Teoria da ressonância: história e ensino
1 Introdução
Linus Pauling dedicou grande parte de sua carreira ao estudo da natureza da ligação química, em suas palavras, “o mais valioso conceito da química”. (PAULING, 1992a, p. 521) Seu trabalho influenciou fortemente o pensamento químico e parte de suas idéias permanecem válidas, com tantos anos passados, compondo os atuais conteúdos de química dos níveis médio e superior de ensino.
Neste artigo, focaremos em uma das construções intelectuais mais importantes de Pauling, a teoria da ressonância, que costuma ser ensinada de modo pouco significativo, pela falta de apresentação da origem o do desenvolvimento das ideias que a compõem. Assim é que, no que se refere à teoria da ressonância, há uma grande diferença entre o os textos didáticos, em todos os níveis, e as ideias originais de Pauling.
Na primeira seção descreveremos, de modo breve, a elaboração histórica da teoria da ressonância. Em seguida, apresentaremos as críticas que lhe foram formuladas pelos contemporâneos de Pauling e suas réplicas. Passaremos, então, à análise de textos didáticos de importância reconhecida pela comunidade dos químicos e professores de química e, por fim, apresentaremos nossas considerações finais.
2 Teoria da ressonância
Pauling tornou-se adepto da ideia de ligação química como partilha de elétrons, em 1919, ao ler os artigos de Lewis e Langmuir que tratavam do assunto(PAULING, 1984). Estes autores propunham um modelo atômico em que os elétrons mantinham posições definidas em torno do núcleo. (LANGMUIR, 1919; LEWIS, 1916) Pauling, porém, viria a adotar uma visão dinâmica do átomo e da ligação química: em 1926, ele publicou artigo onde procurou conciliar as propostas para a ligação química de Lewis (par eletrônico compartilhado), Bohr (elétrons em movimento) e Knorr (os elétrons participantes de uma ligação orbitam em torno dos dois núcleos que ligam), no que denominou teoria da ligação química como elétrons compartilhados em órbita. (PAULING, 1926)
No período de abril de 1926 a setembro de 1927, Pauling viajou como bolsista para a Europa, com o projeto de estudar a estrutura eletrônica de átomos e moléculas e a natureza da ligação química. (PAULING, 1992b) Ali esteve em meio aos acontecimentos que geraram a nova mecânica quântica. Particularmente importante para suas ideias foi o conceito de ressonância quantum-mecânica, que viria a se tornar central no desenvolvimento de sua concepção acerca da ligação química:
[...] a principal contribuição da mecânica quântica para a química tem sido a sugestão de novas idéias, tais como a ressonância de moléculas entre várias estruturas eletrônicas com um associado aumento de estabilidade. (PAULING, 1945, p. ix)
2.1 Ressonância quantum-mecânica
O termo ressonância foi introduzido na mecânica quântica em 1926 por Werner Heisenberg que, para explicar os espectros de emissão dos átomos de hélio (singlete e triplete), elaborou um tratamento matemático correspondente ao de dois osciladores quânticos acoplados que trocam energia entre si, análogo à situação clássica de dois pêndulos em ressonância. Heisenberg intitulou seu trabalho Mehrkörpenproblem und Resonanz in der Quantenmechanik (Ressonância em Mecânica Quântica e o Problema de Muitos Corpos). (MEHRA; RECHENBERG, 1982)
Para se compreender o significado atribuído por Pauling à ressonância quantum-mecânica é preciso considerar o processo de resolução do problema do átomo de hélio excitado, que é o átomo que emite a radiação. Como primeira aproximação, considera-se o sistema com um elétron no nível fundamental de energia, 1s, e outro no estado excitado mais próximo, 2s, cada elétron interagindo com o núcleo, mas não com o outro elétron. Como o hélio possui dois elétrons, existem duas possibilidades: (a) elétron 1 no estado fundamental (1s) e elétron 2 no estado excitado (2s); e (b) elétron 2 no estado fundamental e elétron 1 no estado excitado, aos quais correspondem as seguintes funções de onda:
ψI(1, 2) = 1s(1) 2s(2) e ψII(1, 2) = 2s(1) 1s(2)
onde (1) e (2) representam as coordenadas dos elétrons. Ambas as representações do átomo de hélio excitado correspondem à mesma energia, E0, pois os elétrons são indistinguíveis. São soluções aproximadas, pois é evidente que os elétrons constituintes de um átomo estão próximos e interagem.
O segundo passo do método consiste em melhorar a representação do átomo, considerando a interação dos elétrons. Esta é introduzida como uma perturbação no sistema anterior e determinam-se os estados perturbados do átomo, considerados reais, que resultam ser combinações lineares das funções de onda não-perturbadas, ψI e ψII:
ψs(1, 2) = 2-1/2 (ψI + ψII) = 2-1/2 { 1s(1) 2s(2) + 2s(1) 1s(2) }
e
ψa(1, 2) = 2-1/2 (ψI - ψII) = 2-1/2 { 1s(1) 2s(2) - 2s(1) 1s(2) }.
Na análise de Pauling, estes resultados mostram que ψI e ψII contribuem igualmente para os estados perturbados do sistema (ψs e ψs). Assim, não há razão para crer que no átomo de hélio excitado um elétron esteja permanentemente localizado no nível 1s e o outro no 2s, mas que “[...] cada elétron ressoa entre uma órbita 1s e uma 2s”. (PAULING; WILSON JR, 1935, p. 324)
Interpretações similares são encontradas em contemporâneos de Pauling, tais como Herzberg (1945) e White (1934).
Aos dois estados perturbados do átomo de hélio excitado correspondem distintos valores de energia,
Ec = E0 + J + K e Ed = E0 + J - K
e assim, Heisenberg pode explicar a diferença de energia entre os estados singlete e triplete, observados experimentalmente. Nestas expressões, J e K representam as parcelas de energia devidas à interação dos elétrons, dadas, respectivamente, por:
J = ∫∫ 1s(1) 2s(2) (e2/ r12) 1s(1) 2s(2) dτ1 dτ2
e
K = ∫∫ 1s(1) 2s(2) (e2/ r12) 2s(1) 1s(2) dτ1 dτ2
A primeira expressão é denominada integral de Coulomb e representa a energia da interação coulombiana entre o elétron 1 no estado 1s e o elétron 2 no estado 2s.
A segunda integral, onde cada elétron está relacionado às duas funções de onda, representa a energia resultante da transição do elétron 1 do nível 1s para o 2s, ao mesmo tempo que o elétron 2 realiza a transição em sentido inverso e, por isso, é denominada integral de ressonância ou integral de troca. (PAULING, 1928a)
De acordo com a interpretação de Pauling, o fenômeno da ressonância quantum-mecânica manifesta-se através da forma da função de onda do sistema, como combinação linear das funções ψI e ψII (forma similar à da equação de movimento dos pêndulos acoplados), bem como pela expressão da energia de interação eletrônica, a integral de ressonância. Embora admita a impossibilidade de verificação experimental da ressonância, Pauling defende a importância de tal interpretação para uma “compreensão intuitiva segura e produtiva das equações da mecânica quântica e dos resultados de sua aplicação.” (PAULING; WILSON JR, 1935, p. 314-315)
No entender de Pauling (PAULING; WILSON JR, 1935), o caso dos estados do hélio excitado encaixa-se no problema mais geral de como descrever o estado resultante da interação de dois sistemas com igual energia. Um procedimento para encontrar a solução é o método de perturbação e as soluções resultam ser combinações lineares das funções não-perturbadas do sistema considerado. Nesses casos, é possível interpretar o estado do sistema com interação, como ressoando entre os estados sem interação.
2.2 Ressonância e ligação química
Em 1927, Heitler e London resolveram o problema da molécula do hidrogênio, cujo estado é descrito como uma combinação de dois estados não-perturbados de mesma energia, cada qual constituído por dois átomos de hidrogênio. Em artigo de revisão, Pauling (1928a) rediscutiu a molécula do hidrogênio e outros problemas deste tipo, sempre concluindo pelo importante papel da ressonância na explicação da ligação química.
A discussão da molécula do hidrogênio começa por considerar os estados não-perturbados, que seriam dois átomos de hidrogênio sem interação mútua, ou seja, cada próton interagindo com apenas um elétron. Há duas possibilidades, com igual energia: se o elétron 1 está ligado ao núcleo A e o elétron 2 está ligado ao núcleo B, a função de onda é dada por ψA(1) ψB(2); se o elétron 2 está ligado ao núcleo A e o elétron 1 está ligado ao núcleo B, a função de onda é dada por ψB(1) ψA(2). Ao considerar a ação de todas as quatro partículas entre si (dois elétrons e dois prótons) a resolução do problema leva às seguintes funções de onda:
Ψs = Cs { ψA(1) ψB(2) + ψB(1) ψA(2) }
e
Ψ = C { ψ(1) ψ(2) - ψ(1) ψ(2) }
onde Cs e C a são constantes. Note-se que, à semelhança do átomo de hélio excitado, os estados calculados para a molécula do hidrogênio são compostos pelos estados não-perturbados — ψA(1) ψB(2) e ψB(1) ψA(2) —podendo ser interpretados, à moda de Pauling, como estados em que há ressonância dos elétrons entre os núcleos A e B.
A formulação da ligação química proporcionada pela mecânica quântica possibilitou Pauling (1928b, p. 359) interpretar os pares eletrônicos de Lewis em seus termos:
Com o desenvolvimento da mecânica quântica tornou-se evidente que os fatores responsáveis pela valência química são, essencialmente, o princípio da exclusão de Pauli e o fenômeno da ressonância de Heisenberg-Dirac. Mostrou-se que no caso de dois átomos de hidrogênio no estado normal, trazidos à proximidade um do outro, a autofunção que é simétrica nas coordenadas de posição dos dois elétrons corresponde a um potencial que conduz a que os dois átomos se combinem para formar uma molécula. Este potencial é principalmente devido a um efeito de ressonância que pode ser interpretado como envolvendo uma troca na posição dos dois elétrons que formam a ligação, de modo que cada elétron está parcialmente associado com um e outro núcleo. (Tradução nossa).
Pauling (1928b, p. 359) admite, de acordo com London, que “a energia de troca de dois elétrons, um pertencendo a cada um de dois átomos, é a energia da ligação não-polar, em geral” e interpreta que “o par de elétrons de Lewis consiste, agora, de dois elétrons que estão em estados idênticos exceto que seus spins são opostos”.
Pauling chegou a considerar o trabalho de Heitler e London como a maior contribuição isolada para o conceito de valência, desde a noção de par eletrônico compartilhado, proposta por Lewis. (BROCK, 2000)
2.3 Ressonância entre estruturas e ligações
À ideia de ressonância de elétrons seguiu-se a de ressonância entre estruturas eletrônicas das moléculas. Para tanto foi necessário um passo decisivo: postular que a toda configuração eletrônica corresponde uma função de onda. Ocorre que, se uma estrutura é explicada como resultante de mais de uma configuração eletrônica, a autofunção correspondente a esta estrutura deverá ser uma combinação das autofunções das configurações contribuintes. (PAULING, 1932a) Uma vez que, a cada configuração eletrônica corresponde uma fórmula química, com este postulado Pauling estabeleceu a relação entre as representações química e quantum-mecânica de uma molécula.
Pauling tinha clareza de que, para várias substâncias, não havia como conciliar suas propriedades com uma única fórmula química construída pelo sistema usual de ligações covalentes (simples, duplas e triplas). Em lugar de condenar a representação usual das estruturas moleculares e criar outra, Pauling advogou em favor de sua manutenção, como uma atitude prática, introduzindo a ideia de que a estrutura correta seria híbrida das várias estruturas convencionais possíveis.
O caso mais difundido é o do benzeno, que apresenta várias possibilidades de representação:
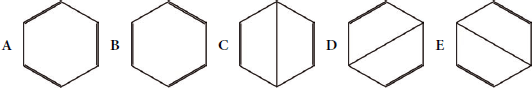
Figura 1 - Formas ressonantes do benzeno, de acordo com Pauling (1945)
De acordo com a teoria da ressonância, a estrutura do benzeno seria um híbrido de 5 estruturas possíveis, representadas pelas 5 fórmulas de ligação de valência da Figura 1, acima. Em outras palavras, estas 5 estruturas em ressonância representariam a verdadeira estrutura do benzeno.
Pauling estendeu a noção de ressonância às ligações químicas, explicando porque distâncias interatômicas medidas situavam-se entre os valores calculados para ligações simples e duplas, como nos casos da grafita, do benzeno e de outros compostos. A ideia é que as ligações “estão ressoando entre ligações simples e duplas.” (PAULING, 1932a, p. 295) Ainda nesse artigo, Pauling empregou a ressonância entre ligações para explicar qualitativamente o caráter ácido ou básico de compostos orgânicos de diversos tipos.
Nos trabalhos subsequentes da série The nature of the chemical bond, o modelo de estruturas e ligações ressonantes foi aprimorado e consolidado. Foi estabelecida uma escala de eletronegatividades associada à polaridade das ligações, resultante da ressonância entre uma estrutura iônica e outra covalente. (PAULING, 1932b) Esta escala de eletronegatividades constituiu-se num importante instrumento na análise da distribuição de cargas em estruturas ressonantes. Foram desenvolvidos procedimentos de cálculo da energia de ressonância, a partir de dados termoquímicos, e refinada a explicação da estrutura de vários compostos: dióxido de carbono, isocianatos de alquila, ácidos carboxílicos, ésteres, amidas, hidrocarbonetos aromáticos (benzeno e derivados, naftaleno, antraceno), compostos heterocíclicos, sistemas conjugados em geral etc. (PAULING; WHELAND, 1933)
A conceituação básica da teoria da ressonância estava estabelecida e explicava vários fenômenos químicos com base no movimento entre estruturas eletrônicas e no movimento de ligações no interior dessas estruturas.
De modo conciso e direto (PAULING, 1938, p. 1857-1858, tradução nossa):
A idéia de ressonância, em sua aplicação à química, é a seguinte. Se for possível escrever para uma molécula (ou outro sistema) duas ou mais estruturas eletrônicas correspondendo a aproximadamente a mesma energia e satisfazendo outras condições, então, nenhuma das estruturas isoladamente pode ser considerada como representante do estado normal da molécula que, em vez disso, é representado essencialmente por uma média de todas elas; e, além disso, a molécula é mais estável (possui um menor conteúdo energético) que poderia ser se tivesse qualquer uma das estruturas isoladas. A molécula é descrita como ressoando entre as várias estruturas e a energia de estabilização da molécula é denominada energia de ressonância [grifos do autor].
(Em termos quantum-mecânicos, diz-se que a função de onda que representa o estado normal da molécula não é qualquer uma das funções de onda correspondentes às várias estruturas eletrônicas, mas é uma combinação linear destas.).
3 Críticas à teoria da ressonância
O trabalho de Pauling sobre ligação química foi bem aceito pela comunidade científica. A primeira edição da obra que resumiu a teoria, The Nature of the Chemical Bond, lançada em 1939, esgotou-se rapidamente e já no ano seguinte foi lançada uma segunda edição, revista e atualizada. Este livro foi considerado pela crítica como a apresentação mais clara e unificada sobre estrutura molecular (FRENCH, 1940), um marco na história da teoria de valência (MULLIKEN, 1940) e recomendado a quem desejasse um entendimento mais profundo sobre ligação química. (KISTIAKOWSKY, 1940) Entre os reparos feitos por seus revisores, constam a ressalva à ideia de estruturas ressoando em alta frequência, (KISTIAKOWSKY, 1940) e a ausência de referências à teoria do orbital molecular. (MULLIKEN, 1940) As principais divergências e críticas à teoria da ressonância referem-se a três aspectos inter-relacionados: a analogia entre ressonância quântica e clássica, a realidade das estruturas constituintes de um sistema ressonante e a relação entre ressonância e tautomeria (equilíbrio químico).
3.1 Problemas da analogia entre ressonância quântica e ressonância clássica
Como visto na primeira parte deste artigo, Pauling entendia a ressonância quantum-mecânica como movimento, uma troca de posição de elétrons entre os átomos participantes da ligação. Seus escritos mostram que a analogia entre ressonância clássica e quântica tinha por base a troca de energia dos sistemas em ressonância, sugerindo uma visão clássica da ressonância quântica.
Embora cientistas importantes da época, como Herzberg (1945) e White (1934), houvessem concordado com Pauling, outros igualmente importantes, como Coulson (1952), Heitler (1956), Slater (1963), Fermi (1966), divergiram dessa interpretação.
Um crítico especial da ressonância como fenômeno foi George W. Wheland, já que foi um dos químicos que mais contribuiu para seu desenvolvimento. Entre 1932 e 1936 Wheland realizou um estágio de pós-doutorado no Caltech, onde colaborou com Pauling no desenvolvimento da teoria da ressonância. Publicaram juntos três artigos, inclusive o quinto trabalho da série The Nature of the Chemical Bond, que tratou do cálculo de energia de ressonância do benzeno e outros compostos aromáticos. (PAULING; WHELAND, 1933) Em 1936-1937, Wheland esteve como bolsista na Inglaterra, onde trabalhou com o grupo de Ingold, em Londres, e serviu de elo de ligação entre Pauling e os físico-químicos orgânicos ingleses. Posteriormente, esteve com Hinshelwood, em Oxford e realizou visitas a Hückel, em Munique, e a Lennard-Jones, em Cambridge (PARK, 1999), o que revela seu interesse pelos aspectos mais centrais da físico-química, além de sua aplicação aos compostos orgânicos.
Park (1999) atribui ao contato de Wheland com os físico-químicos orgânicos o desenvolvimento da percepção das críticas que poderiam ser dirigidas à teoria da ressonância. Por isso, quando da publicação de The Theory of Resonance and Its Application to Organic Chemistry, em 1944, e de seu sucessor, Resonance in Organic Chemistry (WHELAND, 1955), buscou resolver o problema de como situar-se equilibradamente entre a exposição físico-matemática e a exposição química da ressonância.
Tanto Wheland quanto Pauling reconheciam na ressonância uma ampliação da teoria estrutural clássica (pré-quântica). Entretanto, enquanto Pauling percebia a ressonância como fenômeno, Wheland considerava-a como um artifício, um método aproximativo para tratar moléculas (WHELAND, 1955, p. 25, tradução nossa, grifo do autor):
Um de tais métodos aproximativos que é conveniente, embora não seja o único, para resolver a equação de onda é baseado na consideração de que a função de onda ψ pode ser expressa como uma combinação linear de funções conhecidas, do seguinte modo:
ψ = k1φ1 + k2φ2 + ... +knφn (1) onde φ1, φ2, ..., φn são as funções conhecidas e k1, k2, ..., kn são constantes numéricas. [...] Quando este método de aproximação é empregado e quando as funções φ1, φ2, [...], φn podem ser correlacionadas de algum modo com definidas estruturas da molécula em questão, então, diz-se que a molécula é um híbrido de ressonância dessas estruturas, ou, diz-se que as estruturas contribuem para o estado da molécula. As grandezas relativas dos coeficientes ki são medidas qualitativas dos pesos relativos atribuídos às respectivas estruturas.
Wheland considerava que o termo ressonância havia sido introduzido por Heisenberg na mecânica quântica, com a finalidade de estabelecer uma analogia formal com a mecânica clássica, mais familiar, possuindo significado “[...] apenas com referência a um particular método de aproximação da situação real”. (WHELAND, 1955, p. 29, grifo do autor) Em vista disso, a ressonância seria
[...] um conceito artificial em um sentido mais fundamental que na maioria das outras teorias físicas. Ele não corresponde a nenhuma propriedade intrínseca da molécula, mas, em vez disso, é apenas um expediente matemático deliberadamente inventado pelo físico ou químico para sua própria conveniência. Inclusive, se o problema quantum-mecânico pudesse ser rigorosamente resolvido ou mesmo se um método aproximativo diferente fosse empregado, a idéia de ressonância não apresentar-se-ia. Portanto, se um conjunto diferente de φ's fosse usado na equação 1 [combinação linear, acima] a molécula cuja função de onda fosse aproximada por ψ poderia ser descrita como um híbrido de um conjunto de estruturas inteiramente diferente. (WHELAND, 1955, p. 28-29, tradução nossa)
Para Wheland (1955), os análogos mecânicos clássicos das estruturas componentes do sistema seriam os movimentos independentes dos pêndulos acoplados porque, tanto uns quanto outros, podem ser empregados como artifícios para representar os estados de sistemas compostos — pêndulos acoplados ou híbridos de ressonância — através de combinações lineares.
3.2 Estatuto ontológico das estruturas constituintes de um sistema ressonante
Para Wheland, a ressonância era um método para descrever o estado das moléculas, de modo que, as estruturas constituintes de um sistema ressonante eram meras ficções, não podiam existir realmente. Em sua opinião, o termo estrutura não deveria ser aplicado às fórmulas utilizadas para descrever o híbrido de ressonância. Wheland tinha restrições à terminologia empregada pelos químicos da época pela imprecisão de linguagem que, acreditava, poderia gerar equívocos no entendimento do conceito de ressonância. Por isso, dedicou uma seção inteira de Resonance in Organic Chemistry (WHELAND, 1955) à discussão de termos que considerava impróprios pela ideia de fenômeno que vinculava à ressonância. Advogou contra o emprego do verbo “ressoar” e outras expressões que pudessem sugerir que
[...] as moléculas de um híbrido de ressonância estejam oscilando de um lado para o outro entre as várias estruturas e, por isso, que estas estruturas devam possuir significado físico real. (WHELAND, 1955, p. 7)
Coulson (1952) também pensava que as estruturas componentes do sistema ressonante não tinham realidade no sentido objetivo do termo, porque as autofunções associadas não correspondem a estados estacionários permitidos. Sendo assim, seria inexato falar de ressonância entre estruturas no sentido de oscilação temporal.
Christopher Ingold foi um dos químicos britânicos a reconhecer imediatamente o valor da teoria da ressonância porque, em 1926, havia elaborado o conceito de efeito mesomérico para descrever o estado de moléculas, cuja fórmula seria intermediária entre duas fórmulas de ligação de valência, conceito similar à ressonância entre estruturas proposta por Pauling em 1931-1932. Por isso, reconheceu na ressonância “a propriedade ondulatória fundamental na teoria da valência”, considerando a mesomeria como um caso particular que recebeu denominação própria em função de sua importância na química orgânica. (INGOLD, 1938, p. 314) Entretanto, como Wheland e Coulson, Ingold considerava que a analogia entre ressonância quântica e ressonância clássica era de ordem matemática apenas. Por isso desenvolveu uma crítica severa à ideia de que as estruturas ressonantes tivessem existência real. Seu argumento se baseia em que os estados não-perturbados empregados na descrição da molécula sob estudo, embora estejam relacionados a fórmulas químicas clássicas, não correspondem à realidade; é através da perturbação destes estados que se encontra a o correto estado da substância: “[...] apenas o estado mesomérico é real.” (INGOLD, 1934, p. 946)
O estatuto ontológico das estruturas componentes de um sistema ressonante esteve no centro da crítica à teoria da ressonância elaborada na União Soviética. Na discussão sobre a teoria da ressonância, o princípio de Butlerov — a cada substância uma fórmula estrutural — foi sistematicamente empregado para contrapor-se à idéia de várias fórmulas para representar uma estrutura e a teoria da ressonância foi condenada como irreal. (GRAHAM, 1964)
3.3 Confusão entre ressonância, mesomeria e tautomeria
A confusão entre ressonância e tautomeria (equilíbrio químico) perdurou por um intervalo de tempo razoável. Bachelard (1990) conta que os membros de um congresso sobre mesomeria realizado na França, em 1947, foram consultados a respeito de a mesomeria ser um fenômeno real ou um método de investigação e resultou que as opiniões se dividiram. Tal situação seria corroborada na crítica de Dewar, que apontava a dificuldade dos químicos em distinguir os conceitos intuitivamente. (RUSSELL, C., 1971) Para Ingold (1934), que considerava as estruturas ressonantes como irreais, não fazia sentido confundir mesomeria com tautomeria, porque a última pressupõe que as moléculas se encontrem em um determinado intervalo de tempo, em um ou outro dos estados moleculares separados, ao passo que na ressonância isso não ocorre. Por isso, o termo estado não teria o mesmo sentido ao tratar-se de formas tautoméricas e ressonantes.
Outro cientista a criticar a identificação da ressonância com o tautomerismo foi Nevil Sidgwick, presidente da Chemical Society of London (atual Royal Society of Chemistry), nos anos de 1936-1937. Considerando a ressonância como o desenvolvimento mais importante desde os dias de van't Hoff, acreditava, contudo, que o termo era “ruim porque sugere que a molécula está oscilando entre dois estados, o que não é verdade”. Propunha o emprego de algum outro nome não-comprometido, a exemplo do mesomerismo. (SIDGWICK, 1936, p. 535)
De acordo com Wheland (1955), ressonância e mesomeria seriam equivalentes. A ressonância distinguia-se da tautomeria porque as estruturas ressonantes não têm existência real, ao passo que as estruturas tautoméricas existem, podendo as substâncias ser isoladas e identificadas. No caso do benzeno, por exemplo, em momento algum o benzeno toma a forma das estruturas de Kekulé. Apesar de virtuais, as propriedades das estruturas componentes de um sistema ressonante, como o “cicloexatrieno”, podem ser inferidas a partir de estruturas reais que possuam elementos componentes similares, como o cicloexeno e, então, serem utilizadas na construção do híbrido de ressonância.
4 Réplicas de Pauling
Pauling não aceitava a sinonímia (e consequente identificação) da ressonância com a mesomeria. A única crítica à teoria da ressonância que ele admitia abertamente era um elemento de arbitrariedade na escolha das estruturas componentes de um sistema ressonante, considerado de pouca importância, em vista da contribuição do conceito de ressonância na discussão de problemas químicos. (PAULING, 1945) Portanto, na conferência realizada durante a entrega do prêmio Nobel de Química de 1954 (PAULING, 1954), procurou responder às críticas à teoria da ressonância; tais réplicas foram posteriormente desenvolvidas. (PAULING, 1956)
Ressaltando a conveniência e a utilidade do conceito de ressonância na resolução dos problemas químicos, Pauling procura mostrar que a teoria da ressonância é tão arbitrária quanto a teoria estrutural clássica da química orgânica, pois ambas empregam idealização. Na descrição do benzeno pela teoria da ressonância, as estruturas de Kekulé devem ser consideradas como hipotéticas porque não é possível sintetizar moléculas com essas estruturas. De modo similar, na descrição do propano, como tendo ligações simples carbono-carbono e carbono-hidrogênio, as ligações simples são idealizações porque é impossível isolar uma porção da molécula do propano e identificá-la como uma dessas ligações. Portanto, conclui, o valor das teorias independe das idealizações e da arbitrariedade envolvidas.
Considerando a teoria da ressonância como essencialmente idêntica à teoria estrutural clássica da química orgânica, concluiu que, se a crítica fosse consistente, seria necessário abandonar ambas as teorias, pois a teoria clássica também emprega idealizações na descrição das moléculas. (PAULING, 1956)
Pauling acreditava que a teoria da ressonância perduraria e seria desenvolvida, apesar das críticas que lhe eram feitas. Contudo, prevaleceu a posição dos seus críticos e se a ressonância tornou-se um instrumento indispensável ao trabalho dos químicos, é como um método de investigação e não como fenômeno. Prova disso é que o March's Advanced Organic Chemistry (SMITH; MARCH, 2001), um dos mais conceituados manuais de química orgânica na atualidade, empregado como referência para a pesquisa, trata a teoria da ressonância como método da ressonância.
5 Ensino da teoria da ressonância
Há toda uma problemática a cercar o conceito de ressonância que remete às concepções de molécula e representação do mundo microscópico. São questões delicadas porque a passagem do território da matéria macroscópica para o dos modelos microscópicos não é tranquila; requer um modo de raciocínio que relacione grandezas observáveis e características dos modelos, algo difícil, porém necessário a todo químico e professor de química.
Defendemos o emprego da história das ciências como coadjuvante do ensino de ciências, uma vez que pode esclarecer a formação dos conceitos científicos. (SILVA, 2002) Sendo assim, a transposição didática do conhecimento científico ao ambiente escolar deve considerar os problemas que geraram os conceitos, bem como seu desenvolvimento. (SILVA; ROQUE, 2004) Nesta seção, analisamos como a ressonância é apresentada aos químicos em materiais de estudo empregados nos diversos níveis de ensino, da pós-graduação ao ensino médio. Os materiais didáticos de ciências do ensino fundamental não incluem a discussão desse conceito.
Se Pauling passou à história como o criador da ressonância, as ideias de Wheland terminaram por definir seu significado entre os químicos. No March's Advanced Organic Chemistry (2001), que contém um texto didático introdutório de cada capítulo, a ressonância é introduzida como um modo de representar estruturas de substâncias que possuem ligações deslocalizadas. A referência apontada como definitiva é o livro de Wheland (1955).
Ainda em nível de pós-graduação, o Advanced Organic Chemistry (CAREY; SUNDBERG, 2001) segue na mesma linha: um método útil para representar a deslocalização de elétrons e discutir a estabilidade das estruturas de substâncias. Toda a problemática que cerca o conceito é ignorada. A importância do método se revela pela aplicação a casos exemplares.
Pontos comuns aos livros universitários de pós-graduação e de graduação são: (a) a introdução da ressonância pela citação do problema histórico original: a limitação das fórmulas de Lewis para representar estruturas de substâncias; (b) a ausência de explicação para emprego do termo ressonância e sua controversa polissemia; (c) a ausência de justificativa para a afirmação de que as fórmulas de ressonância não possuem realidade. Enquanto na pós-graduação a ressonância é discutida em termos gerais, os livros de química orgânica básica da graduação recorrem à discussão de casos: radical alila (MORRISON; BOYD, 1996); íon carbonato (ALLINGER et al., 1976; SOLOMONS, 1996); benzeno. (AMARAL, 1981; CAMPOS, 1976, 1980) Nos dois níveis de ensino, a representação estrutural é considerada resolvida pelo método da ressonância e a problemática em torno da ideia de ressonância não é claramente exposta.
Na química geral de ensino superior, a ressonância é também introduzida a partir de casos particulares: ozônio e benzeno (BROWN; LEMAY; BURSTEN, 1999; CHANG, 1994; RUSSELL, J., 1994), óxidos de enxofre (BRADY; HUMINSTON, 1983), nitrato e benzeno (ATKINS; JONES, 2001), óxidos de nitrogênio, nitrato, carbonato, ozônio, benzeno. (ROZEMBERG, 2002) São apresentações curtas onde é explícita a ideia de método de representação estrutural, sem explicações da elaboração conceitual.
Nos livros didáticos tradicionais de química do ensino médio, os resultados da transposição didática da ressonância são variados. Peruzzo e Canto (2002) procuram defini-la de modo simples, como “o termo usado para descrever uma situação na qual, sem mudar a posição dos átomos, podemos escrever mais de uma fórmula estrutural diferente, mudando apenas a posição de alguns elétrons”, considerando híbrido de ressonância como “um misto” de estruturas. A distinção entre ressonância e equilíbrio resume-se à diferença dos tipos de setas usadas na representação, sem qualquer discussão. Não distingue entre fenômeno e método.
Há quem apresente a ressonância como movimento eletrônico. Para Feltre (2001) “[...] as ligações duplas saltam espontaneamente de suas posições” nas fórmulas de Kekulé do benzeno. Para Fonseca (1993) os elétrons “mudam de lugar o tempo todo”.
Outros livros didáticos de química do ensino médio não discutem o conceito de ressonância. Mortimer e Machado (2002) tratam o benzeno do ponto de vista da teoria do orbital molecular, ao passo que o livro do Gepeq (2000) emprega fórmulas de Kekulé.
6 Considerações finais
A história do conceito de ressonância mostra que, embora a ideia tenha nascido com Pauling, com um cunho realista, associada ao movimento de elétrons, ligações e estruturas químicas, a controvérsia que se desenvolveu finalizou com a vitória da noção de instrumento teórico útil para representar estruturas.
Verificamos que os manuais didáticos empregados no ensino de química, sejam de pós-graduação, graduação e voltados para a educação básica, espelham o resultado da disputa científica, mas escondem o processo de elaboração conceitual do método da ressonância. Essa atitude equipara a ciência a outras formas de conhecimento dogmático, o que contraria a prática da pesquisa científica.
Por outro lado, várias obras fortalecem uma visão de ciência muito difundida, a de que o conhecimento provém da experiência particular e depois é generalizado. (ATKINS; JONES, 2001; FELTRE, 2001) Esta posição está em desacordo com as principais correntes da epistemologia de base histórica, que têm explicitado a construção dos conceitos científicos.
Por fim, os livros didáticos examinados apresentam alguns traços de realismo ingênuo, produzindo um ensino que abre mão da criticidade que caracteriza as ciências.
Concluímos, portanto, por uma necessidade de maior aproximação entre o ensino de química e a história da teoria quântica, de modo a produzir um ensino mais significativo e esclarecedor, tanto quanto aos conceitos, como quanto à atividade científica.
Agradecimentos
Queremos agradecer aos árbitros que, com suas críticas, contribuíram para a melhoria do artigo.
Referências
ALLINGER, Norman L. et al. Química orgánica. 2. ed. Rio de Janeiro: LTC, 1976. 2v.
AMARAL, Luciano do. Química orgânica. São Paulo: Moderna: Edusp, 1981.
ATKINS, Peter W.; JONES, Loretta. Princípios de química. Porto Alegre: Bookman, 2001.
BACHELARD, Gaston. O materialismo racional. Lisboa: Edições 70, 1990.
BRADY, James E.; HUMINSTON, Gerard E. Química geral. Rio de Janeiro: LTC, 1983.
BROCK, William H. The chemical tree: a history of chemistry. New York: W. W. Norton, 2000.
BROWN, Theodore L.; LEMAY, H. Eugene; BURSTEN, Bruce H. Química ciência central. Rio de Janeiro: LTC, 1999.
CAMPOS, Marcello de Moura (Coord.). Fundamentos de química orgânica. São Paulo: Edgard Blücher: Edusp, 1980.
______. (Coord.). Química orgânica. São Paulo: Edgard Blücher: Edusp, 1976. 4v.
CAREY, Francis A.; SUNDBERG, Richard J. Advanced organic chemistry. 4th ed. New York: Plenum, 2001.
CHANG, Raymond. Química. 5. ed. Lisboa: McGraw-Hill, 1994.
COULSON, Charles A. Valence. Oxford: Claredon, 1952.
FELTRE, Ricardo. Fundamentos da química. 3. ed. São Paulo: Moderna, 2001.
FERMI, Enrico. Molecule, crystals, and quantum statistics. New York: W. A. Benjamin, 1966.
FONSECA, Martha REIS Marques. Química integral. São Paulo: FTD, 1993.
FRENCH, Sidney J. Nature of the chemical bond. Journal of Chemical Education, v.17, n.7, p.551, 1940.
GEPEQ. Interações e Transformações. São Paulo: Edusp, 2000.
GRAHAM, Loren R. A soviet marxist view of structural chemistry. Isis, v. 55, n.179, p. 20-31, 1964.
HEITLER, Walter. Elementary wave mechanics with applications to quantum chemistry. 2nd ed. Oxford: Clarendon, 1956.
HERZBERG, Gerhard. Atomic spectra and atomic structure. New York: Dover, 1945.
INGOLD, Christopher K. Mesomerism and tautomerism. Nature, v.133, p. 946, 1934.
______. Resonance and mesomerism. Nature, v. 141, n. 3564, p. 314-318, 1938.
KISTIAKOWSKY, G. B. Journal of the American Chemical Society, v. 62, p.457, 1940.
LANGMUIR, Irving. The arrangement of electrons in atoms and molecules. Journal of the American Chemical Society. v. 41, p. 868-934, 1919.
LEWIS, Gilbert N. The atom and the molecule. Journal of the American Chemical Society. v.38, p.762-785, 1916.
MEHRA, Jagdish; RECHENBERG, Helmut. The Historical Development of Quantum Theory. New York: Springer, 1982.
MORRISON, Robert T.; BOYD, Robert N. Química Orgânica. Lisboa: Calouste Gulbenkian, 1996.
MORTIMER, Eduardo; MACHADO, Andréa Horta. Química para o ensino médio. São Paulo: Scipione, 2002.
MULLIKEN, Robert S. Nature of the chemical bond. Journal of Physical Chemistry, v.44, n.3, p.827-828, 1940.
PARK, Buhn S. Chemical translators: Pauling, Wheland and their strategies for teaching the theory of resonance. British Journal of History of Science, v. 32, p. 21-46, 1999.
PAULING, Linus C. The application of the quantum mechanics to the structure of the hydrogen molecule and hydrogen molecule-ion and to related problems. Chemical Reviews, v. 5, p.173-213, 1928a.
______. The dynamical model of the chemical bond and its application to the structure of benzene. Journal of the American Chemical Society, v. 48, p.11321143, 1926.
______. Lewis and the chemical bond. Journal of the Chemical Education, v. 61, n. 3, p.201-203, 1984.
______. Modern structural chemistry. Science, v.123, n.3190, p. 255-258, 1954.
______. The nature of the Chemical Bond - 1992. Journal of Chemical Education, v. 69, n. 6, p. 519-521, jul. 1992a.
______. The nature of the Chemical Bond. III: the transition from one extreme bond type to another. Journal of the American Chemical Society, v. 54, p. 9881003, mar. 1932a.
______. The nature of the Chemical Bond. IV: the energy of single bonds and the relative electronegativity of atoms. Journal of the American Chemical Society, v. 54, p. 3570-3582, 1932b.
______. The nature of the Chemical Bond and the structure of molecules and crystals. 2nd ed. Ithaca-NY: Cornell University, 1945.
______. The nature of the theory of resonance. In: TODD, Alexander (Ed.). Perspectives in organic chemistry. New York: Interscience, 1956. p.1-8.
______. The shared-electron chemical bond. Proceedings of the Academy of Sciences of the United States, v.14, p.359-362, 1928b.
______. The significance of resonance to the nature of chemical bond and the structure of molecules. In: GILMAN, Henry et al. (Ed.). Organic chemistry: an advanced treatise. New York: John Wiley, 1938. p.1857-1858.
______. The value if rough quantum mechanical calculations. Foundations of Physics, v. 22, n. 6, p. 829-838, 1992b.
PAULING, Linus C.; WILSON JR, E. Bright. Introduction to quantum mechanics with applications to chemistry. New York: Dover, 1935.
PAULING, Linus C.; WHELAND, G. W. The nature of the chemical bond. V. The quantum-mechanical calculation of the resonance energy of benzene and naphthalene and the hydrocarbon free radicals. Journal of Chemical Physics, v.1, p.363-374, 1933.
PERUZZO, Francisco Miraglia; CANTO, Eduardo Leite do. Química: na abordagem do cotidiano. 2. ed. São Paulo: Moderna, 2002.
ROZEMBERG, I. M. Química geral. São Paulo: Edgard Blücher, 2002.
RUSSELL, Colin A. The history of valence. Leicester: Leicester University, 1971.
RUSSELL, John Blair. Química geral. 2. ed. São Paulo: Makron Books, 1994. 2v.
SANTOS, Wildson Luiz Pereira et al. Química e sociedade. São Paulo: Nova Geração, 2005.
SIDGWICK, Nevil V. Structural chemistry. Journal of the Chemical Society, p.535-538. p.335, apr. 1936.
SILVA, José Luis P. B. O valor pedagógico da história das ciências. Ideação, n.9, p.109-124, 2002.
SILVA, José Luis P. B.; ROQUE, Nídia Franca. Ordens de transposição didática. In: ENCONTRO NACIONAL DE PESQUISA EM EDUCAÇÃO EM CIÊNCIAS, 4., 2003, Bauru-SP. Atas... . Bauru: Unesp, 2004. 1 CD.
SLATER, John C. Quantum theory of molecules and solids. New York: McGraw-Hill, 1963. v. 1
SMITH, Michael B.; MARCH, Jerry. March's advanced organic chemistry. New Yoprk: Wiley-Interscience, 2001.
SOLOMONS, T. W. Graham. Química Orgânica. 6. ed. Rio de Janeiro: LTC, 1996.
WHELAND, George W. Resonance in organic chemistry. New York: John Wiley, 1955.
WHITE, Harvey E. Introduction to atomic Spectra. Tokyo: McGraw-Hill/Kogakusha, 1934. p. 210.