyes and a shaky voice, June says, “When that child survived it was a course. An amazing event.” Emma survived and her cancer is in remission. Today, she is a beautiful little girl with a head of brown, wavy hair, and still loves purple.
The gene therapy Emma received was informed by research trying to replicate Timothy’s cure. Timothy’s experience has influenced not just the path of HIV cure research but also that of cancer gene therapies. This is because Carl June was able to refine Emma’s therapy based on lessons learned from modifying T cells in HIV gene therapy trials targeting CCR5.
• • •
Carl June’s draft number was 50. It wasn’t a good number. Any American man born between 1944 and 1950 with a draft number less than 195 was classified as 1-A and required to report for service. June knew what that number meant, that he would be fighting in the frontlines of the Vietnam War. It was 1971 and he had just graduated from high school. Two years later, the war was over, but June’s career in the military was just beginning. He went into the United States Naval Academy in Annapolis, and then, in an effort to “control his destiny,” June decided to go to medical school. It was a way to exert control over what the long years of his military obligation would be like. By the early 1980s, he was among a small group of physicians chosen to learn a complicated procedure in Japan: bone marrow transplantation.
For the military, this move was not about treating cancer. In the 1950s, as fears of nuclear warfare grew, the military considered radiation poisoning to be a considerable threat to the public. During the Manhattan Project, a group of scientists observed that the spleen seemed to offer protection against radiation poisoning. Building on these observations, in 1951 they tested the first bone marrow transplant in mice. Their results were remarkable: The procedure, infusing the stem cells taken from the bone marrow back into the mouse, was able to rescue the animal from a lethal dose of radiation.
June would learn bone marrow transplantation from the master: E. Donnall Thomas, the man who performed the first bone marrow transplant in a human in 1956. His work, which drastically improved the survival of those with blood cancers like Timothy’s, won him the Nobel Prize in Medicine in 1990. With June’s interest in cancer research now piqued, he was disappointed to learn that the program in Japan was being discontinued. There was simply no cancer research being done in the military. And since he couldn’t work on cancer, he decided to work on an infectious disease program in which the Navy was beginning to invest heavily: HIV. Although he would never have chosen such diverse training in cancer and HIV if left to his own devices, thanks to the military, June had a perfect background for developing a completely novel approach to both diseases.
In the mid-1990s, June was working at what is now the Walter Reed National Military Medical Center in Bethesda, Maryland, across the street from the NIH, when a new postdoctoral fellow came to interview for a position in his lab. Bruce Levine, from a family of scientists, had been working in labs since high school. As he interviewed for June’s lab, he sat two floors above the maternity ward where he first entered the world. It felt like a sign. Levine joined June’s lab, excited as a PhD to be part of the hospital environment.
Levine and June began looking at how to make T cells grow outside the body. At the time, growing T cells in the lab was a major challenge, involving either a complicated mix of cell-signaling molecules or the use of dendritic cells. As you can imagine, this was not good for those researching HIV. Researchers needed a simple way to model the virus in human cells. June and Levine took on this problem by creating an artificial dendritic cell. Dendritic cells are immune cells formed all over the body. They’re called dendritic because of their funny, treelike appearance, where the edges of the cell branch out like roots of a tree. In addition to other functions, they provide signals to the T cells that tell them to mature. June and Levine developed an artificial dendritic cell, a tiny magnetic bead that could induce the same effect in T cells. The artificial cells were a success; by adding them to the cultured T cells biweekly, the T cells could be grown in the incubator easily. But something odd happened when they tested their technique in T cells taken from HIV-positive individuals. The T cells that once harbored virus were suddenly resistant to HIV infection. Here was a mystery: How were the artificial dendritic cells able to confer resistance to HIV infection?
It wasn’t until after they published their paper in 1996 that the answer became clear: The T cells growing in the incubator didn’t express CCR5. The artificial dendritic cells, in addition to telling the T cells to mature, were also clearing CCR5 off the cell surface. Without CCR5, the cells couldn’t be infected with HIV.
These observations stuck with June and Levine as they continued their research. Moving their lab to the University of Pennsylvania, in 2004, June received a visit from an old friend: Dale Ando. Ando had bounced around several different biotech companies before landing at Sangamo BioSciences that year. He came to June with a crazy idea: a “Star Wars approach” to treating HIV. “What if,” Ando postulated, they “could knock out the co-receptor HIV needs to enter T cells?” The idea sounded crazy. Even more crazy was the data Ando had. Their efficacy at knocking out co-receptors was less than 1 percent. It was madness to spend time and money on such an inefficient technology. If it had been anyone else but his friend Ando, June would have likely dismissed the whole concept. As it was, he told Levine about the idea and then added dismissively, “Yeah, right, like that’s going to work.”
• • •
Edward Lanphier founded Sangamo BioSciences in 1995. Intensely interested in gene therapy, Lanphier had been working for Somatix, one of a host of start-ups that had jumped on the gene therapy bandwagon. It was a challenging time for gene therapy. Almost every gene Lanphier wanted to acquire was already owned. Restrictions based on intellectual property were weakening the new industry. Lanphier says that “it was not optimal; you would get what you could get.”
In the midst of negotiating these complicated deals, Lanphier became interested in the work of Srinivasan Chandrasegaran. Chandra, as his friends call him, was a postdoc in Jeremy Berg’s lab at Johns Hopkins University when he created and patented zinc finger nucleases (ZFNs), which are small gene-editing machines. To make them, Chandra put together two mechanisms, both borrowed from the natural world. The first are zinc fingers. Zinc fingers were first discovered when looking at the RNA of the African clawed frog. Scientists wondered how the RNA of this creature was able to bind both strongly and specifically to a protein. They discovered that the secret was due to the unique structure of a protein, which had elongated fingerlike structures held together in the center by a zinc ion. Here, in nature, was a perfect example of how to specifically target and grab a piece of DNA: a zinc finger. Chandra stitched together several zinc finger proteins and attached them to an enzyme that cuts DNA. These enzymes, called restriction enzymes, were first discovered in bacteria. Amusingly, the bacteria use the enzymes to fight off viruses. The enzymes cut out the intruder DNA from the native DNA. The enzymes are a powerful tool in molecular biology and cloning, allowing scientists to cut up the DNA they’re working with and rearrange it. By combining the DNA-gripping nature of a zinc finger with the DNA-cutting ability of a restriction enzyme, Chandra had created a novel tool.
Somatix, however, wasn’t interested in zinc finger nucleases. That didn’t deter Lanphier, who knew he had found a powerful new technology. After being tangled in the intellectual property laws that were crippling gene therapy, Lanphier appreciated this fresh approach to modifying genes. He decided to take a chance and form his own company. Going to family and friends, he raised $750,000 to start the business. When remembering those years, Lanphier says he “should have been scared.” But instead those early years were exhilarating.
The idea to tackle HIV would come years later when Dale Ando joined the company. Between him and Philip Gregory, the chief scientific officer at Sangamo, they devised a plot to use the specificity of the zinc finger nucleases to attack the co-receptor HIV needs to enter T cells.
Each zinc finger is designed to bind only one specific part of a gene. The zinc finger nucleases Sangamo designed for HIV are specifically created to target CCR5 and only CCR5. The zinc finger matches up to twelve building blocks of DNA, the A, T, G, and C that make up the DNA alphabet. To knock out the CCR5 gene, it’s not enough to take out one strand of DNA, which the cell can repair. Instead, both strands of DNA that encode CCR5 have to be cut because a double-stranded break is not repaired well by the cell. Our repair enzymes need the information contained in the complementary strand in order to reconstruct the gene. It’s similar to when a building is demolished, if only one wall is taken down, matching up the structural work to the remaining walls can likely repair it. But if we take down all the walls, the building is done for.
For this reason, two zinc finger nucleases are delivered to the nucleus of the cell. Each zinc finger makes its way to its specific target: the single strands of DNA that encode the CCR5 gene (see the illustration on page 230). The zinc fingers bind to the DNA, gripping the molecule strongly. Only after the DNA is in its grasp does the nuclease portion of the ZFN engage. Alone, each nuclease doesn’t have the ability to cut DNA. But, when two ZFNs line up perfectly, they form a dimer, two halves making a whole, creating a precise break in both strands of DNA. Here was the way to tackle HIV. Using the CCR5-specific ZFNs, they could wipe out expression of CCR5 on the surface of T cells, keeping HIV from entering the cells.
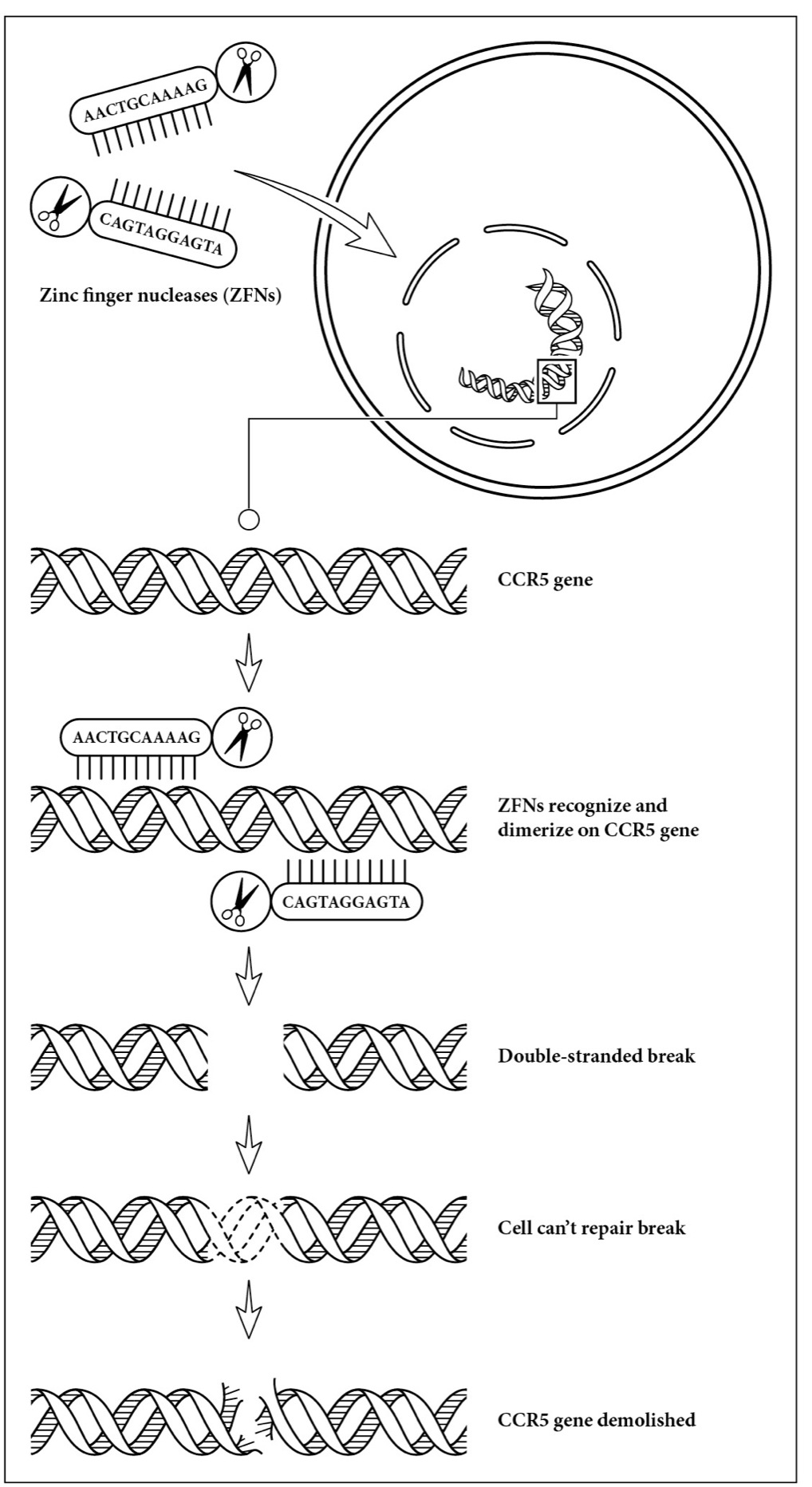
How zinc finger nucleases (ZFNs) take out CCR5. Two ZFNs are delivered to a cell. Each contains a region that binds to a part of the CCR5 gene. When they are both attached to the gene, their accompanying restriction enzymes combine and cause both DNA strands to break. Unable to make the CCR5 protein, the ZFN-treated cell locks out HIV.
The gene therapy could potentially work as both a vaccine, keeping healthy people exposed to the virus from ever becoming infected, or as a cure, eradicating the virus from the body of those harboring the virus. It was a highly original approach, perhaps too original. For most scientists hearing it for the first time, it seemed crazy.
• • •
Carl June was no exception. Nevertheless, by the twists of fate, June had a background tailored to pursue the unusual project. He had experience working with CCR5 and HIV from his days in the 1990s culturing T cells. He was actively pursuing gene therapy for cancer patients and was interested in how it could be applied to HIV. He had experience in engineering and transplanting blood cells. All these components combined to give him the exact experience needed to tackle such a seemingly crazy and complex project. In fact, it would be hard to imagine any other researcher able to grapple with the amalgam of research experience needed.
June and his lab grudgingly took the CCR5 ZFNs from Sangamo and began testing them in T cells taken directly from patients. Behind closed doors, they laughed at the method, calling it the Star Wars approach for HIV. Working with the finicky human cells, they were able to optimize the disruption of CCR5. They then took the cells and injected them in a mouse model. But it wasn’t just any mouse model. June and his lab chose a humanized mouse model.
Humanized mice are the latest trend to hit the animal model world. The problem with modeling disease in animals is that it can never completely mimic pathogens in humans. Human diseases simply behave differently in a mouse than they do in a human. For HIV, the problem is severe. Mice can’t be infected with HIV. So instead, we turn to monkey models of infection. But even here, in our primate cousins, we run into problems. Monkeys can’t be infected with HIV, with the exception of chimpanzees, an animal model no longer used because of their endangered status in the wild. Instead, the monkey models we use are infected with SIV, or simian immunodeficiency virus. SIV, a close relative of HIV, behaves a lot like its human counterpart, but is certainly not identical. There are over 40 strains of SIV, native to different primate species. Despite the diversity of strains to choose from, genetically, only 50 percent of the SIV strain most commonly used in research studies matches that of HIV.
Perhaps the biggest difference between HIV and SIV is that most SIVs don’t cause disease. Monkeys live out their lives with the virus multiplying inside them with little effect. The virus and monkey coexistence has been tweaked over time, evolution smoothing the path to a stable truce between virus and host. The reason monkeys have this and we don’t is because they’ve lived with SIV longer than we have, likely over 32,000 years. Compare this to HIV, which has had only one hundred years to adapt to us. SIV has had the time to work out the kinks.
For this reason, most SIV models aren’t very good for studying HIV. Thus, researchers developed SIVmac, an SIV taken from a sooty mangabey and put into a rhesus macaque monkey. Rhesus macaques are a species of monkey that aren’t naturally infected with SIV in the wild, so they haven’t had time to adapt to the virus, especially a virus from a different monkey species. This is why SIVmac behaves more like HIV: It causes disease, even death.
All the HIV vaccines that have made their way to clinical trials have done so through the SIVmac model. The path to these unsuccessful vaccines, although numerous factors resulted in their failure, was paved in macaque monkeys. Although the vaccines were able to protect the monkeys from getting SIV, researchers were unable to translate this success to humans. There are simply too many differences between the two viruses and between monkeys and us.
In humans, it typically takes between seven to ten years to develop AIDS. In the SIVmac model, AIDS is usually reached within six months. Extreme levels of virus and deteriorating CD4+ T cells characterize these turbulent months. In comparing the disease courses of the two, SIVmac bears only a resemblance to HIV.
Other problems that arise from using the monkey model are its expense—housing and experimentation on primates requires large primate centers with ample funding—and the sheer impracticality of obtaining enough monkeys. This is a sticking point in science, where statistics hinge on the size of a study. That said, SIVmac was the only game in town. It might not be perfect, but it was the only animal model on which to test HIV approaches.
That is, until the humanized mouse model came along. Mice have long been a favorite in animal studies because it’s easy to obtain large numbers of them and they’re not expensive to maintain. The problem with mice, however, is that they’re not very much like humans. The strain of mouse most commonly used in research studies, called Black 6, has an 85 percent similarity to the human genome. Compare this to macaque monkeys, which are 95 percent similar. The differences don’t lie just in the genes; they also lie in the expression of those genes, many of which, particularly for the immune system, are dissimilar. This was shown in a study looking at inflammatory disease, in which the expression of genes between human and mice didn’t match up. These differences had clinical consequences: Of 150 drugs developed in mice to treat sepsis, not one was effective in human trials.
If only there was an animal model that combined the ease and cost of the mouse model with the clinical relevance of the monkey model. As no example existed in nature, researchers made their own: the humanized mouse model. In simple terms, this model takes a mouse genetically altered to have no immune system of its own and transplants human cells or tissues into the animal. Because the mouse has no immune system, it can’t reject the human tissue. Instead, the human cells multiply, forming a stable human immune system inside the mouse.
This model is sometimes made using stem cells. The human stem cells home to the mouse bone marrow and take up residence. From here, they form all the cells of the human immune system, creating elaborate tissue networks throughout the blood, kidney, liver, thymus, gut, lymph nodes, and even brain. In some mice, they create a tissue where none existed before. In mice that have no thymus, for example, stem cells can form the organ themselves, a tight cluster of brand-new human tissue that pumps out mature T cells.
As you can imagine, these mice are fragile. Several debilitating mutations give them the ability to be engrafted with human cells. A typical mutation is SCID, more commonly known as the boy-in-the-bubble disease. Babies born with this disease have no functioning immune system and usually die in their first year of life. Those who do survive are kept in sterile conditions. The same rules apply for mice with this disease; they must be kept in sterile conditions and treated gently.
So when Carl June wanted to test the CCR5-cutting ZFNs he got from Sangamo, he decided to use humanized mice.
June and his team took human commander T cells and treated them with Sangamo’s CCR5-cutting ZFNs. He then expanded the T cells in culture and transplanted them into a humanized mouse model. When he challenged the mice by infecting them with HIV, he found a new world of natural selection was happening inside the mice. The HIV was killing off the T cells untouched by the ZFNs, while the T cells treated with the ZFNs survived. The efficiency took a giant leap, from only about 10 percent of the CCR5 gene being knocked out, to over 50 percent. Finally, here were reasonable numbers to fight off the virus.
After a month of HIV infection, mice that received cells treated with ZFNs had much lower levels of virus compared to those receiving the control cells. The average viral load in the treated population was 8,300 copies per milliliter, while the controls had 60,100 copies per milliliter. In addition, the CD4+ T cells were substantially higher in those mice that received the unique treatment. All signs pointed to ZFNs as a new gene therapy for HIV.
From his work developing gene therapies, June knew what he needed to do to bring a new drug into clinical trials. But what he found was that the systems for funding cancer and HIV were quite different. It wasn’t easy to convince funding and regulatory agencies to take the leap of faith on such an unconventional therapy to treat HIV. At an NIH RAC (Recombinant DNA Advisory Committee) meeting, he remembers the challenge of convincing the group that they could find the right patient population. However, once news broke of the Berlin patient Timothy Brown, attitudes began to change. Suddenly, there was a real-life example of how changing someone’s genes could cure HIV. Gene therapy had gotten a shot in the arm from an unlikely source. June calls Timothy’s story the “tipping point,” saying, “After Tim, you could talk about gene therapy in public. You could get funding for it.” The timing couldn’t have been better. The news had come just as June was trying to organize the first clinical trial to use CCR5 ZFNs as a therapy for HIV. Of Timothy’s story, he says that if “you define a miracle as being a very rare event it was certainly that.”
June’s CCR5 ZFN clinical trial began in 2009. The Phase I clinical trial tested the experimental therapy in several different treatment groups. The first group, or cohort, was six patients who had failed two different drug regimens. Here was a group of HIV-positive people who were in need; antiviral drugs weren’t working for them. Perhaps a gene therapy could swoop in and save the day. This group would receive a single dose of 5–30 billion of their own T cells that had been modified by the ZFNs to no longer express CCR5. Hopefully, once reinfused in the body, those cells would have a selective advantage in the face of HIV and, like Timothy’s cells, keep the virus at bay.
The second cohort of patients would be more typical of those infected with HIV. This group of six people would be on normal suppressive therapy. Unlike the first cohort, they were doing fine on antiviral drugs. They, too, would receive 5–30 billion of their own cells engineered to be genetically capable of fighting off HIV. However, this group would experience a treatment interruption, a twelve-week period when they would stop taking their drugs. The idea here was that, for the gene therapy to work, the researchers had to put genetic pressure on the virus. Just as the mice experienced a rise in genetically modified cells only in the face of the virus, the same conditions had to be replicated in humans in order to see any effect. They had to create the right environment for selective pressure, and that meant they needed the virus.
The third cohort of patients was on normal suppressive therapy, but, although the drugs had banished their virus, it had not brought their T cells back to optimal levels. This group of six would be given the same dose of their engineered cells but, unlike the second cohort, they would not be put to the test by stopping their therapy. It was too risky in a group like this.
The 18 patients came to the clinic twice to have their T cells extracted, a harmless procedure, similar to a regular blood draw. They also had a rectal biopsy performed both before and after therapy to assess whether the engineered cells would make their way to their tissues. Five weeks after they first came in to give blood, they would receive the new and improved T cells, infused back into their veins. All the patients would be closely monitored. Four weeks after receiving the infusion, the second cohort would stop taking antiviral drugs for twelve weeks, a treatment interruption designed to give the engineered cells a selective advantage.
This clinical trial, as it was the first to use ZFNs, was primarily designed to assess the safety of the new technology, not to test the efficacy of the gene therapy. For this reason, participants wouldn’t be completely stopping their antiviral drugs, the only way to fully test the gene therapy. June and his group found that the ZFNs were safe; the gene-editing machines didn’t target any genes they weren’t supposed to and didn’t cause any adverse reactions. In addition, the engineered cells had made their way to the mucosa in the gut, an important part of any therapy attempting to cure HIV.
When it came to treating HIV, the results mimicked the findings in the humanized mouse model. The cells that were vulnerable to HIV were killed. The cells modified by the ZFNs to be resistant to HIV survived. This was similar to Timothy’s case, in which the virus killed off his cells but spared the new ones that didn’t express CCR5. It was also similar to the mice, where the presence of HIV was needed to expand the genetically modified cells. When the modified cells expanded, they were able to reduce virus and boost T cells. These results were seen only in the patients who temporarily stopped taking their drugs, the second cohort. This is probably because the gene therapy needs the selective pressure of the virus itself in order to exert control over it. The point is made more apparent in the case of one member of June’s clinical trial, who, like Timothy, is hetrozygous for the Δ32 mutation with one functional copy of CCR5 and one copy that doesn’t work. This man, called the Trenton patient, already had a leg up, genetically. He was able to completely control the virus without antiviral therapy. Like Timothy, his CCR5-negative cells controlled HIV. Unlike Timothy, he’d been given this advantage from a gene therapy.
The findings were unprecedented, the first successful gene therapy trial for HIV, prompting June to use the once forbidden c-word, when he said, “The data obtained in our treatment interruption studies are very exciting and represent significant progress toward a ‘functional cure’ for HIV/AIDS.” A gene therapy like the one June did is not without risk. However, this type of therapy presents fewer fiscal and medical challenges than a bone marrow transplant. The cost of this approach is approximately $300,000 less than the lifetime cost of taking antiviral drugs. Because it involves manipulation of one’s own cells, this type of procedure can be performed safely in a clinic.
June is excited about the future of this study. The next step is obvious; June knows they have to take patients off antiviral drugs. The more engineered cells a patient has, the better he does at controlling the virus on his own. The only way to get more engineered cells is to lengthen the time the patients are off therapy, with the possibility of ending therapy altogether. The patients for this Phase II clinical trial, the first with the real possibility of curing HIV through gene therapy, enrolled in 2013.
June sees his cancer and HIV clinical trials as constantly informing each other; he sees the data from his diverging trials as a “cross-pollination and fertilization of ideas.” Despite this, he says, cancer and HIV trials are not treated the same; it’s far more difficult to get approval and funding for new HIV therapies. June says this is the “best and worst time to be in science.” It’s the best because of how promising the science is, and the worst because of the lack of funding to pursue that science.
In this time of shrinking funding for science, June worries about the investment needed to translate his promising clinical trial results into a tangible therapy for HIV-positive people everywhere. June has noticed a lack of motivation from big pharma, the typical powerhouse that brings new therapies to market. At a time when pharmaceutical companies are bringing in considerable profits from their portfolio of antiviral drugs capable of keeping HIV at bay, there is little drive to make the larger investment needed to bring a cure for the disease to the marketplace.
June is optimistic. He believes that “attitudes can change on a dime with a few successful patients” and thinks he has those patients. He hopes that private investors will take the first leap of faith needed to bring the therapy to a wider group of patients. Once this happens, June reasons that the pharmaceutical industry will follow. Sadly, he knows that the NIH, the government source of medical research funding, simply doesn’t have the budget to invest in a cure for HIV. The question remains: We can cure HIV, but will anyone fund it?
In 2012, Timothy visited June’s lab at the University of Pennsylvania. On a wall of the lab, Timothy’s picture is displayed. June says it’s “almost like a religious thing.” He sees Timothy as “raised from the dead.” As Timothy moved about the maze of lab benches and tissue culture rooms, his very presence seemed to inspire the students and technicians around him. In June’s words, “An n=1 is an incredible thing.” In science, n=x, means the number (n) of participants (x) in a study; in a field driven by data, the higher the number of participants, the more convincing the results. But Timothy’s case is the exception. The power of his story rises above statistical significance. Scientists are only human. Sometimes the sway of a great story is as powerful as the most comprehensive dataset. We can’t underestimate the impact a story has on the course of science.