Billions of dollars are spent every year by companies trying to create new drugs to treat human diseases. They hope to find ways to tackle unmet medical needs, a situation that is becoming ever more urgent with the increasing age profile of the global population. The breakthroughs in the understanding of the impact of junk DNA on gene expression and disease progression are triggering a slew of new companies seeking to exploit this field. Specifically, most of the new efforts are in using non-protein-coding RNAs as drugs in themselves. The basic premise is that junk RNA – long non-coding, smallRNAs or another form called antisense – will be given to patients, to influence gene expression and control or cure disease.
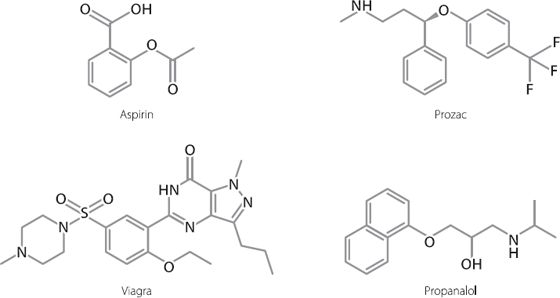
This is very different from the way we treat diseases at the moment. Historically, most drugs have been of a type known as small molecules. These are chemically created and are relatively simple in shape. Examples of some common small molecule drugs are shown in Figure 19.1.
More recently, we have learnt how to use proteins as drugs. Probably the most famous is insulin, the hormone that diabetics use to regulate their blood sugar levels. Antibodies are another very successful type of protein drug. These are engineered versions of the molecules we all produce to fight infections. Drug companies have found ways of adapting these so that they will bind to over-expressed proteins and neutralise their activities. The bestselling antibody is one that treats rheumatoid arthritis very effectively, but there are others that treat conditions as diverse as breast cancer and blindness.1
Figure 19.1 Structures of some commonly used small molecule drugs.
Small molecules and antibodies have advantages and drawbacks. Small molecules are usually relatively cheap to synthesise and easy to administer, frequently just needing to be swallowed. Their drawback is that they don’t last very long in the body, which is why we need to take them on a regular basis. Antibodies can last for weeks or even months in the body, but they have to be injected by a medical professional, and they are very expensive to manufacture.
There are some other drawbacks too. Antibodies are only effective against molecules that are in body fluids such as blood, or are on the surface of cells. These drugs can’t get inside cells to do their work. Depending on their structure, small molecules can get inside cells if necessary. But they may be limited in the kinds of proteins that they can control.
Small molecules act like a key in a lock. If you are inside your house, the easiest way to stop someone coming in is to lock your door and leave the key in it. If you wanted to stop anyone else from ever entering, you could even lock the door using a slightly defective key, which jams the lock for ever.
This works because the key fits into the lock really snugly. But what you can’t do is use a key to block one of those old-fashioned external sliding bolts. There is nowhere for the key to fit in this, it will just keep slipping around on the surface. This is also true of our cells. There are lots of proteins inside our cells that we would like to control but we can’t create small molecules against them, because of the protein structure. They don’t have nice neat clefts or pockets that we can fit drugs into. Instead, they have large flat surfaces, and there is nowhere for a small molecule to lodge.
We could try to make bigger molecules that can cover the whole flat surface. The problem with this is that once we get above a certain size with our drugs they don’t circulate well around the body, and they can’t get into the cells to do their job.
There’s also another problem. It’s hard enough to create drugs that will successfully get inside cells, bind to a specific protein, and stop that protein from working. But it’s incredibly difficult to create drugs that will get inside cells, bind to a specific protein and then make that protein work harder, or faster, or better. And it’s practically impossible to make traditional drugs that will drive up the expression of one specific protein, or switch on one and only one gene.
Could junk DNA save us?
This is why there is so much interest in finding new approaches to drug therapies, and why the increasing knowledge of junk DNA is so important. By using long non-coding RNAs or smallRNAs, it is theoretically possible to target pathways that can’t be tackled using traditional small molecule or antibody drugs. It won’t matter that the targets are inside cells and have large flat surfaces. It won’t matter that we need to increase expression or activity of a protein or gene. We can use this new approach to tackle any type of target.
Theoretically.
That’s the word to focus on. Theoretically. Ideas are common, success is rare. So it’s worth taking a good look at where the reality is before we all cash in our pensions to invest in the latest biotech company working in this space. There is a lot of activity going on,2 so it’s worth concentrating our analysis on a few leading examples.
There is a protein produced in the liver that is responsible for transporting some other molecules around the body. Globally, there are about 50,000 people who have inherited a mutation in the gene that produces this protein. There are lots of different mutations, but they all seem to have a similar effect. They all change the activity of the protein so that it starts to transport the wrong molecules.*3
When this happens, deposits, which include a mixture of normal and mutated protein, begin to build up in tissues. Patients may have a range of symptoms, depending on the tissues in which the deposits build up. In about 80 per cent of known cases, the heart is the main organ that is affected, and this leads to potentially lethal cardiac defects. In many of the other 20 per cent of cases, the deposits build up in the nerves and spinal cord. This can lead to debilitating problems with a range of organs, including abnormal and painful sensory responses to mild stimuli.
A company called Alnylam has created a smallRNA, attached to sugar molecules, which can be injected into patients. The small-RNA binds to the untranslated region at the end of the messenger RNA that codes for the protein that is mutated in this disease. This targets the messenger RNA for destruction.
In 2013 the company released data from a phase II clinical study of their drug. They found that when they injected the drug into patients, there was a rapid and sustained drop in the circulating levels of the mutated and normal versions of the protein.4 This is encouraging, but not yet a cure. The assumption is that a drop in circulating levels will lead to a slowdown in the build-up of tissue deposits. This in turn should lead to at least a slowdown in the progress of the disease. But we won’t know if that is the case until a bigger trial is carried out, in which the actual symptoms and disease progression are monitored. Only if the drug impacts on these will it be considered a success.
A different company, called Mirna Therapeutics, has created a smallRNA which mimics one known to be important in cancer. The endogenous smallRNA is a tumour suppressor, and its overall effect is to hold back cell proliferation. It does this by negatively regulating the expression of at least twenty other genes that try to push the cells into division. Expression of this smallRNA is often lost or decreased in cancer patients, removing the brakes on cell division. The hope is that by reintroducing it into cells, the normal pattern of gene regulation will be restored, and the cells will stop proliferating so quickly.
The company has tested their mimic in patients with liver cancer. So far the trials have just been designed to see what doses of the drug the patients are able to tolerate. It will be some time before we will know if this approach is going to result in clinical benefit.5
There is a clever, although not immediately obvious, angle to the products being developed by both Alnylam and Mirna. One of the biggest problems that companies have faced in the past when trying to develop drugs around nucleic acids has been the body’s own detoxification abilities. This is also often a problem for traditional drug discovery as well. Essentially, when a new chemical of any type enters the body, there is a very high likelihood that it will go to the liver. One of the main jobs of this vastly energetic organ is to detoxify anything it doesn’t like the look of. For all of our evolutionary history, this has served us well, protecting us from toxins in food. But the problem is that the liver has no means of distinguishing between toxins we want to avoid, and drugs we are trying to use. It will just drag them in, and try to destroy them.
To use an old rubric, Alnylam and Mirna are making a virtue from a necessity. Alnylam is targeting expression of a protein that is produced in the liver. Mirna is trying to develop treatments for liver cancer. Their molecules will be taken up by exactly the organ they want them to reach. The companies have adapted the structure or packaging of their molecules to try to ensure that once they are in the liver, they will survive long enough in the cells to do their job. SmallRNA approaches have been put forward for a number of other conditions, and the preliminary cellular and animal experiments often look good. But for a condition such as amyotrophic lateral sclerosis, where the nucleic acids will have to avoid the liver and be taken up by the brain,6 it’s not clear yet how successful the industry will be in capitalising on this technology.
In Chapter 17, we saw how hopes of a promising new approach to treat Duchenne muscular dystrophy may be receding, after unexpectedly disappointing late-stage clinical trial failure. The methodology used in this approach was an example of a particular kind of junk DNA, known as antisense.
Antisense junk RNAs are probably a widespread feature of our genome, and it’s because of the double-stranded nature of DNA. We touched on this in Chapter 7, where the actual biological example we used was of Xist and its antisense counterpart, Tsix. We also used the analogy of the word DEER, which can be read backwards as the word REED. It just depends if the enzymes that make RNA copies of DNA reads one strand from left to right, or the opposite strand from right to left.
However, most words can’t be read in both directions. If we read the word BIOLOGY backwards, we get YGOLOIB, which doesn’t have a meaning. In the same way, messenger RNA from one direction in the genome may code for a protein, but the same region copied backwards simply codes for a junk RNA that cannot be translated into a protein. Sometimes this creates auto-regulatory loops in our cells, limiting expression of certain genes. An example of this is shown in Figure 19.2.
Researchers have reported that about a third of protein-coding genes also produce junk RNA from the antisense strand. However, the antisense is usually produced at lower levels, often no more than 10 per cent.7 Sometimes the antisense is just a short internal section of the gene. Other times the sense and antisense may start and end in different places so that they overlap but also have unique regions. Sometimes the machinery copying the sense DNA strand into sense RNA crashes into the machinery moving in the other direction to create the antisense RNA. Both sets of proteins fall off the DNA, and both RNA molecules are abandoned. There are even antisense strands for some long non-coding RNAs.

Figure 19.2 In some parts of the genome, both strands of DNA can be copied into RNA, in opposite directions. These are known as sense (creating RNAs that code for protein sequences) and antisense (which don’t code for protein sequences). The antisense RNA molecule can bind to the sense RNA molecule and affect its activity, in this example by inhibiting production of protein from the sense messenger RNA template.
The effects of an antisense RNA binding to its sense RNA partner can vary. Figure 19.2 shows an example where this binding prevents the sense messenger RNA from being translated into protein. But there are other situations where the binding stabilises the messenger RNA, ultimately leading to higher protein expression.8
In the Duchenne muscular dystrophy trials that originally held such promise, the patients were treated with an antisense molecule that could recognise and bind to messenger RNA for dystrophin. The antisense molecule was chemically modified to prevent it from being broken down too quickly in the body. When the antisense molecule bound to the dystrophin messenger RNA, it prevented the splicing machinery from binding in the normal way. This altered the way the messenger RNA was spliced together, and got rid of the region that caused the most problems in mutant protein production.
There are some happy endings
The Duchenne muscular dystrophy trial ultimately failed but we shouldn’t take this as meaning the entire antisense field is tainted. In fact, it’s had its successes. In 1998 an antisense drug was licensed for use in immunocompromised patients who had developed a viral infection in the retina* that threatened their sight. The antisense molecule bound to a viral gene, and prevented the virus from reproducing.9 It was an effective drug, which raises two questions. Why did this drug work so well? And given that it worked so well, why did the manufacturer stop selling it in 2004?
Both answers are quite straightforward. The drug worked well because it was injected straight into the eye. There was never a problem about it being scooped up by the liver, because it didn’t go via the liver. It was also targeting a virus, and only in one selfcontained part of the body, so there wasn’t much risk of widespread interference with human genes.
All of which sounds peachy, so why did the manufacturer stop selling it in 2004? This drug was developed for severely immunocompromised patients, of whom the vast majority were people suffering from AIDS. By 2004, there were drugs available that were pretty good at keeping HIV, the causative virus, under control. The patients’ immune systems were in much better shape, and they simply weren’t succumbing to viral infections in the retina anymore.
More recent developments have also shown that there is still life in the use of antisense junk DNA for therapy. There is a serious condition called familial hypercholesterolaemia. In the UK it is predicted that there are about 120,000 people with this disorder, although many of them may not have been diagnosed. These people have genetic mutations that prevent their cells from taking up bad cholesterol and dealing with it properly. As a consequence, between a third and half of all such patients will have serious coronary artery disease by their mid-50s.10
For some patients with this condition the standard lipidlowering drugs, known as statins, work really well to lower their risk of cardiovascular disease. This is often the case for people who have one mutant copy of a particular gene, but in whom the other matching copy is normal. But there are some severely affected patients, especially those in whom both copies of the specific gene are mutated, for whom statins are ineffective. These patients often have to undergo plasmapheresis once or twice a week, where their blood is passed through a machine and the dangerous cholesterol is removed.
If you want to stop a bathtub from overflowing, you have two options. You can keep letting water out via the drain, or you can turn down the taps to stop adding more water.
A company called Isis developed an antisense molecule which targets the primary protein in low-density lipoproteins, the so-called ‘bad cholesterol’.* This antisense therapy for familial hypercholesterolaemia works by turning off the taps. The antisense drug binds to the messenger RNA for the bad cholesterol protein and suppresses it, resulting in lower expression and lower levels of bad cholesterol. Isis licensed this to a larger company called Genzyme, in a deal costing hundreds of millions of dollars.
This antisense drug** was licensed for use by the US Food and Drug Administration in January 2013. It is only licensed for patients who suffer from the most severe form of familial hypercholesterolaemia. One of the reasons this drug has been successful enough to reach the market (albeit at the eye-watering cost of over $170,000 per year per patient11) is because the gene that it targets is expressed in – yes, you guessed it – the liver. A downside to this, however, is that there have been liver toxicities reported with the use of this drug. The Food and Drug Administration has demanded that Sanofi (who bought Genzyme) must monitor liver function of all patients.12 The European Medicines Agency refused to license the drug at all, citing safety concerns.13
The hundreds of millions of dollars that Isis received from Genzyme for its antisense therapy is a lot of money. Yet consider this. It took over twenty years to move from the basic research to a marketed drug, and the whole process cost over $3 billion.14 That’s an awfully big investment to recoup.
Of course, pioneering drugs, especially those which use a relatively untried type of molecule, would be expected to take a long time and a lot of money to develop. The hope is always that later programmes are able to run faster and more smoothly. Certainly, clinical trials for therapies based around junk DNA are building in number. There is a human smallRNA that is co-opted by a virus to help it infect cells. In an example of using junk to fight junk, an antisense drug is in phase II clinical trials, targeting this smallRNA.15
But here’s an odd thing to consider. In 2006 the pharmaceutical giant Merck paid over a billion dollars for a company that was developing smallRNAs as therapeutics. In 2014 it sold the company on, for a fraction of what it paid.16 Another company, Roche, stopped its own efforts in this research area in 2010.
There has recently been a big upsurge in investment into biotech companies working on smallRNAs. RaNA Therapeutics, which is believed to be developing RNA-based drugs that will prevent the interaction of long non-coding RNAs with the epigenetic machinery, raised over $20 million in 2012.17 Dicerna, which is developing smallRNAs against some rare diseases and oncology indications, raised $90 million in 2014.18 That’s the third set of financing it has received, despite having no programmes that have reached clinical trials yet.19
Yet here’s the weird thing. Literally as I write this chapter, in spring 2014, an alert comes up in my email account and tells me that Novartis has decided to slow down dramatically its research on this topic.20 The pharma giant mainly cited the ongoing problems with working out how to deliver smallRNAs to the right tissues. This has been the biggest issue with these therapeutics since companies first started trying to develop them. Many of the companies in the junk RNA field have been set up by brilliant scientists, but that doesn’t mean that any of the basic drug delivery problems will just disappear overnight. Not all the companies will fail. But quite a lot of them probably will. There haven’t been any major breakthroughs on this problem, and certainly nothing that would explain why investors are pouring money into new biotechs in this area.
One day science will probably be able to interpret all the possible epigenetic modifications that are found in the genome and predict precisely what their consequences will be for gene expression. We’ll work out how to capture carbon, and how to establish colonies on Mars. Tuberculosis will be a distant memory and we’ll all have a good grasp of the Higgs boson. But unravelling the reasons behind the triumph of hope over experience in the investment community? Be realistic.