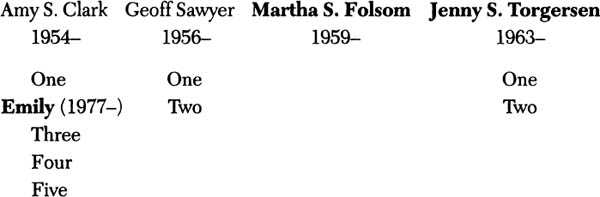
There are two likely explanations for why antibiotic therapy has proven so effective in treating the rheumatoid forms of arthritis and other connective tissue diseases. The more obvious rationale is Dr. Brown’s still very plausible hypothesis that an infectious agent is fundamental to the onset of the disease, and perhaps even to its chronicity. But Dr. Brown was also aware that that hypothesis probably did not stand alone, and today it is coupled in all ongoing research with the fact that antibiotic therapy is also anti-inflammatory, the latter effect being independent of any antimicrobial activity. Several years ago, after reading The Road Back, Dr. Rothschild wrote, “This is the first time that I realized the tetracycline was being provided for its long-term impact. As tetracycline also has immunomodulatory effects, this is quite fascinating.”
A number of the current therapies that are generally characterized as anti-inflammatory drugs, including steroids, methotrexate, gold, and all the standard agents that have migrated into the field of rheumatology, are equally mysterious in the way they achieved their effects. “We just don’t know how they work,” Dr. Trentham says. “They work partially in some patients; we can’t predict in which patients they have this partial effect. Obviously they still are too toxic to be highly popular, and because of that toxicity we’re still seeking alternative therapies. That’s a lot of the rationale for further evaluating antibiotic therapy, because of its inherent safety, at least with the tetracycline drugs.”
Meanwhile, Dr. Trentham defended the principle of using other drugs, even when it is obvious they won’t affect an infectious agent, simply because they might still help the patient overall. He offered the analogy of the patient with pneumonia. It’s usually a type of bacteria that causes the disease, and doctors can effectively cure pneumonia by giving penicillin, an antibiotic. But regardless of whether the patient is treated with an antibiotic at the outset, it’s well established that giving aspirin will at least make the patient feel better; it doesn’t get to the heart of the matter, but it still provides a benefit.
Like Tom Brown, Dr. Trentham uses cortisone very sparingly and only to the extent that it eases suffering and increases the patient’s tolerance for the sometimes long period of waiting for the antibiotic therapy to show its results.
“I think there is a lot of truth in Dr. Brown’s feeling that the use of steroids, overall, is not a good approach to the disease. They buy short-term efficacy, but longer exposure does lead to deleterious side effects that he was well aware of. It’s a two-edged blade that we all have to play with, but he provided a real service in voicing his concern and caution.I think that today all rheumatologists appreciate that concept.”
Collectively, most of those second-line agents Dr. Trentham cited are called DMARDs, an acronym for disease modifying anti-rheumatic drug; another name is slow-acting drug. With any of them, but particularly with the old standard gold, rheumatologists realized it could take six months for patients to respond, if they were going to respond at all, and during that time it was commonplace, when patients were suffering acute pain, to give them a short course of steroids for immediate help. The dosage was kept as low as possible; for most rheumatologists, the cutoff would be 10 milligrams or less of prednisone a day. However, that kind of restraint was still hardly a universal standard. As recently as 1990, a lupus sufferer in New York lost his sight to cataracts after being treated for months with prednisone in dosage levels of twenty times that amount.
Another development in the early ’90s was the growing body of new evidence about the potential role of heredity. Tom Brown had recognized the strong hereditary predisposition to the disease, and from time to time all rheumatologists encounter striking instances in which rheumatoid arthritis permeates the family—although today most rheumatologists accept Dr. Brown’s thesis that this predisposition is not the cause per se. Trentham chronicled one such case from his own experience.
Robert and Nancy Sawyer lived in Greenfield, Massachusetts, and by late 1992 Nancy, a travel agent, had been Trentham’s patient for fifteen years. He described the entire family as being highly intelligent, motivated people.
Nancy’s mother, Helen Caldwell Thayer, was born in 1894. Her husband was a doctor and, although there was always a reluctance to say she had rheumatoid arthritis, surviving family members recalled that she suffered from some form of joint disease most of her adult life. Nancy was born in 1928 and suffered from severe rheumatoid arthritis since the late 1970s, when she was almost fifty. They have four children: Amy Clark, born in 1954; Geoff, born in 1956; Martha Folsom, born in 1959; and Jenny Torgersen, born in 1963. Amy has been spared the disease, but one of her five children, Emily, was diagnosed with juvenile rheumatoid arthritis in 1989. At the time Mrs. Sawyer offered the family’s medical history, Emily was thirteen and she had only been treated with aspirin. The disease wasn’t at a point where her doctor wanted to risk stirring things up by using anything stronger. Nancy’s son, Geoff, had been spared as well, as had his two children. Martha had rheumatoid arthritis, but no children. Jenny had it as well, although her children so far did not.
Family members diagnosed with (or suspected of having) RA in bold
Helen Caldwell Thayer
1894–1954
Nancy Thayer Sawyer
1928–
“Of course,” Mrs. Sawyer cautioned, “it would be premature to say the picture is complete: rheumatoid arthritis waited fifty years in my case, and it could take as long for any of the others. But we know it has been present in at least three generations of the family, and probably four.”
Given that hereditary liability, what happens next is still the subject of speculation and debate. One theory is that a bacterial or other infectious agent comes into the host and, because a portion of that organism has a shape similar to a tissue constituent in the joint the body first mounts an immune attack on the foreign organism that may or may not get rid of it. The attack is targeted to the structure of the invading organism, and because the structure mimics or is identical to perhaps a host protein or tissue constituent, it may be that there is then a secondary response directed against the body. This is the so-called auto-immune attack, or anti-self reactivity, in which the nature of the process turns inward and attacks the patient in a pattern that propagates rheumatoid arthritis. The theory is termed molecular mimicry. Many of these infectious agents are quite old phylogenetically, going back many millennia. These ancient, highly conserved organisms arose at a time in evolution when their structure was very close to the structure of other living organisms, including mammals.
If indeed rheumatoid arthritis is caused by such an infectious agent, there are several promising candidates. One is a tuberculosis-like organism, a mycobacterial species that still hasn’t been identified but is suspected to be a cousin to the bug that causes tuberculosis. Another strong possibility is that it could be a virus, perhaps some very primitive type of retrovirus that can hide itself completely—or it could be similar to the virus that causes AIDS, which again would affect a certain cell, the T cell, and initially remain very quiet and elusive of detection. Or it still could be a mycoplasmal organism, as Tom Brown suspected. Others now suspect mycoplasmas as well. Up until very recently, however, despite Tom Brown’s pioneering efforts at the Rockefeller Institute sixty years ago, mycoplasmas remained amazingly elusive to scrutiny. The wall would crack dramatically in 1995, with the breakthrough in Bordeaux (see “Evidence Accumulated,” page 24).
Although mycobacteria, viruses, and mycoplasmas are the three leading infectious suspects, a fourth possibility may be nothing more complex than a fairly common bacterial invader of the sort we’re all exposed to from time to time, such as a streptococcus, the organism that causes sore throats and still occasionally rheumatic fever. If that’s the cause, then it would appear that only in people who are allergic or sensitive to the organism, based on their hereditary makeup, does the process go on to develop rheumatoid arthritis.
Another feature of what many physicians call the second phase of this disease, frequently identified as the “auto-immune” phase, is an attack on the joints by the body’s white blood cells. The original invader may or may not be present when the attack takes place, but most scientists are now convinced that the type of cell that’s producing the problem in rheumatoid arthritis is indeed the T cell mentioned earlier, and it seems to be the very same T cell that becomes infected in AIDS.
The natural role of the T cell is to kill foreign cells or those that have become infected by virus. It does this by helping other cells respond to antigens, or by suppressing the activity of specific populations of cells. The role of T cells in these two diseases became dramatically apparent in those cases where a patient with rheumatoid arthritis also contracts AIDS, and the arthritis simply disappears. Conversely, in patients who already have active AIDS, their bodies are not able to generate sufficient numbers of these T cells to develop rheumatoid arthritis. This phenomenon has been under intense investigation for the better part of a decade and is now recognized throughout the world. By the time of the MIRA trial not a single case of active rheumatoid arthritis had been reported in an AIDS patient, nor had an AIDS patient been known to develop any form of the disease.
“All of this background fits very nicely into the context of Dr. Brown’s thinking, and from that standpoint he really was quite provocative and innovative at the time,” Dr. Trentham said. “He took it as far as he could, given the existing knowledge. AIDS did not exist when he developed his theory. There was a little bit of evidence that rheumatoid arthritis was a tuberculosis-like disease, and certainly there was a great interest in infectious-disease candidates such as mycoplasma and streptococci that continues today. But this is not a theory that died and now may be coming back to life again; it’s really a logical continuum in the evolution of Dr. Brown’s thinking.”
Although Dr. Trentham and his coworkers were among the first to recognize the importance of the T cell in rheumatoid arthritis, he is quick to credit many other researchers for equally pivotal roles in the research that eventually produced the shift back toward the infectious theory.
The first true pioneer in the field was Harold Paulus, the UCLA rheumatologist whose editorial in Annals of Internal Medicine would introduce the MIRA study in 1995. Back in the 1960s, Paulus was the first person to hook up patients with rheumatoid arthritis to a plastic tube that went into their thoracic duct, the structure in the chest where lymph first enters the blood to commence its trip throughout the body. The major cell constituent in lymph is the T cell. By eliminating these cells in the thoracic duct, Paulus made the striking observation that patients with rheumatoid arthritis would get remarkably better within two or three days after starting this intervention, and they stayed better as long as this tube continued to drain their lymph.
This avenue of research received another major boost some years later when Dr. van Boxel of the NIH first reported that almost all the cells in the joints in rheumatoid arthritis were T cells.
In about 1981, a group at Harvard headed by Trentham and another group at Stanford Medical School led by Dr. Samuel Strober, both showed that one could treat rheumatoid arthritis using a radiation therapy technique identical to that developed to combat Hodgkin’s disease, a lymphatic cancer. Because Hodgkin’s also triggers the production of T cells, and since patients improved after radiation therapy, it became all the more plausible that the T cell was indeed central to the disease. This research in radiation therapy may also have given an unintended boost to the popularity of exposure to radon gas and visits to abandoned uranium mines, a folk remedy among arthritis sufferers in the Southwest. For many years, mild radiation has also been an accepted treatment for rheumatoid arthritis in the former Soviet Union.
The importance of the T cell was further reinforced when, in 1987, Dr. Trentham showed that an antibody against T cells, an anti-thymocyte globulin, seemed to alleviate rheumatoid arthritis. Shortly thereafter it was widely agreed that AIDS and rheumatoid arthritis were mutually exclusive diseases.
So that’s pretty much where matters stood up to the time of the MIRA study. There was general recognition of the important role played by the T cell, and continued mystery as to what specifically initiates the disease. Prior to the use of antibiotics, most therapies attempted to attack the disease by focusing on this aberrant immune response, provided mainly by T cells. This response can, to some degree, be held in check by certain conventional agents. Scientists were divided on how antibiotics worked, but they were equally in the dark about the mechanism of methotrexate, gold, or other drugs.
What is the connection between these discoveries and treatment that uses the minocycline form of tetracycline? How did they provide part of the rationale for its further study in the MIRA trials?
Minocycline may help patients with rheumatoid arthritis because it eliminates an infectious agent. Alternatively, arthritis research in animal systems has shown that minocycline affects calcium metabolism or pathways in T cells; and calcium pathways are a critical component of the ways in which T cells activate themselves or function. T cells exposed to minocycline take up too much calcium internally, and then become sick or malfunctional. They seem to quiet down, and the animals go into complete remission or are at least visibly improved by the therapy. This new information favors the possibility that some antibiotic drugs may be beneficial in rheumatoid arthritis because they eliminate these T cell autoimmune responses, and not just because they home in on an infectious agent.
A few years ago, a specific request was issued from NIH headquarters in Bethesda for grant applications that might relate to the infectious theory, and that has been cited as tangible evidence of NIH interest in Dr. Brown’s original thesis. Some reasoned the response was slow because many scientists felt at the time that applications would result in little more than a fishing trip. Even at the time of the MIRA trials, most arthritis researchers felt they were still working in the dark. Many of those interested in the disease lacked the aptitude, training, or credentials to explore viruses or related phenomena. Dr. Trentham suggested it would require a scientific consortium to attack the problem, but with the funding difficulties in the present economy it was not a particularly viable prospect.
In the meantime, he predicted, the next generation of research would take its direction from the NIH-sponsored clinical trials of minocycline therapy and other such clinical studies.
What has happened in the field of rheumatology since the direction has finally started to change from the dogma that inflammatory arthritis was a metabolic failure? It was an error that stagnated both scientific inquiry and clinical treatment for a long, long time.
In the preface to a special edition of Rheumatic Disease Clinics of North America devoted entirely to “New Directions in Antirheumatic Therapy,” Trentham wrote in 1992, “Rheumatology is at a crossroads. It is confronted by a precipitous decline in entrants into the subspecialty. Is the problem solely attributable to the modest economic remuneration inherent to the discipline? I think it is also reflective of past therapeutic patterns of the craft! . . . In the upcoming decades, a shift from dogma to diversity is likely to occur.” It went on to list some of the specific articles of past faith that now were being abandoned: “about immune complexes being the cause of autoimmune disease, about the effects of gold, about the role of rheumatoid factor in the etiology of the disease, about the notion that non-steroidal anti-inflammatory drugs (NSAIDs) were all alike, about scleroderma being inevitably fatal. . . .”
Another article he had written for the same publication titled “Rheumatologic therapy for the 1990s: Evolution or Revolution?” notes: “. . . the care of the patient with rheumatic disease in the upcoming decade can be easily perceived as the result of a therapeutic revolution as an orderly succession of new approaches.” It was hard to imagine a mainstream medical journal publishing any one of these heresies even as recently as five years earlier. Clearly, the shift was already well under way by the time of the MIRA study.
The drugs that had been considered the standards, even recently, for treating the disease were also being reassessed. A prime mover was a friend and colleague of Trentham at the University of California in San Francisco named Wally Epstein. Using novel epidemiologic methodology, Epstein demonstrated that gold therapy in rheumatoid arthritis actually does nothing to change the outlook for patients with rheumatoid arthritis over the long term. “Our feeling that gold has been so beneficial in the past has been actually incorrect,” Trentham said.
Of all the drugs Tom Brown had hated, gold was at the top of the list. If the patient dies of some other cause, then the fact that gold has temporarily masked the symptoms is beneficial. But most patients don’t die of some other cause.
Trentham said that Epstein’s work represented another change of direction that would be provocative and useful for patients to know about, and in many cases they would learn it from this book before they heard about it from their physicians. “All these people have a little bit of the maverick in them,” he said. “Yet Wally is a very well respected professor of medicine and has been in the field for years. He is currently taking some flak for his work, but it was published in one of the most respected journals, Annals of Internal Medicine, and he’s a first-rate person. It’s scientifically legitimate. It’s newsworthy. And it has patient-information value. So far he’s only dealt with gold; he should go to the others—it’s a natural.”
One of those other drugs, Trentham knew, was very likely to be methotrexate. He was one of the authors of the lead article when it was first reported in The New England Journal of Medicine as an arthritis treatment, and he had had as much experience as anyone in its prescription for connective tissue diseases. Despite the fact that its ancestry as a potent anticancer agent still frightened a lot of potential patients, Trentham pointed out that the levels used in arthritis are a small fraction of the dosage for cancer, and the side effects had, over time, proven to be minimal.
Unlike its use in cancer patients, methotrexate doesn’t function as a toxin in arthritis, and Trentham didn’t consider its Achilles heel to be toxicity. He viewed it as an appropriate drug for most people with severe rheumatoid arthritis, but not one that doctors should prescribe for mild cases. He also acknowledged that because it only works for two or three years there is a finite limitation in its usefulness.When methotrexate is no longer recommended, it isn’t because side effects kick in, but because arthritis is an amazingly adaptive disease: there’s a rewiring of the circuits, and the disease simply outsmarts it.
Another class of compound, the non-steroidal anti-inflammatory drugs, also entered the arthritis market relatively recently. Buoyed up by claims for safety as a low-risk alternative to prednisone, they soon were being consumed on a daily basis by some nine million arthritics in America alone. It turned out that some of those same people were dying each year from so-called “silent ulcers,” and in December of 1988, the Food and Drug Administration estimated the actual NSAID annual mortality rate was as high as 20,000. While a new drug, Cytotec, has apparently solved the ulcer problem and NSAIDs are reportedly much safer now, their debut into the market took a heavy toll.
In the years since I first met Tom Brown, I have been able to share, sometimes in intimate detail, in two sets of perspectives on what happens to people who have to function in an environment where progress is held hostage by culture. The first was that of Tom’s patients, who speak for themselves in the book; the second was the viewpoint of the doctors. Tom was considerably less revealing on this process; he was inclined to view his long struggle more as a phenomenon of nature than as something to be examined closely for the good or the bad in it. He was as clear-eyed as anyone I ever met, but I think he had decided long ago that introspection in adversity could be a handicap, and that it is far less productive to acknowledge personal pain than to be merely relentless. Furthermore, Tom Brown’s unique position in his profession was a deliberate choice—and liberating.
Tom hadn’t told me much about his own feelings, and I hoped Dr. Trentham might be more accessible. For all his quiet, even self-effacing style, he was clearly a maverick himself, and I suspected his career contained some useful clues on how to break out of the rut of dogma
He said he became involved in his current work out of serendipity. He was trying to make antibodies against joint tissues and discovered that the process re-created rheumatoid arthritis in animals. That was to become his central focus through the years: the possibility that the aberrant immune response is directed to a collagen or cartilage protein.
David Trentham went to college at the University of Tennessee and then stayed there through medical school. He had almost no money, but the school had a fast track program and he was able to become a doctor by the time he was twenty-three, which he acknowledged “is a little unusual.” Tennessee had an advanced placement plan in the college and he combined that with the honors program to get through in three years, then did medical school in three as well.
Since 1974 he had been studying rheumatoid arthritis, initially from the viewpoint that collagen is a pivotal auto-antigen in the disease. His research in that area started in Tennessee, but he wanted to look deeper, and that led him to Harvard in 1976. “I came because I felt I could really be in a hotbed of science, that there was a critical mass here.” Initially, he joined the staff of the Peter Bent Brigham Hospital, at that time the only free-standing arthritis hospital in the country. There was a lot of science going on there as well, and he continued his research at Brigham for the next ten years.
Along the way, he became interested in learning—assuming the crucial influence in rheumatoid arthritis was collagen activity—the mechanism to which it was subservient. That led him into the field of T cells and involvement in a number of therapeutic trials, which, in turn, brought him to his present interest in minocycline. Trentham’s work with T cells in rheumatoid arthritis pre-dated T cell theory in AIDS research by several years. Largely based on the work Paulus had done with thoracic duct drainage and Boxel’s observations, it began in 1976 and 1977.
Trentham got a fair share of the right kind of encouragement. Larry Shulman at the NIH and Claude Bennett, chief of medicine at the University of Alabama, supplied the link between antibiotic therapy and rheumatoid arthritis. Back in the ’60s and early ’70s, when he was very active as an investigator, Bennett had worked on mycoplasma and rheumatoid arthritis. Bennett and Shulman were both extremely interested in antibiotics and prodded Trentham to take a closer look in that direction.
For all the similarities in their approach to the disease, Trentham was largely unaware of Thomas McPherson Brown. Back in 1975, Trentham’s knowledge of Brown’s work was at best peripheral, limited almost entirely to disparaging remarks by other colleagues. “The fact is, I really felt he must have been some kind of a crazy, a charlatan, and what he was doing at that point was of no interest to me whatsoever.”
Shulman and Bennett urged him to submit a grant application to the NIH. They felt the whole field needed further inquiry. They informed Trentham of Larry Golub, a dental researcher at SUNY in Stony Brook, whose research proved that minocycline was a calcium chelator (a binding agent that suppresses chemical activity), which, in tests, suppressed some deleterious enzyme release in collagenates. Collagenase is known to the laity as a meat tenderizer, and it is probable that this tendency for breaking down tissue is, in part, responsible for the destruction of the joints in rheumatoid arthritis patients.
Trentham reasoned that Golub’s findings helped to explain scattered anecdotal claims that minocycline was useful in treating rheumatoid arthritis. He sought out a few of the authors of these reports, like Lee Bartholomew, who at that time was at the medical college at Albany and by the time of the MIRA trials was semiretired in upstate New York. Bartholomew also had studied mycoplasma in rheumatoid arthritis and published a paper in about 1965 on isolating it from synovial fluid. When he tried again he couldn’t reproduce his results, however, and he began to have doubts, principally that his first success might have actually been a contaminant. But Bartholomew still felt there was something there, and he, too, encouraged Trentham to take a further look into the area.
Up to then, Trentham had been unwilling to explore the infectious theory in animals because he thought the model would be so distant from humans that the information would not be extrapolatable, and he also felt he lacked the training or background to pursue infectious agents himself. But in combination, the new incentives proved irresistible. He was excited about the possibility that collagen reactivity appeared to play an important role in the disease; he had reason to believe that minocycline might work in a way that hadn’t been explored before, and he had been interested in T cells for over a decade. A study of animal models of rheumatoid arthritis produced by collagen, determining whether minocycline really produced the benefits reported in the anecdotes, could have great predictive value for human treatment. Along the way, he would focus on whether minocycline also influenced T cells in the same model. In 1968 he was awarded a grant from the NIH for three years.
Clarence Kassens
of Colville, Wash., will be eighty on his next birthday. He suffered from rheumatoid arthritis for twenty years, but after an acute flareup he heard of antibiotic therapy in 1992.

Since then, I have felt very good—literally have not even had a cold or a sniffle. I am exercising regularly and my strength and endurance are improving. The permanent damage to my right hip socket is still there, so I no longer do any jogging. However, I am still bicycling and hiking with a moderate pack. I feel very fortunate in being able to get started on the right treatment. If it had not been for this, by now I would probably be crippled or bedridden.
It did everything he had hoped for. He showed that tetracyclines are effective in animals, just as Tom Brown had demonstrated when he had used the same antibiotics to cure Tomoka, the gorilla at the National Zoo. He also identified the changes in calcium metabolism that might explain the event.
David Trentham was forty-seven years old when the code was broken on the double-blind clinical trials of minocycline in 1993. The MIRA study was scheduled as the lead presentation at that year’s scientific meeting of the American College of Rheumatology, and when the moment came the San Antonio auditorium was filled to its 5,000-seat capacity. Floating high in the void above the darkened stage, massive televised images of the speakers were projected onto three enormous screens, and impeccable acoustics carried their voices with face-to-face clarity into every corner of the hall. With the president of the ACR serving as emcee, the statistician from Detroit, Barbara Tilley, the only non-medical doctor on the study team and an unlikely Moses, took her place in the circle of light.
Her presentation was limited to only a few minutes, but for those in the audience who had been waiting for this moment for years before the MIRA trials were conceived, the results (see “Let’s Start with the Proof”) were electrifying. As questioners lined up in long queues behind two microphones in the aisles, the name of Dr. Thomas McPherson Brown was invoked repeatedly. The emcee, no doubt in the spirit of fairness and on a tight schedule, began responding to such mentions of the ACR’s old nemesis by cutting in and requesting the speaker to state his question.
By the third or fourth time this happened, one of the participants who knew I had been Tom Brown’s co-author grinned at me from across the aisle and rolled his eyes in amusement.
It would be another fifteen months before the MIRA study would at last appear in print.